
Organocatalytic enantioselective direct alkylation of phloroglucinol derivatives: asymmetric total synthesis of (+)-aflatoxin B2†
Zhonglei
Wang
and
Liansuo
Zu
 *
*
School of Pharmaceutical Sciences, Key Laboratory of Bioorganic Phosphorus, Chemistry & Chemical Biology (Ministry of Education), Beijing Frontier Research, Center for biological Structure, Tsinghua University, Beijing 100084, China. E-mail: zuliansuo@tsinghua.edu.cn
First published on 8th April 2019
Abstract
The organocatalytic enantioselective Friedel–Crafts alkylation of phloroglucinol derivatives with enals is reported, providing general access to the benzylic chiral centers shown in a variety of phloroglucinol natural products. The synthetic utility is demonstrated by the very concise asymmetric total synthesis of aflatoxins B2.
The naturally occurring molecules containing phloroglucinol moieties represent an important class of natural products with a vast array of biological activities.1 About 700 phloroglucinol natural products have been isolated from various natural resources with broad structural diversity. One subset of phloroglucinol molecules that has received significant attention features the presence of benzylic chiral centers (Fig. 1). Representative examples include the 4-aryl-3,4-dihydrocoumarin natural product calomelanol F (1),2 semisynthetic clinical candidates flavopiridol (2) and riviciclib (3),3 bullataketals (4)4 and the carcinogenic substances aflatoxin B1 (5) and B2 (6).5 Progress in the asymmetric construction of the benzylic chiral centers and total synthesis of related natural products has been elegantly achieved, providing valuable insights and inspiration from different aspects for the chemical synthesis of this subset of phloroglucinol molecules.6
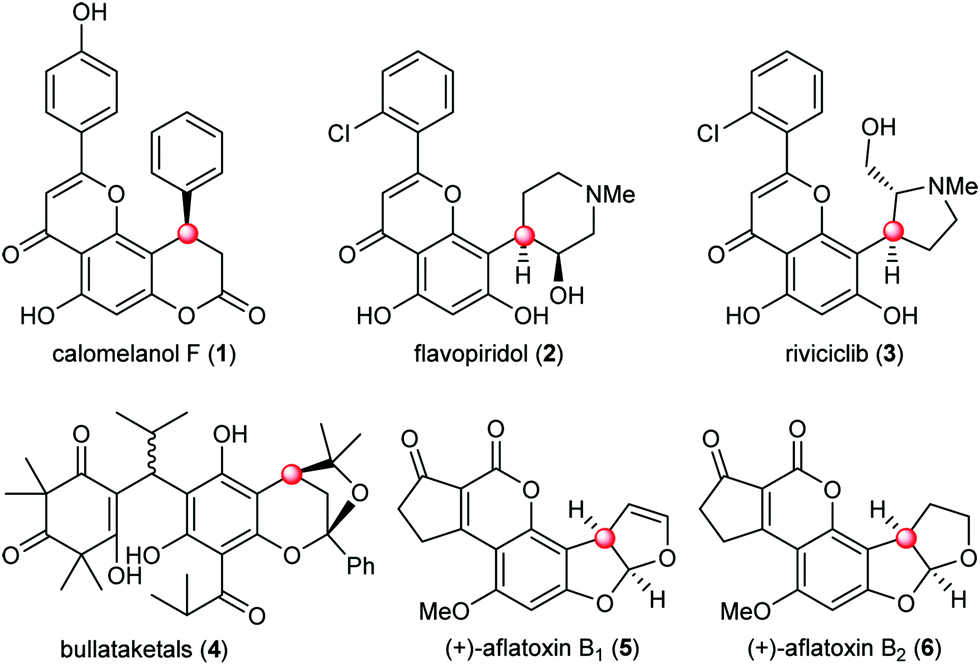 | ||
| Fig. 1 Representative phloroglucinol natural products and drug candidates. | ||
The examination of the chemical structures of the molecules in Fig. 1 revealed some common structural patterns, including the benzylic chiral centers, the eastern heterocyclic rings and the western decoration of the phloroglucinol phenyl rings. Based on these shared structural features and lessons learned from the previous synthetic work, we envisioned a general strategy to address the synthesis of this subset of phloroglucinol molecules at three different phases. Specifically, as depicted in Scheme 1, we envisioned that the direct asymmetric para-selective alkylation of phloroglucinol derivatives7 with enals could build up the benzylic chiral center and introduce several functional groups into the key intermediate 9. Subsequent synthetic manipulations and pairing of the functional groups (R, CHO and OPG) in 9 could furnish the eastern heterocyclic rings. Finally, the late stage decoration could install the required moieties in the western parts. Herein, we report our efforts to explore this concept in the context of developing a total synthesis of aflatoxins B2.
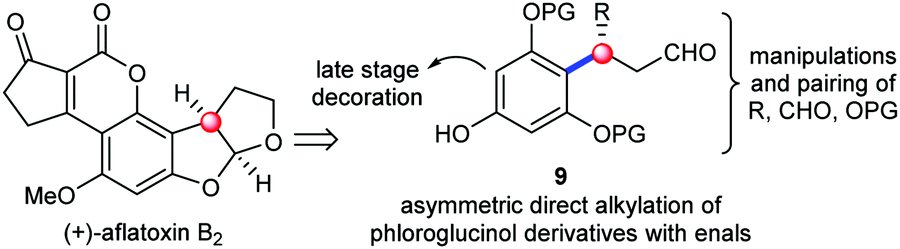 | ||
| Scheme 1 Designed strategy to synthesize phloroglucinol natural products. | ||
One of the key elements in our design was to utilize iminium catalysis8 to control the benzylic chiral center in 9. While the LUMO-lowering activation of enals via iminium ions has become a general strategy for asymmetric catalysis, the Friedel–Crafts alkylation of phenols with enals represents a significant challenge. Notable progress on the Friedel–Crafts alkylations of naphthols with enals has been realized by the groups of Wang, Jørgensen and others,9 but very limited success has been achieved on phenols with sesamol as the sole reported substrate.9b,c Of note, all these reported reactions are ortho-selective and accompanied with subsequent cascade reactions due to the close proximity of the OH and other functional groups. In order to render the alkylation process amenable to natural product synthesis, two fundamental problems have to be addressed: (1) applying the phloroglucinol derivatives as the substrates; (2) obtaining para-selectivity to avoid the uncontrolled pairing of the OH and neighboring functional groups.
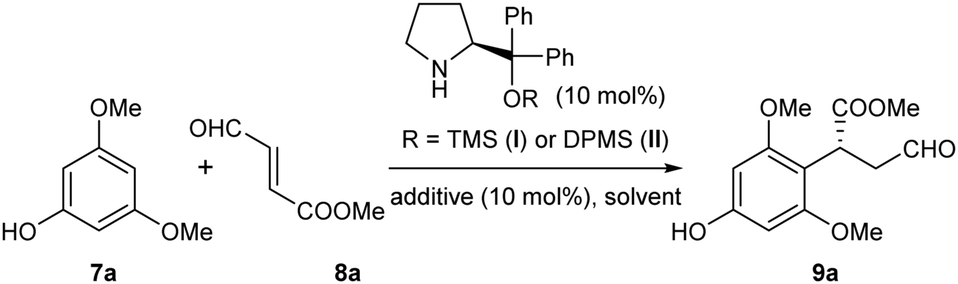
With these concerns in mind, a model reaction of 7a and 8a was carried out first to identify the optimized reaction conditions for the Friedel–Crafts alkylation (Table 1). The diphenylprolinol TMS ether I was selected as the initial organocatalyst10 due to its exceptional ability in activating enals. To our delight, in the presence of benzoic acid as the additive, this catalyst I could indeed enable the desired para-selective alkylation in various solvents with moderate yields and enantioselectivities (entries 1–6). These results validated the feasibility of the direct alkylation of the phloroglucinol derivatives with enals and identified chloroform as the choice of solvent. Further optimizations of the yield and ee were then carried out. Surprisingly, improvements in both the reaction yield and ee were observed without using benzoic acid as the additive (entry 7). Then, we observed that the reaction temperature had a marked effect on the enantioselectivity of the reaction, affording 96% ee at −20 °C (entries 8 and 9). Finally, by switching to a bulkier organocatalyst II, excellent results in terms of both reaction yield and enantioselectivity were achieved (entry 10).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | T (°C) | Yielda | eeb |
| a Isolated yield (%). b Determined by the chiral HPLC analysis. | ||||||
| 1 | I | PhCOOH | EtOH | rt | 51 | 40 |
| 2 | I | PhCOOH | THF | rt | 46 | 48 |
| 3 | I | PhCOOH | Toluene | rt | 25 | 62 |
| 4 | I | PhCOOH | DCM | rt | 76 | 77 |
| 5 | I | PhCOOH | DCE | rt | 71 | 62 |
| 6 | I | PhCOOH | CHCl3 | rt | 79 | 78 |
| 7 | I | None | CHCl3 | rt | 88 | 82 |
| 8 | I | None | CHCl3 | 0 | 90 | 93 |
| 9 | I | None | CHCl3 | −20 | 89 | 96 |
| 10 | II | None | CHCl3 | −20 | 95 | 97 |
With the optimized reaction conditions identified, the substrate scope of the Friedel–Crafts alkylation was next examined (Scheme 2). The method turned out to be a general approach for the generation of the benzylic chiral centers with good to excellent yields and enantioselectivities. The structural variations of the enals 8 were well tolerated: the R group could be esters, aryl and alkyl groups with varied steric and electronic properties (9a–9i). The absolute configuration of 9b was determined by single-crystal X-ray diffraction.11 The different O-alkylated phloroglucinol derivatives also proved to be efficient substrates (9j–9m). Of note, the sterically hindered MOM (methoxymethyl) protecting group could be tolerated, thus generating products amenable to subsequent synthetic transformations (9k–9m). In addition to the phloroglucinol derivatives, several other polyphenolic derivatives could also participate in the alkylation processes, significantly expanding the scope of the phenolic partners (9n–9q). When R1 = H, hemiacetals 9r–9u were obtained through subsequent internal interactions between the OH and aldehyde. Several substrates that could not participate in the process are listed in the bottom of Scheme 2. It seemed that the presence of at least one electron-donating oxygen at the meta position of phenols was essential for the reaction to occur. Whether such structural requirement also dictated the para-selectivity of the Friedel–Crafts alkylation was not clear at this moment.
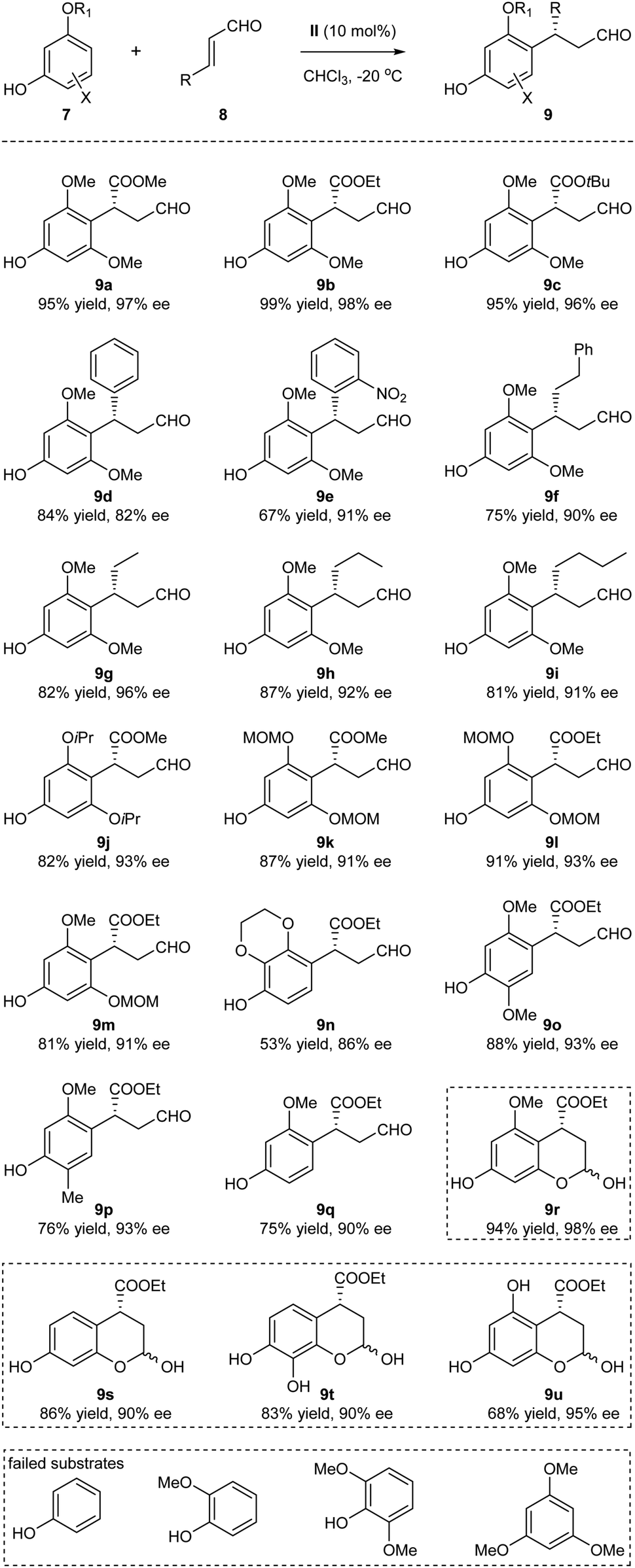 | ||
| Scheme 2 Substrate scope of the organocatalytic Friedel–Crafts alkylation. | ||
Finally, we turned to the chemical synthesis of aflatoxin B2, which is a representative member of the aflatoxin natural products. While significant progress in the total synthesis of racemic aflatoxins has been achieved since the 1960s,12 relatively limited success in the catalytic enantioselective total synthesis of this class of natural products has been realized.6 In 2003, the group of Trost successfully achieved the asymmetric total synthesis of aflatoxin B1 in 9 steps utilizing the palladium catalyzed transformation of γ-acyloxybutenolides.6a Later, in 2005, the group of Corey completed the asymmetric total synthesis of aflatoxin B2 in 8 steps by the application of an elegant oxazaborolidium catalyzed [3+2] cycloaddition.6b Most recently, the group of Hong reported an organocatalytic approach for enantioselective synthesis of the aflatoxin system.6c In this work, remarkable efficiency could be achieved by telescoping several synthetic operations into a one-pot reaction, and thus the concise formal total synthesis of aflatoxin B2 was realized.
Our developed organocatalytic Friedel–Crafts alkylation of the phloroglucinol derivatives with enals offered a direct approach for the efficient generation of the benzylic chiral center shown in aflatoxin B2. Furthermore, the method successfully introduced several functional groups into the alkylation product, thus providing synthetic handles for the rapid generation of the furobenzofuran ring system through manipulations and pairing of these functional groups. As depicted in Scheme 3, from the alkylation product 9l, methylation of the OH produced intermediate 12. Reduction of the aldehyde and partial reduction of the ester could then generate hemiacetal 13 and the equilibrated hydroxyl aldehyde 13′ as inseparable mixtures,13 which were treated with triflic acid (TfOH) to afford 10via cleavage of the MOM groups and subsequent cyclization to the acetal. At this stage, the remaining task of aflatoxin B2 was to install the annulated cyclopentanocoumarin ring. The single step transformation of 10 to aflatoxin B2 reported by Corey6b through Buchi's Pechmann type annulation12d using the brominated precursor 15 represents by far the most efficient known approach. The bromine served as the activating group for the annulation and later a leaving group for the generation of the aromatic coumarin system. While the annulation was very concise, the process proceeded with low yield (36%).6b To increase the overall synthetic efficiency of the late stage annulation, we envisioned to use a simpler precursor 14. We surmised that a similar Pechmann type annulation followed by spontaneous aerobic oxidation would generate the cyclopentanocoumarin ring. After the systematic screening of a variety of Lewis acids, we could successfully realize the desired transformation in 77% yield using Sc(OTf)3 as the promoter, thus completing the total synthesis of (+)-aflatoxin B2 (6) in only five steps from the MOM-protected phloroglucinol. The newly developed annulation protocol for the incorporation of the coumarin ring utilizing a simpler precursor and proceeding with better yield represents a marked improvement when compared to previous methods.
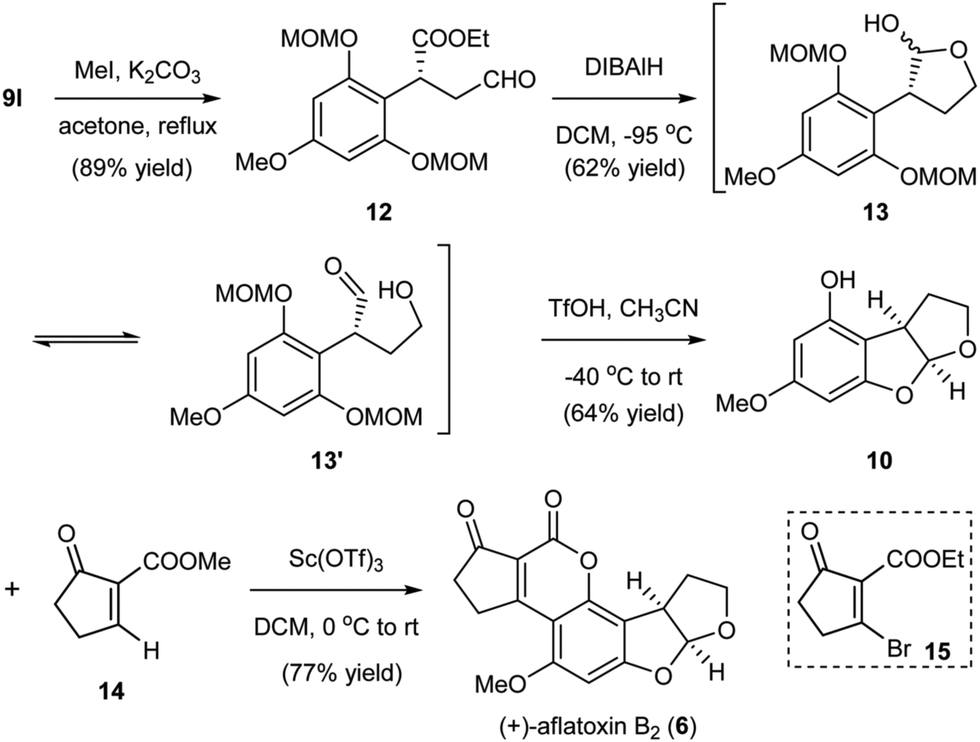 | ||
| Scheme 3 Asymmetric total synthesis of aflatoxin B2. | ||
In summary, we have developed an organocatalytic enantioselective direct alkylation of the phloroglucinol derivatives with enals for the rapid generation of the benzylic chiral centers existing in a variety of phloroglucinol natural products. The synthetic manipulations and pairing of the functional groups of an alkylation product along with a newly developed late-stage annulation protocol have enabled the very concise asymmetric total synthesis of aflatoxins B2, demonstrating the synthetic potential of the developed chemistry in preparing phloroglucinol natural products.
This work is supported by the National Natural Science Foundation of China (Grant No. 21871161) and the Drug Innovation Major Project (Grant No. 2018ZX09711-001).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) I. P. Singh and S. B. Bharate, Nat. Prod. Rep., 2006, 23, 558–591 RSC; (b) R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed; (c) I. P. Singh, J. Sidana, P. Bansal and W. J. Foley, Expert Opin. Ther. Pat., 2009, 19, 847–866 CrossRef CAS; (d) I. P. Singh, J. Sidana, S. B. Bharate and W. J. Foley, Nat. Prod. Rep., 2010, 27, 393–416 RSC; (e) S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouysegu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed; (f) J. M. Estrela, S. Mena, E. Obrador, M. Benlloch, G. Castellano, R. Salvador and R. W. Dellinger, J. Med. Chem., 2017, 60, 9413–9436 CrossRef CAS PubMed.
- F. Asai, M. Inuma, T. Tanaka, M. Takenaka and M. Mizuno, Phytochemistry, 1992, 31, 2487–2490 CrossRef CAS.
- (a) B. A. Christian, M. R. Grever, J. C. Byrd and T. S. Lin, Curr. Opin. Oncol., 2007, 19, 573–578 CrossRef CAS PubMed; (b) N. P. Shirsath, S. M. Manohar and K. S. Joshi, Mol. Cancer, 2012, 11, 77–88 CrossRef CAS.
- (a) L. Larsen, M. H. Benn, M. Parvez and N. B. Perry, Org. Biomol. Chem., 2005, 3, 3236–3241 RSC; (b) H. Tan, H. Liu, X. Chen, Y. Yuan, K. Chen and S. Qiu, Org. Lett., 2015, 17, 4050–4053 CrossRef CAS PubMed.
- For reviews, see: (a) R. E. Minto and C. A. Townsend, Chem. Rev., 1997, 97, 2537–2556 CrossRef CAS PubMed; (b) S. Brase, A. Encinas, J. Keck and C. F. Nising, Chem. Rev., 2009, 109, 3903–3990 CrossRef CAS PubMed.
- For the asymmetric total synthesis of aflatoxins, see: (a) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 3090–3100 CrossRef CAS PubMed; (b) G. Zhou and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 11958–11959 CrossRef CAS PubMed; (c) W. Huang, A. Raja, B. Hong and G. Lee, Org. Lett., 2017, 19, 3494–3497 CrossRef CAS PubMed . For the asymmetric formal total synthesis of aflatoxins, see: ; (d) J. P. Marino, Pure Appl. Chem., 1993, 65, 667–674 CAS; (e) E. R. Civitello and H. Rapoport, J. Org. Chem., 1994, 59, 3775–3782 CrossRef CAS; (f) T. Bando and K. Shishido, Synlett, 1997, 665–666 CrossRef CAS; (g) J. P. Marino, K. A. Kieler and M.-W. Kim, Tetrahedron, 2011, 67, 837–841 CrossRef CAS . For the selected asymmetric total synthesis of other phloroglucinol natural products, see: ; (h) M. Charpentier, M. Hans and J. Jauch, Eur. J. Org. Chem., 2013, 4078–4084 CrossRef CAS; (i) M. Charpentier and J. Jauch, Tetrahedron, 2017, 73, 6614–6623 CrossRef CAS; (j) M. Cheng, J. Cao, X. Yang, L. Zhong, L. Hu, X. Lu, B. Hou, Y. Hu, Y. Wang, X. You, L. Wang, W. Ye and C. Li, Chem. Sci., 2018, 9, 1488–1495 RSC; (k) Y. Guo, Y. Zhang, M. Xiao and Z. Xie, Org. Lett., 2018, 20, 2509–2512 CrossRef CAS PubMed.
- For recent ortho-selective asymmetric transformations of phloroglucinol derivatives, see: (a) L. Albrecht, L. K. Ransborg, V. Lauridsen, M. Overgaard, T. Zweifel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 12496–12500 CrossRef CAS PubMed; (b) Z. Yang, Y. He and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 9775–9778 CrossRef CAS PubMed; (c) G. Li, Z. Li, Q. Gu and S. You, Org. Lett., 2017, 19, 1318–1321 CrossRef CAS PubMed; (d) X. Han, C. Ye, F. Chen, Q. Chen, Y. Wang and X. Zeng, Org. Biomol. Chem., 2017, 15, 3401–3407 RSC.
- For selected reviews on iminium catalysis, see: (a) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79–87 CAS; (b) A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed; (c) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed; (d) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748–9770 CrossRef CAS PubMed; (e) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821–3832 RSC.
- For ortho-selective reactions of naphthols and sesamol with enals, see: (a) L. Hong, L. Wang, W. Sun, K. Wong and R. Wang, J. Org. Chem., 2009, 74, 6881–6884 CrossRef CAS PubMed; (b) P. H. Poulsen, K. S. Feu, B. M. Pza, F. Jensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 8203–8207 CrossRef CAS PubMed; (c) B. M. Paz, L. Klier, L. Nasborg, V. H. Lauridsen, F. Jensen and K. A. Jørgensen, Chem. – Eur. J., 2016, 22, 16810–16818 CrossRef CAS PubMed; (d) M. Giardinetti, J. Marrot, C. Greck, X. Moreau and V. Coeffard, J. Org. Chem., 2018, 83, 1019–1025 CrossRef CAS PubMed . For the organocatalytic cross-coupling-like reactions of bromo-phenols with enals, see: ; (e) X. Song, A. Song, F. Zhang, H. Li and W. Wang, Nat. Commun., 2011, 2, 524 CrossRef PubMed; (f) H. Zhang, C. Ma, Z. Zheng, R. Sun, X. Yu and J. Zhao, Chem. Commun., 2018, 54, 4935–4938 RSC.
- For the pioneering works on the diarylprolinol silyl ethers, see: (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed; (c) H. Gotoh, R. Masui, H. Ogino, M. Shoji and Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 6853–6856 CrossRef CAS PubMed.
- CCDC 1893156 (9b)†.
- For selected racemic chemical synthesis of aflatoxins, see: (a) G. Buchi, D. M. Foulkes, M. Kurono and G. F. Mitchell, J. Am. Chem. Soc., 1966, 88, 4534–4536 CrossRef; (b) G. Buchi, D. M. Foulkes, M. Kurono, G. F. Mitchell and R. S. Schneider, J. Am. Chem. Soc., 1967, 89, 6745–6753 CrossRef CAS; (c) J. C. Roberts, A. H. Sheppard, J. A. Knight and P. Roffey, J. Chem. Soc. C, 1968, 22–24 RSC; (d) G. Buchi and S. M. Weinreb, J. Am. Chem. Soc., 1971, 93, 746–752 CrossRef CAS; (e) A. J. Castellino and H. Rapoport, J. Org. Chem., 1986, 51, 1006–1011 CrossRef CAS; (f) G. Weeratunga, S. Home and R. Rodrigo, J. Chem. Soc., Chem. Commun., 1988, 721–722 RSC; (g) S. Home, G. Weeratunga and R. Rodrigo, J. Chem. Soc., Chem. Commun., 1990, 39–41 Search PubMed; (h) M. Koreeda, L. A. Dixon and J. D. Hsi, Synlett, 1993, 555–556 CrossRef CAS; (i) M. C. Pirrung and Y. R. Lee, Trtrahedron Lett., 1996, 37, 2391–2394 CrossRef CAS; (j) L. K. Casillas and C. A. Townsend, J. Org. Chem., 1999, 64, 4050–4059 CrossRef CAS; (k) T. L. Graybill, E. G. Casillas, K. Pal and C. A. Townsend, J. Am. Chem. Soc., 1999, 121, 7729–7746 CrossRef CAS; (l) W. E. Noland and B. L. Kedrowski, Org. Lett., 2000, 2, 2109–2111 CrossRef CAS PubMed; (m) D. W. Udwary, L. K. Casillas and C. A. Townsend, J. Am. Chem. Soc., 2002, 124, 5294–5303 CrossRef CAS; (n) S. A. Eastham, S. P. Ingham, M. R. Hallett, J. Herbert, A. Modi, T. Morley, J. E. Painter, P. Patel, P. Quayle, D. C. Ricketts and J. Raftery, Tetrahedron, 2008, 64, 936–948 CrossRef CAS.
- The hemiacetal 13 and the opened hydroxyl aldehyde 13′ should be in equilibrium with each other and could not be separated by chromatography. The yield was reported for the mixtures, and the mixtures were used for the next step.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1893156 (9b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc01833f |
| This journal is © The Royal Society of Chemistry 2019 |