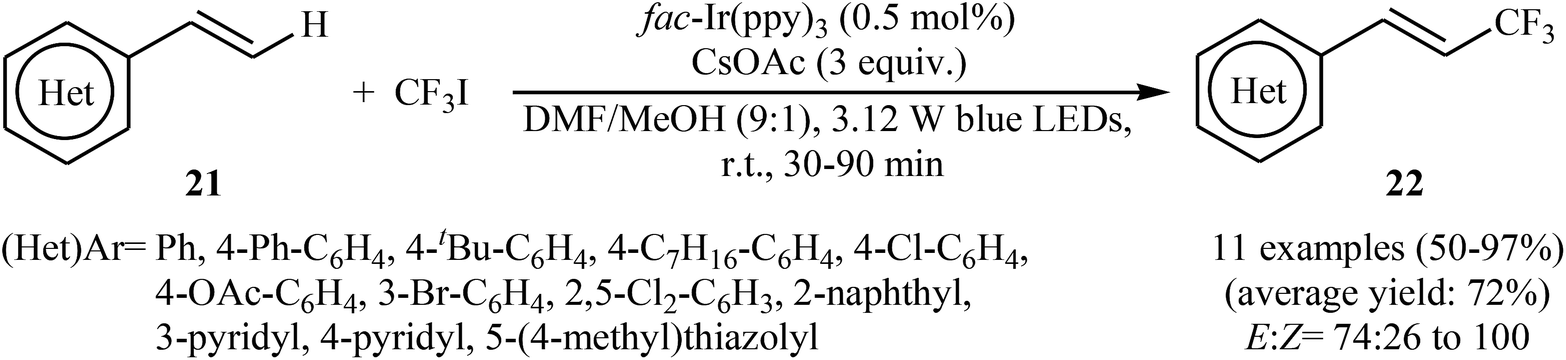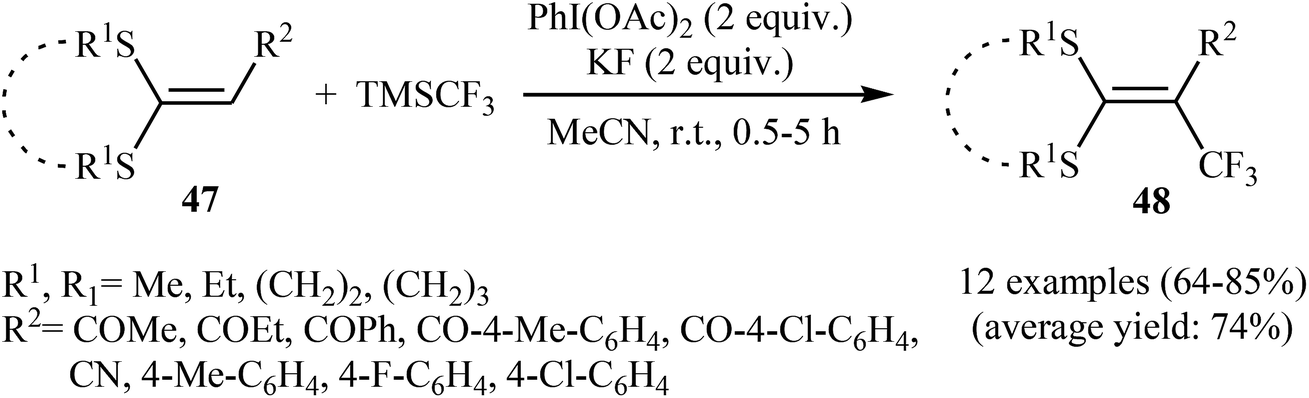DOI:
10.1039/C9RA04170B
(Review Article)
RSC Adv., 2019,
9, 27625-27639
Recent advances in direct trifluoromethylation of olefinic C–H bonds
Received
2nd June 2019
, Accepted 21st August 2019
First published on 3rd September 2019
Abstract
The aim of this review is to provide a comprehensive overview of the direct trifluoromethylation of olefinic C–H bonds with special attention on the mechanistic aspects of the reactions. The review is divided into two major sections. The first focuses exclusively on trifluoromethylation of terminal alkenes, while the second will cover trifluoromethylation of internal alkenes. Literature has been surveyed until the end of April 2019.
 Akbar Hassanpour | Akbar Hassanpour was born in Tabriz, Iran, in 1978. He received his B.S. degree in pure chemistry from Tabriz Azad University, Iran, and his M.S. degree in organic chemistry from Tabriz University, Tabriz, Iran, in 2004 under the supervision of Prof. Kazem D. Safa. He completed his PhD degree in 2008 under the supervision of Prof. Kazem D. Safa. He was a postdoctoral fellow in the lab of Prof. W. A. Stanczyk at the Centre of Molecular and Macromolecular Studies in Łódź, Poland. Now he is working at Azad University as assistant professor. His research interests include organometallic and organic synthesis, the and new methodologies in nano material synthesis. |
 Khatereh Ghorbanpour | Khatereh Ghorbanpour was born in Miandoab, Iran, in 1984. She received her B.S. degree in pure chemistry from Tabriz University, Iran, and her M.S. degree in organic chemistry from Tabriz University, Tabriz, Iran, in 2009 under the supervision of Prof. Kazem D. Safa. She completed her PhD degree in 2013 under the supervision of Prof. Kazem D. Safa. Her research interests include organometallic and organic synthesis, the and new methodologies in nano science. |
 Shahrzad Abdolmohammadi | Shahrzad Abdolmohammadi was born in Iran, in 1976. She received her BSc. degree in chemistry from Alzahra University (AU), Tehran, Iran, in 1999, her MSc. degree in organic chemistry from the Tarbiat Modarres University (TMU), Tehran, Iran under the supervision of Professor Issa Yavari in 2002 and her PhD degree in organic chemistry from the Tehran University (TU), Tehran, Iran, under the supervision of Professor Hooshang Pirelahi and Professor Saeed Balalaie, in 2008. She is associate professor in East Tehran Branch, Islamic Azad University, Tehran, Iran. Her research interests include organic synthesis, heterocyclic synthesis, multicomponent reactions, nanocatalysis, organocatalysis, and synthetic methodology. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Today, fluorine-containing organic compounds are prominent in the various branches of the chemical sciences, particularly in medicinal, agricultural, and materials chemistry.1 Approximately 25% of modern pharmaceuticals and 40% of agrochemicals2 contain fluorine atoms in their structure, because fluorinated compounds exhibit a high chemical stability, bioavailability and lipophilicity over their non-fluorinated analogues.3 Particularly, molecules bearing a trifluoromethyl (–CF3) group show a wide range of pharmacological and biological properties.4 Although a variety of efficient and practical methodologies for the construction of Calkyl–CF3 and Caryl–CF3 bonds have been developed over the last few years,5 much less reports are available for the fabrication of Calkenyl–CF3 bonds. Moreover, alkenes are key intermediates in many organic cyclocondensation reactions.6–10 As evidenced in the literature, C–CF3 bond formation in alkenes usually relied on the use of prefunctionalized substrates, such as vinyl halides,11 vinylboronic acids,12 vinyl borates,13 vinyl sulfonates,14 vinyl carboxylic acids,15 and nitro olefins.16 Unfortunately, most of these strategies suffer from limited substrate scope, multistep synthesis, and/or toxic waste stream.
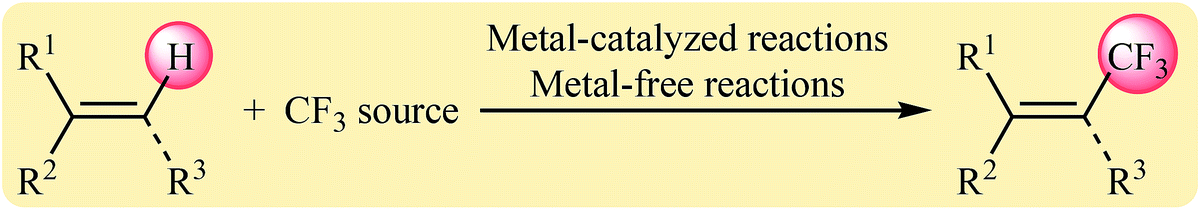
An alternative protocol for the preparation of CF3-substituted alkenes involves the direct trifluoromethylation of corresponding olefins via C–H activation. As this synthetic strategy avoid wasteful pre-functionalization of starting materials, it is undoubtedly more cost effective, atom-economical, practical, and eco-friendly alternative compared to the traditional procedures. To the best of our awareness, a comprehensive review has not appeared on the direct trifluoromethylation of olefinic C–H bonds in the literature as yet. In continuation of our recent reviews on the direct C(X)–H bonds functionalization,17 cross-coupling reactions and heterocyclic synthesis,18 herein, we will highlight recent discoveries and developments on the direct C–H trifluoromethylation of alkenes (Scheme 1), with special attention on the mechanistic aspects of the reactions.
 |
| | Scheme 1 Direct trifluoromethylation of olefinic C–H bonds. | |
2. Terminal alkenes
2.1. Transition-metal-catalyzed reactions
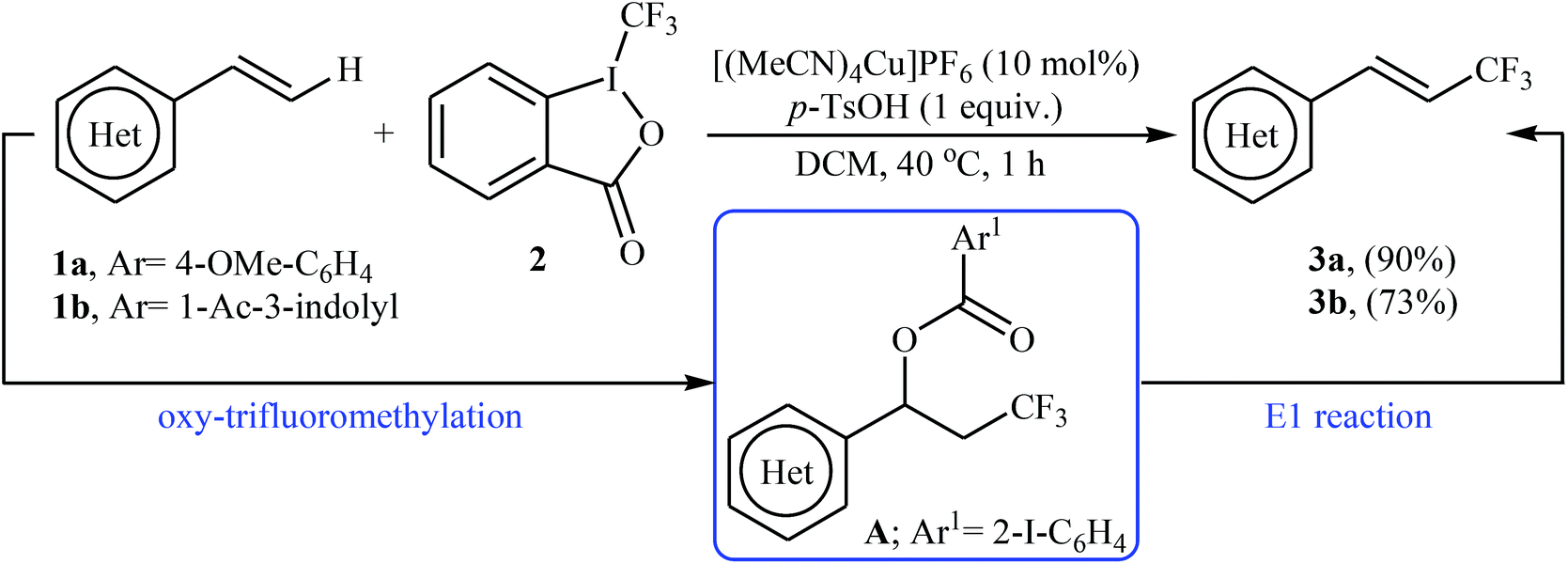
In 2012, Sodeoka and colleagues reported that a combination of cationic copper catalyst ([(MeCN)4Cu]PF6), Brønsted acid (para-toluenesulfonic acid), and Togni's reagent 2 (1-trifluoromethyl-1,2-benziodoxol-3-(1H)-one) enables direct trifluoromethylation of olefinic C–H bonds of styrene derivatives 1 giving β-trifluoromethyl styrenes 3 in high yields and outstanding (E)-selectivity (Scheme 2).19 Although only two examples were disclosed, this paper represents the first example of direct trifluoromethylation of alkenyl C–H bonds. It should be noted that the presence of a Brønsted acid was crucial for the success of this transformation. In the absence of p-TsOH, the reaction failed to give the desired trifluoromethylated styrenes 3, providing instead oxy-trifluoromethylated products. The authors suggested that the reaction proceeded through the formation of oxy-trifluoromethylated intermediate A via an oxy-trifluoromethylation process followed by E1 reaction.
