
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic probes for super-resolution microscopy
Eszter
Kozma
* and
Péter
Kele
 *
*
Chemical Biology Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok krt. 2, 1117 Budapest, Hungary. E-mail: kele.peter@ttk.mta.hu; kozma.eszter.erika@ttk.mta.hu
First published on 4th December 2018
Abstract
Fluorogenic probes efficiently reduce non-specific background signals, which often results in highly improved signal-to-noise ratios. Although this implies improved resolution, fluorogenic probes in the context of super-resolution microscopy are somewhat overlooked. Several excellent reviews summarize recent developments in SRM techniques, labeling techniques or different aspects of small synthetic fluorophores, however there is no comprehensive review on fluorogenic probes suitable for super-resolution microscopy. Herein we wish to fill this gap by providing the readers with an up-to-date summary of fluorogenic probes applied to super-resolution imaging of cellular structures.
Introduction
Recent developments in super-resolution microscopy (SRM) techniques have transformed life sciences in the past decade (for detailed explanation of the respective super-resolution techniques the readers are directed to review papers compiled in ref. 1). Breaking Abbé's law enabled non-invasive visualization of cells at unprecedented resolutions giving us new insights into subcellular structures and the dynamics of biomolecular events. Due to the recent hardware developments, it is the unavailability of fluorescent markers suitable for site-specific intracellular labeling schemes that can currently be considered as the biggest limitation in SRM techniques.1b During the past few decades, several labeling techniques have emerged.2,3 Fusion of engineered fluorescent proteins (FP), for example, is routinely accomplished, however their relatively large size (2–5 nm) often impairs precise localization of the protein of interest (POI) in SRM, not to mention their possible interference with the function of the POI.1b,3a Synthetically tailored organic fluorophores have a much smaller size than FPs, allow greater flexibility in terms of labelling sites and often provide much better photophysical properties such as a wider spectral range, greater photostability, and, in many cases, higher brightness. Not to mention that some FPs have the tendency to polymerize.1b,2c,4 Furthermore, the use of FPs and other fusion tags is limited to the (mostly terminal) labeling of proteins only.3a Synthetic fluorophores in contrast, can be applied to virtually any biomolecular structures (e.g. by means of bioorthogonal chemistry).3b,c,4a Techniques using small organic fluorophores covering the whole range of the visible spectrum in terms of excitation and emission colors represent the state-of-the-art methods for fluorescence labeling in SRM.4a Besides suitable brightness and photostability, synthetic probes should also address problems such as auto- and background fluorescence in order to achieve further improvements in resolution.4b,c With careful selection of fluorescent cores the first three criteria are easily met. Suppression of the background fluorescence of non-specifically adsorbed dyes, however, often requires extensive washing cycles. An alternative approach to overcome this problem uses so-called fluorogenic (or sometimes also referred to as smart) probes.5a,b Fluorescent cores can be made fluorogenic by several means but they share one thing in common: they exist in a quenched, non- or weakly emissive form, which is transformed into a fluorescent species at a given wavelength solely upon specific conjugation/binding to target structures. The extent of fluorogenicity, i.e. the ratio of the fluorescence of the bound and unbound forms, can reach even four orders of magnitudes.5c Although the larger the increase the higher the contrast and the less it is required to wash off unreacted probes, experience suggests that even one order of magnitude difference is enough to tell specifically reacted and freely diffusing or non-specifically bound probes apart.5d,e Irreversibly switched-on fluorogenic probes offer low-background, thus high-contrast images. Reversibly switchable fluorogenic probes can be used to induce random fluorescent events (better known as stochastic blinking), which can be used in single-molecule-localization microscopy (SMLM) based super-resolution technologies, such as stochastic optical reconstruction microscopy (STORM) (for detailed discussion of the technique see ref. 2c and 5f). Yet, relatively few examples are found in the literature that demonstrate the robustness of fluorogenic probes in the context of super-resolution imaging.Although the set of small-molecule fluorogenic probes is much larger, herein we restrict our discussion to fluorogenic systems that found applications in super-resolution imaging of biological structures. The arbitrary classification that we follow distinguishes structural change induced, (photo)chemically modulated systems and energy transfer based fluorogenicity.
Structural fluorogenicity
This category covers the examples of fluorophores that undergo certain structural changes upon specific conjugation/binding events resulting in an increase of fluorescence intensity. Herein we list instances for probes with fluorogenic behavior due to tautomeric changes, reduction of rotational freedom or planarization of out-of-plane twisted frames, in the context of SRM imaging of cellular structures.2′-Carboxy-rhodamines and congeners
Xanthene dyes possess several favorable (photo)physical characteristics with respect to bioimaging applications. These include high water solubility, fluorescence quantum yields and molar extinction coefficients. Consequently, xanthene probes are widely utilized as fluorescent markers in protein labeling, ion sensing, etc.6 The emission maxima of rhodamines can be chemically tailored on the one hand by varying the substituents around the framework. On the other hand, the excitation and emission maxima are also efficiently modulated, i.e. shifted towards the red end of the spectrum by incorporating group-14 (X = C, Si, Ge, Sn) elements within the rhodamine frame.7 The reason is the low LUMO energy of these fluorophores which is explained by conjugation between the σ* orbitals of exocyclic X–R bonds and the π*-system of the X-containing tricyclic system. The extent of the red shift is in the order of Si > Ge > Sn > C (although Sn-rhodamines are unstable), due to the decreasing efficiency of the σ*–π* overlap with the increasing atomic radius of X and C–X bond length. Regardless of their elemental composition, rhodamines carrying a carboxylic function at their 2′ position have the unique feature of existing in two forms due to valence tautomerism, depending on the polarity of the microenvironment (Fig. 1).8 In less polar media they are mostly present in a non-fluorescent, spirolactone form, while in polar environments a fluorescent, zwitterionic form is the major contributor. The polarity or pH dependency of the equilibria between the two forms is readily fine-tuned by electron withdrawing/donating substituents. In general, group-14 element modified carboxyl-rhodamine derivatives are more prone to spirocyclization, which makes them excellent polarity-sensitive fluorogenic probes. Such polarity-driven fluorogenicity of rhodamines can be harvested in fluorescence imaging as non-specific adsorption of the probes onto apolar surfaces forces the molecules to remain dark.9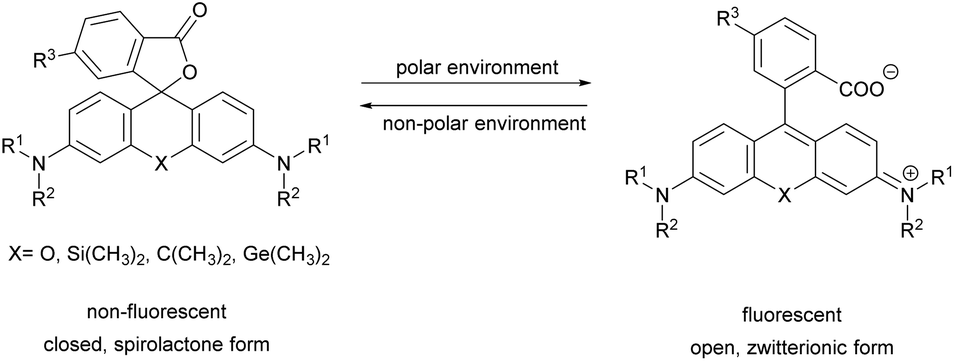 | ||
| Fig. 1 General structure of 2′-carboxy-rhodamines and their spirolactonization based on the environment polarity. | ||
The first report on membrane-permeable siliconrhodamine (carboxy-SiR, 1) derivatives was published by Johnsson et al. (Scheme 1).10 The dye has excitation and emission maxima in the far red region (λabs max = 645 nm; λem max = 661 nm) with a spirocyclization D0.5 value of 65. The D0.5 value allows the estimation of the sensitivity of the dye to polarity and is equal to the dielectric constant, where the dye has half of the maximum absorbance. In physiologically relevant environments low D0.5 values suggest non-fluorogenic dyes, while high values of D0.5 (e.g. 60–70) indicate fluorogenic behavior. They discovered superior performance of SiR-SNAP (2), SiR-CLIP (3) and SiR-Halo (4) derivatives in different types of super-resolution microscopy and performed live cell direct stochastic optical reconstruction microscopy, (dSTORM) with a 640 nm laser power of ∼1 kW cm−2. Labeling of H2B histone proteins with SiR-SNAP (2) allowed the localization of ∼140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules in 10000 frames, at a time rate of 50 Hz, yielding a super-resolved image in just over three minutes (for an overview of dSTORM technology and key parameters of the acquisition, see ref. 2d). SiR-SNAP performed well in live cell STED imaging of centrosomal protein Cep41 as well. STED microscopy revealed that SNAP-Cep41 forms a ring-like structure along the axis of the centriole, a structural feature previously hidden in confocal microscopy (Fig. 2a). Further derivatives of SiR-carboxyl were made for visualization of microtubules (SiR-docetaxel derivative-SiR-tubulin, 5) and actin (SiR-desbromo-desmethyl-jasplakinolide derivative-SiR-actin, 6) in human fibroblasts and also in primary rat hippocampal neurons using live-cell structured illumination microscopy (SIM) and stimulated emission depletion (STED) super-resolution techniques.11 Importantly, the authors proved that a higher resolution (39 ± 10 nm) can be achieved with SiR-tubulin (5) compared to SiR-SNAP (2) (77 ± 3 nm) in microtubule imaging, which underlines the importance of using small molecule probes that directly target the structure of interest since it enhances the resolution substantially. Mishin et al. used SiR-actin (6) to visualize endogenous actin within tissues with dSTORM and revealed that the normal tissue actin pattern differs substantially in comparison to cancer cells.12 SiR-Hoechst (7) was applied to nucleus staining enabling non-toxic, SRM-compatible visualization of nuclei in various cell types. This derivative showed a 50-fold increase in fluorescence upon DNA binding.13 A bioorthogonally applicable SiR-tetrazine (8) was also developed and applied in STORM imaging of cytoskeletal proteins (actin, vimentin) previously modified genetically with a bicyclononyne-derivatized non-canonical amino acid allowing 92 nm lateral resolution.14 Just very recently, SiR-Halo (4) was used in studying the distribution of abundant scaffolding protein, PSD95, at the postsynaptic membrane of excitatory synapses in living mice with STED microscopy at ≤25 μm depth revealing the previously unseen morphologies of PSD95 distribution.15 Modifications of the original SiR-core provides further derivatives with superb photophysical properties. Carboxy-SiR-700 (9) has excitation and emission maxima at 690 nm and 715 nm, respectively. This probe allows live cell dual-colour STED microscopy imaging in combination with carboxy-SiR (1) (Fig. 2b).16 Fluorination of the SiR core induces a further red-shift of both excitation and emission wavelengths (∼15–20 nm).17 However, fluorine substitution drastically increases lipophilicity, which in turn impairs water solubility. As a consequence, incorporation of hydrophilic moieties, such as a sulfonic acid, was necessary to compensate for this effect of fluorines. Furthermore, incorporation of fluorine atoms can also significantly reduce quantum yields and fluorescent lifetimes.18
