
Molar mass and composition effects on the thermal stability of functional P(S-r-MMA) random copolymers for nanolithographic applications†
Diego
Antonioli
ab,
Valentina
Gianotti
 ab,
Katia
Sparnacci
ab,
Michele
Laus
*ab,
Marco
Clericuzio
ab,
Tommaso Jacopo
Giammaria
abc,
Gabriele
Seguini
c and
Michele
Perego
*c
ab,
Katia
Sparnacci
ab,
Michele
Laus
*ab,
Marco
Clericuzio
ab,
Tommaso Jacopo
Giammaria
abc,
Gabriele
Seguini
c and
Michele
Perego
*c
aDipartimento di Scienze e Innovazione Tecnologica (DISIT), Università del Piemonte Orientale “A. Avogadro”, Viale T. Michel 11, 15121 Alessandria, Italy. E-mail: michele.laus@uniupo.it
bINSTM, UdR, Alessandria, Italy
cLaboratorio MDM, IMM-CNR, Via C. Olivetti 2, 20864 Agrate Brianza, MB, Italy. E-mail: michele.perego@mdm.imm.cnr.it
First published on 28th September 2017
Abstract
Functional hydroxyl-terminated poly(styrene-r-methyl methacrylate) (P(S-r-MMA)) copolymers (RCPs) are becoming key materials to control the substrate wetting characteristics not only for the conventional polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA) block copolymers (BCPs) but also for high χ BCP systems. This paper reports the thermal stability of RCPs at fixed composition as a function of molar mass and at fixed molar mass as a function of composition. Distinct and sometimes opposite dependencies were observed when the samples were analyzed in bulk, in the form of thermally untreated thin films featuring different thicknesses and in the form of brush layers after the grafting process performed at different temperatures. The overall picture of these effects is useful for optimizing the thermal processing conditions of the various steps involved in BCP-based pattern transfer technology.
Design, System, ApplicationBlock copolymer (BCP) films for pattern transfer applications are promising candidates for developing nanolithographic techniques. The use of random copolymer (RCP) brushes of poly(styrene-r-methyl methacrylate) (P(S-r-MMA)) is the preferred method to control the BCP orientation. By tuning the RCP composition and molar mass, neutral surfaces can be obtained not only for PS-b-PMMA but also for high χ BCPs. A processing key factor is represented by the time to obtain a suitable neutralized surface compatible with the tight ITRS requirements. Accordingly, the thermal stability of functional P(S-r-MMA) is important to develop high-temperature/short-time grafting procedures. The present paper addresses this topic by preparing and studying the thermal stability of P(S-r-MMA) RCPs both at fixed composition as a function of molar mass and at fixed molar mass as a function of composition. Distinct and sometimes opposite dependencies were observed when the samples were analyzed in bulk, in the form of thermally untreated ultrathin films and in the form of grafted brush layers. The overall picture of data indicates that the RCP-based approach to surface modification provides a powerful technological platform for the development of a BCP-based technology which can be easily integrated into an industrial processing line. |
1 Introduction
In the last few years, a great deal of interest was addressed to nano-manufacturing strategies based on block copolymer (BCP) thin films.1–5 The propensity for BCPs to self-assemble in a diversity of morphologies, with periodicities spanning over a wide size range, was exploited to produce tunable templates.6,7 Many successful studies devoted to the fabrication of precisely controlled patterns with a high degree of complexity and functionality suggest that nanostructured BCP films for pattern transfer applications can be suitable candidates for integrating with or even replacing time-consuming and costly conventional lithographic techniques.8The orientation control of BCP morphologies represents a key success factor, with the perpendicular orientation being preferred because of the sharp sidewalls and the high aspect ratio of the resulting template. Although surface modification using self-assembled monolayers of low molar mass alkyl chlorosilane9 or ethylene glycol10 was found efficient in controlling the BCP orientation, a substantial technological step forward was represented by the concept introduced by Mansky11 involving the use of random copolymer (RCP) brushes of poly(styrene-r-methyl methacrylate) (P(S-r-MMA)) tethered to the substrate surface to control the orientation behaviour of polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA) copolymers.12 Taking inspiration from this seminal paper, a great variety of functional RCPs, having the ability to promote not only end-chain but also side-chain grafting reactions, was introduced.13,14 However, functional hydroxyl end-chain P(S-r-MMA) copolymers represent the most commonly employed materials to control the substrate surface characteristics on both flat15 and prepatterned samples for directed self-assembly (DSA).16–19 In addition, the use of the P(S-r-MMA) copolymers was recently extended to mixtures of low molar mass PS-b-PMMA and ionic liquids20 as well as to high χ polystyrene-b-poly(propylene carbonate) (PS-b-PPC) BCPs21 and poly(styrene)-block-poly(D,L-lactide).22 Accurate tuning of the P(S-r-MMA) composition allows the substrate surface to become neutral for both blocks, leading to the perpendicular orientation of BCP features. These results clearly indicate that functional P(S-r-MMA) can be employed to control the substrate wetting characteristics also for high χ BCP systems, although some further modification is needed.
At variance with the original nitroxide-mediated polymerization (NMP) employed by Mansky, ARGET-ATRP was recently used for the preparation of a RCP with a styrene unit fraction (XS) of 0.58 (Mn = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, PDI = 1.36) suitable for promoting the perpendicular orientation of lamella-forming BCPs.23 The use of 2-hydroxyethyl(2-bromoisobutyrate) (HEBIB) as a polymerization initiator results in a RCP terminated on one end by a moiety containing a hydroxyl (OH) group and on the other end by a bromine atom in place of the TEMPO group.24 This substitution provided a significant increase in the thermal stability.23,25,26 Accordingly, this copolymer can be grafted to the activated substrate surface by a high-temperature thermal process in a very short time.23 In turn, BCP ordering can be performed at high temperatures without deterioration of the neutralized substrate. Consequently, the overall effects of the increased thermal stability of the RCP are the accelerated grafting process and the extended temperature window for the processing of BCPs, thus improving the performance of the BCP-based technology and facilitating its integration into production lines.
200 g mol−1, PDI = 1.36) suitable for promoting the perpendicular orientation of lamella-forming BCPs.23 The use of 2-hydroxyethyl(2-bromoisobutyrate) (HEBIB) as a polymerization initiator results in a RCP terminated on one end by a moiety containing a hydroxyl (OH) group and on the other end by a bromine atom in place of the TEMPO group.24 This substitution provided a significant increase in the thermal stability.23,25,26 Accordingly, this copolymer can be grafted to the activated substrate surface by a high-temperature thermal process in a very short time.23 In turn, BCP ordering can be performed at high temperatures without deterioration of the neutralized substrate. Consequently, the overall effects of the increased thermal stability of the RCP are the accelerated grafting process and the extended temperature window for the processing of BCPs, thus improving the performance of the BCP-based technology and facilitating its integration into production lines.
In addition, to assess the RCP layer thickness effect on the orientation of BCP features, several hydroxyl-terminated P(S-r-MMA) copolymers with number average molar mass (Mn) from 1700 to 69000 g mol−1, narrow Mn distribution and a styrene unit fraction of approximately 61% were prepared by ARGET-ATRP and grafted onto a silicon oxide surface up to the inherent grafting thickness limit.27 It was demonstrated that extremely thin RCP films (approximately 2 nm) are also able to induce the perpendicular orientation of BCPs, thus further decreasing the etching time and increasing the pattern transfer fidelity.27,28
Along this line, the present paper describes the thermal stability of functional P(S-r-MMA) RCPs at fixed composition (XS approximately 61%) with Mn ranging from 3400 to 69000 g mol−1 and at fixed Mn (approximately 14000 g mol−1) with XS between 23.5 and 61%. To obtain a complete picture, the thermal stability of the RCPs was investigated in bulk, in the form of thermally untreated thin films featuring different thicknesses and after the grafting process performed at different temperatures.
2 Experimental
2.1 Materials
2-Hydroxyethyl(2-bromoisobutyrate) (HEBIB), tris(2-(dimethylamino)ethyl)amine (Me6TREN), copper(II) bromide (CuBr2), tin(II) 2-ethylhexanoate (Sn(EH)2) and solvents were purchased from Aldrich and used as received. Styrene and methyl methacrylate were purchased from Aldrich and purified by passing through an inhibitor removal column (Aldrich) before use.α-Hydroxyl ω-Br RCPs with different Mn values, identified with the acronym Rn where n is the Mn of the sample in kg mol−1, were prepared as previously reported.27 The α-hydroxyl ω-Br functional P(S-r-MMA) RCPs with different compositions, identified with the acronym RSn where n is the styrene unit fraction, were synthesized by ARGET-ATRP copolymerization of styrene and methyl methacrylate as described in the following section.
2.2 ARGET-ATRP copolymerization of styrene and methyl methacrylate (RSn)
The α-hydroxyl ω-Br functional RCPs were obtained by ARGET-ATRP of styrene and methyl methacrylate initiated by HEBIB and catalyzed by a CuBr2/Me6TREN complex in the presence of Sn(EH)2 as the reducing agent.29 By varying the molar ratio between styrene and methyl methacrylate, keeping all other reaction parameters constant, copolymers with different compositions were obtained.In detail, 7.0 mg CuBr2 (31.2 μmol) and 8.3 μL Me6TREN (31.2 μmol) were dissolved in 9.0 mL degassed anisole, and transferred via degassed syringes to a dry Schlenk flask, purged by flushing with nitrogen. Then, appropriate amounts of degassed styrene and degassed methyl methacrylate (see Table 1 for details) and 120.0 μL HEBIB (0.78 mmol) were added and the mixture was degassed for three freeze–thaw cycles. Then, a purged solution of Sn(EH)2 (0.31 mmol) and Me6TREN (0.31 mmol) in degassed anisole (1.0 mL) was added and the reaction mixture was sealed under nitrogen. The polymerization was carried out at 90 °C for 4 hours, then the polymer was recovered by precipitation in methanol, purified by repeated precipitation from THF solution in methanol, and finally dried under vacuum at room temperature. Table 2 reports the molar mass and composition characteristics of copolymers RSn and, for comparison purposes, Rn.27
| Sample | M/I | STY/MMA | t R (h) | Conv. (%) |
|---|---|---|---|---|
| a T = 90 °C in anisole (0.5 vol equiv. vs. monomers). | ||||
| RS23.5 | 200/1 | 20/80 | 4 | 36.3 |
| RS30.0 | 200/1 | 28/72 | 4 | 38.5 |
| RS32.0 | 200/1 | 30/70 | 4 | 33.8 |
| RS39.9 | 200/1 | 40/60 | 4 | 34.8 |
| RS44.7 | 200/1 | 50/50 | 4 | 39.5 |
| RS57.1 | 200/1 | 65/35 | 4 | 42.6 |
| Sample | Mn (g mol−1) | PDI | XS % |
|---|---|---|---|
| R3.4 | 3400 | 1.20 | 61.6 |
| R5.4 | 5400 | 1.17 | 59.6 |
| R8.7 | 8700 | 1.19 | 62.3 |
| R14.2 | 14200 |
1.25 | 61.0 |
| R19.9 | 19900 |
1.29 | 59.1 |
| R69.0 | 69000 |
1.19 | 61.0 |
| RS23.5 | 13100 |
1.36 | 23.5 |
| RS30.0 | 15700 |
1.46 | 30.0 |
| RS32.0 | 12500 |
1.27 | 32.0 |
| RS39.9 | 14400 |
1.33 | 39.9 |
| RS44.7 | 14800 |
1.52 | 44.7 |
| RS57.1 | 13200 |
1.36 | 57.1 |
2.3 Random copolymer characterization
The copolymer composition was evaluated by 1H NMR spectroscopy employing a Jeol Eclipse ECP300 spectrometer. The obtained styrene unit fractions are reported in Table 2. The SEC analysis was performed on THF solutions of the RCPs using a Waters 590 chromatograph equipped with refractive index and ultraviolet detectors and using a column set consisting of Waters HSPgel HR3 and HR4 with a flow rate of 0.5 mL min−1. The column set was calibrated against standard PS samples. The obtained values of Mn and PI are reported in Table 2.2.4 Substrate preparation
Substrates were cut from (100)-oriented Si wafers with native oxide. Samples with a surface area of 1 cm2 were cleaned and activated using piranha solution (conc. H2SO4/30% H2O2 with a 3/1 vol. ratio, warning: piranha solution reacts strongly with organic compounds and should be handled with extreme caution) at 80 °C for 40 min. Then, the substrates were rinsed with H2O, dried in nitrogen and finally treated in an ultrasonic bath using 2-propanol.2.5 Deposition of the random copolymers with different layer thicknesses
Rn and RSn were spin-coated onto the substrates for 30 s at 3000 rpm. The RCP film thickness was tuned from 30 to 500 nm by changing the BCP concentration in the toluene solution.2.6 Random copolymer grafting
Rn and RSn (18.0 mg in solution with 2.00 mL of toluene) were spin-coated onto the substrates for 30 s at 3000 rpm. The samples were then thermally treated to promote the grafting reaction in a rapid thermal processing (RTP) Jipelec JetFirst Series system, for 900 s at 250 and 310 °C in a nitrogen atmosphere with a heating ramp of 18 °C s−1. An ultrasonic treatment in toluene was then performed to remove the non-grafted material. Finally, the grafted substrates were dried under a nitrogen flow.2.7 Film characterization
The thickness of the RCP films was measured using an M-200U spectroscopic ellipsometer (J. A. Woollam Co. Inc.) using a xenon lamp at an incident angle of 70°. Contact angle measurements were performed using an optical tensiometer (Theta model).2.8 TGA-GC-MS copolymer characterization
All TGA and TGA-GC-MS analyses were performed with a Mettler TGA/SDTA 851e with a steady flow of inert gas to purge the system. The temperature scanning rate was 20 °C min−1 from room temperature to 1100 °C. For bulk materials, each sample was placed in an open alumina crucible. The polymeric films on the substrate were directly placed on a thermo-balance plate. All the measurements were replicated three times to assess the reproducibility of the experimental results.GC-MS analysis was performed using a FINNIGAN TRACE GC-ULTRA and TRACE DSQ. GC separation was carried out using a Phenomenex DB5-5ms capillary column (30 m, 0.25 mm i.d., 0.25 μm thickness). The details of the hyphenation of TGA with GC-MS as well as the details of the analytical method are reported elsewhere.25 The acquisition of data was performed both in full-scan mode in the 20–350 m/z range and in single-ion monitoring (SIM) mode by acquiring the signals corresponding to styrene (S) at m/z = 104 and methyl methacrylate (M) at m/z = 100.
3 Results and discussion
3.1 Molar mass effect – stability in bulk and thin films
Fig. 1 reports the TGA and DTG curves of the Rn samples in bulk recorded at a heating rate of 20 °C min−1 under a nitrogen atmosphere. A single broad loss peak, extending from 300 to about 450 °C, is observed. The maxima in the DTG loss curves progressively shift from 406 to 423 °C as Mn decreases.30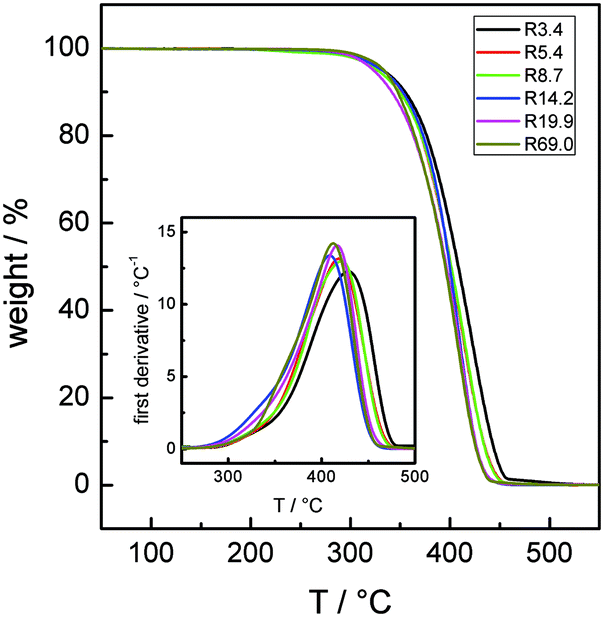 | ||
| Fig. 1 TGA (continuous lines) and DTG (inset) curves at a heating rate of 20 °C min−1 for the Rn samples in bulk under a nitrogen atmosphere. | ||
Fig. 2 illustrates the TGA-GC-MS chromatograms of R8.7 as a typical example, with specific reference to the mass peaks at m/z = 100 and 104, corresponding to methyl methacrylate and styrene, respectively. The sampling of the evolved gas occurs every 30 s and the loss profile is delineated by the dashed lines joining the gas-chromatographic peaks.
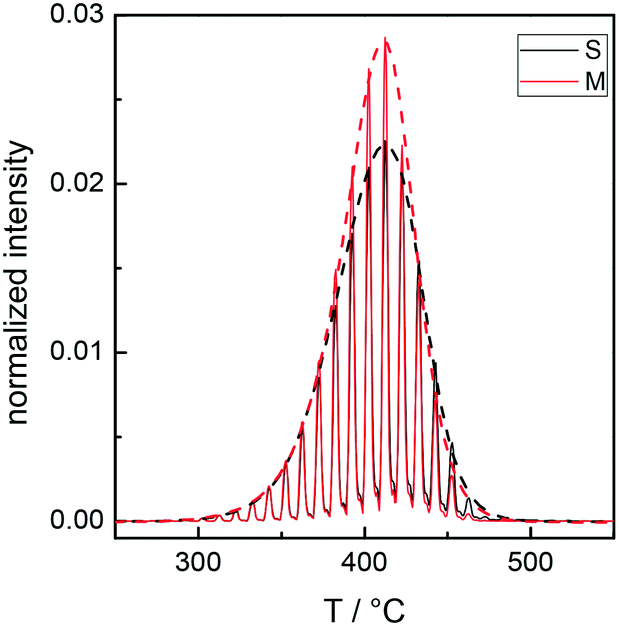 | ||
| Fig. 2 TGA-GC-MS chromatograms for the bulk R8.7 with specific reference to the mass peaks of methyl methacrylate at m/z = 100 (M, red curve) and styrene at m/z = 104 (S, black curve). The signal intensity of methyl methacrylate is multiplied by five to allow a direct comparison with styrene.31 | ||
As the mass detector sensitivity to styrene is definitely higher31 than that to methyl methacrylate, for a direct comparison, the signal intensity of the latter is multiplied by five. TGA-GC-MS indicates that the loss observed in the TGA corresponds to the evolution of styrene and methyl methacrylate monomers, which represent more than 99% of the released species. Signals corresponding to dimers or trimers are extremely small31 and the losses of methyl methacrylate and styrene occur in a closely parallel fashion, at temperatures between 300 and 460 °C. This experimental evidence suggests that the degradation of the RCPs occurs through an unzipping process, in agreement with previous results26 showing that a close similarity between the thermal behavior of P(S-r-MMA) RCPs and that of the PMMA homopolymer was noticed. Consequently, considering the higher sensitivity of the mass detector to styrene, to fully assess the thermal profile of the Rn samples, only the evolution of styrene was monitored.
Fig. 3A and B collectively report the TGA-GC-MS styrene evolution profiles of the Rn samples in bulk and in 30 nm thick films. The dashed lines connect the maximum of the various curves to guide the eyes. The TGA-GC-MS analysis confirms that in bulk the thermal stability decreases as the Mn of the copolymers increases, whereas the opposite effect is observed in thin films. To better understand this divergent behavior, the thermal stability was studied as a function of the film thickness for samples R3.4, R14.2 and R69.0, which were considered representative of low, medium and high Mn samples. Fig. 4 illustrates the TGA-GC-MS chromatograms of R14.2. The temperature corresponding to the maximum in the styrene evolution peak increases from the bulk to the 200 nm thick film and then levels off. The same effect is observed also for R3.4 and R69.0, as illustrated in Fig. 5, which reports the maximum temperature of the styrene evolution peak as a function of the film thickness. The temperature of the degradation peak levels off at a film thickness of approximately 200 nm. It is also interesting to observe that the R14.2 and R69.0 curves approach the same limiting value.
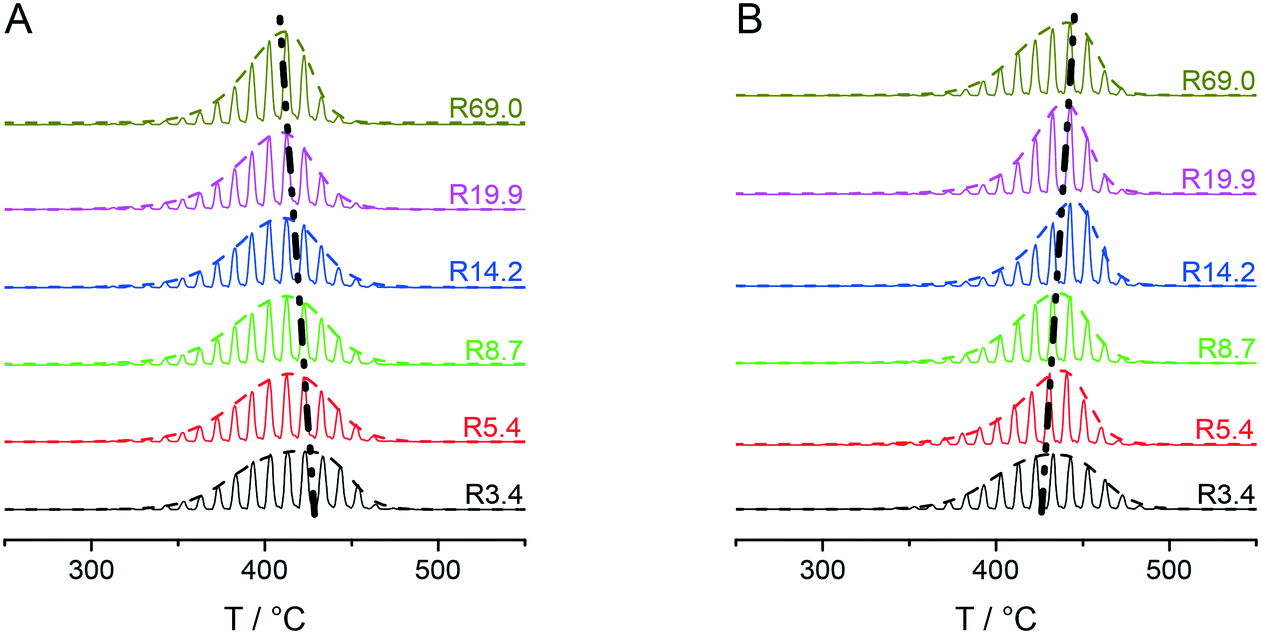 | ||
| Fig. 3 TGA-GC-MS chromatograms of the Rn samples with reference to the mass peaks at m/z = 104 (styrene) in bulk (A) and as 30 nm films, spun on silicon wafer substrates (B). | ||
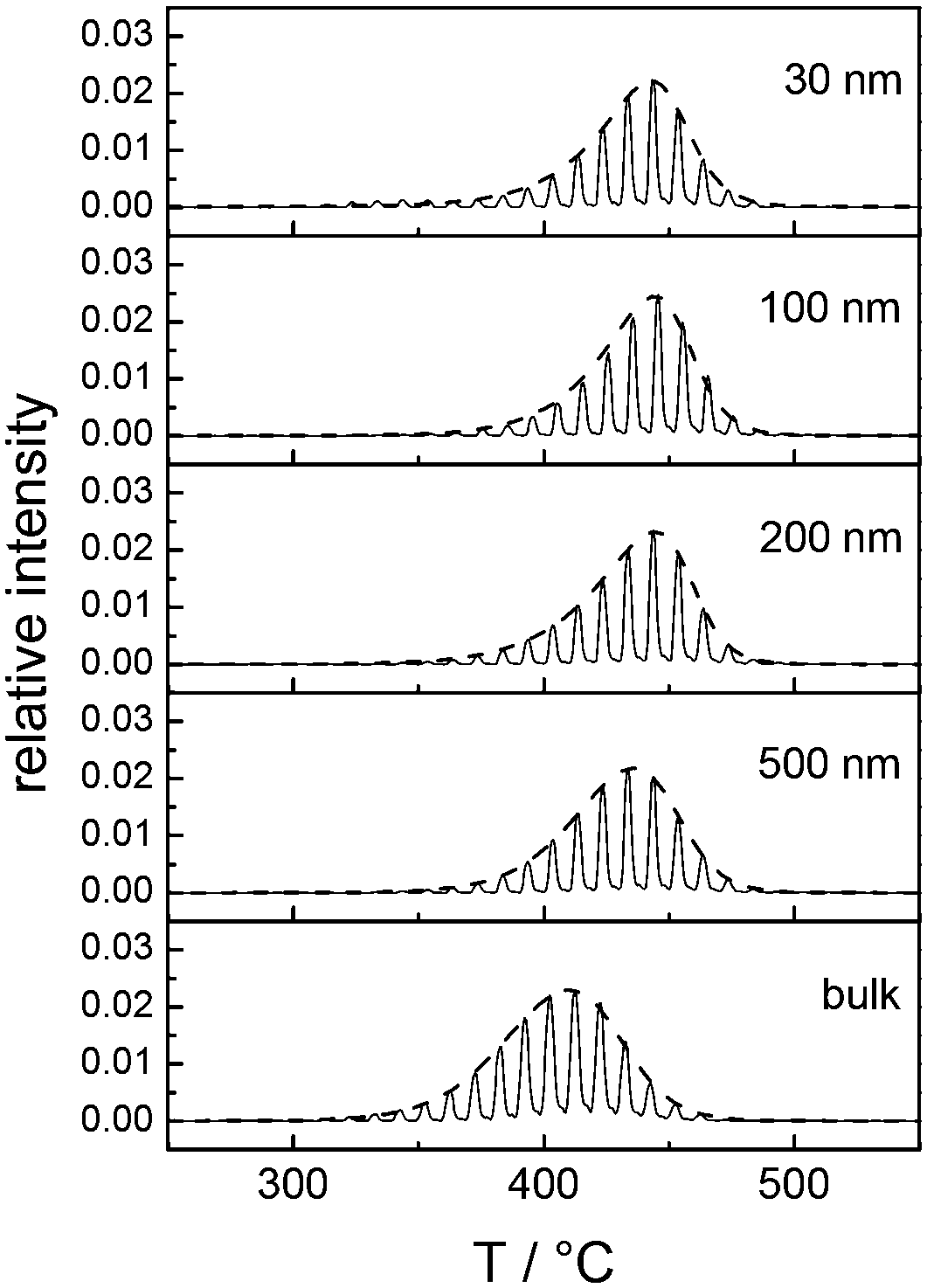 | ||
| Fig. 4 TGA-GC-MS chromatograms of R14.2 with reference to the mass peaks at m/z = 104 (styrene) in bulk and in films with different thicknesses spun on silicon substrates. | ||
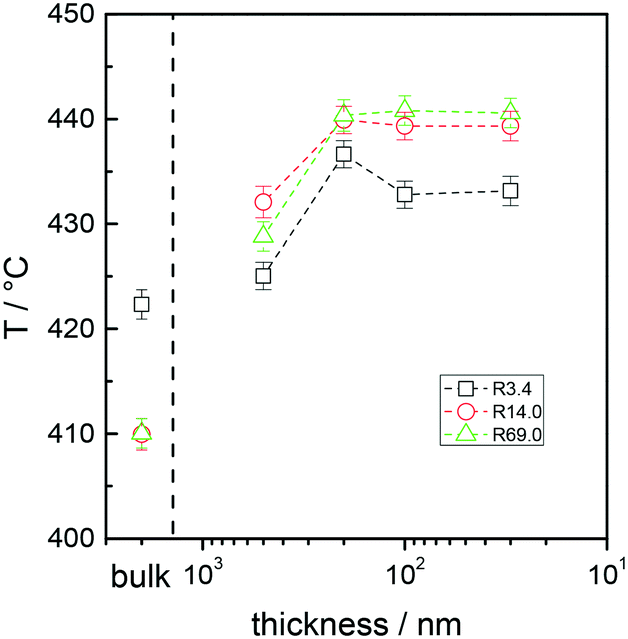 | ||
| Fig. 5 Maximum temperature of the styrene evolution peaks in the TGA-GC-MS chromatograms of samples R3.4, R14.2 and R69.0 in bulk and in films with different thicknesses. | ||
In general, the thermal degradation process consists of initiation reactions, by chain-end initiation or by initiation at random positions by chain scission, depropagation reactions, termination and transfer reactions, as well as transport of the decomposition products from the inside of the sample to the outside.32 This latter process is particularly relevant because the time for diffusion of volatile radicals out of the system is proportional to the sample thickness. As the average degree of polymerization (N) of all the Rn samples is lower than the unzipping length33 (N < 200), each initiation event results in the complete loss of the chain. According to theory,34 for monodisperse systems, long zipping conditions and chain-end initiation, the monomer evolution rate is independent of N, whereas in the case of chain scission initiation, a first-order dependence of the monomer evolution rate on the polymerization degree is expected. Moreover, in chain-end initiation,35 volatile radicals are obtained from the chain-end degradation and these radicals are able to transfer the active site to neighboring chains, thus starting the chain scission process. In bulk, at temperatures higher than 300 °C, a certain amount of volatile radicals can be formed from chain-end initiation and can induce chain scission initiation in the next chains via the activation transfer process. As the depolymerization by chain scission initiation has a deeper effect on the samples with a high N, the monomer evolution occurs at lower temperatures with respect to the samples with a low N, thus explaining the higher thermal stability of the lower Mn samples. In contrast, in the case of thin films, the efficiency of the chain transfer decreases due to the radical loss by evaporation and becomes negligible when the film thickness is lower than 200 nm. In this case, the thermal stability of the samples with high N is similar to or slightly higher than that of the samples with a lower N with a lower amount of chain ends.
3.2 Molar mass effect – stability after grafting at 250 and 310 °C
The effect of the grafting treatment was investigated by TGA-GC-MS under dynamic conditions. The samples were prepared by spinning a thin film (about 30 nm) of Rn on the Si substrate activated with piranha solution. Subsequently, the samples were annealed in the RTP system by heating the samples at 18 °C s−1 from room temperature to the grafting temperatures of 250 or 310 °C. Once the grafting temperature was reached, the samples were maintained at this temperature for 900 s. The isothermal annealing time corresponds to the time at which the grafted layer thickness reaches the plateau values, as previously demonstrated.27 As the plateau thickness corresponds11,27 to approximately twice the radius of gyration of the grafted chains, the layer thickness increases as the molar mass of the grafted random copolymer increases. Finally, a washing step with toluene was performed to remove all the non-grafted material. The relevant film thicknesses are reported in Table 3.| Sample | Thickness at 250 °C (nm) | Thickness at 310 °C (nm) |
|---|---|---|
| R3.4 | 4.3 ± 0.3 | 3.5 ± 0.4 |
| R5.4 | 5.8 ± 0.2 | 5.0 ± 0.3 |
| R8.7 | 7.3 ± 0.2 | 7.3 ± 0.3 |
| R14.2 | 8.6 ± 0.3 | 9.0 ± 0.2 |
| R19.9 | 10.9 ± 0.3 | 10.7 ± 0.3 |
| R69.0 | 19.0 ± 0.2 | 17.8 ± 0.2 |
Fig. 6 describes the evolution of the thermal profiles of samples R3.4, R14.2 and R69.0 in bulk, in the form of 30 nm films and after grafting at 310 °C leading to films with thicknesses of 3.5, 9.0 and 17.8 nm, respectively. In the case of R3.4, i.e. the lowest Mn sample, the thermal profile of the 30 nm film after grafting at 310 °C is shifted by a few degrees to higher temperatures with respect to the bulk and is quite similar to that of the bulk, although in the case of the grafted sample, the maximum temperature is slightly higher and the thermal profile is sharper. For sample R14.2, the profiles of the 30 nm film and the grafted layer are clearly separated from that of the bulk. However, the thermal profiles of the 30 nm thick film and the grafted sample are substantially overlapped. Finally, for R69.0, the various profiles are clearly separated and the width of the thermal profiles is very narrow.
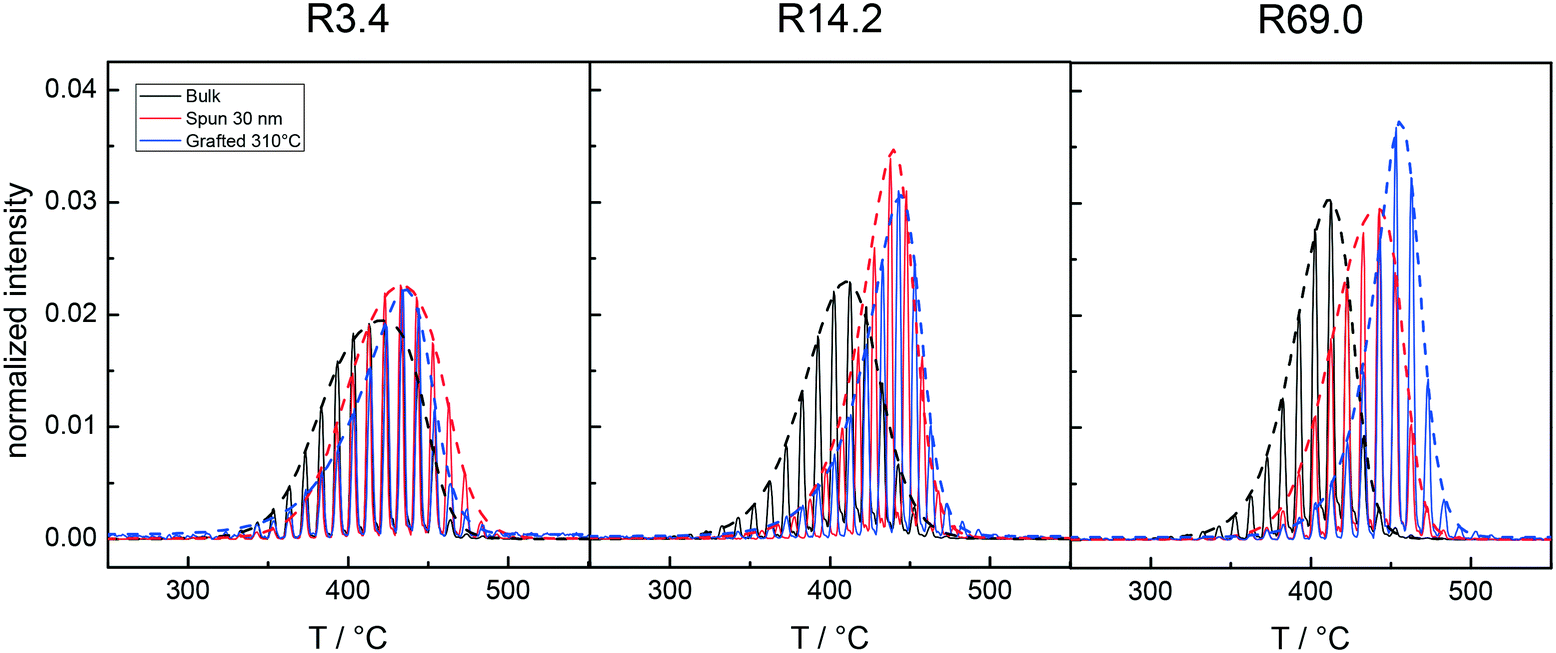 | ||
| Fig. 6 TGA-GC-MS chromatograms of the bulk (black curve), the 30 nm films (red curve) and the layers grafted at 310 °C (blue curve) for samples R3.4, R14.2 and R69.0 with specific reference to the mass peaks at m/z = 104 (styrene). | ||
Fig. 7 collectively reports the maximum temperature of the monomer evolution profiles for the bulk, 30 nm films, and layers grafted at 250 and 310 °C as a function of the Mn of the Rn samples.
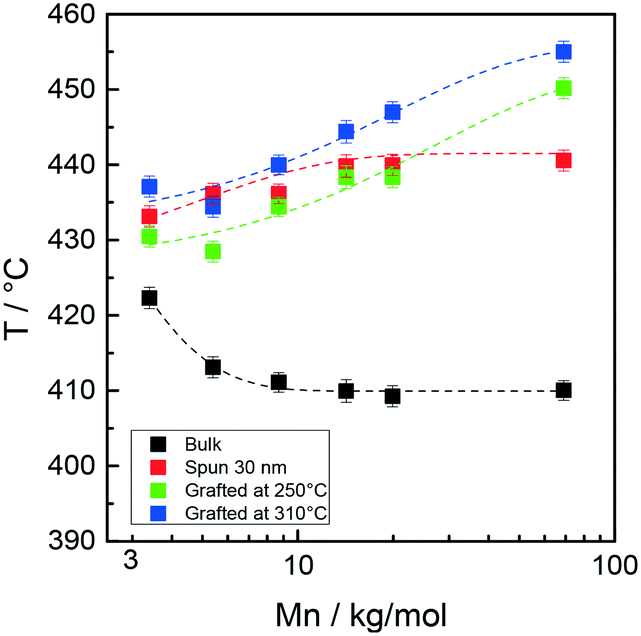 | ||
| Fig. 7 Peak maximum temperature of styrene evolution in the TGA-GC-MS chromatograms of samples Rn in bulk, as 30 nm films and after grafting at 250 and 310 °C as a function of Mn. | ||
In the low Mn region, the thermal stability of the samples grafted at 250 °C is similar for the grafted and ungrafted 30 nm thick films, whereas in the high Mn region, a definite increase in the thermal stability is observed upon grafting at 250 °C. This trend is even more evident at 310 °C. In turn, the shapes of the thermal stability curves of the samples grafted at 250 and 310 °C are very similar, with the latter being vertically shifted toward higher temperatures by approximately 15 °C.
A similar behavior was observed in a previous study in which a comparison was made between the thermal stabilities of statistical copolymers prepared by ATRP and NMP.26 The thermal stability of the grafted samples increased with increasing grafting temperature. It was suggested that during the high-temperature thermal treatment, homolytic cleavage of the end groups occurred. It is reasonable to suppose that a sequence of homolytic dissociation of these weak bonds, evolution of some monomers and radical recombination occurs, possibly facilitated by a radical cage effect. This mechanism would ultimately lead to the prevalence of the more stable styrene–styrene linkages over the styrene–(methyl methacrylate) and (methyl methacrylate)–(methyl methacrylate) linkages, thus in turn improving the overall thermal stability of the statistical copolymer chains. In addition, the degree of prevalence should increase as the grafting temperature increases. In the present context, this mechanism explains both the increased stability of the samples treated at 310 °C over those grafted at 250 °C and their narrower monomer evolution profiles. In this perspective, the increase in the thermal stability observed for the highest molar mass sample is due to the minimum number of weak linkages derived from the above described radical recombination process.
3.3 Composition effect – stability and surface characteristics in bulk, as 30 nm films and after grafting at 250 and 310 °C
The thermal stability of the RSn samples was studied by TGA-GC-MS as described for the Rn samples. The RSn samples were analyzed in bulk, as 30 nm thin films and after grafting at 250 and 310 °C. The film thickness for all the RSn samples after grafting at both temperatures was 8.8 ± 0.4 nm according to their close Mn values (approximately 14–15 kg mol−1). Irrespective of the copolymer composition, in the TGA-GC-MS chromatograms, only the evolution of styrene and methyl methacrylate monomers is observed and the losses of methyl methacrylate and styrene occur in a closely parallel fashion, at temperatures between 300 and 460 °C, thus further confirming that under the present conditions, the thermal degradation of the RCPs occurs through an unzipping process.Fig. 8 collectively reports the maximum temperatures of the styrene evolution profiles for the RSn samples in bulk, as 30 nm films and after grafting at 250 and 310 °C as a function of the styrene unit content in the various copolymers. The thermal stability increases linearly as the styrene unit content increases, due to the blocking action of the styrene unit segments for which the depolymerization occurs at higher temperatures than that for methyl methacrylate unit segments.36 Upon going from the bulk to the film with 30 nm thickness, a stability increase of about 10 °C is observed for sample RS23.5, whereas a more pronounced increase of 30 °C is observed for sample RS57.1, thus indicating that the increase in styrene units and styrene unit sequences reduces the efficiency of chain transfer reactions. Finally, the samples grafted at 310 °C show the highest thermal stability, which increases as the styrene unit content increases, thus further indicating that the radicals formed at high temperatures undergo recombination or disproportionation processes, ultimately leading to the formation of carbon–carbon bonds where the prevalence of the more stable styrene–styrene linkages is more and more probable as the styrene unit content increases.
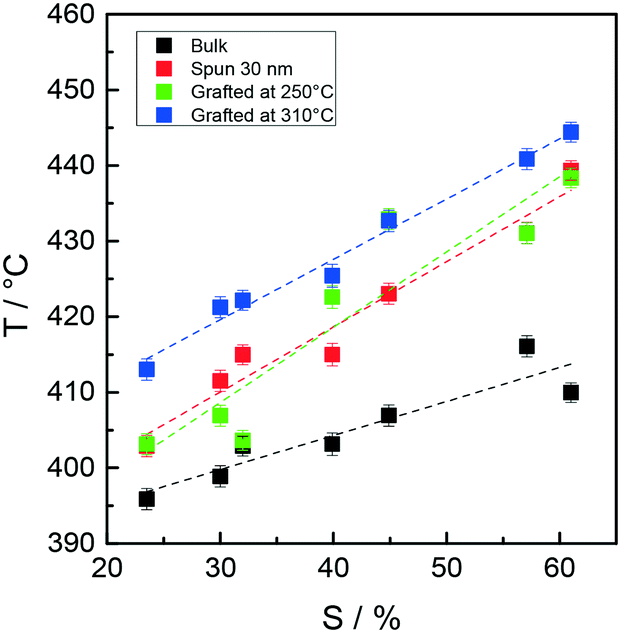 | ||
| Fig. 8 Peak maximum temperature of styrene evolution in the TGA-GC-MS chromatograms of samples RSn in bulk, as 30 nm films and after grafting at 250 and 310 °C as a function of the styrene unit content in the copolymers. | ||
The relative hydrophilicity of the substrates modified by RSn random copolymers was estimated using a static WCA (Fig. 9). Since the WCA for PS is higher than that for PMMA, the WCA for the substrates modified by random copolymers gradually increases as the PS mole fraction increases. WCA data for the 30 nm films and the samples after grafting at 250 and 310 °C are identical within the experimental error, which is consistent with data in the literature.11–13 Interestingly, the WCA data indicate that the high-temperature thermal processes do not change the surface characteristics of the grafted substrates, whose characteristics are essentially determined by the chemical composition of the RSn random copolymers.
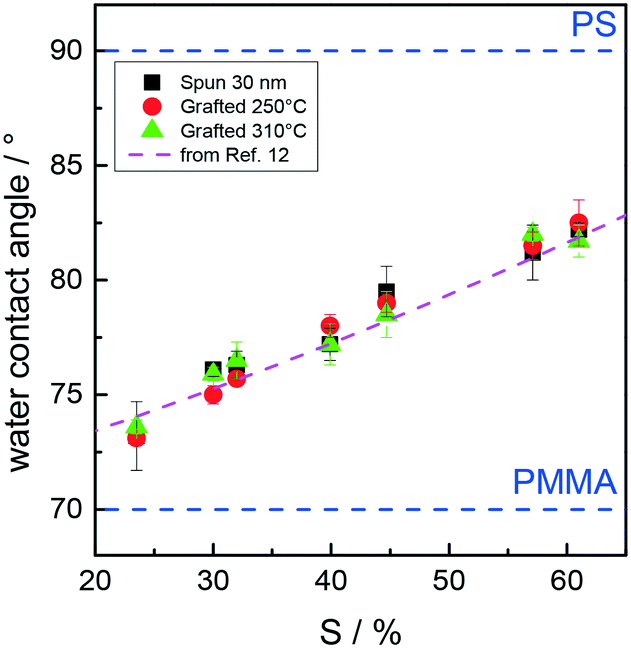 | ||
| Fig. 9 Water contact angle (WCA) of the substrates modified by RSn random copolymers for the 30 nm films and the samples after grafting at 250 and 310 °C as a function of the styrene unit content in the copolymers. The pink dashed line was obtained from the polynomial fit of WCA data reported in ref. 12. | ||
4 Conclusions
The thermal stability of hydroxyl-terminated poly(styrene-r-methyl methacrylate) (P(S-r-MMA)) RCPs at fixed composition as a function of Mn and at fixed Mn as a function of composition was investigated. In all cases, the samples degrade following an unzipping mechanism. The molar mass and the composition play distinct roles in determining the thermal stability of the samples. Concerning the molar mass effect, it was shown that in bulk, the thermal stability decreases as Mn increases, whereas the opposite trend is observed for ultrathin films. This effect is ascribed to the different incidence in bulk and in ultrathin films of volatile radicals obtained from chain-end degradation. The thermal stability of the grafted samples increases with increasing grafting temperature, thus indicating that, during the grafting process, radical recombinations occur, leading to the formation of more stable linkages.Concerning the effect of the copolymer composition, the thermal stability increases linearly as the styrene unit content increases, irrespective of the sample nature, either bulk, thin or grafted films. The highest thermal stability observed corresponds to the sample with the highest styrene unit content grafted at the highest temperature, thus suggesting that high-temperature radical recombination or disproportionation processes favor the formation of the more stable styrene–styrene linkages.
From a processing point of view, these data indicate that the approach based on the surface modification by the formation of a grafted polymer layer is extremely robust. The high-temperature processed brush layers exhibit a very high thermal stability irrespective of the Mn and composition of the specific RCP. The surface energy of the substrate can be finely tuned over a broad range of values by proper selection of the RCP composition without introducing any significant limitation in terms of accessible temperatures during the subsequent processing of the samples. From this point of view, the proposed approach provides a valuable solution to properly neutralize the substrate and promote the perpendicular orientation of nanodomains in self-assembled BCP films. Moreover, the high thermal stability of these brush layers guarantees the possibility of operating over a large window of temperatures and optimizing the thermal processing conditions that are necessary to achieve highly ordered self-assembled BCP templates in a short time. As the thermal stability is relatively high across the entire composition range, surface neutralization to promote orientation control of nanodomains in thin films of block copolymer systems as those introduced in ref. 20–22 can be achieved by substrate modification via high-temperature thermally induced grafting of P(S-r-MMA) copolymers that can be easily integrated into an industrial production line. Consequently, this extended approach provides a powerful technological platform for the development of a BCP-based pattern transfer technology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is original and received financial support from Università del Piemonte Orientale.Notes and references
- K. Koo, H. Ahn, S. W. Kim, D. Y. Ryu and T. P. Russell, Soft Matter, 2013, 9, 9059–9071 RSC.
- M. Luo and T. H. Epps III, Macromolecules, 2013, 46, 7567–7579 CrossRef CAS.
- C. M. Bates, M. J. Maher, D. W. Janes, C. J. Ellison and C. G. Willson, Macromolecules, 2014, 47, 2–12 CrossRef CAS.
- D. J. C. Herr, J. Mater. Res., 2011, 26, 122–139 CrossRef CAS.
- N. Collaert, A. Alian, H. Arimura, G. Boccardi, G. Eneman, J. Franco, T. Ivanov, D. Lin, R. Loo, C. Merckling, J. Mitard, M. A. Pourghaderi, R. Rooyackers, S. Sioncke, J. W. Sun, A. Vandooren, A. Veloso, A. Verhulst, N. Waldron, L. Witters, D. Zhou, K. Barla and A. V. Thean, Microelectron. Eng., 2015, 132, 218–225 CrossRef CAS.
- J. Y. Cheng, D. P. Sanders, H. D. Truong, S. Harrer, A. Friz, S. Holmes, M. Colburn and W. D. Hinsberg, ACS Nano, 2010, 4, 4815–4823 CrossRef CAS PubMed.
- G. Aprile, F. Ferrarese Lupi, M. Fretto, E. Enrico, N. De Leo, L. Boarino, F. G. Volpe, G. Seguini, K. Sparnacci, V. Gianotti, M. Laus, J. Garnæs and M. Perego, ACS Appl. Mater. Interfaces, 2017, 9, 15685–15697 CAS.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- R. D. Peters, X. M. Yang, T. K. Kim and P. F. Nealey, Langmuir, 2000, 16, 9620–9626 CrossRef CAS.
- R. D. Peters, X. M. Yang, T. K. Kim, B. H. Sohn and P. F. Nealey, Langmuir, 2000, 16, 4625–4631 CrossRef CAS.
- P. Mansky, Y. Liu, E. Huang, T. P. Russell and C. J. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- S. Ham, C. Shin, E. Kim, D. Y. Ryu, U. Jeong, T. P. Russell and C. J. Hawker, Macromolecules, 2008, 41, 6431–6437 CrossRef CAS.
- E. Han, K. O. Stuen, Y. H. La, P. F. Nealey and P. Gopalan, Macromolecules, 2008, 41, 9090–9097 CrossRef CAS.
- E. Han and P. Gopalan, Langmuir, 2010, 26, 1311–1315 CrossRef CAS PubMed.
- M. Perego, F. Ferrarese Lupi, M. Ceresoli, T. J. Giammaria, G. Seguini, E. Enrico, L. Boarino, D. Antonioli, V. Gianotti, K. Sparnacci and M. Laus, J. Mater. Chem. C, 2014, 2, 6655–6664 RSC.
- H. Kang, G. S. W. Craig, E. Han, P. Gopalan and P. F. Nealey, Macromolecules, 2012, 45, 159–164 CrossRef CAS.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS PubMed.
- F. Ferrarese Lupi, T. J. Giammaria, G. Seguini, M. Laus, E. Enrico, N. De Leo, L. Boarino, C. K. Ober and M. Perego, J. Mater. Chem. C, 2014, 2, 2175–2182 RSC.
- R. Ruiz, N. Ruiz, Y. Zhang, R. L. Sandstrom and C. T. Black, Adv. Mater., 2007, 19, 2157–2162 CrossRef CAS.
- A. Kawaue, T. Matsumiya, T. Seo, T. Maehashi, T. Seshimo, H. Yamano, K. Miyagi, T. Dazai, X. Chen, P. Rincon-Delgadillo, R. Gronheid, P. F. Nealey and K. Ohmori, J. Photopolym. Sci. Technol., 2016, 29, 667–670 CrossRef CAS.
- G. W. Yang, G. P. Wu, X. Chen, S. Xiong, C. G. Arges, S. Ji, P. F. Nealey, X. B. Lu, D. J. Darensbourg and Z. K. Xu, Nano Lett., 2017, 17, 1233–1239 CrossRef CAS PubMed.
- I. Keen, A. Yu, H. Cheng, K. S. Jack, T. M. Nicholson, A. K. Whittaker and I. Blakey, Langmuir, 2012, 28, 15876–15888 CrossRef CAS PubMed.
- M. Ceresoli, F. Ferrarese Lupi, G. Seguini, K. Sparnacci, V. Gianotti, D. Antonioli, M. Laus, L. Boarino and M. Perego, Nanotechnology, 2014, 25, 275601 CrossRef PubMed.
- F. Ferrarese Lupi, T. J. Giammaria, G. Seguini, M. Ceresoli, M. Perego, D. Antonioli, V. Gianotti, K. Sparnacci and M. Laus, J. Mater. Chem. C, 2014, 2, 4909–4917 RSC.
- V. Gianotti, D. Antonioli, K. Sparnacci, M. Laus, T. J. Giammaria, F. Ferrarese Lupi, G. Seguini and M. Perego, Macromolecules, 2013, 46, 8224–8234 CrossRef CAS.
- K. Sparnacci, D. Antonioli, V. Gianotti, M. Laus, G. Zuccheri, F. Ferrarese Lupi, T. J. Giammaria, G. Seguini, M. Ceresoli and M. Perego, ACS Appl. Mater. Interfaces, 2015, 7, 3920–3930 CAS.
- K. Sparnacci, D. Antonioli, V. Gianotti, M. Laus, F. Ferrarese Lupi, T. J. Giammaria, G. Seguini and M. Perego, ACS Appl. Mater. Interfaces, 2015, 7, 10944–10951 CAS.
- K. Sparnacci, D. Antonioli, M. Perego, T. J. Giammaria, G. Seguini, F. Ferrarese Lupi, G. Zuccheri, V. Gianotti and M. Laus, Polym. Int., 2017, 66, 459–467 CrossRef CAS.
- W. Jakubowski, K. Min and K. Matyjaszewski, Macromolecules, 2006, 39, 39–45 CrossRef CAS.
- H. Nishizaki and K. Yoshida, J. Appl. Polym. Sci., 1981, 26, 3503–3504 CrossRef CAS.
- V. Gianotti, D. Antonioli, K. Sparnacci, M. Laus, T. J. Giammaria, M. Ceresoli, F. Ferrarese Lupi, G. Seguini and M. Perego, J. Chromatogr. A, 2014, 1368, 204–210 CrossRef CAS PubMed.
- T. Kashiwagi, T. Hirata and J. E. Brown, Macromolecules, 1985, 18, 131–138 CrossRef CAS.
- L. E. Manring, Macromolecules, 1989, 22, 2673–2677 CrossRef CAS.
- Thermal stability of polymers, ed. R. H. Boyd, Marcel Dekker, 1970 Search PubMed.
- L. E. Manring, Macromolecules, 1991, 24, 3304–3309 CrossRef CAS.
- V. Kovacevic, Polym. Eng. Sci., 1999, 39, 600–608 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00064b |
| This journal is © The Royal Society of Chemistry 2017 |