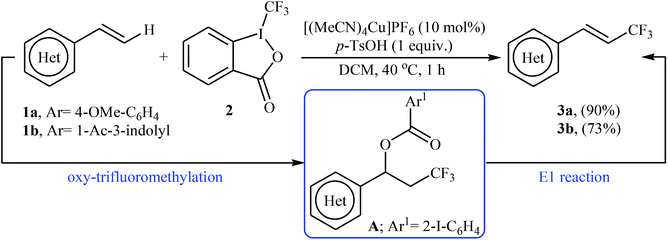 |
| | Scheme 2 Cu-catalyzed trifluoromethylation of styrenes 1 using Togni's reagent. | |
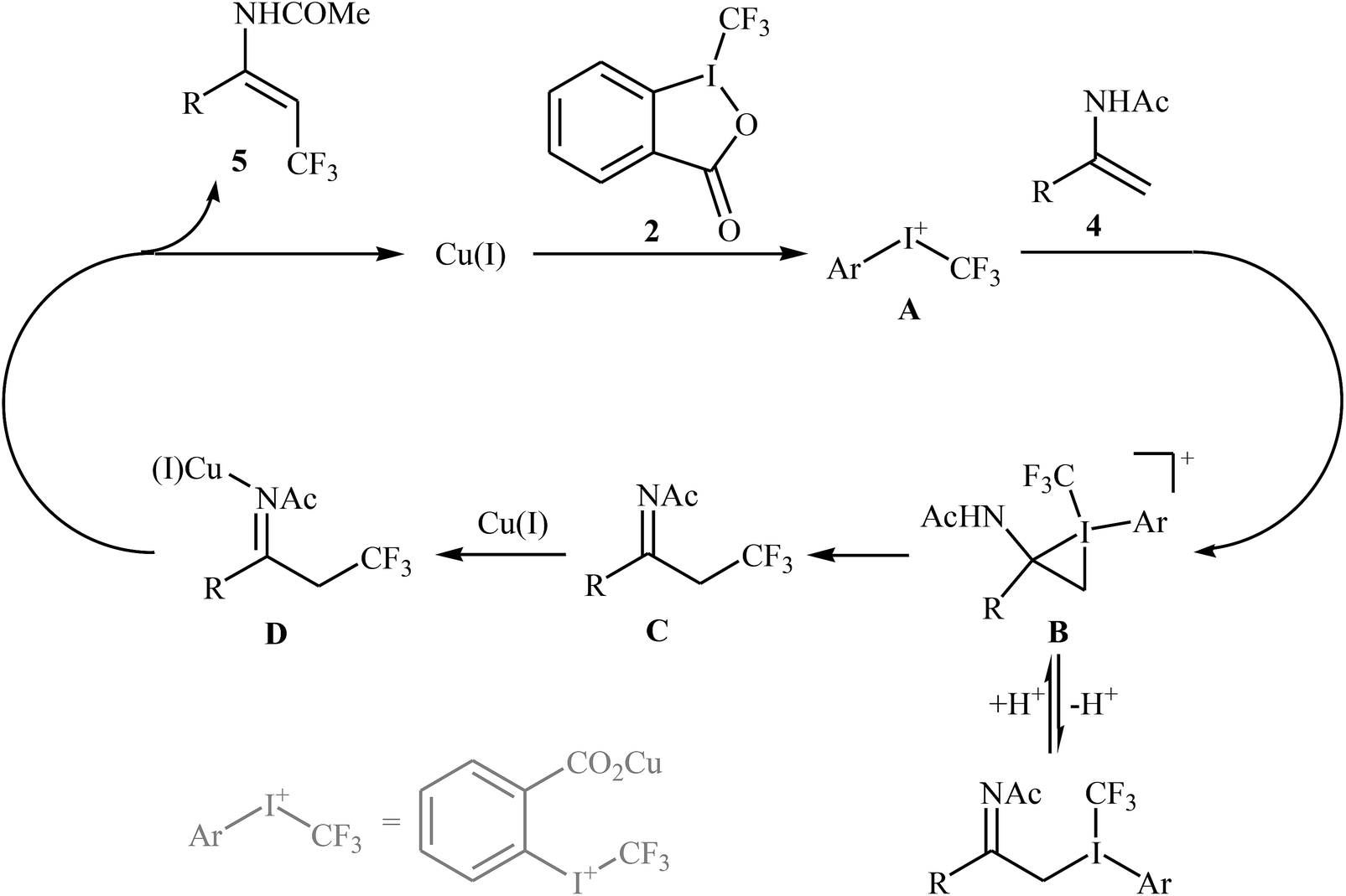
At the same time, with the objective of designing a general procedure to the direct olefinic C–H bonds trifluoromethylation, Feng and Loh were able to revealed that a diverse set of enamides 4 can undergo Cu-catalyzed (E)-selective trifluoromethylation using Togni's reagent 2 as the source of CF3.20 The reaction were carried out in the presence of 10 mol% of [(MeCN)4Cu]PF6 as the catalyst in THF under an inert atmosphere and provided the target trifluoromethyl-substituted olefins 5 in moderate to excellent yields, ranging from 40% to 92% (Scheme 3). It should be mentioned that the amido moiety introduced onto the olefin moiety had a dual role: (i) stabilizing the putatively formed α-carbonium; and (ii) inducing the subsequent proton elimination or a migration process during the reaction. The outcomes of the radical trapping experiments by addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 2,6-di-tert-butyl-pcresol (BHT) to the reaction mixture and electron paramagnetic resonance (EPR) spectroscopy studies suggested that the mechanism of this reaction cannot be involved a radical pathway. A plausible mechanism is shown in Scheme 4. This transformation starts with the reaction between Togni's regent 2 and Cu(I), leading to the formation of the reactive intermediate A, which after the reaction with enamide 4 affords the iodo(III) cyclopropane intermediate B. Subsequently, reductive elimination of intermediate B produces the α-trifluoromethyl imine intermediate C that, after coordination with Lewis acidic Cu(I) generates complex D. Finally, elimination (or transfer) of α-proton affords the final olefinic trifluoromethylation product 5 and concomitantly regenerates the Cu(I) catalyst for the next catalytic cycle.
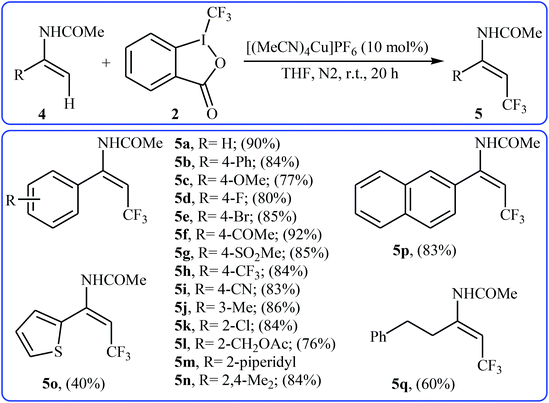 |
| | Scheme 3 Direct trifluoromethylation of enamides 4 with Togni's regent. | |
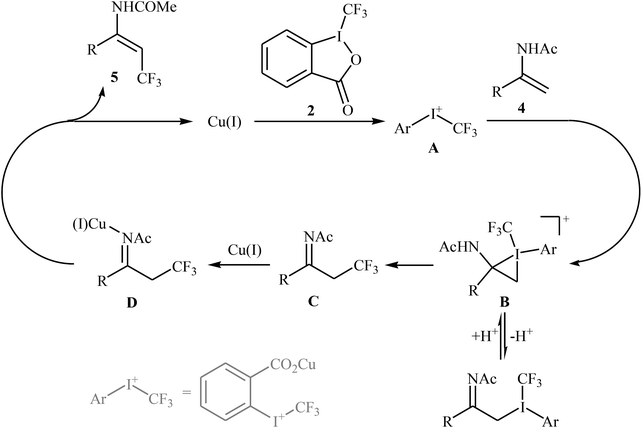 |
| | Scheme 4 Mechanism for the trifluoromethylation of enamides 4. | |
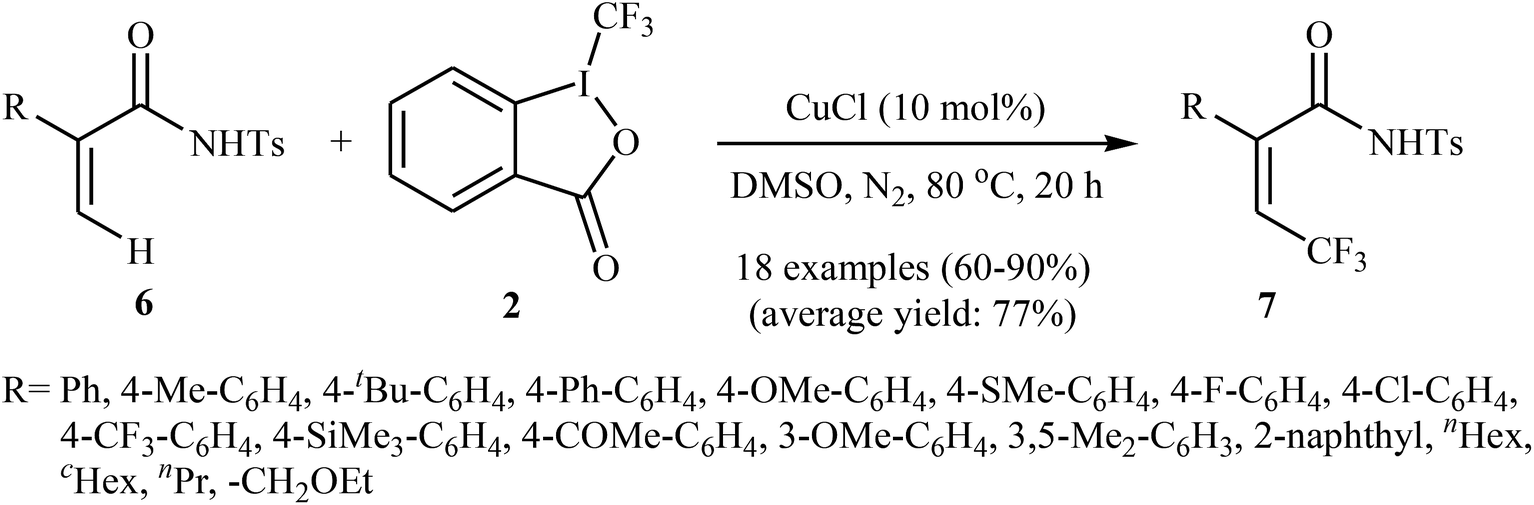
Shortly afterwards, the same authors extended the substrate scope of their methodology and reported the direct olefinic C–H bond trifluoromethylation of acrylamide derivatives 6 with Togni's regent 2.21 Thus, a library of β-CF3-functionalized acrylamides 7 were obtained in up to 90% yields employing 10 mol% of commercially available and inexpensive CuCl as a catalyst and DMSO as the solvent in the absence of any additive (Scheme 5). In this system, the reaction shows a remarkable degree of stereospecificity and occurs selectively in the cis position with respect to the Ts-protected amide directing group. Of note, other Cu-catalysts such as CuBr, CuI, CuOAc, Cu(OAc)2, Cu2O, and [(MeCN)4Cu]PF6 were also found to promote this trifluoromethylation reaction, albeit with reduced efficiencies.
