
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards environmentally acceptable synthesis of chiral α-hydroxy ketones via oxidase-lyase cascades†
Sandy
Schmidt
 *a,
Tiago
Pedroso de Almeida
a,
Dörte
Rother
b and
Frank
Hollmann
*a
*a,
Tiago
Pedroso de Almeida
a,
Dörte
Rother
b and
Frank
Hollmann
*a
aDepartment of Biotechnology/Biocatalysis Delft University of Technology Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl; s.schmidt@tudelft.nl
bInstitute of Bio- and Geosciences, IBG-1 Biotechnology, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
First published on 31st January 2017
Abstract
The one-pot multistep enzymatic oxidation of aliphatic and benzylic alcohols to the corresponding aldehydes combined with their subsequent carboligation to chiral α-hydroxy ketones has been exemplarily evaluated in terms of being a “green” biocatalytic approach. Besides the potential to start from bio-derived alcohols, this concept avoids the direct use of the reactive aldehyde intermediates, enables addition of high substrate concentrations in one liquid phase while maintaining enzyme activity and enables a simplified product isolation with diminished waste formation.
Chiral α-hydroxy ketones are valuable building blocks especially in fine chemistry and pharmaceutical applications.1–6 They can conveniently be produced from simple aldehydes with thiamine-dependent carboligases catalysing the biocatalytic version of the well-known benzoin condensation (albeit enantioselectively and omitting the use of cyanides).7–10
There are, however, still some practical issues related to the use of aldehydes as starting materials. First, aldehydes are reactive molecules prone to undesired side-reactions such as aerobic oxidation and hydroperoxide formation. More importantly, reactive aldehydes can form Schiff-bases with lysine residues of the biocatalysts thereby blocking the access channel to the enzymes’ active sites and/or modulating the polarity of the enzymes (neutral imines instead of positively charged lysine residues).11
Furthermore, aldehydes are relatively hydrophobic with generally poor solubility in aqueous reaction media. This issue is often addressed by adding water-soluble cosolvents enhancing the aqueous concentration of the starting materials.
Inspired by earlier work of Domínguez de María and coworkers,12 we evaluated a bi-enzymatic cascade comprising alcohol oxidase from Pichia pastoris (PpAOX) and benzaldehyde lyase from Pseudomonas fluorescens (PfBAL) for the formation of chiral benzoins from the corresponding alcohols (Scheme 1).
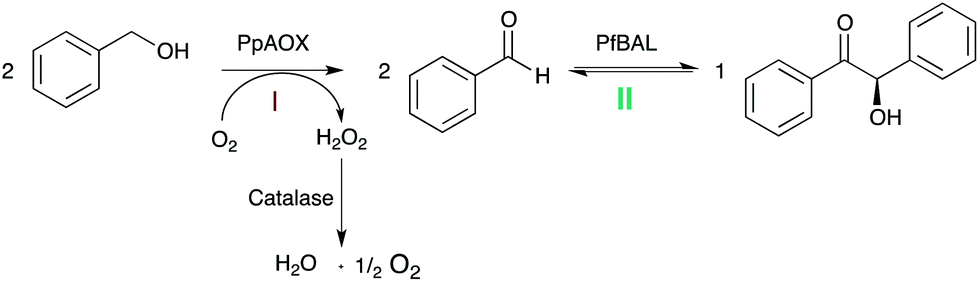 | ||
| Scheme 1 The envisioned synthetic enzyme cascade consisting of alcohol oxidase (PpAOX) from Pichia pastoris and benzaldehyde lyase (PfBAL) from Pseudomonas fluorescens for the stereoselective synthesis of (R)-benzoin. To circumvent the hazardous effect of H2O2 (formed in the alcohol oxidation step) catalase is added. | ||
Due to the significantly higher solubility of alcohols compared to aldehydes (benzyl alcohol for example is at pH 7.0 and 25 °C approximately 20 times more soluble than benzaldehyde), we envisioned a buffered reaction system without cofactor addition exhibiting substrate concentrations >350 mM in one liquid phase. Also, the inhibitory effects of the intermediate aldehyde should be minimized due to the further conversion to the corresponding benzoin in the one-pot approach. Furthermore, the poor water-solubility of the benzoin products should efficiently pull the equilibrium of the carboligation reaction and thereby avoid unnecessary molar surpluses of the starting material to shift the equilibrium. Finally, the absence or at least minimal use of co-solvents should facilitate the work-up of the reaction mixtures by simple filtration avoiding solvent-dependent extraction procedures.
In a first set of experiments, PpAOX and PfBAL were combined under arbitrarily chosen conditions (based on the standard oxidase-lyase reaction conditions as reported by Perez-Sanchez et al.) in the same pot in aqueous solution without co-solvent in the presence of 20 mM benzyl alcohol yielding 56% of the desired enantiopure (R)-benzoin. Encouraged by these promising results, we further investigated the parameters influencing the efficiency of the cascade.
PpAOX has been reported to preferentially oxidize short-chain, aliphatic alcohols.13,14 Indeed, PpAOX activity towards benzyl alcohol was only about 1% compared to the rate observed with methanol (Fig. S21†), which to some extend can be attributed to a comparably high KM value around 140 mM against benzyl alcohol (Fig. S22†). Nevertheless, thanks to a very high intrinsic activity of PpAOX, this was still sufficient to convert benzylic alcohols into the corresponding aldehydes. PpAOX was mildly inhibited by aldehydes showing reduction of its activity above 50 mM benzaldehyde (Fig. S23†). This inhibition, however, should not negatively influence PpAOX performance in the envisaged cascade in which the concentration of the aldehyde should be low due to the direct ligation of the intermediate (see also Fig. 1). H2O2 proved to be more detrimental to PpAOX (Fig. S24†) as already 10 min incubation with 10 mM lead to complete inactivation of the oxidase. Therefore, all subsequent experiments were performed in the presence of at least 1 g L−1 of bovine catalase. The optimal pH value for PpAOX was around 7.5 (Fig. S25†), whereas PfBAL performs best at pH 9.5 for ligation.8 Therefore, as compromise, we chose a pH of 8.5 for all subsequent experiments. The ratio of PpAOX and PfBAL had a significant influence on the performance of the cascade with an optimal performance at a molar ratio of approx. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. S29†).
:2 (Fig. S29†).
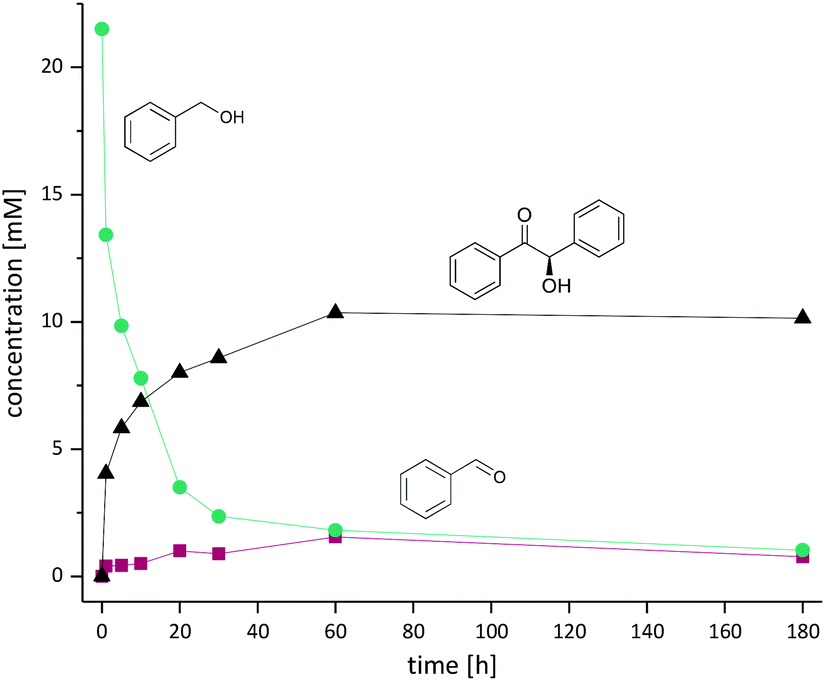 | ||
| Fig. 1 Kinetic profile obtained for the oxidase-lyase one-pot cascade reaction using benzyl alcohol as substrate. The green line shows benzyl alcohol, the red line benzaldehyde and the black line (R)-benzoin. Reaction conditions: Alcohol substrate (20 mM), PpAOX (6.3 μM), crude cell extract containing PfBAL (39.13 μM), MgCl2 (2.5 mM), ThDP (0.15 mM), catalase from bovine liver (2.5 mg mL−1), phosphate buffer (50 mM, pH 8.5), 30 °C. Due to the precipitation of the product, product profile was determined by HPLC and 1H NMR. | ||
Using the partially optimised reaction conditions, we performed a time-course experiment using 20 mM of benzyl alcohol (Fig. 1).
Within the first 10 h more than 60% conversion was achieved with significantly longer reaction time needed to achieve full conversion (10 mM) of the desired benzoin. We attribute this to the comparably high KM values for both enzymes (KM value for benzoin formation of PfBAL is 10 ± 1.5 mM).8 Notably, the concentration of the intermediate aldehyde remained low during the whole reaction low (0.4–1.5 mM). In agreement with previous literature, PfBAL showed an outstanding enantioselectivity of >99%.8,12,15,16
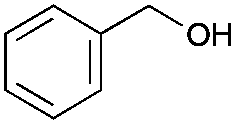
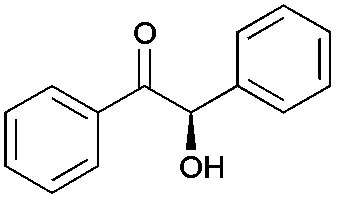
We next investigated the conversion of further benzylic alcohol substrates (Tables 1 and 2 as well as Tables S2 and S3†). To circumvent kinetic limitations observed at low substrate concentrations and to show that high product concentrations at high substrate loadings can be achieved, we formally applied 350 mM of the alcohol starting material.
| Substratea | Product | Aldehyde [%] | Yieldb [%] | eec [%] |
|---|---|---|---|---|
| Reaction conditions: The reaction mix contained the liquid alcohol substrate (350 mM), phosphate buffer (50 mM, pH 8.5) containing MgSO4 (2.5 mM), ThDP (0.15 mM) and catalase from bovine liver (2.5 mg mL−1). Crude cell extract containing PfBAL (around 5 mg, 62.72 μM, 8 U), and PpAOX (17.75 μL, 788.89 nM, 25 U), oxygen-supply, 800 rpm, 30 °C, 24 hours.a Non-soluble alcohols formed a two-liquid phase system.b Determined from 1H NMR spectra.c Enantioselectivities were determined by HPLC using enantiopure product standards or, if not available, literature data17 was used. | ||||
|
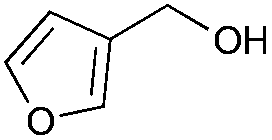
|
4 | 96 | >99 |
|
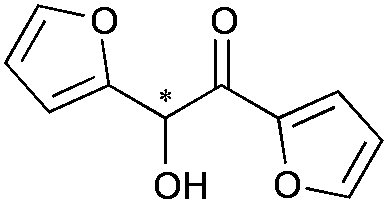
|
13 | 84 | >99 |
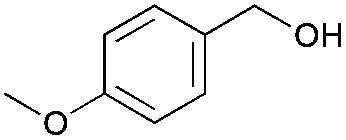
|
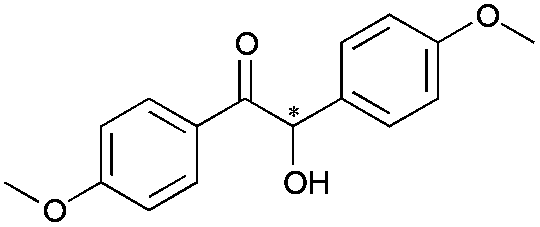
|
2 | 94 | >99 |
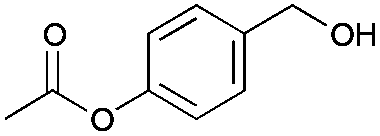
|
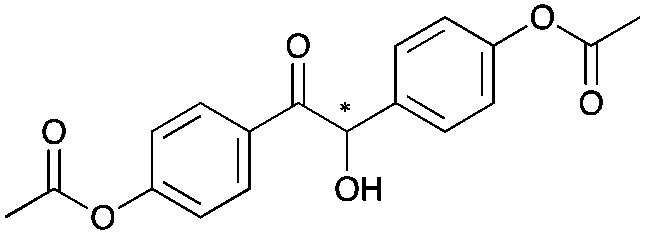
|
24 | 19 | 93 |
| Substratea | Product | Aldehyde [%] | Yieldb [%] | eec [%] |
|---|---|---|---|---|
| Reaction conditions: The reaction mix contained the solid alcohol substrate (350 mM), phosphate buffer (50 mM, pH 8.5) containing MgSO4 (2.5 mM), ThDP (0.15 mM) and catalase from bovine liver (2.5 mg mL−1). Crude cell extract containing PfBAL (5 mg, 62.72 μM, 8 U), and PpAOX (17.75 μL, 788.89 nM, 25 U), oxygen-supply, 800 rpm, 30 °C, 24 hours.a Alcohols were directly used in their solid form due to their low solubility.b Determined from 1H NMR spectra.c Enantioselectivities were determined by HPLC using enantiopure product standards or, if not available, literature data17 was used. | ||||
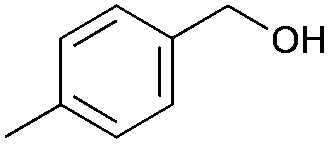
|
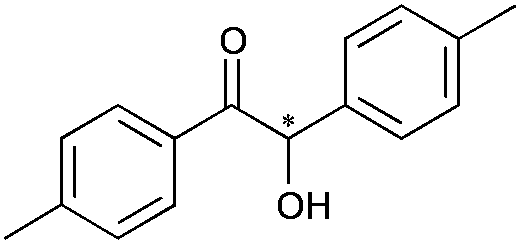
|
8 | 81 | 99 |
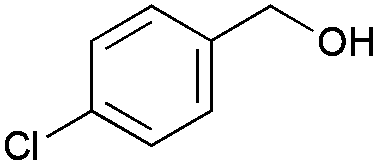
|
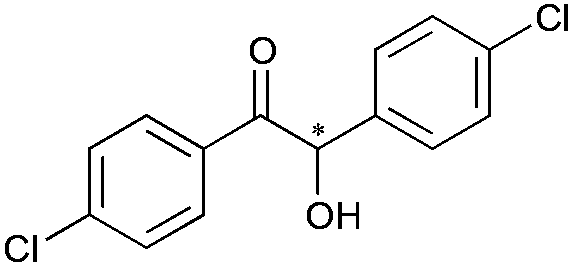
|
4 | 96 | 92 |
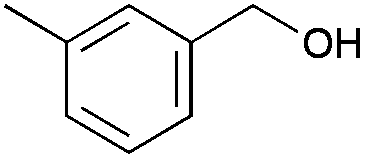
|
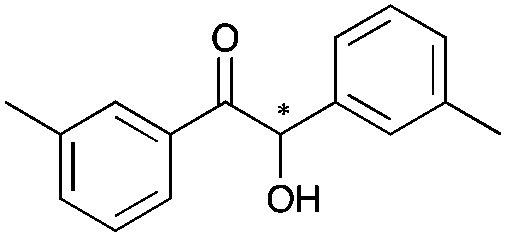
|
6 | 15 | 94 |
Liquid alcohols (Tables 1 and S2†) were added directly to the aqueous solution of the biocatalysts whereas in case of solid alcohols (Tables 2 and S3†) 5% (v/v) of 2-methyl tetrahydrofuran was added to facilitate the dissolution of the alcohols.
Especially non- or p-substituted benzyl alcohols were converted to the enantiomerically pure benzoins in good to excellent yields. Other substitution patterns lead to significantly reduced conversions and optical purities of the final products. While the poor stereoselectivity cannot be ascribed to PfBAL (always high optical purities were reported),17,18 it is interesting to note that in these cases also the concentration of the intermediate aldehyde was low indicating that the oxidation step was overall rate-limiting. Also, racemisation of some products was observed under the current reaction conditions. In case of vanillyl alcohol for instance, the ee after 72 hours of reaction is only 43%, whereas after 18 hours the ee is >99%. Optimized reaction setups (particularly adjusting the molar ratio of PpAOX and PfBAL) will enable higher conversion. It is worth mentioning here, that in all cases the final product precipitated from the reaction mixture and could be removed easily via filtration.
The same selectivity pattern was also observed in case of solid starting materials; particularly p-substituted benzyl alcohols excelled in terms of conversion and enantioselectivity. Our first attempts to perform these reactions under ‘cosolvent-free’ conditions resulted in slow reactions. Most probably, this is due to slow dissolution kinetics of the starting material. Therefore, 5% (v/v) 2-methyl tetrahydrofuran was added to accelerate the process (Table 2).
Interestingly, the presence of 2-Me-THF did not impair the facile downstream processing by simple filtration. Finally, we performed the semi-preparative scale conversion of benzyl alcohol with up to 500 mM substrate. Due to the limited solubility of benzyl alcohol and to avoid possible substrate inhibition on PpAOX, benzyl alcohol was added to the reaction mixture in 10 portions á 50 mM (adding up to a nominal final concentration of 500 mM or 54 g L−1). Almost instantaneously, the desired (R)-benzoin precipitated from the reaction mixture (Fig. S2†). After filtration, the raw product contained 5.4 mM of the intermediate aldehyde, which was easily removed by evaporation under vacuum. Overall, enantiomerically pure (R)-benzoin was obtained in 83% isolated yield (4.6 g).
Finally, we were interested on the environmental impact of the established cascade comprising PpAOX and PfBAL for the stereoselective synthesis of α-hydroxyketones. To assess the waste generation in the cascading oxidase-lyase reactions, we performed an E-factor analysis including downstream processing of the product (Table 3) and the catalyst preparation (Table S4†).19,20 Additionally, the waste generation in a similar cascade reaction reported by Perez-Sanchez et al. were calculated for comparison reasons.12
| Contribution | Enzymatic reaction with soluble producta [g] | Cascade reaction with precipitating productb [g] |
|---|---|---|
| Perez-Sanchez et al.12 | This work | |
| a Calculations were performed for 1-butanol and benzaldehyde as substrates assuming the same conversion (91%) and substrate addition (6 mM) on large scale based on the work of Perez-Sanchez et al. (2013).12 b Calculations were performed for the amount of crude product obtained (4.6 g). c Only the catalyst itself, not the cell mass which is not the catalyst was included in the calculation. | ||
| Reaction water | 250 | 250 |
| Wash water | 2000 | 150 |
| Buffer (phosphate) | 3 | 3 |
| Substrate | 30 | 2 |
| Catalase | 0.09 | 0.6 |
| PpAOXc | 0.09 | 0.2 |
| PfBALc | 0.25 | 0.1 |
| MgCl2 | 0.05 | 0.05 |
| ThDP | 0.01 | 0.01 |
| 2-Me-THF | — | 0.06 |
| Brine | 50 | — |
| EtOAc | 1500 | — |
| Sum | 3835 | 406 |
As shown in Table 3, solvents constitute the main contributors to the E-factor. First, water as the reaction medium contributes by 25 kg kg−1 and the water as the solvent for the preparation of the biocatalysts roughly by 14 kg kg−1 to the final product (Table S4†). Secondly, ethyl acetate and 2-methyl tetrahydrofuran used as co-solvents in the reaction and/or for product removal (work by Perez-Sanchez et al.)12 also significantly contribute to the overall waste formed. Nevertheless, by exploiting the poor solubility of the products, problematic solvents such as ethyl acetate for extraction were avoided simply by filtration. Furthermore, preliminary results indicate that the filtrate (containing the biocatalysts and traces of the reagents) can be re-used, thereby also reducing the overall waste-contribution of the fermentation step.
Conclusions
Enzymatic cascade reactions have gained an enormous interest in recent years as alternative synthesis strategy, which overall bears a great potential to make organic syntheses environmentally more benign. The mere use of enzymes, however, does not automatically render the reaction ‘green’ or ‘environmentally benign’ and a critical evaluation of the environmental impact of a given reaction is important to identify bottlenecks and to adapt the whole reaction setup en route to a greener process.In this study, we have demonstrated that reaction design exploiting the reagents’ properties can significantly simplify product isolation and thereby reduce or even omit organic solvents. Particularly, solid products not necessarily need to be solubilized throughout the process. In contrary, product precipitation can shift reaction equilibria and simplify product isolation. By emphasizing problems like waste generation, the search for simplified downstream processing and the need of integrated reactors our work may trigger others to think along those premises to be able to develop more examples on how biocatalysis should move on to establish greener alternatives. Especially the applicability of this systems for larger-scale reactions may also render this system attractive for greener syntheses in the future.
Acknowledgements
We thank the Netherlands Organisation for Scientific Research for financial support through a VICI grant (no. 724.014.003) TPdA is carrying out his PhD project as a Dual Degree PhD project under the agreement between UNICAMP, BE-Basic and Delft University of Technology.Notes and references
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcantara and P. Domínguez de María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS PubMed.
- J. Streuff, Synlett, 2013, 276–280 CAS.
- O. B. Wallace, D. W. Smith, M. S. Deshpande, C. Polson and K. M. Felsenstein, Bioorg. Med. Chem. Lett., 2003, 13, 1203–1206 CrossRef CAS PubMed.
- Q. K. Fang, Z. X. Han, P. Grover, D. Kessler, C. H. Senanayake and S. A. Wald, Tetrahedron: Asymmetry, 2000, 11, 3659–3663 CrossRef CAS.
- T. Tanaka, M. Kawase and S. Tani, Bioorg. Med. Chem., 2004, 12, 501–505 CrossRef CAS PubMed.
- C. Palomo, M. Oiarbide and J. M. Garcia, Chem. Soc. Rev., 2012, 41, 4150–4164 RSC.
- P. P. Giovannini, O. Bortolini and A. Massi, Eur. J. Org. Chem., 2016, 4441–4459 CrossRef CAS.
- E. Janzen, M. Muller, D. Kolter-Jung, M. M. Kneen, M. J. McLeish and M. Pohl, Bioorg. Chem., 2006, 34, 345–361 CrossRef CAS PubMed.
- H. C. Hailes, D. Rother, M. Muller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- R. Westphal, C. Vogel, C. Schmitz, J. Pleiss, M. Muller, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2015, 54, 4699–4699 CrossRef CAS.
- B. Franken, T. Eggert, K. E. Jaeger and M. Pohl, BMC Biochem., 2011, 12, 10 CrossRef CAS PubMed.
- M. Pérez-Sánchez, C. Müller and P. Domínguez de María, ChemCatChem, 2013, 5, 2512–2516 CrossRef.
- G. Dienys, S. Jarmalavicius, S. Budriene, D. Citavicius and J. Sereikaite, J. Mol. Catal. B: Enzym., 2003, 21, 47–49 CrossRef CAS.
- M. Pickl, M. Fuchs, S. M. Glueck and K. Faber, Appl. Microbiol. Biotechnol., 2015, 99, 6617–6642 CrossRef CAS PubMed.
- P. Domínguez de María, T. Stillger, M. Pohl, S. Wallert, K. Drauz, H. Gröger, H. Trauthwein and A. Liese, J. Mol. Catal. B: Enzym., 2006, 38, 43–47 CrossRef.
- P. Domínguez de María, M. Pohl, D. Gocke, H. Groger, H. Trauthwein, T. Stillger, L. Walter and M. Muller, Eur. J. Org. Chem., 2007, 2940–2944 CrossRef.
- A. S. Demir, O. Sesenoglu, E. Eren, B. Hosrik, M. Pohl, E. Janzen, D. Kolter, R. Feldmann, P. Dunkelmann and M. Muller, Adv. Synth. Catal., 2002, 344, 96–103 CrossRef CAS.
- T. Hischer, D. Gocke, M. Fernandez, P. Hoyos, A. R. Alcantara, J. V. Sinisterra, W. Hartmeier and M. B. Ansorge-Schumacher, Tetrahedron, 2005, 61, 7378–7383 CrossRef CAS.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc00020k |
| This journal is © The Royal Society of Chemistry 2017 |