
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence-based tools to probe G-quadruplexes in cell-free and cellular environments
Sudeshna Manna
and
Seergazhi G. Srivatsan
 *
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER), Pune, Dr. Homi Bhabha Road, Pune 411008, India. E-mail: srivatsan@iiserpune.ac.in
First published on 17th July 2018
Abstract
Biophysical and biochemical investigations provide compelling evidence connecting the four-stranded G-quadruplex (GQ) structure with its role in regulating multiple cellular processes. Hence, modulating the function of GQs by using small molecule binders is being actively pursued as a strategy to develop new chemotherapeutic agents. However, sequence diversity and structural polymorphism of GQs have posed immense challenges in terms of understanding what conformation a G-rich sequence adopts inside the cell and how to specifically target a GQ motif amidst several other GQ-forming sequences. In this context, here we review recent developments in the applications of biophysical tools that use fluorescence readout to probe the GQ structure and recognition in cell-free and cellular environments. First, we provide a detailed discussion on the utility of covalently labeled environment-sensitive fluorescent nucleoside analogs in assessing the subtle difference in GQ structures and their ligand binding abilities. Furthermore, a detailed discussion on structure-specific antibodies and small molecule probes used to visualize and confirm the existence of DNA and RNA GQs in cells is provided. We also highlight the open challenges in the study of tetraplexes (GQ and i-motif structures) and how addressing these challenges by developing new tools and techniques will have a profound impact on tetraplex-directed therapeutic strategies.
 Sudeshna Manna | Sudeshna Manna obtained her B. Sc. in Chemistry from Jadavpur University in 2010 and M. Sc. in Chemistry from Indian Institute of Technology, Guwahati, in 2012. She is pursuing her Ph. D. degree in the group of Dr S. G. Srivatsan at the Indian Institute of Science Education and Research, Pune. Her doctoral research is focused on the development of biophysical tools to probe noncanonical nucleic acid structures and their recognition properties in cell-free and cellular environments. |
 Seergazhi G. Srivatsan | S. G. Srivatsan obtained a Ph. D. in Bioorganic Chemistry from the Indian Institute of Technology, Kanpur. He was an Alexander von Humboldt postdoctoral fellow at the University of Bonn, Germany. After completing his second postdoctoral fellowship at the University of California, San Diego, he joined the Indian Institute of Science Education and Research, Pune in November 2008. He is currently an associate professor and a Wellcome Trust-DBT India Alliance Senior Fellow. His research interests lie in the area of nucleic acid chemistry and biophysics, particularly, in the development of nucleoside probes and imaging tools to study nucleic acid structure and function. |
1. Introduction
The ability of nucleic acids to function as a genetic material, catalyst and regulatory element emanates from their ability to adopt complex secondary and tertiary structures. In addition to the classical double helical structure of nucleic acids, they form structural motifs like hairpin, pseudoknot, bulge, internal loop, cruciform, triplex, tetraplex (G-quadruplex and i-motif), Z-DNA, etc.1–3 Unlike the chemical diversity in proteins, nucleic acids use only four nucleosides (A, G, C, and T/U) to form these structures. Formation of such diverse structures is aided by canonical and non-canonical base pairing interactions, stacking interaction and interactions of nucleobase, sugar and phosphate groups with various metal ions.4 The structure thus formed has intrinsic conformation dynamics, and interconverts between different states depending on the environment and cognate protein factors and small molecule metabolites.5–8 This structural reorganization not only controls the ensuing function of the nucleic acid sequence but also has therapeutic implication as aberrations in the structure often lead to disease states.9–11Among the various structural motifs, GQ structure formed by guanine-rich sequences has received much of the attention owing to its (i) abundance in the genome,12,13 (ii) structural polymorphism14 and (iii) important cellular functions.15,16 Bioinformatics suggest that putative GQ-forming sequences are abundantly found in the telomeric region of the chromosomes, and in the promoter DNA regions and untranslated regions of mRNA of several proto-oncogenes.17,18 Further, the conservation and location of such sequences among various eukaryotes suggest that GQ structures could serve as important structural elements with concrete roles in cellular processes.19,20 With respect to the structure, GQ exhibits multiple folding topologies in vitro, which depend on the sequence, ionic conditions and the surrounding environment like molecular crowding and confinement.21–24 The functions of GQs are associated with their location in the genome. For example, there is compelling evidence to connect the GQ structure of the telomeric repeat with its function namely, protecting the chromosome from fusion and maintaining the stability of genome.25–27 Mutating the G-rich sequences in the promoter region of certain oncogenes and by using GQ-binders, it has been inferred that GQ could potentially serve as a regulatory element to control the expression of genes at the transcriptional level.28,29 Similar biochemical analysis on G-rich motifs present in the UTR of mRNAs also suggests GQ-specific gene expression control at the translation level.29,30 Owing to the diversity in structure and function of GQ and its potential role in diseases, there has been a flurry of interest in developing small molecule ligands that bind and stabilize GQ structures.31–33 Several such ligands have been used as biophysical tools to probe the structure and function of GQ motifs and also have been evaluated as potential chemotherapeutic agents.34–40 Recent investigations elucidating the existence of DNA and RNA GQ structures in cells of various organisms, including mammals, have further bolstered the interest in the study of GQs.41–44
Analogous to the GQ structures formed by G-rich DNA sequences, C-rich sequences form four-stranded structures called i-motifs under slightly acidic or even at near neutral pH by intercalation of hemiprotonated C·CH+ base-paired strands.45–48 While putative GQ- and i-motif-forming sequences mostly coexist as partners in the genome, the majority of studies have been focused on GQs. This is because there is clear evidence for the presence and function of GQs in vivo, while the existence of i-motif structure in cellular environment remains ambiguous. However, protein factors have been identified, which preferentially bind to C-rich sequences and transcriptionally activate the expression of certain oncogenes.49–52 In one such example, hnRNP LL has been shown to bind to BCL2 promoter i-motif-forming sequence and activate BCL2 expression.53 The role of i-motif as a molecular switch in controlling the expression of an oncogene has been shown using small molecule binders.53,54 These findings suggest that i-motif and GQ structures could be cooperatively targeted by using structure-specific small molecule binders.55 Hence, i-motif as a potential therapeutic target is gaining interest in recent years.56
Although several GQ-binding ligands have been developed, which are known to influence the levels of oncogenes in protein expression assays, discovery of clinically suitable candidates has remained elusive.57 The reasons are the following. While the structural diversity of GQ is fairly understood in cell-free conditions, development of methods and tools to identity the GQ topology adopted by a given sequence in cellular environment remains a major challenge. This is because, most of the currently available chemical probes (i) poorly distinguish between different GQ, i-motif and duplex structures, and (ii) cannot be effectively implemented in complementing assays that will provide a direct correlation between cell-free and in-cell structural data. For example, NMR technique is commonly used to study the GQ structures of short oligonucleotides in vitro. However, in-cell NMR analysis often fails to provide unambiguous structural information due to severe signal broadening in the complex environment of the cell.58 Structural polymorphism and abundance of tetraplex-forming sequences in the genome, and lack of efficient screening platforms to identify topology-specific binders further increases the complexity of targeting a specific GQ motif using small molecule ligands.
To address these challenges, much of the recent efforts are aimed towards the development of biophysical tools and techniques that (i) would enable the determination of the topology adopted by individual tetraplex-forming motifs in conditions closely resembling the cell and (ii) are compatible with discovery assays capable of estimating the binding of ligands to a specific topology. Here, we review recent studies on the design of chemical probes, which provide new means to study GQ topologies and their recognition properties in cell-free and cellular environments. First, we discuss in detail the utility of responsive fluorescence probes in detecting the formation and ligand binding ability of GQs. Following this, a discussion on the development of structure-specific antibodies and small molecule probes to visualize DNA and RNA GQs in cells is provided.
2. Structural polymorphism of GQ
The four-stranded GQ assembly is composed of stacks of two or more G-tetrads formed by multiple H-bonds and stabilized by metal ions (e.g., K+, Na+, etc.).14,21 In general, intramolecular GQ is formed by sequences having four or more G-tracts, which involve in tetrad formation. The intervening nucleotides, which form the loops, connect the tetrads. While tetrad stacks are almost exclusively formed by guanines, the bases involved in loop formation are not limited. GQ exhibits wide variety of folding topologies, which can be broadly classified as parallel stranded, antiparallel stranded and hybrid type mixed parallel-antiparallel stranded conformations (Fig. 1B). These structures differ in the (i) strand orientation, (ii) loop type (propeller, diagonal and lateral) and length, (iii) groove width (wide, medium and narrow), and (iv) guanosine glycosidic conformation (syn and anti) of the tetrads. The ability of a given G-rich region to adopt one or more GQ structures primarily depends on the nucleotide sequence itself.21 Simulation and experimental data suggest that parallel conformation is formed when the loops are made of one residue.59 Whereas, when the loops are longer than two residues or made of a combination of one or more residues, then different GQ structures are formed. In addition, surrounding environment and assay conditions can considerably influence the conformation of the GQs.23 In the following section, an overview of the conformation of different GQ structures formed under different conditions is provided by taking examples of some well studied G-rich sequences. The readers may refer to the following reviews for detailed discussion on the structural features of various DNA and RNA GQs obtained by using various techniques.3,9,21,60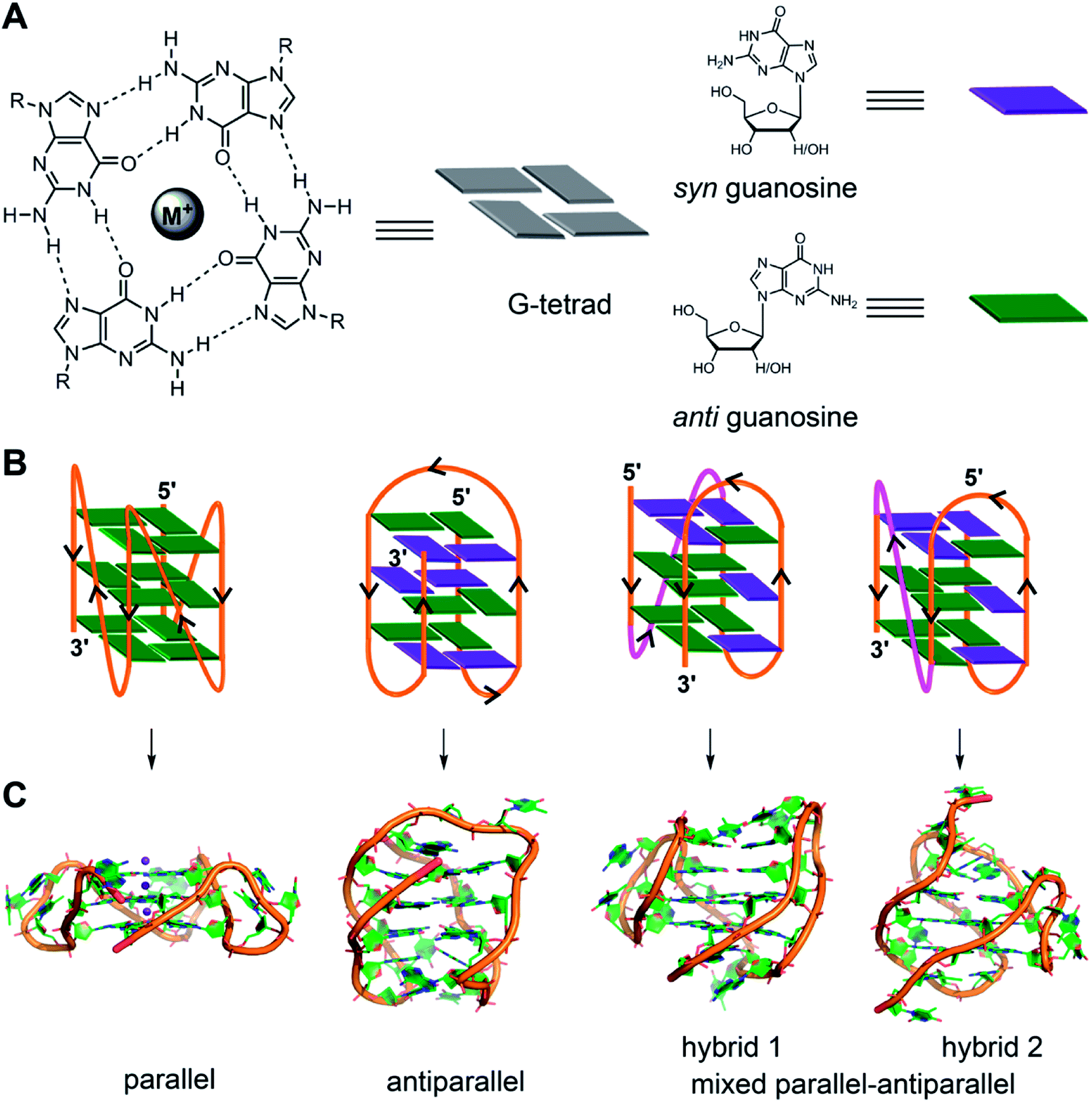 | ||
| Fig. 1 (A) Schematic representation of a G-tetrad where four coplanar guanines are involved in Hoogsteen and Watson-Crick hydrogen bonding. Metal ions stabilize the tetrad. For representation, guanines are shown as rectangular box. syn and anti glycosidic conformations of guanosine are represented in violet and green colour, respectively. (B) Loop orientation and tetrads of different GQ structures, namely parallel, antiparallel, hybrid 1 and hybrid 2 are shown. (C) Crystal/NMR structure of parallel (PDB ID: 1KF1),65 antiparallel (PDB ID: 143D),61 hybrid 1 (PDB ID: 2GKU)63 and hybrid 2 (PDB ID: 2JPZ)64 conformations of H-Telo DNA ON. | ||
2.1. DNA GQs