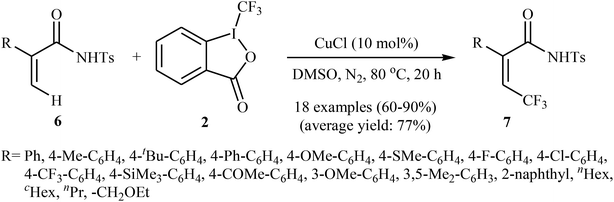 |
| | Scheme 5 Cu(I)-catalyzed trifluoromethylation of acrylamide derivatives 6 with Togni's regent 2. | |
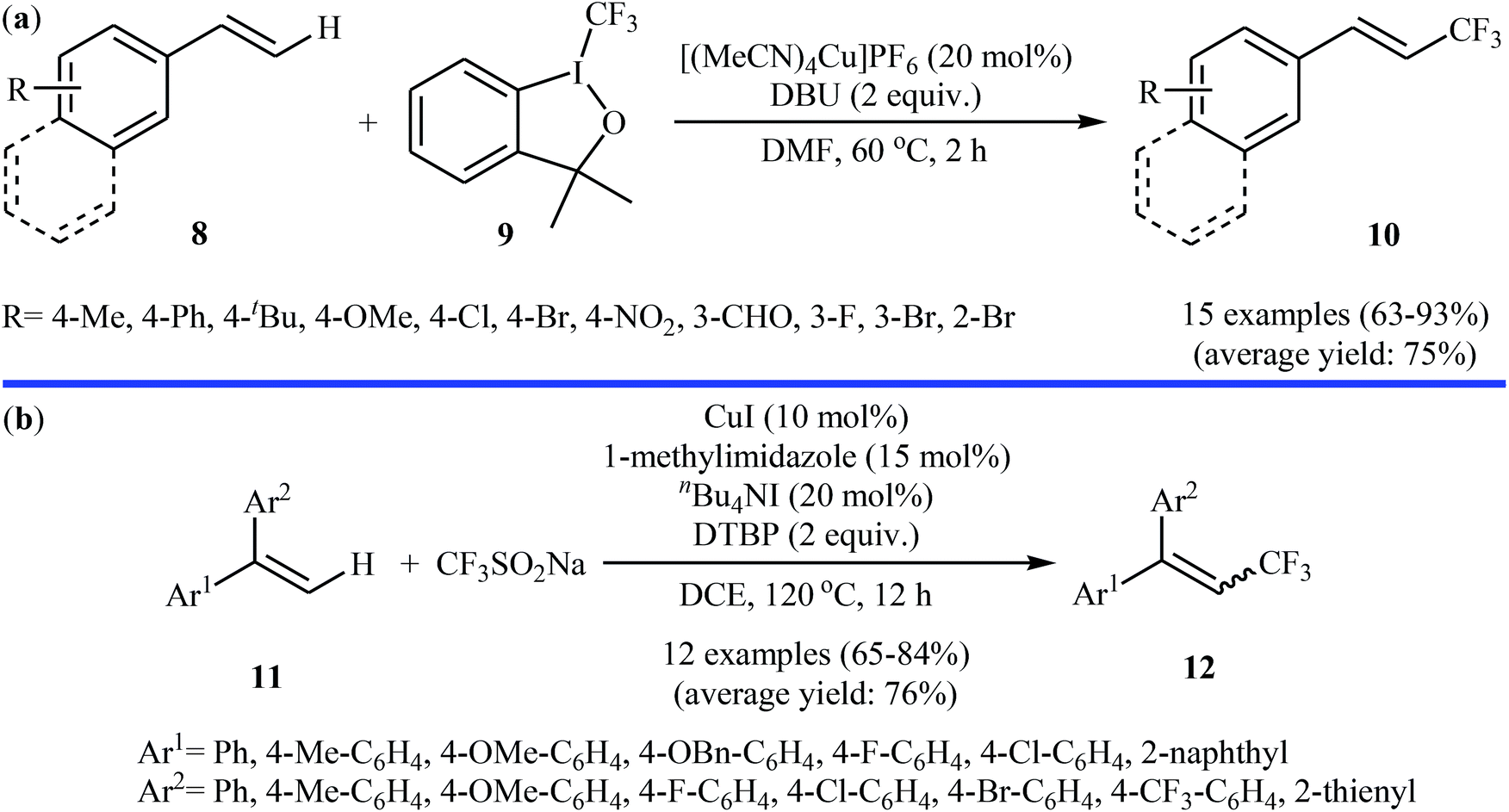
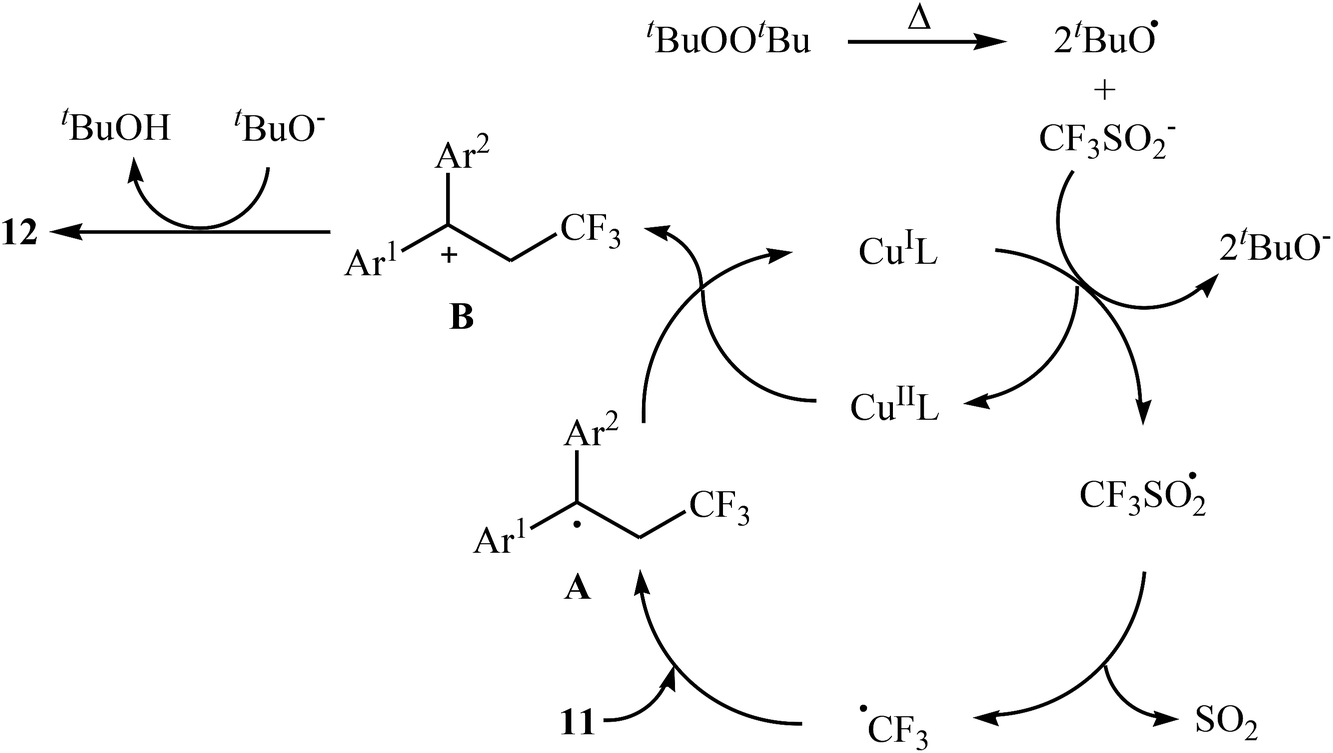
In related work, Xiao and co-workers have reported the synthesis of β-trifluoromethylated styrenes 10 through the direct trifluoromethylation of corresponding styrene derivatives 8 with Togni's reagent 9 utilizing [(MeCN)4Cu]PF6 as catalyst and 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) as base in DMF (Scheme 6a).22 The scope of this reaction was broad, and various important functional groups (e.g., OMe, CHO, F, Cl, Br, NO2) were well tolerated. The reaction was also shown to be compatible with terminal heteroaromatic alkenes. Noteworthy, all products were obtained with excellent stereoselectivity (E/Z > 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Recently, Shen and Loh along with their co-workers reported the fabrication of 1,1-diaryl-2-trifluoromethylethenes 12 shown in Scheme 6b.23 These compounds were formed by reaction of corresponding diarylethenes 11 with the Langlois reagent (CF3SO2Na) as an easy-to-handle and inexpensive trifluoromethylating agent through a radical process. The reaction carried out in the presence of CuI/1-methylimidazole/nBu4NI/DTBP combination as a catalytic system in DCE at 120 °C resulted in relatively slow trifluoromethylthiolation to give good to high isolated yields of trifluoromethylated products 12 with a strong preference for (E)-isomers. The results demonstrated that substrates bearing an electron-donating group afforded better yields than those with an electron-withdrawing group. The mechanism proposed by the authors to explain the title reaction is depicted in Scheme 7.
:3). Recently, Shen and Loh along with their co-workers reported the fabrication of 1,1-diaryl-2-trifluoromethylethenes 12 shown in Scheme 6b.23 These compounds were formed by reaction of corresponding diarylethenes 11 with the Langlois reagent (CF3SO2Na) as an easy-to-handle and inexpensive trifluoromethylating agent through a radical process. The reaction carried out in the presence of CuI/1-methylimidazole/nBu4NI/DTBP combination as a catalytic system in DCE at 120 °C resulted in relatively slow trifluoromethylthiolation to give good to high isolated yields of trifluoromethylated products 12 with a strong preference for (E)-isomers. The results demonstrated that substrates bearing an electron-donating group afforded better yields than those with an electron-withdrawing group. The mechanism proposed by the authors to explain the title reaction is depicted in Scheme 7.
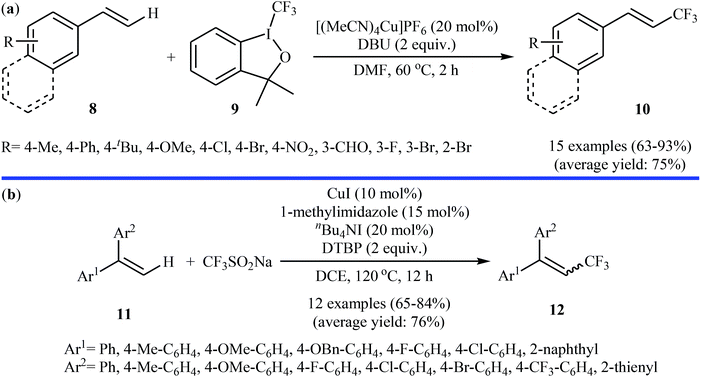 |
| | Scheme 6 (a) Xiao's synthesis of β-trifluoromethylated styrenes 10; (b) direct trifluoromethylation of diarylethenes 11 with the Langlois reagent. | |
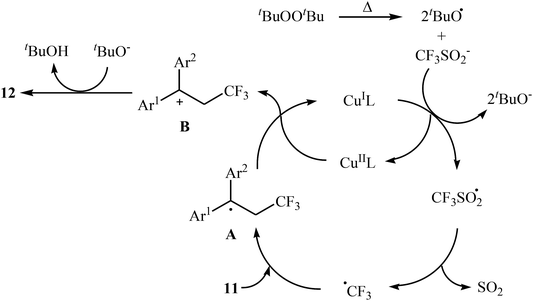 |
| | Scheme 7 Mechanistic proposal for the reaction in Scheme 6a. | |
2.2. Visible-light-mediated photoredox-catalyzed reactions
In 2012, the group of Cho developed a mild and general approach for visible light-mediated direct trifluoromethylation of terminal alkenes 13 with CF3I using commercially available Ru(Phen)3Cl2 as a photocatalyst and DBU as an additive.24 The reaction proceeded smoothly in MeCN under a 14 W household light bulb at room temperature, giving generally high to excellent yields of trifluoromethyl-substituted olefins 14 with only (E)-stereochemistry (Scheme 8). Although the reactions of terminal alkenes are regio- and stereoselective, those of internal alkenes generated a mixture of isomers. Of note, aromatic systems were unreactive under the reaction conditions. Interestingly, in this reaction DBU played a dual role; the base and the reductant.
 |
| | Scheme 8 Visible light-mediated direct trifluoromethylation of terminal alkenes 13 developed by Cho. | |
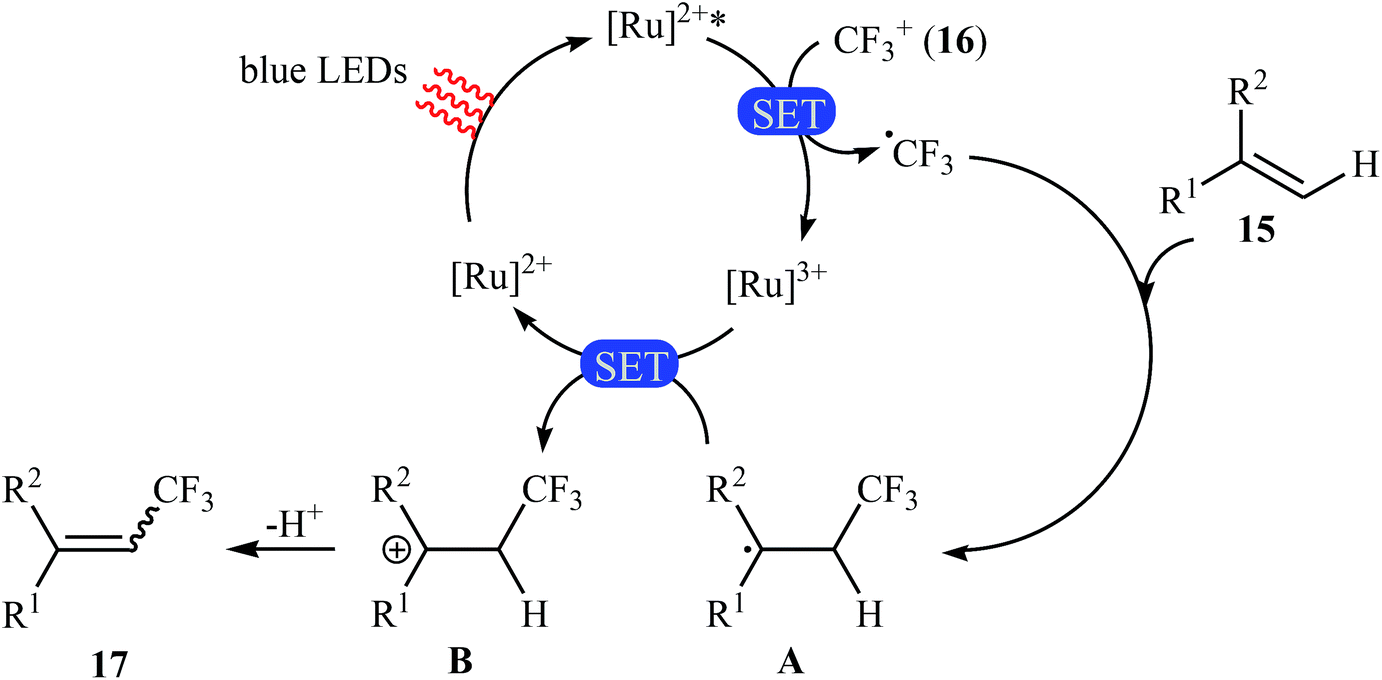
Not long after this report, Akita and co-workers disclosed that 1,1-disubstituted-2-trifluoromethylethenes 17 could be successfully synthesized from the reaction of corresponding 1,1-disubstituted alkenes 15 with Umemoto's reagent 16, as a CF3 source in the presence of [Ru(bpy)3](PF6)2 (bpy = 2,2′-bipyridine) as the photocatalyst under visible light irradiation.25 As shown in Scheme 9, a series of sensitive substituents (e.g., NO2, NHBoc, Cl, Br) and heterocycles were all compatible under the conditions. In addition, this system also worked for direct trifluoromethylation of trisubstituted aryl alkenes, and the target products were isolated in moderate to high yields. Additionally, use of an excess amount of trifluoromethylating agent (4 equiv.) induced double C–H trifluoromethylation to afford geminal bis(trifluoromethyl)alkenes. A plausible mechanistic pathway was proposed by the authors for this reaction (Scheme 10) that involves the initial formation of the trifluoromethyl radical (˙CF3) via an one-electron-reduction of electrophilic Umemoto's reagent 16 by the photoactivated Ru catalyst, [Ru(bpy)3]2+*, which after reaction with alkene 15 leads to the benzyl radical-type intermediate A. Subsequently, one-electron-oxidation of this intermediate by highly oxidizing Ru species, [Ru(bpy)3]3+ gives β-CF3 carbocation intermediate B. Finally, elimination of the olefinic proton from this intermediate affords the observed trifluoromethylated alkene 17.
