
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual chirality transfer from silica to metallic nanoparticles with formation of distorted atomic array in crystal lattice structure†
Seiji
Tsunega
a,
Toyokazu
Tanabe
b and
Ren-Hua
Jin
 *a
*a
aDepartment of Material and Life Chemistry, Kanagawa University, 3-2-7 Rokkakubashi, Yokohama 221-8686, Japan. E-mail: rhjin@kanagwa-u.ac.jp
bDepartment of Materials Science and Engineering, National Defense Academy, 1-10-20 Hashirimizu, Yokosuka 239-8686, Japan
First published on 10th October 2018
Abstract
Transfer of chirality from chiral organic molecules to metallic nanoparticles (NPs) is a very attractive field of research and some unique approaches to obtaining chiral metallic NPs have been developed. However, to date, there has been no report in the literature that the chiral information of silica can be transferred into metallic NPs. In this work, a new chirality transfer system to metallic NPs from chiral silica has been achieved. The chiral transfer was performed by simple two steps: (1) trapping metal cations of silver (Ag) and gold (Au) in chiral silica of nano fibrous bundles embedding poly(ethyleneimine) inside and (2) thermoreducing the metal ions into metallic NPs. The metallic NPs of Au and Ag grown around a silica frame, using a thermo-reduction (calcination) process, showed a spherical shape with a size of about 30 nm. Interestingly, the metallic NPs detached or isolated from the silica via crushing and/or hydrolysis of the silica showed remarkable circular dichroism activity in their plasmon absorption band with an exciton coupling feature. Using an atomic resolution scanning transmission protocol, it was found that the chiral metallic NPs have a definite distortion in the atomic array in their crystal lattice structures. In comparison, achiral metallic NPs, which were prepared using a similar method around achiral silica bundles, showed a precisely ordered atomic line without distortion.
Introduction
Chiral inorganic materials have attracted considerable attention for over 15 years because of their unique optical activity and potential applications in many fields, such as chiral catalysis, optical separation, and chiral sensors.1–6 Among them, particularly, the preparation and application of chiral materials in relation to metallic nanoparticles (NPs) helped to clarify the potential of the plasmonic feature of the NPs. There are several approaches to imparting the chiroptical property to the metallic NPs. For example, arranging achiral metallic NPs along chiral configurations such as helical structures of DNA can originate strong chiroptical properties and display exciton coupling effects around the plasmon absorption band although the individual metallic NPs are achiral.7–10 A similar method was also used in the presence of templates of helical silica (SiO2) where achiral metallic NPs are arranged along the helical structure appearing in the silica.11–13 In contrast, generation of metallic NPs in the presence of chiral ligand molecules (containing thionic groups) could give chirality on the individual NPs with intrinsic chiral structures.14–19 In this ligand molecule scenario, very interesting recent reports found in the literature revealed that achiral ligands coordinated to metals were able to promote the growth of racemic metallic clusters with definite numbers of gold (Au) metal atoms such as Au28, Au38, Au40, and Au102 which can be resolved into optically active enantiomers using a chiral column.20–23 This means that at least the chiral domain can be easily generated on the precisely structured metallic cluster surface as long as the organic ligands effectively coordinate onto the growing cluster surface, whether the ligand molecules are chiral or achiral. In other words, achiral ligands could not initiate homo-chirality but are able to prompt the generation of a pair of enantiomeric metals. However, up to now, there have been no success in directly generating the intrinsic chiroptically active metallic NPs without the assistance of chiral organic molecules. In this research, the aim in the construction of metal chirality is to avoid the issues from chiral ligand molecules and to reveal that the generation of chirality on metallic NPs is not limited to the system of organisation between organic ligands and metallic NPs.Recently, a simple and effective method of preparation of silica nanofiber-based chiral materials was established, which were produced using biomimetic silicification of alkoxysilane around chiral catalytic templates self-organized from linear poly(ethyleneimine) (PEI) and chiral tartaric acid (tart).24 In this system, a PEI containing secondary amine group can easily interact with a guest chiral tartaric acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of NH/COOH to form nanoscale crystalline complexes, which can serve as a template/catalyst/scaffold in a biomimetic silicification process. The nanofiber-based silica products mediated by PEI/tart complexes have not helices in their elementary morphology, but exhibits remarkable CD optical signals with mirror relationship in the absorption band lower than 200 nm wavelength due to Si–O bond and can make the achiral molecules adsorbed on the chiral silica to become optically active in the CD spectra. The characteristic feature of the chiral silica is its high-temperature resistance even over 900 °C.24,25 It was assumed that silica chirality is assigned to the silica frame of the Si–O–Si bond which may have asymmetric/helical structures similar to the molecular scale configuration and conformation with accompanying deviation of the bond of Si–O from the regular (symmetrical) tetrahedron of orthosilicate (SiO4). For accessing to this opinion, recently, destruction and/or pulverization of nanofibrous chiral silica into silica sols smaller than 10 nm using hydrothermal or chemical pulverizing methods was performed and it was found that the sub-10 nm silica sols with D- and L-forms showed a sharp chiroptical mirror relationship with exciton coupling around the Si–O–Si vibration regions in their vibration circular dichroism (VCD) spectra, although the sols did not possess any specific outwards shape.26,27 This strongly motivated the exploration of the chirality transfer from silica to other solid products.25–29 For example, lanthanide oxides prepared in chiral silica exhibited a remarkable circular polarized luminescent property after thermo-oxidation at 900 °C (ref. 25) whereas direct reduction of the chiral SiO2 using magnesium at 500 °C could result in silicon (Si) with a sustained chirality.26 Also, the chiral transfer from the inorganic silica to crosslinked polymeric materials was examined and it was found that the chiral silica could act as a chiral matrix for the formation of optically active chiral phenolic resins when using a polycondensation of resorcin and formaldehyde on the chiral silica surface.28 In addition, silver NP encapsulated chiral silica (Ag@SiO2) were also prepared using concentration of the cationic silver ions in chiral silica and which was followed by an in situ thermo-reduction of silver at over 500 °C, and then these were used in the reaction between nano silver and L-cysteine (L-Cys). Interestingly, it was found that chiral Ag@SiO2 preferentially reacted with L-Cys to give oligomeric complexes of [Ag–L-Cys]n but achiral Ag@SiO2 did not show reactivity.29 Compared to achiral Ag@SiO2, our chiral Ag@SiO2 exhibited excellent catalytic activity in the [3 + 2] cycloaddition reaction of azomethine ylides with 2′-hydroxychalcone derivatives with high yield and high regio-selectivity.30 However, it was not possible to elucidate whether the silver NPs themselves showed chirality although the silver NPs encapsulated in chiral Ag@SiO2 showed a strong CD signal in their plasmon resonance.
:1 molar ratio of NH/COOH to form nanoscale crystalline complexes, which can serve as a template/catalyst/scaffold in a biomimetic silicification process. The nanofiber-based silica products mediated by PEI/tart complexes have not helices in their elementary morphology, but exhibits remarkable CD optical signals with mirror relationship in the absorption band lower than 200 nm wavelength due to Si–O bond and can make the achiral molecules adsorbed on the chiral silica to become optically active in the CD spectra. The characteristic feature of the chiral silica is its high-temperature resistance even over 900 °C.24,25 It was assumed that silica chirality is assigned to the silica frame of the Si–O–Si bond which may have asymmetric/helical structures similar to the molecular scale configuration and conformation with accompanying deviation of the bond of Si–O from the regular (symmetrical) tetrahedron of orthosilicate (SiO4). For accessing to this opinion, recently, destruction and/or pulverization of nanofibrous chiral silica into silica sols smaller than 10 nm using hydrothermal or chemical pulverizing methods was performed and it was found that the sub-10 nm silica sols with D- and L-forms showed a sharp chiroptical mirror relationship with exciton coupling around the Si–O–Si vibration regions in their vibration circular dichroism (VCD) spectra, although the sols did not possess any specific outwards shape.26,27 This strongly motivated the exploration of the chirality transfer from silica to other solid products.25–29 For example, lanthanide oxides prepared in chiral silica exhibited a remarkable circular polarized luminescent property after thermo-oxidation at 900 °C (ref. 25) whereas direct reduction of the chiral SiO2 using magnesium at 500 °C could result in silicon (Si) with a sustained chirality.26 Also, the chiral transfer from the inorganic silica to crosslinked polymeric materials was examined and it was found that the chiral silica could act as a chiral matrix for the formation of optically active chiral phenolic resins when using a polycondensation of resorcin and formaldehyde on the chiral silica surface.28 In addition, silver NP encapsulated chiral silica (Ag@SiO2) were also prepared using concentration of the cationic silver ions in chiral silica and which was followed by an in situ thermo-reduction of silver at over 500 °C, and then these were used in the reaction between nano silver and L-cysteine (L-Cys). Interestingly, it was found that chiral Ag@SiO2 preferentially reacted with L-Cys to give oligomeric complexes of [Ag–L-Cys]n but achiral Ag@SiO2 did not show reactivity.29 Compared to achiral Ag@SiO2, our chiral Ag@SiO2 exhibited excellent catalytic activity in the [3 + 2] cycloaddition reaction of azomethine ylides with 2′-hydroxychalcone derivatives with high yield and high regio-selectivity.30 However, it was not possible to elucidate whether the silver NPs themselves showed chirality although the silver NPs encapsulated in chiral Ag@SiO2 showed a strong CD signal in their plasmon resonance.