Human telomeric (H-Telo) DNA overhang composed of a (TTAGGG)n repeat is the most studied GQ-forming sequence. It forms an antiparallel structure in Na+ solution and multiple conformations in the presence of K+ ions (Fig. 1).61,62 In a buffer solution containing K+ ions, telomeric repeat mainly adopts hybrid 1 and hybrid 2 conformations in which the double chain reversal loop is located at the 5′- and 3′-end, respectively.63,64 However, under crystallization conditions in the presence of K+ ions and polyethylene glycol (PEG), the same sequence adopts a parallel GQ conformation.24,65 Structural analysis performed in the presence of crowding agents (PEG), highly viscous cosolutes (deep eutectic solvents) and polysaccharides, which are commonly used synthetic models mimicking the confinement and molecular crowding of cells, show that parallel GQ structure is the preferred structure of the telomeric repeat.23,66–68 Further, studies in these systems reveal a slower folding kinetics with reduced stability and ligand binding ability of GQ as compared to in dilute aqueous buffers. On the contrary, a recent study using optical tweezers indicate that telomeric DNA repeat in the confined space of DNA nanocages forms hybrid type or anti-parallel structure, depending on the size of the confinement.69 Further, these studies indicated that the GQs fold faster and have higher stability in the confined environment. Though these studies provide information on the folding behaviour and recognition properties of GQs in different environments, the results obtained under these conditions have to be taken with a word of caution. For example, PEG used as a synthetic crowding agent in buffers and in crystallization experiments, due to dehydrating effect, bias telomeric repeat to adopt the parallel GQ structure.70Unlike telomeric DNA repeat, the GQ-forming sequences present in the promoter region of oncogenes have varying numbers of G-tracts with unequal number of guanines and intervening loop residues.21 Hence, these motifs can potentially form multiple GQ structures by using different combinations of the G-tracts and loop residues. G-rich motif (NHE III1) present in the promoter region of the c-MYC gene is one of the well studied promoter GQ-forming motifs. It has five tracts of guanine residues each separated by a nucleotide residue. The I–IV tracts and II–V tracts both form parallel GQ structure with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 loop-size arrangement (Fig. 2A).71,72 Other promoter sequences, which form parallel GQ structure are VEGF, HIF-1α, c-Kit, RET and PDGF-A.73–77 A common feature of these promoter GQs is the presence of G3NG3 arrangement in the first and third loops and variable residues in the middle loop. Notably, parallel GQ of PDGF-A is made of four G-tetrads.77 Variants of GQ like parallel-stranded topology with a broken strand and a heptad plane is adopted by human PDGFR-β and c-MYB promoters, respectively.78,79
:2:1 loop-size arrangement (Fig. 2A).71,72 Other promoter sequences, which form parallel GQ structure are VEGF, HIF-1α, c-Kit, RET and PDGF-A.73–77 A common feature of these promoter GQs is the presence of G3NG3 arrangement in the first and third loops and variable residues in the middle loop. Notably, parallel GQ of PDGF-A is made of four G-tetrads.77 Variants of GQ like parallel-stranded topology with a broken strand and a heptad plane is adopted by human PDGFR-β and c-MYB promoters, respectively.78,79
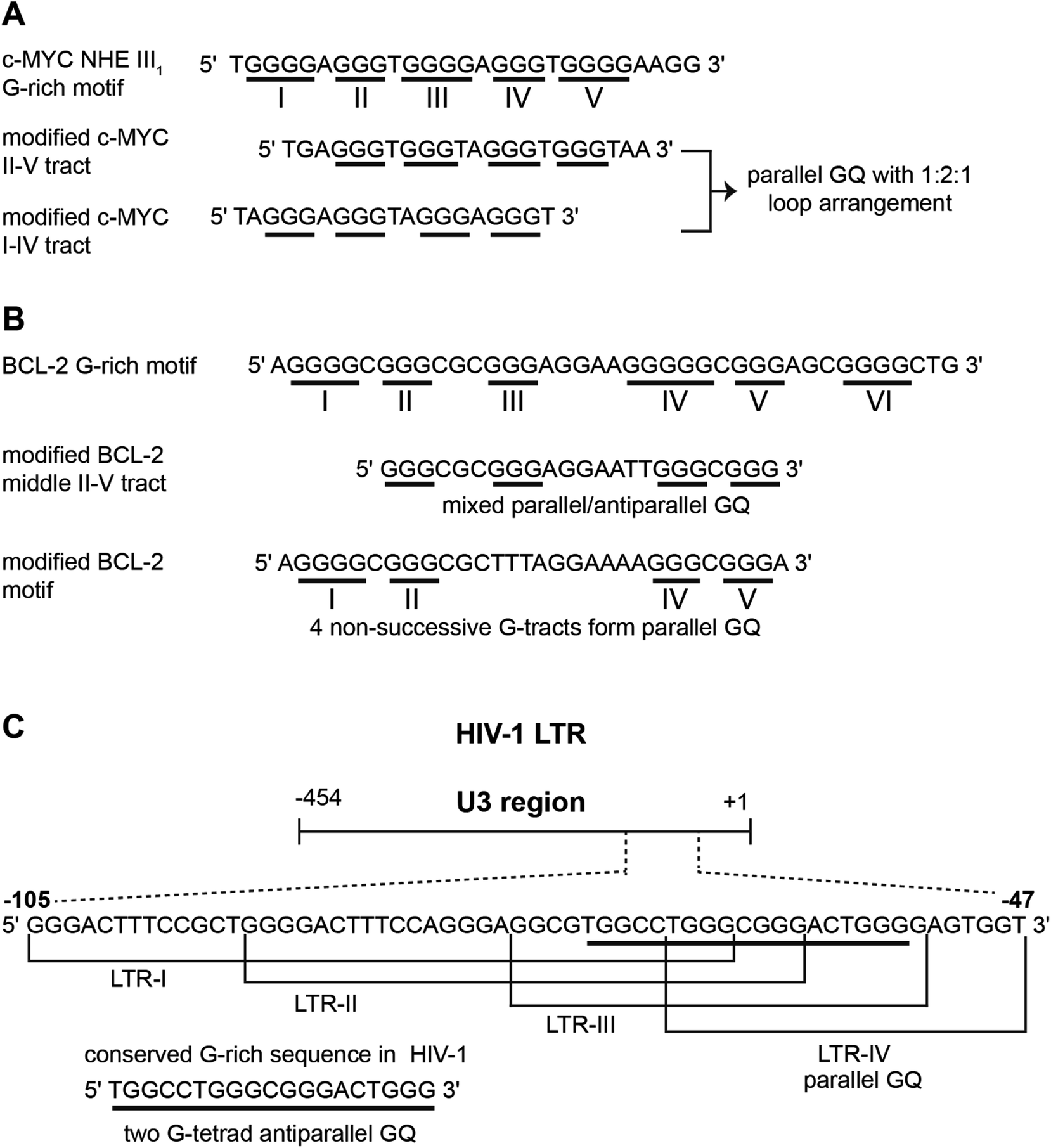 | ||
| Fig. 2 (A) The G-rich NHE III1 sequence with five G-tracts in the promoter region of c-MYC gene. G-tracts are underlined. Modified c-MYC sequences, which are used for structural analysis is shown. (B) The sequence of GQ-forming BCL-2 promoter with six G-tracts is shown. Different GQs are formed by different G-tract combinations. (C) The dynamic GQ-forming sequence in the U3 region of 5′ LTR of HIV-1 is shown. It has four overlapping GQ-forming segments. | ||
Upstream of P1 promoter region of the human BCL-2 gene is a 39-bp GC rich sequence containing six G-tracts.80 Inhibiting the expression of this gene reduces cell proliferation, thereby improving chemotherapy efficacy.81 Notably, the P1 promoter region, including the GC rich segment, has been shown to play a significant role in regulating the transcription of BCL-2 gene.82 This G-rich sequence can form three GQs involving different G-tracts (Fig. 2B). The middle segment (four consecutive G-tracts) forms a stable mixed parallel–antiparallel-stranded hybrid type GQ structure with two lateral loops (3 nucleotides and 7 nucleotides, respectively) and one single nucleotide double chain reversal loop.80 However, recent studies indicate that four nonsuccessive G-tracts (I, II, IV and V) fold into a parallel GQ structure containing three loops, which is unexpectedly more stable than the GQ of middle segment.83
Recently, a dynamic GQ forming segment has been identified in the U3 domain of the 5′ long terminal repeat (LTR) of proviral HIV-1 genome (Fig. 2C).84 The U3 region contains a stretch of eight G-tracts upstream of the transcription start site and overlaps with the binding site of important transcription activators, Sp1 and NF-κB. This G-rich sequence (−105 to −47) is sub-divided into four overlapping G-tract segments (LTR I–IV), each containing four G-tracts. When individually studied, LTR-II and LTR-III produced circular dichroism (CD) signatures resembling a parallel-type GQ structure, whereas, CD profile of LTR-I did not match with any of the GQ forms. Similarly, LTR-IV formed a parallel-stranded GQ structure containing a single-thymidine bulge.85 However, studies with full-length G-rich sequence showed the presence of multiple GQs. In another study, the central part of the G-rich region, which shows high level of conservation, adopted a two G-tetrad antiparallel GQ structure.86 Further, mutations that disrupt GQ formation and by using ligands that stabilize GQ structures, it was inferred that the LTR of HIV-1 could serve as a new target for anti-HIV-1 drug screening.
2.2. RNA GQs
Unlike the structural diversity exhibited by DNA GQs, G-rich RNA sequences generally form all-parallel GQ structure, which is thermodynamically more stable and less hydrated than equivalent DNA GQs.87,88 For example, telomeric repeat-containing RNA (TERRA) (UUAGGG)n, an equivalent sequence of the human telomeric DNA repeat, adopts a parallel GQ structure irrespective of the type of metal ions and surrounding environment.89,90 GQ-forming motif has been identified in different positions of UTR of mRNAs, which includes Zic-1, NRAS, BCL-2, TRF2, and IRES element of human of VEGF.30,87 These motifs form a parallel GQ structure with different loop configurations in vitro. The conformational constraint and higher stability of RNA GQs have been attributed to the presence of 2′-OH groups.91 The 2′-OH group restricts all the guanosines in the tetrad to adopt an anti-glycosidic conformation and imparts C3′ endo puckering to the sugar, which in fact is the preferred geometry for the formation of a parallel GQ structure. Modification of 2′-OH groups suggests that the hydroxyl groups are involved in intramolecular interactions, which further stabilize the GQ structure.923. Techniques to study GQ structure and ligand binding
The structure, stability, folding dynamics and ligand binding ability of tetraplex-forming ON sequences are commonly studied in vitro by using CD, NMR, X-ray crystallography and fluorescence techniques. CD provides a qualitative understanding of the various GQ structures, and can be used to distinguish between GQs, i-motifs and duplex forms. This is because, the CD signal is highly sensitive to strand orientation of G-tract and anti/syn-glycosidic conformation of guanosines of the G-tetrads.93 A G-rich–C-rich ON duplex exhibits a typical CD signature of a B-form of the DNA duplex with a positive band at ∼265 nm and a negative band at ∼240 nm.94 All anti-glycosidic conformation of the tetrads in the parallel GQ structures of DNA and RNA ONs typically exhibit a strong positive band at ∼260 nm and a shallow negative band at ∼240 nm. A CD spectrum containing a positive peak at ∼295 nm, a negative peak at ∼265 nm and a smaller positive peak at ∼245 nm is characteristic of a chair- or basket-type antiparallel GQ structure having alternating syn- and anti-glycosidic conformation.62,93 Hybrid type mixed parallel-antiparallel GQ structures show a positive band (∼290 nm) with a shoulder near 265 and a smaller negative band at ∼240 nm. On the other hand, i-motifs typically display a positive band at ∼288 nm and a negative band at ∼256 nm.95 While CD analysis is highly useful in studying individual structures, the spectrum is not easy to interpret when multiple conformations are present. For example, telomeric DNA repeat is known to fold into multiple GQs in K+ ionic conditions. However, the CD spectrum resembles more like hybrid type structures.62 Determination of conformation of the sequences containing longer G-tracts, which can fold into multiple structures, is also not straight forward by CD.96Thermal melting is another convenient method to evaluate the stability of different GQ structures.97,98 The melting temperature of GQ by UV method is mainly calculated by monitoring the changes in absorbance at 295 nm due to larger variations (50–80%) in absorbance at 295 nm compared to at 260 nm (4%). As GQ structures have higher absorbance at 295 nm, the GQ to random coil transition shows a typical inverted melting profile as compared to the classical duplex to single-strand transition profile. Apart from UV absorption, temperature-dependent CD and FRET assays have also been used to study the formation as well as binding of ligands to tetraplexes.99,100 However, sequences showing multiple conformations pose challenges in deconvoluting the signal from individual structures. Hence, CD and thermal melting are used as complementing techniques along with fluorescence, NMR or X-ray crystallography in determining the structure and ligand binding properties of tetraplexes.101,102 Some of the prominent examples of ligand–GQ interaction studied include TMPyP4-c-MYC, RHPS4-Telo DNA, quindoline-c-MYC, telomestatin derivative-Telo DNA, Phen-DC3-c-MYC, 2,4,6-triarylpyridine-bound to H-Telo DNA (Fig. 3). In addition, surface plasmon resonance (SPR), isothermal titration calorimetry (ITC) and laser tweezers have been used in studying the folding kinetics, ligand binding specificity and energetic aspects of G-quadruplex–ligand interactions.103–109
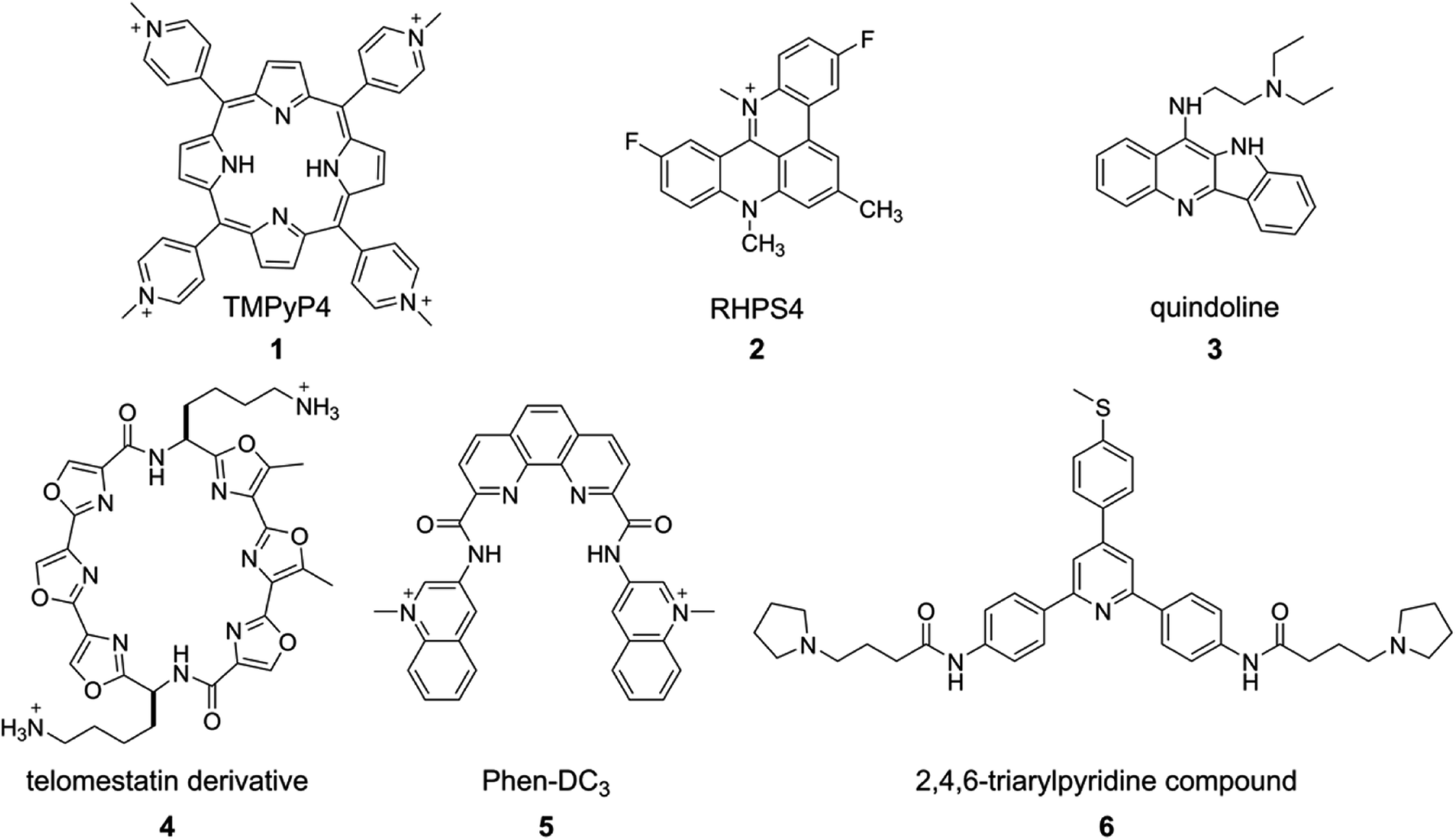 | ||
| Fig. 3 Representative examples of GQ-binding ligands. | ||
Among the various biophysical methods discussed, fluorescence based methods are advantageous as they provide convenient tools to study the structure, folding dynamics and recognition of tetraplexes in real time with a high level of sensitivity.110 Since the native nucleosides are practically non-emissive, the study of four-stranded structures is accomplished by using non-covalent fluorescent binders or by labeling ONs with fluorescent nucleoside reporters.110,111 These probes show changes in fluorescence properties like emission maximum, intensity, quantum yield, anisotropy and lifetime upon changes in conformation. These tools not only provide means to study the formation of tetraplexes in vitro and in vivo, but also have enabled the establishment of screening-compatible assays to identify efficient binders. Excellent reviews detailing the development and applications of non-covalent fluorescent ligands, metal complexes and FRET-based systems in GQ and i-motif studies in vitro are available in literature.110–112 Hence, in the following sections, we review recently developed fluorescence-based approaches to study the GQs and their interaction with small molecule ligands in cell-free and cellular environments. In particular, the utility of covalently labeled environment-sensitive fluorescent nucleoside analogs in distinguishing different GQ topologies and estimating binding of ligands to different topologies is presented in detail. Following this, the development of structure-specific antibodies and small molecule ligands, which have been successfully implemented in fluorescence microscopy assays to prove the occurrence of GQ in cells, is discussed.