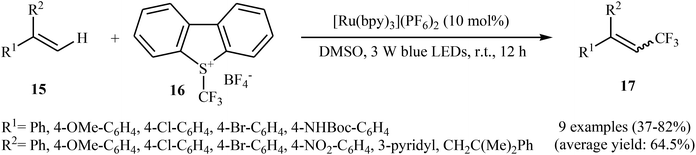 |
| | Scheme 9 Akita's synthesis of trifluoromethylated alkenes 17. | |
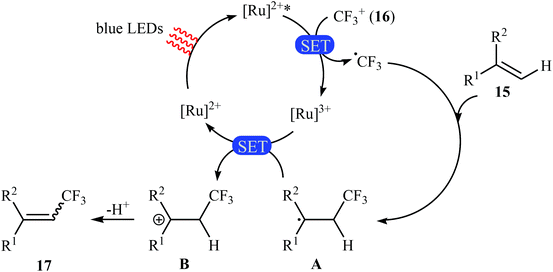 |
| | Scheme 10 Mechanism that accounts for the formation of trifluoromethylated alkenes 17. | |
In 2014, Lin, Xu, and Qing developed an interesting stereocontrolled direct trifluoromethylation of olefinic C–H bonds through an appropriate combination of the photoredox catalyst and of the CF3-containing substrate.26 They showed that trifluoromethylation of styrene derivatives 18 with Togni's reagent 9 in the presence of Ru(bpy)3Cl2·6H2O as the photocatalyst under visible light irradiation gave exclusively more thermodynamically stable (E)-trifluoromethylated styrenes 19, whereas reaction with Umemoto's reagent 16 in the presence of Ir(ppy)3 lead to less thermodynamically stable (Z)-trifluoromethylated alkenes 20 with high to excellent stereoselectivity (Scheme 11). According to the authors, the mechanism of (E)-selective reaction is analogous to the one depicted in Scheme 10. The putative mechanism for the (Z)-selective trifluoromethylation is based on the initial formation of a thermodynamically stable (E)-isomer, which after the triplet–triplet energy transfer (TTET) process converts into (Z)-isomer.
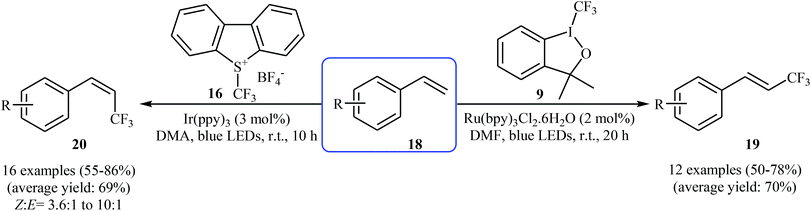 |
| | Scheme 11 Chemo-, regio-, and stereoselective trifluoromethylation of styrenes 18 developed by Qing. | |
Another innovative example on direct C–H trifluoromethylation of alkenes is shown in Scheme 12, where fac-Ir(ppy)3 was used as heterogeneous photocatalyst.27 Mono-trifluoromethylated vinylarenes 22 were easily formed in moderate to excellent yields and high E/Z selectivity under continuous-flow conditions and visible light irradiation via the treatment of the corresponding alkenes 21 with CF3I in the presence of CsOAc as a base in DMF. The authors found that transferring continuous-flow conditions to batch reactions also provided high yield for the desired compounds, albeit with considerably reduced E/Z ratio. This fact can be explained by the shorter reaction times in flow.
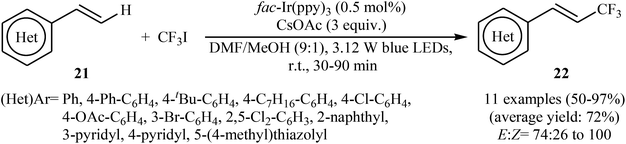 |
| | Scheme 12 Photocatalytic trifluoromethylation of terminal alkenes 21 with CF3I. | |
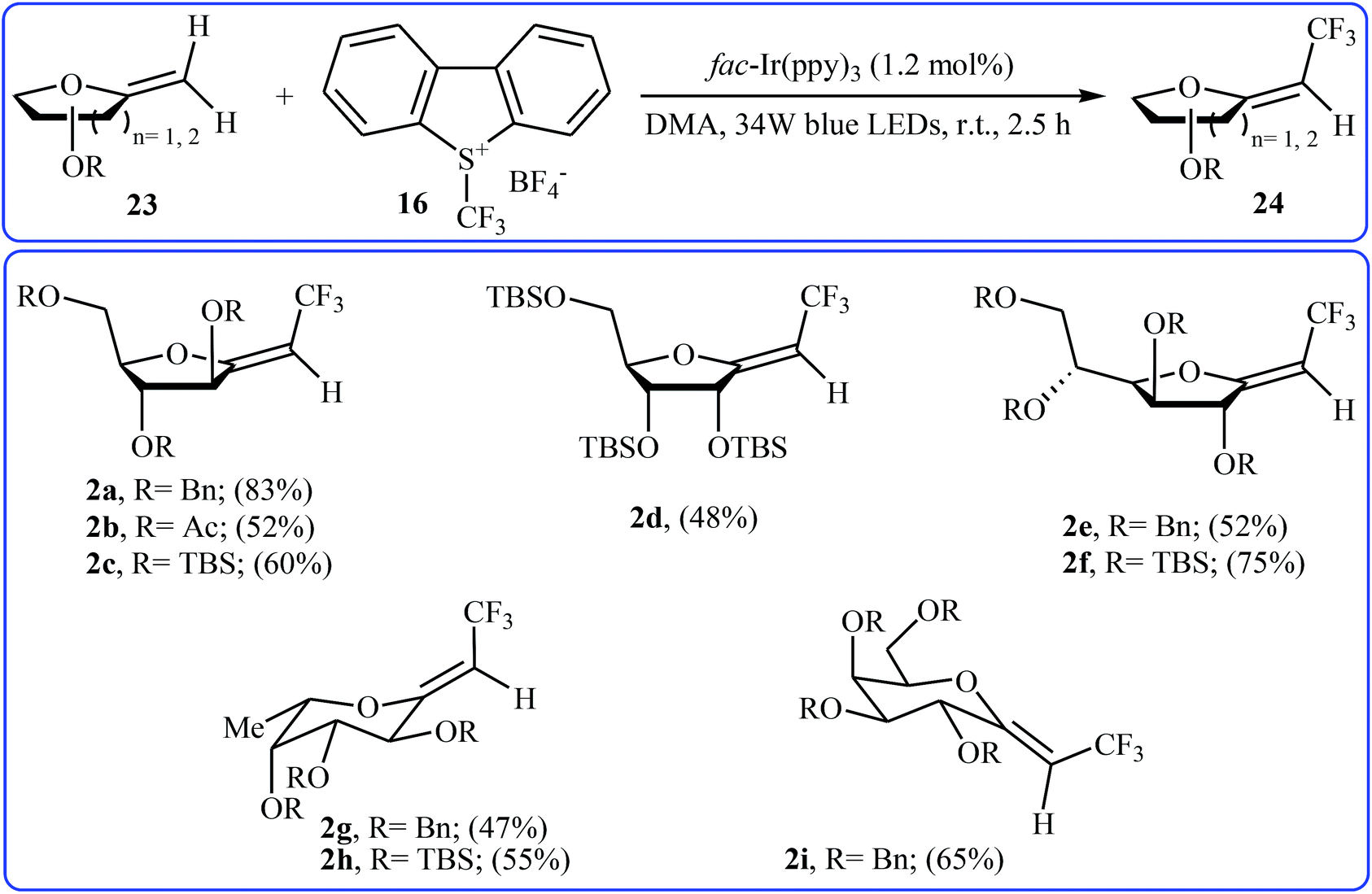
Recently, an elegant and efficient protocol for photoredox-catalyzed (Z)-diastereoselective trifluoromethylation of methylene exo-glycals at room temperature using the Umemoto's reagent 16 was explored by Vincent et al.28 By this procedure, a diverse array of unsubstituted exo-glycals 23 were successfully converted to the corresponding mono-trifluoromethyl analogues 24 with yield ranging from 47% to 83% and excellent (Z)-stereoselectivity (Scheme 13). The authors also showed that the synthesis of the same products were possible by performing the reaction in the presence of a catalytic amount of CuI in chloroform at 120 °C under microwave irradiation. The results demonstrated that the photoredox-catalyzed reaction is in general more efficient compared to the latter reaction.
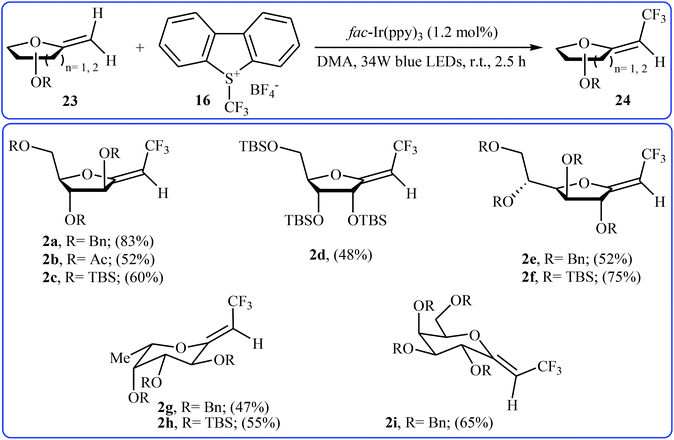 |
| | Scheme 13 Photoredox-catalyzed (Z)-diastereoselective trifluoromethylation of methylene exo-glycals 23 using the Umemoto's reagent. | |
2.3. Transition-metal-free reactions
In 2014, Tan and Liu along with their co-workers reported an interesting iodide-induced metal-free direct C–H trifluoromethylation of α,β-unsaturated carbonyl compounds 25 with Togni's reagent 2 using over-stoichiometric amounts of nBu4Nl as an initiator and NaOAc as an additive in MeCN at 80 °C (Scheme 14).29 The reaction proceeds with outstanding regioselectivity for β-site and stereoselectivity to the E-isomers and afforded the corresponding (E)-β-trifluoromethyl α,β-unsaturated hydroxamic acid derivatives 26 in moderate to good yields. The reaction could also be extended for β-trifluoromethylation of acrylamide derivatives. The mechanism of this trifluoromethylation reaction probably involves the generation of the highly reactive iodine(III) intermediate A from the reaction of nBu4NI with Togni's reagent, followed by reaction with activated alkene 25 to form intermediate B, which after an elimination and deprotonation sequential process furnishes the desired product 26 (Scheme 15).
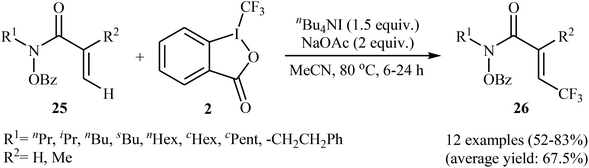 |
| | Scheme 14 Iodide-induced direct C–H trifluoromethylation of α,β-unsaturated carbonyl compounds 25 with Togni's reagent. | |
 |
| | Scheme 15 Mechanism proposed to explain the formation of (E)-β-trifluoromethyl α,β-unsaturated hydroxamic acids 26. | |
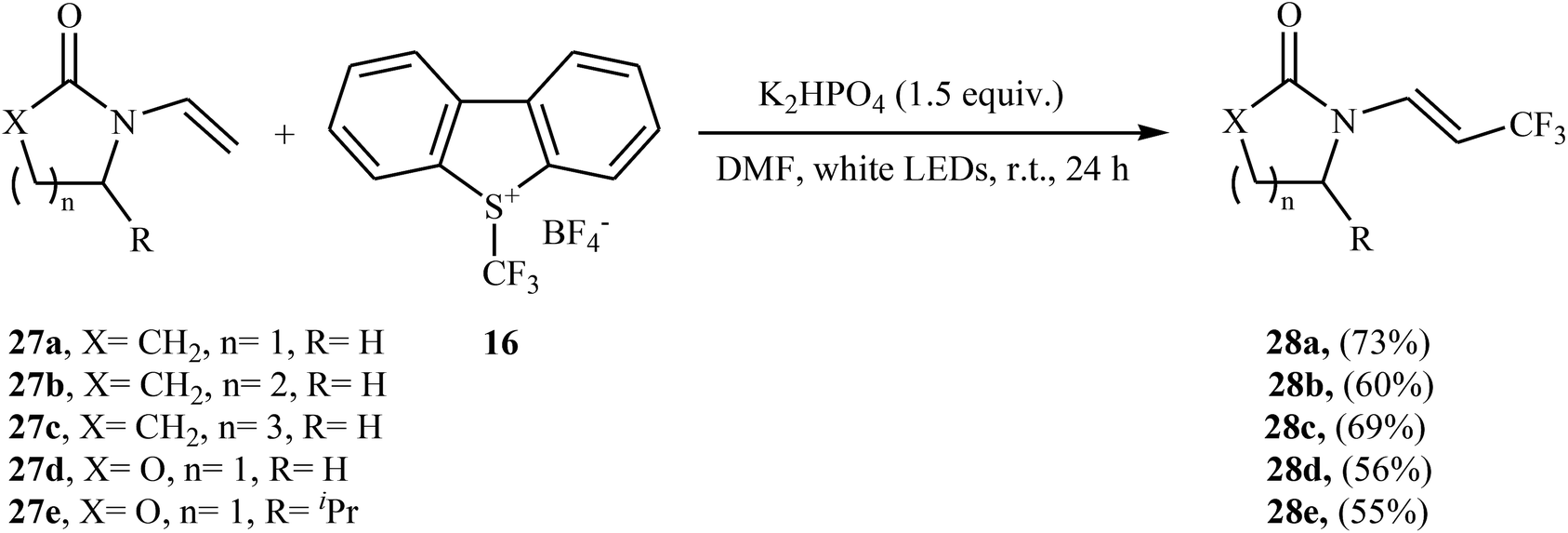
Later, an interesting protocol for the stereoselective trifluoromethylation of enamides 27 with the Umemoto's reagent 16 was reported by Yu et al.30 This photochemical, metal-free reaction was proceeded under visible light irradiation without any photocatalyst at room temperature and only inexpensive K2HPO4 is needed to promote this transformation. Both cyclic and acyclic substituents are well-tolerated and all the trifluoromethylated enamides were isolated in (E)-configuration exclusively. Some of the reported examples are shown in Scheme 16.
