
Cobalt oxide and N-doped carbon nanosheets derived from a single two-dimensional metal–organic framework precursor and their application in flexible asymmetric supercapacitors†
Cao
Guan
 *a,
Wei
Zhao
b,
Yating
Hu
a,
Zhuangchai
Lai
b,
Xin
Li
a,
Shijing
Sun
c,
Hua
Zhang
b,
Anthony K.
Cheetham
*ac and
John
Wang
*a
*a,
Wei
Zhao
b,
Yating
Hu
a,
Zhuangchai
Lai
b,
Xin
Li
a,
Shijing
Sun
c,
Hua
Zhang
b,
Anthony K.
Cheetham
*ac and
John
Wang
*a
aDepartment of Materials Science and Engineering, National University of Singapore, 117574 Singapore. E-mail: msegc@nus.edu.sg; msewangj@nus.edu.sg
bSchool of Materials Science and Engineering, Nanyang Technological University, 639798 Singapore
cDepartment of Materials Science and Metallurgy, University of Cambridge, Cambridge, CB2 3QZ, UK. E-mail: akc30@cam.ac.uk
First published on 17th January 2017
Abstract
Based on a new “one for two” strategy, a single two-dimensional (2D) metal–organic framework (MOF) precursor has been transformed into both electrodes (i.e., a Co3O4 cathode and a N-doped carbon anode) for a flexible asymmetric supercapacitor. The device demonstrated not only highly robust mechanical flexibility but also outstanding electrochemical performance. The “one for two” concept can significantly ease the fabrication process and has great potential to be extended to other functional materials for different applications.
Conceptual insightsSupercapacitors with high energy density and flexibility are highly desired for the development of advanced flexible electronics. Most of the previous reports on asymmetric supercapacitors need two different precursors for the respective anode and cathode, thus the fabrication process is rather complicated. Herein, an effective “one for two” strategy is used to construct the “two” nanoporous electrodes of a flexible asymmetric supercapacitor from just “one” precursor. To specify, both electrode materials, i.e. a porous cobalt oxide nanosheets cathode (CC@Co3O4) and a porous nitrogen-doped carbon nanosheets anode (CC@NC) can be transformed from a single 2D cobalt MOF. Due to the advantages of the MOF-derived nanoporous electrode materials, and their direct contact with the flexible substrates, the asymmetric supercapacitor expressed high specific energy densities with excellent mechanical stability. The “one for two” strategy can greatly ease the fabrication process of a full device and can be extended to design other advanced integrated devices. |
MOFs represent a new class of hybrid nanoporous materials with crystalline architectures, highly tunable porosities and different functionalities for a wide variety of applications.1–7 Recently, MOFs have been utilized for electrical energy storage (EES) devices, taking the advantage of their intrinsically large surface areas with sufficient reaction sites for redox reactions.8–10 They can be directly used as electrode materials for lithium-ion batteries and supercapacitors,11 and through proper post-synthesis treatment, MOFs can also be readily transformed to porous carbons, metal oxides, and metal sulfides as active materials for energy storage.12–20 In particular, two-dimensional nanostructured MOFs have attracted considerable scientific and technological interest, because of their low dimensionality with unique physical and chemical properties.21–26 However, most of the previous research work on MOF-based materials has focused only on one of the electrodes (e.g. anode for lithium ion batteries or cathode for supercapacitors), thus to assemble a full cell additional efforts are needed on a second material for the other electrode. Furthermore, most of the MOF or MOF-derived active materials are in powdered form, where the usage of polymer binders will increase the “dead mass” for the electrodes and limit the gravimetric/volumetric capacity of the full device.27–31 Last but not least, with the rapid development of portable and flexible electronics, such as roll-up displays and curved smartphones, it is of considerable value to explore flexible and environmental friendly EES devices.32–35
For EES devices, batteries and electrochemical capacitors are playing dominant roles.36 Compared with lithium-ion batteries, supercapacitors can offer much higher power densities (∼10 kW kg−1) with shorter charging times (within 1 min), and the cycle life (>100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) is also much longer.37 However, the energy densities of supercapacitors are normally low (∼15 W h kg−1 to 35 W h kg−1), which limits their potentially wide applications in areas such as hybrid electric vehicles and portable electronics.38 To address this issue, one direction for high-energy-density supercapacitors is to develop flexible binder-free electrodes, which can avoid the “dead” mass of binders and provide direct mechanical and electrical contacts between the active materials and substrates.39–44 In addition, constructing hybrid supercapacitors is also a promising way to achieve high energy densities, as the voltage window can be significantly expanded.45,46 Generally, in an asymmetric supercapacitor, the two electrodes are quite different, thus it would be intriguing and also promising if the two electrodes could be derived from a single-source precursor. In this way, the two electrodes will not only share similar properties (e.g. porosity and flexibility), but also offer possible synergetic effects with different physiochemical properties. The “one for two” and “one for all” strategy can also simplify the fabrication process to reduce the cost, and thus has great potential for the development of various functional materials for different integrated devices. Based on the above discussion, it would be of considerable interest to develop different functional materials from a single MOF precursor and assemble them into a well-integrated device.
000 cycles) is also much longer.37 However, the energy densities of supercapacitors are normally low (∼15 W h kg−1 to 35 W h kg−1), which limits their potentially wide applications in areas such as hybrid electric vehicles and portable electronics.38 To address this issue, one direction for high-energy-density supercapacitors is to develop flexible binder-free electrodes, which can avoid the “dead” mass of binders and provide direct mechanical and electrical contacts between the active materials and substrates.39–44 In addition, constructing hybrid supercapacitors is also a promising way to achieve high energy densities, as the voltage window can be significantly expanded.45,46 Generally, in an asymmetric supercapacitor, the two electrodes are quite different, thus it would be intriguing and also promising if the two electrodes could be derived from a single-source precursor. In this way, the two electrodes will not only share similar properties (e.g. porosity and flexibility), but also offer possible synergetic effects with different physiochemical properties. The “one for two” and “one for all” strategy can also simplify the fabrication process to reduce the cost, and thus has great potential for the development of various functional materials for different integrated devices. Based on the above discussion, it would be of considerable interest to develop different functional materials from a single MOF precursor and assemble them into a well-integrated device.