4. Environment-sensitive fluorescent nucleoside probes
FRET based assays are widely used in investigating the formation, stability and dynamics of GQs at the ensemble as well as single-molecule level.113–116 ONs labeled with appropriate FRET pairs are used in assessing the selectivity of tetraplex binding ligands,117–119 and also in devising fluorometric methods to detect metal ions and proteins.120–122 Similarly, several structure-specific responsive fluorescent ligands and metals complexes that display changes in their photophysical properties in the bound and unbound states serve as good sensors to detect the formation of various GQ and i-motif structures.123–127 While the utility of these tools are arguably undeniable, probing the conformational differences in different four-stranded structures at the nucleotide level is not straightforward. A detailed understanding of the tetraplex structures at the nucleotide level is important for the following reasons. The sequence and structural diversity of GQ-forming motifs, though a gold mine for research, is truly overwhelming. While biochemical and biophysical investigations indicate that ligands bind to different domains of a quadruplex, estimating their affinity to different domains and different GQs formed by different sequences remains a major challenge as many of the chemical probes fail to distinguish subtle differences in the structure. In this context, conformation-sensitive fluorescent nucleoside analogs that can report subtle difference in GQ structures via changes in fluorescence properties have been highly useful. Some of these probes are potentially suitable for designing screening platforms to identify GQ-specific binders of therapeutic relevance.4.1. Isomorphic fluorescent nucleoside probes
Majima and coworkers demonstrated the usefulness of 2-aminopurine (2AP), a widely used environment-sensitive fluorescent adenine analog, in distinguishing GQ structure from duplex form (Fig. 4A). The H-Telo DNA ON repeat AGGG(TTAGGG)3 was used as the test system.128 The three loop adenine residues were individually replaced with 2AP. The modification, as confirmed by thermal melting and CD studies, had little effect on the formation and stability of an antiparallel GQ structure in a buffer containing Na+ ions. The fluorescent label reported the formation of GQ structure with up to 30-fold enhancement in fluorescence intensity as compared to the complementary duplex. The lifetime measurements also indicated a similar trend, wherein the GQ form exhibited nearly 14-fold longer lifetime than the duplex for a sequence containing the modification in the first loop. The enhancement in fluorescence intensity and longer lifetime was attributed to the more solvent exposed and less stacked 2AP in GQ structure as compared to in the duplex structure. The sensing ability of 2AP was further utilized in monitoring the binding of TMPyP4 to the GQ.129 Interestingly, modification at diagonal loop produced the maximum response upon binding to the ligand, which was found to be consistent with the binding mode of TMPyP4 to GQ, i.e., ligand inserted between the diagonal loop and G-tetrad (Fig. 4A).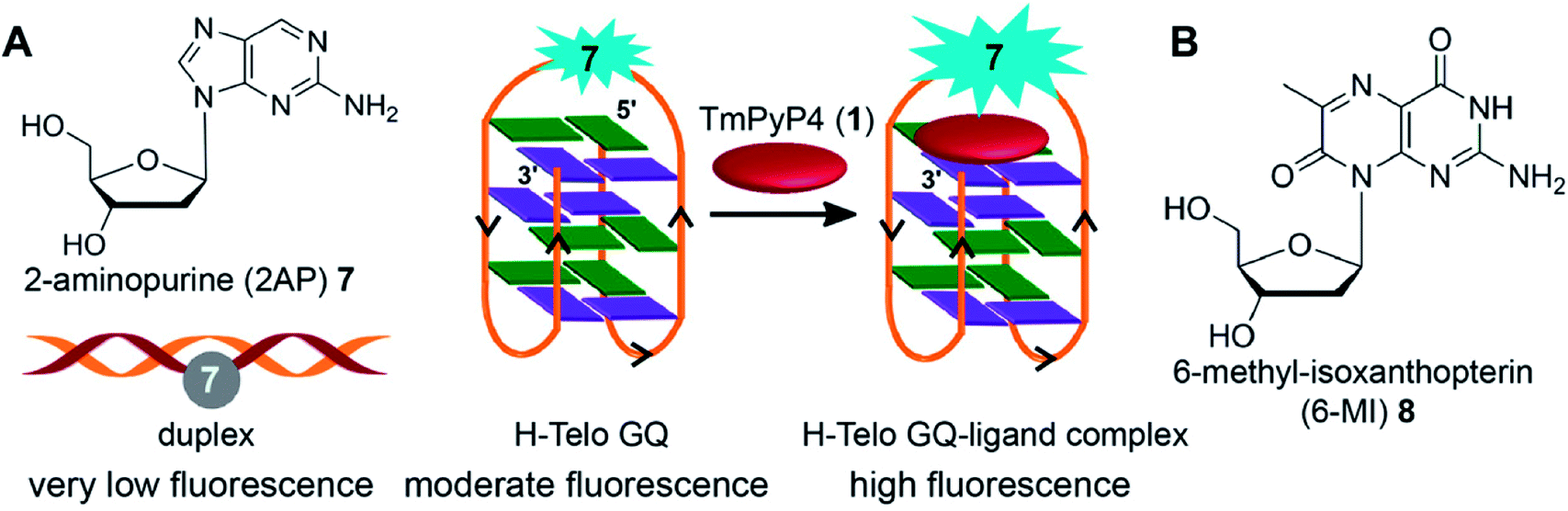 | ||
| Fig. 4 (A) 2AP displays different fluorescence properties in duplex, GQ and GQ–ligand complex.128,129 (B) Structure of fluorescent 6-MI 2′-deoxyribonucleoside is shown. | ||
Chaires and coworker utilized 2AP-modified H-Telo DNA ONs to study the formation of GQ structures in different ionic conditions. The results obtained from these studies indicated that GQ structure adopted by telomeric repeat in solution containing K+ ions and in solid-state are not same.130 Following this, Chaires and coworker used stopped-flow and fluorescence polarization methods to study the ion-induced conformational change in the 2AP-labeled H-Telo DNA ON.131 The folding kinetics of GQ in the presence Na+ or K+ ions as determined by fluorescence experiments revealed at least three steps with time scale ranging between milliseconds to several hundred seconds. Importantly, K+-dependent folding curves indicated the presence of multiple GQ conformations. This observation is in contrast to the parallel structure obtained under X-ray crystallography conditions,65 which in a way suggest that crystal environment may favour a particular conformation.
In another study conducted by Sugiyama and Xu, 2AP-substituted G-rich and C-rich sequences present in the 5′ end of retinoblastoma susceptibility genes (Rb) were employed in probing the competition between duplex, GQ and i-motif formation.132 Both GQ and i-motif exhibited considerably higher fluorescence intensity as compared to the duplex form. Addition of complementary C-rich ON sequence to the pre-formed 2AP-labeled GQ resulted in quenching in fluorescence intensity at neutral pH as a consequence of slow conversion of GQ to duplex form. At acid pH the formation of i-motif structure facilitated the formation of GQ, thereby providing a competitor for the formation of duplex relative to the random coil of the C-rich ON sequence at neutral pH. However, the general drawback of 2AP is that it has excitation and emission maximum in the UV region (Table 1), and importantly, the quantum yield of 2AP incorporated into ONs and into duplexes is very low. For example, the quantum yield of free 2AP is 0.68, and when incorporated into GQ-forming sequences and corresponding duplexes the quantum yield decreases dramatically (up to 0.06 and 0.005, respectively), which compromises the sensitivity of the probe.128 Further, replacement of a guanine residue with 2AP results in considerable destabilization (ΔTm = −28 °C) of the GQ structure, which is understandable as it can disrupt the tetrad formation.133 In a recent study, Kankai and coworkers used 2AP to study the folding of thrombin and G3T aptamers into antiparallel and parallel GQ structure, respectively.134 2AP was introduced in the loop regions of thrombin aptamer d(GGTTGGTGTGGTTGG) and G3T aptamer d(GGGTGGGTGGGTGGG), which binds to HIV-1 integrase. In the unfolded state the fluorescence of all ONs was low. The formation of respective GQ structures in the presence of K+ ions was signaled with enhancement in fluorescence intensity. In particular, 2AP-labeled G3T exhibited remarkable enhancement in fluorescence intensity, which was equal to the fluorescence of the free nucleoside analog.
| Nucleoside probe | Solvent | λmax (nm)a | λem (nm) | Φb | τ (ns)c |
|---|---|---|---|---|---|
| a The lowest energy maximum is given.b Φ = Quantum yield.c τ = Lifetime or average lifetime of the nucleoside. | |||||
| 2-AP (7)153 | Water | 303 | 370 | 0.68 | 7.0 |
| 6-MI (8)153 | Buffer | 340 | 431 | 0.70 | 6.4 |
| 2-PyG (9)139 | Water | 300 | 415 | 0.02 | — |
| Acetonitrile | 300 | 415 | 0.71 | — | |
| StG (10)139 | Water | 340 | 450 | 0.49 | — |
| Acetonitrile | 340 | 450 | 0.74 | — | |
| 4PVG (11)139 | Water | 355 | 490 | 0.16 | — |
| Acetonitrile | 355 | 490 | 0.57 | — | |
| VdG (12)140 | Buffer | 277 | ∼400 | 0.72 | — |
| FurdG (13)143 | Buffer | 292 | 384 | 0.49 | — |
| PyrdG (14)143 | Buffer | 292 | 379 | 0.10 | — |
| ThdG (15)143 | Buffer | 284 | 414 | 0.79 | — |
| BfurdG (16)143 | Buffer | 323 | 405 | 0.76 | — |
| InddG (17)143 | Buffer | 321 | 390 | 0.78 | — |
| BthdG (18)143 | Buffer | 315 | 419 | 0.46 | — |
| CNPhdG (19)145 | Water | 308 | 468 | 0.04 | — |
| FurdU (20)141 | Water | 316 | 431 | 0.03 | — |
| 23a150 | Water | 322 | 447 | 0.21 | 2.55 |
| Methanol | 322 | 423 | 0.15 | 0.94 | |
| Acetonitrile | 322 | 410 | 0.06 | 0.33 | |
| Dioxane | 322 | 404 | 0.10 | 0.43 | |
| 24a149 | Water | 318 | 458 | 0.04 | 1.04 |
| Methanol | 321 | 446 | 0.05 | 0.60 | |
| Acetonitrile | 321 | 443 | 0.03 | 0.49 | |
| Dioxane | 322 | 435 | 0.06 | 0.45 | |
Mergny and coworkers systematically studied the influence of substitution of guanine residues on the formation of GQ structure with different base analogs.135 In this study, each of the guanine residues of the central tetrad of parallel GQs was substituted with twelve types of bases. The results revealed that most guanine substitutions perturb the GQ structure. Notably, 8-bromoguanine and 6-methyl-isoxanthopterin (6-MI), a fluorescent purine analog, significantly accelerated the formation of the GQ structure when placed at the 5′ end (Fig. 4B). However, the fluorescence of 6-MI was found to be drastically quenched in the GQ structure due to stacking interaction with a neighboring purine. The responsiveness of 6-MI was used by Shamoo and coworkers in estimating the binding of unwinding protein (UP1) to the H-Telo repeat.136 Upon addition of increasing concentration of UP1, the probe-labeled telomeric repeat DNA ON exhibited a dose-dependent increase in fluorescence intensity as a result of unstacking of 6-MI in the complexed state. The affinity of UP1 for the telomeric repeat was determined to be ∼5 nM, and the results also indicated that the protein destabilized the GQ structure.
4.2. 8-Substituted fluorescent purine nucleoside probes
8-Substituted fluorescent guanosines incorporated into G-tetrads have provided efficient tools to study GQs.137 Luedtke and coworkers developed a turn-on fluorescent GQ sensor, 8-(2-pyridyl)-2′-deoxyguanosine (2PyG), and incorporated it in different tetrad positions of H-Telo repeat and c-Kit DNA ONs.138 The probe placed in the position where native guanine residue prefers to adopt syn glycosidic conformation did not affected the global GQ structure and stability. However, replacement of anti-guanine residue with 2PyG had significant impact on the native GQ topology and thermal stability. This is because, 2PyG nucleoside prefers the syn glycosidic conformation as has been observed with many other guanosine analogs containing bulky modification at 8 position. Nevertheless, 2PyG enabled the fluorescence detection of GQs and study the energy transfer in GQs. H-Telo DNA ON labeled with 2PyG in different ionic conditions (Na+, K+, NH4+ and Rb+) showed considerable enhancement in fluorescence as compared to random structure in Li+ ionic conditions and free nucleoside (Fig. 5A). However, the quantum yields were not very different in Na+ (0.08) and K+ (0.09) ionic conditions, which favor antiparallel and multiple GQ structures, respectively. Interestingly, the excitation peak at 260 nm indicated energy transfer between the DNA and probe. Energy transfer efficiencies were found to be 70–350% higher for the GQ structures of telomeric DNA repeat ONs and a c-Kit ON formed in aqueous buffers containing Na+, K+, NH4+ and Rb+ as compared to the random structure of the same labeled DNA ONs in Li+ conditions. On contrary, the overall fluorescence efficiency of 2AP incorporated into GQ-forming sequences is very low and also shows very low energy transfer efficiencies with no discernible difference among different structures.133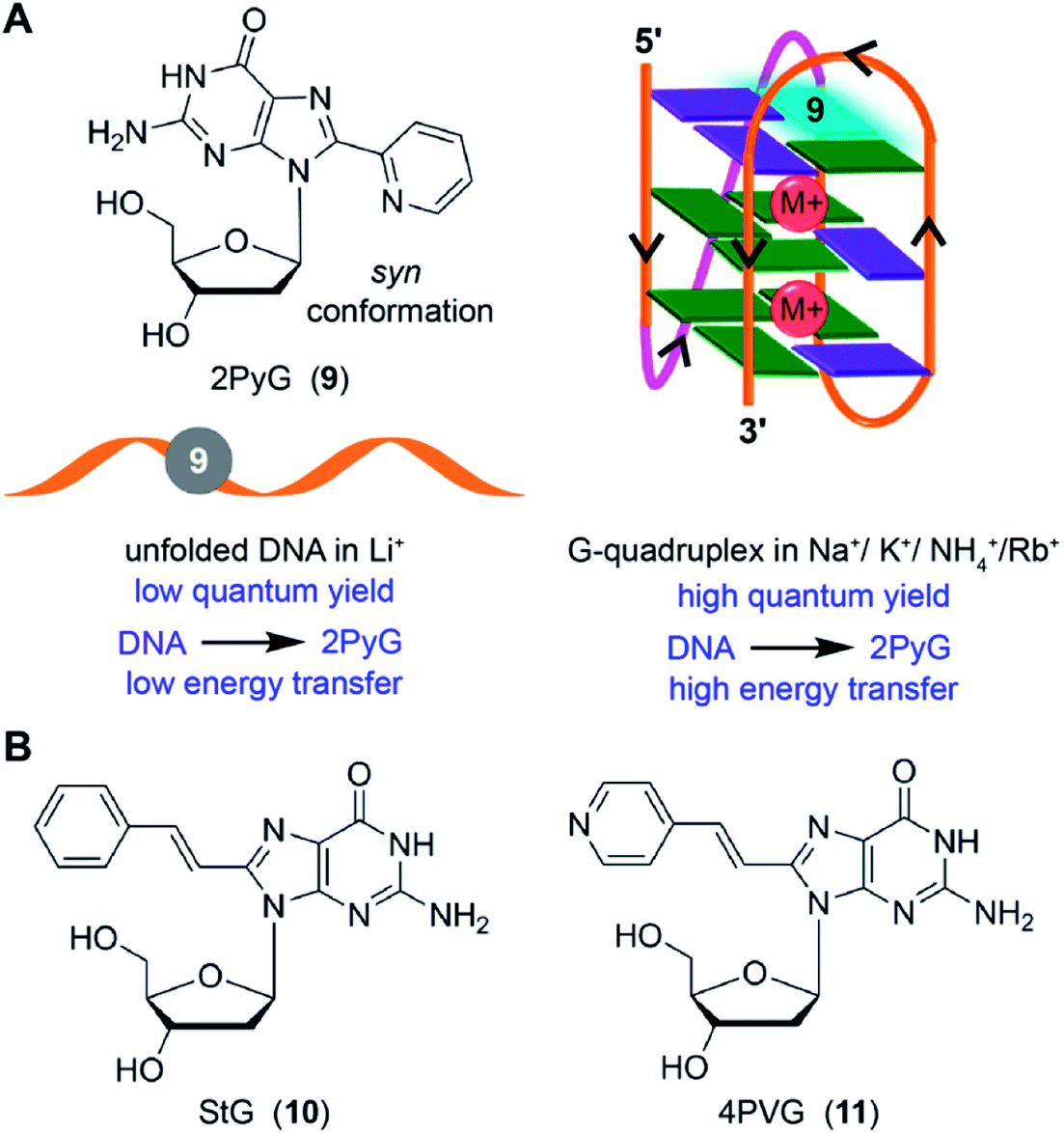 | ||
| Fig. 5 (A) 2PyG in syn glycosidic conformation is shown. GQ-forming H-Telo and c-Kit ONs containing 2PyG in the folded GQ state exhibits higher quantum yield and energy transfer efficiency compared to unfold state.138 (B) Structure of 8-(2-phenylethenyl)-2′-deoxyguanosine (StG, 10) and 8-[2-(pyrid-4-yl)-ethenyl]-2′-deoxyguanosine (4PVG, 11) is shown.139 | ||
Luedtke and Dumas introduced second generation GQ sensors based on the 2PyG scaffold. 8-(2-Phenylethenyl)-2′-deoxyguanosine (StG) and 8-[2-(pyrid-4-yl)-ethenyl]-2′-deoxyguanosine (4PVG) with extended conjugation were incorporated into H-Telo DNA ON repeat (Fig. 5B).139 Unlike 2PyG, StG and 4PVG prefer the anti glycosidic conformation, which make them structurally less perturbing in both quadruplex and duplex structures. Among these probes, GQ structures of StG-modified telomeric ONs exhibited very high quantum yields as compared to the GQs of 4PVG- and 2PyG-modified ONs. Importantly, high quantum yield and high molar extinction coefficient of ONs labeled with StG enabled the detection of GQ structures using conventional fluorescence spectrophotometer at concentrations as low as 0.25 nM. However, these probes may not be useful in distinguishing different GQ topologies as they show similar quantum yields for different GQ structures formed under different ionic conditions.