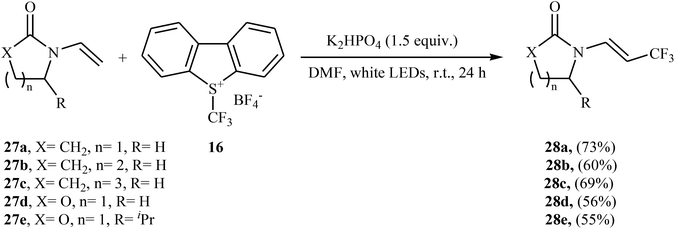 |
| | Scheme 16 Yu's trifluoromethylation of enamides 27. | |
2.4. Miscellaneous reactions
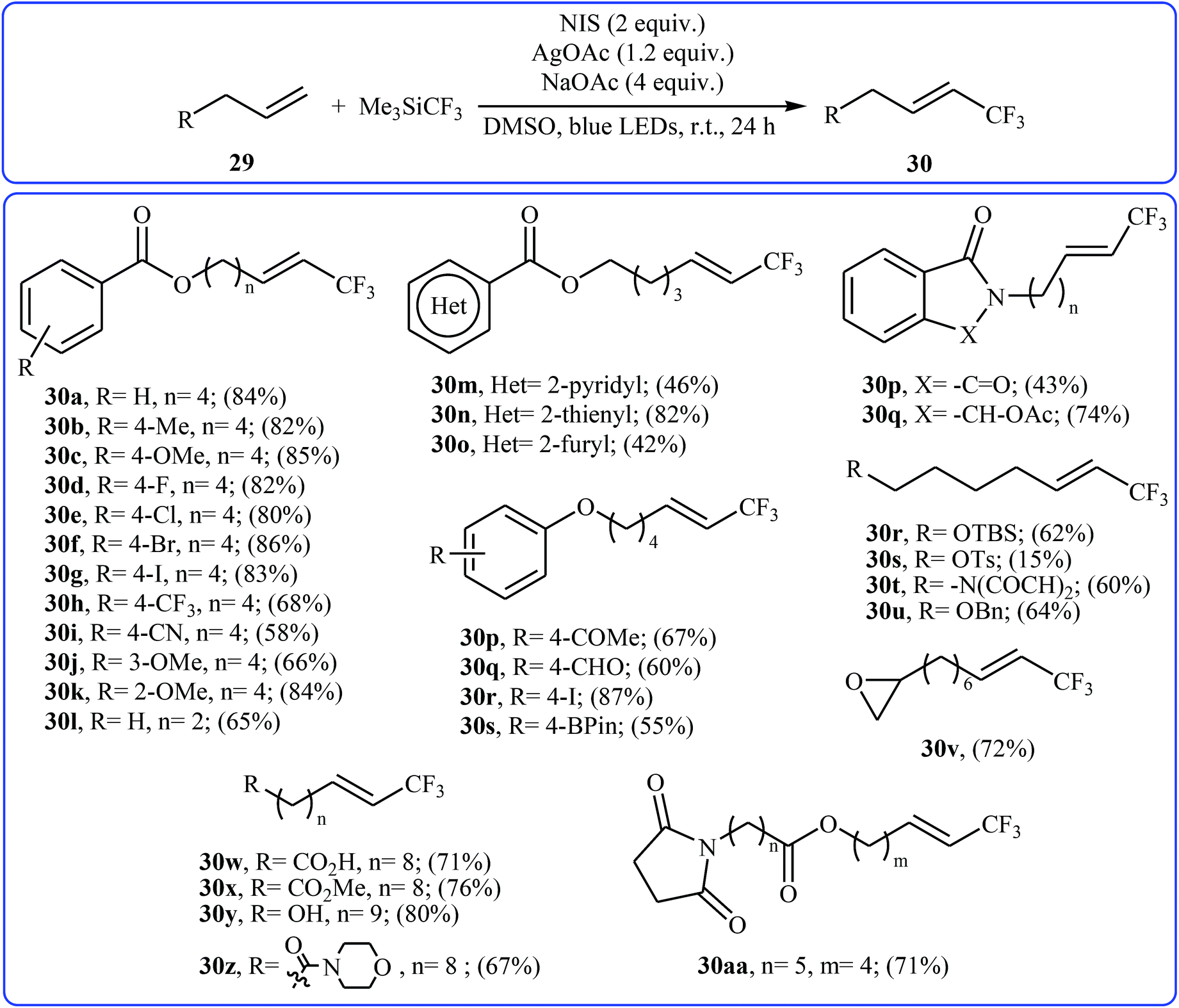
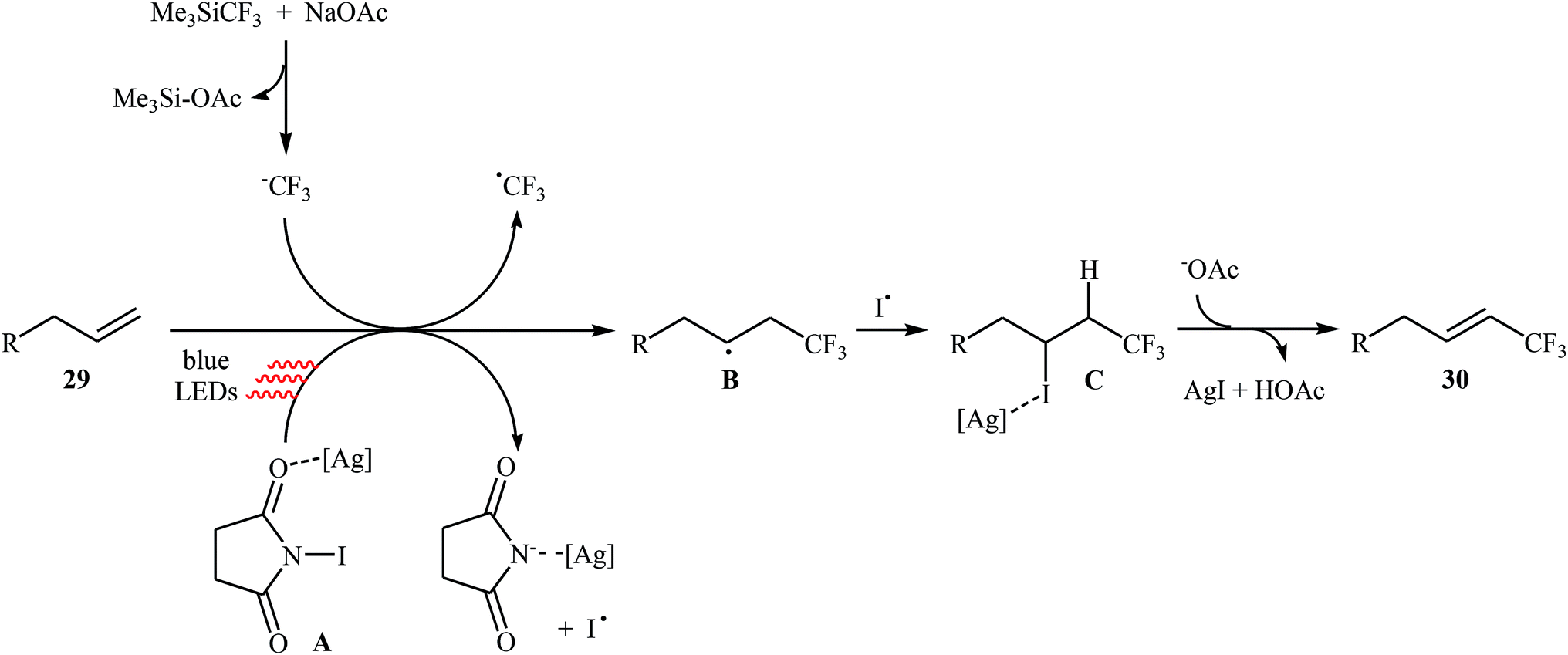
Very recently, Yang and Tsui presented a very elegant method for the synthesis of trifluoromethylated alkenes using easy-to-handle and commercial Me3SiCF3 as the CF3 source.31 They showed that the trifluoromethylation of unactivated aliphatic alkenes 29 under a combination of N-iodosuccinimide (NIS) and visible light in the absence of any photocatalyst afforded the corresponding trifluoromethylated (E)-alkenes 30 in moderate to high yields, ranging from 42% to 86% (Scheme 17). The reaction was shown to be remarkably tolerant toward a large number of functional groups such as fluoro, chloro, bromo, iodo, cyano, hydroxyl, acid, ester, ether, ketone, and aldehyde functionalities. However, 2-vinylnaphthalene did not take part in this trifluoromethylation and therefore no other aromatic terminal alkenes were examined in the protocol. The procedures could also be adapted to gram-scale trifluoromethylation of a series of natural product derivatives (e.g., estrone, umbelliferone, flavone, and quinine cores). The putative mechanism for this NIS-promoted reaction is depicted in Scheme 18.
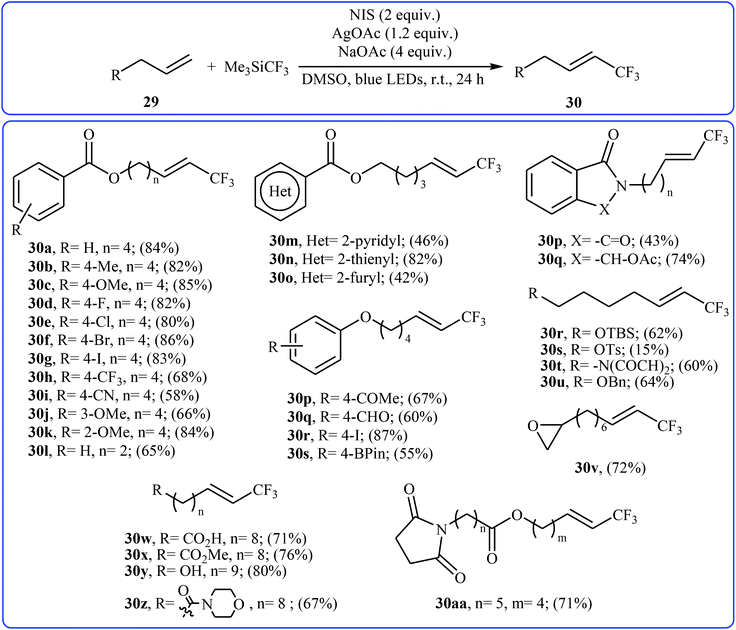 |
| | Scheme 17 NIS-promoted trifluoromethylation of unactivated alkenes 29 with Me3SiCF3. | |
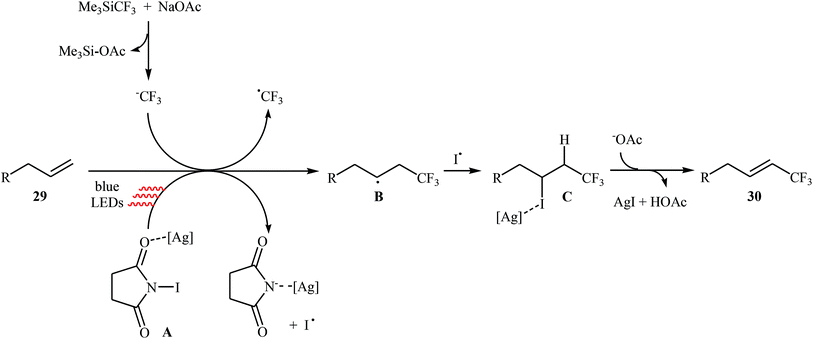 |
| | Scheme 18 Mechanism that accounts for the formation of trifluoromethylated (E)-alkenes 30. | |
3. Internal alkenes
3.1. Cyclic internal alkenes
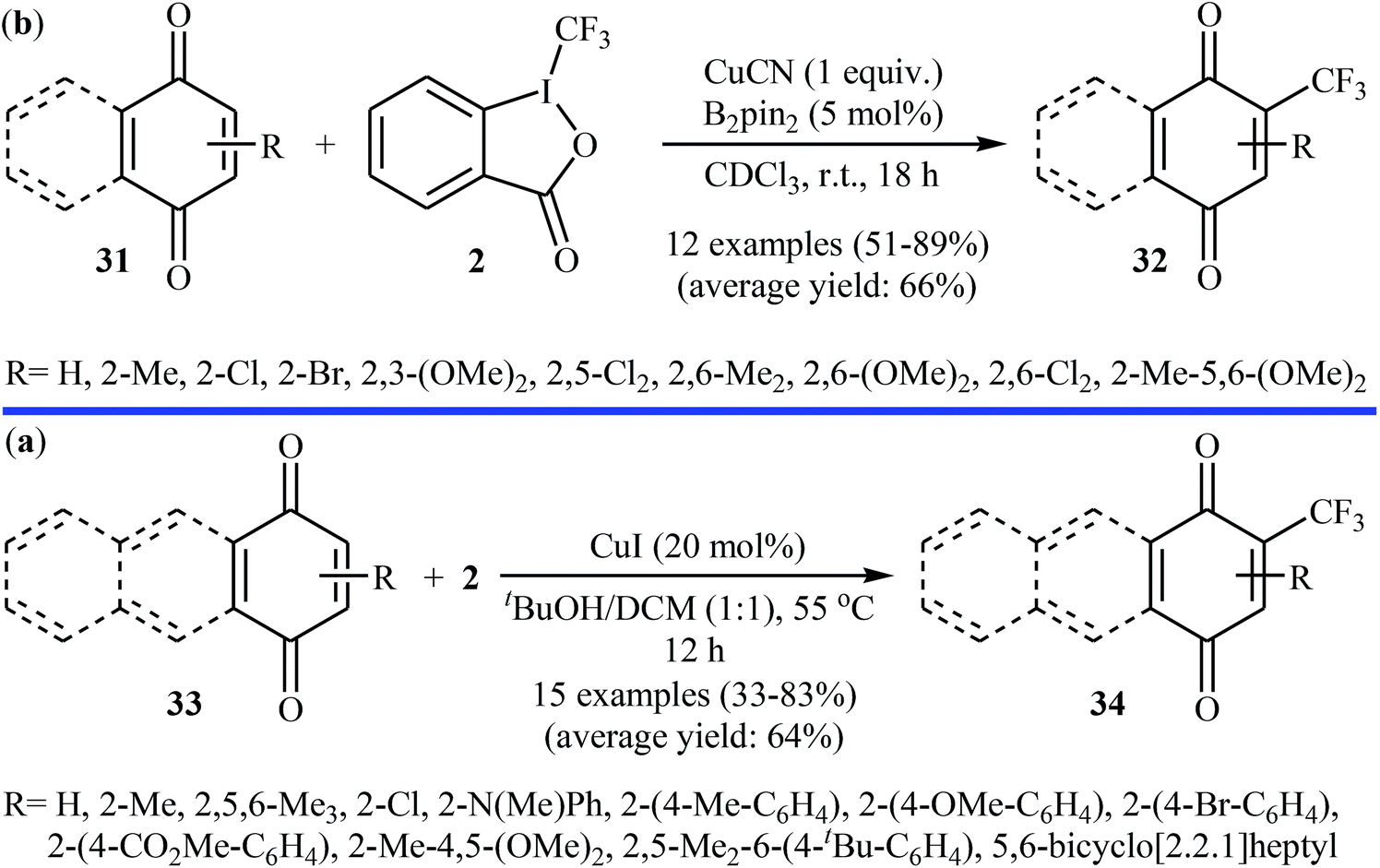
3.1.1. Transition-metal-catalyzed reactions. In 2013, Szabó and co-workers showed that functionalized (benzo)quinone derivatives 31 can undergo direct C–H mono-trifluoromethylation with Togni's reagent 2 in the presence of Cu-salts, according to Scheme 19a.32 After the optimization of the reaction conditions, the use of a stoichiometric amount of CuCN and 5 mol% of bis(pinacolato)diboron (B2pin2) as the mediator and as radical activator, respectively, in CHCl3 at room temperature were required for high product yields. Unfortunately, almost all of the tested substrates for this reaction were exclusively electron-rich derivatives. At the same time, a closely related study was published by Wang et al.33 Here, (benzo)quinones 33 were trifluoromethylated, using 20 mol% of CuI as the catalyst in a 1:1 mixture of tBuOH/DCM at 55 °C. The desired CF3-substituted quinones 34 were obtained with yield ranging from 33% to 83% (Scheme 19b). Both groups suggested that the CF3 radical is involved in the transformation.
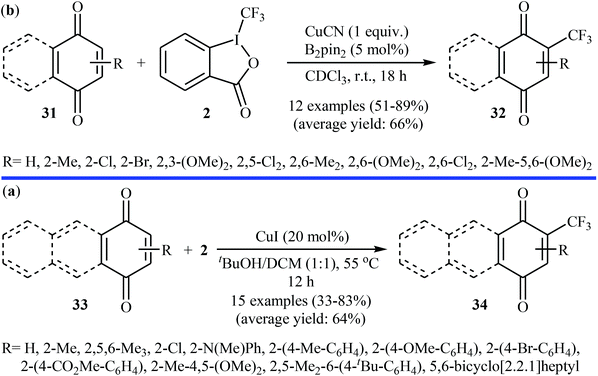 |
| | Scheme 19 (a) Cu-catalyzed trifluoromethylation of (benzo)quinones 31 with Togni's reagent; (b) Wang's synthesis of CF3-substituted quinones 34. | |
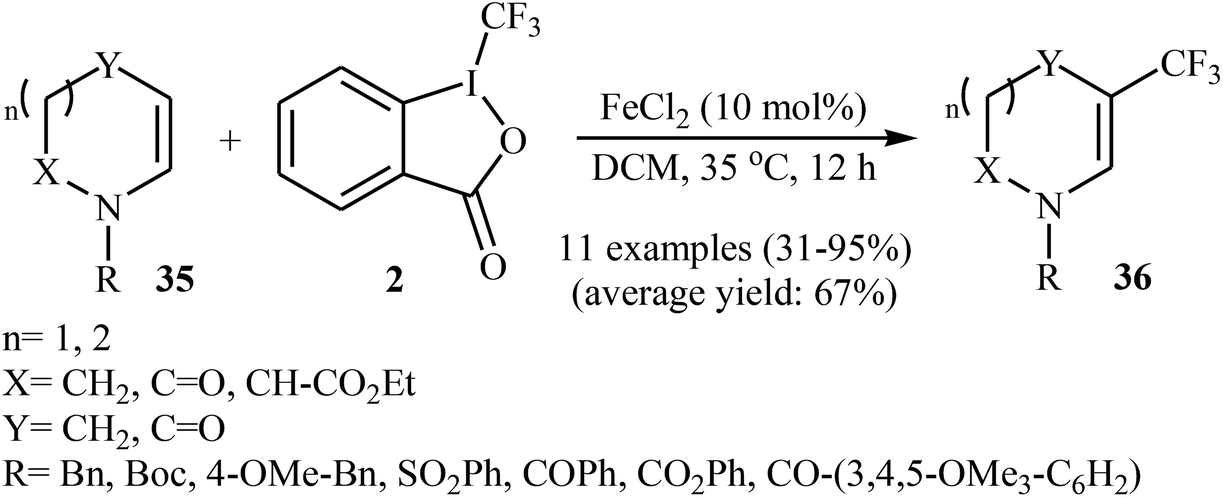
Two years later, Gillaizeau and colleagues have demonstrated that, in the presence of a catalytic amount of FeCl2 in DCM, various cyclic enamides 35 undergo a mild and regioselective trifluoromethylation at the C3 position with the Togni's reagent 2 to afford the corresponding β-CF3-enamides 36 in modestly to excellent yields.34 Some reported examples are shown in Scheme 20. Other metal catalysts such as SnCl2, AlCl3, CuCl, and Cu(OAc)2 displayed lower reactivity compared to FeCl2 in this transformation and Sc(OTf)3 and Cu2O proved to be completely ineffective. It should be mentioned that the reaction was also applied to acyclic enamides and indole derivatives.
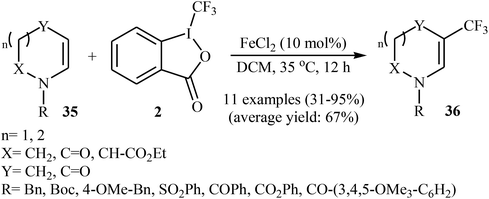 |
| | Scheme 20 Fe-catalyzed regioselective trifluoromethylation of cyclic enamides 35. | |
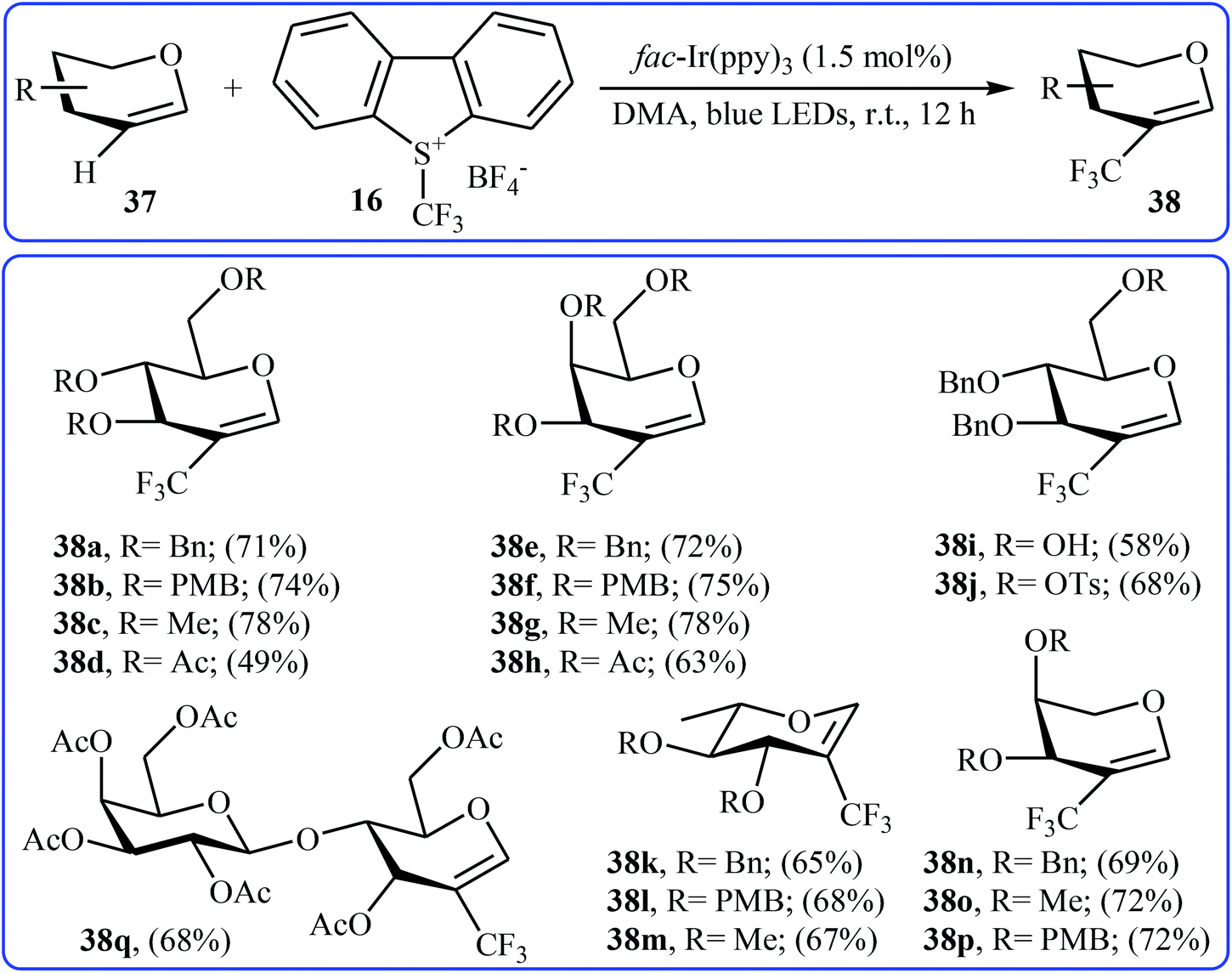
Another direct C–H trifluoromethylation method for cyclic alkenes was described by Wang, Xiong, and Ye in 2015 with Umemoto's reagent under visible light irradiation.35 They showed that the reaction of glycals 37 with Umemoto's reagent 16 in the presence of fac-Ir(ppy)3 as catalyst under the household blue LED irradiation smoothly afforded the corresponding trifluoromethylated glycals 38 in moderate to good yields (Scheme 21). Glycals bearing both electron-withdrawing and electron-donating protective groups were compatible with the reaction conditions. More significantly, the reaction can be applied to the trifluoromethylation of the biologically important 2,3-unsaturated N-acetylneuraminic acid (Neu2en) derivatives.
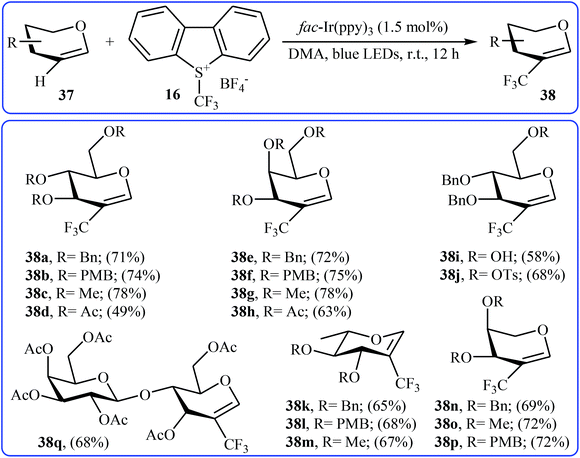 |
| | Scheme 21 Photoredox-catalyzed direct trifluoromethylation of glycals 37 with Umemoto's reagent. | |
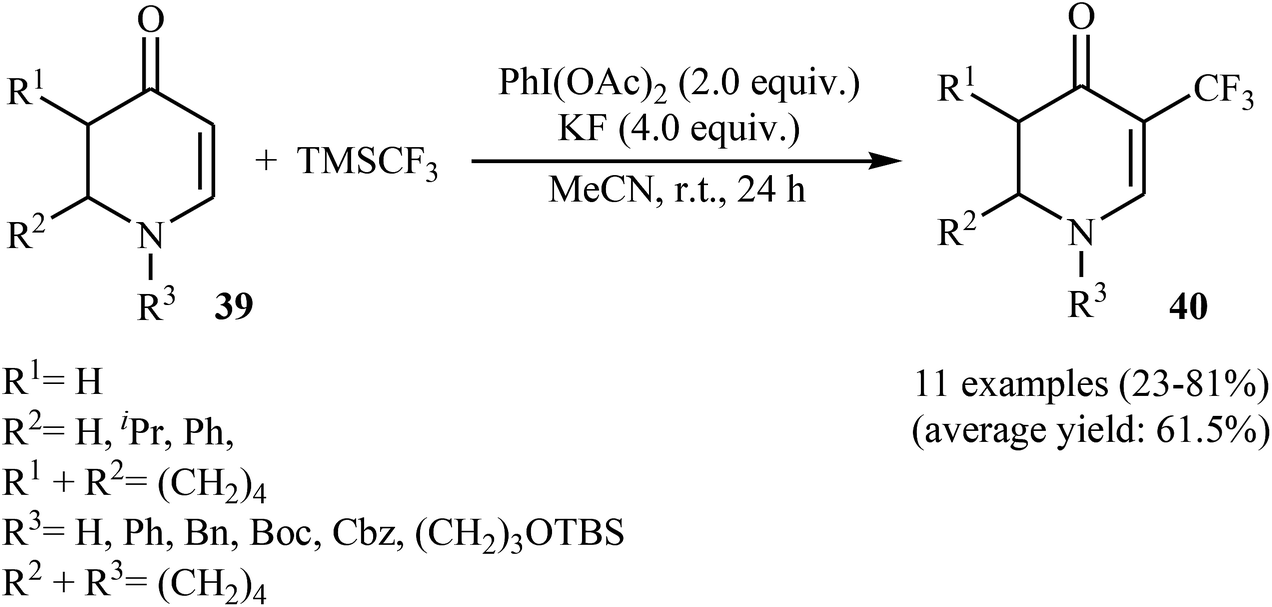
3.1.2. Transition-metal-free reactions. The first and only study on metal-free direct trifluoromethylation of cyclic alkenes was reported by Georg and co-workers in 2014.36 It described the regioselective introduction of a CF3 group at the C-5 position of 2,3-dihydropyridin-4(1H)-ones (cyclic enaminones) using PhI(OAc)2 as an oxidant and the Ruppert's reagent. A screening of reaction conditions proved that KF and MeCN were the most effective base and solvent, respectively. With these optimized reaction conditions, a series of electron-rich and electron-deficient 2,3-dihydropyridin-4(1H)-ones 39 was successfully trifluoromethylated (Scheme 22). This strategy was also successfully used for the trifluoromethylation of pyridin-4(1H)-one, quinolin-4(1H)-one, and 3-aminocyclohex-2-enone derivatives. To the best of our awareness, this is only example dealing with the metal-free direct trifluoromethylation of cyclic alkenes thus far.
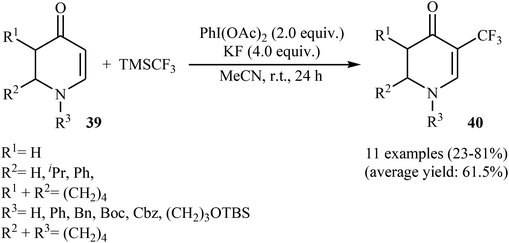 |
| | Scheme 22 Metal-free direct trifluoromethylation of cyclic alkenes 39 developed by Georg. | |
3.2. Acyclic internal alkenes
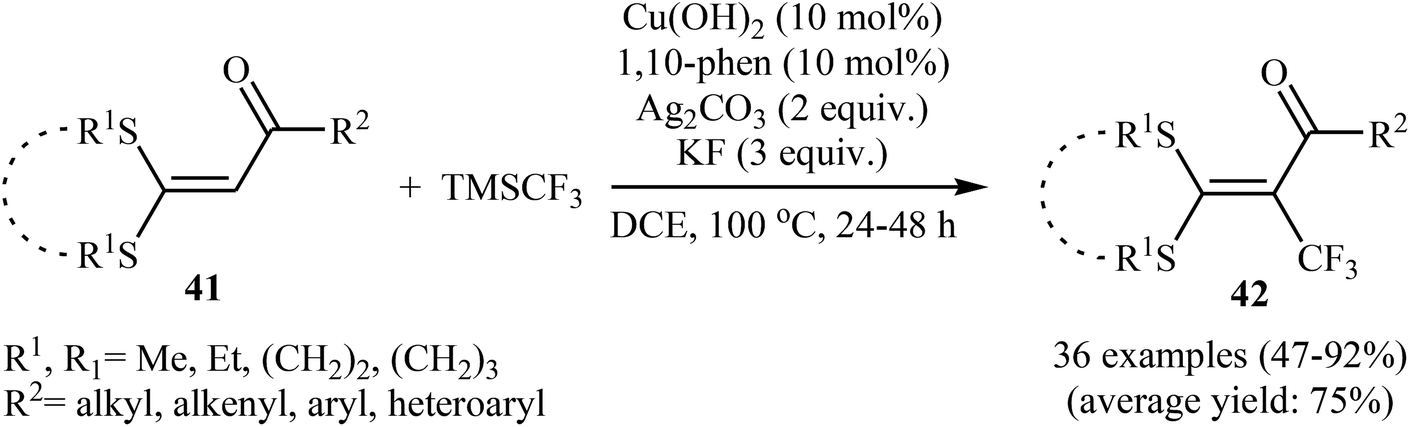
3.2.1. Transition-metal-catalyzed reactions. In 2014, Yu and colleagues developed an efficient copper-catalyzed direct trifluoromethylation of internal olefinic C–H bonds by using TMSCF3, which provides a practical method to obtain trifluoromethylated tetrasubstituted olefins.37 The optimization of the reaction conditions employing α-oxoketene dithioacetal as the model reactant, indicated that Cu(OH)2 was more effective than other Cu catalysts (e.g., CuI, CuOAc, Cu(OAc)2) and compared to other oxidants Ag2CO3 was the best choice for the transformation. The results also showed that KF was the best base over CsF, K2CO3, NaOAc, and KOtBu. Under the optimized conditions, various cyclic and acyclic dithioalkyl α-oxoketene acetals 41 were tolerated well and provided the expected trifluoromethylated products 42 in moderate to excellent yields (Scheme 23). Interestingly, no kinetic isotope effect (KH/KD = 1) was observed with deuterium-labelled compounds under the standard conditions, signifying that the cleavage of the internal olefinic C–H bond was not involved in the rate-determining step of this reaction. The radical-trapping experiments clearly indicated that these reactions proceed via a radical mechanism.
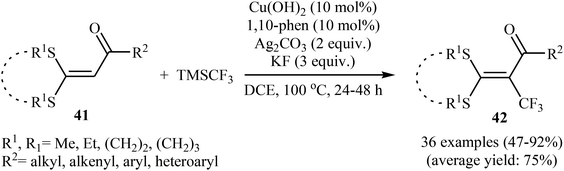 |
| | Scheme 23 Cu-catalyzed trifluoromethylation of internal olefinic C–H bonds by using TMSCF3. | |
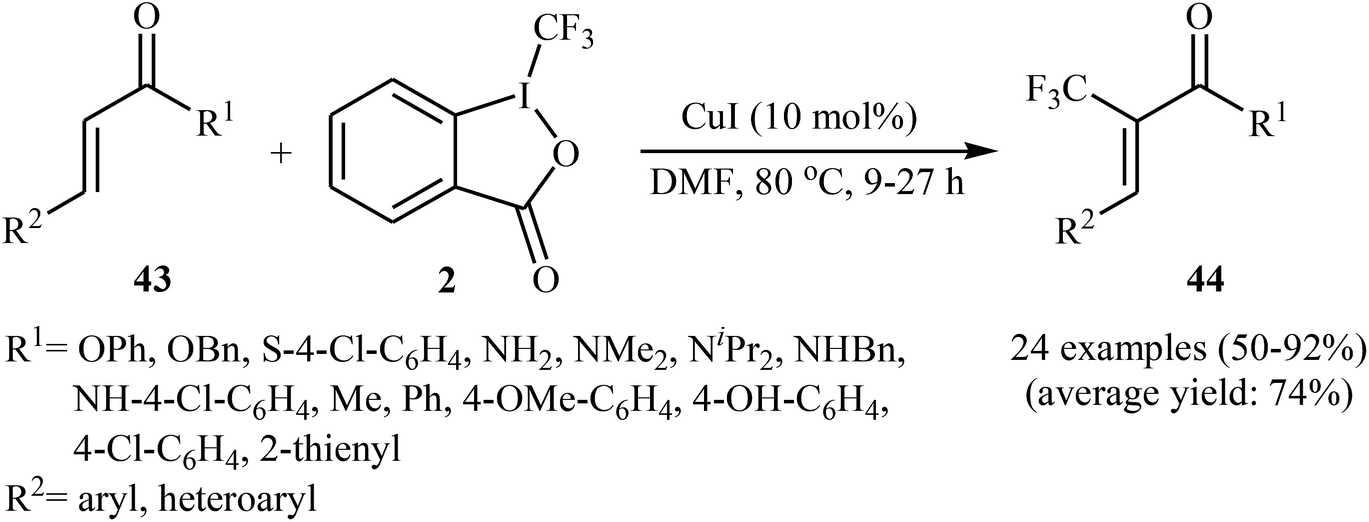
Concurrently, the group of Bi accomplished regioselective C–H α-trifluoromethylation of α,β-unsaturated carbonyl compounds 43 through the employment of Togni's reagent 2 in DMF as the solvent, with the intervention of CuI as catalyst in the absence of any oxidant and base (Scheme 24).38 Interestingly, a diverse array of α,β-unsaturated carbonyl compounds including enones as well as α,β-unsaturated esters, thioesters, and amides were applicable to this reaction. It is noteworthy to mention that this procedure was applied to the high yielding C–H trifluoromethylation of commercialized drugs such as tranilast, hymecromone, flavone, and 2′-deoxyuridine. In a related investigation, Monteiro–Bouyssi and collaborators have reported the direct β-trifluoromethylation of α,β-unsaturated aldehyde N,N-dibenzylhydrazones with Togni's reagent employing CuCl as catalysts and chloroform as solvent at room temperature.39 Moderate to good yields were obtained and excellent (E)-stereoselectivity were observed.
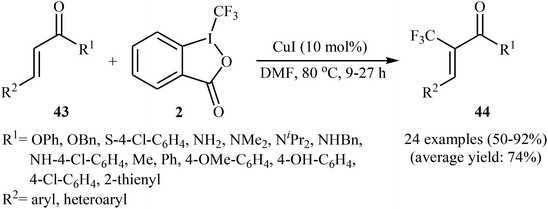 |
| | Scheme 24 Regioselective C–H α-trifluoromethylation of α,β-unsaturated carbonyl compounds 43 using CuI as catalyst. | |
Very recently, Sun, Xu, and Dai have reported the copper-catalyzed α-selective C–H trifluoromethylation of acrylamides 45 with TMSCF3.40 Employment of the Cu(OAc)2/Ag2CO3/KH2PO4/pyridine combination as the catalytic system in DMSO at 120 °C produce satisfactory results providing the corresponding (E)-trifluoromethylated products 46 in moderate yields (Scheme 25). Interestingly, the reaction proceeded very fast (30 min) and was compatible with the presence of a wide range of arenes and heteroarenes at the β-position of acrylamides. The authors proposed mechanism for this reaction is outlined in Scheme 26.
 |
| | Scheme 25 Copper-catalyzed α-selective C–H trifluoromethylation of acrylamides 45 with TMSCF3. | |
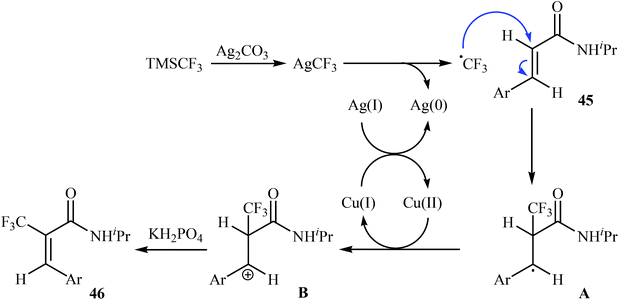 |
| | Scheme 26 Plausible mechanism for reaction in Scheme 25. | |
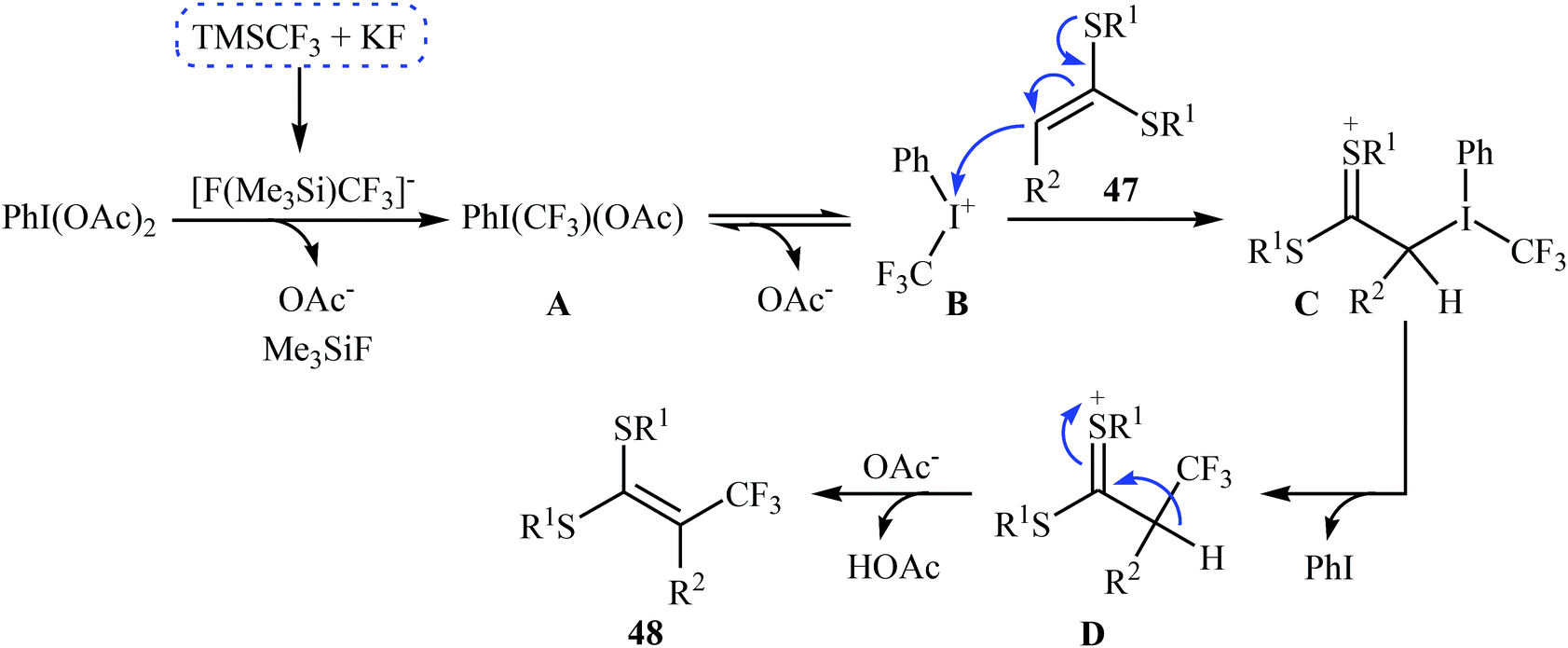
3.2.2. Transition-metal-free reactions. In 2013, Wang, Liu and co-workers described the direct sp2 C–H trifluoromethylation of ketene dithioacetals by in situ generated hypervalent iodide trifluoromethylating species [PhICF3]+ under mild transition metal-free conditions.41 Various substituted cyclic as well as acyclic ketene dithioacetals 47 are readily α-trifluoromethylated by adding TMSCF3 to a premixed mixture of PhI(OAc)2 and KF in MeCN, affording the corresponding 3,3,3-trifluoro-1,1-bis(alkylthio)prop-1-ene derivatives 48 in good to high yields (Scheme 27). Likewise, 2-substituted indoles were trifluoromethylated selectively at the 3-position by [PhICF3]+ species. In accord to the presumed reaction mechanism, trifluoromethylation of ketenes proceeds through the following key steps (Scheme 28): (i) initial formation of PhI(CF3)(OAc) (A) via the reaction of PhI(OAc)2 with TMSCF3 in the presence of KF; (ii) cleavage of the I–O bond of the intermediate A to give phenyl(trifluoromethyl)iodonium B; (iii) nucleophilic attack of the α-carbon of ketene dithioacetal 47 at cation B to afford the thionium intermediate C; (iv) reductive elimination of PhI from intermediate C to produce intermediate D; and (v) abstraction of the acidic proton of D to form the final products 48.
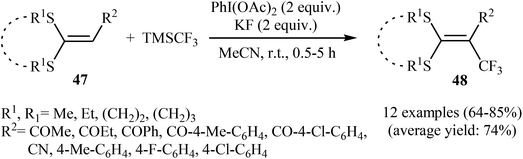 |
| | Scheme 27 Metal-free direct C–H trifluoromethylation of ketene dithioacetals 47 with TMSCF3. | |
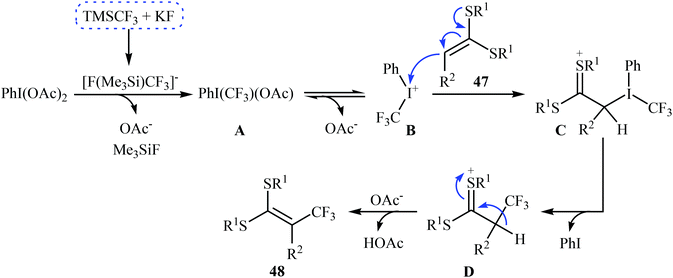 |
| | Scheme 28 The possible radical reaction mechanism for trifluoromethylation of ketene dithioacetals 47. | |
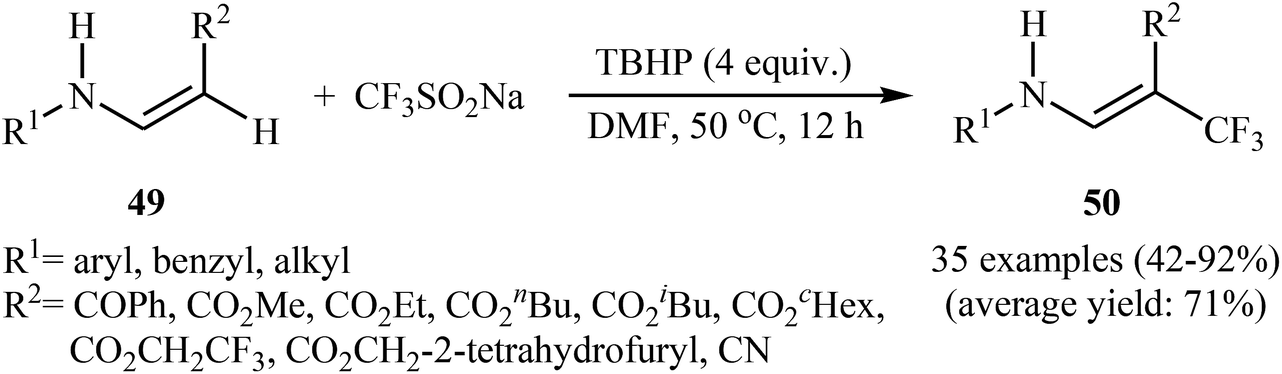
Afterwards, the group of Jiang–Wu reported a mild transition-metal-free synthesis of a wide range of β-trifluoromethyl substituted enamines 50 by treatment of activated enamines 49 with Langlois reagent in the presence of tert-butyl hydroperoxide (TBHP) as an oxidant and initiator in DMF (Scheme 29).42 Moderate to excellent yields were obtained and only (E)-isomers were observed. Noteworthy, the reaction is insensitive to air and water.
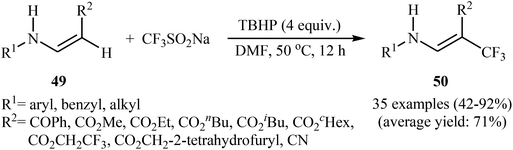 |
| | Scheme 29 Synthesis of β-trifluoromethyl substituted enamines 50 by trifluoromethylation of corresponding enamines 49 with Langlois reagent. | |
4. Conclusion
Needless to say that the introduction of the trifluoromethyl group (–CF3) into organic molecules usually enhances their chemical stability, bioavailability and lipophilicity. Consequently, considerable effort has been directed towards the development of practical and efficient methods for the synthesis of CF3-substituted compounds. Recently, direct trifluoromethylation of organic compounds via C–H bond activation has drawn significant attention from synthetic chemists because of its high atom, step, and pot economy. Along this line, a variety of catalytic system has been developed to promote the direct trifluoromethylation of olefinic C–H bonds using various trifluoromethylthiolating reagents. As illustrated, trifluoromethylation of both terminal and internal alkynes were successfully achieved through this new page of CF3-substituted organic compounds synthesis. Interestingly, almost all of the trifluoromethylation reactions covered in this review showed a remarkable degree of regio- and stereospecificity. We hope that this review will contribute to stimulate further thinking and research in this important and interesting research field.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed;
(b) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS;
(c) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC.
-
(a) V. V. Grushin, Acc. Chem. Res., 2009, 43, 160–171 CrossRef PubMed;
(b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS.
-
(a) C. Zhang, Adv. Synth. Catal., 2014, 356, 2895–2906 CrossRef CAS;
(b) S.-M. Wang, J.-B. Han, C.-P. Zhang, H.-L. Qin and J.-C. Xiao, Tetrahedron, 2015, 71, 7949–7976 CrossRef CAS;
(c) B. Lantano, M. R. Torviso, S. M. Bonesi, S. Barata-Vallejo and A. Postigo, Coord. Chem. Rev., 2015, 285, 76–108 CrossRef CAS;
(d) H. Guyon, H. Chachignon and D. Cahard, Beilstein J. Org. Chem., 2017, 13, 2764–2799 CrossRef CAS PubMed.
- A. Hafner and S. Braese, Adv. Synth. Catal., 2011, 353, 3044–3048 CrossRef CAS.
- S. Abdolmohammadi, M. Mohammadnejad and F. Shafaei, Zeitschrift für Naturforschung B, 2013, 68, 362–366 CAS.
- S. Abdolmohammadi, M. Afsharpour and S. Keshavarz-Fatideh, South African Journal Of Chemistry-Suid-Afrikaanse Tydskrif Vir Chemie, 2014, 67, 203–210 Search PubMed.
- S. Abdolmohammadi and Z. Aghaei-Meybodi, Comb. Chem. High Throughput Screening, 2015, 18, 911–916 CrossRef CAS PubMed.
- S. Abdolmohammadi, R. Ghiasi and S. Ahmadzadeh-Vatani, Zeitschrift für Naturforschung B, 2016, 71, 777–782 CAS.
- S. Khalilian, S. Abdolmohammadi and F. Nematolahi, Lett. Org. Chem., 2017, 14, 361–367 CrossRef CAS.
- Y. Li, L. Wu, H. Neumann and M. Beller, Chem. Commun., 2013, 49, 2628–2630 RSC.
- A. T. Parsons, T. D. Senecal and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 2947–2950 CrossRef CAS PubMed.
- J. Xu, D.-F. Luo, B. Xiao, Z.-J. Liu, T.-J. Gong, Y. Fu and L. Liu, Chem. Commun., 2011, 47, 4300–4302 RSC.
- A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS.
- S. P. Midya, J. Rana, T. Abraham, B. Aswin and E. Balaraman, Chem. Commun., 2017, 53, 6760–6763 RSC.
-
(a) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef;
(b) A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 565–585 CrossRef CAS;
(c) A. Hosseinian, S. Arshadi, S. Sarhandi, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 289–311 CrossRef CAS;
(d) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(e) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(f) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC.
-
(a) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(b) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(c) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(d) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(e) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(f) A. Monfared, R. Mohammadi, S. Ahmadi, M. Nikpassand and A. Hosseinian, RSC Adv., 2019, 9, 3185–3202 RSC;
(g) S. Arshadi, S. Ebrahimiasl, A. Hosseinian, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 8964–8976 RSC;
(h) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed.
- H. Egami, R. Shimizu and M. Sodeoka, Tetrahedron Lett., 2012, 53, 5503–5506 CrossRef CAS.
- C. Feng and T.-P. Loh, Chem. Sci., 2012, 3, 3458–3462 RSC.
- C. Feng and T. P. Loh, Angew. Chem., Int. Ed., 2013, 52, 12414–12417 CrossRef CAS PubMed.
- X.-P. Wang, J.-H. Lin, C.-P. Zhang, J.-C. Xiao and X. Zheng, Beilstein J. Org. Chem., 2013, 9, 2635–2640 CrossRef PubMed.
- L.-H. Wu, K. Zhao, Z.-L. Shen and T.-P. Loh, Org. Chem. Front., 2017, 4, 1872–1875 RSC.
- N. Iqbal, S. Choi, E. Kim and E. J. Cho, J. Org. Chem., 2012, 77, 11383–11387 CrossRef CAS PubMed.
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Beilstein J. Org. Chem., 2014, 10, 1099–1106 CrossRef PubMed.
- Q.-Y. Lin, X.-H. Xu and F.-L. Qing, J. Org. Chem., 2014, 79, 10434–10446 CrossRef CAS PubMed.
- N. J. Straathof, S. E. Cramer, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2016, 55, 15549–15553 CrossRef CAS PubMed.
- C. J.-M. Frédéric, J. Cornil, M. Vandamme, L. Dumitrescu, A. Tikad, R. Robiette and S. P. Vincent, Org. Lett., 2018, 20, 6769–6773 CrossRef PubMed.
- L. Li, J.-Y. Guo, X.-G. Liu, S. Chen, Y. Wang, B. Tan and X.-Y. Liu, Org. Lett., 2014, 16, 6032–6035 CrossRef CAS PubMed.
- H. Wang, Y. Cheng and S. Yu, Sci. China: Chem., 2016, 59, 195–198 CrossRef CAS.
- X. Yang and G. C. Tsui, Org. Lett., 2019, 21, 1521–1525 CrossRef CAS PubMed.
- N. O. Ilchenko, P. G. Janson and K. J. Szabó, Chem. Commun., 2013, 49, 6614–6616 RSC.
- X. Wang, Y. Ye, G. Ji, Y. Xu, S. Zhang, J. Feng, Y. Zhang and J. Wang, Org. Lett., 2013, 15, 3730–3733 CrossRef CAS PubMed.
- R. Rey-Rodriguez, P. Retailleau, P. Bonnet and I. Gillaizeau, Chem.–Eur. J., 2015, 21, 3572–3575 CrossRef CAS PubMed.
- B. Wang, D.-C. Xiong and X.-S. Ye, Org. Lett., 2015, 17, 5698–5701 CrossRef CAS PubMed.
- Y. Y. Yu, A. R. Ranade and G. I. Georg, Adv. Synth. Catal., 2014, 356, 3510–3518 CrossRef CAS.
- Z. Mao, F. Huang, H. Yu, J. Chen, Z. Yu and Z. Xu, Chem.–Eur. J., 2014, 20, 3439–3445 CrossRef CAS PubMed.
- Z. Fang, Y. Ning, P. Mi, P. Liao and X. Bi, Org. Lett., 2014, 16, 1522–1525 CrossRef CAS PubMed.
- A. Prieto, E. Jeamet, N. Monteiro, D. Bouyssi and O. Baudoin, Org. Lett., 2014, 16, 4770–4773 CrossRef CAS PubMed.
- S.-Z. Sun, H. Xu and H.-X. Dai, Chin. Chem. Lett., 2019, 30, 969–972 CrossRef CAS.
- C. Xu, J. Liu, W. Ming, Y. Liu, J. Liu, M. Wang and Q. Liu, Chem.–Eur. J., 2013, 19, 9104–9109 CrossRef CAS PubMed.
- H. Jiang, W. Huang, Y. Yu, S. Yi, J. Li and W. Wu, Chem. Commun., 2017, 53, 7473–7476 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d
d