In this paper, a new concept of chirality transfer from chiral silica (inorganic) to metallic NPs (inorganic) is given. The metallic NPs, which were grown in the matrices of a chiral siliceous network using a high temperature thermo-reduction procedure, showed remarkable chiroptical activity and possessed atomic distortion in their atomic packing lattice.
Experimental
Synthesis of PEI/tart@SiO2
In the typical procedure for preparing PEI/tart complexes, PEI·2H2O (0.316 g, 4.0 mmol –NH– groups; here one unit of (NHCH2CH2) possesses two molecules of water) was dissolved in 100 mL of water at about 80 °C. Then, 100 mL of an aqueous solution (at approximately 80 °C) containing 0.300 g of L- or D-tartaric acid (2.0 mmol carboxyl groups) was added to the hot PEI solution, stirred for a few minutes and allowed to cool until it reaches room temperature. Subsequently, the pH of the mixture was adjusted to pH 4 using ammonia (NH3) and was left standing at 4 °C overnight. The crystalline complexes of PEI/tart formed, were collected using centrifugation and then washed with water. Then, the obtained products were re-dispersed in 40 mL of water, and to this dispersion 6 mL of tetramethoxysilane (TMOS) as a silica source was added. The mixture was stirred for 2 h at room temperature. Finally, the white solid was collected, washed with water and acetone, and then air dried.Synthesis of ex-PEI@SiO2
The component of tartaric acid from PEI/tart@SiO2 can be easily removed using hydrochloric acid (HCl) solution. In a typical method, PEI/tart@SiO2 powders (1–2 g) were added into HCl (aq) (2.5 M, 50 mL), stirred for 1 h and then the product was collected using centrifugation. The same treatment was repeated five times. Then, the powders obtained were washed with 0.1 M NH3 (aq) to remove the HCl associated with PEI. Finally, the product was washed with water and acetone, and then dried at room temperature.Synthesis of PEI/tart@SiO2@Ag and SiO2@Ag composites
An aqueous solution of silver acetate (AgOAc) was prepared by dissolving 0.20 g of AgOAc in 35 mL of water in a 50 mL brown bottle at room temperature. Next, 0.30 g of PEI/tart@SiO2 hybrid was added into this solution and the mixture was stirred for 24 h under ambient conditions. The color of all the dispersions turned black. The black powders (Ag/PEI/tart@SiO2) were collected using centrifugation and washed five times with water and acetone and then dried in ambient conditions. Then, the black powders were calcined (thermo-reduction) at specific temperatures (500, 600, 700 or 800 °C) for 3 h in ambient conditions to obtain metal/silica composites, namely, Ag@SiO2-500, Ag@SiO2-600, Ag@SiO2-700 and Ag@SiO2-800.Synthesis of ex-PEI@SiO2@Au and SiO2@Au composites
Au aqueous solution of hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O) was prepared by dissolving 0.10 g of HAuCl4·4H2O in 30 mL of water in a 50 mL bottle at room temperature. Next, 0.20 g of ex-PEI@SiO2 hybrid was added into this solution and the mixture was stirred for 24 h under ambient conditions. The yellow powders (Au/PEI@SiO2) were collected using centrifugation, washed with water and acetone, and then dried at room temperature. Then, the yellow powders were calcined (thermo-reduction) at specific temperatures (500, 600, 700 or 800 °C, respectively) for 3 h in ambient conditions to obtain metal/silica composites, namely, Au@SiO2-500, Au@SiO2-600, Au@SiO2-700 and Au@SiO2-800.Preparation of Au/sol and Ag/sol using a hydrothermal reaction
Chiral (D-,L-) Au@SiO2-500 (50–80 mg) and distilled water (35 mL) were added into a 50 mL Teflon bottle. The Teflon bottle was tightly sealed up and hydrothermally treated in a stainless-steel autoclave at 180 °C for 5 h. After cooling, the unreacted sample was separated by centrifugation (4000 rpm, 5 min), and the supernatants containing SiO2 sol and Au NPs were collected and subjected to CD and VCD determination.Chiral (D-,L-) Ag/sol were prepared in the same way from Ag@SiO2-600 and subjected to CD and VCD determination.
Preparation of isolated chiral Au and Ag NPs from the hydrolysis of Au@SiO2 and Ag@SiO2 using NaOH (aq)
A portion of Au@SiO2-500 powder (0.02 g) and 30 mL of sodium hydroxide (NaOH) (aq) (5 wt%) were added in to a plastic bottle and stirred for at least 4 h. The silica was completely hydrolyzed into silicate and dissolved in the solution. The free Au NPs were collected using centrifugation (4000 rpm, 5 min) and washed with water. Then, Au NPs were re-dispersed in water and subjected to CD determination.Chiral (D-,L-) Ag NPs were prepared in the same way from Ag@SiO2-600 and subjected to CD and VCD determination.
Preparation of achiral cPEI@SiO2 and aAu@SiO2-500
Using the typical procedure of preparing achiral PEI@SiO2 as shown below in the next section. PEI (0.474 g, 6.0 mmol basic secondary amino groups) was dissolved in 20 mL of water at about 80 °C. Then, 20 g of crushed ice was added into the hot solution of PEI and left to stand at room temperature for 2 h to obtain a white dispersion (cPEI: crystalline aggregates of PEI). The aggregates were collected using centrifugation and washing with water. Then, the aggregates were re-dispersed in 40 mL of water to give white dispersions, and then 3 mL of TMOS as a silica source was added to the dispersion. The mixture was stirred for 2 h at room temperature. Finally, the white solid (cPEI@SiO2) was collected, washed with water and acetone, and then air dried.Then, the cPEI@SiO2 was used for preparation of an Au/silica composite (aAu@SiO2-500) using the following procedure. An aqueous solution of HAuCl4·4H2O was prepared by dissolving 0.10 g of HAuCl4·4H2O in 30 mL of water in a 50 mL bottle at room temperature. Next, 0.20 g of cPEI@SiO2 hybrid was added into this solution and the mixture was stirred for 24 h under ambient conditions. The yellow powders (Au/cPEI@SiO2) were collected using centrifugation, washed with water and acetone, and then dried at room temperature. Then, the yellow powders were calcined at 500 °C for 3 h in ambient conditions to obtain the inorganic composite (aAu@SiO2-500).
Preparation of achiral aAg@SiO2-500
Similar to the method described in the previous section in the preparation of achiral Au/silica system, the cPEI@SiO2 was used for the preparation of an Ag/silica composite (aAg@SiO2-500) using the following procedure. An aqueous solution of silver nitrate (AgNO3) was prepared by dissolving 0.08 g of AgNO3 in 10 mL of water containing 1 mL of NH3 (aq) solution (100 mM). Next, 0.10 g of cPEI@SiO2 hybrid was added into this solution and the mixture was stirred for two days under ambient conditions. The dark powder (Ag/cPEI@SiO2) was collected using centrifugation, washed with water and acetone, and then dried at room temperature. Next, the powder was heated at 500 °C for 3 h in ambient conditions to obtain the inorganic composite (aAg@SiO2-500).Characterization
The X-ray diffraction (XRD) patterns were collected on a Rigaku RINT Ultima III X-ray diffractometer with Cu Kα radiation (λ = 0.1540 nm). The scanning electron microscopy (SEM) images were taken on a Hitachi SU8010 SEM equipped with an energy dispersive spectrometer. The transmission electron microscopy (TEM) analysis was performed on a Hitachi HT7700 instrument with an acceleration voltage of 200 kV. The spectra of solid-state diffuse reflectance circular dichroism (DRCD) and ultraviolet-visible (UV-vis) absorption of the solid products (40 wt%) dispersed in potassium chloride were recorded simultaneously on a JASCO J-820 spectropolarimeter equipped with a DRCD-466L integrating unit. The thermogravimetric (TG) analysis was conducted on a Seiko Instruments Exstar6000 instrument. The UV-vis absorption spectra of liquid samples were acquired on a Shimadzu UV-2500 PC spectrophotometer. The VCD spectra and infrared (IR) spectra were obtained simultaneously on a JASCO FVS-6000 VCD spectrometer. The suspension of SiO2 debris was prepared, by homogenizing the methanol solution containing chiral silica powder (calcined PEI/tart@SiO2 at 600 °C) at for at least 20000 rpm for 2 min, and dropping the resulting suspension onto a silicon wafer for VCD testing. The Au/SiO2 and Ag/SiO2 sols were dispersed in 0.25 mL of methanol containing 0.002 g of poly(vinylpyrrolidone) K90 (PVP). After stirring for several minutes, the solution was dropped onto a silicon wafer and then subjected to VCD testing. Scanning transmission electron microscopy (STEM) images were obtained using a Jeol JEM ARM200F atomic resolution analytical electron microscope with an operating voltage at 200 kV. A Jeol JED-2300T X-ray energy-dispersive spectrometer was used for compositional analysis. The samples for the STEM analysis were prepared by dropping an ethanol suspension of the sample powders onto a commercial copper microgrid coated with a polymer film. The samples were thoroughly dried under vacuum prior to observation.
Results and discussion
Usually, templating dependent chirality transfer is carried out from chiral organic objects to inorganic silica. But it is rare that examination of the successive chiral transfer from the chiral silica prepared in advance to the other materials. Realization of chiral transfer relay from organics to silica and then from silica to inorganics and/or organics is a great challenge for understanding the rules of chirality transfer and for developing new chiral materials. In this research, as shown in Scheme 1, nanofibrous bundles of two chiral composites of Ag@SiO2 and Au@SiO2 were first prepared, using a chiral transfer system started with tartaric acid and then the Ag and Au NPs were detached and/or isolated by removing the silica bundles using hydrothermal treatment and/or hydrolysis of the silica frames. Firstly, nanofibrous silica bundles were transformed into SiO2 sol and then the Ag and Au NPs encapsulated in the silica bundles were released, while in the last part of the synthesis, the silica was completely dissolved and metallic NPs were isolated as a free-state solution. All the samples obtained using two different approaches were analyzed using CD and VCD and it was found that the isolated Ag and Au NPs intermediates on the chiral matrices of D- and L-form silica showed remarkable exciton coupling chiroptical properties with mirror relations although there were no outward helical appearances on the metallic NPs. As far as is known, this is first time that it has been found that the chirality information of silica was able to be transferred directly to the metallic NPs without the assistance of chiral organic compounds.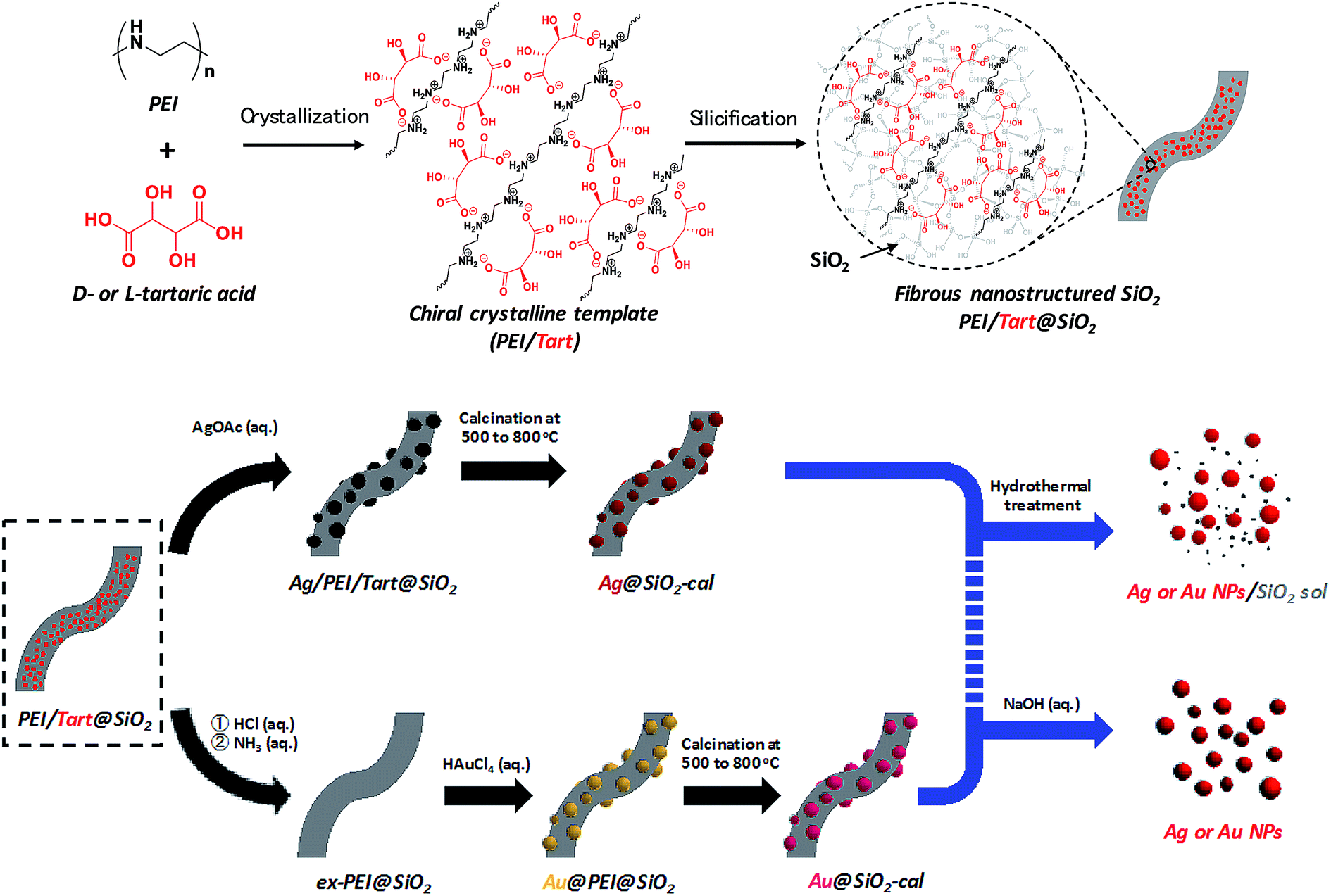 | ||
| Scheme 1 Representation of the procedure for obtaining chiral SiO2 and metal@SiO2 composites and two approaches for isolation of metal NPs from the metal@SiO2 composites. | ||
As shown in Scheme 1, a PEI containing a basic secondary amine group in the main chain easily interacts with D- or L-tartaric acid in a 1:1 molar ratio of NH/COOH to form bundles of nanofiber-based crystalline complexes (PEI/tart) (Fig. S1, ESI†). These bundles spontaneously catalyzed the silicification of TMOS around the bundles to give morphology-transcribed hybridized bundles of PEI/D-tart@SiO2 and PEI/L-tart@SiO2. The detailed features of the D- and L-forms of PEI/tart@SiO2 based on the results of Fourier-transform-IR (FT-IR), SEM, TG-differential thermal analysis (DTA), and CD are indicated in Fig. S2–S5 (ESI†). To directly clarify the chirality of the silica, the fibrous bundles of silica (calcined at 600 °C) were crushed using a homogenizer and the broken silica pieces were analyzed using VCD. Fig. 1a–c shows SEM images before and after crushing. It is clearly observable that the D- and L-form silica bundles were densely aggregated with nanofibers which have not any helical outwards and the crushed pieces appeared as irregular forms. Interestingly, the crushed silica showed very clear VCD activity with exciton chirality around the Si–O–Si stretches at 980–1200 cm−1. The D-form appeared with a positive and then a negative spectral line whereas the L-form appeared with a negative and then a positive spectral line, which just satisfied the requirements for the non-superposable mirror image relationship with each other (Fig. 1d). This result was consistent with previous results for chiral silica sols prepared using a hydrothermal reaction of silica bundles, indicating that the silica skeletons of supporting the bundles have chirality.
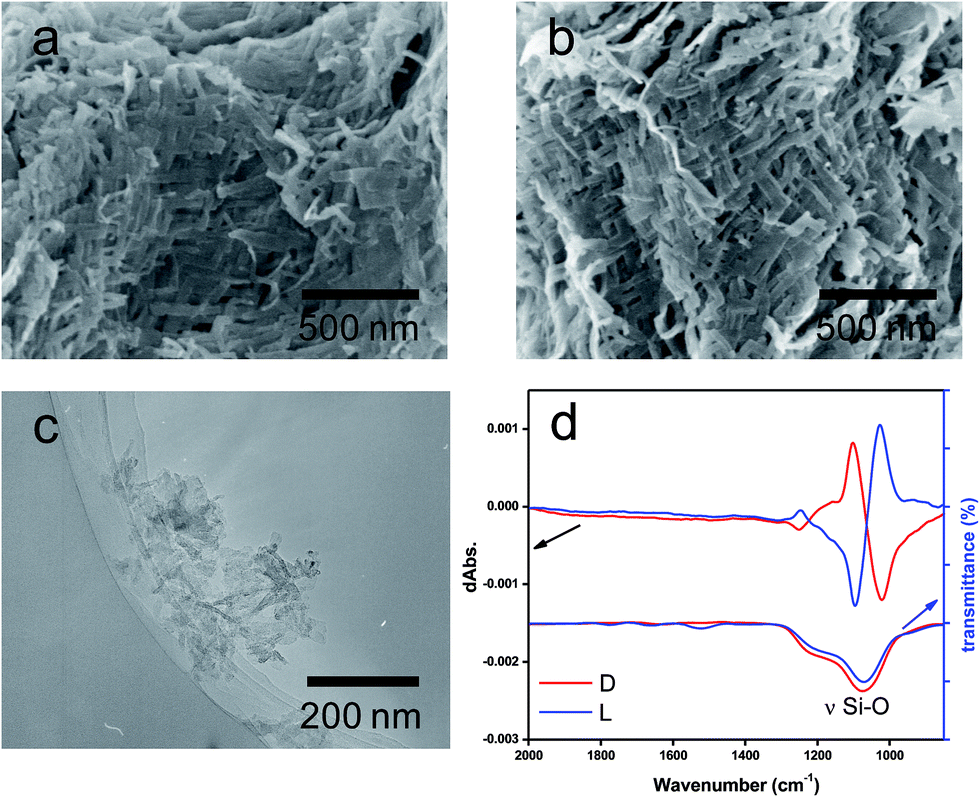 | ||
| Fig. 1 SEM images of calcined silicas, (a) D-SiO2-600 and (b) L-SiO2-600. TEM images of (c) the piece of L-SiO2 crushed by homogenizing the methanol solution containing chiral silica bundles (calcined PEI/tart@SiO2) for at least 20000 rpm for 2 min. (d) FT-IR/VCD spectra of the crushed SiO2, red lines for the D-form, and blue lines for the L-form. | ||
The hybrids of PEI/tart@SiO2 themselves are effective scavengers for trapping silver ions (AgNO3, AgOAc, and so on) via anion exchange to form complexes Ag/tart/PEI in silica. Therefore, the precursors of Ag/PEI/tart@SiO2 were synthesized by simply mixing PEI/tart@SiO2 with AgOAc. However, the tartaric acid component was excluded from PEI/tart@SiO2 by using alternate treatment with HCl (aq) and NH3 and the ex-PEI@SiO2 obtained was mixed with an aqueous solution of HAuCl4 for the preparation of the precursor of Au/PEI@SiO2. In this process, the PEI encapsulated in silica can act as a reductant to reduce a moderate amount of silver and gold ions to metallic NPs.24,29,31 The two metallic precursors of Au/PEI@SiO2 and Ag/PEI/tart@SiO2 were further thermo-reduced (calcination) under an air atmosphere at a temperature above 500 °C and under these conditions the metal ions were totally transformed into metallic NPs to form hybrids of Au@SiO2 and Ag@SiO2. Fig. 2a–d show TEM images of D- and L-Au@SiO2 and D- and L-Ag@SiO2. It is obvious that the fibrous nanosilica remained unchanged after thermo-reduction for both Ag and Au, and the metallic NPs smaller than 50 nm were distributed around the silica bundles. The powders were analyzed before and after thermo-reduction (calcination) using DRCD spectroscopy and the results of this were compared with their CD spectra. As shown in Fig. 3, the precursors of the D- and L-form of Ag/PEI/tart@SiO2 as-prepared at room temperature showed strong CD activity at the plasmon resonances at longer wavelengths of over 600 nm. However, the powders of D- and L-Ag@SiO2 after thermo-reduction exhibited CD at plasmon resonances below 600 nm. For the gold NP system, the precursor of D- and L-Au/PEI@SiO2 prepared at room temperature, had a yellow color indicating that a large amount of Au was trapped, as the anionic form of AuCl4−, on the silica hybrids because of the binding to the protonated PEI but showed chiroptical mirror relationship CD lines corresponding to the absorption band below 400 nm. In comparison, the samples of D- and L-Au@SiO2 obtained after thermo-reduction at 500 °C appeared as wine-red color and their plasmon resonance appeared in the longer visible wavelength region. Corresponding to the characteristic electronic absorbance around 500–800 nm, the samples of D- and L-Au@SiO2 showed chiroptical mirror relationship CD lines like a lying “∫” shape which were tugged by the accompanying strong CD signals within 200–500 nm.
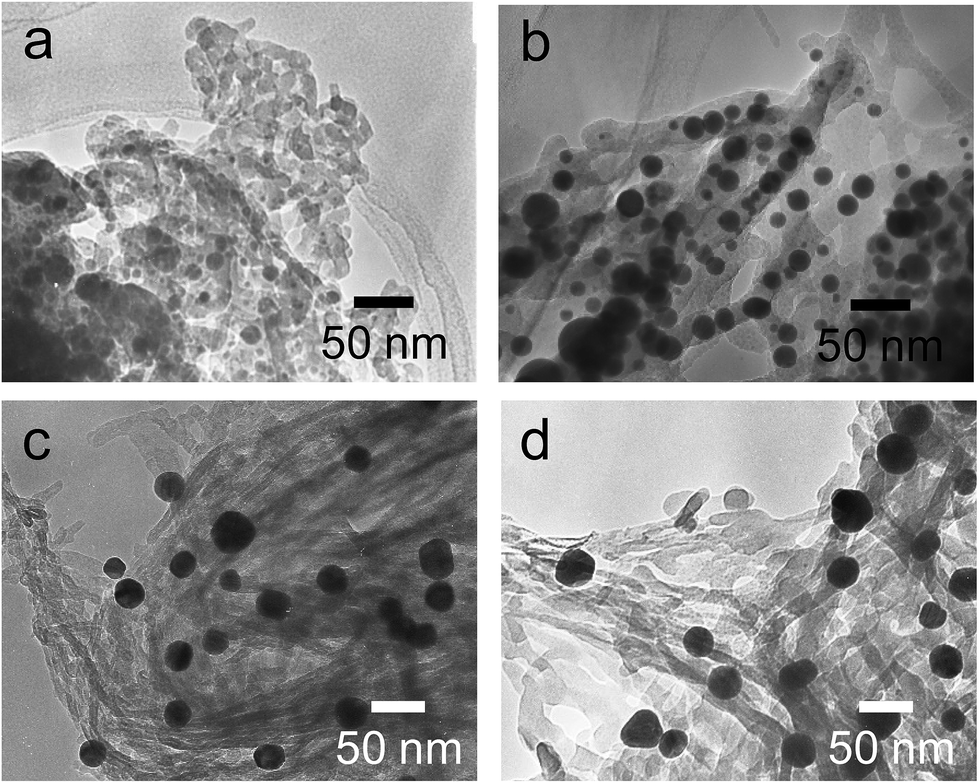 | ||
| Fig. 2 TEM images of (a) D-, (b) L-Ag@SiO2-500 and (c) D-, (d) L-Au@SiO2-500. | ||
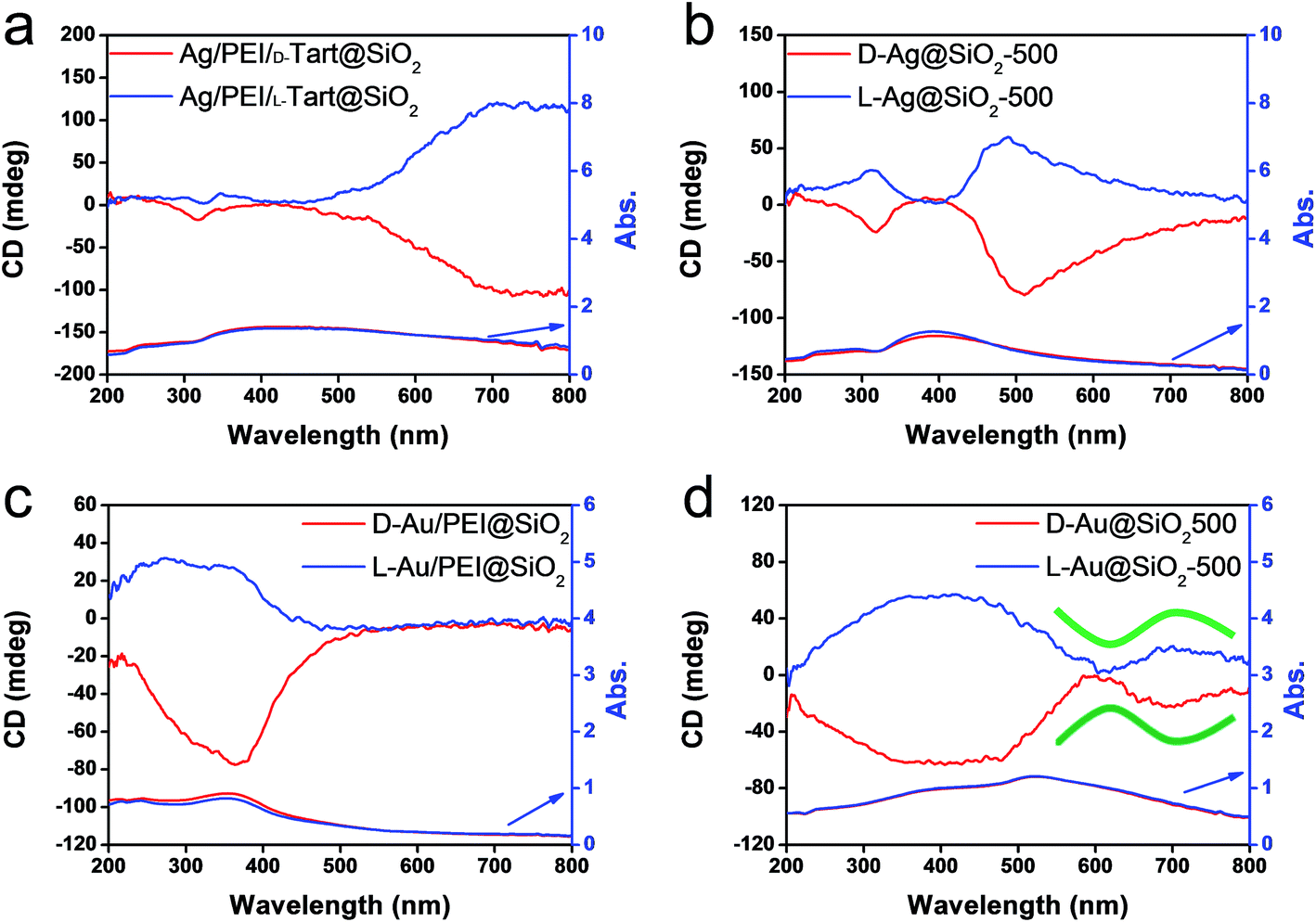 | ||
| Fig. 3 DRCD and UV-vis spectra of (a) Ag/PEI/tart@SiO2, (b) Ag@SiO2-500, (c) Au/PEI@SiO2 and (d) Au@SiO2-500, red lines for D-form and blue lines for L-form. | ||
To clarify the effects of the thermo-reduction temperature on the features of the metallic NPs, the as-prepared samples of Ag/PEI/tart@SiO2 and Au/PEI@SiO2 were treated at different temperatures of 600, 700 or 800 °C and the crystallite size, morphology and CD activities of the resulting metallic NPs were compared using XRD, TEM and DRCD. As shown in the XRD results (Fig. S6 and Table S1, ESI†), the Ag NPs had a fcc lattice crystalline structure and their averaged crystallite sizes trended to increase from 20 nm to near 40 nm as the temperature increased from 600 °C to 800 °C. In parallel, the sizes of the Ag NPs observed from the TEM images became larger (most of them up to 100 nm; individual NPs) with the spherical morphology remaining as the temperature increased (Fig. S7, ESI†). Nevertheless, the fibrous silica structure remained intact in this thermo-reduction (calcination) process. These results indicated that the Ag NPs can easily gather and merge together under a higher temperature environment. Unexpectedly, the size alteration of the Ag NPs caused dramatic changes in the DRCD spectral lines (see Fig. S8, ESI†). Compared to the as-prepared Ag/PEI/D-tart@SiO2, which showed a negative Cotton effect in the plasmon resonance region of the Ag NPs, the Cotton effect of the composites D-Ag@SiO2-800 became completely positive via a stepwise sign inversion of bisignate CD signals exhibited from minus to plus accompanying a blue shift in the course of increasing temperature. Such a phenomenon was also the same for the L-form samples with an opposite signature to form chiroptical mirror relationships to the D-forms.
Slightly different to the Ag system, the Au/SiO2 samples obtained using thermo-reduction at 500, 600, 700 or 800 °C did not change their crystallites sizes which altered around 35–45 nm (see Table S2 and Fig. S9, ESI†). The absorption spectra based on the plasmon resonance were also similar to each other for the D- and L-forms obtained at different temperatures and the corresponding DRCD spectra did not show CD line inversion (Fig. S10, ESI†). However, around longer wavelengths over 550 nm, the CD lines seems to be changed somewhat with the appearance of bisignate CD signals for the samples Au@SiO2-800 after treatment of Au/PEI@SiO2 at 800 °C. However, it is not known what causes the strong and broad CD lines ranged within 200–500 nm for the samples of Au@SiO2. It is well known that the Au NPs should have plasmon resonance at a visible wavelength region larger than 450 nm, and thus, the CD line below 450 nm did not relate to Au NPs. In some ways, the CD line shape below 450 nm should be attributed to side-products transformed from PEI. Possibly, the side-products are of relevance with Au ions and/or NPs which act as catalysts to promote the formation of a few carbonaceous products that have an electronic absorption property in the range of 200–500 nm. A similar phenomenon was also observed in recent work when a precursor of PEI/SiO2/terbium was sintered at high temperature25 (but this is not an argument for this paper).
As has been mentioned previously, it is thought that the hybrids of PEI/SiO2 are effective matrices to prepare metallic NPs on SiO2 and the formed metallic NPs behave as if they have chirality. The interest here is to make clear whether the metallic NPs themselves have chirality. For this purpose, two approaches were introduced to detach or isolate the metallic NPs from the SiO2 silica bundles: one is detachment of Ag and/or Au from the silica bundles using a hydrothermal treatment of the powders of Ag@SiO2-600 and Au@SiO2-500, and another is isolation of Ag and/or Au from the silica bundles using complete hydrolysis of the SiO2 frame into sodium silicate (Na2SiO3) using NaOH (aq). These two approaches could detach or isolate the metallic NPs from the SiO2 bundles and they then form metallic NPs dispersed aqueous solutions in which the former one (Ag + sol and Au + sol) was a mixture of metallic NPs and silica sols that was still chiroptically active whereas the latter one (Ag and Au) was free metallic NPs without a SiO2 component. The hydrothermal reaction was performed by heating the mixture of chiral SiO2@metal composites and distilled water at 180 °C. This reaction produced colored transparent aqueous solutions (Fig. 4a1 and b1) containing free metallic NPs and SiO2 sol (M/sol) indicating that the SiO2 bundles were broken into SiO2 sol and metallic NPs which were well dispersed in water. Subjecting the Ag/sol and Au/sol (Fig. 4a1, a2 and b1, b2) to TEM visualization, spherical and/or elliptical metallic nanoparticles smaller than 50 nm for Au and Ag were observed. In addition, silica sols smaller than 10 nm were also observed from these solutions. To determine the chiroptical properties of these aqueous solutions, their CD and electronic absorption spectra were investigated. Interestingly, both Au and Ag showed bisignates in the wavelengths of their plasmon absorption regions (Fig. 4c and d, Ag/sol and Au/sol). D-Form Au NPs showed the CD line from negative to positive whereas the L-form Au NPs just appeared in the opposite order, exhibiting a well-paired mirror relationship to each other. Similarly, the D- and L-form Ag NPs also showed bisignates but with a different sign order compared to the Au samples. Previously, it was reported that the chiral silica sols with an average size of 7 nm, obtained using a hydrothermal reaction of the chiral silica bundles, exhibited remarkable VCD activity.26 Therefore, there would be a question about whether the chiral SiO2 sols cause the induced chirality of the Ag NPs or Au NPs.
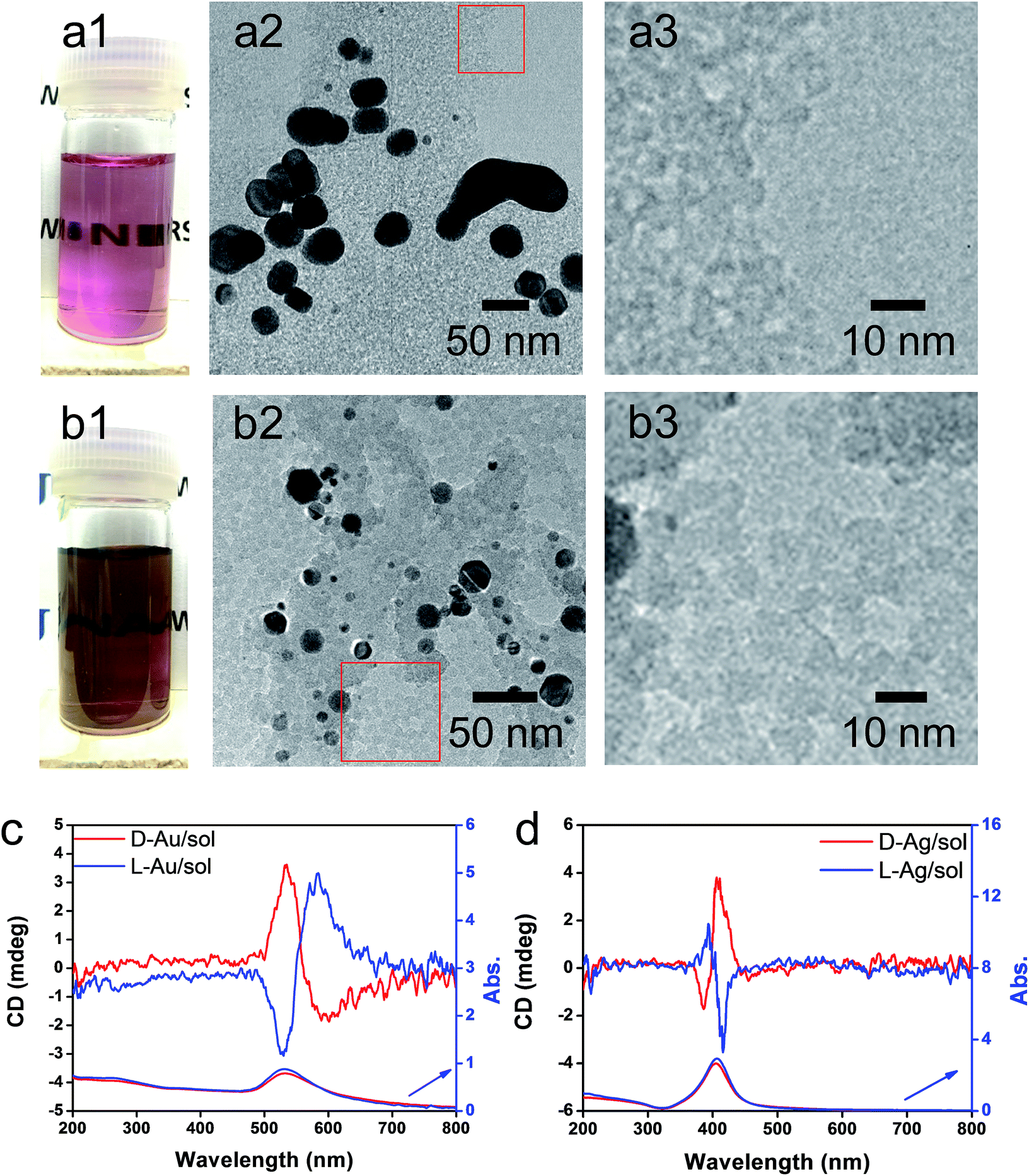 | ||
| Fig. 4 Aqueous solution of L-Au/sol (a1) and L-Ag/sol (b1). TEM images at different magnifications of L-Au/sol (a2 and a3) and L-Ag/sol (b2 and b3) (taken from a highly diluted solution containing Au/sol or Ag/sol). CD and UV-vis spectra of Au/sol (c) and Ag/sol (d) (both samples were diluted from their stock solutions), red lines for the D-form and blue lines for the L-form. | ||
To elucidate the chirality originating from the metallic NPs, the optical properties of Au/sol and Ag/sol were further investigated using VCD by introducing a labelling probe for the metallic NPs. From previous work on chiral Si NPs in which it was learned that the adsorbed PVP was made active in VCD spectra because of the induced effect from Si although the silica sol could not cause such an effect.26 It is well known that PVP interacts effectively with the surface of the metallic NPs because of the strong capping power of the amide groups on the metallic NPs, and thus, it is often used as a stabilizing agent for the dispersion of metallic NPs in organic/aqueous/liquid ionic media.32,33 For this reason, in this research, PVP was used as a convenient probe in VCD to determine the chiroptical features of the metallic NPs. First of all, casting films were prepared on a silicon wafer using a mixture of PVP/Au/sol and PVP/Ag/sol and then they were analyzed using VCD measurements (Fig. 5). In this case, two chiral factors, which would be derived from metallic NPs and SiO2 sol, can be considered. If the chiral domain exists in the metallic NPs, PVP should exhibit the induced VCD signals in the region of PVP absorption. As shown in Fig. 5c and d, FT-IR spectra of PVP/Au/sol and PVP/Ag/sol showed vibration signals of PVP in the range of 1000–1800 cm−1 (the bands located at 1658, 1427 and 1287 cm−1 were assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, CH2 bending and C–N vibration, respectively). As was expected, the VCD spectra indicated the Cotton effects corresponding to the vibrations of PVP, particularly to the stretching vibration of the CO group with a remarkable chiroptical mirror relationship (see Fig. 5a and b) both for D/L-Au and D/L-Ag systems. This strongly suggests that the metallic NPs have sufficient chiral structures to interact with the amide groups of PVP, thus causing the induced VCD activity from the achiral polymer. In comparison, PVP adsorbed achiral Ag and Au NPs did not show VCD signals although they have strong stretching peaks around 1660 cm−1 because of the CO group (see Fig. S11†).
O stretching, CH2 bending and C–N vibration, respectively). As was expected, the VCD spectra indicated the Cotton effects corresponding to the vibrations of PVP, particularly to the stretching vibration of the CO group with a remarkable chiroptical mirror relationship (see Fig. 5a and b) both for D/L-Au and D/L-Ag systems. This strongly suggests that the metallic NPs have sufficient chiral structures to interact with the amide groups of PVP, thus causing the induced VCD activity from the achiral polymer. In comparison, PVP adsorbed achiral Ag and Au NPs did not show VCD signals although they have strong stretching peaks around 1660 cm−1 because of the CO group (see Fig. S11†).
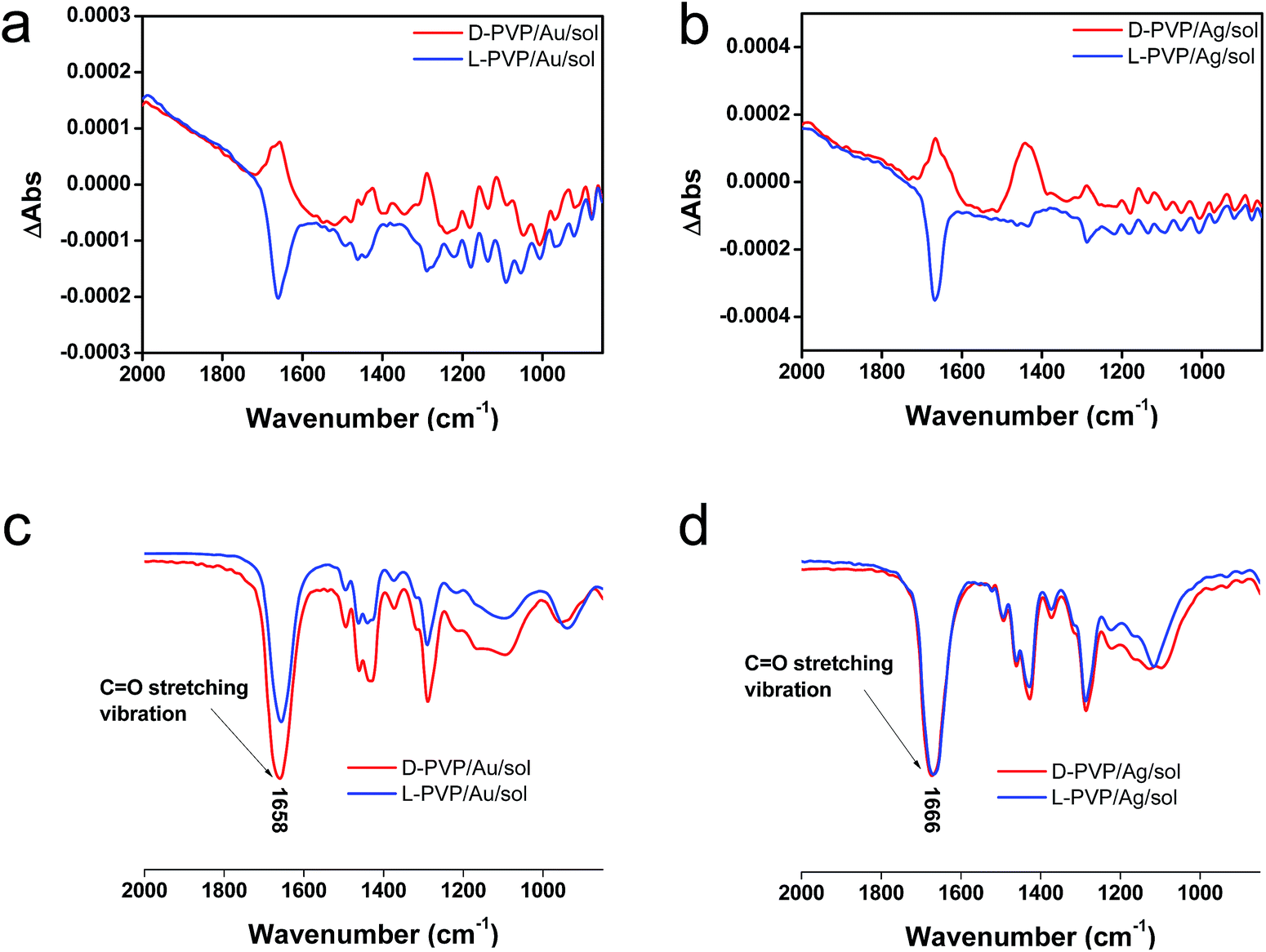 | ||
| Fig. 5 VCD and IR spectra of (a), (c) PVP/Au/sol and (b), (d) PVP/Ag/sol (the Au/sol and Ag/sol were dispersed in 0.25 mL of methanol containing 0.002 g of PVP K90), after stirring for several minutes, the solution was dropped onto the silicon wafer and then subjected to VCD testing. | ||
From the above results, it is conclusive that Au and Ag NPs formed using thermo-reduction in chiral silica have chirality. However, it is still need to further evaluate the behaviors of the isolated metallic NPs via destroying and removing the silica. For this purpose, the hydrolysis of silica from Au@SiO2-500 and Ag@SiO2-600 was performed by use of NaOH (aq) and all the water-soluble Na2SiO3 was removed using centrifugal washing with water (twice). The Au NPs precipitated were re-dispersed in water before the characterizations. To confirm the state of removal of the SiO2 component, the D- and L-Au samples were subjected to FT-IR. As can be seen from Fig. S12 (ESI†), the strong stretching vibration around 980–1150 cm−1 from the Si–O–Si bond had completely disappeared from the isolated samples. In addition, the isolated D- and L-Au still had the same shape – either a spherical and/or elliptical shape smaller than 50 nm (Fig. S13, ESI†). For Ag NPs, however, some fused nanoparticles were observed in the TEM image (Fig. S14, ESI†) indicating that the Ag NPs were less stable than the Au NPs. The solution containing the isolated Au NPs was examined using CD and UV-vis spectroscopies. As shown in Fig. 6a, surprisingly, the D- and L-Au NPs showed remarkable bisignate CD signals with mirror relationships in the plasmon resonance region around 480–800 nm. These spectral lines were identical to those shown for Au/sol shown in Fig. 4c, that is, both chiral signs from isolated Au and detached Au/sol were attributed to the Au NPs themselves. However, the Ag NPs obtained in the same way also showed CD activity (Fig. 6b), but the bisignate CD lines were insufficient to satisfy a clear mirror relationship unlike the Au NPs, and this was probably because of lower chemical durability of Ag NPs towards the NaOH (aq) treatment.
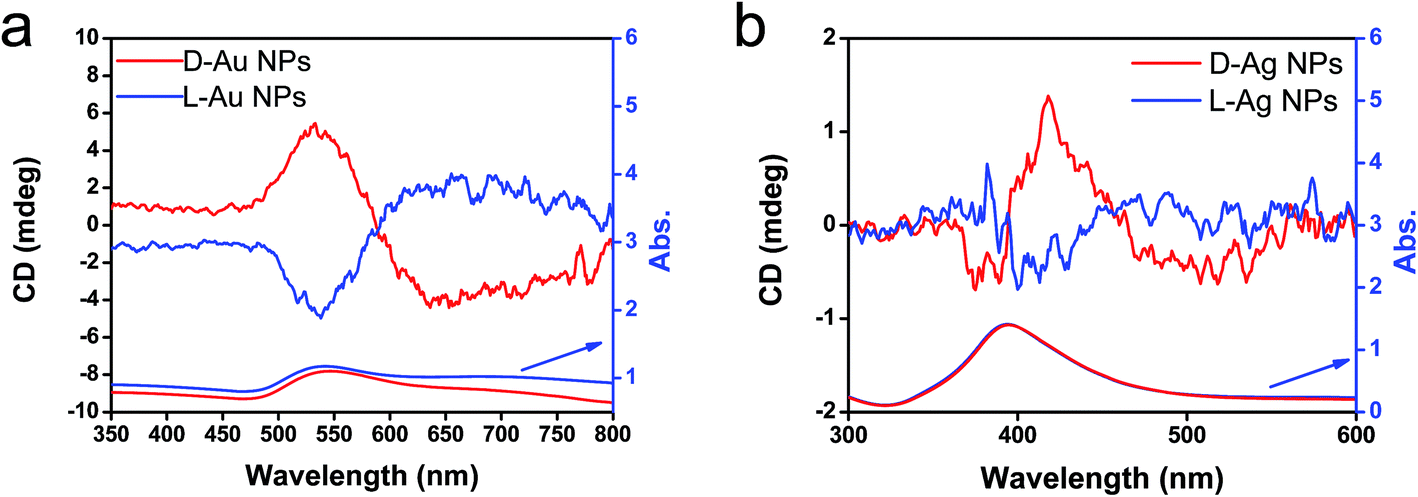 | ||
| Fig. 6 CD and UV-vis spectra of (a) chiral Au NPs and (b) Ag NPs dispersed in water obtained from the hydrolysis (by using 5 wt% NaOH) of chiral Au@SiO2-500 and Ag@SiO2-600, respectively. | ||
The previous results unambiguously indicated that the silica with chiral domains in the siliceous frames was capable of transferring chiral information through space to metallic NPs grown in the siliceous matrices. This is the first time that inorganic based chiral transfer systems from silica (inorganics) to nano metals (inorganics) has been achieved. The metallic NPs generated from chiral siliceous matrices were less than 30 nm with almost spherical morphology so that there was no doubt that there was no shape chirality. Therefore, it was expected that the chiral domain of the metallic NPs might be derived from defects or distortions of crystalline structure including point, line and plane, which would lead to the asymmetric arrangement of metal atoms.34–36 That is, in this system, chirality might originate at the atomic scale through crystalline structures. To support this hypothesis, effort was concentrated on determining the fine crystalline structure from the metallic NP-loaded silica samples using high-angle annular dark-field (HAADF)-STEM. Fortunately, clear nanocrystal lattices were visualized with atomic orientation along the {111} direction of fcc from samples of D-SiO2@Au-500 (Fig. 7a1 and a2). Surprisingly, the atomic arrangement line, which was magnified from the edge (region in the square in Fig. 7a1) of a nearly 30 nm Au particle, distorted exactly through several nanometer lengths in which atoms do not occupy the straight line. This was an uncommon phenomenon in Au NPs. To address the novelty of such distortion, achiral Au NPs were prepared on the matrix of achiral nanofibrous bundles of cPEI@SiO2 using a 500 °C thermo-reduction process similar to the chiral system and the two types of AU NPs were compared. As shown in Fig. S15 (ESI†), the Au NPs were well distributed around the SiO2 nanofibers. This achiral aAu@SiO2-500 sample was subjected to HAADF-STEM to compare it with the chiral D-Au@SiO2-500. As can be seen in Fig. 7b, in this nanocrystal lattice surface, no distortions of atomic array along the {111} direction were observable. All the atoms on the edge (square region in Fig. 7b) of a nearly 20 nm sized Au NP occupying a straight orientation. Quite obviously, there are definite differences between chiral and achiral Au NPs when viewed in the same direction of {111} fcc nanocrystal lattice surface although their preparation process is the same. In the chiral Au nanocrystal, the atoms tend to pack in a distorted fashion whereas in achiral one, the atoms pack in a precisely ordered form. Such distinct differences of atomic arrangement also vividly appeared between chiral Ag@SiO2-500 and achiral aAg@SiO2 (see Fig. 8). The atoms packing along the {111} direction seemed disordered in chiral D-Ag@SiO2 whereas it was precisely ordered in the achiral aAg@SiO2. Therefore, it was thought that the atomic distortion would be the reason why the chiral D-Ag@SiO2 showed a high catalytic activity and high regio-selectivity in the [3 + 2] cycloaddition reaction of azomethine ylides with 2′-hydroxychalcone derivatives unlike that of achiral Ag@SiO2.30 From these comparative atomic lattice results, it was believed that the origin of chirality in the metallic NPs was in the atomic packing distortion in their nanocrystalline structure. However, the relationship between the atomic distortion and the chirality is not clear at present, and it needs to be solved in the future.
 | ||
| Fig. 7 STEM analysis and crystal models (insets) for chiral (a) and achiral (b) Au NPs. (a1 and b1) Low magnification HAADF-STEM images of chiral D-Au@SiO2-500 and achiral aAu@SiO2-500. (a2 and b2) The atomic resolution HAADF-STEM images of Au NPs of green square regions in (a1) and (b1), respectively, viewed along the direction of {111} for chiral and achiral Au NPs. Insets: model for the atomic arrangement of the fcc structure viewed along the {111} direction for chiral and achiral Au NPs; the incident: {110} for (a2) and {112} for (b2) with the unit cells (gray regions). (a3) Enlarged image of (a2) for visualization of the non-straight arrangement of the atomic array. Similar images from other areas are also shown in Fig. S16 (ESI†). | ||
 | ||
| Fig. 8 STEM analysis and crystal models (insets) for chiral (a) and achiral (b) Ag NPs. (a1 and b1) Low magnification TEM images of chiral D-Ag@SiO2-500 and achiral aAg@SiO2-500. (a2 and b2) Low magnification HAADF-STEM images of one Ag particle from the green marked regions in (a1) and (b1), respectively. (a3 and b3) The atomic resolution HAADF-STEM images of Ag NPs from the red marked regions in (a2) and (b2), respectively, viewed along the direction of {111} for chiral and achiral Au NPs. Insets: model for the atomic arrangement of the fcc structure viewed along the {111} direction for chiral and achiral Ag NPs; the incident: {110} for (a3) and (b3) with the unit cells (gray regions). Similar images from other areas are also shown in Fig. S17 (ESI†). | ||
Conclusions
The chiral silica with nanofibrous bundles templated using crystalline complexes of PEI/tart has molecular scale chiral domains throughout its siliceous frame and is able to transfer their chiral information through restricted space to the metallic nanoparticles grown around the silica matrices. Notably, the interior of the as-prepared chiral silica retaining PEI in the silica wall is a favourable place to self-capture Au+ and Ag+ ions. This is an ideal ground to allow the captured metal ions to be reduced and grew along a relay-like chiral track given and ruled by the silica. So, the chiroptically active metallic NPs in SiO2 can easily be prepared, and thus, dual chirality composites consisted of chiral silica and chiral nanometals. Such a chemical reduction process without assistance of asymmetric organic molecules is a new method for endowing metallic NPs with chirality, although using physical processing, for example, a so-called glancing angle deposition technique, it is possible to prepare helical nanometals without the assistance of molecules.37 In particular, the Au NPs and Ag NPs exhibited intrinsic CD signals with exciton chirality in the plasmonic resonance of nano Ag and Au indicating the existence of the distorted atomic orientation in their crystalline structures. It is expected that the chiral metallic NPs with irregular lattice structures will be able to be used in developing and designing thermo-resistant plasmonic metallic materials and catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by MEXT-Supported Program for the Strategic Research Foundation at Private Universities: “Creation of new fusion materials by integration of highly-ordered nano inorganic materials and ultra-precisely controlled organic polymers” (2013–2017) and by the JSPS KAKENHI Grant Number JP16H06515 (Coordination Asymmetry). We thank Mr K. Shinoda for his help with the STEM measurements at National Institute for Materials Science (NIMS) Battery Research Platform.References
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- Y. Wang, J. Xu, Y. Wang and H. Chen, Chem. Soc. Rev., 2013, 42, 2930–2962 RSC.
- A. G. Mark, J. G. Gibbs, T.-C. Lee and P. Fischer, Nat. Mater., 2013, 12, 802–807 CrossRef CAS PubMed.
- Y. Okazaki, T. Buffeteau, E. Siurdyban, D. Talaga, N. Ryu, R. Yagi, E. Pouget, M. Takafuji, H. Ihara and R. Oda, Nano Lett., 2016, 16, 6411–6415 CrossRef CAS PubMed.
- M. V. Mukhina, V. G. Maslov, A. V. Baranov, A. V. Fedorov, A. O. Orlova, F. Purcell-Milton, J. Govan and Y. K. Gunko, Nano Lett., 2015, 15, 2844–2851 CrossRef CAS PubMed.
- A. Gabashvili, D. D. Medina, A. Gedanken and Y. Mastai, J. Phys. Chem. B, 2007, 111, 11105–11110 CrossRef CAS PubMed.
- A. J. Mastroianni, S. A. Claridge and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 8455–8459 CrossRef CAS PubMed.
- A. Guerrero-Martínez, B. Auguie, J. L. Alonso-Gomez, Z. Dzolic, S. Gmez-Graa, M. Zinic, M. M. Cid and L. M. Liz-Marzn, Angew. Chem., Int. Ed., 2011, 50, 5499–5503 CrossRef PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Hogele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- K. W. Smith, ACS Nano, 2016, 10, 6180–6188 CrossRef CAS PubMed.
- H. Qi, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, J. Am. Chem. Soc., 2011, 133, 3728–3731 CrossRef CAS PubMed.
- J. Xie, Y. Duan and S. Che, Adv. Funct. Mater., 2012, 22, 3784–3792 CrossRef CAS.
- J. Cheng, G. L. Saux, J. Gao, T. Buffeteau, Y. Battie, P. Barois, V. Ponsinet, M. Delville, O. Ersen, E. Pouget and R. Oda, ACS Nano, 2017, 11, 3806–3818 CrossRef CAS PubMed.
- A. Ben-Moshe, et al. , Chem. Soc. Rev., 2013, 42, 7028–7041 RSC.
- F. Lu, Y. Tian, M. Liu, D. Su, H. Zhang, A. O. Govorov and O. Gang, Nano Lett., 2013, 13, 3145–3151 CrossRef CAS PubMed.
- T. Hu, B. P. Isaacoff, J. H. Bahng, C. Hao, Y. Zhou, J. Zhu, X. Li, Z. Wang, S. Liu, C. Xu, J. S. Biteen and N. A. Kotov, Nano Lett., 2014, 14, 6799–6810 CrossRef CAS PubMed.
- G. Cyrille and B. Thomas, J. Am. Chem. Soc., 2008, 130, 7077–7084 CrossRef PubMed.
- H.-E. Lee, H.-Y. Ahn, J. Mun, Y. Lee, M. Kim, N. Cho, K. Chang, W. Kim, J. Rho and K. Nam, Nature, 2018, 556, 360–365 CrossRef CAS PubMed.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Burgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- S. Knoppe, I. Dolamic, A. Dass and T. Burgi, Angew. Chem., Int. Ed., 2012, 51, 7589–7591 CrossRef CAS PubMed.
- S. Knoppe, O. A. Wong, S. Malola, H. Hakkinen, T. Burgi, T. Verbiest and C. J. Ackerson, J. Am. Chem. Soc., 2014, 136, 4129–4132 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- H. Matsukizono and R.-H. Jin, Angew. Chem., Int. Ed., 2012, 51, 5862–5865 CrossRef CAS PubMed.
- M. Sugimoto, X.-L. Liu, S. Tsunega, E. Nakajima, S. Abe, T. Nakashima, T. Kawai and R.-H. Jin, Chem.–Eur. J., 2018, 24, 6519–6524 CrossRef CAS PubMed.
- X.-L. Liu, S. Tsunega and R.-H. Jin, Nanoscale Horiz., 2017, 2, 147–155 RSC.
- X.-L. Liu, S. Tsunega and R.-H. Jin, ACS Omega, 2017, 2, 1431–1440 CrossRef CAS.
- X.-L. Liu, S. Tsunega, T. Ito, M. Takanashi, M. Saito, K. Kaikake and R.-H. Jin, Chem. Lett., 2017, 46, 1518–1521 CrossRef CAS.
- D.-D. Yao, H. Murata, S. Tsunega and R.-H. Jin, Chem.–Eur. J., 2015, 21, 15667–15675 CrossRef CAS PubMed.
- S. Madhavana and S. Okamoto, ChemCatChem, 2018, 10, 2014–2018 CrossRef.
- J.-J. Yuan, P.-X. Zhu, N. Fukazawa and R.-H. Jin, Adv. Funct. Mater., 2006, 16, 2205–2212 CrossRef CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzan, Langmuir, 2002, 18, 2888–2894 CrossRef CAS.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- S. Jin, M. J. Bierman and S. A. Morin, J. Phys. Chem. Lett., 2010, 1, 1472–1480 CrossRef CAS.
- Y. K. A. Lau, D. J. Chernak, M. J. Bierman and S. Jin, J. Am. Chem. Soc., 2009, 131, 16461–16471 CrossRef CAS PubMed.
- R. S. Wagner, M. J. Bierman, Y. K. Lau, A. V. Kvit, A. L. Schmitt and S. Jin, Science, 2008, 320, 1060–1063 CrossRef PubMed.
- J. Liu, Y. Lin, H. Zhang, J. Wang and Z. Huang, Small, 2017, 13, 1701112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00159f |
| This journal is © The Royal Society of Chemistry 2019 |