In this work, we develop a facile approach to directly grow 2D MOFs on flexible substrates; through different thermal treatments they can then be transformed to metal oxides and carbon, suitable for the cathode and anode of an asymmetric supercapacitor, respectively. As a proof-of-concept demonstration of the “one for two” strategy, a solid-state flexible supercapacitor is assembled based on the single MOF precursor-derived cathode and anode, and excellent electrochemical performance, such as high energy density and high power density, is achieved. The solid-state supercapacitor also demonstrates good mechanical stability with long-time cycling stability. The proposed “one for two” strategy for the development of porous 2D metal oxides and carbon can be extended to other MOF-based functional materials for different integrated devices.
The fabrication process of the electrode materials and the architecture of the flexible asymmetric supercapacitor are schematically illustrated in Fig. 1. Firstly, though a facile reaction between Co2+ and 2-methylimidazole (MIM, C4H6N2) in aqueous solution, a 2D sheet-like Co-MOF was directly grown on the surface of carbon macro-fiber of the carbon cloth (noted as CC@Co-MOF). X-ray powder diffraction data, analyzed using Pawley refinement with the Topas program (Fig. S1, ESI†), showed that the Co-MOF has the same 2D layered crystal structure as its known zinc analogue.16,47 (Pawley refinement details are in the ESI.†) Then, through different thermal treatment processes, the Co-MOF precursor can be either oxidized to Co3O4 nanosheets (CC@Co3O4) or transformed to N-doped porous carbon nanosheets (CC@NC) (see Experimental in ESI† for details). In a final step, a flexible asymmetric supercapacitor is assembled using the CC@Co3O4 cathode and the CC@NC anode with a poly(vinyl alcohol) (PVA)–KOH gel-electrolyte. Note that the MOF coating is very uniform and that the facile fabrication process can thus be easily scaled up on different substrates such as FTO, graphite foam, Ti foil, and nickel foam (refer to Fig. S2 and S3, ESI†), here we just focus on the carbon cloth substrate as a demonstration of principle.
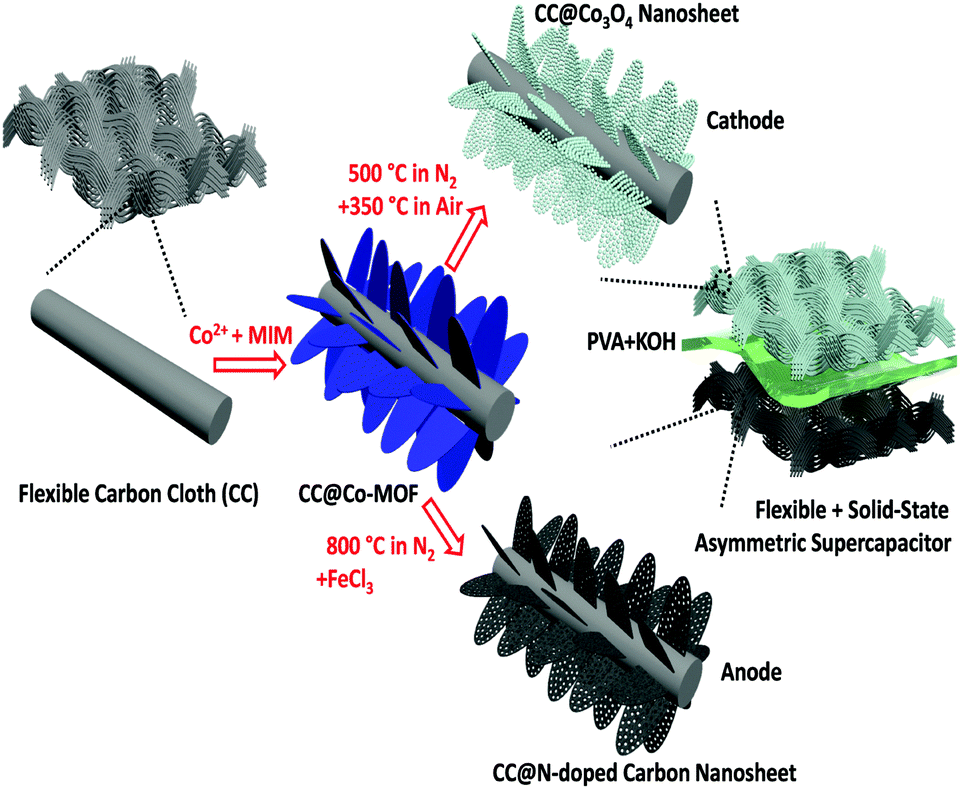 | ||
| Fig. 1 Schematic illustration of the “one for two” fabrication process: 2D Co3O4 nanosheets cathode and N-doped carbon nanosheets anode are obtained from the same Co-MOF precursor and assembled into a flexible asymmetric supercapacitor. | ||
The scanning electron microscopy (SEM) image in Fig. S4 (ESI†) shows that Co-MOFs with a typical 2D morphology (length ∼1.5 μm and thickness of ∼100–150 nm) have been uniformly grown on the carbon cloth. To transform the Co-MOF into Co3O4, the CC@Co-MOF was stabilized in N2 and then oxidized in air. Digital images and large-scale SEM images of CC@Co3O4 in Fig. S5 (ESI†) shows the uniform coverage of Co3O4 after thermal treatment. The MOF precursor will decompose and react with O2 releasing gas molecules such as NH3, H2O and CO2 during the annealing process, thus the resultant Co3O4 nanosheets become much thinner compared with the precursor (Fig. 2a), yet the 2D vertical aligned structure can be largely kept with no apparent collapse. Although the Co3O4 nanosheets are slightly tilted, the vertical alignment with cross-linked features was well retained, which could facilitate the electrolyte penetration. The enlarged SEM image in Fig. 2b and the transmission electron microscopy (TEM) image in Fig. 2c clearly show that the 2D Co3O4 nanosheets are highly porous and consist of many tightly interconnected nanoparticles (less than 10 nm). A top view of 2D Co3O4 indicates a typical thickness of the nanoflakes of ∼50 nm (Fig. S5f, ESI†). The porous structure of the Co3O4 nanosheets is believed to provide high electrode–electrolyte contact, reduced ion diffusion length, and effectively buffer the volume change during electrochemical reactions.48 The selected area electron diffraction (SAED) pattern (Fig. 2d) reveals the cubic crystalline structure of the Co3O4 nanosheets (JCPDS # 42-1467), and the HRTEM image in Fig. 2e shows that the spacings of the lattice fringes (0.244 nm and 0.286 nm) match well with the d-spacings of the (311) and (220) planes of Co3O4.49 The chemical composition of the Co3O4 nanosheets is further illustrated by the high-angle annular dark-field (HAADF)-scanning transmission electron microscopy (STEM) image (Fig. 2f) with the corresponding elemental mapping images (Fig. 2g and h). From the mapping result, the elements of Co and O are uniformly distributed, further confirming the successful formation of porous Co3O4 nanosheets.
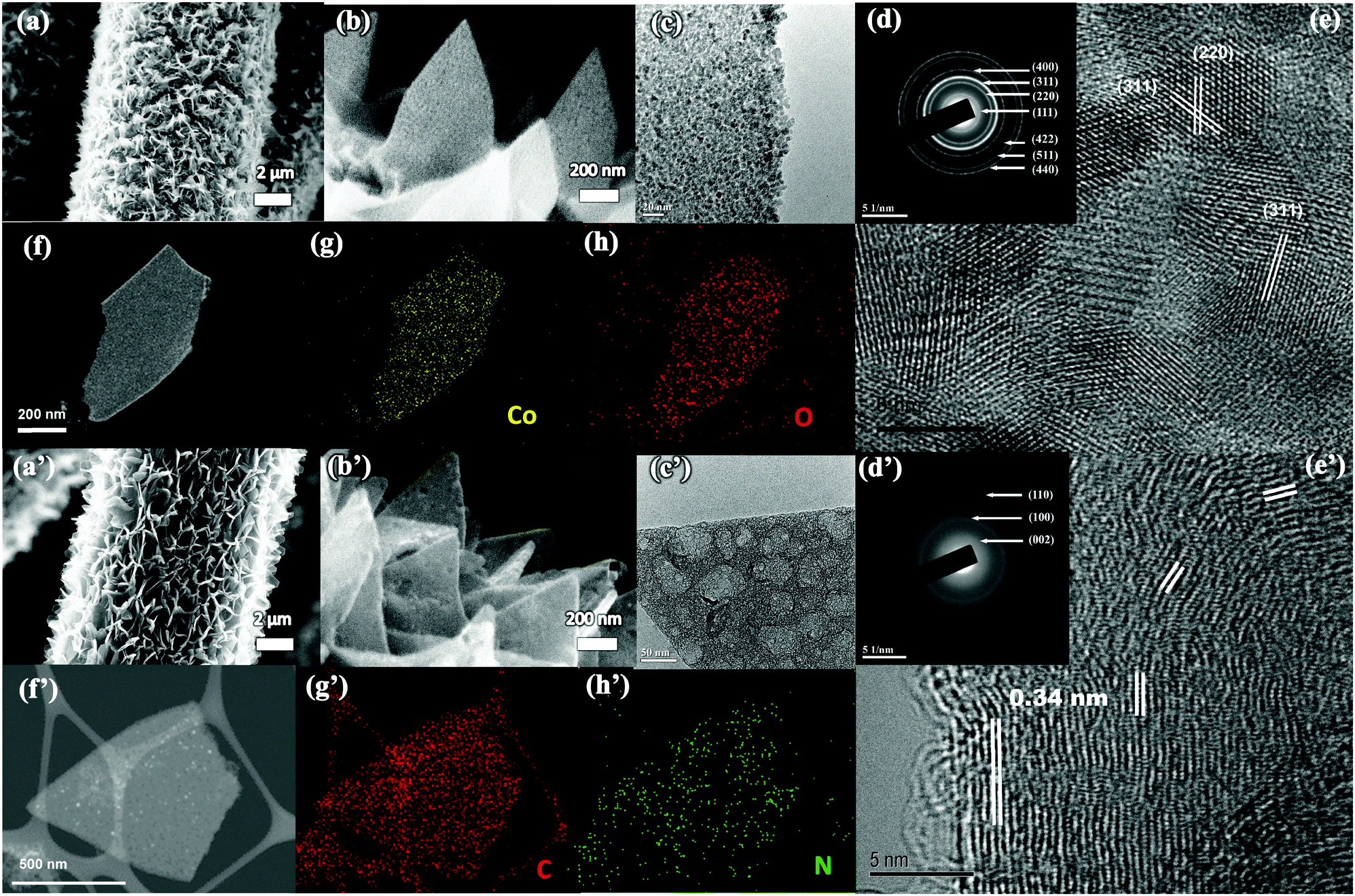 | ||
| Fig. 2 (a and b) SEM images, (c) TEM image, (d) SAED pattern, (e) HRTEM image, (f) HAADF-STEM image, (g and h) EDX mapping result of Co3O4 nanosheets deposited on CC. (a′ and b′) SEM images, (c′) TEM image, (d′) SAED pattern, (e) HRTEM image, (f′) HAADF-STEM image, (g′ and h′) EDX mapping result of porous N-doped carbon nanosheets deposited on CC. | ||
The Co-MOFs can be also easily transformed to N-doped carbon nanosheets through N2 thermal treatment with the following FeCl3 etching process. As shown in Fig. 2a′ and b′, after the transformation, the N-doped carbon nanosheets maintained uniform coverage on the carbon cloth, and the thickness (∼70 nm) of the carbon nanosheets is a little larger than that of the Co3O4 (Fig. S5g, ESI†). Fig. 2c′ shows a representative TEM image of a porous carbon nanosheet, in which both meso- and macro-pores can be observed. The porous feather is believed to facilitate the electrode–electrolyte contact.50 The SAED pattern in Fig. 2d′ reveals the graphitic feature of the carbon nanosheets, and the lattice spacing in Fig. 2e′ can be assigned to the (200) plane of graphitic carbon.51–53 The HAADF-STEM image (Fig. 2f′) with the corresponding elements (N and C) mapping results in Fig. 2g′ and h′ also confirm the uniform distribution of N and C, and proves the formation of N-doped carbon nanosheets. Note that the N2 thermal treatment of the Co-MOF results in a Co/carbon composite (refer to TEM images in Fig. S6, ESI†), and in the current work we use FeCl3 aqueous solution (instead of hazardous HF) to successfully etch away most of the Co.
XRD, Raman, and X-ray photoelectron spectroscopy (XPS) have been carried out to investigate the compositions of CC@Co3O4 and CC@NC. As shown in Fig. 3a, excluding the peaks from the carbon cloth substrate (CC), all the other diffraction peaks obtained from CC@Co3O4 can be well indexed as Co3O4 (JCPDS # 42-1467).49,54 From the Raman spectrum in Fig. 3b, in addition to the D band and G band signals from the carbon support, the Raman bands located at 478, 519, 616 and 685 cm−1 can be assigned to the Eg, F12g, F22g, and A1g modes of Co3O4, respectively. Fig. 3c shows the Co 2p XPS spectrum collected from CC@Co3O4, in which the two main peaks located at ∼780.5 and 795.9 eV, each accompanied by a shake-up satellite peak with low intensity, can be attributed to Co 2p3/2 and Co 2p1/2 of pure Co3O4, respectively.55–58 From the XPS result in Fig. 3d, the signals of pyridinic-N, graphitic-N, and terminal N–O bonding further confirms the nitrogen doping of the carbon nanosheets, which is in accordance with previous reports on ZIF derived N-doped carbon.59,60 The wide survey XPS spectra of CC@Co3O4 and CC@NC can be found in Fig. S7 (ESI†).
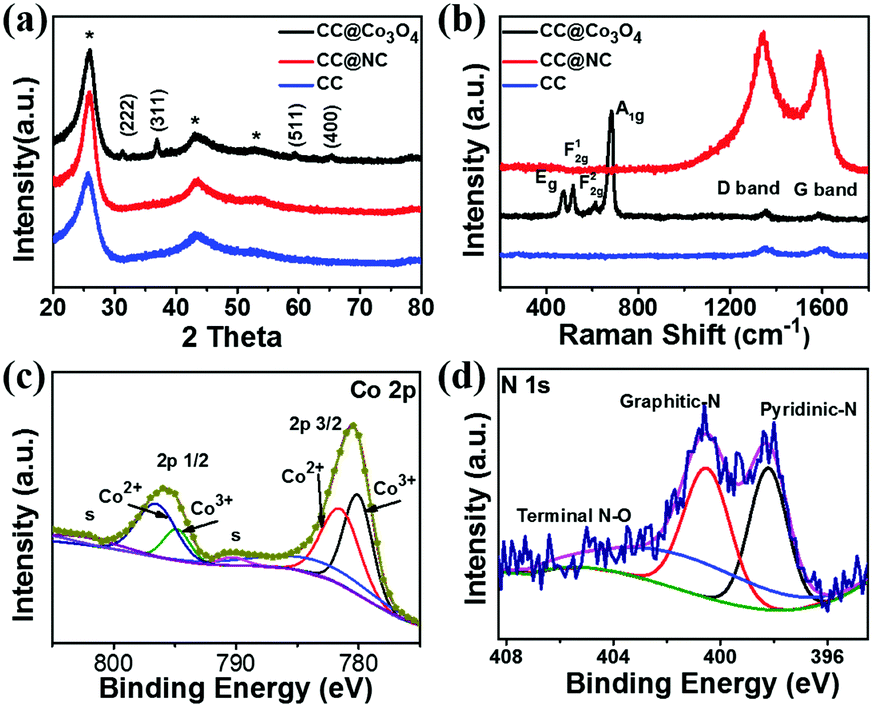 | ||
| Fig. 3 (a) XRD and (b) Raman results of CC, CC@Co3O4 and CC@NC. XPS spectra of (c) Co 2p in CC@Co3O4 and (d) N 1s in CC@NC. | ||
Based on the above discussion, a single 2D MOF precursor can be successfully transformed to both porous Co3O4 nanosheets and nitrogen-doped porous carbon nanosheets, which are promising electrode materials for supercapacitors. To evaluate the electrochemical performance of the CC@Co3O4 cathode and CC@NC anode as an asymmetric supercapacitor, electrochemical tests were first conducted using a three-electrode system in KOH aqueous electrolyte, with Pt as the counter electrode and Hg/HgO as the reference electrode. Fig. 4a compares the cyclic voltammetry (CV) curves of the CC@Co3O4 at different scan rates from 10 to 100 mV s−1. One pair of apparent oxidation (at ∼0.55 V) and reduction (at ∼0.45 V) peaks can be observed, suggesting reversible Faradaic reaction between Co2+ and Co3+ associated with OH−.61–63 Note that the symmetric redox peaks do not shift much when the scan rate increases from 10 to 100 mV S−1, showing that the electrode has good reaction kinetics. Noting the voltage window at 100 mV s−1 is enlarged a little to fully express the oxidation peak. The reversible reaction of CC@Co3O4 is further confirmed by the galvanostatic charge–discharge (GCD) curves in Fig. S8a (ESI†), in which a charge plateau at ∼0.51 V and a discharge plateau at ∼0.47 V can be observed, reflecting a small voltage hysteresis of only ∼0.04 V. Based on the GCD curves, the capacitances at different current densities are readily determined. It is found that the CC@Co3O4 shows the highest specific capacity of ∼900 F g−1 (corresponding to ∼0.225 mF cm−2) at 10 A g−1, and 58.7% of this value can be maintained when the current density is increased 16 times (Fig. S8b, ESI†), demonstrating the excellent rate performance. Electrochemical impedance spectroscopy (EIS) in Fig. S8c (ESI†) further demonstrates a small charge transfer resistance (∼0.3 Ω) of the electrode. The high capacity and excellent rate performance of the CC@Co3O4 electrode are due to the highly porous Co3O4 nanosheets with ultrathin thickness and their direct electrical contact with the conductive carbon cloth support.
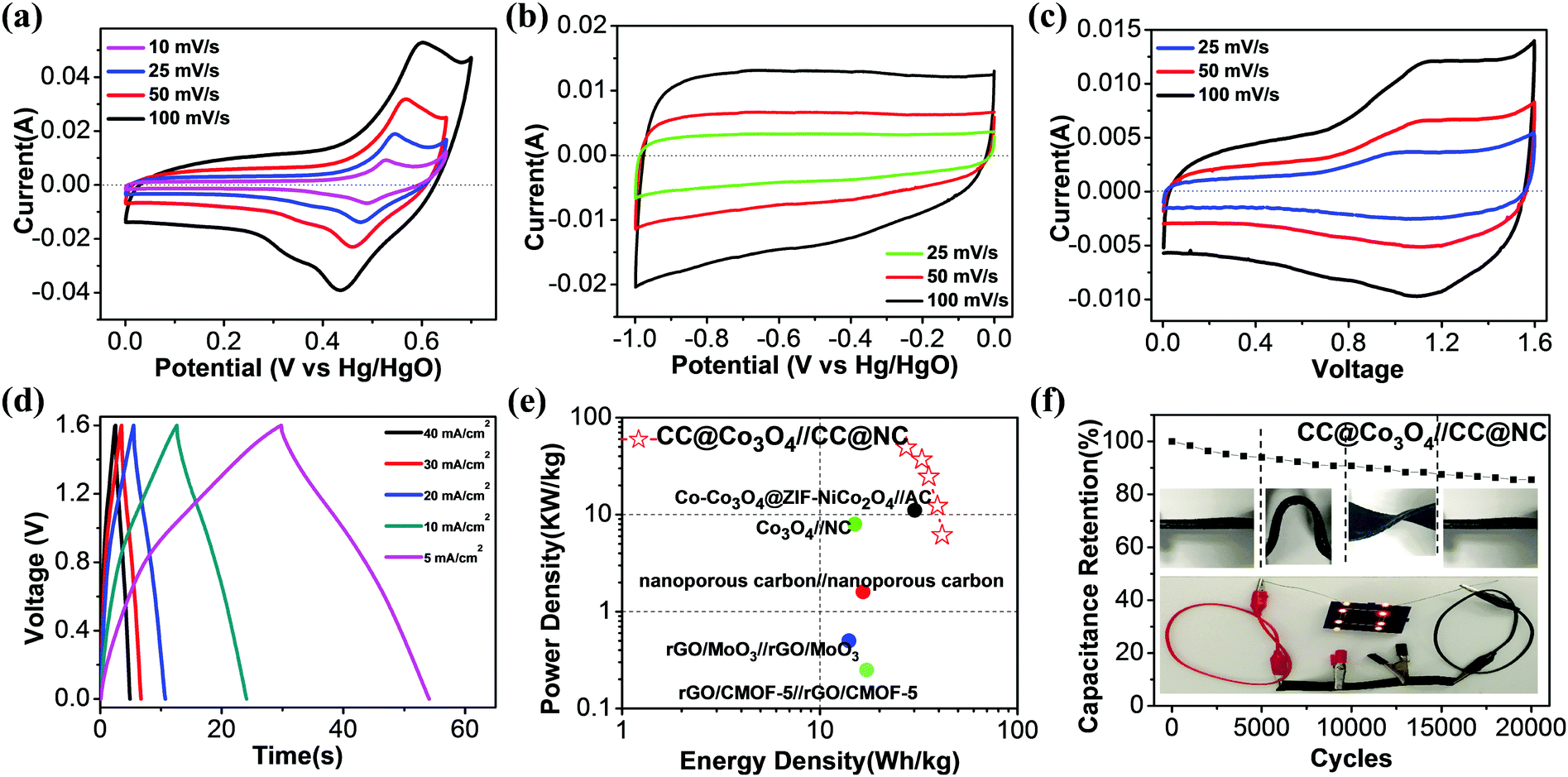 | ||
| Fig. 4 CV curves of (a) CC@Co3O4, (b) CC@NC, (c) CC@Co3O4//CC@NC; (d) charge–discharge curves, (e) Ragone plots, and (f) cycling test result of the asymmetric supercapacitor with PVA–KOH gel electrolyte. Insert in (f) shows digital images of the flat, bending, and twisting states of the cell during cycling test (upper) and a demo that three full cells (each size of 1 × 2.5 × 0.08 cm3) can light up 8 orange LEDs. | ||
Similarly, the electrochemical performance of the CC@NC anode was studied in the same three-electrode system. From the near-rectangular CV curves in Fig. 4b (results at higher scan rates are shown in Fig. S9a, ESI†) and triangular GCD curves in Fig. S9b (ESI†), the CC@NC electrode shows a typical non-Faradaic double-layer behavior with good charge transfer.64 At a current density of 6.25 A g−1, the CC@NC shows a specific capacitance of 321.9 F g−1, which is evidently higher than those of powdered porous carbon materials.65–67 Note that 80.8% of the capacitance can be maintained when the current is increased 16 times (Fig. S9c, ESI†), and that the EIS curve in Fig. S9d (ESI†) demonstrates the excellent conductivity of the CC@NC electrode. The good electrochemical performance of the NC probably arises from the N-doping and porous features, together with its vertical aligned 3D structure which has direct contact with the carbon support.68
A flexible asymmetric supercapacitor using the CC@Co3O4 cathode, CC@NC anode and a PVA–KOH electrolyte was assembled and typical CV curves are shown in Fig. 4c. The CV curves are quasi-rectangular and symmetrical in shape without sharp redox peaks, even at the high scan rate of 100 mV, indicating that CC@Co3O4//CC@NC has ideal capacitive properties with good reversibility. Fig. 4d shows typical charge and discharge curves of the asymmetric supercapacitor at different current densities from 7.7 to 61.5 A g−1; all the curves demonstrate almost linear charge–discharge characteristics, confirming the ideal capacitive properties of the full-cell. At a current density of 7.7 A g−1, the asymmetric supercapacitor delivers a capacity of ∼116.8 F g−1, and it only decreases to ∼76.9 F g−1 when the current density is increased 8 times, showing fast kinetic behavior. The high rate properties of the asymmetric supercapacitor could arise from: (i) the nanoporous structure of the electrodes which provide high surface areas and short ion diffusion lengths, and (ii) the direct contact between the active materials and the carbon support, which provides a good electron transport highway. A Ragone plot (power (P) vs. energy (E) density) of the CC@Co3O4//CC@NC is given in Fig. 4e, with data obtained from other MOF-based solid-state supercapacitors inserted for comparison. The CC@Co3O4//CC@NC device demonstrated performance comparable to the previously reported MOF-based supercapacitors;13,27,69–71 it delivers a maximum energy density of 41.5 W h kg−1 at a power density of 6.2 kW kg−1, and it maintains 27.4 W h kg−1 at a peak power density of 49.2 kW kg−1. When the mass of the whole system (including the carbon cloth substrate) is calculated, the maximum energy and power density decrease to 1.3 W h kg−1 and 1.55 kW kg−1, respectively, due to the heavy carbon cloth substrate. Here we just use carbon cloth as a typical prototype demonstration, and the energy and power density of the full cell can be further improved with an optimized flexible and light-weight support. The long-term cycling stability and stable mechanical flexibility of the asymmetric supercapacitor were also tested. As shown in Fig. 3f, even when the CC@Co3O4//CC@NC full-cell was under different bending/twisting conditions (insert in Fig. 4f) during the cycling test, it still retains 90.8% and 85.5% of the initial capacity after 10000 and 20000 cycles, respectively, showing structural stability with excellent cycling durability. The capacity fading can be from the structure change, volume expansion during the cycling, however, this retention of performance compares favorably with results that have been reported for other CoOx based supercapacitors.57,62,72–74 The potential usage of the solid-state full-cell for flexible/bendable electronics is further demonstrated, as shown in the insert of Fig. 3f, whereby eight orange LEDs can be lit by three connected asymmetric supercapacitors (3 × 2.5 × 0.08 cm3) under bending states.
Conclusions
To conclude, based on a new “one for two” strategy, a 2D MOF has been directly grown on conductive substrates, and through different thermal treatment it can be steadily transformed to porous metal oxide and carbon, suitable for both electrodes in an asymmetric supercapacitor configuration. Due to the highly porous electrodes and their direct contact with the flexible substrates, the flexible solid-state supercapacitor demonstrated both high electrochemical performance and high mechanical stability. The “one for two” strategy can be extended to other functional materials for different integrated devices, e.g., MOFs for different porous metal oxides/sulfides and carbon materials for energy and environment applications.Acknowledgements
This work is supported by the Ministry of Education, Grant number: MOE2015-T2-2-094, conducted at the National University of Singapore. This work was supported by MOE under AcRF Tier 2 (ARC 26/13, No. MOE2013-T2-1-034; ARC 19/15, No. MOE2014-T2-2-093; MOE2015-T2-2-057) and AcRF Tier 1 (RG5/13), and NTU under Start-Up Grant (M4081296.070.500000) in Singapore. This research is supported by the National Research Foundation, Prime Minister's Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) programme. The authors also thank Dr Yue Wu from University of Cambridge for fruitful discussions and help on XRD analysis.References
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- I. Stassen, M. Styles, G. Grenci, H. V. Gorp, W. Vanderlinden, S. D. Feyter, P. Falcaro, D. D. Vos, P. Vereecken and R. Ameloot, Nat. Mater., 2015, 15, 304–310 CrossRef PubMed.
- S. Bai, X. Liu, K. Zhu, S. Wu and H. Zhou, Nat. Energy, 2016, 1, 16094 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- J. Long, K. Shen, L. Chen and Y. Li, J. Mater. Chem. A, 2016, 4, 10254–10262 CAS.
- W.-J. Li, M. Tu, R. Cao and R. A. Fischer, J. Mater. Chem. A, 2016, 4, 12356–12369 CAS.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307(Part 2), 361–381 CrossRef CAS.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 CAS.
- Y. Zhao, Z. Song, X. Li, Q. Sun, N. Cheng, S. Lawes and X. Sun, Energy Storage Mater., 2016, 2, 35–62 CrossRef.
- X.-W. Liu, T.-J. Sun, J.-L. Hu and S.-D. Wang, J. Mater. Chem. A, 2016, 4, 3584–3616 CAS.
- F.-S. Ke, Y.-S. Wu and H. Deng, J. Solid State Chem., 2015, 223, 109–121 CrossRef CAS.
- D. Yu, B. Wu, L. Ge, L. Wu, H. Wang and T. Xu, J. Mater. Chem. A, 2016, 4, 10878–10884 CAS.
- H. Hu, J. Zhang, B. Guan and X. W. Lou, Angew. Chem., 2016, 128, 9666–9670 CrossRef.
- G. Zhang, S. Hou, H. Zhang, W. Zeng, F. Yan, C. C. Li and H. Duan, Adv. Mater., 2015, 27, 2400–2405 CrossRef CAS PubMed.
- G. Fang, J. Zhou, C. Liang, A. Pan, C. Zhang, Y. Tang, X. Tan, J. Liu and S. Liang, Nano Energy, 2016, 26, 57–65 CrossRef CAS.
- X. Cao, B. Zheng, X. Rui, W. Shi, Q. Yan and H. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1404–1409 CrossRef CAS PubMed.
- K. Xi, S. Cao, X. Peng, C. Ducati, R. Vasant Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192–2194 RSC.
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- N. Campagnol, R. Romero-Vara, W. Deleu, L. Stappers, K. Binnemans, D. E. De Vos and J. Fransaer, ChemElectroChem, 2014, 1, 1182–1188 CrossRef CAS.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. Zhang, J. Am. Chem. Soc., 2016, 138, 6924–6927 CrossRef CAS PubMed.
- J.-C. Tan, P. J. Saines, E. G. Bithell and A. K. Cheetham, ACS Nano, 2012, 6, 615–621 CrossRef CAS PubMed.
- S. C. Junggeburth, L. Diehl, S. Werner, V. Duppel, W. Sigle and B. V. Lotsch, J. Am. Chem. Soc., 2013, 135, 6157–6164 CrossRef CAS PubMed.
- T. De Baerdemaeker, M. Feyen, T. Vanbergen, U. Müller, B. Yilmaz, F.-S. Xiao, W. Zhang, T. Yokoi, X. Bao, D. E. De Vos and H. Gies, Chem. Mater., 2015, 27, 316–326 CrossRef CAS.
- C. Hermosa, B. R. Horrocks, J. I. Martinez, F. Liscio, J. Gomez-Herrero and F. Zamora, Chem. Sci., 2015, 6, 2553–2558 RSC.
- A. Gallego, C. Hermosa, O. Castillo, I. Berlanga, C. J. Gómez-García, E. Mateo-Martí, J. I. Martínez, F. Flores, C. Gómez-Navarro, J. Gómez-Herrero, S. Delgado and F. Zamora, Adv. Mater., 2013, 25, 2141–2146 CrossRef CAS PubMed.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS PubMed.
- R. Li, Z. Lin, X. Ba, Y. Li, R. Ding and J. Liu, Nanoscale Horiz., 2016, 1, 150–155 RSC.
- R. Li, Y. Wang, C. Zhou, C. Wang, X. Ba, Y. Li, X. Huang and J. Liu, Adv. Funct. Mater., 2015, 25, 5384–5394 CrossRef CAS.
- J. Liu, C. Guan, C. Zhou, Z. Fan, Q. Ke, G. Zhang, C. Liu and J. Wang, Adv. Mater., 2016, 28, 8732–8739 CrossRef CAS PubMed.
- C. Wang, L. Wu, H. Wang, W. Zuo, Y. Li and J. Liu, Adv. Funct. Mater., 2015, 25, 3524–3533 CrossRef CAS.
- Z. Liu, J. Xu, D. Chen and G. Shen, Chem. Soc. Rev., 2015, 44, 161–192 RSC.
- X. Lu, M. Yu, G. Wang, Y. Tong and Y. Li, Energy Environ. Sci., 2014, 7, 2160–2181 Search PubMed.
- Y. Zhang, W. Bai, X. Cheng, J. Ren, W. Weng, P. Chen, X. Fang, Z. Zhang and H. Peng, Angew. Chem., Int. Ed., 2014, 53, 14564–14568 CrossRef CAS PubMed.
- Y. Meng, Y. Zhao, C. Hu, H. Cheng, Y. Hu, Z. Zhang, G. Shi and L. Qu, Adv. Mater., 2013, 25, 2326–2331 CrossRef CAS PubMed.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- M. F. El-Kady, Y. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- J. Ni, S. Fu, C. Wu, J. Maier, Y. Yu and L. Li, Adv. Mater., 2016, 28, 2259–2265 CrossRef CAS PubMed.
- L. Wang, X. Feng, L. Ren, Q. Piao, J. Zhong, Y. Wang, H. Li, Y. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- D. Ye, G. Zeng, K. Nogita, K. Ozawa, M. Hankel, D. J. Searles and L. Wang, Adv. Funct. Mater., 2015, 25, 7488–7496 CrossRef.
- D. Kong, W. Ren, C. Cheng, Y. Wang, Z. Huang and H. Y. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 21334–21346 CAS.
- P. Xiong, J. Zhu, L. Zhang and X. Wang, Nanoscale Horiz., 2016, 1, 340–374 RSC.
- A. L. M. Reddy, S. R. Gowda, M. M. Shaijumon and P. M. Ajayan, Adv. Mater., 2012, 24, 5045–5064 CrossRef CAS PubMed.
- R. Chen, J. Yao, Q. Gu, S. Smeets, C. Baerlocher, H. Gu, D. Zhu, W. Morris, O. M. Yaghi and H. Wang, Chem. Commun., 2013, 49, 9500–9502 RSC.
- M.-H. Sun, S.-Z. Huang, L.-H. Chen, Y. Li, X.-Y. Yang, Z.-Y. Yuan and B.-L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC.
- Q. Liao, N. Li, S. Jin, G. Yang and C. Wang, ACS Nano, 2015, 9, 5310–5317 CrossRef CAS PubMed.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2013, 26, 849–864 CrossRef PubMed.
- H. Tang, S. Cai, S. Xie, Z. Wang, Y. Tong, M. Pan and X. Lu, Adv. Sci., 2016, 3 DOI:10.1002/advs.201500265.
- Y. Cheng, L. Huang, X. Xiao, B. Yao, L. Yuan, T. Li, Z. Hu, B. Wang, J. Wan and J. Zhou, Nano Energy, 2015, 15, 66–74 CrossRef CAS.
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed.
- Y. M. Chen, L. Yu and X. W. Lou, Angew. Chem., 2016, 128, 6094–6097 CrossRef.
- Y. Fan, N. Zhang, L. Zhang, H. Shao, J. Wang, J. Zhang and C. Cao, J. Electrochem. Soc., 2013, 160, F218–F223 CrossRef CAS.
- X. Wang, F. Wang, L. Wang, M. Li, Y. Wang, B. Chen, Y. Zhu, L. Fu, L. Zha, L. Zhang, Y. Wu and W. Huang, Adv. Mater., 2016, 28, 4904–4911 CrossRef CAS PubMed.
- Y. Wang, T. Zhou, K. Jiang, P. Da, Z. Peng, J. Tang, B. Kong, W.-B. Cai, Z. Yang and G. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- G. Huang, F. Zhang, X. Du, Y. Qin, D. Yin and L. Wang, ACS Nano, 2015, 9, 1592–1599 CrossRef CAS PubMed.
- J. Wei, Y. Hu, Y. Liang, B. Kong, J. Zhang, J. Song, Q. Bao, G. P. Simon, S. P. Jiang and H. Wang, Adv. Funct. Mater., 2015, 25, 5768–5777 CrossRef CAS.
- M. Jiang, X. Cao, D. Zhu, Y. Duan and J. Zhang, Electrochim. Acta, 2016, 196, 699–707 CrossRef CAS.
- D. Kong, C. Cheng, Y. Wang, J. I. Wong, Y. Yang and H. Y. Yang, J. Mater. Chem. A, 2015, 3, 16150–16161 CAS.
- C. Feng, J. Zhang, Y. He, C. Zhong, W. Hu, L. Liu and Y. Deng, ACS Nano, 2015, 9, 1730–1739 CrossRef CAS PubMed.
- C. Guan, X. Qian, X. Wang, Y. Cao, Q. Zhang, A. Li and J. Wang, Nanotechnology, 2015, 26, 094001 CrossRef CAS PubMed.
- H. Wang, Y. Zhang, H. Ang, Y. Zhang, H. T. Tan, Y. Zhang, Y. Guo, J. B. Franklin, X. L. Wu, M. Srinivasan, H. J. Fan and Q. Yan, Adv. Funct. Mater., 2016, 26, 3082–3093 CrossRef CAS.
- J. Xu, Z. Tan, W. Zeng, G. Chen, S. Wu, Y. Zhao, K. Ni, Z. Tao, M. Ikram, H. Ji and Y. Zhu, Adv. Mater., 2016, 28, 5222–5228 CrossRef CAS PubMed.
- J. Tang, P. Yuan, C. Cai, Y. Fu and X. Ma, Adv. Energy Mater., 2016, 6, 1600813 CrossRef.
- C. Long, D. Qi, T. Wei, J. Yan, L. Jiang and Z. Fan, Adv. Funct. Mater., 2014, 24, 3953–3961 CrossRef CAS.
- X. Xu, M. Wang, Y. Liu, Y. Li, T. Lu and L. Pan, Energy Storage Mater., 2016, 5, 132–138 CrossRef.
- R. R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, N. Kobayashi and Y. Yamauchi, Chem. Commun., 2016, 52, 4764–4767 RSC.
- X. Cao, B. Zheng, W. Shi, J. Yang, Z. Fan, Z. Luo, X. Rui, B. Chen, Q. Yan and H. Zhang, Adv. Mater., 2015, 27, 4695–4701 CrossRef CAS PubMed.
- P. Wen, Z. Li, P. Gong, J. Sun, J. Wang and S. Yang, RSC Adv., 2016, 6, 13264–13271 RSC.
- H. Hu, B. Guan, B. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- Z. Bi, M. P. Paranthaman, P. A. Menchhofer, R. R. Dehoff, C. A. Bridges, M. Chi, B. Guo, X.-G. Sun and S. Dai, J. Power Sources, 2013, 222, 461–466 CrossRef CAS.
- J. Yang, F. Wei, Y. Sui, J. Qi, Y. He, Q. Meng and S. Zhang, RSC Adv., 2016, 6, 61803–61808 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, additional XRD pattern, SEM images, TEM images, digital photos, XPS results of MOF precursor and the derived products, and additional electrochemical characterization. See DOI: 10.1039/c6nh00224b |
| This journal is © The Royal Society of Chemistry 2017 |