000 molecules in 10000 frames, at a time rate of 50 Hz, yielding a super-resolved image in just over three minutes (for an overview of dSTORM technology and key parameters of the acquisition, see ref. 2d). SiR-SNAP performed well in live cell STED imaging of centrosomal protein Cep41 as well. STED microscopy revealed that SNAP-Cep41 forms a ring-like structure along the axis of the centriole, a structural feature previously hidden in confocal microscopy (Fig. 2a). Further derivatives of SiR-carboxyl were made for visualization of microtubules (SiR-docetaxel derivative-SiR-tubulin, 5) and actin (SiR-desbromo-desmethyl-jasplakinolide derivative-SiR-actin, 6) in human fibroblasts and also in primary rat hippocampal neurons using live-cell structured illumination microscopy (SIM) and stimulated emission depletion (STED) super-resolution techniques.11 Importantly, the authors proved that a higher resolution (39 ± 10 nm) can be achieved with SiR-tubulin (5) compared to SiR-SNAP (2) (77 ± 3 nm) in microtubule imaging, which underlines the importance of using small molecule probes that directly target the structure of interest since it enhances the resolution substantially. Mishin et al. used SiR-actin (6) to visualize endogenous actin within tissues with dSTORM and revealed that the normal tissue actin pattern differs substantially in comparison to cancer cells.12 SiR-Hoechst (7) was applied to nucleus staining enabling non-toxic, SRM-compatible visualization of nuclei in various cell types. This derivative showed a 50-fold increase in fluorescence upon DNA binding.13 A bioorthogonally applicable SiR-tetrazine (8) was also developed and applied in STORM imaging of cytoskeletal proteins (actin, vimentin) previously modified genetically with a bicyclononyne-derivatized non-canonical amino acid allowing 92 nm lateral resolution.14 Just very recently, SiR-Halo (4) was used in studying the distribution of abundant scaffolding protein, PSD95, at the postsynaptic membrane of excitatory synapses in living mice with STED microscopy at ≤25 μm depth revealing the previously unseen morphologies of PSD95 distribution.15 Modifications of the original SiR-core provides further derivatives with superb photophysical properties. Carboxy-SiR-700 (9) has excitation and emission maxima at 690 nm and 715 nm, respectively. This probe allows live cell dual-colour STED microscopy imaging in combination with carboxy-SiR (1) (Fig. 2b).16 Fluorination of the SiR core induces a further red-shift of both excitation and emission wavelengths (∼15–20 nm).17 However, fluorine substitution drastically increases lipophilicity, which in turn impairs water solubility. As a consequence, incorporation of hydrophilic moieties, such as a sulfonic acid, was necessary to compensate for this effect of fluorines. Furthermore, incorporation of fluorine atoms can also significantly reduce quantum yields and fluorescent lifetimes.18
 | ||
| Scheme 1 Structure of carboxy-rhodamine derivatives mentioned in the text. | ||
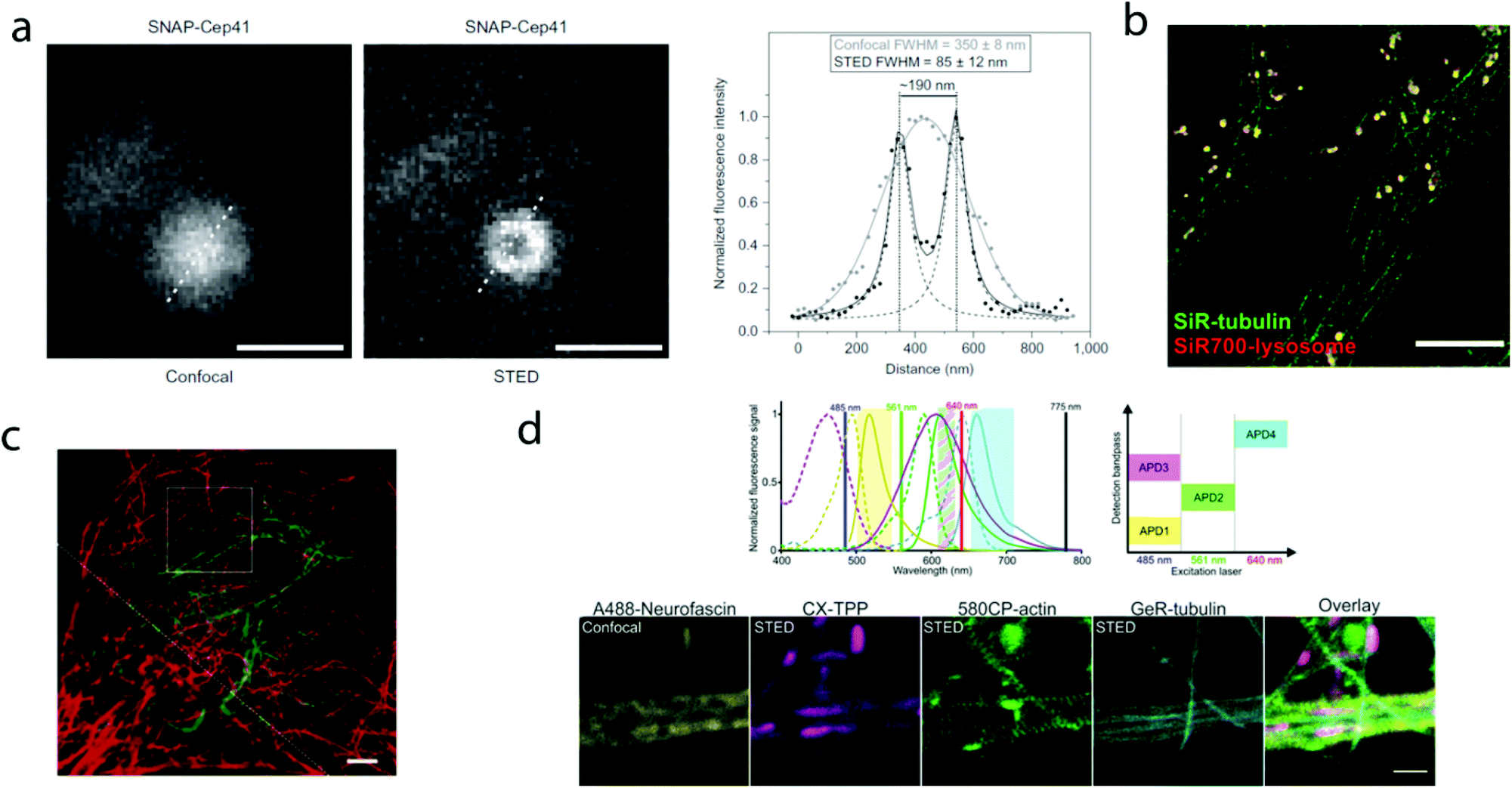 | ||
| Fig. 2 Superresolution microscopy images with SiR and other carboxy-rhodamine derivatives. (a) Comparison of confocal and STED images of SiR-SNAP-Cep41 localized at the centrosome with an intensity line profile (right) along the white dotted line showing two separated Lorentz-distribution on the STED image (reprinted partly from ref. 10 with permission. Copyright 2013 Springer Nature). (b) Two-color STED nanoscopy of human primary fibroblasts stained with SiR(1)-tubulin (green) and SiR-700(9)-lysosome (red). Scale bar: 5 μm (adapted from ref. 16 with permission. Copyright 2016 American Chemical Society). (c) Two-color STED image of vimentin (green) and tubulin (red) in a living HeLa cell. Vimentin was labelled with rhodamine 580R (13) applied as a Halo-tag probe, tubulin was directly labelled with SiR(1)-tubulin. Scale bar: 2 μm. Source: ref. 20. (d) Four-colour microscopy with CX-TPP (22). Upper panel: normalized absorption and emission of Alexa Fluor 488 (yellow), CX (22) (magenta), 580CP (15) (green) and GeR (16) (cyan), related laser lines and detection windows. Lower panel: living rat hippocampal neurons stained with 580CP-actin, GeR-tubulin, CX-TPP (targeting mithochondria) and anti-neurofascin and anti-mouse AlexaFluor 488. Scale bar: 1 μm. (Adapted from ref. 29 with permission. Copyright 2017 American Chemical Society.) | ||
Carborhodamines (also known as carbopyronines, CP) have somewhat blue-shifted spectral and very similar spirocyclization properties to SiRs. The first detailed synthesis of the carborhodamine scaffold was described by Hell et al. in 2010.19 Compound 10 possessed sufficient brightness and signal-to-noise ratio to provide high resolution STED images in immunofluorescence labeling studies of the tubulin network of PtK2 cells. The same authors also reported on a set of fluorescent rhodamine and fluorogenic carbopyronine derivatives and elaborated the relationship between structure and fluorogenicity.20 As they observed, introduction of strongly electron withdrawing substituents, like fluorine or N-trifluoroethyl groups, onto the carbopyrone core favored lactone ring formation (e.g. 630CP, 11). The authors also stated that replacement of the carboxyphenyl moiety with a carboxythiophene in siliconrhodamines (SiR-thiophene, 12) leads to the loss of polarity-sensitive fluorogenicity as lactonization into the colorless form becomes less favorable due to the increased angle strain. Representatives of their compound set (i.e. 580R, 13 and carboxy-SiR, 1) allowed two-color STED imaging as both probes could be depleted with a single 775 nm STED laser (Fig. 2c).
The loss of charges upon spirolactonization often induces aggregation of the probes, which further decreases quantum yields (QY), probably due to homo-FRET processes. Careful examination of taxane conjugated 510R – 14, 580CP – 15, GeR – 16, and SiR – 1 probes revealed several key issues.21 As the authors speculated, aggregation could facilitate membrane trafficking via endocytosis pathways since a direct correlation was found between the aggregated dye fraction and the cytotoxicity of the conjugated taxane derivatives. Cabazitaxel conjugates of 510R (14) and SiR (1) were used to stain microtubule filaments of living human fibroblasts. As a result of high-contrast images obtained with the fluorogenic probes, an apparent diameter of 35 ± 13 nm was obtained corresponding to 29 ± 11 nm optical resolution after taking into account the real microtubule diameter of ∼20 nm in STED microscopy.
The Hell group has extended two-channel imaging to an 810 nm STED setup. In their account a carbopyronine derivative (610CP, 17) and a siliconrhodamine (680SiR, 18) ensured minimal crosstalk and clear color separation. At the same time both probes were found to be efficiently depleted at 810 nm. The two dyes were used for near-IR live cell imaging of vimentin and tubulin filaments labeled with 610 CP and 680SiR, respectively using two distinct excitation lasers (i.e. 600/20 nm for 610 CP and 676/10 nm for 680 SiR) and one 810 nm de-excitation (STED) laser enabling apparent optical resolution values ca. 100 and 70 nm, respectively.22
When a biologically applicable fluorescent or fluorogenic probe comes to intracellular live-cell applications, membrane permeability is always an issue. Although, as just discussed above, sometimes aggregation may facilitate cross-membrane trafficking, it may also impair the efficiency of labeling. Rhodamines conjugated to covalent (e.g. SNAP-, Halo-Tag) or non-covalent recognition units (e.g. taxanes) efficiently stain intact cell structures, however their affinity and specificity largely depend on the recognition unit, the probe or the linker applied for conjugation. To address some of these limitations, Butkevich and Hell introduced hydroxylated rhodamines, together with group-14 derivatives (e.g. 530RH, 19). While hydroxylation of the fluorescent cores made these probes hydrophilic without compromising their cell permeability the photophysical properties remained mostly unaffected. These probes were indeed cell- and nucleus-permeant that enabled one and two-color STED imaging in live cells with high contrast. However, the authors found that all probes possessed lower Halo-tag binding affinity, and thus required increased dye loadings. Consequently, the study suggests that moderate or high values of D0.5 are required but not sufficient to obtain desirable fluorogenic probes.23
Single molecule localization microscopy, SMLM, generally requires intense laser irradiation (∼1 kW cm−2) and additives such as thiols (10–100 mM) in the form of so-called blinking buffers to induce on–off switching of the fluorophore, limiting live-cell applications of these techniques. Spontaneously blinking fluorophores that are able to switch on/off stochastically without high power laser irradiation or any additives allow super-resolved localization microscopy of living cells.24,25 Urano and co-workers developed siliconrhodamines with an appending internal nucleophile at the 2′-position, e.g. HMSiR (20). Similar to 2′-carboxyrhodamines, such structures tend to form a non-fluorescent spirocyclic form, which is in thermal equilibrium with its fluorescent open form in the ground state (Fig. 3).26 For SMLM applications it is required that at physiological pH only a small subset of probes exist in the fluorescent form and that this state should last for several camera frames in order to enable the detection of sufficiently enough photons for accurate localization. These two requirements are dependent on the intramolecular spirocyclization equilibrium constant (pKcycl) and the lifetime of the open form (τ). They found that pKcycl is strongly dependent on the electrophilicity of the fluorophore core and the nucleophilicity of the pendant group. Following the evaluation of these two properties for several fluorophore–pendant nucleophile combinations, a SiR-derivative, HMSiR (20), with an appending 2′-hydroxymethyl group (–CH2OH), was found to be ideal with a pKcycl of 5.8 and a τ of 0.245 s. Halo and SNAP substrate derivatives of HMSiR (20) showed spontaneous blinking and enabled live-cell time-lapse SMLM of microtubules in Vero-2 cells in sodium phosphate buffer (pH 7.4) with laser irradiation at 647 nm with an intensity of 40 W cm−2. In multichannel SMLM imaging, using conventional probes with redox-active blinking buffers, the composition of the imaging buffer has to be carefully optimized to each fluorophore. Thus it is particularly difficult to find optimal conditions that induce blinking of multiple fluorophores to visualize two different targets at the same time. Spontaneously blinking fluorophores require only regular buffers without any additives therefore suitable candidates for multi-color SMLM imaging. Urano et al. developed further probes based on the rhodamine scaffold. A screening process involving differently substituted rhodamines in combination with 2′-hydroxyalkyl chains of varying length resulted in the identification of a green-light emitting probe, HEtetTFER (21). HMSiR (20) in combination with HETetFER (21) allowed dual-color SMLM of mitochondria and the microtubular network in PBS solution without the use of any redox-buffer or photomodulation.27
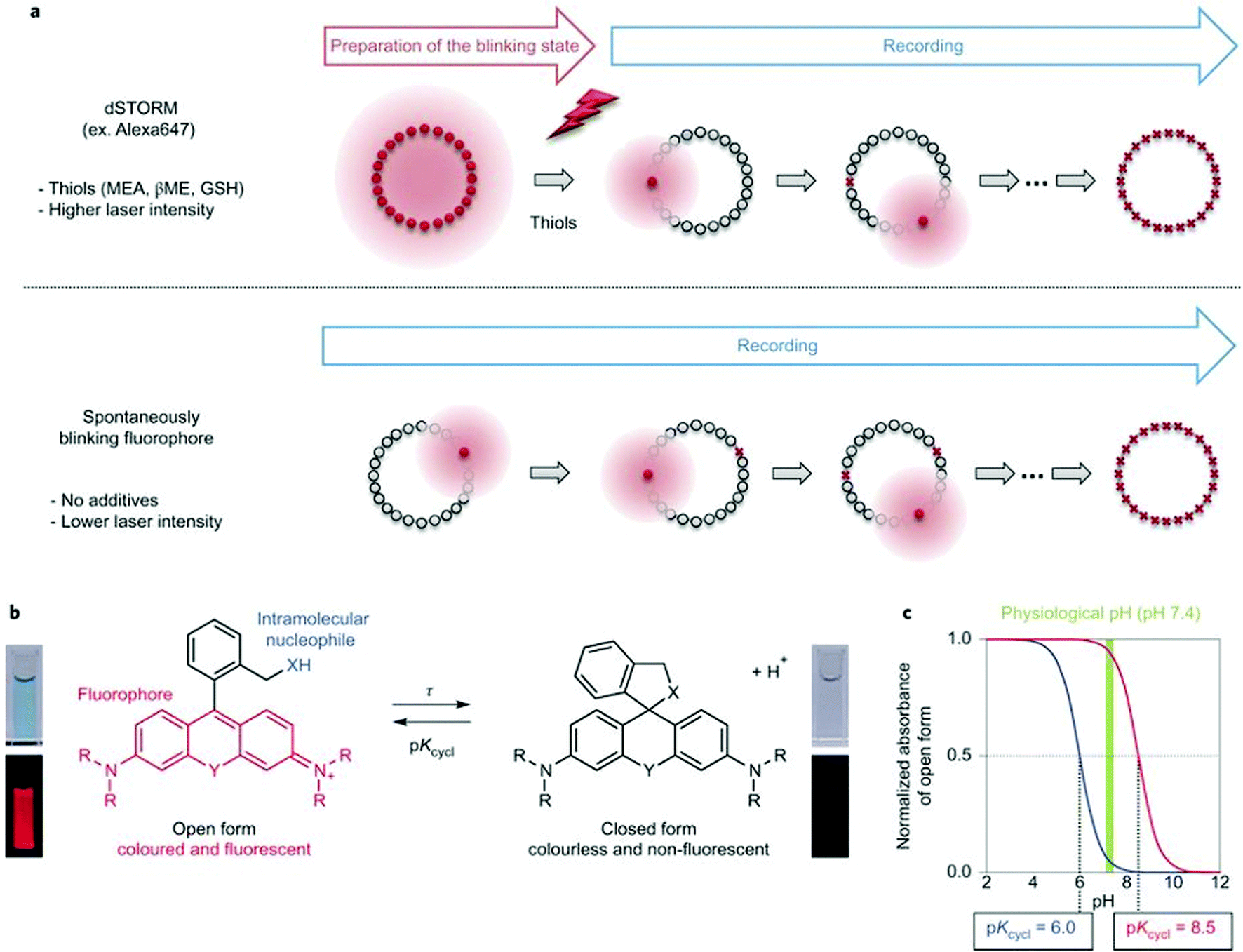 | ||
| Fig. 3 (a) Single molecule localization sequence using a conventional fluorophore and the spontaneously blinking fluorophore. (b) Thermal equilibrium of intramolecular spirocyclization between the fluorescent open form and the non-fluorescent closed form. pKcycl is the equilibrium constant for intramolecular spirocyclization and τ is the lifetime of the open form (the duration until the open form reverts to the closed form). (c) Schematic pH titration curves of rhodamine derivatives with a pKcycl of 6.0 (blue) and 8.5 (red). For SMLM, most of the individual fluorophores should be in the closed form at the physiological pH of 7.4 to avoid overlapping signals. (Reprinted from ref. 26 with permission. Copyright 2014 Springer Nature.) | ||
In a comprehensive review paper Hell et al. highlighted the importance of large Stokes-shift dyes in multi-color STED microscopy.28 With the use of such large Stokes-shift dyes multichannel imaging becomes possible without the need for sophisticated techniques such as hyperspectral detection or fluorescence lifetime recording. In a separate account, the same group realized such multi-channel imaging using 9-iminoanthrone dyes possessing Stokes shifts between 140 and 165 nm. These probes were not fluorescent in apolar media but showed protonation-based fluorogenic behavior in polar-protic environments (CX-TPP, 22) (Fig. 4).29
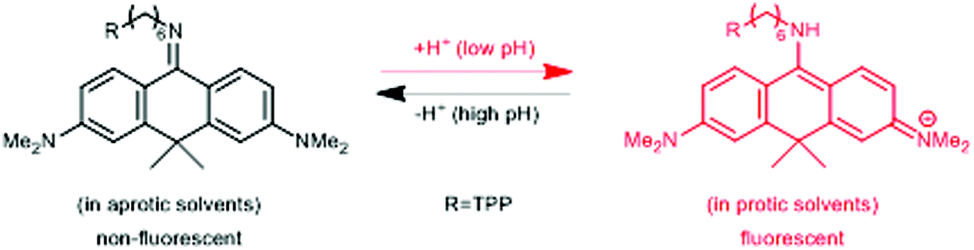 | ||
| Fig. 4 Protonation–deprotonation mechanism responsible for the environment sensitivity of CX-TPP (22). | ||
Such a set of cell-permeant dyes enabled two-, three- and even four-color imaging (3× STED, 1× confocal) of cytoskeletal frameworks (GeR-tubulin – 16, 580CP-actin – 15) and mitochondria (CX-TPP, 22, TPP: triphenylphosphonium) in human fibroblasts or living rat hippocampal neurons with a single 775 nm STED laser (Fig. 2d).