Diederichsen and coworker came up with one of the most conservatively modified fluorescent nucleoside analogs, 8-vinyl-2′-deoxyguanosine (VdG).140 The fluorescence of this analog, when placed in-between different bases in single-stranded DNA ONs and duplexes, is highly sensitive to its neighboring base environment. Encourage by these results, VdG was incorporated into the human telomeric DNA ON repeat 5′-d[AGGG(TTAGGG)3T] at positions G3, G4 or G15. CD analysis indicated that the modifications in these positions did not affect the formation of respective topologies in different ionic conditions. Interestingly, telomeric ONs with VdG at G3 and G15 positions, which participate in the formation of the middle tetrad, were able to discriminate the GQ topology formed in NaCl (antiparallel) and in KCl (multiple conformations) with significant enhancement in fluorescence intensity (Fig. 6). However, these ONs show almost similar fluorescence intensity in duplex and quadruplex states in KCl with only a slight difference in the emission maximum. On the other hand, modification at G4 position, which is part of the terminal tetrad, although, reported the formation of GQ structure with enhancement in fluorescence intensity as compared to the duplex form, it failed to discriminate between different GQ topologies. Apart from fluorescence detection, an important advantage of this nucleoside analog is that it can adopt both syn and anti glycosidic conformations required for the formation of different GQ structures.
 | ||
| Fig. 6 8-Vinyl-2′-deoxyguanosine VdG adopts both syn and anti glycosidic conformations. When VdG is placed in the middle tetrad it shows higher fluorescence intensity for the antiparallel GQ in NaCl compared to the multiple GQs in KCl and duplex form.140 | ||
Taking cue from Tor's isomorphic analogs based on furan-modified fluorescent nucleosides,141,142 Manderville and coworkers developed a series of fluorescent C8-heteroaryl-2′-deoxyguanosine analogs – furyl (FurdG), pyrrolyl (PyrdG), thienyl (ThdG), benzofuryl (BfurdG), indolyl (InddG), and benzothienyl (BthdG) (Fig. 7A).143 The modifications were designed such that they have varying (i) ring size, (ii) degree of twist angle between aryl and nucleobase rings, and (iii) electron donor–acceptor character.144 These analogs exhibited moderate to very high quantum yields (0.1–0.8, Table 1), and importantly, were found to be sensitive to solvent polarity and viscosity changes. Cyclic voltammetry analysis indicated that the heteroaryl moiety enhanced the electron donating potential of guanosine with thienyl and benzothienyl substituents being the least electron-donating group.143
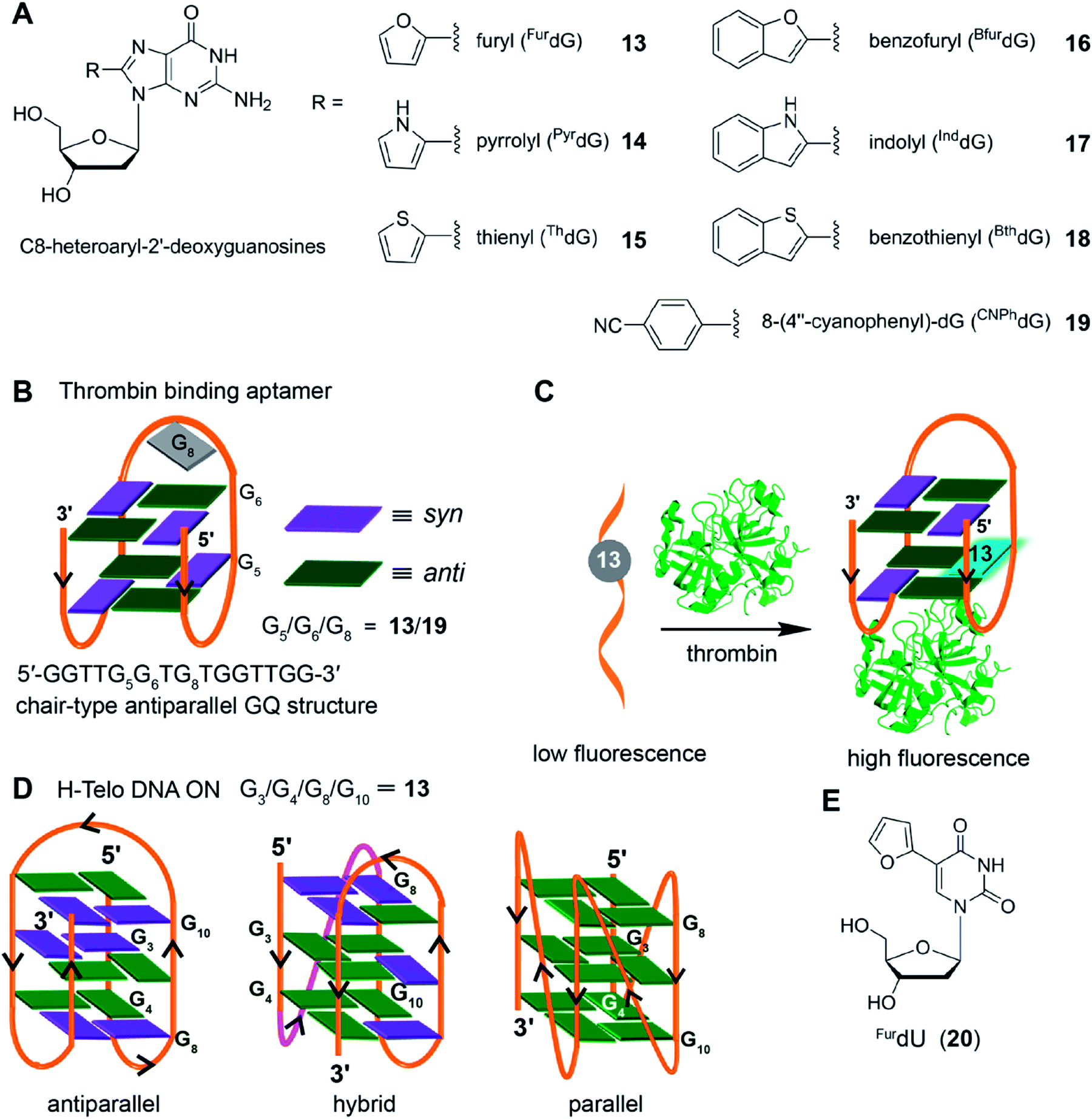 | ||
| Fig. 7 (A) C8-heteroaryl-2′-deoxyguanosine fluorescent analogs.143 (B) Site of incorporation of FurdG (13)/CNPhdG (19) into thrombin binding aptamer GQ at position G5 (syn)/G6 (anti)/G8 (loop reside) is shown. (C) FurdG (13) modified thrombin aptamer shows higher fluorescence upon thrombin-mediated GQ formation compared to the aptamer in the unfolded state.146 (D) Site of incorporation of FurdG into different tetrads (G3/G4/G8/G10) of H-Telo DNA ON repeat.147 (E) Chemical structure of solvent polarity- and viscosity-sensitive fluorescent nucleoside analog (FurdU) used in studying the interaction between GQ of an thrombin aptamer and thrombin protein.148 | ||
The group of Wetmore and Manderville utilized 8-furyl-dG (FurdG) and 8-(4′′-cyanophenyl)-dG (CNPhdG) as tools to detect the GQ folding of a 15-mer (5′-GGTTGGTGTGGTTGG) thrombin-binding aptamers.145 The aptamer forms a two tetrad chair-type antiparallel GQ structure. The guanosine analogs, which adopt a syn-conformation, were placed at G5 (syn), G6 (anti) and G8 (TGT) loop positions of the aptamer. FurdG and CNPhdG incorporated at G5 (syn) position were found to increase the stability of GQ structure, while destabilizing the duplex structure formed with complementary strand of the thrombin aptamer (Fig. 7B). Incorporation of FurdG at G6 (anti) resulted in the destabilization of both duplex and quadruplex structures. In the case of G8 loop modification, CNPhdG modification had lesser destabilization effect as compared to FurdG modification due to better stacking interaction of CNphdG with the G-tetrad. Importantly, GQ form of FurdG- and CNPhdG-modified thrombin aptamer sequences and the corresponding duplexes show distinct fluorescence properties. The G5 FurdG-modified sequence, which forms a stable GQ, shows significantly higher fluorescence intensity as compared to its duplex form. The enhancement in fluorescence intensity was ascribed to the energy-transfer bands (∼250 nm and ∼290 nm) from native guanosine residues of the tetrad, which were observed in the excitation spectrum. In contrast, the push–pull donor–acceptor probe CNPhdG placed at G8 position exhibited very high fluorescence intensity in the duplex state, whereas its fluorescence was considerably quenched in the GQ form. Taken together, FurdG placed at G5 and CNPhdG placed at G8 serve as a turn-on and turn-off fluorescence probe for duplex-quadruplex transition, respectively. Further, these probes were used in estimating the thrombin-mediated GQ folding of thrombin aptamer.146 Upon increasing the thrombin concentration, FurdG-modified aptamer displayed gradual increase in fluorescence intensity, whereas, CNPhdG-modified aptamer exhibited gradual quenching in fluorescence intensity (Fig. 7C). The association constant (Ka) of thrombin binding to the aptamer determined using fluorescence titration experiments revealed a trend; FurdG (G6) < FurdG (G8) < FurdG (G5), which correlated with the trend in GQ stabilization by the modification.
The use of FurdG was extended in the study of GQ structures of the H-Telo DNA repeat.147 The emissive analog was incorporated into four different positions within the three G-tetrads, and the GQ formation was evaluated by using a combination of UV-vis thermal melting, CD and fluorescence techniques (Fig. 7D). The folding of telomeric repeat was studied under different ionic conditions and in the presence of additives and cosolutes (e.g., CH3CN, PEG and N-methyl mesoporphyrin IX). The analog placed in the middle tetrad, though showed considerable enhancement in fluorescence intensity as compared to the duplex, the fluorescence intensity for antiparallel GQ in Na+ and hybrid type GQs in K+ was similar. When the nucleoside probe was placed in the terminal tetrads (G4, G8 and G10 positions), it displayed distinct fluorescence intensity for duplex, hybrid GQ and antiparallel GQ structures. Further, the positional impact on the formation of parallel GQ topology in the presence additives and cosolutes was also studied. These studies indicated that the modification at G8 position was structurally noninvasive, and gave the best response in terms of enhancement in fluorescence upon GQ formation. In another report, Manderville and coworker used the responsiveness of FurdU (20)141,142 to solvent polarity and viscosity changes to distinguishing thymine residues that are solvent-exposed in a GQ structure of a thrombin aptamer and identify the thymine residues that bind specifically to thrombin protein (Fig. 7E).148 Collectively, these studies with 8-aryl nucleoside analogs suggest that, when placed in an appropriate position, these analogs could serve as good diagnostic tools to study the conformations of GQs in vitro.
4.3. 5-Heterocycle-conjugated pyrimidine nucleoside probes
Although the fluorescent nucleoside analogs discussed above provide efficient systems to study GQ structure and recognition, their ability to distinguish different GQ structures based on topology and nucleic acid type (DNA versus RNA) and estimate topology-specific binding of ligands to different GQs is either not well explored or not possible. In this context, Srivatsan and coworkers introduced new fluorescent nucleoside analogs derived by attaching heterobicycles like indole, benzofuran and benzothiophene at the 5- and 8-position of pyrimidine and purine nucleosides, respectively.149–152 The design of heterobicycle-conjugated nucleosides is based on the intrinsically fluorescent indole core of the amino acid tryptophan. The microenvironment-sensitive of tryptophan has been used in devising several fluorescence based assays to study protein structure, dynamics and recognition. Therefore, it was hypothesized that attaching heterobicycles to the otherwise non-emissive native nucleosides would expand the conjugation and impart favorable fluorescence properties. Based on this design strategy, a series of ribonucleoside analogs was developed by conjugating indole, N-methyl indole, benzothiophene and benzofuran at the 5-position of uridine (Fig. 8).149,150 Unfortunately, indole- and N-methyl indole-modified nucleosides were found to be very weakly fluorescent for any practical applications. Rewardingly, 5-benzothiophene- and 5-benzofuran-modified uridine analogs were found to be moderately emissive. Importantly, both the nucleoside analogs displayed excellent fluorescence solvatochromism and viscochromism, meaning their fluorescence properties like emission maximum, quantum yield, lifetime and anisotropy were highly sensitive to the surrounding solvent polarity and viscosity. The sensitivity of nucleoside analogs to viscosity changes was due to the presence of a molecular rotor element in the form of a rotatable aryl–aryl bond between uridine and heterocycle moieties. In the viscous medium, due to rigidification of the fluorophore, the fluorescence quantum yield, lifetime and anisotropy were found be significantly higher as compared to in a medium of low viscosity. It is important to mention here that 5-benzothiophene- and 5-benzofuran-modified uridine analogs in solvents of different polarity show distinctly different trend. While 5-benzothiophene uridine shows high fluorescence efficiency in a non-polar solvent (dioxane) as compared to in a polar solvent (water), 5-benzofurn uridine shows high fluorescence efficiency in a polar solvent (water) as compared to in a non-polar solvent (dioxane, Table 1). However, the emission maximum of both the nucleosides is blue-shifted as the polarity of the medium is lowered from water to dioxane. Encouraged by these results, Srivatsan group synthesized the complete set of benzofuran-modified purine and pyrimidine nucleosides.151 While the 8-substituted purine nucleosides were highly emissive, they were not very sensitive to solvent polarity and viscosity changes.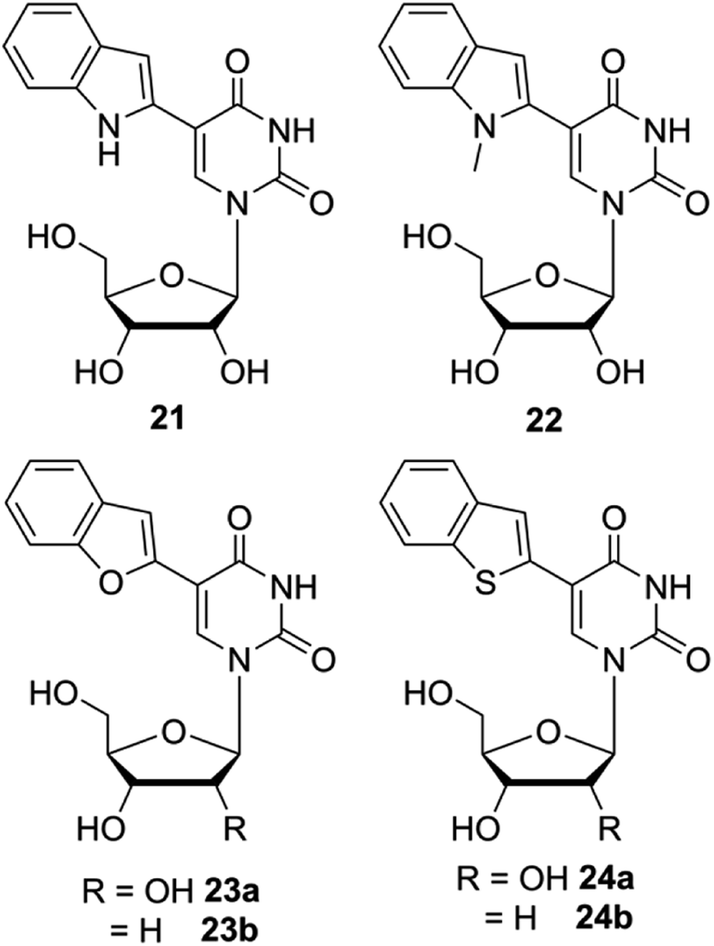 | ||
| Fig. 8 Chemical structure of indole (21), N-methylindole (22), benzofuran (23a and 23b) and benzothiophene (24a and 24b) modified 2′-deoxyuridine and or uridine analogs. | ||
Subsequently, 5-benzothiophene- and 5-benzofuran-modified 2′-deoxyuridine and uridine analogs, which exhibited similar photophysical properties, were incorporated into DNA and RNA ONs by either solid-phase ON synthesis protocol or by enzymatic method using triphosphate substrates. The nucleoside analogs placed in different nucleobase environment are highly sensitive to flanking bases and base pair substitutions. Based on the fluorescence outcome, these analogs were appropriately utilized in designing assays to detect abasic sites (depurinated site) in DNA and RNA ONs,150,154,155 and in studying the ON dynamics in cell-like confined environment.156
The non-perturbing nature and conformation-sensitivity of the heterobicycle-modified nucleoside analogs was put to use in addressing the challenges associated with probing GQ topologies and recognition. Srivatsan and Tanpure used 5-benzofuran-modified 2′-deoxyuridine and uridine nucleosides to study the formation of GQ structures of H-Telo DNA and RNA (TERRA) repeats (Fig. 9).157 Since the loop residues of different GQ topologies of telomeric repeats show considerable differences in their conformation, one of the dT/U residues in the middle loop was replaced with corresponding nucleoside analog. CD and thermal melting studies confirmed that the modification in the loop position did not affect the formation as well as the stability of respective DNA and RNA GQ structures in different ionic conditions. The labeled telomeric DNA ON repeat, which forms hybrid-type GQ structures in KCl, displayed nearly 4-fold higher fluorescence intensity than the duplex (Fig. 9A). Interestingly, H-Telo ON in NaCl, which promotes the formation of an antiparallel GQ structure, displayed nearly 9-fold enhancement in fluorescence intensity as compared to the duplex. A similar trend in quantum yield and lifetime was observed. The average lifetime of antiparallel GQ is 3.0 ns, hybrid-type GQs is 1.4 ns and duplex is ∼0.64 ns. On the other hand, TERRA ON adopts a parallel GQ topology irrespective of the ionic conditions, and hence, displayed a similar fluorescence quantum yield (∼0.19) and average lifetime (∼2.80 ns) in NaCl and KCl (Fig. 9B). These results clearly indicate that the benzofuran-modified nucleosides photophysically report the formation as well as distinguish different DNA and RNA GQ topologies. This feature was further successfully utilized in determining the topology-specific binding of ligands like pyridostatin (PDS) and BRACO19 to telomeric DNA and RNA GQs (Fig. 10). Titration of telomeric DNA and RNA repeats with increasing concentration of the ligands resulted in a dose-dependent quenching in fluorescence intensity. The dissociation constant determined using the fluorescence data revealed that the ligands bind to GQ topologies with different affinities. For example, PDS has higher affinity for the antiparallel structure compared to hybrid-type GQs. However, BRACO19 has higher affinity for hybrid-type GQs. PDS and BRACO19 show similar binding affinities for the parallel conformation of the TERRA ON. The binding of ligands to biologically relevant higher-order GQ structures was also evaluated using the nucleoside probe. Unlike short telomeric ON repeat, PDS and BRACO19 binding to a longer telomeric repeat, which could form two consecutive GQs, revealed similar binding constants in Na+ and K+ ionic conditions. This observation and literature precedence point out that the ONs containing flanking sequences along with the GQ-forming motif would be better models in the study of GQs.158
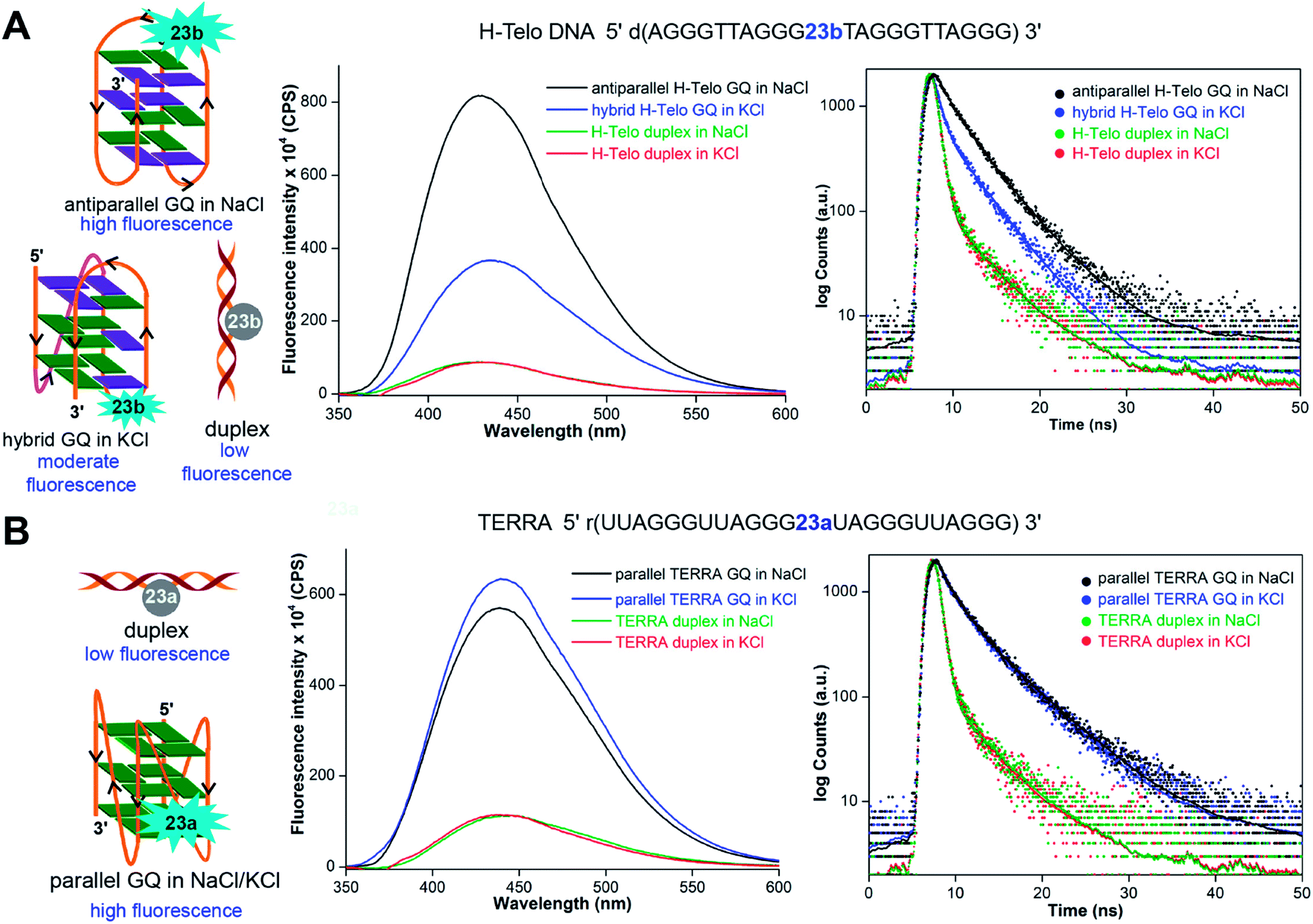 | ||
| Fig. 9 (A) 5-Benzofuran-modified 2′-deoxyuridine (23b) incorporated into a H-Telo DNA ON fluorescently distinguishes different GQ structures. The antiparallel GQ in NaCl, mixed hybrid-type GQs in KCl and duplex show distinct fluorescence quantum yield and average lifetime. (B) Equivalent RNA repeat, TERRA, labeled with 5-benzofuran-modified uridine (23a) reports the formation of a parallel GQ structure, irrespective of the ionic conditions. The fluorescence intensity and average lifetime of TERRA GQ in NaCl and KCl are similar. In steady-state fluorescence study, all samples were excited at 330 nm. The λem of H-Telo DNA in NaCl and KCl is ∼430 nm and λem of corresponding duplexes is ∼440 nm. In both the salt conditions, λem of TERRA RNA and corresponding duplex is ∼440 nm. In lifetime study, samples were excited using a 339 nm light source and decay was measured at respective emission maximum. This figure has been adapted with the permission of Nucleic Acids Research: Oxford Journals.157 | ||
 | ||
| Fig. 10 The quenching in fluorescence intensity as a function of increasing ligand (PDS and BRACO19) concentration enabled the determination of dissociation constant. A representative titration plot of 23b-labeled H-Telo DNA ON with increasing concentration of PDS.157 | ||
Srivatsan and coworkers used benzofuran-modified nucleosides to establish a fluorescence-based platform to probe the GQ structure and ligand binding of H-Telo DNA and RNA repeats in a commonly used cellular model AOT (aerosol OT, bis(2-ethylhexyl) sodium sulfosuccinate) reverse micelles (RM, Fig. 11).159 This was essentially possible because the nucleoside analogs faithfully reported the microenvironment of different domains of the confined water in RM via changes in fluorescence properties. The fluorescence detection of various GQ structures in aqueous buffer and in the confined water pool of RM was studied by using telomeric DNA repeat ONs containing benzofuran modification in different loops. In aqueous buffers containing NaCl and KCl, the probe clearly reported the formation GQ structures with a distinct fluorescence profile for the antiparallel and hybrid-type GQs, respectively. The fluorescence intensity and lifetime for the antiparallel GQ was the highest, followed by hybrid-type GQs and duplex. While in aqueous buffer, the nucleoside probe fluorescently distinguished different GQ topologies based on ionic conditions, the labeled telomeric ONs displayed almost similar fluorescence intensity and lifetime in AOT RM, irrespective of added Na+ and K+ ions. Comparing the fluorescence data and CD profiles in buffer and AOT RM it was confirmed that the telomeric repeats preferred the antiparallel GQ structure in RM. In this study, the sodium salt of AOT (200 mM) was used to form stable RM. Hence, it was inferred that the exchange of K+ ions with Na+ ions of AOT head group would have biased the telomeric ONs to adopt an antiparallel GQ structure. In case of TERRA, the probe reported the formation of a parallel GQ structure in both aqueous buffer and AOT RM with significantly higher fluorescence intensity and lifetime compared to the duplex form. Notably, the binding affinities of PDS to DNA and RNA GQs obtained by fluorescence assay in aqueous buffer and AOT RM were comparable. Collectively, these studies underscore the potential of benzofuran-modified nucleoside analogs in not only detecting the formation of GQs but also estimating the binding of ligands to various GQs in a cell-like confined environment.
 | ||
| Fig. 11 Fluorescence detection of GQ structures in RM by using nucleoside analogs 23a and 23b. The probe labeled H-Telo DNA and RNA ONs provided a simple platform to compare the GQ formation as well as ligand binding in aqueous buffer and in the confined water pool of RM, which is a commonly used membrane model. This figure has been adapted with the permission of ChemBioChem: John Wiley and Sons.159 | ||
In another study, the environment sensitivity of 5-benzofuran uracil peptide nucleic acid (PNA) base analog in combination with the strand-invasion ability of PNA oligomer was utilized in devising a fluorescence turn-on assay to detect the H-Telo DNA repeat (TTAGGG)n.160 The detection platform was based on graphene oxide (GO), an excellent fluorescence quencher, which binds single-stranded DNA ONs significantly better compared to structured ONs. 5-Benzofuran uracil PNA base analog incorporated into an 18-mer PNA oligomer complementary to the H-Telo DNA repeat was synthesized by solid-phase PNA synthesis protocol. This oligomer upon hybridization with telomeric DNA repeat exhibited discernible enhancement in fluorescence intensity. In order to increase the sensitivity of the PNA probe, it was complexed with GO, which resulted in a significant quenching in fluorescence intensity as compared to the free PNA probe. Upon addition of the human telomeric DNA repeat, the PNA probe diffused out of the GO surface and formed a duplex by invading the GQ structure of the telomeric DNA repeat, thereby exhibiting huge increase in fluorescence as compared to the PNA–GO complex. Using this platform, as low as ∼30 nM concentration of the H-Telo repeat could be detected.
Following this report, Srivatsan and Sabale developed another environment sensitive PNA building block made of 5-(benzothiophen-2-yl)-uracil core.155 Both benzofuran and benzothiophene PNA analogs were incorporated into a series of PNA oligomers and hybridized to complementary and mismatched DNA ONs to study the responsive of the emissive bases to flanking bases and base pair substitutions. While both the analogs were highly sensitive to changes in neighboring base environment, benzothiophene PNA base analog placed in-between C residues selectively reported the presence of a complementary nucleobase (dA) in a GAG ON sequence with significant enhancement in fluorescence intensity. This observation enabled the development of PNA hybridization probes against GQ-forming oncogenic sequences containing GAG base set like c-MYC and c-Kit2.155 Complementary PNA oligomers containing the benzothiophene-modified uracil base were incubated with pre-formed GQ structures of c-MYC and c-Kit2 ONs. The hybridization of PNA oligomers with GQ ONs places the emissive base opposite to dA in the GAG base set and leads to enhancement in fluorescence intensity. Interestingly, the PNA probe complementary to c-Kit2 was able to invade the GQ structure and show considerable enhancement in fluorescence intensity due to the formation of a more emissive PNA-c-Kit2 duplex. However, the PNA probe against c-MYC was not very effective in invading the strand as c-MYC is known to form a highly stable GQ structure as compared to c-Kit2. In a similar study, Diederichsen and coworkers used a fluorescent PNA analog based on 8-vinyl guanine to monitor the formation of PNA-DNA hybrid GQ structures.161 It has been proposed that such environment-sensitive PNA probes could be very useful in detecting as well as in studying the functional role of individual GQ-forming sequences.
It is important to mention here that the nucleoside analogs described above are not suitable for in-cell analysis of GQs as their excitation maximum are in the UV region (Table 1). Further, the cellular autofluorescence covers the fluorescence of many of these nucleoside-based probes, and hence, may not be suitable for imaging purpose. Alternatively, antibody-based immunofluorescence staining and fluorescent binders, which are compatible to fluorescence microscopy, have been developed to visualize GQs in cells.
5. Structure-specific tools to probe GQs in cells
5.1. Antibody-based tools
Although biochemical and biophysical in vitro analysis have long confirmed the formation of different tetraplex structures, the evidence for the occurrence and function in vivo is fast accumulating. Seminal contributions from the laboratories of Plückthun and Balasubramanian have provided strong evidence for the occurrence of GQs in ciliates and mammalian cells. Plückthun and coworkers, for the first time selected high-affinity single-chain antibody fragments (scFvs) for the GQs formed by the Stylonychia lemnae telomeric repeat d(T4G4)2 by ribosome display.162 Among the selected fragments, Sty3 had an affinity in the picomolar range for the parallel-stranded GQ structure. A second scFv (Sty49) was found to bind to both parallel and antiparallel GQs with similar affinities in the nanomolar range. Among the fragments, Sty49 produced a clear signal in the macronuclei in immunofluorescence staining experiments, indicating the occurrence of GQ structure. Further, this antibody was used in studying the accumulation of GQs in the replication band and the unfolding of GQs by telomerase.163Balasubramanian and coworkers developed an elegant method to pull-down GQ-forming sequences from mammalian cells by using a derivative of PDS (Fig. 12).164 Derivative 26 was attached to biotin, an affinity tag, which did not adversely affect the affinity of the ligand to GQs. Initial assessment in vitro in the presence of a pool of structured DNA ONs indicated that 26 could be used to pull-down longer genomic DNA fragments capable of forming GQs. This method was applied in pulling down telomeric repeats of HT1080 human cancer cells, which upon high throughput sequencing (ChIP-Seq) provided further evidence for the formation of GQs in human cells. Based on this approach, a PDS derivative 27 containing a clickable alkyne group was developed. Cells treated with 27 for 12 h were click-stained with an Alexa-azide, which showed a fluorescent pattern resembling the staining of nucleoli containing GQ-forming sequences.165
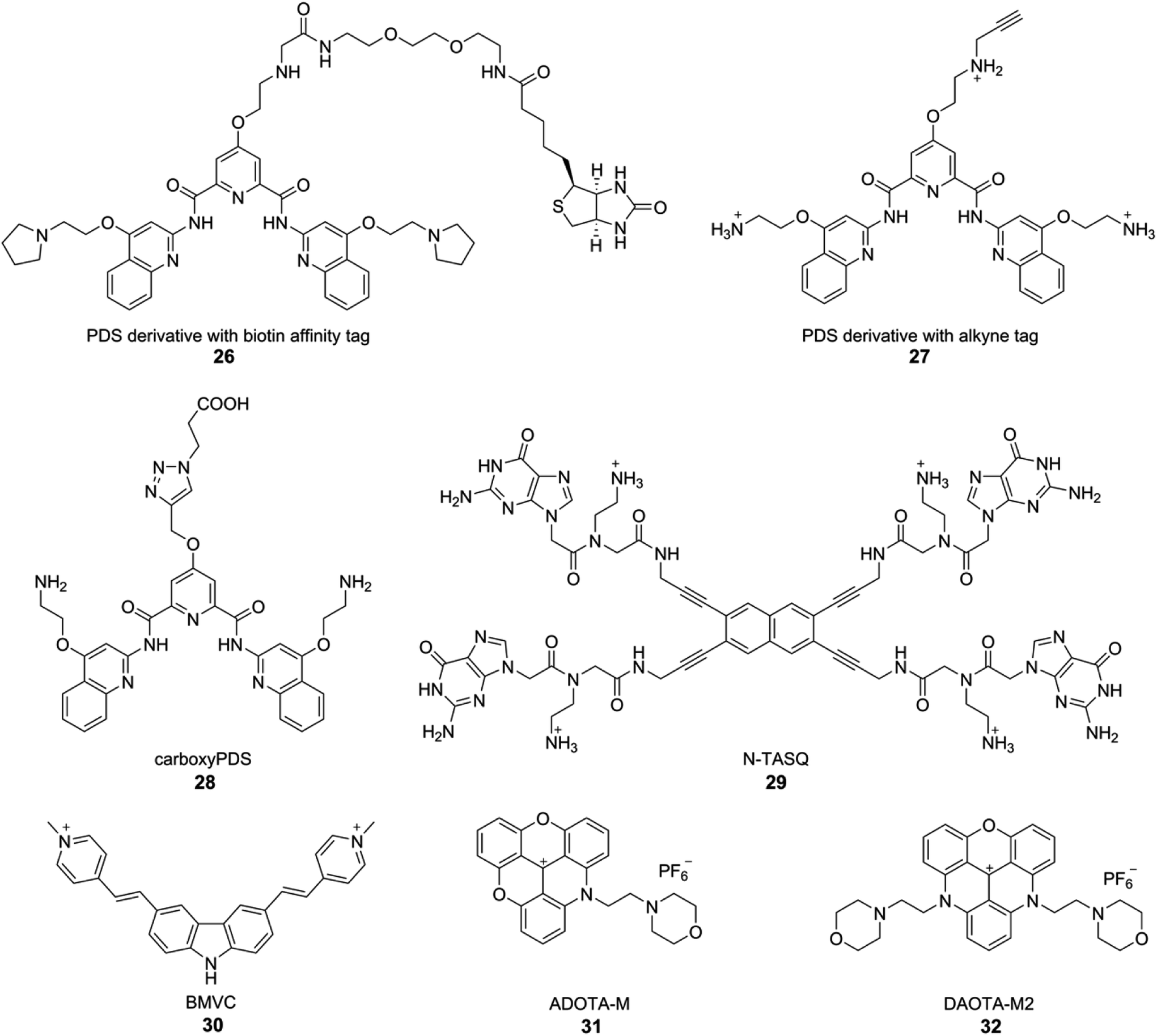 | ||
| Fig. 12 GQ-structure-specific small molecule ligands used in the visualization of DNA and RNA GQs in cells. | ||
Although these small-molecule GQ binders provide indirect evidence for the presence of GQs in cells, Balasubramanian's group developed a GQ-specific antibody to directly visualize DNA GQs in human cells.42 By employing phage display composed of 2.3 × 1010 different single-chain antibody clones, antibody called BG4 with nanomolar affinity for intramolecular and intermolecular GQ structures was indentified. Preliminary binding studies with various sequences capable of forming different GQ structures indicated that BG4, though highly GQ structure-specific, does not have a preference for a particular GQ topology. Cells treated with BG4 were visualized by using a secondary antibody and then a tertiary fluorochrome-labeled antibody. Punctate nuclear staining in all cell lines tested along with appropriate control experiments confirmed the presence of DNA GQs in human cells. Further, it was identified that BG4 also binds to RNA GQs of TERRA, BCL2 and NRAS with affinities in the nanomolar range. Encouraged by this observation, the presence of endogenous RNA GQs was tested in human normal endothelial, immortalized and cancer cell lines.43 Fixed and permeabilized cells were first incubated with BG4, and were immunofluorescence stained using secondary antibody and tertiary fluorochrome-labeled antibody as mentioned above. The presence of cytoplasmic foci, which disappeared upon treating the fixed cells with RNase A prior to incubating with BG4 indicated that the fluorescence signal emanating from the cytoplasm is due to RNA GQs present in the human transcriptome. Interestingly, a derivative of PDS, carboxyPDS 28, which binds strongly to RNA GQs as compared to DNA GQs, increased the number of GQs in the cytoplasm.43 These results suggest that structure-specific small molecule ligands can be used to target as well as trap GQs in cells.
Lansdorp and coworkers identified a new murine monoclonal antibody specific for GQ structures by immunizing spleen cells from mice with stable DNA GQs.44 The antibody designated as 1H6 displayed a strong nuclear signal from human and murine cells in immunofluorescence microscopy studies. Addition of a GQ stabilizing ligand (TMPyP4) increased the number of nuclear foci. Further, cells depleted with FANCJ, a GQ DNA-specific helicase, displayed pronounced fluorescence signal from nuclei as compared to control cells. Apart from providing evidence for the occurrence of GQs in mammalian cells, these results strongly support the notion that GQ in cells can be targeted by using small molecule ligands. While BG4 and 1H6 are useful in GQ visualization experiments, they fail to provide information on the topology as these antibodies don't have preference for a certain GQ topology. In this context, Huang and coworkers recently developed a new scFv antibody, D1, which exhibits very high binding affinity and selectivity for the parallel GQ structure as opposed to other GQ forms.166 Genome-wide identification of consensus sequences for parallel GQ structure was performed by chromatin immunoprecipitation using D1, followed by deep sequencing. This analysis also revealed that G-rich sequences containing a shorter loop (<3 nt) are prone to form parallel GQ structure. The antibody enabled the visualization of parallel GQ structure at the human telomere in fixed cells. Importantly, using ligands like NMM and QPB-15e, which are known to induce parallel GQ conformation, the formation of parallel GQ structure in cells could be modulated. Further, immunofluorescence experiments with fixed cells incubated with both BG4 and D1 antibodies indicated that the human telomeric GQs predominantly (77%) exist in the parallel form. However, immunofluorescence detection of GQs using currently available antibodies has its own limitations, which include elaborate assay setup and restricted application to fixed and permeabilized cells.167
5.2. Small molecule ligand-based tools
Alternatively, structure specific light-up and fluorogenic small molecule probes have been developed to visualize GQs in live cells. In one of the first efforts, Komiyama and coworkers used a light-switching pyrene probe, which forms excimers, to study the structure of TERRA RNA in living cells.168 ONs containing TERRA RNA sequence were labeled with a pyrene moiety at both 5′ and 3′ ends. In the unfolded state, only the fluorescence emission (∼400 nm) from pyrene monomer was observed. GQ formation brings pyrenes closer to form an excimer, which shows a red-shifted emission at ∼480 nm. This change in emission colour was used to study the formation of GQ in cells. HeLa cells in culture were incubated with a dual-pyrene labeled ON containing TERRA RNA repeat in the presence of a transfecting agent. Cells imaged using fluorescence microscopy clearly revealed the excimer emission from pyrene due to the GQ folded state of the ON. Further, colocalization experiments indicated that the TERRA RNA GQ is restricted to nuclei. These observations indicate that RNA GQ can be potentially stabilized in vivo.Monchaud and coworkers developed a GQ ligand called NaphthoTASQ (N-TASQ), which serves as a multi-photon fluorescence probe to directly visualize RNA GQs in live cells (Fig. 12).167 N-TASQ binding to TERRA GQ resulted in 22-fold improvement in fluorescence of the probe. The probe also showed preference for RNA GQs as compared to DNA GQs. In cell based experiments, N-TASQ stained cytoplasmic and nucleolar compartments. Further, experiments with nucleases and BG4 antibody confirmed the GQ staining ability of the multi-photon probe.
In another report, the GQ specific fluorogenic N-TASQ probe was observed to exhibit red-edge effect, which was utilized in visualizing DNA and RNA in human cell.169 The unique red-edge fluorescence properties of N-TASQ–GQ complex enabled it to absorb light upto near infrared region and the emission maximum was observed to be linearly dependent on the excitation wavelength. These properties made N-TASQ–GQ complex compatible to be imaged by both confocal and two-photon microscopy. Further, the quadruplex specific red edge probe (G4-REP) N-TASQ was utilized to quantify BRACO-19 and TmPyP4 induced GQ in HeLa cells.170 Additionally, the confocal imaging of N-TASQ treated live cancerous and non-cancerous cells displayed different staining pattern in cytoplasm, which proved that N-TASQ can enter both type of cells but the cellular response to N-TASQ is different in cancerous cell than non-cancerous cell. Most interestingly, a microplate-compatible LED based microscope housed inside a cell incubator was utilized in real time analysis of N-TASQ treated live MDA-MB 231 breast cancer cells for longer periods (∼120 h).
Chang and coworkers employed a fluorogenic GQ-binding ligand BMVC (3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide) to investigate the cellular uptake and localization of various exogenously added GQ-forming DNA sequences (Fig. 12).171 The G-rich sequences, which form a parallel conformation, were found to accumulate in the lysosome, and the G-rich sequences, which form non-parallel GQ structures, were found to accumulate in the mitochondria of CL1-0 lung cancer cells. Further, o-BMVC and derivatives of BMVC, which have longer fluorescence decay time when bound to GQs than when bound to duplex structure, enabled the development of a method to visualize GQs in live cells by using fluorescence lifetime imaging microscopy (FLIM).172,173 In a similar approach, Vilar and coworkers developed small molecule optical probes, which exhibit fluorescence enhancement and significant difference in lifetime depending on the nucleic acid topology.174 They developed new triangulenium derivatives, ADOTA-M and DAOTA-M2 (Fig. 12). In particular, DAOTA-M2 upon binding to GQs served as a turn-on fluorescence probe, and importantly, exhibited distinctly different lifetimes for GQ, duplex and single-stranded nucleic acids. This feature was further utilized in visualizing GQs and also in studying the interaction of small molecule ligands with GQs in cells by using FLIM.
Manet and Freccero reported the theranostic application of a fluorescent water-soluble tetracationic quaternary ammonium naphthalene diimide (NDI), which showed high binding affinity for different GQ topologies in vitro.175 However, NDI was not very selective to only GQs as it was also found to bind to duplex DNA. Emission in red-NIR region aided its visualization in the nuclei of different types of tumor cells. NDI was not cytotoxic to the human tumor cells but upon irradiation of NDI-loaded cells with red light generated singlet oxygen species, which reduced the cell viability and induced DNA damage foci in cell nuclei. The fluorescence lifetime confocal imaging of NDI treated fixed tumor cell and lifetime analysis of an area of cell's nuclei revealed two lifetimes-one corresponding to the free NDI and other NDI bound to duplex DNA. These observation matches with the in vitro binding studies, which showed that NDI can bind also to the duplex DNA.
Richter and Freccero group developed a red-NIR core-extended NDI probe, which showed quenching in fluorescence intensity in water due to aggregation compared to monomer emission in organic solvent.176 Interestingly, the probe was observed to bind GQ structure selectively compared to duplex and single stranded DNA. The presence of H-Telo DNA made the probe monomeric, and enhanced the fluorescent intensity significantly. Further, the location of NDI probe in the nucleus was visualized by confocal microscopy, and good colocalization of the NDI foci and GQ-specific antibody 1H6 proved that the probe indeed targets GQs in cells.
Chow and coworker examined the efficiency of different pyridinium-based fluorescent dyes with varying symmetry and different styrene like side groups in detecting GQs.177 Pyridinium molecule with C2 symmetry and indolyl side group, which was the best GQ-specific fluorescent probe was used in live cell imaging. The highest enhancement of fluorescence intensity in S phase of the cell cycle supported the replication-dependent GQ formation in cell.
Zhang and coworkers reported a GQ sensor based on the triphenylamine-quinolinium dye, which exhibited remarkable enhancement (∼180 times) in fluorescence intensity selectively in the presence of HRAS GQ compared to other GQs and nucleic acid structures.178 Using this probe, as low as 1 nM of HRAS GQ could be detected. Further, confocal imaging of triphenylamine-quinolinium dye treated cell and co-staining with DAPI displayed the accumulation of the dye in cell nucleoli, suggesting that this dye could be useful in live cell imaging.
In another study, Tan and coworkers developed hybridization ON probe, composed of a fluorogenic reporter ISCH-1, to identity GQs in cells.179 ISCH-1, which shows remarkable enhancement in fluorescence efficiency, was click-tagged to an ON sequence complementary to the sequence adjacent to the GQ-forming motif of NRAS UTR. Since the target mRNA concentration in a single cell is less, the NRAS RNA ON was transfected into the cells followed by annealing with the hybridization probe. Under these conditions the cells displayed intense fluorescence spots in the cytoplasm. However, mutated version of the NRAS did not produce any signal. While this method confirms that an exogenous RNA ON of NRAS can adopt GQ structure in cellular environment, detection of endogenous GQ-forming sequences expressed in low concentrations remains to be explored.
6. Conclusions and perspectives
Structure-specific tools that use fluorescence readout to detect the formation of GQ structures and their interaction with small molecule ligands have greatly advanced our understanding of these structures in cell-free and cellular environments. Internal modification strategy using nucleoside analogs has provided an alternative means to investigate local conformational differences in the loop and tetrad domains of different GQs under equilibrium conditions. The differences in physico-chemical properties in these domains essentially dictate the selectivity and affinity of a ligand to a certain GQ structure. In this context, recently developed fluorescent nucleoside probes, which distinguish different DNA and RNA GQ topologies and report topology-specific binding of ligands to GQs could be very useful in establishing screening platforms to indentify functional binders of clinical potential. While GQ-specific antibodies have been very useful in establishing the occurrence of DNA and RNA GQs in fixed and permeabilized cells, cell-compatible small molecule fluorescent probes, which show remarkable changes in fluorescence properties upon interacting with GQs, have been very useful in studying the structure and localization of exogenously added GQ-forming ON sequences.Nevertheless, the diversity in GQ-forming sequences in the genome and the ability of GQ to adopt different topologies depending on the sequence and microenvironment pose important challenges in the study of tetraplexes. Despite extensive studies, our understanding of the tetraplex structures in cellular environment is inadequate. For example, it is not clear what structure or structures a G-rich sequence adopts inside the cell, and which one is responsible for its function. In this context, in-cell NMR techniques are being developed to determine the structure of individual GQ- and i-motif-forming ON sequences in an environment very close to the cell. It is also not easy to conceive how to specifically target a GQ motif among various endogenous GQ-forming sequences by using small molecule ligands. Further, probing the formation and completion between duplex, GQ and i-motif structures of G-rich and C-rich strands coexisting in the promoter region is not straight forward as tools that can unequivocally distinguish these structures are rare. Hence, development of new probes that can distinguish different non-canonical four-stranded structures in cell-free and cellular environments, improvements in in-cell structural and function analysis, accessibility to wide varieties of labeled ON sequences, and newer screening approaches will have a profound impact on GQ and i-motif directed therapeutic strategies. Since the scientific community interested in these aspects of nucleic acids is fast expanding, it is only a matter of time that the core challenges associated with the study of GQs and i-motifs will be addressed. This is quite evident from a very recent report, which has provided the first evidence for the occurrence of i-motifs in mammalian cells.180
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Pramod Sabale and Dr Ashok Nuthanakanti for their suggestions and for carefully proofreading the manuscript. S. M. is grateful to the UGC, India for a graduate research fellowship. S. G. S thanks DST, India (EMR/2014/000419) and Wellcome Trust-DBT India Alliance (IA/S/16/1/502360) for research grants.Notes and references
- B. Tian, P. C. Bevilacqua, A. Diegelman-Parente and M. B. Mathews, Nat. Rev. Mol. Cell Biol., 2004, 5, 1013–1023 CrossRef PubMed.
- N. Saini, Y. Zhang, K. Usdin and K. S. Lobachev, Biochimie, 2013, 95, 117–123 CrossRef PubMed.
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef PubMed.
- P. Belmont, J.-F. Constant and M. Demeunynck, Chem. Soc. Rev., 2001, 30, 70–81 RSC.
- T. Hermann and D. Patel, Structure, 2000, 8, R47–R54 CrossRef PubMed.
- K. B. Hall, Curr. Opin. Chem. Biol., 2008, 12, 612–618 CrossRef PubMed.
- H. M. Al-Hashimi and N. G. Walter, Curr. Opin. Struct. Biol., 2008, 18, 321–329 CrossRef PubMed.
- J. Choi and T. Majima, Chem. Soc. Rev., 2011, 40, 5893–5909 RSC.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 RSC.
- A. Cammas and S. Millevoi, Nucleic Acids Res., 2017, 45, 1653–1668 Search PubMed.
- A. Theisen and L. G. Shaffer, Appl. Clin. Genet., 2010, 3, 159–174 Search PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2005, 33, 2908–2916 CrossRef PubMed.
- A. K. Todd, M. Johnston and S. Neidle, Nucleic Acids Res., 2005, 33, 2901–2907 CrossRef PubMed.
- S. Burge, G. N. Parkinson, P. Hazel, A. K. Todd and S. Neidle, Nucleic Acids Res., 2006, 34, 5402–5415 CrossRef PubMed.
- D. Rhodes and H. J. Lipps, Nucleic Acids Res., 2015, 43, 8627–8637 CrossRef PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- J. L. Huppert and S. Balasubramanian, Nucleic Acids Res., 2007, 35, 406–413 CrossRef PubMed.
- A. Bedrat, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2016, 44, 1746–1759 CrossRef PubMed.
- J. Meyne, R. L. Ratliff and R. K. Moyzis, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7049–7053 CrossRef.
- V. K. Yadav, J. K. Abraham, P. Mani, R. Kulshrestha and S. Chowdhury, Nucleic Acids Res., 2008, 36, D381–D385 CrossRef PubMed.
- Y. Chen and D. Yang, Curr. Protoc. Nucleic Acid Chem., 2012, 50, 17.5.1–17.5.17 Search PubMed.
- S. Zhang, Y. Wu and W. Zhang, ChemMedChem, 2014, 9, 899–911 CrossRef PubMed.
- D. Miyoshi, T. Fujimoto and N. Sugimoto, Top. Curr. Chem., 2013, 330, 87–110 CrossRef PubMed.
- B. Heddi and A. T. Phan, J. Am. Chem. Soc., 2011, 133, 9824–9833 CrossRef PubMed.
- Y.-C. Tsai, H. Qi and L. F. Liu, J. Biol. Chem., 2007, 282, 18786–18792 CrossRef PubMed.
- T. M. Fletcher, IUBMB Life, 2003, 55, 443–449 CrossRef PubMed.
- J. Cuesta, M. Read and S. Neidle, Mini-Rev. Med. Chem., 2003, 3, 11–21 CrossRef PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef PubMed.
- D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429–7455 CrossRef PubMed.
- A. Bugaut and S. Balasubramanian, Nucleic Acids Res., 2012, 40, 4727–4741 CrossRef PubMed.
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef PubMed.
- Y. Xu and M. Komiyama, Methods, 2012, 57, 100–105 CrossRef PubMed.
- A. Ohnmacht and S. Neidle, Bioorg. Med. Chem. Lett., 2014, 24, 2602–2612 CrossRef PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef PubMed.
- A. Laguerre, L. Stefan, M. Larrouy, D. Genest, J. Novotna, M. Pirrotta and D. Monchaud, J. Am. Chem. Soc., 2014, 136, 12406–12414 CrossRef PubMed.
- A. C. Bhasikuttan and J. Mohanty, Chem. Commun., 2015, 51, 7581–7597 RSC.
- D. Zhao, X. Dong, N. Jiang, D. Zhang and C. Liu, Nucleic Acids Res., 2014, 42, 11612–11621 CrossRef PubMed.
- T. Agarwal, M. K. Lalwani, S. Kumar, S. Roy, T. K. Chakraborty, S. Sivasubbu and S. Maiti, Biochemistry, 2014, 53, 1117–1124 CrossRef PubMed.
- D. Panda, P. Saha, T. Das and J. Dash, Nat. Commun., 2017, 8, 16103 CrossRef PubMed.
- H. J. Lipps and D. Rhodes, Trends Cell Biol., 2009, 19, 414–422 CrossRef PubMed.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef PubMed.
- G. Biffi, M. Di Antonio, D. Tannahill and S. Balasubramanian, Nat. Chem., 2014, 6, 75–80 CrossRef PubMed.
- A. Henderson, Y. Wu, Y. C. Huang, E. A. Chavez, J. Platt, F. B. Johnson, R. M. Brosh Jr, D. Sen and P. M. Lansdorp, Nucleic Acids Res., 2013, 42, 860–869 CrossRef PubMed.
- M. Guéron and J.-L. Leroy, Curr. Opin. Struct. Biol., 2000, 10, 326–331 CrossRef.
- S. Benabou, A. Aviñó, R. Eritja, C. González and R. Gargallo, RSC Adv., 2014, 4, 26956–26980 RSC.
- E. P. Wright, J. L. Huppert and Z. A. E. Waller, Nucleic Acids Res., 2017, 45, 2951–2959 CrossRef PubMed.
- A. M. Fleming, Y. Ding, R. A. Rogers, J. Zhu, J. Zhu, A. D. Burton, C. B. Carlisle and C. J. Burrows, J. Am. Chem. Soc., 2017, 139, 4682–4689 CrossRef PubMed.
- L. Lacroix, H. Liénard, E. Labourier, M. Djavaheri-Mergny, J. Lacoste, H. Leffers, J. Tazi, C. Hélène and J.-L. Mergny, Nucleic Acids Res., 2000, 28, 1564–1575 CrossRef PubMed.
- J.-F. Cornuel, A. Moraillon and M. Guéron, Biochimie, 2002, 84, 279–289 CrossRef PubMed.
- D. J. Uribe, K. Guo, Y.-J. Shin and D. Sun, Biochemistry, 2011, 50, 3796–3806 CrossRef PubMed.
- Y. M. K. Yoga, D. A. K. Traore, M. Sidiqi, C. Szeto, N. R. Pendini, A. Barker, P. J. Leedman, J. A. Wilce and M. C. J. Wilce, Nucleic Acids Res., 2012, 40, 5101–5114 CrossRef PubMed.
- H.-J. Kang, S. Kendrick, S. M. Hecht and L. H. Hurley, J. Am. Chem. Soc., 2014, 136, 4172–4185 CrossRef PubMed.
- Y. Chen, K. Qu, C. Zhao, L. Wu, J. Ren, J. Wang and X. Qu, Nat. Commun., 2012, 3, 1074 CrossRef PubMed.
- Y. Cui, D. Koirala, H. Kang, S. Dhakal, P. Yangyuoru, L. H. Hurley and H. Mao, Nucleic Acids Res., 2014, 42, 5755–5764 CrossRef PubMed.
- H. A. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407–4418 CrossRef PubMed.
- D. Drygin, A. Siddiqui-Jain, S. O'Brien, M. Schwaebe, A. Lin, J. Bliesath, C. B. Ho, C. Proffitt, K. Trent, J. P. Whitten, J. K. C. Lim, D. Von Hoff, K. Anderes and W. G. Rice, Cancer Res., 2009, 69, 7653–7661 CrossRef PubMed.
- R. Hänsel, L. M. Luh, I. Corbeski, L. Trantírek and V. Dötsch, Angew. Chem., Int. Ed., 2014, 53, 10300–10314 CrossRef PubMed.
- P. Hazel, J. Huppert, S. Balasubramanian and S. Neidle, J. Am. Chem. Soc., 2004, 126, 16405–16415 CrossRef PubMed.
- J. L. Huppert, FEBS J., 2010, 277, 3452–3458 CrossRef PubMed.
- Y. Wang and D. J. Patel, Structure, 1993, 1, 263–282 CrossRef PubMed.
- A. Ambrus, D. Chen, J. Dai, T. Bialis, R. A. Jones and D. Yang, Nucleic Acids Res., 2006, 34, 2723–2735 CrossRef PubMed.
- K. N. Luu, A. T. Phan, V. Kuryavyi, L. Lacroix and D. J. Patel, J. Am. Chem. Soc., 2006, 128, 9963–9970 CrossRef PubMed.
- J. Dai, M. Carver, C. Punchihewa, R. A. Jones and D. Yang, Nucleic Acids Res., 2007, 35, 4927–4940 CrossRef PubMed.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef PubMed.
- Z. Chen, K.-W. Zheng, Y.-H. Hao and Z. Tan, J. Am. Chem. Soc., 2009, 131, 10430–10438 CrossRef PubMed.
- M. C. Miller, R. Buscaglia, J. B. Chaires, A. N. Lane and J. O. Trent, J. Am. Chem. Soc., 2010, 132, 17105–17107 CrossRef PubMed.
- F. M. Lannan, I. Mamajanov and N. V. Hud, J. Am. Chem. Soc., 2012, 134, 15324–15330 CrossRef PubMed.
- P. Shrestha, S. Jonchhe, T. Emura, K. Hidaka, M. Endo, H. Sugiyama and H. Mao, Nat. Nanotechnol., 2017, 12, 582–588 CrossRef PubMed.
- R. Buscaglia, M. C. Miller, W. L. Dean, R. D. Gray, A. N. Lane, J. O. Trent and J. B. Chaires, Nucleic Acids Res., 2013, 41, 7934–7946 CrossRef PubMed.
- A. Ambrus, D. Chen, J. Dai, R. A. Jones and D. Yang, Biochemistry, 2005, 44, 2048–2058 CrossRef PubMed.
- R. I. Mathad, E. Hatzakis, J. Dai and D. Yang, Nucleic Acids Res., 2011, 39, 9023–9033 CrossRef PubMed.
- K. Guo, V. Gokhale, L. H. Hurley and D. Sun, Nucleic Acids Res., 2008, 36, 4598–4608 CrossRef PubMed.
- R. De Armond, S. Wood, D. Y. Sun, L. H. Hurley and S. W. Ebbinghaus, Biochemistry, 2005, 44, 16341–16350 CrossRef PubMed.
- H. Fernando, A. P. Reszka, J. Huppert, S. Ladame, S. Rankin, A. R. Venkitaraman, S. Neidle and S. Balasubramanian, Biochemistry, 2006, 45, 7854–7860 CrossRef PubMed.
- K. Guo, A. Pourpak, K. Beetz-Rogers, V. Gokhale, D. Sun and L. H. Hurley, J. Am. Chem. Soc., 2007, 129, 10220–10228 CrossRef PubMed.
- Y. Qin, E. M. Rezler, V. Gokhale, D. Sun and L. H. Hurley, Nucleic Acids Res., 2007, 35, 7698–7713 CrossRef PubMed.
- Y. Chen, P. Agrawal, R. V. Brown, E. Hatzakis, L. Hurley and D. Yang, J. Am. Chem. Soc., 2012, 134, 13220–13223 CrossRef PubMed.
- S. L. Palumbo, R. M. Memmott, D. J. Uribe, Y. Krotova-Khan, L. H. Hurley and S. W. Ebbinghaus, Nucleic Acids Res., 2008, 36, 1755–1769 CrossRef PubMed.
- J. Dai, D. Chen, R. A. Jones, L. H. Hurley and D. Yang, Nucleic Acids Res., 2006, 34, 5133–5144 CrossRef PubMed.
- T. Oltersdorf, S. W. Elmore, A. R. Shoemaker, R. C. Armstrong, D. J. Augeri, B. A. Belli, M. Bruncko, T. L. Deckwerth, J. Dinges, P. J. Hajduk, M. K. Joseph, S. Kitada, S. J. Korsmeyer, A. R. Kunzer, A. Letai, C. Li, M. J. Mitten, D. G. Nettesheim, S. Ng, P. M. Nimmer, J. M. O'connor, A. Oleksijew, A. M. Petros, J. C. Reed, W. Shen, S. K. Tahir, C. B. Thompson, K. J. Tomaselli, B. Wang, M. D. Wendt, H. Zhang, S. W. Fesik and S. H. Rosenberg, Nature, 2005, 435, 677–681 CrossRef PubMed.
- R. L. Young and S. J. Korsmeyer, Mol. Cell. Biol., 1993, 13, 3686–3697 CrossRef PubMed.
- P. Agrawal, C. Lin, R. I. Mathad, M. Carver and D. Yang, J. Am. Chem. Soc., 2014, 136, 1750–1753 CrossRef PubMed.
- R. Perrone, M. Nadai, I. Frasson, J. A. Poe, E. Butovskaya, T. E. Smithgall, M. Palumbo, G. Palù and S. N. Richter, J. Med. Chem., 2013, 56, 6521–6530 CrossRef PubMed.
- B. D. Nicola, C. J. Lech, B. Heddi, S. Regmi, I. Frasson, R. Perrone, S. N. Richter and A. T. Phan, Nucleic Acids Res., 2016, 44, 6442–6451 CrossRef PubMed.
- S. Amrane, A. Kerkour, A. Bedrat, B. Vialet, M.-L. Andreola and J.-L. Mergny, J. Am. Chem. Soc., 2014, 136, 5249–5252 CrossRef PubMed.
- P. Agarwal, S. Pandey and S. Maiti, Org. Biomol. Chem., 2015, 13, 5570–5585 RSC.
- A. Arora and S. Maiti, J. Phys. Chem. B, 2009, 113, 10515–10520 CrossRef PubMed.
- Y. Xu, K. Kaminaga and M. Komiyama, J. Am. Chem. Soc., 2008, 130, 11179–11184 CrossRef PubMed.
- H. Martadinata and A. T. Phan, J. Am. Chem. Soc., 2009, 131, 2570–2578 CrossRef PubMed.
- C.-F. Tang and R. H. Shafer, J. Am. Chem. Soc., 2006, 128, 5966–5973 CrossRef PubMed.
- B. Sacca, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2005, 33, 1182–1192 CrossRef PubMed.
- M. Vorlíčková, I. Kejnovská, J. Sagi, D. Renčiuk, K. Bednářová, J. Motlová and J. Kypr, Methods, 2012, 57, 64–75 CrossRef PubMed.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlíčková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef PubMed.
- W. Li, P. Wu, T. Ohmichi and N. Sugimoto, FEBS Lett., 2002, 526, 77–81 CrossRef PubMed.
- H.-Q. Yu, D. Miyoshi and N. Sugimoto, J. Am. Chem. Soc., 2006, 128, 15461–15468 CrossRef PubMed.
- P. A. Rachwal and K. R. Fox, Methods, 2007, 43, 291–301 CrossRef PubMed.
- J.-L. Mergny and L. Lacroix, Curr. Protoc. Nucleic Acid Chem., 2009, 37, 17.1.1–17.1.15 Search PubMed.
- C. M. Olsen and L. A. Marky, Methods Mol. Biol., 2010, 608, 147–158 CrossRef PubMed.
- A. De Cian, L. Guittat, M. Kaiser, B. Saccà, S. Amrane, A. Bourdoncle, P. Alberti, M.-P. Teulade-Fichou, L. Lacroix and J.-L. Mergny, Methods, 2007, 42, 183–195 CrossRef PubMed.
- M. Adrian, B. Heddi and A. T. Phan, Methods, 2012, 57, 11–24 CrossRef PubMed.
- G. W. Collie, N. H. Campbell and S. Neidle, Nucleic Acids Res., 2015, 43, 4785–4799 CrossRef PubMed.
- S. Lago, M. Nadai, M. Rossetto and S. N. Richter, Biochim. Biophys. Acta, 2018, 1862, 1276–1282 CrossRef PubMed.
- N. Deng, L. Wickstrom, P. Cieplak, C. Lin and D. Yang, J. Phys. Chem. B, 2017, 121, 10484–10497 CrossRef PubMed.
- G. Piraux, L. Bar, M. Abraham, T. Lavergne, H. Jamet, J. Dejeu, L. Marcélis, E. Defrancq and B. Elias, Chem.–Eur. J., 2017, 23, 11872–11880 CrossRef PubMed.
- B. Pagano, C. A. Mattia and C. Giancola, Int. J. Mol. Sci., 2009, 10, 2935–2957 CrossRef PubMed.
- A. Funke and K. Weisz, J. Phys. Chem. B, 2017, 121, 5735–5743 CrossRef PubMed.
- M. Bončina, Č. Podlipnik, I. Piantanida, J. Eilmes, M.-P. Teulade-Fichou, G. Vesnave and J. Lah, Nucleic Acids Res., 2015, 43, 10376–10386 Search PubMed.
- S. Selvam, S. Mandal and H. Mao, Biochemistry, 2017, 56, 4616–4625 CrossRef PubMed.
- B. R. Vummidi, J. Alzeer and N. W. Luedtke, ChemBioChem, 2013, 14, 540–558 CrossRef PubMed.
- J. Choi and T. Majima, Photochem. Photobiol., 2013, 89, 513–522 CrossRef PubMed.
- A. Dembska, P. Bielecka and B. Juskowiak, Anal. Methods, 2017, 9, 6092–6106 RSC.
- J.-L. Mergny and J.-C. Maurizot, ChemBioChem, 2001, 2, 124–132 CrossRef PubMed.
- A. Guédin, J. Gros, P. Alberti and J.-L. Mergny, Nucleic Acids Res., 2010, 38, 7858–7868 CrossRef PubMed.
- L. Ying, J. J. Green, H. Li, D. Kleneram and S. Balasubramanian, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14629–14634 CrossRef PubMed.
- J. Y. Lee, B. Okumus, D. S. Kim and T. Ha, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18938–18943 CrossRef PubMed.
- A. D. Rache and J.-L. Mergny, Biochimie, 2015, 115, 194–202 CrossRef PubMed.
- Q. Sheng, J. C. Neaverson, T. Mahmoud, C. E. M. Stevenson, S. E. Matthews and Z. E. Waller, Org. Biomol. Chem., 2017, 15, 5669–5673 RSC.
- P. Maleki, Y. Ma, K. Iida, K. Nagasawa and H. Balci, Nucleic Acids Res., 2017, 45, 288–295 CrossRef PubMed.
- K. Ohtsuka, S. Sato, Y. Sato, K. Sota, S. Ohzawa, T. Matsuda, K. Takemoto, N. Takamune, B. Juskowiak, T. Nagai and S. Takenaka, Chem. Commun., 2012, 48, 4740–4742 RSC.
- J. J. Li, X. Fang and W. Tan, Biochem. Biophys. Res. Commun., 2002, 292, 31–40 CrossRef PubMed.
- S. De Tito, F. Morvan, A. Meyer, J.-J. Vasseur, A. Cummaro, L. Petraccone, B. Pagano, E. Novellino, A. Randazzo, C. Giancola and D. Montesarchio, Bioconjugate Chem., 2013, 24, 1917–1927 CrossRef PubMed.
- E. Largy, A. Granzhan, F. Hamon, D. Verga and M. P. Teulade-Fichou, Top. Curr. Chem., 2013, 330, 111–177 CrossRef PubMed.
- K. Iida, T. Nakamura, W. Yoshida, M. Tera, K. Nakabayashi, K. Hata, K. Ikebukuro and K. Nagasawa, Angew. Chem., Int. Ed., 2013, 52, 12052–12055 CrossRef PubMed.
- N. C. Sabharwal, V. Savikhin, J. R. Turek-Herman, J. M. Nicoludis, V. A. Szalai and L. A. Yatsunyk, FEBS J., 2014, 281, 1726–1737 CrossRef PubMed.
- I. J. Lee, S. P. Patil, K. Fhayli, S. Alsaiari and N. M. Khashab, Chem. Commun., 2015, 51, 3747–3749 RSC.
- L. Xu, S. Hong, N. Sun, K. Wang, L. Zhou, L. Ji and R. Pei, Chem. Commun., 2016, 52, 179–182 RSC.
- T. Kimura, K. Kawai, M. Fujitsuka and T. Majima, Chem. Commun., 2004, 1438–1439 RSC.
- T. Kimura, K. Kawai, M. Fujitsuka and T. Majima, Chem. Commun., 2006, 401–402 RSC.
- J. Li, J. J. Correia, L. Wang, J. O. Trent and J. B. Chaires, Nucleic Acids Res., 2005, 33, 4649–4659 CrossRef PubMed.
- R. D. Gray, L. Petraccone, J. O. Trent and J. B. Chaires, Biochemistry, 2010, 49, 179–194 CrossRef PubMed.
- Y. Xu and H. Sugiyama, Nucleic Acids Res., 2006, 34, 949–954 CrossRef PubMed.
- A. Dumas and N. W. Luedtke, ChemBioChem, 2011, 12, 2044–2051 CrossRef PubMed.
- J. Johnson, R. Okyere, A. Joseph, K. Musier-Forsyth and B. Kankia, Nucleic Acids Res., 2013, 41, 220–228 CrossRef PubMed.
- J. Gros, F. Rosu, S. Amrane, A. De Cian, V. Gabelica, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2007, 35, 3064–3075 CrossRef PubMed.
- J. C. Myers, S. A. Moore and Y. Shamoo, J. Biol. Chem., 2003, 278, 42300–42306 CrossRef PubMed.
- R. A. Manderville and S. D. Wetmore, Chem. Sci., 2016, 7, 3482–3493 RSC.
- A. Dumas and N. W. Luedtke, J. Am. Chem. Soc., 2010, 132, 18004–18007 CrossRef PubMed.
- A. Dumas and N. W. Luedtke, Nucleic Acids Res., 2011, 39, 6825–6834 CrossRef PubMed.
- A. Nadler, J. Strohmeier and U. Diederichsen, Angew. Chem., Int. Ed., 2011, 50, 5392–5396 CrossRef PubMed.
- N. J. Greco and Y. Tor, J. Am. Chem. Soc., 2005, 127, 10784–10785 CrossRef PubMed.
- R. W. Sinkeldam, A. J. Wheat, H. Boyaci and Y. Tor, ChemPhysChem, 2011, 12, 567–570 CrossRef PubMed.
- K. M. Rankin, M. Sproviero, K. Rankin, P. Sharma, S. D. Wetmore and R. A. Manderville, J. Org. Chem., 2012, 77, 10498–10508 CrossRef PubMed.
- D. J. M. Blanchard, K. L. Fadock, M. Sproviero, P. S. Deore, T. Z. Cservenyi, R. A. Manderville, P. Sharma and S. D. Wetmore, J. Mater. Chem. C, 2016, 4, 2915–2924 RSC.
- M. Sproviero, K. L. Fadock, A. A. Witham, R. A. Manderville, P. Sharma and S. D. Wetmore, Chem. Sci., 2014, 5, 788–796 RSC.
- M. Sproviero and R. A. Manderville, Chem. Commun., 2014, 50, 3097–3099 RSC.
- M. Sproviero, K. L. Fadock, A. A. Witham and R. A. Manderville, ACS Chem. Biol., 2015, 10, 1311–1318 CrossRef PubMed.
- T. Z. Cservenyi, A. J. V. Riesen, F. D. Berger, A. Desoky and R. A. Manderville, ACS Chem. Biol., 2016, 11, 2576–2582 CrossRef PubMed.
- M. G. Pawar and S. G. Srivatsan, Org. Lett., 2011, 13, 1114–1117 CrossRef PubMed.
- A. A. Tanpure and S. G. Srivatsan, Chem.–Eur. J., 2011, 17, 12820–12827 CrossRef PubMed.
- P. M. Sabale, A. Nuthanakanti and S. G. Srivatsan, Indian J. Chem., 2013, 52A, 1004–1013 Search PubMed.
- A. A. Tanpure, M. G. Pawar and S. G. Srivatsan, Isr. J. Chem., 2013, 53, 366–378 CrossRef.
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef PubMed.
- A. A. Tanpure and S. G. Srivatsan, ChemBioChem, 2014, 15, 1309–1316 CrossRef PubMed.
- P. M. Sabale and S. G. Srivatsan, ChemBioChem, 2016, 17, 1665–1673 CrossRef PubMed.
- M. G. Pawar and S. G. Srivatsan, J. Phys. Chem. B, 2013, 117, 14273–14282 CrossRef PubMed.
- A. A. Tanpure and S. G. Srivatsan, Nucleic Acids Res., 2015, 43, e149 CrossRef PubMed.
- C. Zhao, L. Wu, J. Ren, Y. Xu and X. Qu, J. Am. Chem. Soc., 2013, 135, 18786–18789 CrossRef PubMed.
- S. Manna, C. H. Panse, V. A. Sontakke, S. Sangamesh and S. G. Srivatsan, ChemBioChem, 2017, 18, 1604–1615 CrossRef PubMed.
- P. M. Sabale, J. T. George and S. G. Srivatsan, Nanoscale, 2014, 6, 10460–10469 RSC.
- S. Müllar, J. Strohmeier and U. Diederichsen, Org. Lett., 2012, 14, 1383–1385 CrossRef PubMed.
- C. Schaffitzel, I. Berger, J. Postberg, J. Hanes, H. J. Lipps and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8572–8577 CrossRef PubMed.
- K. Paeschke, S. Juranek, T. Simonsson, A. Hempel, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2008, 15, 598–604 CrossRef PubMed.
- S. Müller, S. Kumari, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2010, 2, 1095–1098 CrossRef PubMed.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Nat. Chem. Biol., 2012, 8, 301–310 CrossRef PubMed.
- H.-Y. Liu, Q. Zhao, T.-P. Zhang, Y. Wu, Y.-X. Xiong, S.-K. Wang, Y.-L. Ge, J.-H. He, P. Lv, T.-M. Ou, J.-H. Tan, D. Li, L.-Q. Gu, J. Ren, Y. Zhao and Z.-S. Huang, Cell Chem. Biol., 2016, 23, 1261–1270 CrossRef PubMed.
- A. Laguerre, K. Hukezalie, P. Winckler, F. Katranji, G. Chanteloup, M. Pirrotta, J.-M. Perrier-Cornet, J. M. Y. Wong and D. Monchaud, J. Am. Chem. Soc., 2015, 137, 8521–8525 CrossRef PubMed.
- Y. Xu, Y. Suzuki, K. Ito and M. Komiyama, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14579–14584 CrossRef PubMed.
- A. Laguerre, J. M. Y. Wong and D. Monchaud, Sci. Rep., 2016, 6, 32141 CrossRef PubMed.
- S. Y. Yang, S. Amor, A. Laguerre, J. M. Y. Wong and D. Monchaud, Biochim. Biophys. Acta, 2017, 1861, 1312–1320 CrossRef PubMed.
- T.-Y. Tseng, Z.-F. Wang, C.-H. Chien and T.-C. Chang, Nucleic Acids Res., 2013, 41, 10605–10618 CrossRef PubMed.
- T.-Y. Tseng, C.-H. Chien, J.-F. Chu, W.-C. Huang, M.-Y. Lin, C.-C. Chang and T.-C. Chang, J. Biomed. Opt., 2013, 18, 101309 CrossRef PubMed.
- W.-C. Huang, T.-Y. Tseng, Y.-T. Chen, C.-C. Chang, Z.-F. Wang, C.-L. Wang, T.-N. Hsu, P.-T. Li, C.-T. Chen, J.-J. Lin, P.-J. Lou and T.-C. Chang, Nucleic Acids Res., 2015, 43, 10102–10113 Search PubMed.
- A. Shivalingam, M. A. Izquierdo, A. L. Marois, A. Vyšniauskas, K. Suhling, M. K. Kuimova and R. Vilar, Nat. Commun., 2015, 6, 8178 CrossRef PubMed.
- E. Salvati, F. Doria, F. Manoli, C. D'Angelo, A. Biroccio, M. Freccero and I. Manet, Org. Biomol. Chem., 2016, 14, 7238–7249 RSC.
- F. Doria, M. Nadai, M. Zuffo, R. Perrone, M. Freccero and S. N. Richter, Chem. Commun., 2017, 53, 2268–2271 RSC.
- Y.-J. Lu, D.-P. Hu, K. Zhang, W.-L. Wong and C.-F. Chow, Biosens. Bioelectron., 2016, 81, 373–381 CrossRef PubMed.
- M.-Q. Wang, Z.-Y. Wang, Y.-F. Yang, G.-Y. Ren, X.-N. Liu, S. Li, J.-W. Wei and L. Zhang, Tetrahedron Lett., 2017, 58, 3296–3300 CrossRef.
- S.-B. Chen, M.-H. Hu, G.-C. Liu, J. Wang, T.-M. Ou, L.-Q. Gu, Z.-S. Huang and J.-H. Tan, J. Am. Chem. Soc., 2016, 138, 10382–10385 CrossRef PubMed.
- M. Zeraati, D. B. Langley, P. Schofield, A. L. Moye, R. Rouet, W. E. Hughes, T. M. Bryan, M. E. Dinger and D. Christ, Nat. Chem., 2018, 10, 631–637 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |