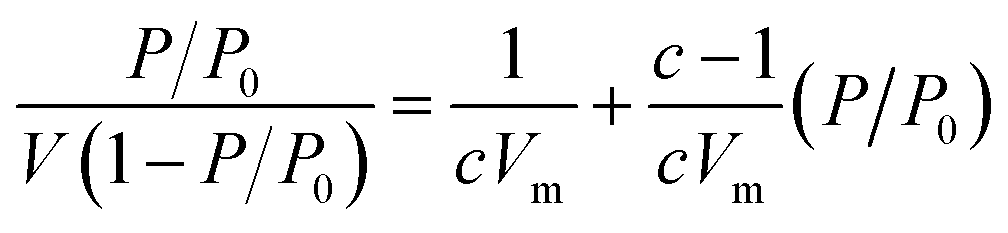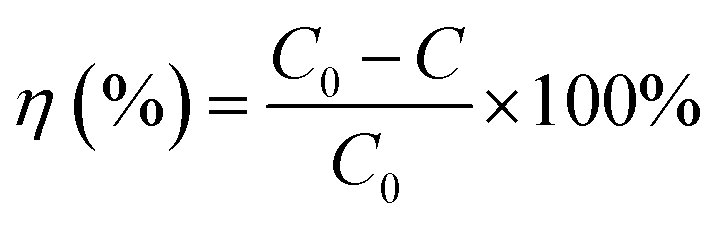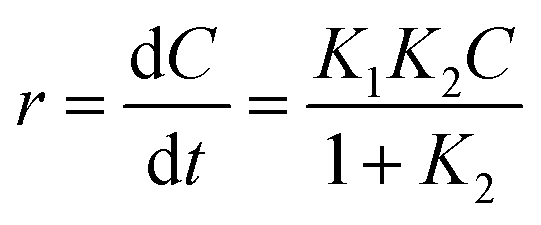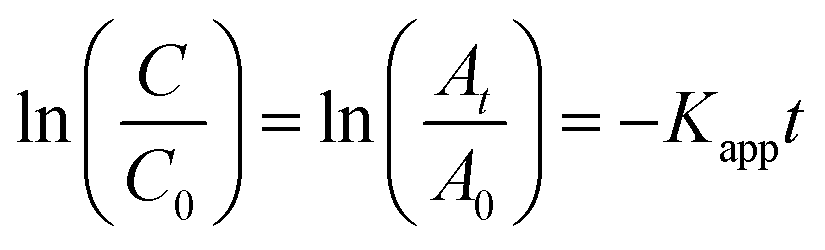DOI:
10.1039/C6RA28640B
(Paper)
RSC Adv., 2017,
7, 8633-8645
Plasmon induced enhanced photocatalytic activity of gold loaded hydroxyapatite nanoparticles for methylene blue degradation under visible light†
Received
23rd December 2016
, Accepted 23rd January 2017
First published on 27th January 2017
Abstract
A facile surfactant free wet-precipitation process was employed to prepare hydroxyapatite (HAp) nanoparticles. Further, a microwave-assisted hydrothermal process was used to synthesize gold-loaded HAp (Au–HAp) nanocomposites with different Au contents. The nanocomposites of mesoporous structures exhibited a high specific surface area. Incorporated gold in the nanocomposites formed Au nanoparticles at the surface of HAp nanoparticles. Apart from characterizing the nanocomposites for their morphology, crystallinity, structural phase and optical behaviors, they were tested for photocatalytic degradation methylene blue under visible-light. Incorporation of Au nanoparticles significantly improved the photocatalytic activity of HAp nanoparticles under visible light irradiation. Fourier transformed infrared (FTIR) spectroscopy analysis confirmed the chemical stability of the catalyst under strong photo-induced oxidation-reaction. The strong dye adsorption capacity of HAp and surface plasmon resonance (SPR) of Au nanoparticles in the visible wavelength range make this novel Au–HAp nanocomposite an effective visible light photocatalyst for the degradation and adsorption of organic dye from their aqueous solutions. Being HAp a nontoxic, bioactive material and gold a nontoxic noble metal, the composite has the potential for utilization as ambiental friendly photocatalyst for wastewater treatment.
1. Introduction
Photocatalytic degradation is an emerging technology, enticing renewed attention for mineralizing organic compounds. The process can be defined as the acceleration of a photoreaction by the presence of a catalyst.1,2 Photocatalysis is best applied when the more common wastewater treatment technologies such as flocculation, sedimentation, biological degradation, adsorption, filtration, centrifugation, reverse osmosis are inadequately effective.3 However, for practical applications, the photocatalyst should be resistant to photocatalytic oxidative degradation and non-toxic. Three components which must be present to take place a heterogeneous photocatalytic reaction are: (i) an emitted photon of appropriate wavelength, (ii) a catalytic material (usually a solid catalyst), and (iii) a strong oxidizing agent which in most cases, is oxygen.
The last few years have witnessed a swift growth of photocatalytic research on a variety of oxide semiconductors such as titanium dioxide,4 zinc oxide,5 and zirconium oxide.6 Efforts have been made to incorporate plasmonic nanoparticles over metal oxide surfaces to fabricate Au/TiO2,7 Au/Fe2O3,8 Au/ZrO2,9 Au/CeO2,10 Au/Al2O3,11 Au/hydroxyapatite12 nanocomposites to enhance their photocatalytic activities, exploiting the strong surface plasmon absorption of metal nanoparticles. Catalytic activity and stability of supported gold depends strongly on the choice of the support material and the specific interaction between the metal and the support.13
Recently, hydroxyapatite (HAp, Ca10(PO4)6(OH)2) has attracted interest for use in a variety of applications such as bio-ceramics,14 drug delivery vector,14 insulator,15 chromatography,16 and also as catalyst.12 The use of HAp as a support for nano-gold has been reported to perform excellent CO oxidation activity and enhancement of stability of gold nanoparticles against sintering at temperatures as high as 600 °C.17 Nano- and micro-structured HAp have also been applied as catalyst for several dehydration and dehydrogenation reactions,18 synthesis of chalcone derivatives,19 gas-phase oxidation reactions20–22 and as photocatalyst for gas-phase photocatalytic processes.23,24 However, only a few studies have evaluated HAp as aqueous phase photocatalyst25,26 in a very limited range of reaction conditions. Due to its high band gap energy, exploitation of HAp for this purpose has been limited only to UV irradiation. Tsukada et al.27 have evaluated the effect of Ti substitution in HAp on its band gap, both experimentally and theoretically. The experimentally obtained optical band gap energies (Eg) of Ti–HAp, HAp and TiO2 powders measured by diffuse reflectance spectroscopy (DRS) were 3.65 eV, >6.0 eV, and 3.27 eV, respectively. Very recently, Bystrov et al.28 have calculated the band gap energy of HAp and its variation on the introduction of oxygen vacancies, using density functional theory (DFT). Both the theoretical and experimental values of band gap energy were close to 5.0 eV for non-catalytic HAp and ∼3.45 eV for photocatalytic (HAp with oxygen vacancies) HAp. On the other hand, a band gap energy of ∼4 eV was estimated for HAp calcinated in air at 1200 °C, which is in the boundary of UVA and UVB regions of solar spectrum. However, Nishikawa29 has reported the production of ˙OH and O2˙−, which are the principal active species for photo-mineralization of organic compounds30 on UV irradiation of 200 °C calcinated HAp dispersed in water.
On the other hand, Mitsionis et al.31 synthesized biphasic photocatalytic powders containing HAp and titanium dioxide (TiO2). Their HAp/TiO2 composite material exhibited excellent activity in photocatalytic NO oxidation. Liu et al.32 prepared HAp modified Ag–TiO2 powders (Ag–TiO2–HAp) by a facile wet-chemical method and observed a marked improvement in the photocatalytic activity (oxidation decomposition of acetone in air) of Ag–TiO2, and TiO2 due to HAp modification.
On the metal–HAp font, Hong et al.33 synthesized a novel Ag3PO4/HAp composite photocatalyst in aqueous solution via wet-impregnation approach. The composite exhibited a strong absorption in both the visible and UV-Vis regions and enhanced photocatalytic activity for methylene blue (MB) degradation. Vukomanovic et al.34 designed and fabricated platinum (Pt0 and Ptn+)–HAp nanocomposites, which are photocatalytically active both under UV and visible irradiation. Substituting calcium (Ca) ions of HAp lattice by vanadium (V) ions (V:HAp), Nishikawa et al.35 could induce photocatalytic activity in HAp under visible light illumination. The substituted pentavalent V ions at bivalent Ca sites generate Ca vacancies in HAp lattice to maintain its electrical neutrality when calcinated at 400 °C. The V ions in the HAp lattice seemed to play a role of electron acceptor and the redox potential of the level due to the V ions was negative enough to reduce O2 molecule into H2O2. Chang et al.36 synthesized HAp supported N-doped carbon quantum dots (N-CQDs) for visible-light photocatalytic application. The formed HAp/N-CQDs composite exhibited a significant performance in the photocatalytic degradation of MB under visible-light.
In the present study, we fabricated gold nanoparticle decorated HAp nanoparticles through wet-precipitation, followed by microwave reduction. Apart from structural and morphological characterizations, fabricated nanocomposites have been tested for the photocatalytic degradation of cationic dye MB under visible light irradiation. MB has been chosen as a model organic dye5,37 in our study as it is one of the principal contaminant in wastewater emanating from textile, wood, leather and food industries. We demonstrate that gold nanoparticle decorated HAp nanostructures, which are non-toxic and bio-degradable, have a great potential for utilizing as visible-light photocatalyst for organic dye degradation.
2. Materials and methods
2.1 Material preparation
Calcium nitrate tetrahydrate [Ca(NO3)2·4H2O, 99%], di-ammonium hydrogen phosphate [(NH4)2HPO4, 98%], ammonium hydroxide [NH4OH, 28%], and gold(III) chloride trihydrate [HAuCl4·3H2O, 99.9%] were purchased from Sigma Aldrich, Mexico. Ethanol [C2H5OH] was purchased from CTR scientific chemicals. Deionized water (DI) of ρ > 18.2 Ω cm (at 25 °C) from a Millipore deionizer was used during the entire experimental study.
2.1.1 Synthesis of HAp nanoparticles. Hydroxyapatite powder was prepared by wet-precipitation method using calcium nitrate tetrahydrate [Ca(NO3)2·4H2O] and di-ammonium hydrogen phosphate [(NH4)2HPO4] as starting materials and ammonia (NH4OH) solution as agent for pH adjustment. A suspension of 0.24 M Ca(NO3)2·4H2O (23.61 g Ca(NO3)2·4H2O in 350 ml DI water) was vigorously stirred in a 500 ml beaker maintaining its temperature at 40 °C. The pH of both Ca(NO3)2·4H2O and (NH4)2HPO4 solutions was adjusted to 11 by drop-wise addition of ammonia solution. Then the solution of 0.29 M (NH4)2HPO4 (7.92 g (NH4)2HPO4 in 250 ml DI water) was drop-wise added to the Ca(NO3)2·4H2O solution. The reaction occurred can be expressed as (eqn (1)):| | |
10Ca(NO3)2·4H2O + 6(NH4)2HPO4 + 8NH4OH → Ca10(PO4)6(OH)2 + 20NH4NO3 + 20H2O
| (1) |
After 1 h of reaction, followed by 24 h of ageing, the formed precipitate was removed from the reaction solution by centrifugation at 5000 rpm for 6 minutes and dried at 80 °C for 1 h. The dry powder was calcinated at 600 °C for 1 h in air.
2.1.2 Synthesis of Au loaded HAp. 200 mg of pre-calcinated (600 °C, 1 h) HAp powder was placed into a Teflon-made autoclave of 40 ml capacity. A mixture of 15 ml of ethanol and 15 ml of DI water was poured into the autoclave. Then a specific volume (0.056 ml, 0.1125 ml, 0.225 ml, and 0.45 ml) of gold(III) chloride solution (2 mg ml−1) was added to the mixture under magnetic stirring. The autoclave was then capped tightly and sonicated for 10 minutes in a commercial ultrasonicator (37 kHz, 360 W). Finally, the autoclave was placed inside a commercial microwave oven (LG-MS0743U, 2450 MHz, and 1000 W) and irradiated for 10 minutes under 20% of its full power. The microwave irradiation was performed in steps (2 minutes on and 15 minutes off) to keep the temperature of the reaction mixture around 130 °C (±4 °C). On finishing the microwave treatment steps, the sample was cooled down to room temperature and separated by centrifugation. For preparing the nanocomposites with different gold loadings, the same procedure was followed, varying only the volume of gold chloride solution.
2.2 Characterization of Au loaded HAp
The structure and crystalline phase of the synthesized samples were studied by powder X-ray diffraction (XRD) analysis (30 keV, 25 mA) using CuKα radiation (λ = 1.5405 Å) of a Bruker 8D diffractometer. To determine the functional groups present in the samples, Fourier transformed infrared spectroscopy (FTIR) was carried out in the 4000–400 cm−1 range using a Perkin Elmer Spotlight 400 FTIR spectrometer. The morphology and chemical composition of the samples were analyzed in a field-emission scanning electron microscope (FE-SEM, JEOL SUPRA 40) coupled with Oxford analytical system. A JEOL 2100 high resolution transmission electron microscope (TEM) was used to detect the incorporation of gold nanoparticles in the composites and analyze their fine structure. An Agilent-Varian Carry 5000 spectrophotometer with diffused reflectance accessories was utilized to record the reflectance spectra of the nanocomposites. To determine the specific surface area of the nanocomposites, their N2 adsorption–desorption isotherms were recorded at 77 K in a Belsorp Mini-II (Belsorp, Japan) sorptometer. All the composite samples were degassed at 300 °C under vacuum for 5 h before recording their adsorption–desorption isotherms.
2.3 Photocatalytic study of Au loaded HAp
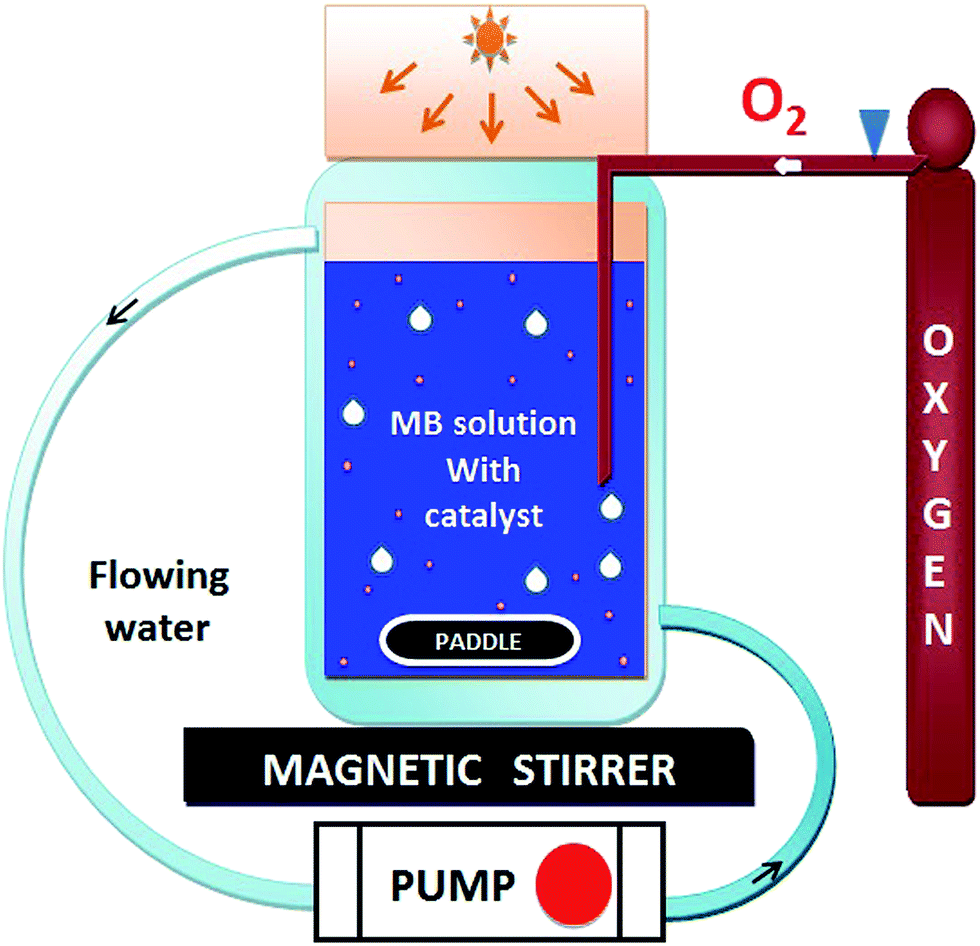
The photocatalytic activity of HAp and gold-loaded HAp samples was studied in a cylindrical glass reactor of 250 ml capacity (50 mm internal diameter and 120 mm height) with water recirculating jacket (Fig. 1). Typically, 40 mg of the sample was added into 80 ml of dye solution (5 ppm) under magnetic stirring and air flow in dark (inside a black box). The mixture was left in dark for 75 min under stirring and air bubbling at 20 °C to reach the adsorption–desorption equilibrium at the surface of the catalyst. The extent of dye adsorption was determined from the decrease of MB concentration in the solution by monitoring the intensity of its principal absorption band (664 nm) in a Shimadzu UV-VIS-NIR 3100PC spectrophotometer. At different intervals of time, about 4 ml of aliquot was withdrawn from of the mixture and filtered by a reusable syringe (z268410) fitted with a nitrocellulose membrane filter of 0.22 μm pore size, to measure the dye concentration in the filtered sample its optical absorption spectrum. Once the dye adsorption–desorption equilibrium is reached, the mixture was illuminated by a 10 W xenon lamp, emitting white light. The concentration of MB in the mixture was monitored through the same way as in the case of dye adsorption under dark. The temperature of the reaction mixture was maintained at 20 °C throughout the experiment.
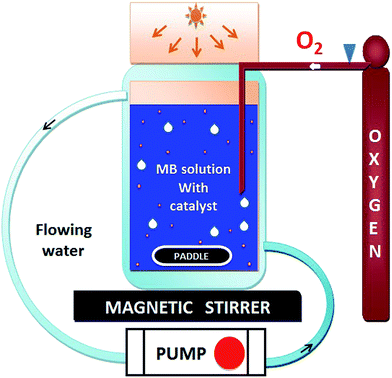 |
| | Fig. 1 Schematic representation of the used photocatalytic reactor setup. | |
3. Results and discussion
3.1 X-ray diffraction (XRD) analysis of HAp and Au-loaded HAp
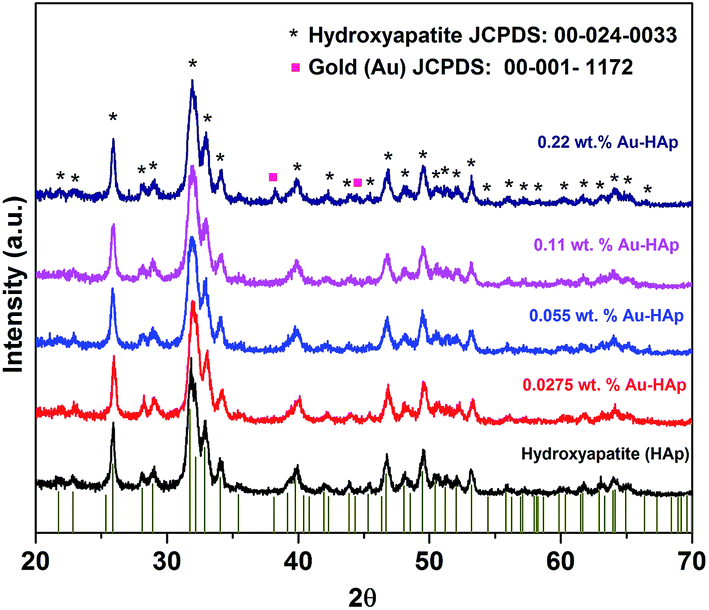
XRD patterns of the synthesized hydroxyapatite, and Au(x)–HAp (x = 0.0275, 0.055, and 0.11 wt%) samples are presented in Fig. 2. The HAp sample prepared in this study revealed characteristic diffraction peaks of pure hydroxyapatite [JCPDS: 00-024-0033] in single hexagonal phase. While the HAp samples loaded with gold in lower concentration did not reveal any diffraction peak associated to gold in their diffraction patterns, diffraction peaks characteristic of gold in bcc phase [JCPDS: 00-001-1172] could be observed at around 38.26 and 44.6° for the Au-loaded HAp samples of higher Au contents. The intensity of the Au peaks increased with the increase of Au content in the samples. While the diffraction peaks associated to Au appeared around 38.26 and 44.6° correspond to the (111) and (200) crystalline planes of Au, the most intense diffraction peak of HAp appeared around 31.77° corresponds to its (211) planes.
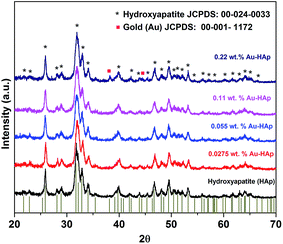 |
| | Fig. 2 XRD spectra of HAp and Au–HAp nanocomposites with different Au contents. The bottom vertical bars show the peak positions of pure hydroxyapatite in hexagonal phase (JCPDS: 00-024-0033). | |
3.2 UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS)
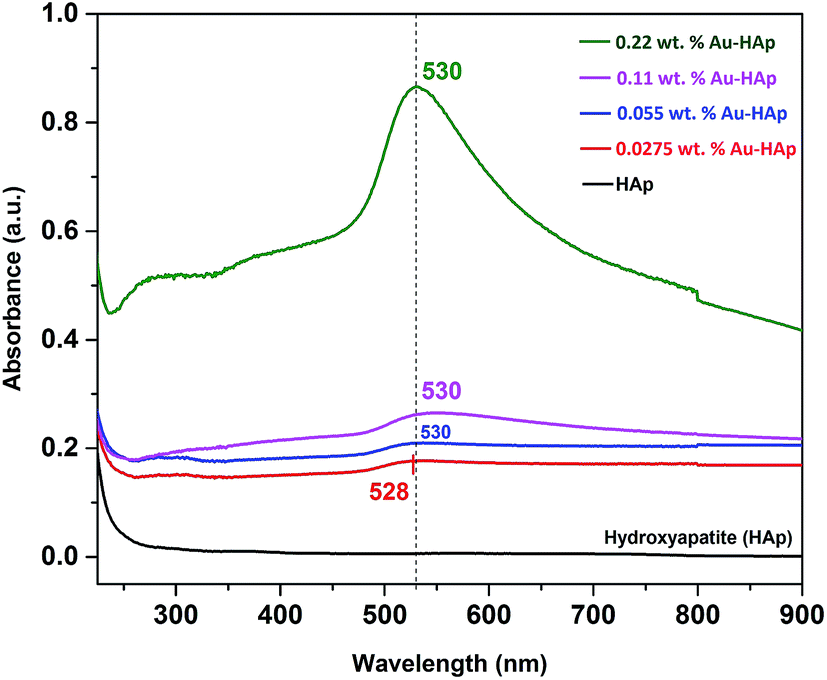
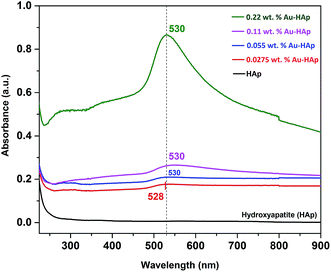
UV-Vis diffuse reflectance spectra of the gold-loaded HAp samples in absorption mode are presented in Fig. 3. The absorption peak appeared in between 528–530 nm for the gold supported HAp samples is attributed to the surface plasmon resonance absorption of Au nanoparticles, which originates from the intraband excitation of 6sp electrons of Au.38,39
 |
| | Fig. 3 UV-Vis DRS spectra of HAp nanoparticles loaded with Au in different weight percentages. | |
The gradual increase of SPR absorption band intensity along with a shift towards longer wavelengths indicates the formation of higher number of Au nanoparticles and an increase of their size with the increase of Au content in the composites.
3.3 N2 adsorption–desorption study
The Brunauer–Emmett–Teller (BET) theory aims to explain the physical adsorption of gas molecules on a solid surface and serves as the basis for an important analysis technique for the measurement of the specific surface area of a material. The average particles pore size is also determined by this adsorption study following BET equation (eqn (2)):| |
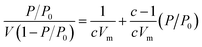 | (2) |
where, P – equilibrium pressure, P0 – saturate vapour pressure of the adsorbed gas at the temperature, P/P0 is called relative pressure, V – volume of adsorbed gas per kg adsorbent, Vm – volume of monolayer adsorbed gas per kg adsorbent. c – constant associated with adsorption heat and condensation heat.
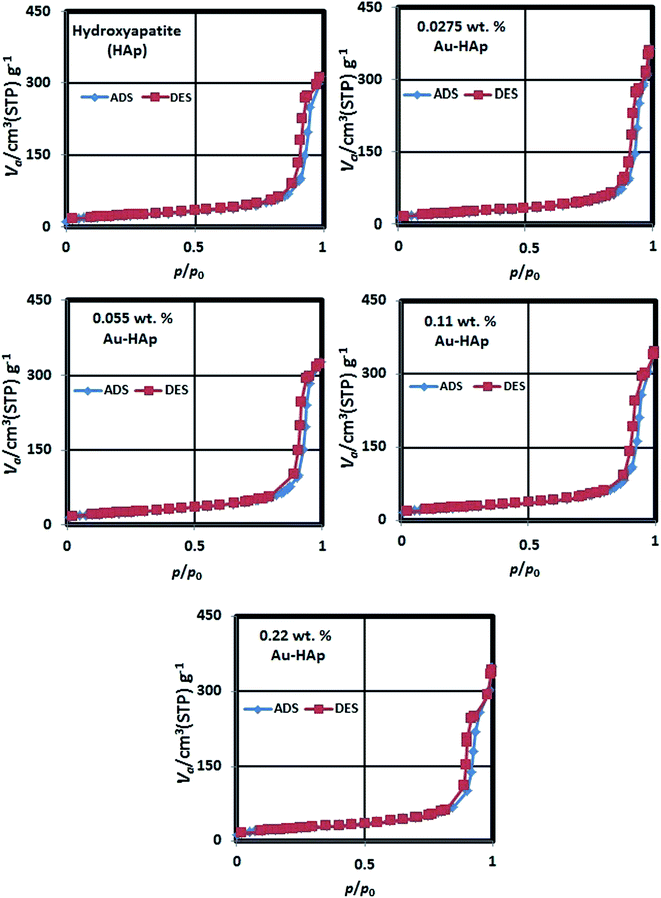
The N2 adsorption–desorption isotherms of the synthesized HAp and Au-loaded HAp samples are presented in Fig. 4. As can be seen, the isotherms of all the samples follow type IV adsorption–desorption characteristics of the IUPAC classification.40 The revealed hysteresis cycles are associated to the capillary condensation of N2 in mesopores. The sharper increase of N2 adsorption at lower relative pressure is attributed to a mono- and multilayer adsorptions at mesoporous surface. The average surface area, average pore size and pore volume in the samples were estimated form their BET analysis and enlisted in Table 1.
 |
| | Fig. 4 N2 adsorption–desorption isotherms of HAp and Au–HAp nanocomposites. | |
Table 1 BET estimated specific surface area and average pore size for the HAp and Au–HAp nanocomposites
| Sample |
Surface area (m2 g−1) |
Avg. pore size (nm) |
| Hydroxyapatite (HAp) |
78.42 |
24.47 |
| 0.0275 wt% Au–HAp |
83.02 |
27.14 |
| 0.055 wt% Au–HAp |
84.97 |
23.53 |
| 0.11 wt% Au–HAp |
85.74 |
23.97 |
| 0.22 wt% Au–HAp |
86.43 |
22.34 |
As it can be noticed from Table 1, the specific surface area of the nanocomposite increases gradually with the increase of gold loading. On the other hand, the average pore size in the nanocomposites increases initially, and then decreases gradually with the increase of gold loading. While the gradual increase of specific surface area of the nanocomposite with the increase of gold loading indicates the formation of higher number of Au nanoparticles at the surface of HAp nanoparticles, which has also been revealed in their DRS spectra, the initial increase of average pore size might be due to the adherence of small Au nanoparticles at their surface. On the other hand, a gradual decrease of average pore size with higher Au loading might be due to the formation of Au nanoparticles at the inter-particle spaces of the nanocomposite.
3.4 SEM and EDS study
SEM study revealed the formation of nearly spherical nanoparticles in the HAp sample (Fig. 5a) prepared by wet-precipitation technique. The size of the formed HAp nanoparticles varied in-between 18 and 29 nm, with average diameter 23.06 ± 2.32 nm. The synthesized gold loaded HAp (0.055 wt% Au–HAp) nanoparticles were also studied by scanning electron microscopy (Fig. 5b). Formation of gold nanoparticles at the surface of HAp particles could be confirmed by the presence of bright spots (Fig. 5b, marked by yellow arrows) in their SEM images. EDS analysis of the 600 °C calcinated HAp nanoparticles revealed the presence of Ca, P, and O in them, with Ca/P atomic ratio of 1.67. A very low intensity emission peak of gold could be detected in the EDS spectra of Au-loaded HAp samples, confirming the presence of gold. With increased gold loading, the atomic% of Au in the nanocomposites increased.
 |
| | Fig. 5 Typical SEM images of (a) 600 °C air annealed HAp, and (b) 0.055 wt% Au–HAp nanocomposites. The inset of (a) presents size distribution histogram of the pristine HAp nanoparticles. A typical EDS spectrum of the Au-loaded sample is presented as inset of (b). Presence of Au nanoparticles at the surface of HAp nanoparticles (indicated by arrows) can be noticed from their brighter contrasts. | |
3.5 TEM study
For a more detailed morphological and microstructural analysis, the HAp and Au-loaded HAp samples were analyzed by transmission electron microscopy (TEM). Typical TEM image of pure HAp, TEM and HRTEM images of 0.055 wt% Au–HAp samples are presented in Fig. 6. In contrary to the SEM images, the TEM images of the HAp sample revealed elongated morphology of the formed nanoparticles; although in general, the particles are near spherical. Formation of tiny Au nanoparticles at the surface of HAp particles could be detected even in their low magnification TEM images (Fig. 6b). The high resolution TEM (HRTEM) image of the 0.055 wt% Au–HAp sample presented in Fig. 6c clearly demonstrates the formation of gold nanoparticles of 2–6 nm size at the surface of HAp nanoparticles and at inter-particle spaces, apart form their highly crystalline structures. Interplaner distances measured from the atomic planes revealed in the HRTEM image of Au–HAp nanocomposite revealed the formation of predominant (111) planes of Au in bcc phase (lattice spacing of 0.235 nm) and (202) planes of HAp (lattice spacing of 0.263 nm). The observations made from the HRTEM image of Au–HAp sample, i.e. the formation of tiny Au nanoparticles at the surface of HAp nanoparticles and at interparticle sites, explain the variation of specific surface area and average pore size of the nanocomposites estimated from their adsorption–desorption isotherms (Table 1).
 |
| | Fig. 6 Typical (a) TEM image of HAp, (b) 0.055 wt% Au–HAp, and (c) HRTEM image of 0.055 wt% Au–HAp samples. Formation of Au nanoparticles of 2 to 6 nm sizes at the surface and inter-particle spaces of the nanocomposite is clear from the HRTEM image. | |
3.6 Photocatalytic degradation of MB by Au–HAp
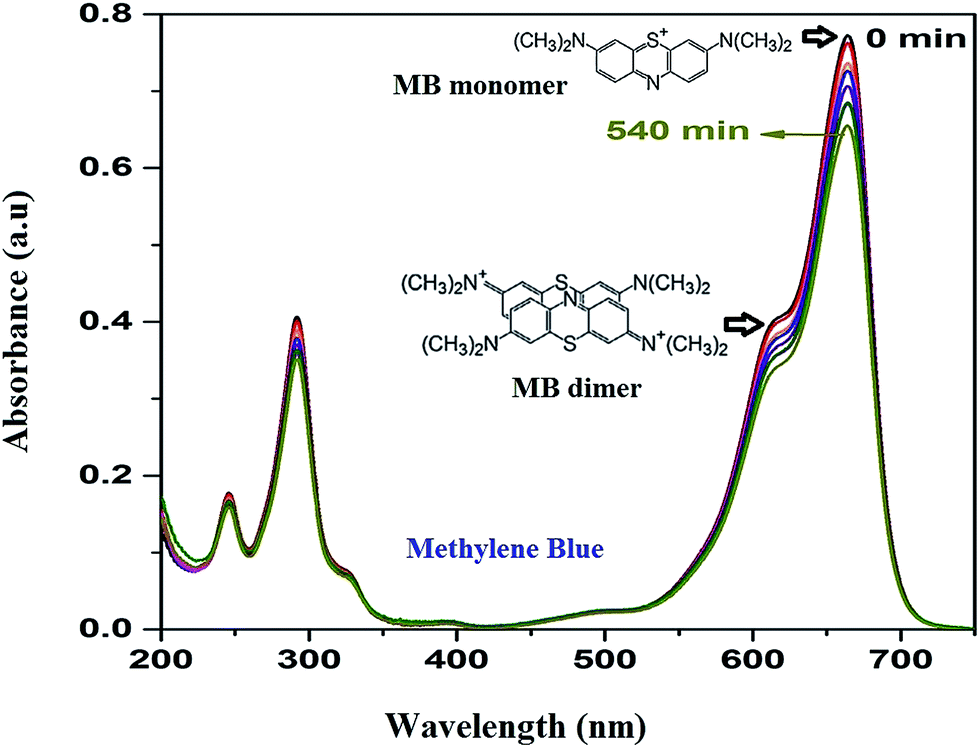
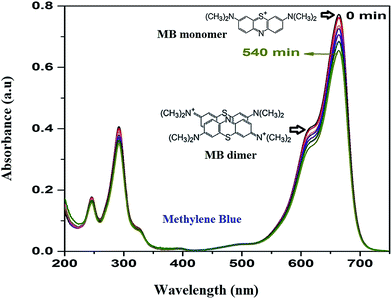
As the adsorption of MB takes place over HAp surface even without UV/Vis irradiation due to its high dye adsorption capacity, both the HAp and Au-loaded HAp catalysts were equilibrated in MB solution under dark for about 75 min. Based on the action spectra (time dependent UV-Vis absorption spectra) of the samples (Fig. 7), possible byproducts of MB degradation were analyzed. As can be seen in Fig. 7, the absorption spectra of MB reveal most intense absorption peak at around 664 nm, with a shoulder at about 612 nm. While the shoulder at 612 nm has been associated to MB dimer [(MB)2],41 the 664 nm band has been associated to MB monomer, the conjugation system between the two dimethylamine substituted aromatic rings through sulfur and nitrogen atoms.42 Additional two bands appeared in the ultraviolet region with peaks around 292 and 245 nm have been associated to substituted benzene rings.42 During photocatalytic degradation, the intensity of all these four absorption bands diminishes gradually, without revealing any new band in the absorption spectra of MB (Fig. 7), which indicates the photocatalytic degradation process does not produce any stable intermediate byproduct.
 |
| | Fig. 7 Time dependent MB absorption spectra for the 0.055 wt% Au–HAp nanocomposite used to determine its photocatalytic activity. | |
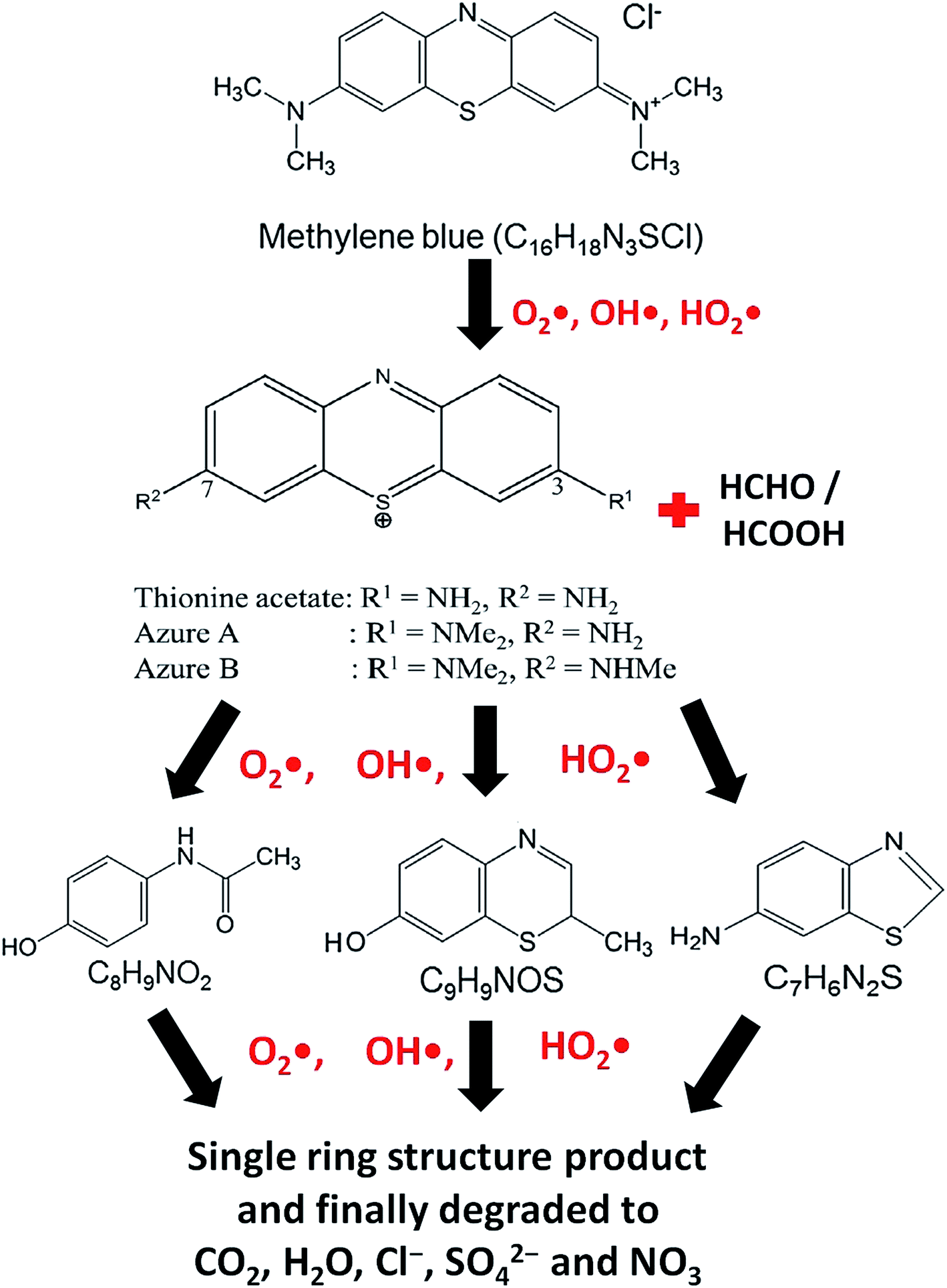
At the time of dissolution of MB molecule in water, the Cl− ion separates from its core structure. The terminal N–CH3 groups which are connected to the core structure at 7C and 12C positions of MB molecular structure, have lowest bond dissociation energy (CH3–N(CH3)C6H5, binding energy = 70.8 kcal mol−1).43 During photocatalytic degradation, the generated active radical species such as OH˙ and HO2˙ first break the N–CH3 bond, and then –CH3 is oxidized to HCHO or HCOOH. The free radicals (OH˙ and HO2˙) then break the C–S and C–N bonds of thionine molecule (Fig. 8) to produce relatively unstable smaller organic byproducts. These oxidization reactions continue until the MB degrades completely to produce smaller inorganic molecules, such as H2O, Cl−, CO2, SO42− and NO3.43 The possible reaction steps involved are schematically presented in Fig. 8.
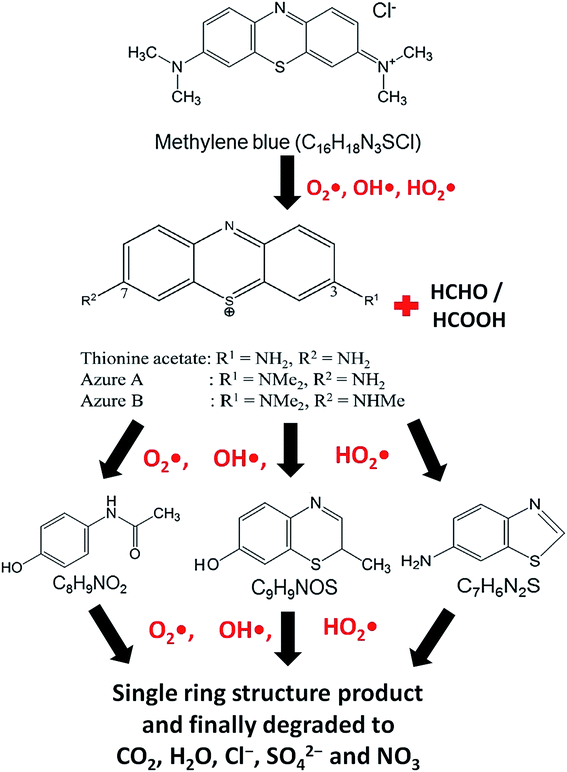 |
| | Fig. 8 Probable reaction steps of MB photocatalytic degradation. | |
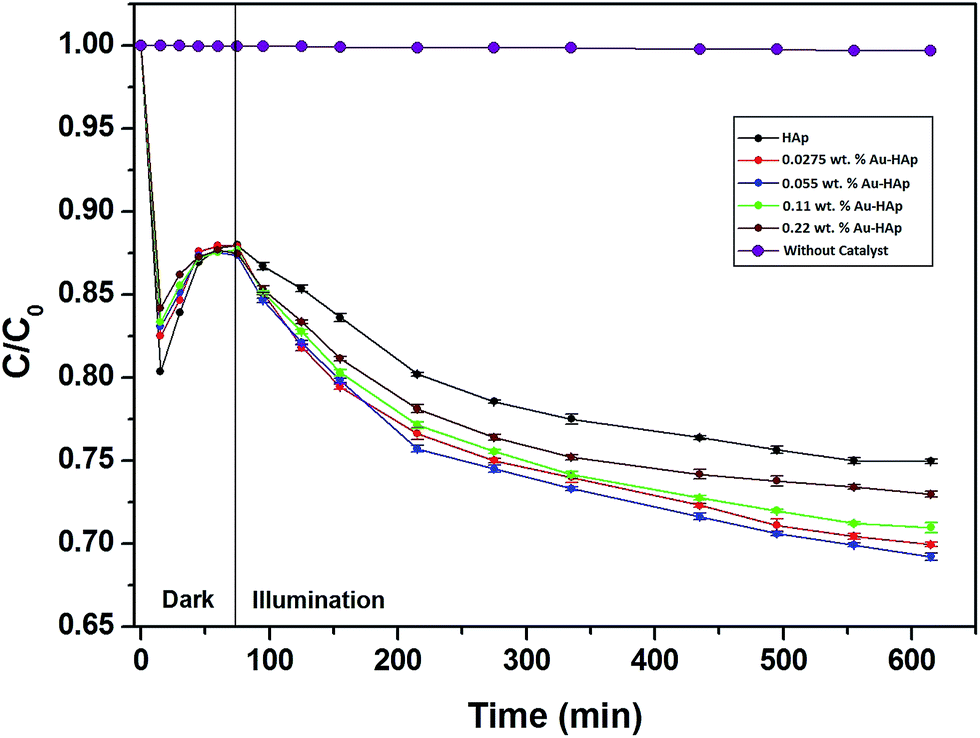
In Fig. 9, the decolorization curves for HAp and Au-loaded HAp samples are presented as C/C0 versus time, where C0 is the initial concentration and C is the concentration of MB at a particular time. As can be seen, both the HAp and Au-loaded HAp strongly adsorb MB under dark. The adsorption–desorption equilibrium reaches in about 75 min. Under prolonged visible light irradiation, the synthesized calcinated HAp degrades about 13% of MB from its aqueous solution in 9 h. The degradation ability of HAp could be enhanced by incorporating Au nanoparticles (Au-loading). The rate of decolorization was calculated as a function of the change in the absorption intensity at λmax (664 nm) of the dye. The decolorization efficiency η (%) was calculated as (eqn (3)):
| |
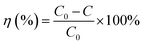 | (3) |
where,
C0 is the initial concentration of the dye and
C is the concentration after photo irradiation.
44 The calculated values of photocatalytic efficiency for the HAp and Au-loaded HAp samples are presented in
Table 2. As can be seen from
Table 2, the total MB removal (adsorption + photocatalytic degradation) efficiency of unmodified HAp nanoparticles (calcinated at 600 °C) is about 25%. It must be noted that previous studies on unmodified HAp
27 have reported no photocatalytic activity for dye degradation under visible light illumination; and the visible light photocatalytic activity in HAp could be induced only after calcinating at 1200 °C in air,
28 which introduces oxygen (V
O) or hydroxyl group (V
OH) vacancies in HAp lattice. However, our 600 °C calcinated HAp manifests significant photocatalytic activity for MB degradation under visible light.
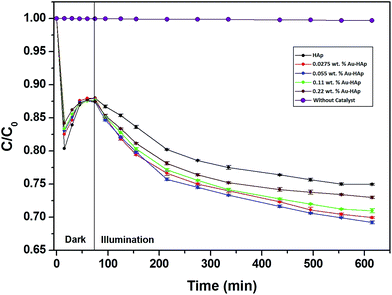 |
| | Fig. 9 Kinetics of MB photodegradation by HAp and Au–HAp nanocomposites. Photodegradation curve of MB solution (without catalyst) has been included to show its stability under visible irradiation. | |
Table 2 Estimated MB removal (adsorption and degradation) efficiencies HAp and Au–HAp photocatalysts at 20 °C and pH 7.2. The results of HAp calcinated at 1200 °C and 0.055 wt% Au–HAp prepared with it are also presented for comparison
| Catalyst |
Adsorption (A) % |
Photo-catalytic degradation (B) % |
Total removal (A + B) % |
| HAp |
11.99 |
13.01 |
25.0 |
| 0.0275 wt% Au–HAp |
12.07 |
17.98 |
30.05 |
| 0.055 wt% Au–HAp |
12.62 |
19.85 |
32.47 |
| 0.11 wt% Au–HAp |
12.30 |
16.71 |
29.01 |
| 0.22 wt% Au–HAp |
12.51 |
14.51 |
27.02 |
| HAp 1200 °C |
13.80 |
10.05 |
23.85 |
| 0.055 wt% Au–HAp (HAp 1200 °C) |
11.21 |
13.79 |
25.0 |
The photocatalytic efficiency of HAp nanoparticles increased significantly after Au-loading. As can be seen from Table 2, the total MB removal efficiency of the HAp nanoparticles increased to 32.47% on 0.055 wt% Au loading. However, on increasing the Au loading further, the MB removal efficiency of the Au-loaded HAp decreased gradually. The decrease of MB removal efficiency of the nanocomposites on higher Au loading might be due to the presence of Au nanoparticles in excess at their surface, which probably act as hole-trappers or recombination centers for photo generated electrons.
The Langmuir–Hinshelwood (L–H) model is the most commonly used kinetic expression to explain the kinetics of heterogeneous catalytic reactions.45 In this model, the reaction rate r can be expressed as (eqn (4)):
| |
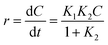 | (4) |
where,
K1 is the apparent rate constant,
K2 is the adsorption constant, and
C is the reactant concentration.
In this study, we used a first order kinetic model as the simplification of the L–H kinetic model, due to dilute concentration of MB in the reaction solution. A semi log data does not construct a single straight line and does not fit first order reaction model for the entire period of the reaction. A consequent sequence of first order reactions is often found to be appropriate for complex oxidation reactions as the degradation can be broken down in to a number of different dominant reaction steps, such as primary degradation of the reactant, followed by several secondary degradation steps corresponding to the final oxidation to stable product or classes of reaction intermediates.7 This kinetic modelling strategy is well accepted for both non-catalytic and heterogeneously catalyzed wet oxidation reactions,46 which also degrades compounds via a free radical oxidation mechanism.47
The L–H kinetic model can be simplified to a pseudo-first order kinetic equation48 for diluted solutions as (eqn (5) and (6)):
| |
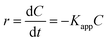 | (5) |
| |
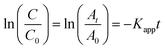 | (6) |
where
C is the concentration of MB at time
t,
C0 is the equilibrium concentration after adsorption and
Kapp is the apparent rate constant, which can be obtained from the decrease of the peak intensity at 664 nm with time. The ratio of absorbance
At of MB at time
t to
A0 measured at
t = 0 must be equal to the concentration ratio
C/
C0 of MB. From the plot of ln(
C/
C0)
vs. irradiation time (
Fig. 10) we could obtain a good linear correlation of the data points, obtaining two linear regressions associated to the pseudo first order rate constant,
Kapp (
k1 and
k2). In general, the first-order kinetics is appropriate for μg L
−1 or few mg L
−1 concentration ranges.
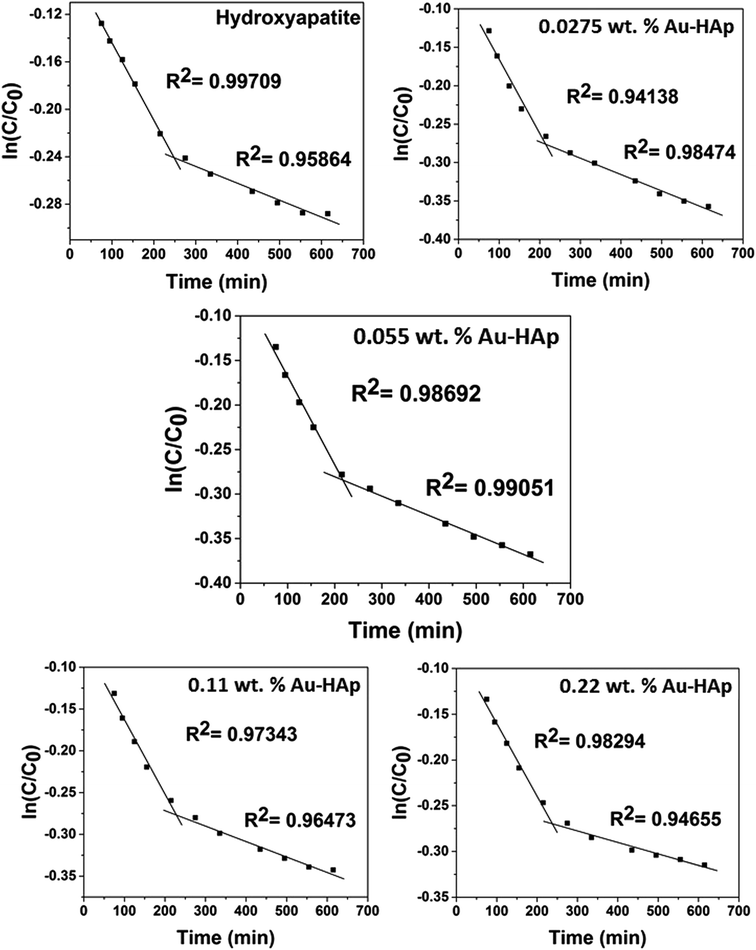 |
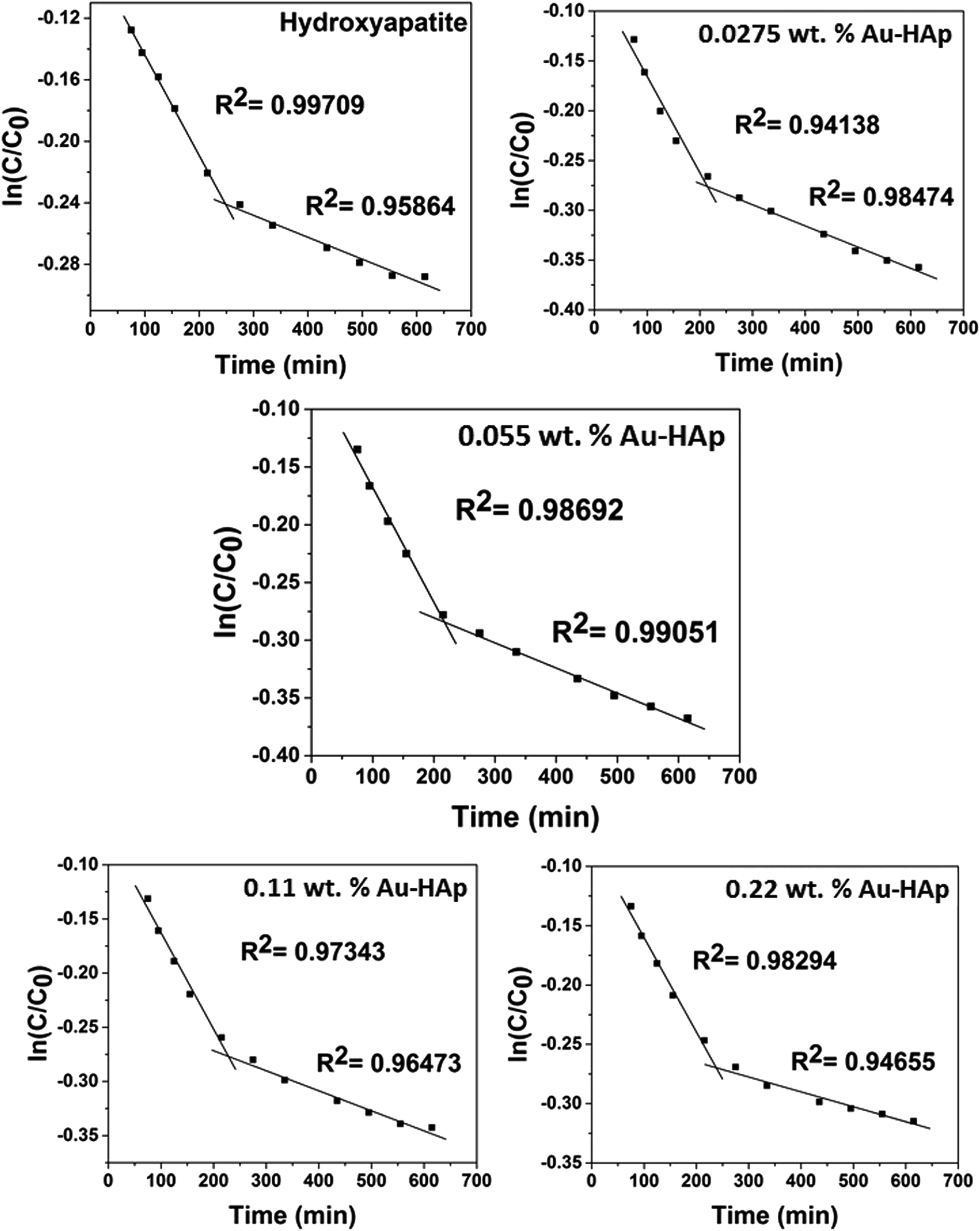
| | Fig. 10 Two stage first order plots for kinetic photodegradation of MB in presence of HAp and different wt% of [Au(x)–HAp, x = 0, 0.0275, 0.055, 0.11, 0.22 wt%] Au–HAp nanocomposites. | |
The kinetic analysis of data for the 600 °C calcinated HAp and Au–HAp photocatalysts is shown in Fig. 10. The degradation of MB and its derivatives (azo dye reaction intermediates) are well modelled by a two-step first order reaction. Such a two-step reaction process indicates that MB degrades first to form azo dye intermediates (which degrade further to smaller, highly oxidizable intermediates, which could be detected by UV-Vis absorption spectroscopy). These processes occur very fast, revealing only one linear fit (the first one) in the semi log plot of the kinetics presented in Fig. 10. Next, these azo dye intermediates probably degrade to form recalcitrant products such as Azure A, Azure B, Azure C and thionine26 resulting the second linear fit in the degradation kinetics.
The estimated first order reaction rate constants for the two first order kinetic regions are presented in Table 3. Variations of the rate constant (k1 and k2) values with increasing Au content in the nanocomposites indicate that 0.055 wt% Au loading is the optimum for visible light photodegradation of MB.
Table 3 Summary of two step first order reaction rate constants on liquid volume basis (k/min−1)
| Samples |
Reaction rate constant k1 (min−1) |
Reaction rate constant k2 (min−1) |
| HAp |
6.57 × 10−4 |
1.42 × 10−4 |
| 0.0275 wt% Au–HAp |
9.67 × 10−4 |
2.14 × 10−4 |
| 0.055 wt% Au–HAp |
9.96 × 10−4 |
2.18 × 10−4 |
| 0.11 wt% Au–HAp |
8.99 × 10−4 |
1.85 × 10−4 |
| 0.22 wt% Au–HAp |
7.94 × 10−4 |
1.28 × 10−4 |
3.7 Fourier transformed infrared (FTIR) spectroscopy
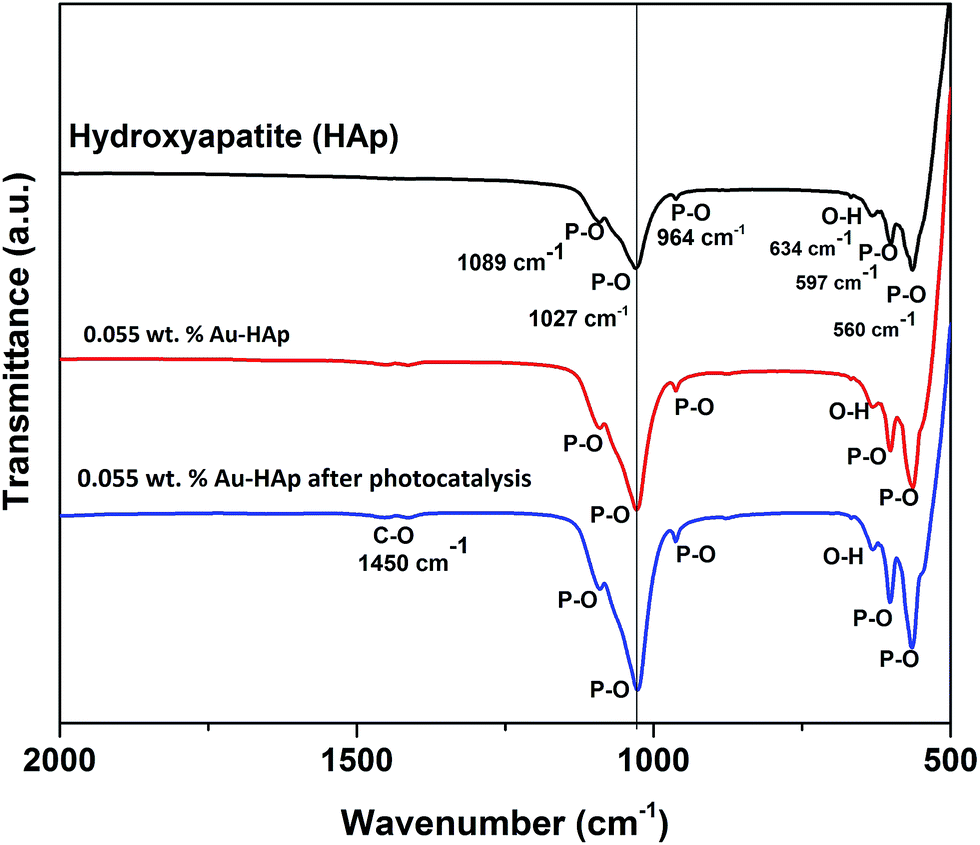
FTIR spectra of the 600 °C calcinated HAp and 0.055 wt% Au loaded HAp were recorded before and after the photocatalytic reaction to observe the possible changes in the catalysts after gold incorporation and due to the photocatalytic reactions (Fig. 11). The unmodified HAp before photocatalytic study revealed absorption peaks associated to PO43− group around 1089, 1027, 964, 597 and 560 cm−1. The spectrum also revealed a broad band centered in between 1000 and 1100 cm−1 associated to the P–O bond of phosphate group.49 The major peaks at 1089 and 1027 cm−1 could be identified as symmetric n3 vibration of PO4 group, which are the principal peaks among the phosphate vibration modes. The other two peaks appeared around 964 cm−1 and in 560–597 cm−1 range are due to n1 and n4 symmetric P–O stretching vibrations of the PO4 group, respectively.50 The distinguishable splitting of the n4 vibration mode of PO4 group is revealed as 560 and 597 cm−1 components. Appearance of these component peaks confirms the presence of more than one distinct site for the phosphate group in HAp lattice. The band assigned to the stretching mode of hydroxyl group (O–H) is observed at 634 cm−1.51
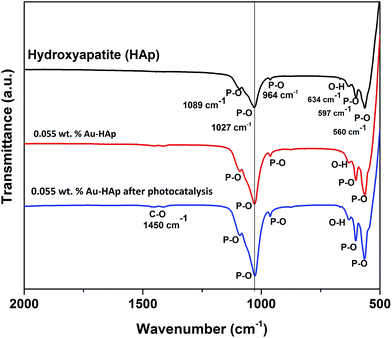 |
| | Fig. 11 FTIR spectra of HAp, 0.055 wt% Au–HAp nanocomposite before and after photocatalysis. | |
In general, the Au-loaded HAp revealed all the FTIR signals appeared for the unmodified HAp before and after their photocatalytic use. However, after photocatalytic use, the Au–HAp revealed a higher signal associated to C–O bond, which generally appear in all FTIR spectra due to ambiental CO2. Appearance of C–O stretching band in higher intensity for the used Au–HAp also indicates the oxidation of MB and generation of its byproducts during photocatalytic degradation.
4. Mechanism of MB degradation
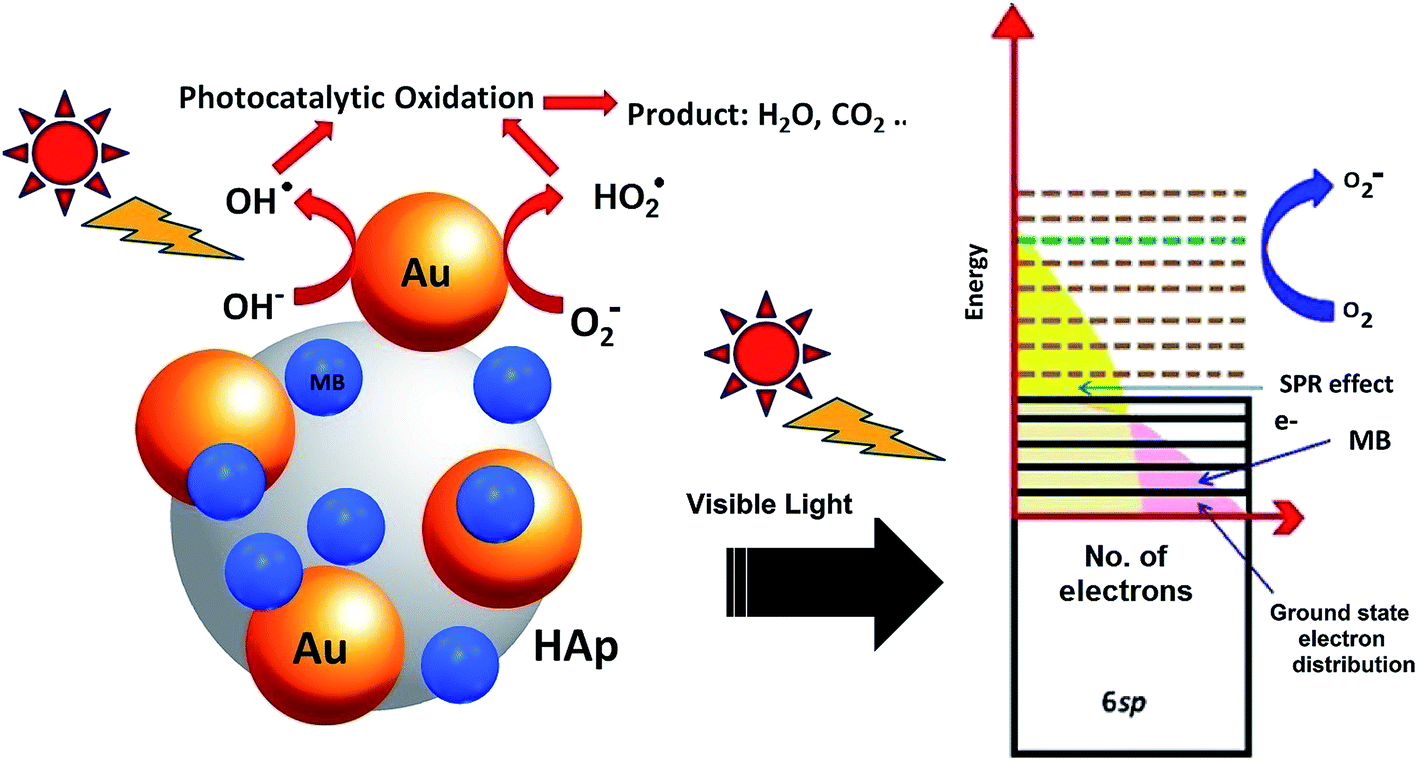
Molecular oxygen acts as oxidant in photocatalytic reactions. A complete oxidation (degradation) of organic compound involves electron transfer from the organic molecules to oxygen. In the photocatalytic reactions of Au-supported HAp nanoparticles, the former (Au nanoparticles) seems to act as initializer and mediator of electron transfer for the oxidation reactions. The strong absorption band appeared around 530 nm in UV-Vis spectra of the Au–HAp nanocomposites (Fig. 3) is the SPR band of the formed Au nanoparticles, originating from the intraband excitation of 6sp electrons39 (Fig. 12). Although all the metallic nanoparticles posse an inherent positive surface charge, the illumination of visible light results in positive charges in gold's 6sp band, which can capture electrons from organic dye molecules to oxidize them. Oxygen molecules on the Au nanoparticles or at HAp (support) – Au nanoparticle interface seize the energetic electrons of the excited energy levels of gold's 6sp band, forming O2˙ species (eqn (7)). The formed O2˙ specie then react with H+ ions generated from water splitting to produce active species like HO2˙, OH˙ (eqn (8)) and other reactive oxygen species such as H2O2 (eqn (9) and (10))39,41 which degrade the MB dye first to azo dye intermediates and then to smaller, highly oxidizable intermediates, and finally to the probable recalcitrant products (eqn (11)). On the other hand, it has been suggested that the HAp supported Au nanoparticles can attract electrons from the organic molecules on the nanoparticles which also helps to catalyze the organic pollutants.52 The possible mechanism of MB degradation by Au supported HAp considered in this study has been illustrated in Fig. 12.| | |
O2˙ + H+ ⇒ HO2˙ + OH˙
| (8) |
| | |
H2O2 + e− ⇒ OH˙ + OH−
| (10) |
| | |
MB + HO2˙ + OH˙ + O2˙ ⇒ MB degradation (CO2 + H2O + NO3 + Cl− + SO2−4)
| (11) |
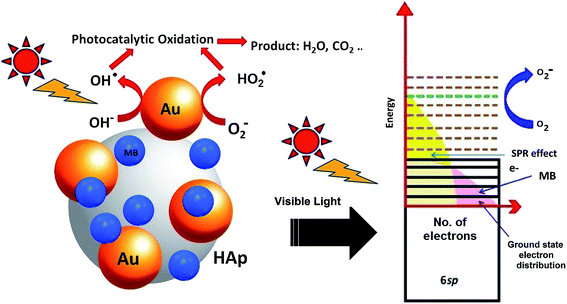 |
| | Fig. 12 Schematic presentation of the photocatalytic degradation process induced by Au–HAp nanocomposites under visible light. | |
It must be noted that previous studies have reported a visible light photocatalytic activity of 1200 °C calcinated HAp induced by oxygen (VO) and/or hydroxyl group (VOH) vacancies generated by thermal annealing.27 However, our 600 °C calcinated HAp manifests a significant photocatalytic activity for MB degradation under visible light, which can also be due to the generation of VO in HAp. On the other hand, by calcinating HAp at 1200 °C, we could not observe any significant enhancement of its photocatalytic activity (Table 2). Rather its photocatalytic activity was a bit lower than the HAp sample calcinated at 600 °C, probably due to the reduction of specific surface area on high temperature calcination. However, a notable enhancement of visible light photocatalytic activity of this HAp sample could be observed after Au loading, which supports the degradation mechanism of organic dye we presented above.
As can be observed from Table 2, a 0.055 wt% Au loading in 1200 °C calcinated HAp resulted a relatively lower percentage (25.0% in 9 h) of MB removal from water in comparison to the 600 °C calcinated HAp (32.47% in 9 h) loaded with 0.055 wt% Au. Such a lower MB removal efficiency of the 1200 °C calcinated Au-loaded HAp might be due to the generation of VOH in its lattice, which generates donor type defect levels in its band gap.28 Those shallow energy levels below the conduction band of HAp probably act as electron trappers for Au–HAp under visible illumination.
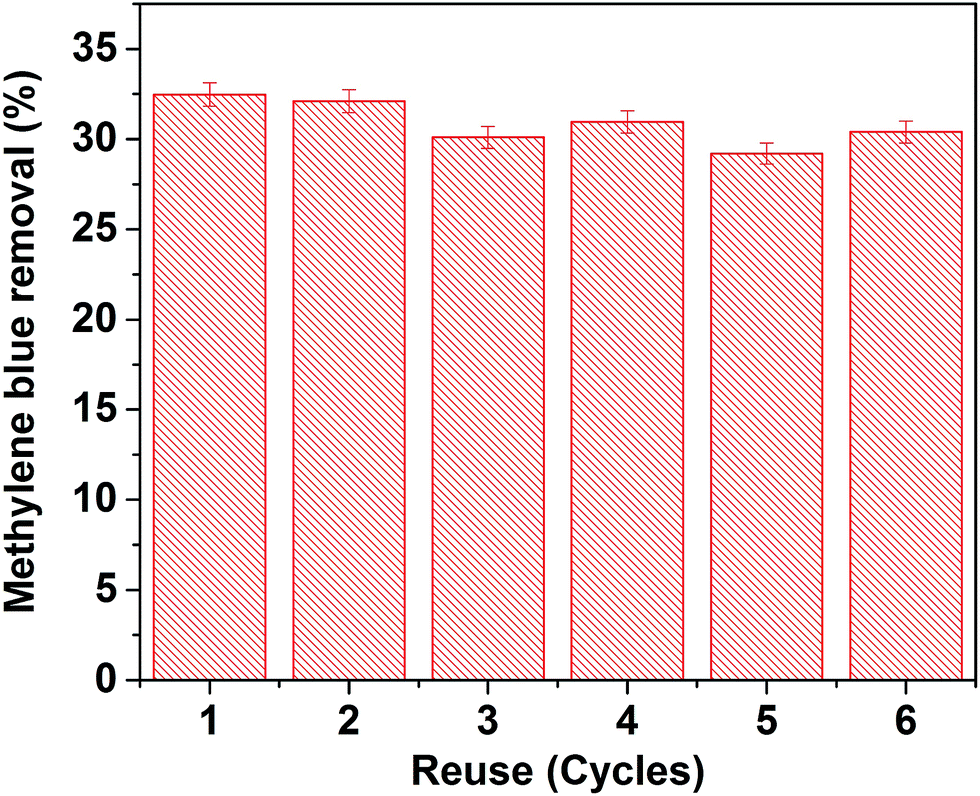
4.1 Reusability performance of Au–HAp catalyst
To verify the reusability of our Au-loaded HAp photocatalysts, repeated photocatalytic tests were performed over the 0.055 wt% Au–HAp nanocomposite, which revealed best MB degradation performance. After each of the photocatalytic cycles, the catalyst was separated from the reaction mixture by decantation, washed with hot (90 °C) DI water H2O2 (5 vol%) mixture and dried at room temperature. The results presented in Fig. 13 demonstrate the photocatalytic performance of the nanocomposite does not change significantly even after 6 cycles of reuse.
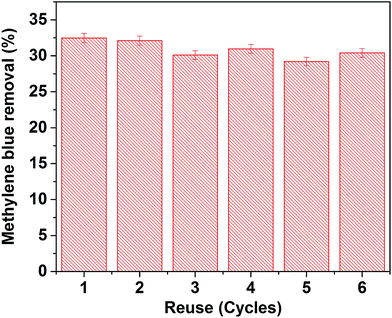 |
| | Fig. 13 Results of reusability tests of the 0.055 wt% Au–HAp nanocomposite for photocatalytic degradation of MB. | |
5. Conclusions
In this study, gold nanoparticles were incorporated in different concentrations at the surface of HAp nanoparticles to fabricate plasmonic nanocomposites, useful for organic dye removal from wastewater. In general, the incorporation of Au nanoparticles at HAp surface increases its specific surface area. Average pore size of the nanocomposite increases up to a certain wt% of Au loading (0.0275%) due to the assembly of Au nanoparticles at their surfaces, and then decreases due to the incorporation of tiny Au nanoparticles at inter-particle spaces. Fabricated nanocomposites show a significant increase of MB removal efficiency under visible light illumination (up to 32.47% in 9 h) when the Au nanoparticles are loaded over 600 °C air calcinated HAp nanoparticles. The enhanced MB removal activity of Au-loaded HAp nanocomposites is ascribed to the generation of energetic electrons form the 6sp orbital of Au through intraband excitation upon visible light illumination. The demonstrated visible light photocatalytic activity of Au-loaded HAp nanoparticles at neutral pH (pH = 7.2) and room temperature (20 °C) in aqueous phase opens up the possibility of utilizing synthetic HAp, which is a nontoxic, biocompatible, eco-friendly ceramic for environmental applications.
Acknowledgements
SM is thankful to Instituto de Física, BUAP, and PRODEP-SEP, Mexico for a postdoctoral fellowship (Of. No. DSA/103.5/15/8164). The work was supported by VIEP-BUAP (Grant # VIEP/EXC-2017) and DITCo-BUAP (Grant # DITCo/2016-13), Mexico.
References
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- G. K. Castello, Handbook of Photocatalysts: Preparation, Structure and Applications, Nova Science Pub. Incorporated. 2009 Search PubMed.
- A. N. Soon and B. H. Hameed, Desalination, 2011, 269, 1–16 CrossRef CAS.
- D. Y. C. Leung, X. Fu, D. Ye and H. Huang, Kinet. Catal., 2012, 53, 239–246 CrossRef CAS.
- I. Boiarkina, S. Pedron and D. A. Patterson, Appl. Catal., B, 2011, 110, 14–24 CrossRef CAS.
- S. Polisetti, P. A. Deshpande and G. Madras, Ind. Eng. Chem. Res., 2011, 50, 12915–12924 CrossRef CAS.
- A. M. Ali, E. A. C. Emanuelsson and D. A. Patterson, Appl. Catal., B, 2011, 106, 323–336 CrossRef CAS.
- C. Li, Y. Shen, M. Jia, S. Sheng, M. O. Adebajo and H. Zhu, Catal. Commun., 2008, 9, 355–361 CrossRef CAS.
- Y. C. Hong, K. Q. Sun, K. H. Han, G. Liu and B. Q. Xu, Catal. Today, 2010, 158, 415–422 CrossRef CAS.
- B. Liu, C. Li, Y. Zhang, Y. Liu, W. Hu, Q. Wang, L. Han and J. Zhang, Appl. Catal., B, 2012, 111–112, 467–475 CrossRef CAS.
- B. B. Chen, X. B. Zhu, M. Crocker, Y. Wang and C. Shi, Catal. Commun., 2013, 42, 93–97 CrossRef CAS.
- Y. Wang, B. B. Chen, M. Crocker, Y. J. Zhang, X. B. Zhu and C. Shi, Catal. Commun., 2015, 59, 195–200 CrossRef CAS.
- D. W. Goodman, Catal. Lett., 2005, 99, 1–4 CrossRef CAS.
- S. S. Syamchand and G. Sony, Microchim. Acta, 2015, 182, 1567–1589 CrossRef CAS.
- O. Kaygili, S. Keser, T. Ates, C. Tatar and F. Yakuphanoglu, Mater. Sci. Eng., C, 2015, 46, 118–124 CrossRef CAS PubMed.
- L. J. Cummings, M. A. Snyder and K. Brisack, Methods Enzymol., 2009, 463, 387–404 CAS.
- K. Zhao, B. Qiao, J. Wang, Y. Zhang and T. Zhang, Chem. Commun., 2011, 47, 1779–1781 RSC.
- M. Khachani, M. Kacimi, A. Ensuque, J. Y. Piquemal, C. Connan, F. B. Verduraz and M. Ziyad, Appl. Catal., A, 2010, 388, 113–123 CrossRef CAS.
- A. Solhy, R. Tahir, S. Sebti, R. Skouta, M. Bousmina, M. Zahouily and M. Larzek, Appl. Catal., A, 2010, 374, 189–193 CrossRef CAS.
- J. H. Jun, T. J. Lee, T. H. Lim, S. W. Nam, S. A. Hong and K. J. Yoon, J. Catal., 2004, 221, 178–190 CrossRef CAS.
- Y. Zhang, Z. Li, W. Sun and C. Xia, Catal. Commun., 2008, 10, 237–242 CrossRef CAS.
- M. I. Domínguez, F. Romero-Sarria, M. A. Centeno and J. A. Odriozola, Appl. Catal., B, 2009, 87, 245–251 CrossRef.
- A. Hu, M. Li, C. Chang and D. Mao, J. Mol. Catal. A: Chem., 2007, 267, 79–85 CrossRef CAS.
- S. Ji, S. Murakami, M. Kamitakahara and K. Ioku, Mater. Res. Bull., 2009, 44, 768–774 CrossRef CAS.
- G. Sheng, L. Qiao and Y. Mou, J. Environ. Eng., 2011, 137, 611–616 CrossRef CAS.
- J. H. Shariffuddin, M. I. Jones and D. A. Patterson, Chem. Eng. Res. Des., 2013, 91, 1693–1704 CrossRef CAS.
- M. Tsukada, M. Wakamura, N. Yoshida and T. Watanabe, J. Mol. Catal. A: Chem., 2011, 338, 18–23 CAS.
- V. S. Bystrov, C. Piccirillo, D. M. Tobaldi, P. M. L. Castro, J. Coutinho, S. Kopyl and R. C. Pullar, Appl. Catal., B, 2016, 196, 100–107 CrossRef CAS.
- H. Nishikawa, Mater. Lett., 2004, 58, 14–16 CrossRef CAS.
- H. Nishikawa, J. Mol. Catal. A: Chem., 2003, 206, 299–311 CrossRef.
- A. Mitsionis, T. Vaimakis, C. Trapalis, N. Todorova, D. Bahnemann and R. Dillert, Appl. Catal., B, 2011, 106, 398–404 CrossRef CAS.
- Y. Liu, C. Y. Liu, J. H. Wei, R. Xiong, C. X. Pan and J. Shi, Appl. Surf. Sci., 2010, 256, 6390–6394 CrossRef CAS.
- X. Hong, X. Wu, Q. Zhang, M. Xiao, G. Yang, M. Qiu and G. Han, Appl. Surf. Sci., 2012, 258, 4801–4805 CrossRef CAS.
- M. Vukomanovic, V. Zunic, M. Otonicar, U. Repnik, B. Turk, S. D. Skapin and D. Suvorov, J. Mater. Chem., 2012, 22, 10571–10580 RSC.
- M. Nishikawa, L. H. Tan, Y. Nakabayashi, T. Hasegawa, W. Shiroishi, S. Kawahara, N. Saito, A. Nosaka and Y. Nosaka, J. Photochem. Photobiol., A, 2015, 311, 30–34 CrossRef CAS.
- Q. Chang, K. K. Li, S. L. Hu, Y. G. Dong and J. L. Yang, Mater. Lett., 2016, 175, 44–47 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- K. Yamada, K. Miyajima and F. Mafuné, J. Phys. Chem. C, 2007, 111, 11246–11251 CAS.
- H. Zhu, X. Chen, Z. Zheng, X. Ke, E. Jaatinen, J. Zhao, C. Guo, T. Xie and D. Wang, Chem. Commun., 2009, 7524–7526 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- N. M. Flores, U. Pal, R. Galeazzi and A. Sandoval, RSC Adv., 2014, 4, 41099–41110 RSC.
- M. A. Rauf, M. A. Meetani, A. Khaleel and A. Ahmed, Chem. Eng. J., 2010, 157, 373–378 CrossRef CAS.
- F. Huang, L. Chen, H. Wang and Z. Yan, Chem. Eng. J., 2010, 162, 250–256 CrossRef CAS.
- Ma. De L. Ruiz Peralta, U. Pal and R. Sanchez Zeferino, ACS Appl. Mater. Interfaces, 2012, 4, 4807–4816 CAS.
- T. López, J. Hernández-Ventura, R. Goómez, F. Tzompantzi, E. Sánchez, X. Bokhimi and A. García, J. Mol. Catal. A: Chem., 2001, 167, 101–107 CrossRef.
- B. Belkacemi, F. Larachi and A. Sayari, J. Catal., 2000, 193, 224–237 CrossRef.
- D. A. Patterson, I. S. Metcalfe, F. Xiong and A. G. Livingston, Ind. Eng. Chem. Res., 2001, 40, 5517–5525 CrossRef CAS.
- Y. Ghayeb and M. M. Momeni, J. Mater. Sci.: Mater. Electron., 2015, 26, 5335–5341 CrossRef CAS.
- S. Mondal, S. Mahata, S. Kundu and B. Mondal, Adv. Appl. Ceram., 2010, 109, 234–239 CrossRef CAS.
- S. Mondal, U. Pal and A. Dey, Ceram. Int., 2016, 42, 18338–18346 CrossRef CAS.
- I. A. Rahman, S. M. Masudi, N. Luddin and R. A. Seikh, Bull. Mater. Sci., 2014, 37, 213–219 CrossRef.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28640b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*