
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reduction chemistry of neptunium cyclopentadienide complexes: from structure to understanding†
Michał S.
Dutkiewicz
ab,
Christos
Apostolidis
a,
Olaf
Walter
 *a and
Polly L.
Arnold
*b
*a and
Polly L.
Arnold
*b
aEuropean Commission, Directorate for Nuclear Safety and Security, Joint Research Centre, Postfach 2340, D-76125, Karlsruhe, Germany. E-mail: Olaf.Walter@ec.europa.eu
bEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: Polly.Arnold@ed.ac.uk
First published on 30th January 2017
Abstract
Neptunium complexes in the formal oxidation states II, III, and IV supported by cyclopentadienyl ligands are explored, and significant differences between Np and U highlighted as a result. A series of neptunium(III) cyclopentadienyl (Cp) complexes [Np(Cp)3], its bis-acetonitrile adduct [Np(Cp)3(NCMe)2], and its KCp adduct K[Np(Cp)4] and [Np(Cp′)3] (Cp′ = C5H4SiMe3) have been made and characterised providing the first single crystal X-ray analyses of NpIII Cp complexes. In all NpCp3 derivatives there are three Cp rings in η5-coordination around the NpIII centre; additionally in [Np(Cp)3] and K[Np(Cp)4] one Cp ring establishes a μ-η1-interaction to one C atom of a neighbouring Np(Cp)3 unit. The solid state structure of K[Np(Cp)4] is unique in containing two different types of metal–Cp coordination geometries in the same crystal. NpIII(Cp)4 units are found exhibiting four units of η5-coordinated Cp rings like in the known complex [NpIV(Cp)4], the structure of which is now reported. A detailed comparison of the structures gives evidence for the change of ionic radii of ca. −8 pm associated with change in oxidation state between NpIII and NpIV. The rich redox chemistry associated with the syntheses is augmented by the reduction of [Np(Cp′)3] by KC8 in the presence of 2.2.2-cryptand to afford a neptunium(II) complex that is thermally unstable above −10 °C like the UII and ThII complexes K(2.2.2-cryptand)[Th/U(Cp′)3]. Together, these spontaneous and controlled redox reactions of organo-neptunium complexes, along with information from structural characterisation, show the relevance of organometallic Np chemistry to understanding fundamental structure and bonding in the minor actinides.
Introduction
Fifty years have passed since the foundation of organometallic neptunium chemistry in the form of cyclopentadienyl chemistry,1 and yet only a handful of complexes have been reported, and even fewer fully characterised.2–4 Yet increasingly, combined synthetic/spectroscopic/computational studies are demonstrating how covalently binding, soft, carbocyclic organometallic ligands that form actinide-ligand σ-, π-, δ- and even φ-(back)bonding interactions provide an excellent platform for advancing the fundamental understanding of the differences in orbital contributions and covalency in f-block metal–ligand bonding.5 Understanding the subtleties are key to the safe handling and separations of the highly radioactive nuclei.6–8 For example, recent quantitative carbon K-edge X-ray absorption spectroscopy (XAS) analyses on the organometallic [An(COT)2] (An = Th, U), “actinocenes” provided the first experimental evidence for extensive φ-orbital interactions in thorocene, and remarkably little in the U analogue, with contrasting trends in orbital mixing.9 Furthermore, a combination of experimental and QTAIM computational comparisons of [M(LAr)X] (M = Sm, U, Np; X = Cl, I; LAr = dianionic arene-bridged trans-calix[2]benzene[2]pyrrole10) showed significant differences (up to 17%) in orbital contributions to M–L bonds between the Ln and An analogues, and that the covalency in the Np–ligand bonding arises from spatial orbital overlap rather than a coincidental energy degeneracy.4 The work also demonstrated differences between U and Np in their reaction chemistry, such as the stability of the NpII formal oxidation state or the reduction of NpIV to NpIII upon complexation.Organoneptunium chemistry has relied heavily on the ubiquitous cyclopentadienyl ligand, Cp = (C5H5)−, as a strongly binding, sterically demanding yet flexible, monoanion, and focused almost exclusively on NpIV complexes. The first evidence for the formation of [Np(Cp)3X] (X = Cl, F) came from a radiochemical synthesis, i.e. the irradiation of a 238U complex with thermal neutrons,1 and [Np(Cp)3X] (X = Cl, F) were subsequently reported from standard chemical routes as thermally robust, volatile complexes.11,12 Baumgärtner et al. reported the first homoleptic organoneptunium complex, tetrakis(η5-cyclopentadienyl)neptunium(IV), [Np(Cp)4], from treatment of NpCl4 with excess KCp in benzene.13 Many of the earliest studies on neptunium cyclopentadienyl complexes also had the aim of exploring covalency in the bonding, using the fact that Np is a Mössbauer active nucleus. However, not all the studies agreed. The first Mössbauer studies on [Np(Cp)4] and [Np(COT)2] suggested less interaction between the central ion and the Cp ligands but appreciable covalency in the Np–COT bonding.14 On the other hand, Bohlander12 reported the isomer shift of the Np nucleus in [Np(Cp)4] closely approaches that of the record-breaking, covalent [Np(COT)2], thus being the most covalent Np–Cp derivative. Finally, from analysis of the isomer shifts Karraker14,15 stated that there were smaller covalent bonding contributions in [Np(Cp)4] than [Np(Cp)3Cl], whereas Adrian16,17 concluded the opposite.
The redox properties of the element play a pivotal role in neptunium chemistry as it conventionally exhibits five oxidation states in compounds, from +3 to +7.18 Recently, we reported that a formally NpII complex Np(LAr)(dme) supported by LAr, the trans-calix[2]benzene[2]pyrrole is accessible; black solutions were sufficiently stable (up to 90 minutes) for spectroscopic analyses but crystals were too small for single crystal diffraction analyses.4 Somewhat surprisingly, given the increased stability of the NpIII oxidation state compared to UIII, this work was also the first to report single crystal structural studies on organometallic NpIII complexes. To date, the only organometallic NpIV complexes characterised by single crystal X-ray diffraction are neptunocene [Np(C8H8)2],2,19 [Np(Cp)Cl3(OPPh2Me)2],20 [Np(Cp)3(OPh)],21 and two examples of the Cp3Np-functionalised adduct [(UO2)(THF)(H2L)] (L = ‘Pacman’ Schiff-base polypyrrolic macrocycle),3 in which we studied the Cp3Np coordination to one oxo group of the uranyl dication to compare the degree of electron transfer via the oxo-bridge between U, Np, and Pu cations. Structurally characterised NpIII organometallics are still limited to the [M(LAr)X] complexes we reported.4 Modern characterising data including 1H NMR spectroscopic data have been reported for just a couple of complexes.
Many routes to solvated and base-free UIII(Cp)3 complexes exist, but only the THF solvate of [NpIII(Cp)3] has been reported to date, and was made from treating [Np(Cp)3Cl] with potassium metal and catalytic naphthalene in refluxing THF for a few days. The product was first assigned as the tris THF solvate [Np(Cp)3(THF)3]14 but subsequent IR, FIR and UV-vis-NIR spectroscopic analyses suggested the constitution of an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Lewis base adduct [Np(Cp)3(THF)] analogously to that of uranium.12 Attempts to desolvate it by heating samples in vacuo led to significant decomposition.14,22
:1 Lewis base adduct [Np(Cp)3(THF)] analogously to that of uranium.12 Attempts to desolvate it by heating samples in vacuo led to significant decomposition.14,22
Herein, we report the synthesis and structural characterisation of a series of NpIII cyclopentadienyl complexes, and their reduction chemistry, both spontaneous and directed. Structural changes are discussed in relation to the neptunium formal oxidation state and nature of the ligands, as the majority of the complexes have been structurally characterised by single crystal X-ray diffraction.
Results
Syntheses
All the syntheses start from NpCl4. This can be directly transformed into [Np(Cp)4], NpCp4 by the reaction of NpCl4 with an excess of KCp in toluene.13 Single crystals of NpCp4 grow in the supernatant during a prolonged extraction of the crude product with pentane. The spectroscopic data agree with the literature reports.12,23NpCp4 itself can be – from a reaction with NH4Cl11,12 – converted to [Np(Cp)3Cl], NpCp3Cl which is an ideal starting material for the synthesis of [Np(Cp)3], NpCp3. The reduction of NpCp3Cl by Na/Hg in Et2O affords NpCp3 as its diethyl ether solvate [Np(Cp)3(Et2O)], Scheme 1. This solvent molecule is labile and can be easily removed in vacuum to afford the solvent free NpCp3 which is only sparingly soluble in non-coordinating solvents due to the polymeric nature of the molecular structure (see below) but dissolves slowly in Et2O, THF, or MeCN, again forming solvates [Np(Cp)3(Et2O)], [Np(Cp)3(THF)], or [Np(Cp)3(NCMe)2], NpCp3(NCMe)2, respectively.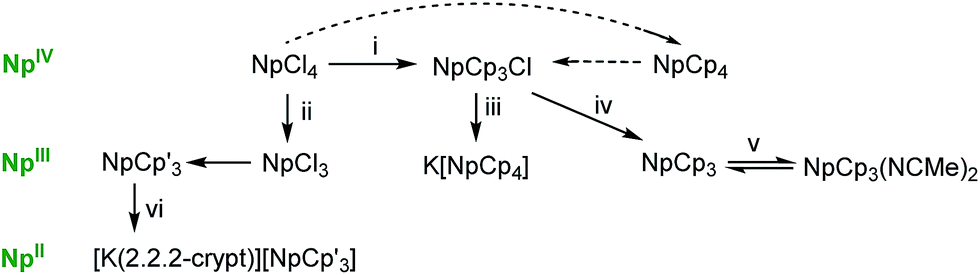 | ||
| Scheme 1 Synthetic routes to neptunium complexes presented in this work. NpCp3 is obtained by reduction from NpCp3Cl and readily forms the MeCN stabilised solvate. The silylated analogue NpCp′3 (Cp′ = C5H4SiMe3) is better obtained from the reaction of NpCl3 with KCp′ and can be reduced to its K salt. Dotted lines indicate literature procedures. Key: (i) excess KCp, PhMe; (ii) Na/Hg, Et2O, –NaCl; (iii) KCp, –KCl; (iv) Na/Hg, Et2O, –NaCl; (v) excess MeCN; (vi) KC8, 2.2.2-cryptand, THF/Et2O, –8C. | ||
Crystals of [Np(Cp)3(Et2O)] could not be analysed via X-ray diffraction as during the crystal mounting procedure they lose coordinated Et2O solvent molecule readily. Similarly, no stable adducts of a lanthanide analogue [Ln(Cp)3(OEt2)] have yet been reported. However, single crystals of the bis-acetonitrile adduct NpCp3(NCMe)2 have been grown from acetonitrile solution at RT and studied by X-ray diffraction.
The reaction of NpCp3Cl with 1.1 equiv. of KCp in THF does not lead to the simple Cl substitution product NpCp4. After 4 d reflux and evaporation of the solvent, n-pentane extraction recovered half of the starting material NpCp3Cl (49%), and a subsequent extraction with Et2O afforded maroon single crystals of the new, reduced, NpIII complex K[Np(Cp)4], K[NpCp4] in 37% yield. When the reaction is repeated without heating the mixture, no soluble, molecular product can be isolated.
For the synthesis of [Np(C5H4SiMe3)3], NpCp′3 another reaction pathway was followed as no starting material [Np(C5H4SiMe3)3Cl] was available: NpCl3 is generated in situ from the reaction between NpCl4 and excess Na(Hg) in diethyl ether at room temperature. The reaction between this NpCl3 and three equivalents of K(C5H4SiMe3) in diethyl ether at room temperature afforded the target NpCp′3. Green, crystalline NpCp′3 is deliquescent under 1 atm of n-pentane vapour at room temperature but single crystals can be isolated reproducibly by evaporation and cooling of pentane solutions.
The reduction of a solution of NpCp′3 with KC8 was carried out similarly to the method originally described for the synthesis of the thermally unstable [K(2.2.2-cryptand)][U(C5H4SiMe3)3] by Evans et al.,24 but the crystallization temperature maintained lower, at −78 °C. A trial reduction of NpCp3 confirmed that no compound could be isolated even at the coldest achievable reaction temperatures. In THF/Et2O the mixture immediately turns very intense dark brown on contact with the solid reducing agent, and small, shiny black crystallites with the assumed composition K(2.2.2-cryptand)[Np(Cp′)3] appear in the filtrate after approx. 1 h of storage at −78 °C. Several potentially single crystals of suitable size for an X-ray diffraction study were analysed but only very weak diffraction was observed as the crystals had degraded during the radiologically protective mounting procedure. Due to the high sensitivity of the compound we were not able to determine the structure of the reduced product from this reaction or to measure any spectra.
Spectroscopy
The 1H and 1H–13C gHMQC NMR spectra of NpCp3 in THF-d8 show only one resonance for the Cp ring protons at δH = −9.65 ppm and the respective 13C shift of δC = 150.4 ppm. The 1H NMR spectrum of NpCp′3 in toluene-d8 solution contains three paramagnetically contact-shifted resonances between −1.38 ppm (the SiMe3 protons) and −9.51 ppm in a 9:2:2 ratio, implying the identical bonding mode of the three ligands. The spectrum acquired in THF-d8 solution appears similar, the resonances are slightly shifted downfield (δH = −0.6 to −9 ppm) suggesting an interaction with the THF solvent. The 1H NMR spectrum of the sparingly soluble K[NpCp4] in THF-d8 contains one broad resonance at −11.9 ppm, again showing the identical coordination behaviour of the Cp rings in solution. The solubility of K[NpCp4] in THF is however too low to observe measurable absorptions in the UV-vis-NIR spectra.
For the NpIII complexes, all the Cp ring proton resonances are observed at ca. −10 ppm suggesting an electronically similar environment in each complex. Where no correlated spectra are reported here then the complex decomposes in the fluoropolymer NMR-tube liners before the spectra could be recorded.25
The ATR spectrum of NpCp3 features several characteristic vibrations of the Cp rings, which correlate well with the previously reported IR data of the complexes [M(Cp)3] (M = U, Pu, Am, and Tm).26 Indeed, the similarity of the values of the entire series shows the very comparable constitution of the complexes. The most characteristic bands are the set of four absorptions at 666, 611, 581 and 519 cm−1.27 ATR-FTIR spectra of K[NpCp4] show very similar Cp ring vibrations to those previously described for NpCp3 with a slight shift to lower energy of the vibrations in K[NpCp4]vs.NpCp3 agreeing with the higher overall negative charge in the complex K[NpCp4].
Molecular structures
The molecular structure of NpCp4 is here reported for the first time. Dark red single crystals were obtained by extraction with pentane over several days. The compound is kinetically stable. X-ray diffraction analyses revealed an ideal tetrahedral environment of the Np centre, shielded with the four Cp rings in an isostructural complex to its Th28 or U29 analogue (Fig. 1).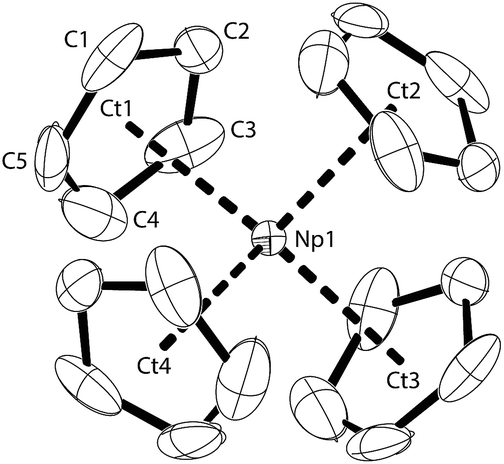 | ||
| Fig. 1 Thermal ellipsoid drawing (50% probability for non-H atoms) of NpCp4 in the solid state. H atoms omitted. Selected bond lengths [Å] and angles [°] for NpCp4: Np1–Ct 2.551(2), Np1–C 2.78(1) to 2.83(1), Ct–Np1–Ct 109.4(2). | ||
Across the row of the isostructural [An(Cp)4] (An: Th, U, Np), in line with the actinide contraction the cell volume decreases from 802 Å3 (Th) to 786 Å3 (U) to 775 Å3 (Np). Furthermore, a shrinking of the entire molecule is expressed by a decreasing An–Cp ring centroid distances. These are found to be 2.606 Å for Th, whereas in the U analogue they are determined to 2.588 Å and for the here presented Np complex they are 2.551 Å, again shorter. The shrinking parallels the decrease of the ionic radii; this implies that the nature of the bonding in the complexes in this row is comparable and even if covalency plays a role it does not affect the bond lengths in the complexes significantly.
Dark-brown, almost black single crystals of NpCp3 suitable for X-ray crystal structure determination were obtained from a 3.5% v/v diethyl ether in n-pentane solution stored at room temperature for 7 d.
The solid-state structure of NpCp3 shows a polymeric structure motif. All the three Cp rings are bound η5 towards the central NpIII atom. Due to its Lewis acidity coordinative saturation is achieved by additional η1-coordination to one of the Cp rings of another NpCp3 unit resulting in one μ-η5,η1-coordinated bridging cyclopentadienyl group, Fig. 2a, establishing an overall polymeric zig–zag structure with a Np1–C1–Np1A angle close to 170, Fig. 2b. This is in agreement with the structures of isostructural [Ln(Cp)3] complexes.30–33 In this coordination environment a distorted tetrahedral geometry around the NpIII centre is established with a strong Np–C interaction to the C-atom to which the η1 coordination is established (Np–C1 2.815(11) Å) whereas the bond distance between the Np1 atom and the carbon atoms C2 (3.266(9) Å) and C5 (3.552(10) Å) may be considered as non-bonding. The geometry around the cation in NpCp3 is more closely aligned with the larger rare earth analogues that have more electron-rich Cp rings, [Ln(C5H4Me)3]4 (Ln = La,34 Ce,35 Nd36). These all show μ-η5,η1-binding for each cyclopentadienyl ligand. However, some of the published data were recorded at room temperature and are less well resolved. In order to discuss fully the differences between the isostructural complexes of the type [M(Cp)3] (M: Ln, An) it will be necessary to re-determine the solid state structures of the corresponding Ln complexes.
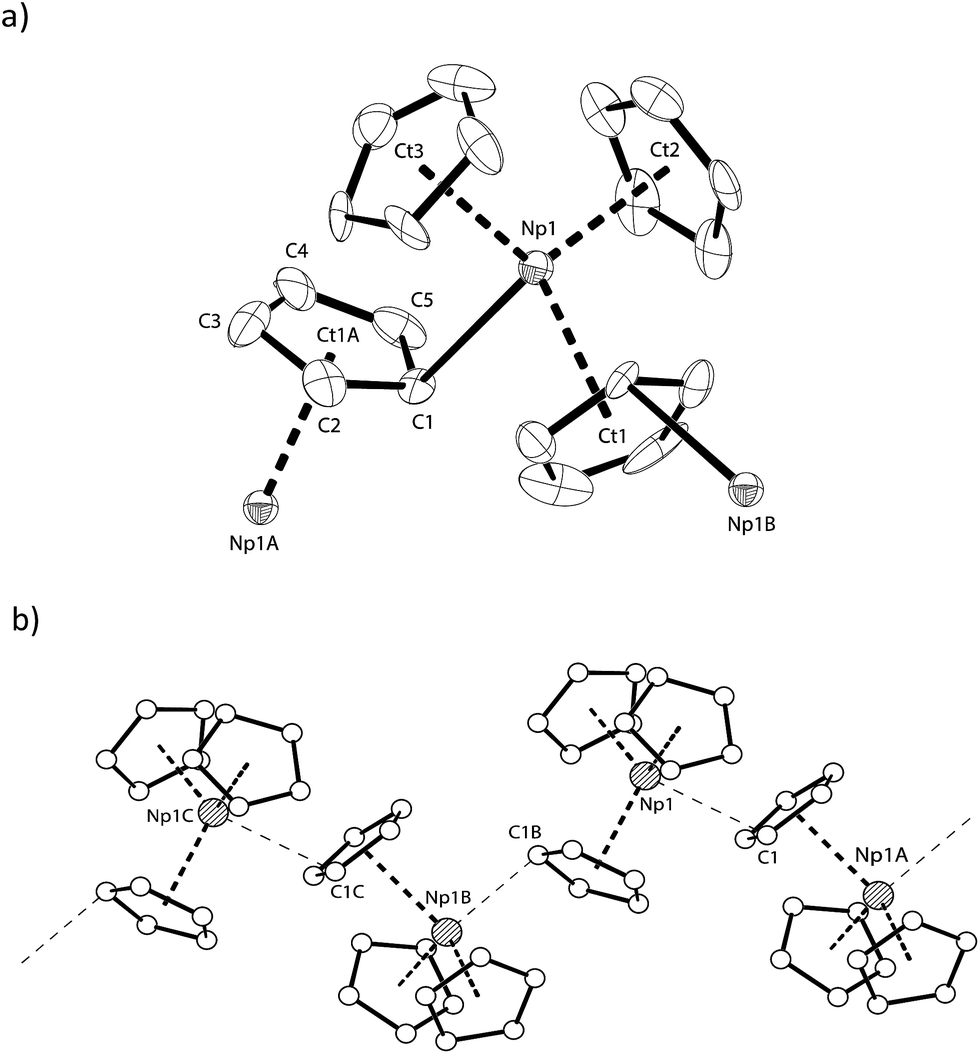 | ||
| Fig. 2 (a) Thermal ellipsoid drawing (50% probability for non-H atoms) of a portion of the polymeric chain formed by [Np(Cp)3] in the solid state. H atoms omitted. Selected bond lengths [Å] and angles [°] for NpCp3: Np1–C1 2.815(11), Np1–Ct1 2.587(5), Np1–Ct2 2.419(6), Np1–Ct3 2.561(10), Ct1–Np1–Ct2 112.23(19), Ct1–Np1–Ct3 113.66(19), Ct2–Np1–C3 115.1(2), Ct1–Np1–C1 94.5(3), Ct2–Np1–C1 102(5), Ct3–Np1–C1 110.7(3), Np1–C1–Np1A 167.5(3), ϕ(C1–Np1–Ct1∠Ct2–Np1–Ct3) 91.30(18), ϕ(C1–Np1–Ct3∠Ct1–Np1–Ct2) 82.9(2); (b) representation of the zig–zag chain arrangement in [Np(Cp)3]. Symmetry generated atom names are labelled with A, B and C. | ||
Red-brown crystals of K[NpCp4] suitable for single crystal X-ray diffraction analysis were grown from a diethyl ether solution stored at room temperature for ca. 100 h. The asymmetric unit consists of 1.5 molecules of K[NpCp4] and 0.5 molecule of a heavily disordered diethyl ether molecule residing on the crystallographic C2 axis. K[NpCp4] is also polymeric in the solid-state, with all Cp ligands forming bridging interactions of either η1 to another Np atom or η5 to a K or Np cation. This results in two different types of Np coordination geometry arising from the bridging modes, Fig. 3. The coordination environment around the first NpIII cation, labelled A, is (η5-Cp)3(η1-Cp), which closely mirrors that of the parent complex NpCp3; that around the second NpIII cation, labelled B, is (η5-Cp)4 comparable to the coordination observed for the four Cp rings in NpCp4. To our knowledge, this is the first instance of a Cp complex showing two different types of metal coordination geometries in the same crystal. This behaviour might be explained by the high coordinative flexibility of the relatively large An cations. Very few f-block complexes have comparable solid state structures. The complex [Ce(C5H4Me)3]4 forms a tetramer in the solid state with (η5,η1-Cp) anions bridging,35 and one uranium complex was published very recently; [K(2.2.2-cryptand)][U(Cp′)4],37 also shows an (η5-Cp)3(η1-Cp) geometry. The AnIII(η1-Cp) M–C distance is 2.752(7) Å for Np and 2.776(2) for U, consistent with the ionic radius difference (six-coordinate Np3+ = 1.01 Å; U3+ = 1.025 Å).38
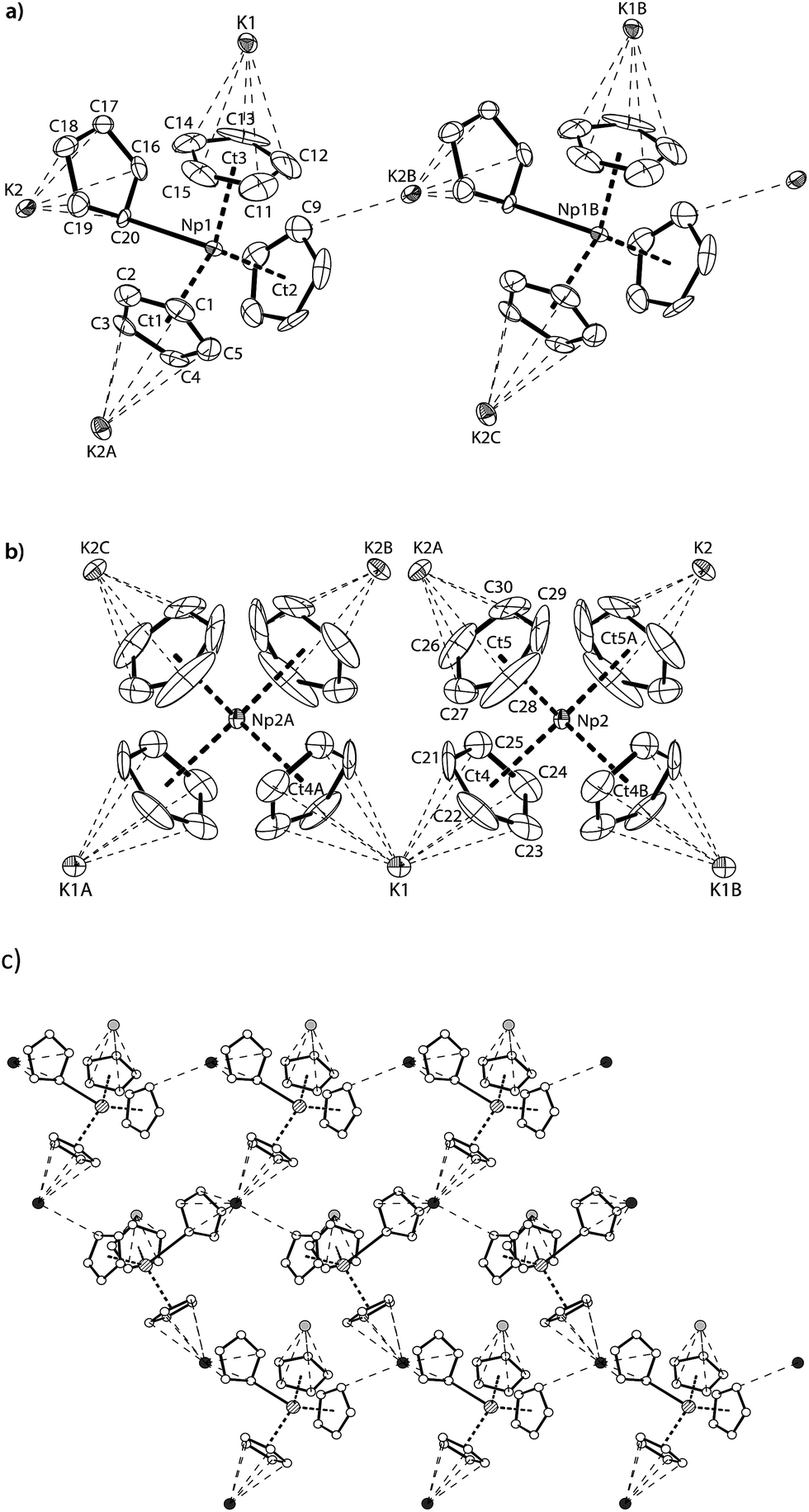 | ||
| Fig. 3 (a) Thermal ellipsoid drawing of a portion of the polymeric structure type K[Np(η5-Cp)3(η1-Cp)] formed by NpIII cations A, and (b) the polymeric sheet-like structure type K[Np(η5-Cp)4] formed by NpIII cations B. Thermal ellipsoids for (a) and (b) are at 50% probability. (c) Ball and stick drawing of part of the 2D-polymeric sheet structure formed by the type A NpIII cations and K2. Atom colours: C (white), K1 (light grey), K2 (dark grey), Np1 (filled with lines). Hydrogen atoms and lattice diethyl ether molecules are omitted for clarity. Symmetry generated atom names are labelled A, B and C. Selected distances [Å] and angles [°] for K[NpCp4]; Np1–C20 2.752(7), Np1–Ct1 2.527(4), Np1–Ct2 2.516(4), Np1–Ct3 2.493(6), K1–Ct3 3.067(6), K1–Ct4 3.013(5), K2–C9 3.197(10), K2–C16 3.049(8), K2–C20 2.976(7), K2A–Ct1 3.003(5), K2–Ct5A 2.884(7), Ct1–Np1–Ct2 115.50(13), Ct1–Np1–Ct3 118.92(16), Ct2–Np1–Ct3 117.19(17), Ct1–Np1–C20 96.53(16), Ct2–Np1–C20 99.01(17), Ct3–Np1–C20 103.51(19), Np2–Ct4 2.631(5), Np2–Ct5 2.645(7), Ct4–Np2–Ct5 109.2(2), Ct4–Np2–Ct4B 109.6(2), Ct4–Np2–Ct5A 110.5(2), Ct5–Np2–Ct5A 107.9(3), Ct4–K1–Ct4A 104.67(19), ϕ(Ct4–Np2–Ct5∠Ct4B–Np2–Ct5A) 88.7(2), ϕ(Ct4–Np2–Ct4B∠Ct5–Np2–Ct5A) 90.9(3). | ||
The Np(A) and (B) type cations both form polymeric chain structures, which connect to each other, and into a 3D network via further K cation interactions. For Np(A), Fig. 3a, there are three distinct Cp binding modes for the four Cp ligands: one K(η5-)Np(η1-); one K(η1-)Np(η5-) (for Ct2); and two K(η5-)Np(η5-) (for Ct1 and Ct3). The second type of Np, type B, Fig. 3b, displays η5 binding of all the Cp ligands arranged in an irregular tetrahedral fashion around the NpIII centre. The average separation between the Np atoms and the η5-Cp ring centroids in these molecules is slightly longer in accordance with the greater steric encumbrance at the Np centre (2.635(1) in B vs. 2.507(5) Å in A-type). Accordingly, the C-atoms for the fourfold η5-coordinated Np centres show larger Np–C distances between 2.835(12) to 2.955(9) Å. The range of Np–C distances to like in NpCp3 coordinated Np centre Np(A) for the η5-bound carbon atoms is with 2.732(13) to 2.842(9) Å comparable.
There are also two different types of potassium coordination. Cation K1 shows only η5-Cp binding, with long K–Cp (centroid) distances of 3.013(5) and 3.067(6) Å. Cation K2 shows both η1-Cp binding (to C9 with a distance of 3.197(10) Å) and much closer η5-Cp binding than K1, with K–Cp (centroid) distances of 2.884(7) and 3.003(5) Å. These latter are more typical K–Cp distances. There are also molecules of diethyl ether present in the lattice, but no close contacts to metal centres are evident, Fig. 3c.
Np(A) in K[NpCp4] exhibit very similar coordination behaviour to NpCp3. The zig–zag chains formed by the A-type NpIII units are only slightly more bent in K[Np(Cp)4] (Np–C–Np 149.8(3)°) than in NpCp3 (156.9(5)°). The mean distance between the Np(A) atoms and the centre of the η5-coordinated Cp rings is in average 2.51(1) Å and absolutely comparable to the one in NpCp3 with 2.52(1) Å. A bigger difference is observed for the bond length C–Np of η1-coordinated C-atoms: in NpCp3 at 2.815(11) Å it is significantly larger than for Np(A) in K[NpCp4] with 2.752(7) Å. The different coordination behaviour of the Cp rings (binding η1 to the Np atoms but η5-coordination to a K cation in K[NpCp4] and to a Np in NpCp3) is attributed to poorer competition by K for coordination than Np, allowing the Cp ring to enable a stronger interaction towards the metal on the opposite site.
Np(B) in K[NpCp4] with its four η5-coordinated Cp rings placed in an identical coordination environment to NpCp4 with the difference of the charge on central Np atoms. According to the higher charge the mean centre of Cp ring to Np distance in NpCp4 is with 2.551(1) Å about 0.08 Å shorter than for Np(B) in K[NpCp4] where it is determined to 2.635(1) Å. This effect can be attributed mainly to the change of the ionic radii from NpIII to NpIV in an otherwise identical coordination environment; in this case four Cp ligands.
Single crystals of NpCp3(NCMe)2 grow from a MeCN solution of NpCp3 as a solution is concentrated under reduced pressure. They are stable enough that they can be transferred on the goniometer and the single crystal solid state structure measured (Fig. 4).
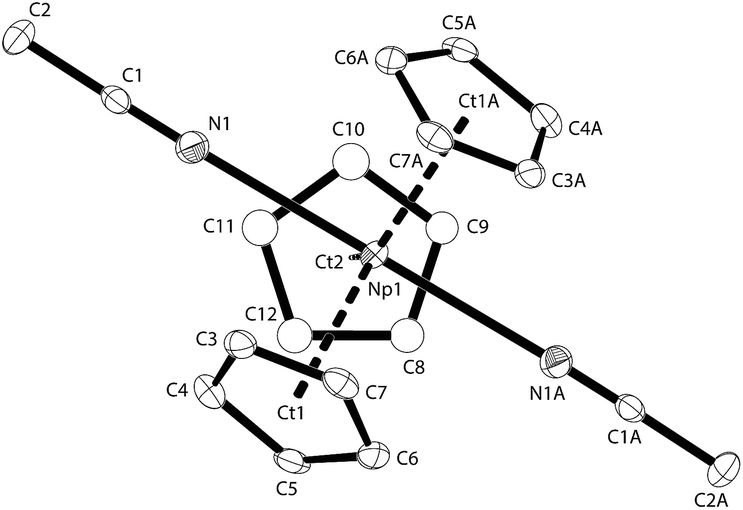 | ||
| Fig. 4 Thermal ellipsoid drawing (50% probability for non-H atoms) of NpCp3(NCMe)2 in the solid state. H atoms omitted. Selected bond lengths [Å] and angles [°] for NpCp3(NCMe)2: Np1–N1 2.665(4), Np1–Ct1(a) 2.539(4), Np1–Ct2 2.540(4), N1–C1 1.135(5), C(1)–C(2) 1.465(5), N1–Np1–N1A 178.4(2), Ct–Np1–Ct: 116.9(2), 119.9(2), 123.5(2). | ||
The geometry around the Np atom in the molecular structure of NpCp3(NCMe)2 can be generally described as distorted trigonal bipyramidal with three Cp in η5-coordination mode exhibiting the trigonal plane around the Np atom whereas the two MeCN ligands are forming occupying its apical positions. In the molecule there is a two-fold axis passing through the Np atom. This results in the nearby linear arrangement of the two acetonitrile ligands exhibiting a Np angle of 178.4(2)°. The low steric demand of the MeCN ligands enables a close to ideal arrangement of the three Cp rings in one plane around the Np centre showing a sum of angle of 360.3° (Ct–Np1–Ct) with a deviation out of the plane consisting of the three centres of the Cp rings plus the Np atom of less than 0.005 Å. Accordingly, the bond distances between the Np atom and the centres of the Cp rings are identical with 2.539(2) and 2.540(2) Å. These findings compare well to the metrics for the series of LnCp3(nitrile)2 complexes that have been structurally characterised.39–43 For the actinides however, only some cationic UIV complexes have been structurally characterised.44–47 In these UIV cationic complexes of the type UCp3(NCR)2+ shorter bond distances are found than here for NpCp3(NCMe)2 (U–N distances <2.6 Å).
All of the NpIII centres described above are coordinatively unsaturated in the absence of ligand bridging, but this situation can be readily changed by the use of sterically more demanding Cp ligands like C5H4SiMe3(Cp′). Olive-green single crystals of NpCp′3 suitable for X-ray diffraction analysis were obtained from cooling a concentrated n-pentane solution to −20 °C. The asymmetric unit consists of a single molecule of NpCp′3. The molecular structure of NpCp′3 is shown in Fig. 5 and consists of the mononuclear NpIII complex containing three η5 bound Cp′ ligands, with all the Ct(η5-Cp′)–Np–Ct(η5-Cp′) angles close to 120° and the average Np–Ct(η5-Cp′) distance of 2.482(3) Å. This means that in NpCp′3 the Cp rings with the bulky substituents are closer to the metal than in the previously described complexes NpCp3(NCMe)2, K[NpCp4], and NpCp3 for which Np–Ct distances of 2.51 Å or 2.54 Å are found. This means that due to the trigonal planar arrangement of the Cp′ ligands with the resulting larger bond angles around the NpIII centre in NpCp′3 the metal is able to establish stronger interactions with the more electron rich Cp′ ligands.
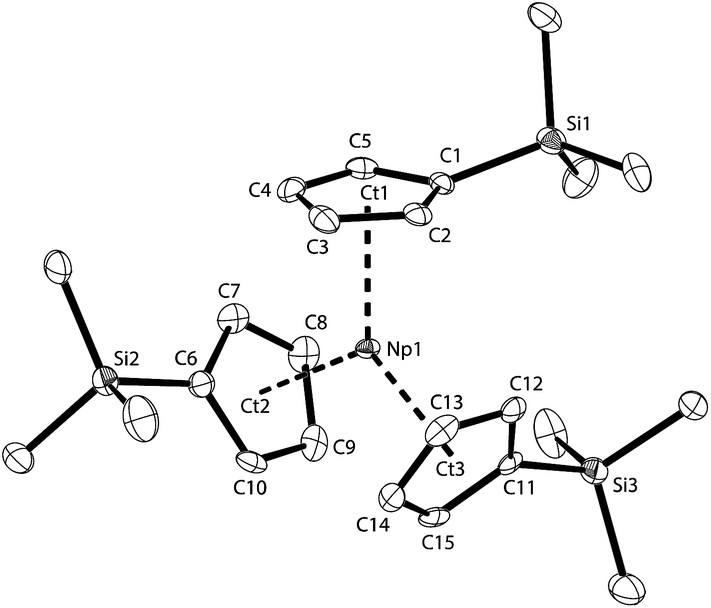 | ||
| Fig. 5 Thermal ellipsoid drawing (50% probability for non-H atoms) of NpCp′3 in the solid state. H atoms omitted. Selected bond lengths [Å] and angles [°]: Np1–Ct1 2.485(2), Np1–Ct2 2.481(2), Np1–Ct3 2.479(2), Np1–Car 2.734(6) to 2.786(4), Si1–(C1–C5) plane −0.382(8), Si2–(C6–C10) plane −0.109(8), Si3–(C11–C15) plane −0.169(8), Ct1–Np1–Ct2 119.86(8), Ct1–Np1–Ct3 120.46(7), Ct2–Np1–Ct3 119.06(8). | ||
The Np1 atom in NpCp′3 lies in plane of the Cp ring centroids with only a minor out-of-plane distortion (0.113(1) Å), affording a nearly trigonal planar (D3h) geometry which is isostructural with the previously reported uranium complex [U(Cp′)3].48 The average An–C distance of 2.78(4) Å (2.76(3) Å for U) and An–Ct(η5-Cp′) of 2.482(3) Å (2.51(3) Å for U) are the same within standard uncertainties. However, the similarity of the values can be taken as a sign that the arrangement of the three Cp′ ligands is dominated more by steric factors than by ionic radii. The ligands take a trigonal planar coordination around the Np centre, which is not only in agreement with the structure of its U analogue, but as well with the solid state structure found for the small lanthanide cation in [Yb(Cp)3]49,50 or U(CpR)3− containing complexes (where R represents a bulky hydrocarbyl group).24,42,48,51–60
Discussion
Redox reactivity
In Scheme 1 there are mainly represented two reaction pathways: nucleophilic substitution at the metal centre or reduction. NpIVCp4 was prepared readily from the reaction of NpIVCl4 with excess of KCp via SN reaction. From this a Cp ligand can be abstracted by protonation and so that even Cl− is able to coordinate to the metal forming NpIVCp3Cl.The homoleptic complexes NpCp3 and NpCp′3 are accessible only by reduction: NpCp3 is best produced by reduction of NpCp3Cl whereas NpCp′3 is well prepared by the in situ formation of NpCl3 from NpCl4 followed by SN reaction at the metal centre leading to NpCp′3. As the silyl-substituted C5H4SiMe3 anion more effectively stabilises lower oxidation state metal cations the synthesis of the first cyclopentadienyl Np(II) complex succeeded by reduction of NpCp′3 with KC8 in parallel with the results of Evans et al.,24 the crystallization temperature was lowered to −78 °C but the small, shiny black crystallites appearing in the filtrate after 1 h of storage at −78 °C showed insufficient diffraction properties.
Although the neutral complex [Np(Cp)4] has been reported several times to be formed in the reaction between NpCl4 and excess KCp in THF,12 benzene,12,13 or toluene61 solution, the reaction reported here between [Np(Cp)3Cl] and KCp affords the NpIII ate product K[Np(Cp)4] giving evidence that in this case Cp− acts in two roles: as reducing agent plus as stabilising ligand for the coordinatively unsaturated NpIII ion, dependent on the reaction conditions.
These observations could provide an explanation for the disagreements in the Mössbauer studies on covalency. Adrian observed that Mössbauer spectra of the [Np(Cp)4] targets provided by Bohlander contained two low intensity bands arising from the unidentified impurities, which may provide an argument for this study.16 The utility of the Cp anion as a reductant is well documented in preparative inorganic chemistry, and an additional equivalent(s) of either NaCp or [MgBr(Cp)] can be conveniently employed to reduce in situ the higher oxidation state transition metal and lanthanide precursors and produce metallocenes of the MII centres i.e. Cr,62,63 V,63,64 Ru,65 Os,66 or Eu.67 In actinide chemistry this reactivity is rarer, and the only reported synthesis to date is of the homoleptic complex [239Pu(Cp)3] from treatment of [Cs2(239PuCl6)] with excess Mg(Cp)2.68 However, it is pertinent to note that the salt metathesis reactions between [Np(Cp)3Cl], and group 1 alkyl- or aryl-anions formed only low yields of [Np(Cp)3(n-Bu)] and [Np(Cp)3Ph] (40–60%) alongside undefined NpIII by-products, presumably due to the homolysis of the NpIV-alkyl bond.12,61
The reported formal potentials summarized in Table 1 show the NpIV/NpIII couple is intermediate in value between U and Pu in the triad, as would be expected. Cyclic voltammetry experiments have demonstrated that [An(Cp)3Cl] (An = U, Np) complexes show reversible one-electron reduction processes at E1/2 = −1.80 V (UIV/UIII) and −1.29 V (NpIV/NpIII) in THF (vs. Fc+/Fc).70 Early actinide elements (An = Th–U) demonstrate a clearer thermodynamic preference for the +4 over +3 oxidation state and in its organometallic chemistry,11 for NpIV/III the preference is more finely balanced. The electrochemical properties of actinide centres in organoactinides are usually considerably affected by ligand environments.71
| Couple | Formal potential, E°′, in V vs. SHE | ||
|---|---|---|---|
| 1 M HClO4 | pH 8 | 1 M NaOH | |
| UIV/UIII | −0.631 | −1.95 ± 0.17 | −2.78 ± 0.35 |
| NpIV/NpIII | 0.155 ± 0.010 | −1.13 ± 0.14 | −1.88 ± 0.24 |
| PuIV/PuIII | 0.9821 ± 0.0005 | −0.39 ± 0.15 | −1.04 ± 0.24 |
The disproportionation of UIII into 0.75 eq. of AnIV and 0.25 eq. of An0 is well-known, and has been reported for NpIII.11,72 We used a variety of techniques to confirm the formal UIII oxidation state in the inverse sandwich complexes [{X2U}2(μ-η6,η6-C6R6)], (in which the arene carries a dianionic charge) (X = bulky aryloxide or amido monoanion, C6R6 = benzene, toluene, naphthalene, and silylated or borylated arene derivatives) that were formed from the disproportionation of UIIIX3 molecules into UIVX4 and the formal intermediate UIIX2.72 More recently, Meyer used computational analyses to confirm the formal UII oxidation state in the arene-supported tris(aryloxide) ate complex [K(2.2.2-crypt)][((Ad,MeArO)3mes)U].73 Following the report of the +2 oxidation state for uranium in a molecular complex [K(2.2.2-cryptand)][U(Cp′)3] by Evans et al.,24,37 and our report of the relatively stable, formally NpII complex Np(LAr)(dme),4 which survives up to 90 minutes in solution and as small near-black crystals, the synthesis of the neptunium homologue K[NpCp′3] of the U ‘ate-’ complex seemed a reasonable target. While a convenient low-temperature route with radiological protection was devised to afford solutions and crystals of a Np(II) complex it was insufficiently thermally stable to enable characterisation of the solutions or X-ray data collection on single crystals.
This situation should be even easier moving from Np to Pu which already shows a much more stable MIII oxidation state in its complexes.
Solid state structures
All the complexes presented here, three NpIII and one NpIVCp4, contain at least three Cp ligands in the coordination sphere of the Np, so that a structural comparison can be performed. In the structures of the NpIII complexes NpCp3(NCMe)2, K[NpCp4], NpCp3 the Np centres are surrounded by three Cp rings in η5-coordination mode. In all these complexes the centre of the Cp ring is placed between 2.51 Å and 2.54 Å distant from the Np atom. However, in K[NpCp4] there is a second coordination mode of the NpIII atoms: besides the coordination known from the NpCp3 (and from the complexes LnCp3) consisting of the three already mentioned η5-coordinated Cp rings plus one bridging Cp ring establishing an additional μ-η-coordination in K[NpCp4] there is a NpCp4 unit with the Np atom surrounded by four Cp rings all in η5-coordination. This situation is comparable to the coordination found in complexes [AnIVCp4], where in the row from Th over U to Np M to centre of ring distances are found of 2.606 (Th), 2.588 (U), and 2.551 Å (Np), respectively. These values compare to the one of 2.635 Å for the four times η5-coordinated Np centres in K[NpCp4]. Thus one can consider the difference in the ionic radii between NpIII and NpIV in an equivalent coordination environment built by in this case four η5-coordinated Cp rings to be equal to (2.635 − 2.551=) 0.08 Å.We note that the CN stretch in the IR spectrum of NpCp3(NCMe)2 is observed at 2262 cm−1, lower than in the corresponding U cationic complexes which have a stronger M–N interaction.
The trigonal planar arrangement of the three Cp′ ligands [Np(Cp′)3] around the NpIII centre, analogous to the corresponding U complex raises the possibility that this complex should be able to show comparable redox chemistry to that of U and Th, where the geometry provides suitable orbitals for an additional valence electron to reside. Therefore, it was used as the starting material for the organometallic Np(II) complex for reduction with KC8.
Conclusions
As could be anticipated, the synthetic chemistry of cyclopentadienyl-supported NpIII and NpIV complexes is comparable to that of uranium, with the differences mainly being caused by the less negative reduction potential of the Np4+ ion. For the first time a solution-based method for the quantitative formation of green, poorly soluble, but high-surface area, and therefore reactive NpCl3 has been demonstrated from reduction of NpCl4, and shown to be synthetically useful in anaerobic reactions, even in the absence of strongly coordinating solvents. Complexes NpCp3 and NpCp′3 were synthesized reproducibly in high yields via salt metathesis routes from this or from more traditional reduction of the known complex NpCp3Cl.One notable example of the greater stability of the NpIII ion with respect to UIII in these complexes is the overlooked reactivity of NpCp3Cl with excess KCp, which results in the isolation of the first actinide(III) tetrakis-cyclopentadienyl complex, K[NpCp4] under the synthetic conditions previously assumed to afford only the neutral complex NpCp4. Remarkably, the solid-state structure of K[NpCp4] exhibits intra-crystal dimorphism; two different types of NpCp4 coordination geometries, half of the NpIII cations are (η1-Cp)(η5-Cp)3 and half are (η5-Cp)4, with the two different types of NpIII forming separate polymeric chains that are bridged by potassium counter-cations to form the extended polymeric structure. Unexpectedly, this structure may answer the concerns expressed by Adrian et al. who reported two similar, but unidentified impurities in samples of NpCp4 that they studied by Mössbauer spectroscopy. Comparison of the structures of K[NpCp4] and NpCp4 enables a differentiation of the ionic radii of NpIII and NpIV in this organometallic environment of 0.08 Å. Complex NpCp′3 shows an even closer contact around the Np atom establishing a trigonal planar coordination environment which is again 0.03 Å smaller but offering further redox chemistry opportunities.
Indeed, potassium reduction at low temperatures of NpCp′3 leads to the formation of very dark-brown crystals of a complex assigned as [K(2.2.2-cryptand)][Np(Cp′)3] K[NpCp′3]; these can be isolated but are less thermally stable than the formally Np(II) complex [Np(LAr)(dme)] previously reported by us;4 single crystals of the putative NpII complex K[NpCp′3] do not survive for long enough to be encapsulated for radiological protection prior to the collection of diffraction data.
The results presented show that neptunium cyclopentadienyl chemistry can show significant deviations from its uranium congeners, in sharp contrast to previous assertions, and the resulting spectroscopic, redox, and structural investigations provide a significant and deeper understanding of minor actinide chemistry.
Acknowledgements
The experiments were supported by Talisman Joint Research Project under contract with the European Commission. M. S. D. acknowledges the European Commission for support in the frame of the Training and Mobility of Researchers programme. P. L. A. also thanks the UK EPSRC (grants EP/N022122/1 and EP/M010554/1).References
- F. Baumgärtner, E. O. Fischer and P. Laubereau, Naturwissenschaften, 1965, 52, 560 CrossRef
.
- N. Magnani, C. Apostolidis, A. Morgenstern, E. Colineau, J.-C. Griveau, H. Bolvin, O. Walter and R. Caciuffo, Angew. Chem., Int. Ed., 2011, 50, 1696–1698 CrossRef CAS PubMed
- P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A.-F. Pécharman, N. Magnani, J.-C. Griveau, E. Colineau, R. Caciuffo, X. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12797–12801 CrossRef CAS PubMed
- M. S. Dutkiewicz, J. H. Farnaby, C. Apostolidis, E. Colineau, O. Walter, N. Magnani, M. G. Gardiner, J. B. Love, N. Kaltsoyannis, R. Caciuffo and P. L. Arnold, Nat. Chem., 2016, 8, 797–802 CrossRef CAS PubMed
- M. J. Tassell and N. Kaltsoyannis, Dalton Trans., 2010, 39, 6719–6725 RSC
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed
- B. E. Bursten, L. F. Rhodes and R. J. Strittmatter, J. Am. Chem. Soc., 1989, 111, 2756–2758 CrossRef CAS
- I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124–131 RSC
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC
- J. L. Sessler, W.-S. Cho, V. Lynch and V. Král, Chem.–Eur. J., 2002, 8, 1134–1143 CrossRef CAS
- E. Dornberger, R. Klenze and B. Kanellakopulos, Inorg. Nucl. Chem. Lett., 1978, 14, 319–324 CrossRef
-
R. Bohlander, Thesis (doctoral), Universität(TH) Karlsruhe, 1986
- F. Baumgärtner, E. O. Fischer, B. Kanellakopulos and P. Laubereau, Angew. Chem., Int. Ed. Engl., 1968, 7, 634 CrossRef
- D. G. Karraker and J. A. Stone, Inorg. Chem., 1972, 11, 1742–1746 CrossRef CAS
-
D. G. Karraker, in Organometallics of the f-Elements: Proceedings of the NATO Advanced Study Institute held at Sogesta, Urbino, Italy, September 11–22, 1978, ed. T. J. Marks and R. D. Fischer, Springer, Netherlands, Dordrecht, 1979, pp. 395–420 Search PubMed
- G. Adrian, Inorg. Chim. Acta, 1987, 139, 323–325 CrossRef CAS
- G. Adrian, H. Appel, R. Bohlander, H. Haffner and B. Kanellakopulos, Hyperfine Interact., 1988, 40, 275–278 CrossRef CAS
-
K. Nash, C. Madic, J. N. Mathur and J. Lacquement, in The Chemistry of the Actinide and Transactinide Elements, ed. J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, 4th edn, 2010, vol. 5, ch. 24, pp. 2622–2798 Search PubMed
- D. J. A. De Ridder, J. Rebizant, C. Apostolidis, B. Kanellakopulos and E. Dornberger, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 597–600 CrossRef
- K. W. Bagnall, G. F. Payne, N. W. Alcock, D. J. Flanders and D. Brown, J. Chem. Soc., Dalton Trans., 1986, 783–787 RSC
- D. J. A. De Ridder, C. Apostolidis, J. Rebizant, B. Kanellakopulos and R. Maier, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1436–1438 CrossRef
- C. Apostolidis, Private communication, 2015.
- B. Kanellakopulos, R. Klenze, G. Schilling and A. H. Stollenwerk, J. Chem. Phys., 1980, 72, 6311–6312 CrossRef CAS
- M. R. MacDonald, M. E. Fieser, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 13310–13313 CrossRef CAS PubMed
- R. D. Fischer, R. V. Ammon and B. Kanellakopulos, J. Organomet. Chem., 1970, 25, 123–137 CrossRef CAS
- B. Kanellakopulos, E. O. Fischer, E. Dornberger and F. Baumgärtner, J. Organomet. Chem., 1970, 24, 507–514 CrossRef CAS
- V. T. Aleksanyan and B. V. Lokshin, J. Organomet. Chem., 1977, 131, 113–120 CrossRef CAS
- R. Maier, B. Kanellakopulos, C. Apostolidis, D. Meyer and J. Rebizant, J. Alloys Compd., 1993, 190, 269–271 CrossRef CAS
- J. H. Burns, J. Organomet. Chem., 1974, 69, 225–233 CrossRef CAS
- S. H. Eggers, W. Hinrichs, J. Kopf, R. D. Fischer and L. Xing-Fu, Private communication to CSD, 1992, CCDC 1262726.
- S. H. Eggers, W. Hinrichs, J. Kopf, R. D. Fischer and L. Xing-Fu, Private communication to CSD, 1992, CCDC 1134714.
- S. H. Eggers, J. Kopf and R. D. Fischer, Organometallics, 1986, 5, 383–385 CrossRef CAS
- W. Hinrichs, D. Melzer, M. Rehwoldt, W. Jahn and R. D. Fischer, J. Organomet. Chem., 1983, 251, 299–305 CrossRef CAS
- Z. Xie, F. E. Hahn and C. Qian, J. Organomet. Chem., 1991, 414, C12–C14 CrossRef CAS
- S. D. Stults, R. A. Andersen and A. Zalkin, Organometallics, 1990, 9, 115–122 CrossRef CAS
- J. H. Burns, W. H. Baldwin and F. H. Fink, Inorg. Chem., 1974, 13, 1916–1920 CrossRef CAS
- C. J. Windorff, M. R. MacDonald, K. R. Meihaus, J. W. Ziller, J. R. Long and W. J. Evans, Chem.–Eur. J., 2016, 22, 772–782 CrossRef CAS PubMed
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- L. Xing-Fu, S. Eggers, J. Kopf, W. Jahn, R. Dieter Fischer, C. Apostolidis, B. Kanellakopulos, F. Benetollo, A. Polo and G. Bombieri, Inorg. Chim. Acta, 1985, 100, 183–199 CrossRef
- H. Schulz, H. Reddmann, H.-D. Amberger, B. Kanellakopulos, C. Apostolidis, J. Rebizant and N. M. Edelstein, J. Organomet. Chem., 2001, 622, 19–32 CrossRef CAS
- M. R. Spirlet, J. Rebizant, C. Apostolidis and B. Kanellakopulos, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2322–2324 CrossRef
- W. J. Evans, T. J. Mueller and J. W. Ziller, Chem.–Eur. J., 2010, 16, 964–975 CrossRef CAS PubMed
- W. J. Evans, T. J. Mueller and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 2678–2686 CrossRef CAS PubMed
- J. Maynadié, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Organometallics, 2006, 25, 5603–5611 CrossRef
- G. Bombieri, F. Benetollo, E. Klahne and R. D. Fischer, J. Chem. Soc., Dalton Trans., 1983, 1115–1121 RSC
- J. Rebizant, C. Apostolidis, M. R. Spirlet and B. Kanellakopulos, Inorg. Chim. Acta, 1987, 139, 209–210 CrossRef CAS
- H. Aslan, K. Yünlü, R. D. Fischer, G. Bombieri and F. Benetollo, J. Organomet. Chem., 1988, 354, 63–76 CrossRef CAS
- A. Zalkin, J. G. Brennan and R. A. Andersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 2104–2106 CrossRef
- S. H. Eggers, J. Kopf and R. D. Fischer, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 2288–2290 CrossRef
- R. G. Denning, J. Harmer, J. C. Green and M. Irwin, J. Am. Chem. Soc., 2011, 133, 20644–20660 CrossRef CAS PubMed
- M. del Mar Conejo, J. S. Parry, E. Carmona, M. Schultz, J. G. Brennann, S. M. Beshouri, R. A. Andersen, R. D. Rogers, S. Coles and M. B. Hursthouse, Chem.–Eur. J., 1999, 5, 3000–3009 CrossRef CAS
- N. A. Siladke, J. W. Ziller and W. J. Evans, Z. Anorg. Allg. Chem., 2010, 636, 2347–2351 CrossRef CAS
- W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed
- J. J. Kiernicki, B. S. Newell, E. M. Matson, N. H. Anderson, P. E. Fanwick, M. P. Shores and S. C. Bart, Inorg. Chem., 2014, 53, 3730–3741 CrossRef CAS PubMed
- W. J. Evans, S. A. Kozimor, G. W. Nyce and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 13831–13835 CrossRef CAS PubMed
- J.-C. Berthet, J.-F. Le Marechal, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 1992, 1573–1577 RSC
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Organometallics, 2005, 24, 3407–3412 CrossRef CAS
- J.-C. Berthet, C. Villiers, J.-F. Le Maréchal, B. Delavaux-Nicot, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Organomet. Chem., 1992, 440, 53–65 CrossRef CAS
- W. J. Evans, K. J. Forrestal and J. W. Ziller, Angew. Chem., Int. Ed. Engl., 1997, 36, 774–776 CrossRef CAS
- W. J. Evans, G. W. Nyce, M. A. Johnston and J. W. Ziller, J. Am. Chem. Soc., 2000, 122, 12019–12020 CrossRef CAS
- D. G. Karraker and J. A. Stone, Inorg. Chem., 1979, 18, 2205–2207 CrossRef CAS
- E. O. Fischer, W. Hafner and H. O. Stahl, Z. Anorg. Allg. Chem., 1955, 282, 47–62 CrossRef CAS
- G. Wilkinson, F. A. Cotton and J. M. Birmingham, J. Inorg. Nucl. Chem., 1956, 2, 95–113 CrossRef CAS
- E. O. Fischer and W. Hafner, Zeitschrift für Naturforschung B, 1954, 9, 503–504 Search PubMed
- D. E. Bublitz, W. E. McEwen and J. Kleinberg, Org. Synth., 1961, 41, 96 CrossRef CAS
- E. O. Fischer and H. Grubert, Chem. Ber., 1959, 92, 2302–2309 CrossRef
- T. D. Tilley, R. A. Andersen, B. Spencer, H. Ruben, A. Zalkin and D. H. Templeton, Inorg. Chem., 1980, 19, 2999–3003 CrossRef CAS
- L. R. Crisler and W. G. Eggerman, J. Inorg. Nucl. Chem., 1974, 36, 1424–1426 CrossRef CAS
-
G. Choppin, J.-O. Liljenzin, J. Rydberg and C. Ekberg, in Radiochemistry and Nuclear Chemistry, Academic Press, Oxford, 4th edn, 2013, pp. 753–788 Search PubMed
- D. C. Sonnenberger and J. G. Gaudiello, Inorg. Chem., 1988, 27, 2747–2748 CrossRef CAS
-
T. J. Marks and R. D. Ernst, in Comprehensive Organometallic Chemistry, ed. F. G. A. Stone and E. W. Abel, Pergamon, Oxford, 1982, pp. 173–270 Search PubMed
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed
- H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158–7162 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, synthetic details, spectroscopic data, X-ray crystallographic data. CCDC 1524162–1524166. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00034k |
| This journal is © The Royal Society of Chemistry 2017 |