Conformationally lockable fluorogenic probes
The probe–target interaction induced conformational confinement of freely rotating or out-of-plane twisted structures often results in a dramatic increase of fluorescence due to forced planarization or rigidificiation of the probe. Formation of conformationally locked, fluorescent forms can be the result of covalent interactions or non-covalent binding events. Such conformational dynamics related fluorescent changes are realized e.g. in covalent binding of biarsenical probes to tetracysteine tag fused proteins or upon intercalation of freely rotating dyes between DNA-bases or binding to DNA grooves (Scheme 2). The same phenomenon can be tracked down during non-covalent binding of quenched systems to engineered fluorogen activating proteins.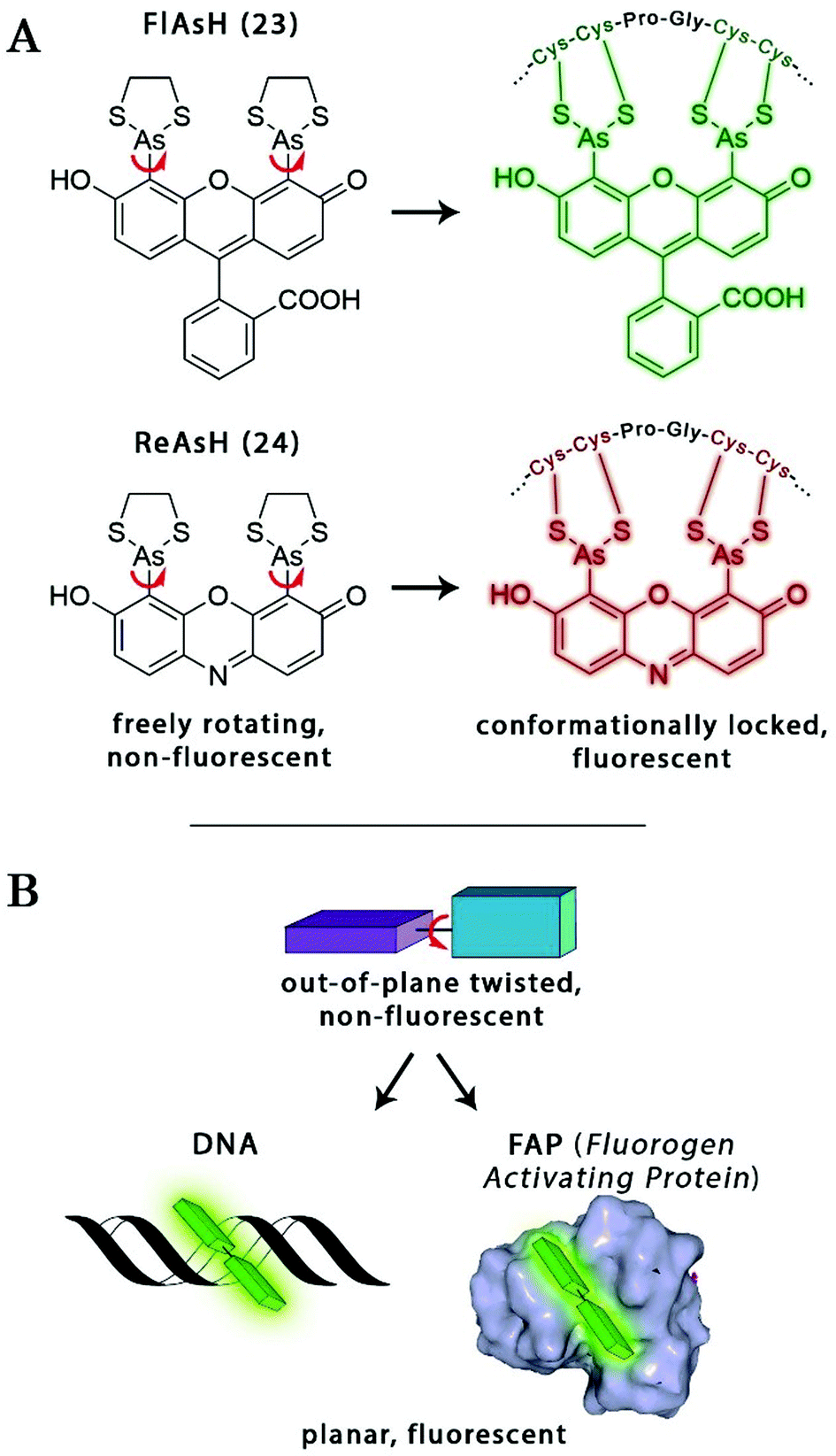 | ||
| Scheme 2 Schematic examples of conformational change driven fluorogenicity. | ||
Biarsenical helix binders for protein imaging
Amongst protein fusion tags, tetracysteine (Cys4) motifs represent the smallest ones (4–15 amino acids) that enable selective labeling of the protein of interest (POI). The non-natural arrangement of these natural functions allows highly specific interactions between target proteins and biarsenical probes.30 The bis-1,2-ethanedithiol (EDT) derivative of the fluorescein arsenical helix binder (Sheme 2, FlAsH-EDT, 23) is weakly fluorescent due to free rotation around the C–As bond (Scheme 2). However, when EDT is replaced by the Cys4 tag, the cyclic product becomes intensely fluorescent.31,32 Since fluorescein can undergo photoswitching, it was a promising candidate to be applied to fluorogenic SRM techniques.33 To confirm this, Zimmer et al. demonstrated FlAsH-based photoactivated localization microscopy (dubbed as FlAsH-PALM) with a spatial resolution ∼30 nm.34,35 They studied Human Immunodefficiency Virus, HIV, by inserting the tetracysteine tag into integrase (IN), an enzyme that mediates the integration of viral DNA into the host genome.FlAsH-PALM recovered subdiffraction virion morphology, imaging HIV-1 as free virions and as intracellular complexes in fixed and living cells (Fig. 5). A substantial change in virus size was detected between cytoplasmic and nuclear complexes. Their data highlighted that FlAsH-PALM preserves the biological function of a protein that otherwise would have been disrupted by larger fusion tags (e.g. GFP, SNAP, etc.) or antibody binding, and furthermore revealed many unexplored steps in the replication cycle of HIV in vivo. A red-emitting variant of FlAsH called ReAsH (24) has also been developed with an emission maximum at around 608 nm (Scheme 3).30 Selvin et al. attached single ReAsH-EDT (Scheme 2, 24) to Cys4-fused calmodulin immobilized on a glass surface at low density.36 They determined the center of the distribution of photons, using TIRF microscopy from the image of a single molecule in order to determine the position of the dye. They also found that by increasing photostability, 2-mercaptoethanesulphonic acid increased the number of collected photons from ReAsH molecules by a factor of two. To the best of our knowledge, live cell applications of ReAsH have not yet been presented.
 | ||
| Fig. 5 FlAsH-PALM imaging of HIV-1 integrase (IN) enzyme in fixed cells. (a) Widefield image, (b) super-resolution reconstruction of the same area, (c) histogram of positions projected on (b). (Reprinted from ref. 34 with permission.) | ||
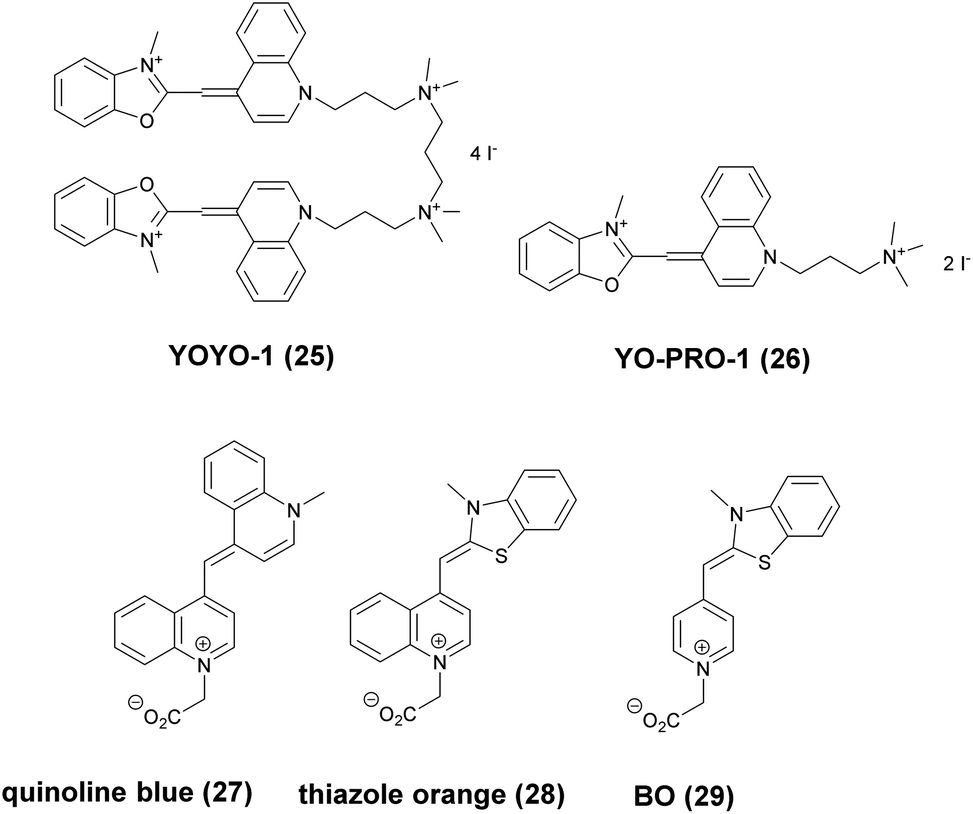 | ||
| Scheme 3 Structure of nucleic acid binding probes. | ||
Non-covalent binder probes for DNA imaging
Minor groove binder or intercalating DNA stains have the key advantage that their fluorescence emission intensity increases typically 100–1000 times upon binding to the DNA.37 Minor groove binding molecules tend to be small and flexible in order to bend and fit into the minor groove, whereas intercalator dyes usually contain out-of-plane twisted heterocyclic systems, which interact with DNA bases. Some cyanines, such as TOTO or YOYO, bind to DNA in a ‘bis-intercalating mode’ meaning that the aromatic part of the cyanine intercalates, whilst the amine ‘arms’ lie in the minor groove.38 DNA binder cyanines adopt an out-of-plane twisted conformation when unbound. The twist makes these dyes non-fluorescent. However, intercalation into DNA induces the confinement of these dyes into a fluorescent planar form.39 YOYO-1 is a DNA-binding cyanine derivative and is the tetracationic homodimer of oxazole yellow. In aqueous solution it is virtually non-fluorescent, however, its emission intensity greatly increases upon bis-intercalation into DNA (3200×).40 In 2009, Flors and co-workers applied YOYO-1 (25), to image λ-DNA topology in switching buffers allowing 38 nm resolution of stained structures using SMLM (Scheme 3).41 The authors suggested that the blinking occurs through an electron transfer reaction of YOYO-1 (25) with the reducing mercaptoethylamine (MEA). YOYO-1 (25) was further applied to single DNA imaging using binding-activated localization microscopy (BALM) in various dye-base pair ratios to analyse the distance between base pairs.42,43 A drawback of YOYO-1 (25) and other dimeric intercalating dyes is that their binding constant to DNA is rather high (K = 108–109 M−1), which influences the DNA topology and its ability to interact with DNA-binding proteins.44Monomeric intercalating dyes with lower affinity towards DNA can address this problem, though their brightness is about half of their dimeric congeners, and their affinity to DNA is about two magnitude lower. In a separate account Flors et al. tested monomeric YO-PRO-1 (26) in DNA staining and achieved 43 nm resolution using SMLM, however, the much lower intensity and staining ability of YO-PRO-1 required higher dye loadings (Scheme 3).45 Given the photophysical properties of YOYO dyes, the deterministic nature of STED microscopy may have an inherent advantage over the stochastic methods in DNA imaging.46 Hell et al. demonstrated that STED can be readily used to identify subtle structures (bends and kinks) along the DNA strand with an average FWHM = 42 nm using YOYO-1 (25) with a 568 nm depletion laser (Fig. 6).46 Although YO-PRO-1 (26) is able to pass the cell membrane under very specific circumstances, bis-intercalating cationic dyes are considered to be impermeant to cell membranes.47 Thus, it is necessary to use membrane permeable alternatives for live cell DNA imaging e.g. SYTO dyes. Like YOYO and its derivatives, SYTO dyes belong to the cyanine family, with the substantial difference of being minor groove binders. Compared to e.g. YOYO they produce dimmer SRM images, therefore in general, they are less efficiently localized in stochastic methods.48 Moreover, SYTO dyes are not DNA-specific and they also bind to RNA, however the staining pattern is dye-dependent.49 Preliminary data suggest that SYTO-13 might be useful for imaging chromatin of fixed mitotic chicken DT40 cells in blinking buffer.50 SYTOX Orange, whose chemical structure is proprietary, is reported to be monomeric and intercalating.51 It has just recently been found to be promising in live cell labeling applications.52 Moerner et al. studied single molecule rotational dynamics using SYTOX Orange and detected the nanoscale deformations of individual λ-DNA strands in vitro.53a By combining image data with thousands of dye molecule orientation measurements, they developed a technique to probe the structure of individual DNA strands. In spite of the promising results, live cell DNA imaging using fluorogenic DNA-binders is still a field full of challenges.
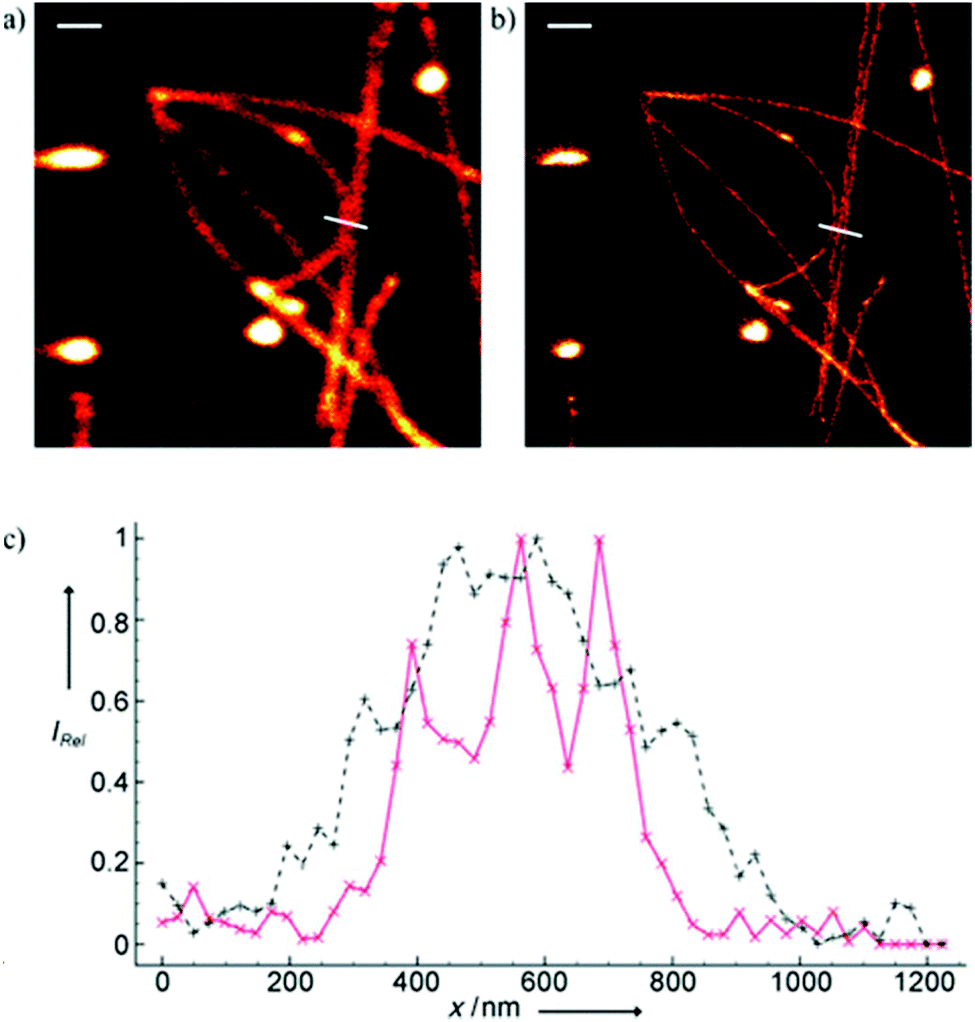 | ||
| Fig. 6 Confocal image of YOYO stained λ-DNA (a) and the corresponding STED image (b). Scale bars: 1 μm. (c) Average of three line profiles from the STED (solid red line) and confocal (dotted black line) images. Line profiles extracted along the white lines in (a) and (b). The three distinct peaks belonging to different DNA molecules are only resolved by STED. (Reprinted partly from ref. 46 with permission. Copyright 2011 John Wiley and Sons.) | ||
Simultaneous imaging of multiple intracellular RNA molecules is also challenging and requires careful optimization of label and probe design. Seitz and co-workers described a set of quencher-free forced intercalation (FIT) probes, in which a single non-symmetric thiazole orange (TO) derived cyanine dye serves as an environment sensitive nucleobase surrogate.53b Hybridization with a complementary target forces the dye to intercalate between predetermined base pairs, resulting in restricted torsions around the methine bridge leading to a prolonged lifetime of the excited state. The authors developed the first red-emitting FIT probe, quinoline blue (27), showing up to 152-fold enhancement in fluorescence upon hybridization with RNA. The combined use of quinoline blue with other FIT probes (e.g. thiazole orange, TO, 28 and benzothiazole orange BO, 29) allowed localization of three different mRNA molecules in developing oocytes of Drosophila melanogaster by means of wash-free in situ hybridization and STED microscopy (Scheme 3).
Fluorogen activating proteins for super-resolution microscopy
A similar approach, involving conformationally lockable, non-covalent binder probes is also applied for super-resolution imaging of proteins in combination with genetically encodable fusion protein tags. Such a group of proteins termed fluorogen activating proteins (FAP) are generated e.g. by screening of yeast surface-displayed libraries for specific binding and efficient activation of a variety of fluorogenic probes.54,55 Binding of the probes results in significant enhancements in the extinction coefficient and fluorescence quantum yields, sometimes in combination with significant absorption band shifting as a result of structural confinements (Fig. 7a).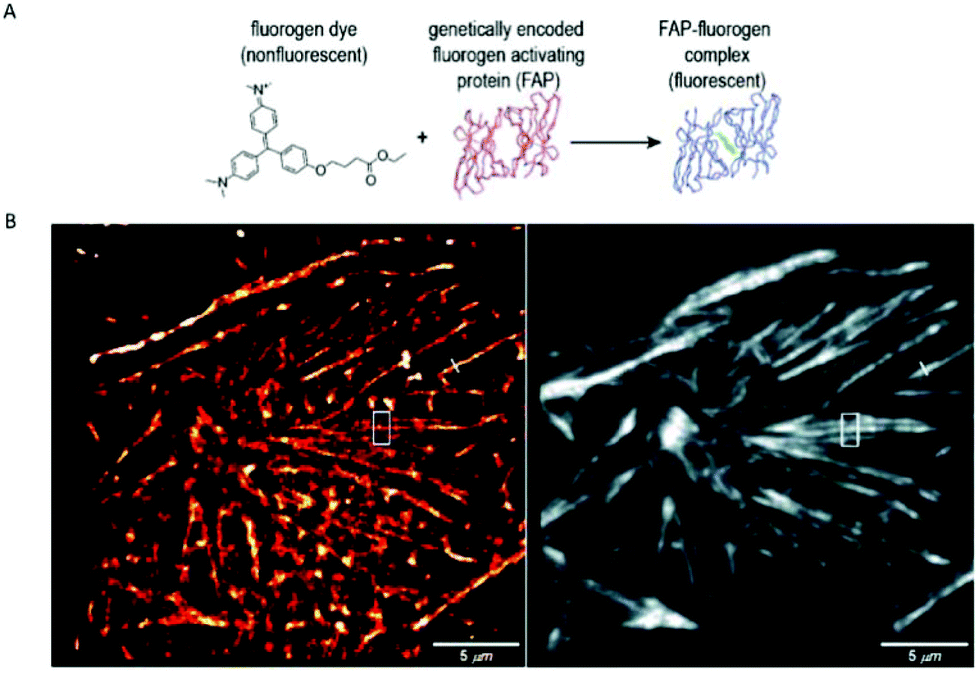 | ||
| Fig. 7 (a) The fluorogen activating protein complex consists of a nonfluorescent dye (fluorogen) and a genetically encoded protein which forms a fluorescent complex upon interaction. (b) Live cell F-BALM imaging of actin in HeLa cells. Left: reconstruction of SMLM of actin, right: wide-field image of phalloidin-Alexa488. (Reprinted partly from ref. 58 with permission. Copyright 2013 John Wiley and Sons.) | ||
These FAPs have been fused to a variety of extra- and intracellular proteins and recently, have been applied in super-resolution microscopy. Bruchez et al. genetically expressed FAP-fused proteins (e.g. yeast cell surface and actin in HeLa cells) that selectively bind a far-red fluorogenic malachite green (30) probe (Scheme 4).56 Free malachite green is essentially non-fluorescent due to rotational relaxation, however an enhanced fluorescence is observed in the presence of cognate FAPs (up to 18000-fold).57 Live-cell STED microscopy revealed a sufficient signal-to-noise ratio resulting in a three-fold resolution enhancement compared to confocal microscopy. More recently, it was reported that with careful control of malachite green (30) concentration a sparse labeling distribution on densely tagged genetically encoded proteins can be achieved.58 Stochastic SRM was enabled with equilibrium binding using concentrations below the dissociation constant (Kd) of the fluorogenic substrate. Spontaneous blinking was produced without the use of any additives or switching buffers. The developed method, dubbed as F-BALM (binding-activated localization microscopy), permitted live cell single molecule imaging of actin in HeLa cells (Fig. 7b).
 | ||
| Scheme 4 Structure of fluorogen activating protein (FAP)-complex ligands used in super-resolution microscopy. | ||
Mishin et al. performed in silico mutagenesis of amino acids within the ligand binding pocket of Blc protein, belonging to the lipocalin family, in order to identify potential protein-chromophore pairs for SMLM.59 Protein complexes (named as Dye in Blc, DIB) capable of efficiently activating M739 (31) were fused to different protein structures (histone H2B, cytokeratin, actin, vimentin) in mammalian cells (Scheme 4). As compared to F-BALM, this complex (in a technique named protein-PAINT) showed increased photostability resulting in enhanced temporal resolution at the single-molecule level. A particular FAP, dubbed as dL5, has been optimized for high binding affinity towards a membrane permeant malachite green (30) derivative with increased photostability and better spectral properties. Moerner and co-workers used this construct in the labeling scheme of fusion proteins in live bacteria C. crescentus and demonstrated long time-scale protein tracking and STED microscopy with a four-fold resolution enhancement compared to diffraction-limited imaging. Furthermore, the authors provided evidence that in comparison with YFP, dL5 fusion induced less perturbation in the structure and function of the intermediate filament protein CreS.60,61 Further attempts to perform SMLM failed since MG-ester activated upon binding to the bacterial cell surface.
Bruchez and co-workers reported on an excitation ratiometric, activatable pH responsive tandem system based on a bichromophoric probe acquired by linking the pH sensitive Cy3 donor to a conformationally activatable malachite green acceptor (TRApHIC) (32) (Scheme 4).62 The complex is activated by its cognate FAP dL5**. In the absence of FAP binding, the malachite green rather acts as a non-fluorescent quencher of the Cy3 fluorescence, however, in the presence of the FAP (fused to target receptor protein) dual emission is generated due to the efficient FRET between the chromophores. Fine changes in the pH due to receptor internalization during endocytosis are sensitively followed by the ratio of the signals. TRAPhIC labeling was found to be compatible with ratiometric STED imaging, enabling tracking of β2 adrenergic G-protein coupled receptors (GPCR) with dual excitation (560 nm and 640 nm) and single depletion at 775 nm.
(Photo)chemically modulated systems
This category consists of fluorogenic systems where the fluorescence of light emitting frames is quenched either by a photochemically cleavable quencher unit or a chemically active function that efficiently modulates the fluorescent signal until it is reacted with a specific target. Furthermore, photochromic systems also belong to this category. Here the photoirradiation does not induce cleavage of quenchers but rather affects the equilibrium between dark and emissive structural isomers. Photoirradiative removal of caging units, chemical transformation of the quencher moiety or photoswitching of photochromic dyes leads to restoration or appearance of the fluorescent signal. Photocaged and photochromic probes are quite popular in super-resolution applications due to their spatiotemporally controllable switch-on characteristics.Photoactivatable and photochromic systems
Photoactivatable fluorophores are non- or weakly fluorescent molecules that can be photochemically converted to intensively emitting species. For PALM imaging, photoactivatable fluorophores bearing photolabile protecting (caging) groups have been developed. Common caging groups are the 2-nitrobenzyl moiety and its derivatives such as 4,5-dimethoxy-2-nitrobenzyl- (DMNB) or ortho-nitroveratryloxycarbonyl-groups (NVOC) that are efficiently removed by UV light irradiation.63,64 Caged synthetic fluorophores have particular advantages in localization microscopy. They exhibit a higher photon count than fluorescent proteins resulting in better localization precision. Additionally, caged compounds are activated by a photochemical reaction, allowing a sparse, controlled turn-on of individual molecules. This eliminates the need of using harsh imaging conditions (such as buffers in dSTORM), however uses a UV-laser, which can potentially be phototoxic.The open-closed equilibrium of 2′-carboxy-xanthene derivatives can also be controlled by installing caging groups onto the oxygens of fluoresceins or the aniline nitrogen of rhodamines, which shifts the equilibrium significantly towards the dark spirolactone form. Removal of the caging groups restores the equilibrium shifted towards the fluorescent zwitterionic form yielding a large increase in fluorescence. Lavis and co-workers used a photocaged carborhodamine NVOC-carborhodamine 110 (33, Scheme 5) derivatized phalloidin for the imaging of actin filaments by means of PALM without any redox buffers.65 The shifted open-close equilibrium observed with carborhodamine derivatives ensures that PALM imaging can be performed even on densely labeled structures. The same authors reported a general synthetic route to access photoactivatable xanthene fluorophores.66 In this scheme, they used reduced “leuco” form as a synthetic intermediate allowing installation of the caging groups using mild conditions. NVOC-caged azide-derivatized Q-rhodamine (34), rhodamine 110 and Oregon Green derivatives were applied for the first time in SRM imaging of DNA in the cellular context (Scheme 5). Using the NVOC-caging strategy, Grimm et al. reported a Si-containing analogue of Q-rhodamine (SiRhQ, 35), which exhibits higher photon counts compared to established localization microscopy dyes, such as Alexa Fluor 647, especially after the addition of oxygen-depriving Trolox.67 The achieved high precision allowed high resolution imaging of actin-rich structures, such as filopodia (FWHM = 45 nm). SiRhQ (35) performed well in multicolor localization microscopy as well, in combination with the fluorescent protein mEos2. SiRhO, 35 and mEos2 have sufficiently different activation cross-sections to allow successive imaging of the two colors (Fig. 8). Johnsson et al. presented a caged rhodamine 110 derivative that can be conjugated to SNAP-tag fusion proteins (BG-cRhod, 36).68 In BG-cRhod (36), the SNAP-tag reactive O6-benzylguanine (BG) is linked via a urea motif to rhodamine 110. The photolabile DMNB group attached to the other amino group of rhodamine 110 forces the probe to its non-fluorescent lactone form until it is irradiated with 365 nm UV light. PALM imaging of mitochondria of U2OS cells resulted in circa twice as better localization as with red fluorescent proteins. Wombacher et al. described new fluorophores for dual color photoactivated SRM in live mammalian cells.69 They synthesized trimethoprim and Halo-tag conjugates of NVOC-Q-Rhodamine and ortho-nitrobenzyl-caged fluorescein (37) and demonstrated dual-color activation following exposure to UV-light using epifluorescent microscopy and iFRAP (inverse fluorescence recovery after photobleaching) as well as PALM imaging with caged RhoQ. It should be noted that the 2-nitrobenzyl group and its surrogates are bulky and generate toxic, colored and reactive 2-nitrosobenzaldehyde or 2-nitrosobenzophenone derivatives upon photolysis.70
 | ||
| Scheme 5 Photoactivatable fluorophores with photolabile groups mentioned in the text. | ||
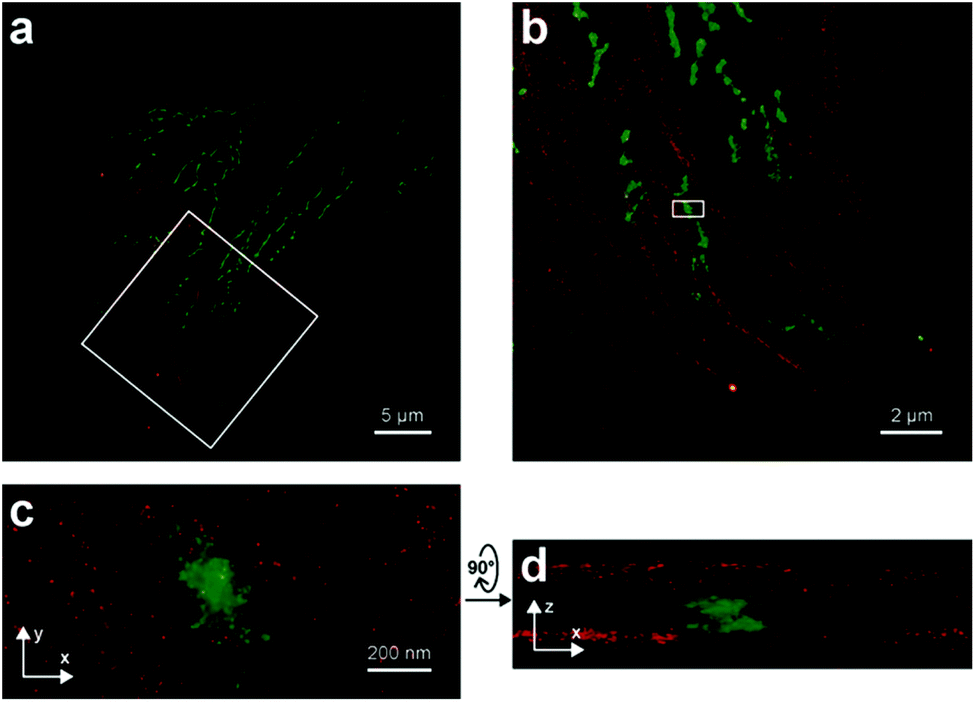 | ||
| Fig. 8 Two-color iPALM images of fixed U2OS cells labelled with SiRhQ(35)-phalloidin conjugate (actin, red) and mEos2 (mitochondria, green). (b) Enlarged image of the boxed area is in (a). (c) is x, y projection of the boxed area in (b). (d) x, z projection of the boxed area in (b). (Reprinted from ref. 67 with permission. Copyright 2015 John Wiley and Sons.) | ||
Dicyanomethylenedihydrofuran (DCDHF) type push–pull fluorophores contain an electron donor amine moiety connected to the DCDHF acceptor via a conjugated π-system. Moerner et al. reported azide caged fluorogen 38 that remains dark until photoactivation. A short burst of 407 nm UV light converts the azide to an amine shifting the absorption to a longer wavelength (i.e. 570 nm). The created photostable compound 39 could be imaged at the single molecule level (Fig. 9a).71 Later, they applied a derivative of this probe in Halo-tag mediated super-resolution imaging of microtubules in fixed mammalian BS-C-1 cells with 85 ± 15 nm resolution.72 The cell-permeable nature of this probe allowed live Caulobacter crescentus cell labeling and revealed protein localization patterns below the diffraction limit (Fig. 9c). In 2013, they replaced the azide with a nitro group, (nitro-DCDHF, 40), and subjected it to treatment with nitroreductase and electron donors (NADH) (Fig. 9b) resulting in the formation of fluorescent products without phototoxic UV irradiation.73 The method, named enzymatic turnover activated localization microscopy (ETALM), enabled live cell SRM imaging of bacterial cells.
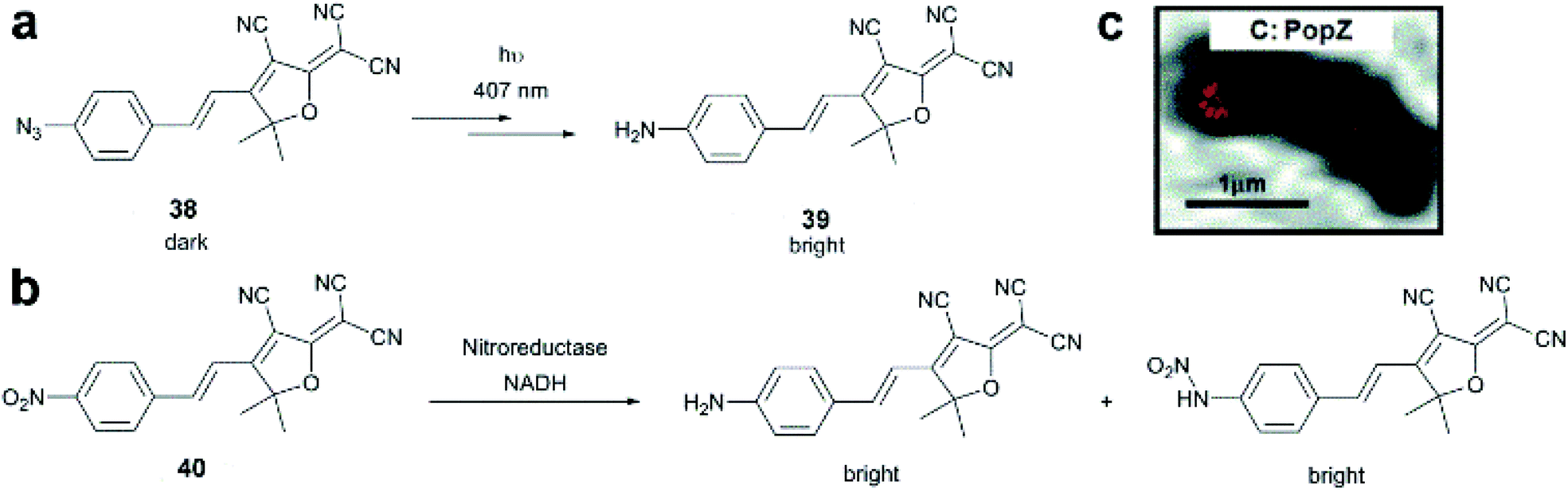 | ||
| Fig. 9 (a–b) Structure of DCDHF class fluorogens and their fluorescent products. (c) SRM imaging of PopZ protein fusions inside live Caulobacter crescentus cells using 38 in a Halo-tag form. (Adapted from ref. 72 with permission. Copyright 2010 American Chemical Society.) | ||
A somewhat distinct group of photoactivatable fluorophores suitable for SRM is represented by so-called photochromic probes. These fluorophores have the characteristics that they can be reversibly interconverted between a dark non-fluorescent and an emissive form. The reversible switching involves either photoirradiation in both directions or a photoirradiation-thermal back isomerization process. Either way, photoswitching does not require the use of blinking buffers. Rhodamine spiroamides exist in a non-fluorescent closed form, which becomes fluorescent upon irradiation with UV light (single photon) or two-photon laser irradiation (Fig. 10a). The open isomer has a lifetime of a few milliseconds in polar solvents before being converted back thermally to the dark form. Such behavior is ideal for SMLM applications.74 Hell et al. developed SRA577 (41), a rhodamine B derivative with an appending 4-aminophthalimide substituent, with a thermal relaxation time of 20–100 ms in polar solvents (Fig. 10a).75 In contrast to caged probes, which are activated and then photobleached, photochromes can be used to localize markers several times, thereby allowing multiple measurements on the same sample. This characteristic is quite beneficial in live-cell imaging applications. In their report, the Hell group tested SRA577 (41) using PALMIRA (PALM with independently running acquisition) on silica nanobeads offering non-invasive optical sectioning. Separate detection of individual molecules enabled assignation of every single molecular event to the corresponding dye class, thus allowing multicolor localization imaging. The same authors later developed four more rhodamine spiroamide derivatives and demonstrated multicolor separation on silica nanobeads with improved resolution (15 nm).76 Two-color labeling of microtubules and the keratin network in Ptk2 cells with SRA552 (42) and SRA577 (41) using secondary antibodies was also presented at the nanoscale. Further rhodamine spiroamides were reported spanning the whole visible spectrum for multicolor SMLM by Belov and Hell.77 Photoswitching of photochromic probes to emitting forms is usually achieved upon UV light irradiation, however these wavelengths may damage cells. To address this, Moerner et al. shifted the photoswitching wavelengths of rhodamine spirolactams towards visible wavelengths >400 nm.78 Additionally, they varied the chromophores on the lactam nitrogen, which facilitated the switching at lower photoactivation energy without substantially altering other photophysical features. An optimized derivative (43) was used to label surface amines of live Caulobacter crescentus bacterial cells for 3D super-resolution imaging (Fig. 11). Low laser intensity purple beam (405 nm, 18 W cm−2) PALM imaging revealed a localization precision of 14–17 nm with enhanced specificity.
 | ||
| Fig. 10 Switching process and structure of photochromic labels discussed in the text: (a) spiroamides, (b) diazoketones, (c) diarylethenes, and (d) oxazine photochromes. | ||
 | ||
| Fig. 11 3D super-resolution reconstruction of surface localization in live Caulobacter crescentus bacterial cells using compound 43. (a) A 75 nm slice is highlighted in yellow, and viewed in the panel of the cell axis (b). (c) Histogram of the radial distances of each of the localizations to the center of the circle fitted to the points in panel B. (Adapted from ref. 78 with permission. Copyright 2014 American Chemical Society.) | ||
In conventional multicolor spectroscopy, the different fluorophores are usually separately excited to induce the corresponding emission wavelength. Such multiple excitations can, however, produce chromatic artefacts and overlaps. Belov, Hell et al. elegantly addressed this and used dyes with similar spectral characteristics but with different switching-on properties.70,79 The authors combined non-caged TMR, diazoketone-caged compound 45 and spiroamide-caged SRA552 (42). Actin, microtubule cytoskeleton, and peroxisomes were labeled using different labeling strategies (phalloidin, antibodies) with the above-mentioned derivatives. First, TMR was imaged and bleached with 546 nm light, and then TMR-NN was uncaged (420/30 nm) and imaged, followed by uncaging of SRA552 (42) (spiroamide of rhodamine S – RhS on the figure) (360/40 nm) (Fig. 12).
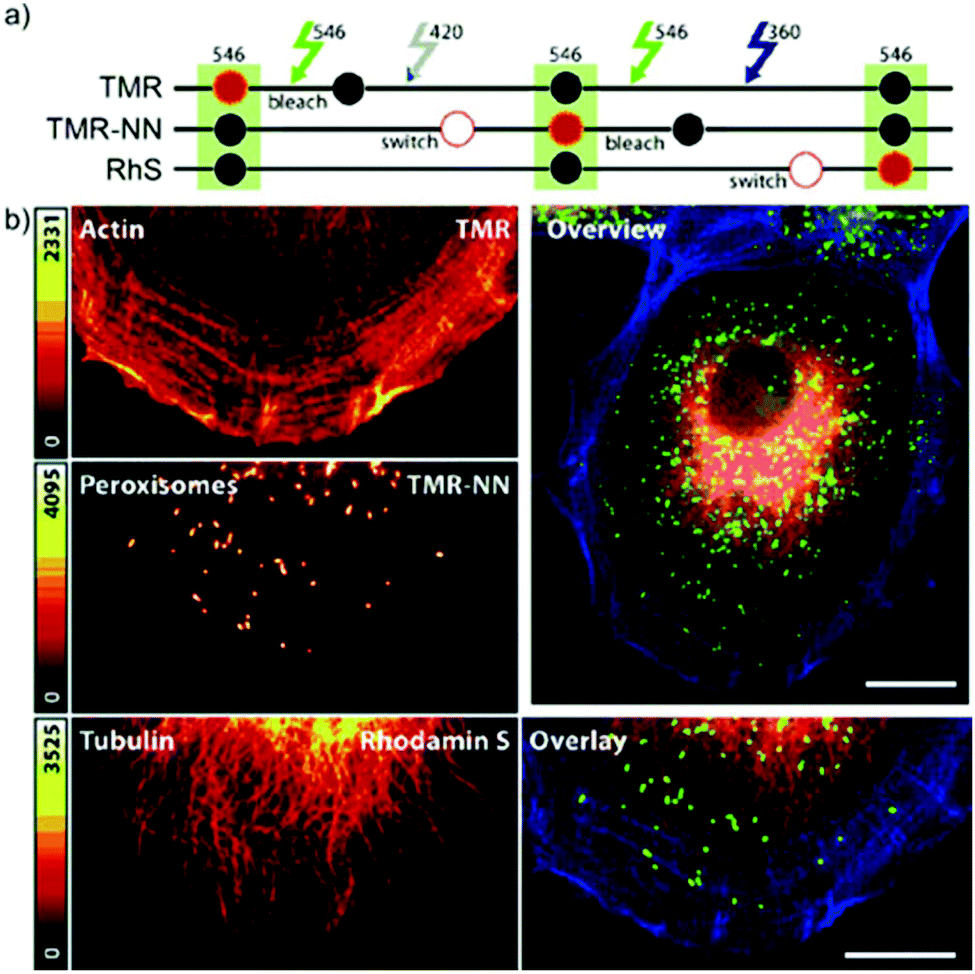 | ||
| Fig. 12 Monochromatic multilabel imaging using photochromic probes TMR-NN (45) and RhS (SRA552, compound 42 in the text) in combination with conventional TMR. See details of imaging scheme (a) in the text. (b) Application on Ptk2 cells. Scale bar: 10 μm. (Reprinted from ref. 70 with permission. Copyright 2010 John Wiley and Sons.) | ||
Rivera-Fuentes and co-workers developed a dual-activatable diazoketone-based probe to sense esterase activity.80 Without enzymatic preactivation, the diazoketone probe (46) has an electron-poor core, which, upon irradiation, undergoes Wolff rearrangement to yield a non-fluorescent ring-expanded xanthene. However, when carboxylesterases activate the fluorogen, the tricyclic core becomes electron-rich, and the photoinduced Wolff rearrangement produces a fluorescent rhodol. This probe was utilized in SMLM to sense esterase activity in live HeLa cells.
As the abovementioned examples show, photoactivatable spiroamides and diazoketones proved to be useful markers recently in SMLM, however their applicability is often limited by aggregation, non-specific adhesion and low reactivity caused by low water-solubility. Hell et al. applied a universal hydrophilizer (a 3-sulfo-L-alanyl – beta-alanine dipeptide) and allylic hydroxyl groups as synthetic modifications to increase the polarity (e.g. compounds 44 and 47).81
The fluorescence of 1,2-bis(2-alkyl-6-phenyl-1-benzothiophene-1,1-dioxide-3-yl)perfluorocyclopentenes (diarylethenes) can be modulated by light in both directions. While UV light facilitates the formation of the fluorescent “closed” form visible light promotes isomerization to the dark “open” form (Fig. 10c). These photoswitches emit bright, green fluorescence with high photostability, however they are generally hydrophobic and non-soluble in aqueous solutions.82 Hell et al. decorated the diarylethene core with four or eight carboxylic groups to access 48 and 49, respectively, to increase the water solubility and prevent the aggregation of the free dyes.83 Derivatives 48 and 49 were tested in immunostaining of tubulin filaments and nucleopore complexes in Vero cells, and robust photoswitching was observed upon irradiation with 355 nm light for 50 μs. Following 200 μs delay irradiation with 488 nm light for 1.2 ms in a doughnut shape resulted in the formation of the dark “open” form at the respective area. By means of Reversible Saturable Optical Fluorescence Transitions (RESOLFT) microscopy the authors achieved an optical resolution of 74 nm (FWHM). Attempts, however, to apply these derivatives in coordinate-stochastic approaches failed since switching properties in aqueous media were inadequate for efficient detection of single molecules. To overcome this limitation, the diarylethene structure was modified with electron donor (p-methoxyphenyl) and electron acceptor substituents in order to increase the push–pull effect to improve the on/off ratio.84 These modifications resulted in octa-acid 50, which showed high aqueous solubility, high emission efficiency and low cycloconversion quantum yield and proved to be suitable for STORM imaging by applying single 488 nm light without additional photoactivation with UV light or further additives. Raymo et al. studied oxazine photochromes in fluorophore-photochrome dyads.85 They designed a set of hemicyanine-based fluorophores involving a photoswitchable oxazine unit. The 2H,4H-[1,3]-oxazine ring of these compounds opens on a subnanosecond time scale to generate a zwitterionic isomer (Fig. 10d). The photogenerated fluorescent forms revert spontaneously to the original, non-emissive species within submicroseconds. They applied derivative 51 for stochastic SRM on amphiphilic copolymer nanoparticles, demonstrating the excellent blinking effect.
Chemically activatable fluorogenic probes
The most important representatives of chemically activatable fluorogenic probes suitable for super-resolution applications possess azide or tetrazine moieties as a reactive quencher unit. Both the azide and tetrazine can participate in highly selective, biocompatible reactions, i.e. bioorthogonal reactions. Thus these functions are two-in-one combinations that make fluorescent cores bioorthogonally applicable and fluorogenic at the same time.86–88 When the azide/tetrazine moiety is transformed in a specific reaction with the target biomolecule into a triazole or dihydropyridazine, respectively, the fluorescence is restored. To the best of our knowledge, up to now, only tetrazine quenched systems were applied to SRM imaging of biological samples. Although the modular design of bioorthogonally active tetrazine quenched fluorogenic probes allows the use of various fluorophores, their potential for site-specific intracellular fluorogenic labeling was just lately exploited in super-resolution imaging techniques.There are three ways tetrazines are able to modulate the fluorescence of aromatic cores: (i) via Förster-type of resonance energy transfer (FRET)88a–c (ii) via a through-bond energy transfer (TBET) process5d,88d,e and (iii) as just recently reported, in directly conjugated, monochromophoric systems.89 In FRET systems the fluorescent core is linked to the tetrazine through a flexible linker. Besides other criteria (e.g. distance, alignment of transition moments etc.), a considerable overlap between the emission band of the fluorophore and the absorption band of the tetrazine (typically falls between 520–540 nm) is necessary. This criterion limits the applicable fluorescent cores to green emitting frames.88a–c Much more efficient energy transfer occurs via TBET fluorogenic probes, where the fluorophore and the tetrazine are attached together via a rigid, conjugated, otherwise electronically decoupled (twisted) linker.5d,88d,e It is important to note that energy transfer via TBET does not require spectral match between the fluorophore and the quencher, which allows the combination of virtually any kind of tetrazines and fluorescent cores with various emission wavelengths. In monochromophoric design where the tetrazine is directly linked to the fluorescent core the presence of an optically inactive n–π* transition is believed to be the reason for quenched fluorescence.89 Despite the many advantages, there are only a few examples of super-resolution applications of tetrazine quenched systems.
We recently applied the TBET strategy to cyanine (Cy3 and Cy5) scaffolds, where the two units were linked either via a phenylene or a vinylene unit.90 All four probes showed fluorogenic behavior, however only one of them, Cy3-derivative 52, produced a fluorescence enhancement over one order of magnitude upon reaction (Scheme 6). The 14-fold intensity increase, however, enabled sufficient distinction between unreacted, non-specific and specifically reacted, turned-on signals even under no-wash conditions. Although indocyanines are not preferred in STED-based super-resolution techniques, we have found compound 52 to be suitable for STED imaging of actin bundles with 168 nm resolution using a 660 nm continuous wave laser for depletion (Fig. 13). It should also be mentioned that no loss of signal-to-noise ratio was observed for three cycles of STED imaging with >90% STED laser intensity.
 | ||
| Scheme 6 Structure of chemically activatable fluorogenic probes. | ||
 | ||
| Fig. 13 Confocal (a, e) and STED images (b, f) of actin filaments tagged with phalloidin-BCN and labeled with compound 52 (3 μM) in mammalian COS7 cells. Images were taken after (a, b) or without washing out (e, f) the dye after 30 min incubation. Enlargement of the boxed areas are shown at (c, g) (confocal, corresponding to a, e) and (d, h) panels (STED, corresponding to (b, f). λexc = 552 nm with a 660 nm depletion laser for STED images. Color encodes the intensity values of each pixel. (Reprinted from ref. 90 with permission. Copyright 2018 American Chemical Society.) | ||
One of the reasons tetrazine quenched fluorogenic probes are not widespread in super-resolved imaging techniques is the fact that the quenching power of the bioorthogonal units (tetrazine and azide) is becoming less and less efficient as the excitation and emission wavelengths approach the biologically preferred red, NIR range. We believe that this is due to the more prominent role of other quenching processes that are present in more conjugated systems and the modulation power exerted by a single unit is less profound. We attempted to address this problem by introducing more than one quencher to cyanine cores and synthesized bisazides and bistetrazines.91–93 It was expected that multiple quenching i.e. more quenchers together with the formation of a constrained cyclic product with biscyclooctynylated targets enhances fluorogenicity. Indeed, some of these probes were quite promising, however, the complementing technique to produce biscyclooctynylated proteins required double-amber suppressed fusion tags, which made the whole process complicated. Thus, in our other attempt to create more efficient fluorogenic probes that are excitable in the red range we rather combined quenching mechanisms and created polarity sensitive 2′-carboxy siliconrhodamines linked to tetrazines via phenylene and vinylene linkers.5e,94 Such polarity and reactivity based fluorogenic scaffolds were found to be quite efficient and resulted in an overall 30–40-fold increase upon conjugation, whereas separate quenching mechanisms are reported to achieve 6–7-fold enhancements.5d,10 The best-performing double-fluorogenic probe, SiR-derivative, 53 was applied in the labeling of intracellular vimentin genetically modified with a cyclooctynylated non-canonical amino acid. GSDIM (ground state depletion microscopy followed by individual molecule return) imaging resulted in an improved, 28 nm lateral resolution.5e,94
FRET-based fluorogenicity
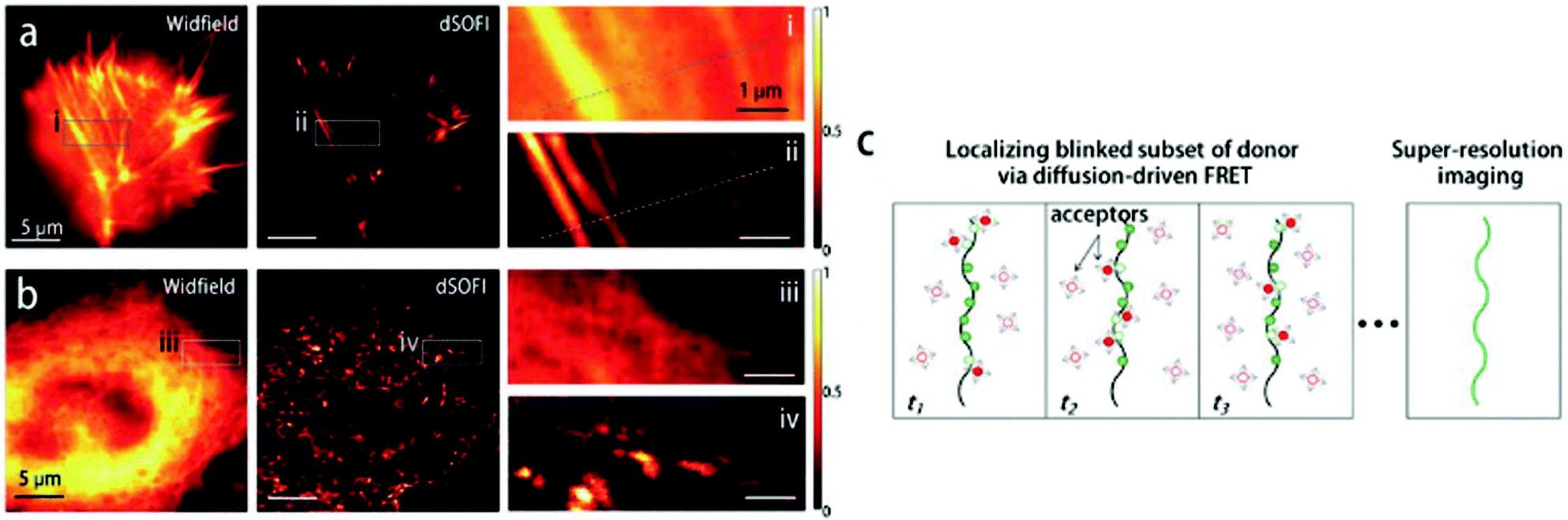
Although Förster resonance energy transfer (FRET) systems are not considered as classical fluorogenic systems, their ability to convert one emission wavelength into a different one justifies their discussion in this context. Indeed, there are few examples where a super-resolved image was acquired through FRET signal. FRET-based SRM was first applied by Park and co-workers based on diffusion-assisted FRET.95 In their setup, fluorescent donor molecules that label target structures could be stochastically quenched by diffusing acceptor molecules, separating temporally otherwise overlapped fluorescence signals, thus allowing SRM. The approach, named dSOFI (direct super-resolution optical fluctuation imaging) is based on SOFI reconstruction and uses general fluorophores in a conventional epifluorescent microscope setup with light-emitting device (LED) illumination.96 Its general utility has been demonstrated in dynamic live-cell imaging of filamentous actin and α-tubulin in NIH 3T3 cells using cerulean and Citrine fluorescent proteins as FRET pairs (Fig. 14).95 | ||
| Fig. 14 Live-cell dynamic imaging using dSOFI. (a) Actin-ceruline expressed in NIH 3T3 cells. Cytoplasmic citrine acts as a FRET acceptor. (i, ii) Magnification of boxed areas. (b) Tubulin-ceruline expressed in NIH 3T3 cells. (iii, iv) Magnification of boxed areas. (c) Principle of dSOFI. (Reprinted from ref. 95. with permission. Copyright 2013 Springer Nature.) | ||
Cui et al. developed a FRET-based dual emission nanoprobe (FREDEN) with improved blinking behaviour for PALM/STORM imaging.97 In their setup, Alexa Fluor 647 was attached to CdSSe/ZnS quantum dots (QD) via glutathione and used as an acceptor in FRET to modulate the 610 nm emission of QDs. The FREDEN conjugate shows two distinct emission bands at 610 nm and 670 nm upon excitation of the QDs at 405 nm due to efficient FRET. The two-channel blinking of this conjugate is explained by the excited state reaction of Alexa Fluor 647 that leads to the formation of a non-fluorescent species. The reaction is reversible and is – coincidentally – induced by irradiation with 405 nm light. Thus, the 405 nm excitation wavelength excites the QDs and reforms the fluorescent form of the Alexa dye. The excited state reaction of the Alexa dye affects the FRET process as well, and thus fluctuations at both channels appear. This led to improved blinking and localization precision, compared to AF647 and QD alone, without the need for blinking buffers. Live cell STORM imaging of FREDEN conjugates inside MRC-5 cells was demonstrated.
DNA point accumulation for imaging in nanoscale topography (DNA-PAINT) was a major breakthrough in recent years in SMLM techniques.98
DNA-PAINT achieves single-molecule switching and localization by applying freely diffusing dye-labeled single-stranded (ss) DNA (imager strand) that transiently binds to a complementary, unlabeled target-bound ss-DNA strand (docking strand) (Fig. 15a). DNA-PAINT offers several advantages over traditional SRM techniques (no bleaching, wide choice of fluorophores, multicolor-imaging up to 9 color, etc.), yet it still has limitations. Since the imager strand is non-fluorogenic, the technique has to rely on optical sectioning methods such as total internal reflection microscopy (TIRF) and limited image acquisition speed in order to eliminate the background fluorescence of freely diffusing imager strands. To address this, Hohng et al. introduced the concept of FRET-PAINT.99 In their work they designed an unlabeled docking strand that could accommodate two labeled imager strands at a time. The imager strands were designed such that they formed a FRET pair either by using Cy3/Cy5 or AlexaFluor 488/Cy5 labeled strands. Furthermore, the imager strand carrying the acceptor fluorophore had higher binding affinity than the donor labeled strand. Transient binding of the imager strands to the docking strand resulted in stochastic blinking of the acceptor probe when exciting the donor, due to FRET. Since there was no need to eliminate the imager strands’ background fluorescence, a higher loading of the freely diffusing imager strands was allowed, enabling faster image acquisition. In a coincident paper, Jungmann and co-workers applied the very same concept, however, in their setup they used an Atto674N labeled docking strand and an Atto488 conjugated imager strand.100 The high efficiency of the energy transfer was proven along with the highly increased acquisition time as low as 28 s. However, the advantages came at the cost of photobleaching, originally one of the main advantages of DNA-PAINT. When the image acquisition exceeded 400 s, the Atto647N got bleached. Thus they also used a non-labeled docking strand with two recognition sites. Transient, simultaneous binding of donor and acceptor labeled imager strands leads to the same results, however, at the price of reduction in the achievable imaging speed (Fig. 15b). They demonstrated the potential of this setup in FRET-PAINT imaging of DNA origami structures. In their work, they also utilized their original setup consisting of labeled docking and imager strands in rapid image acquisition of cellular structures. Microtubules of HeLA cells were resolved with a localization precision of 46 nm (Fig. 15c and d).
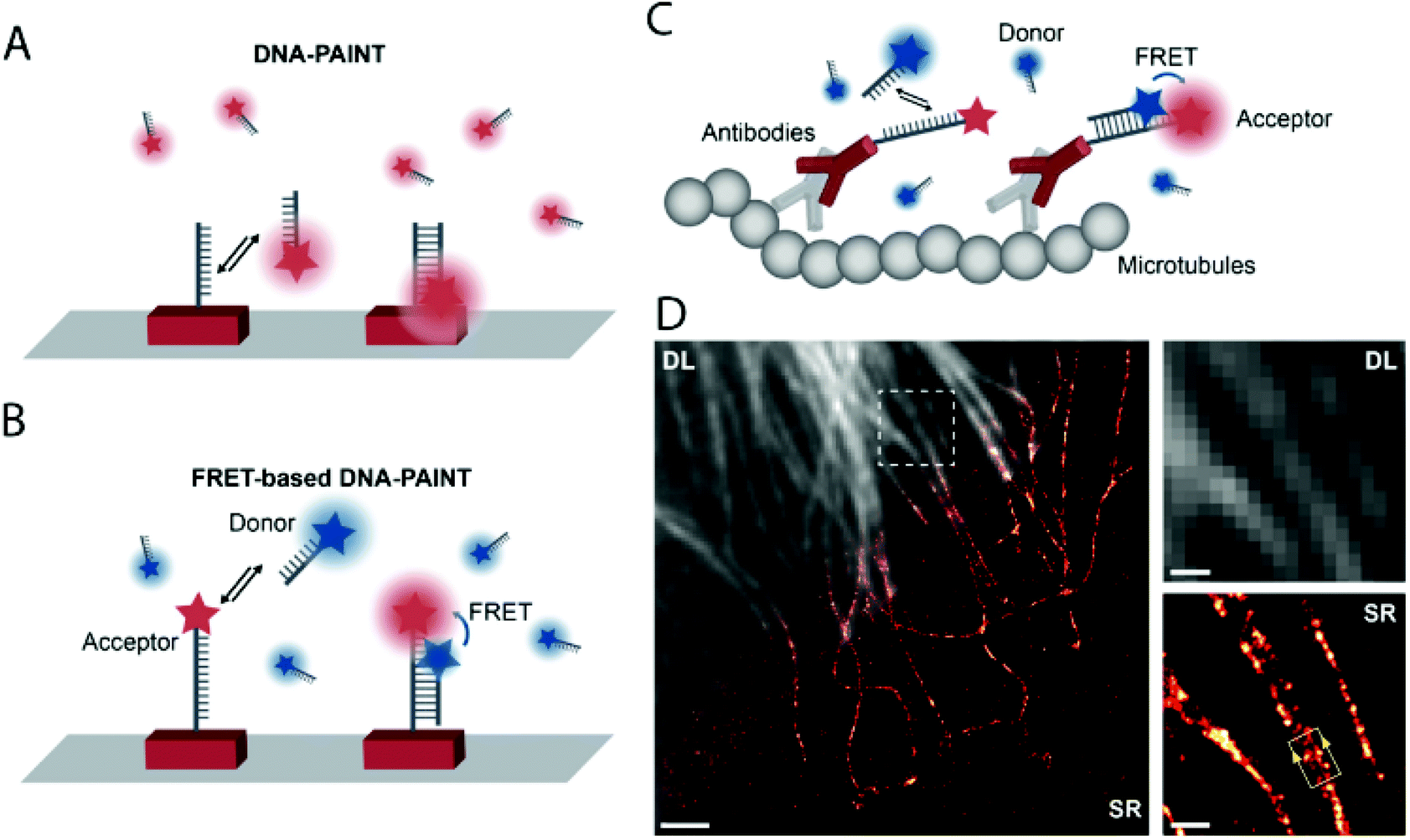 | ||
| Fig. 15 High speed super-resolution imaging with FRET-PAINT. Principle of DNA-PAINT (a) and FRET-PAINT (b). (c) Labeling scheme using fixed acceptor binding strands. (d) Corresponding diffraction limited and SMLM imaging of the microtubule network in HeLa cells. Scale bar: 2 μm. (Adapted from ref. 100 with permission. Copyright 2017 American Chemical Society.) | ||
Heileman and co-workers calculated FRET efficiencies from super-resolved FRET-PAINT images and could assign FRET populations to specific target strands.101 Multiplexed imaging using Atto 674N and Cy3B was also demonstrated on DNA origami with high specificity. FRET-PAINT also allows the measurement of distances in the range of 1–10 nm, extending the spatial resolution of DNA-PAINT near the molecular level.
Conclusions and outlook
Recent developments in super-resolution imaging of biomolecules revealed fine details of cellular structures at sub-diffraction resolutions by means of non-invasive optical methods. The emerged SRM techniques are powerful, yet far from their limits. The biggest constraint that does not allow further improvements is the limited number of suitable probes. High contrast fluorescence imaging of (site)specifically labeled biomolecules by fluorogenic probes is a promising way to address these challenges. The additional level of complexity rationalized in fluorogenic probes that allow site-specific tagging with minimal background and autofluorescence of intracellular targets led to the development of robust labeling schemes with improved resolution. However, the increasing need for multicolor imaging of multiple targets requires the development of further probes with orthogonal fluorogenic and/or distinct spectral properties. Combination of desirable characteristics such as membrane permeability, selective targeting by means of biologically tolerable chemistries and good spectral features make the design and development of such probes quite challenging as implementation of one such feature often comes at the cost of others. Herein we attempted to highlight the few developments that applied small molecular fluorogenic labels in SRM imaging of biologically relevant structures and listed some design principles suitable for making fluorescent frames fluorogenic. In the future it is expected that existing design principles will result in further fluorogenic probes suitable for SRM imaging and substantial developments are expected in the field of DNA-PAINT, with improved acquisition times. Although not listed herein, as no SRM application is reported so far, design principles, which rely on the in situ assembly/generation of fluorescent cores during the conjugation reaction, hold great promise. Such probes possess virtually infinite fluorogenicity and thus provide the users with a superb contrast.Future developments in the field of labeling techniques in combination with new fluorogenic probes should provide us with robust techniques with unprecedented resolutions both spatially and temporally.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was supported by the Hungarian Academy of Sciences within the frames of the “Lendület” Program (LP2013-55/2013). The authors thank Ms Bianka Söveges for her help with the graphical content and Dr Attila Kormos for carefully reading the manuscript.References
- (a) B. Huang, H. Babcock and X. Zhuang, Cell, 2010, 143, 1047 CrossRef CAS PubMed; (b) S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 11, 685 CrossRef PubMed; (c) A. M. Sydor, K. J. Czymmek, E. M. Puchner and V. Mennella, Trends Cell Biol., 2015, 25, 730 CrossRef CAS PubMed; (d) J. Vangindertael, R. Camacho, W. Sempels, H. Mizuno, P. Dedecker and K.P. F. Janssen, Methods Appl. Fluoresc., 2018, 6, 022003 CrossRef CAS PubMed.
- (a) M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929 CrossRef CAS PubMed; (b) Z. Yang, A. Sharma, J. Qi, X. Peng, D. Y. Lee, R. Hu, D. Lin, J. Qu and J. S. Kim, Chem. Soc. Rev., 2016, 45, 4651 RSC; (c) S. van de Linde, S. Aufmkolk, C. Franke, T. Holm, T. Klein, A. Löschberger, S. Proppert, S. Wolter and M. Sauer, Chem. Biol., 2013, 20, 8 CrossRef CAS PubMed; (d) M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172 CrossRef CAS PubMed.
- (a) D. Jung, K. Min, J. Jung, W. Jang and Y. Kwon, Mol. Biosyst., 2013, 9, 862 RSC; (b) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef CAS PubMed; (c) X. Chen and Y. Wu, Org. Biomol. Chem., 2016, 14, 5417 RSC.
- (a) L. D. Lavis, Biochemistry, 2017, 56, 5165 CrossRef CAS PubMed; (b) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2014, 9, 855 CrossRef CAS PubMed; (c) G. B. Cserép, A. Herner and P. Kele, Methods Appl. Fluoresc., 2015, 3, 042001 CrossRef PubMed; (d) C. P. Toseland, J. Chem. Biol., 2013, 6, 85 CrossRef PubMed.
- (a) Y. Hori and K. Kikuchi, Curr. Opin. Chem. Biol., 2013, 17, 644 CrossRef CAS PubMed; (b) C. Li, A. G. Tebo and A. Gautier, Int. J. Mol. Sci., 2017, 18, 1473 CrossRef PubMed; (c) L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531 CrossRef CAS PubMed; (d) A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, J. Chem. Biol., 2017, 8, 1506 CAS; (e) E. Kozma, G. Estrada Girona, G. Paci, E. A. Lemke and P. Kele, Chem. Commun., 2017, 53, 6696 RSC; (f) J. Vogelsang, C. Steinhauer, C. Forthmann, I. H. Stein, B. Person-Skegro, T. Cordes and P. Tinnefeld, ChemPhysChem, 2010, 11, 2475 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC.
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600 CrossRef CAS PubMed.
- Y. Kushida, T. Nagano and K. Hanaoka, Analyst, 2015, 140, 685 RSC.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef CAS PubMed.
- G. Lukinavicius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Correa, Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 139 CrossRef PubMed.
- G. Lukinavicius, L. Reymond, E. D'Este, A. Masharina, F. Göttfert, H. Ta, A. Güther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H. D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731 CrossRef CAS PubMed.
- N. V. Klementieva, L. B. Snopova, N. N. Prodanets, O. E. Furman, V. V. Dudenkova, E. Zagaynova, K. A. Lukyanov and A. S. Mishin, Anticancer Res., 2016, 36, 5287 CrossRef CAS PubMed.
- G. Lukinavicius, C. Blaukopf, E. Pershagen, A. Schena, L. Reymond, E. Derivery, M. Gonzales-Gaitan, E. D'Este, S. W. Hell, D. W. Gerlich and K. Johnsson, Nat. Commun., 2015, 6, 8497 CrossRef CAS PubMed.
- C. Uttamapinant, J. D. Howe, K. Lang, V. Beranek, L. Davis, M. Mahesh, N. P. Barry and J. W. Chin, J. Am. Chem. Soc., 2015, 137, 4602 CrossRef CAS PubMed.
- J. Masch, H. Steffens, J. Fischer, J. Engelhardt, J. Hubrich, J. Keller-Findeisen, E. D'Este, N. T. Urban, S. G. N. Grant, S. J. Sahl, D. Kamin and S. W. Hell, Proc. Natl. Acad. Sci. USA, 2018, 115, E8047 CrossRef CAS PubMed.
- G. Lukinavicius, L. Reymond, K. Umezawa, O. Sallin, E. D'Este, F. Göttfert, H. Ta, S. W. Hell, Y. Urano and K. Johnsson, J. Am. Chem. Soc., 2016, 138, 9365 CrossRef CAS PubMed.
- K. Kolmakov, E. Hebisch, T. Wolfram, L. A. Nordwig, C. A. Wurm, H. Ta, V. Westphal, V. N. Belov and S. W. Hell, Chem. – Eur. J., 2015, 21, 13344 CrossRef CAS PubMed.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290 CrossRef CAS PubMed.
- K. Kolmakov, V. N. Belov, C. A. Wurm, B. Harke, M. Leutenegger, C. Eggeling and S. W. Hell, Eur. J. Org. Chem., 2010, 2010, 3593 CrossRef.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290 CrossRef CAS PubMed.
- G. Lukinavicius, G. Y. Mitronova, S. Schnorrenberg, A. N. Butkevich, H. Barthel, V. N. Belov and S. W. Hell, Chem. Sci., 2018, 9, 3324 RSC.
- A. N. Butkevich, H. Ta, M. Ratz, S. Stoldt, S. Jakobs, V. N. Below and S. W. Hell, ACS Chem. Biol., 2018, 13, 475 CrossRef CAS PubMed.
- A. N. Butkevich, V. N. Belov, K. Kolmakov, V. V. Sokolov, H. Shojaei, S. C. Sidenstein, D. Kamin, J. Matthias, R. Vlijm, J. Engelhardt and S. W. Hell, Chem. – Eur. J., 2017, 50, 12114 CrossRef PubMed.
- H. Takakura, Y. Zhang, R. S. Erdmann, A. D. Thompson, Y. Lin, B. McNellis, F. Rivera-Molina, S. Uno, M. Kamiya, Y. Urano, J. E. Rothman, J. Bewersdorf, A. Schepartz and D. Toomre, Nat. Biotechnol., 2017, 35, 773 CrossRef CAS PubMed.
- P. J. Macdolnald, S. Gayda, R. A. Haack, Q. Ruan, R. J. Himmelbach and S. Y. Tetin, Anal. Chem., 2018, 90, 9165 CrossRef PubMed.
- S. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681 CrossRef CAS PubMed.
- S. Uno, M. Kamiya, A. Morozumi and Y. Urano, Chem. Sci., 2018, 54, 102 CAS.
- M. V. Sednev, V. N. Belov and S. W. Hell, Methods Appl. Fluoresc., 2015, 3, 042004 CrossRef PubMed.
- A. N. Butkevich, G. Lukinavicius, E. D'Este and S. W. Hell, J. Am. Chem. Soc., 2017, 139, 12378 CrossRef CAS PubMed.
- S. R. Adams, R. E. Campbell, L. A. Gross, B. R. Martin, G. K. Walkup, Y. Yao, J. Llopis and R. Y. Tsien, J. Am. Chem. Soc., 2002, 124, 6063 CrossRef CAS PubMed.
- J. Enninga, J. Mounier, P. Sansonetti and G. T. Van Nhieu, Nat. Methods, 2005, 2, 959 CrossRef CAS PubMed.
- B. A. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269 CrossRef CAS PubMed.
- J. Fölling, M. Bossi, H. Bock, R. Medda, C. A. Wurm, B. Hein, S. Jakobs, C. Eggeling and S. W. Hell, Nat. Methods, 2008, 5, 943 CrossRef PubMed.
- M. Lelek, F. Di Nunzio, R. Henriques, P. Charneau, N. Arhel and C. Zimmer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8564 CrossRef CAS PubMed.
- M. Lelek, F. Di Nunzio and C. Zimemr, Methods Mol. Biol., 2014, 1174, 183 CrossRef CAS PubMed.
- H. Park, G. T. Hanson, S. R. Duff and P. R. Selvin, J. Microsc., 2004, 216, 199 CrossRef CAS PubMed.
- C. Flors, J. Miscrosc., 2013, 251, 1 CrossRef CAS PubMed.
- A. I. Dragan, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, Biophys. J., 2010, 99, 3010 CrossRef CAS PubMed.
- A. Larsson, C. Carlsson, M. Jonsson and B. Albinsson, J. Am. Chem. Soc., 1994, 116, 8459 CrossRef CAS.
- H. S. Rye, S. Yue, D. E. Wemmer and M. A. Queseda, Nucleic Acids Res., 1992, 20, 2803 CrossRef CAS PubMed.
- C. Flors, C. N. J. Ravarani and D. T. F. Dryden, ChemPhysChem, 2009, 10, 2201 CrossRef CAS PubMed.
- G. Park, S. K. Chakkarapani, S. Ju, S. Ahn and S. H. Kang, Chin. Chem. Lett., 2018, 29, 505 CrossRef CAS.
- S. K. Chakkarapani, G. Park and S. H. Kang, Chin. Chem. Lett., 2015, 26, 1490 CrossRef CAS.
- X. Meng, W. W. Cai and D. C. Schwartz, J. Biomol. Struct. Dyn., 1996, 13, 945 CrossRef CAS PubMed.
- C. Flors, Photochem. Photobiol. Sci., 2010, 9, 643 RSC.
- F. Persson, P. Bingen, T. Staudt, J. Engelhardt, J. O. Tegenfeld and S. W. Hell, Angew. Chem., Int. Ed., 2011, 50, 5581 CrossRef CAS PubMed.
- T. Idziorek, J. Estaquier, F. De Bels and J. C. Ameisen, J. Immunol. Methods, 1995, 185, 249 CrossRef CAS PubMed.
- H. Miller, Z. Zhou, A. J. M. Wollman and M. C. Leake, Methods, 2015, 88, 81 CrossRef CAS PubMed.
- D. Wlodkowic, J. Skommer and Z. Darzynkiewicz, Cytometry, Part A, 2008, 73, 496 CrossRef PubMed.
- C. Flors, Biopolymers, 2011, 95, 290 CrossRef CAS PubMed.
- X. Yan, R. C. Habbersett, J. M. Cordek, J. P. Nolan, T. M. Yoshida, J. H. Jett and B. L. Marrone, Anal. Biochem., 2000, 286, 138 CrossRef CAS PubMed.
- S. Bakshi, H. Choi, N. Rangarajan, K. J. Barns, B. P. Bratton and J. C. Weisshaar, Appl. Environ. Microbiol., 2014, 80, 4977 CrossRef PubMed.
- (a) A. S. Backer, M. Y. Lee and W. E. Moerner, Optica, 2016, 3 CrossRef CAS PubMed; (b) F. Hövelmann, I. Gáspár, J. Chamiolo, M. Kasper, J. Steffen, A. Ephrussi and O. Seitz, Chem. Sci., 2016, 7, 128 RSC.
- L. Jullien and A. Gautier, Methods Appl. Fluoresc., 2015, 042007 CrossRef PubMed.
- M. P. Bruchez, Curr. Opin. Chem. Biol., 2015, 27, 18 CrossRef CAS PubMed.
- J. A. J. Fitzpartrick, Q. Yan, J. J. Sieber, M. Dyba, U. Schwarz, C. Szent-Gyorgyi, C. A. Woolford, P. B. Berget, A. S. Waggoner and M. P. Bruchez, Bioconjugate Chem., 2009, 20, 1843 CrossRef PubMed.
- C. Szent-Gyorgyi, B. F. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS PubMed.
- Q. Yan, S. L. Schwartz, S. Maji, F. Huang, C. Szent-Gyorgyi, D. S. Lidke, K. A. Lidke and M. P. Bruchez, ChemPhysChem, 2014, 15, 687 CrossRef CAS PubMed.
- N. G. Bozhanova, M. S. Baranov, N. V. Klementieva, K. S. Sarkisyan, A. S. Gavrikov, I. V. Yampolsky, E. V. Zagaynova, S. A. Lukyanov, K. A. Lukyanov and A. S. Mishin, Chem. Sci., 2017, 8, 7138 RSC.
- S. Saurabh, A. M. Perez, C. J. Comerci, L. Shapiro and W. E. Moerner, J. Am. Chem. Soc., 2016, 138, 10398 CrossRef CAS PubMed.
- S. Saurabh, A. M. Perez, C. J. Comerci, L. Shapiro and W. E. Moerner, Curr. Protoc. Cell Biol., 2017, 75, 4.32.1 Search PubMed.
- L. A. Perkins, Q. Yan, B. F. Schmidt, D. Kolodienznyi, S. Saurabh, M. B. Larsen, S. C. Watkins, L. Kremer and M. P. Bruchez, Biochemistry, 2018, 57, 861 CrossRef CAS.
- S. R. Adams and R. Y. Tsien, Annu. Rev. Physiol., 1993, 55, 755 CrossRef CAS PubMed.
- P. Klán, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef.
- J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303 CrossRef CAS PubMed.
- L. M. Wysocki, J. B. Grimm, A. N. Tkachuck, T. A. Brown, E. Betzig and L. D. Lavis, Angew. Chem., Int. Ed., 2011, 50, 11206 CrossRef CAS PubMed.
- J. B. Grimm, T. Klein, B. G. Kopek, G. Shtengel, H. F. Hess, M. Sauer and L. D. Lavis, Angew. Chem., Int. Ed., 2016, 55, 1723 CrossRef CAS PubMed.
- S. Banala, D. Maurel, S. Manley and K. Johnsson, ACS Chem. Biol., 2012, 7, 289 CrossRef CAS PubMed.
- S. Hauke, A. von Appen, T. Quidwai, J. Ries and R. Wombacher, Chem. Sci., 2017, 8, 559 RSC.
- V. N. Belov, C. A. Wurm, V. P. Boyarskiy, S. Jakobs and S. W. Hell, Angew. Chem., Int. Ed., 2010, 49, 3520 CrossRef CAS PubMed.
- S. J. Lord, N. R. Conley, H. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204 CrossRef CAS PubMed.
- H. D. Lee, S. J. Lord, S. Iwanaga, K. Zhan, H. Xie, J. C. Williams, H. Wang, G. R. Bowman, E. D. Goley, L. Shapiro, R. J. Twieg, J. Rhao and W. E. Moerner, J. Am. Chem. Soc., 2010, 132, 15099 CrossRef CAS PubMed.
- M. K. Lee, J. Williams, R. J. Twieg, J. Rao and W. E. Moerner, Chem. Sci., 2013, 4, 220 RSC.
- H. Willwohl, J. Wolfrum and R. Gleiter, Laser Chem., 1989, 10, 63 CrossRef CAS.
- J. Fölling, V. Belov, R. Kunetsky, R. Medda, A. Schönle, A. Egner, C. Eggeling, M. Bossi and S. W. Hell, Angew. Chem., Int. Ed., 2007, 46, 6266 CrossRef PubMed.
- M. Bossi, J. Fölling, V. N. Belov, V. P. Boyarskiy, R. Medda, A. Egner, C. Eggeling, A. Schönle and S. W. Hell, Nano Lett., 2008, 8, 2463 CrossRef CAS PubMed.
- V. N. Belov, M. L. Bossi, J. Fölling, V. P. Boyarskiy and S. W. Hell, Chem. – Eur. J., 2009, 15, 10762 CrossRef CAS PubMed.
- M. K. Lee, P. Rai, J. Williams, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2014, 136, 14003 CrossRef CAS PubMed.
- K. Kolmakov, C. Wurm, M. V. Sednev, M. L. Bossi, V. N. Belov and S. W. Hell, Photochem. Photobiol. Sci., 2012, 11, 522 RSC.
- E. A. Halabi, Z. Thiel, N. Trapp, D. Pinotsi and P. Rivera-Fuentes, J. Am. Chem. Soc., 2017, 139, 13200 CrossRef CAS PubMed.
- B. Roubinet, M. Bischoff, S. Nizamov, S. Yan, C. Geisler, S. Stoldt, G. Y. Mitronova, V. N. Belov, M. L. Bossi and S. W. Hell, J. Org. Chem., 2018, 83, 6466 CrossRef CAS PubMed.
- Y. Shoji, A. Yagi, M. Horiuchi, M. Morimoto and M. Irie, Isr. J. Chem., 2013, 53, 303 CrossRef CAS.
- B. Roubinet, M. L. Bossi, P. Alt, M. Leutenegger, H. Shojaei, S. Schnorrenberg, S. Nizamov, M. Irie, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 15429 CrossRef CAS PubMed.
- B. Roubinet, M. Weber, H. Shojaei, M. Bates, M. L. Bossi and V. N. Belov, J. Am. Chem. Soc., 2017, 139, 6611 CrossRef CAS PubMed.
- E. Deniz, M. Tomasulo, J. Cusido, I. Yildiz, M. Petriella, M. L. Bossi, S. Sortino and F. M. Raymo, J. Phys. Chem. C, 2012, 116, 6058 CrossRef CAS.
- G. B. Cserép, A. Herner and P. Kele, Methods Appl. Fluoresc., 2015, 3, 042001 CrossRef PubMed.
- (a) A. Herner, I. Nikic, M. Kállay, E. A. Lemke and P. Kele, Org. Biomol. Chem., 2013, 11, 3297 RSC; (b) P. Shieh, M. S. Siegrist, A. J. Cullen and C. R. Bertozzi, Proc. Natl. Acad. Sci. USA, 2014, 111, 5456 CrossRef CAS PubMed.
- (a) J. Yang, J. Seckute, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476 CrossRef CAS PubMed; (b) N. K. Devaraj, S. Hildebrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869 CrossRef CAS PubMed; (c) D. S. Liu, A. Tangpeerachaikul, R. Selvaraj, M. T. Taylor, J. M. Fox and A. Y. Ting, J. Am. Chem. Soc., 2012, 134, 792 CrossRef CAS PubMed; (d) J. C. T. Carlson, L. G. Meimetis, S. A. Hildebrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917 CrossRef CAS PubMed; (e) G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, Chem. – Eur. J., 2016, 22, 8972 CrossRef CAS PubMed.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974 CrossRef CAS PubMed.
- G. Knorr, E. Kozma, J. M. Schaart, K. Németh, G. Török and P. Kele, Bioconjugate Chem., 2018, 29, 1312 CrossRef CAS PubMed.
- O. Demeter, E. A. Fodor, M. Kállay, G. Mező, K. Németh, P. T. Szabó and P. Kele, Chem. – Eur. J., 2016, 22, 6382 CrossRef CAS PubMed.
- O. Demeter, A. Kormos, C. Koehler, G. Mező, K. Németh, E. Kozma, L. Takács, E. A. Lemke and P. Kele, Bioconjugate Chem., 2017, 28, 1552 CrossRef CAS PubMed.
- A. Kormos, C. Koehler, E. A. Fodor, Z. R. Rutkai, M. E. Martin, G. Mező, E. A. Lemke and P. Kele, Chem. – Eur. J., 2018, 24, 8841 CrossRef CAS PubMed.
- E. Kozma, G. Paci, G. Estrada Girona, E. A. Lemke and P. Kele, Methods Mol. Biol., 2018, 1728, 337 CrossRef CAS PubMed.
- S. Cho, J. Jang, C. Song, H. Lee, P. Ganesan, T. Yoon, M. W. Kim, M. C. Choi, H. Ihee, W. D. Heo and Y. Park, Sci. Rep., 2013, 3, 1208 CrossRef PubMed.
- T. Dertinger, R. Colyer, G. Iyer, S. Weiss and J. Enderlein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22287 CrossRef CAS PubMed.
- S. Zong, X. Jiang, Z. Wang, C. Chen, J. Lu, L. Wang, D. Zhu and Y. Cui, Nanoscale, 2016, 8, 19110 RSC.
- (a) J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Scheuder and R. Jungmann, Nat. Protoc., 2017, 12, 1198 CrossRef CAS PubMed; (b) R. Jungmann, C. Steinhauer, M. Schieble, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756 CrossRef CAS PubMed.
- J. Lee, S. Park, W. Kang and S. Hohng, Mol. Brain, 2017, 10, 63 CrossRef PubMed.
- A. Auer, M. T. Strauss, T. Schlichthaerle and R. Jungmann, Nano Lett., 2017, 17, 6428 CrossRef CAS PubMed.
- N. S. Deubner-Helfmann, A. Auer, M. T. Strauss, S. Malkusch, M. S. Dietz, H. D. Barth, R. Jungmann and M. Heilemann, Nano Lett., 2018, 18, 4626 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |