
Supramolecular catalysis and dynamic assemblies for medicine
Zhaoqianqi
Feng
a,
Tengfei
Zhang
b,
Huaimin
Wang
 a and
Bing
Xu
*a
a and
Bing
Xu
*a
aDepartment of Chemistry, Brandeis University, 415 South Street, Waltham, MA 02453, USA
bDepartment of Oncology, The First Affiliated Hospital of Zhengzhou University, 1 Jianshe East Road, Zhengzhou, Henan 420052, China. E-mail: bxu@brandeis.edu
First published on 29th August 2017
Abstract
In this review, supramolecular catalysis refers to the integration of the catalytic process with molecular self-assembly driven by noncovalent interactions, and dynamic assemblies are the assemblies that form and dissipate reversibly. Cells extensively employ supramolecular catalysis and dynamic assemblies for controlling their complex functions. The dynamic generation of supramolecular assemblies of small molecules has made considerable progress in the last decade, though the disassembly processes remain underexplored. Here, we discuss the regulation of dynamic assemblies via self-assembly and disassembly processes for therapeutics and diagnostics. We first briefly introduce the self-assembly and disassembly processes in the context of cells, which provide the rationale for designing approaches to control the assemblies. Then, we describe recent advances in designing and regulating the self-assembly and disassembly of small molecules, especially for molecular imaging and anticancer therapeutics. Finally, we provide a perspective on future directions of the research on supramolecular catalysis and dynamic assemblies for medicine.
 Zhaoqianqi Feng | Zhaoqianqi Feng received her BS degree in pharmacy at Nankai University in 2014. During her undergraduate study, she studied bioactive supramolecular hydrogels under the supervision of Prof. Zhimou Yang. She is currently in her fourth year as a graduate student in Chemistry with Prof. Bing Xu at Brandeis University. Her current research focuses on designing small molecules to modulate cellular responses via enzyme-instructed self-assembly. |
 Tengfei Zhang | Tengfei Zhang obtained his MD degree in Peking Union Medical College in July 2012. From January 2011 to September 2016, he worked at Brigham Women Hospital, Beth Israel Deaconess Medical Center & Harvard Medical School as a research fellow. He is currently an oncologist and associate researcher in the Department of Oncology at the First Affiliated Hospital of Zhengzhou University. His research interests include nanostructure-mediated strategy development and related clinical trials of novel treatment for cancer patients. |
 Huaimin Wang | Huaimin Wang received his BS degree from Tianjin University in 2008. After obtaining his PhD degree in 2015 from Nankai University, he moved to the Brandeis University as a Postdoctoral-Fellow in the group of Prof. Bing Xu. His current work focuses on the bioinspired assembly of small molecules in cell milieu and small bioactive small molecule induced hydrogelation. |
 Bing Xu | After receiving his BS and MS degrees from Nanjing University in 1987 and 1990, Bing Xu obtained his PhD in 1996 from the University of Pennsylvania. Before starting his independent research at the Hong Kong University of Science and Technology (HKUST) in Aug 2000, he was an NIH postdoctoral fellow at Harvard University. He was tenured as an associated professor in Jan 2006 and became a full professor in July 2008 at HKUST. Bing Xu currently is a professor in the Department of Chemistry, Brandeis University. His research focuses on the applications of molecular engineering in materials, biology, and medicine. |
Key learning points1. Supramolecular catalysis and dynamic assemblies are ubiquitous in biology2. How to control the self-assembly and disassembly of small molecules in the context of cells 3. The applications of self-assembly and disassembly for molecular imaging 4. The applications of supramolecular catalysis and dynamic assemblies for anticancer therapeutics |
Introduction
Self-assembly and its reverse process (disassembly) are ubiquitous in nature. The self-assembly of biomolecules, driven by weak, noncovalent intermolecular interactions, generates multicomponent complexes with a high level of structural hierarchy. Many biological processes are initiated by the cooperative assembly of such supramolecular complexes, including cell locomotion,1 transcription,2 immune response,3 and apoptosis.4 A common key feature of these processes is that disassembly or dissociation, the reverse process of assembly, plays an indispensable role in modulating and terminating the actions of these supramolecular complexes. To regulate sophisticated biological functions, the assembly and disassembly processes are tightly controlled in living organisms via multiple and interconnected mechanisms. One prominent process for achieving such control is supramolecular catalysis.Supramolecular catalysis, in this review, refers to the integration of the catalytic process (e.g., enzymatic reactions) with molecular self-assembly driven by noncovalent interactions. As catalysis increases the rate of a chemical reaction, the coupling of catalysis with self-assembly affords unique kinetic control over the systems, which is highly efficient and versatile, as shown in nature. Moreover, most of the endogenous assemblies involving cellular processes are dynamic. That is, they are constantly forming and dissociating. A well-established example is the dynamics of cytoskeletons, such as constant assembling and disassembling of actin filaments. Such dynamics mediate multiple cell behaviours, including growth, motility and membrane internalization.1 For example, actin regulatory proteins bind to actins and facilitate their disassembly/assembly, like the actin depolymerizing factor (ADF)/cofilin protein family that promotes dissociation at the pointed end and the recycle of actin monomers and the Arp2/3 complex that nucleates the assembly of filaments near ruffling membranes to create branch points.5 Besides these regulatory proteins, direct catalytic processes also serve as regulatory mechanisms. A recent study demonstrates that, after activation by F-actin, the multidomain redox enzyme (Mical) oxidizes the methionine residue in actin, thus disrupting actin–actin associations and resulting in disassembly.6 Likewise, the tubulin assembly–disassembly cycle, being fueled by the hydrolysis of β-tubulin-bound GTP, plays multifaceted roles in cell division and intracellular transport,7 and is regulated by microtubule polymerases (i.e., XMAP215) and depolymerases (i.e., MCAK). Besides microtubules and actin filaments, emerging evidence reveals that inflammasomes, as a class of supramolecular organizational centers,8 form upon danger signals and disassemble when the inflammation phase ends. The rapid assembly of inflammasomes indicates that, besides enzymatic reactions, ligand–receptor interactions (or molecular recognition) are able to catalyze molecular self-assembly. These fundamental facts in cell biology reveal that assembling or oligomerization to form multicomponent complexes is the forward process to initiate many biological events, and disassembly or dissociation is the reverse process to modulate or to terminate the actions of these complexes.
The rich examples of supramolecular catalysis and dynamic assemblies of proteins in biology have stimulated research activities that are exploring the supramolecular catalysis and dynamic assemblies of small molecules, which is a new frontier of supramolecular chemistry for medicine. Over the last decade, considerable progress has been made in the field, which promises a fundamentally new approach for developing therapeutics for ameliorating human illness. Thus, it is worthwhile to review these studies for the future development of supramolecular medicine, which is an intention of this tutorial review. While the broad perspective in supramolecular biomaterials has already been covered by recent reviews,9,10 this review will mainly focus on the recent development of supramolecular catalysis and dynamic control over the assemblies. We arrange this review in the following manner: first, we provide an overview of strategies for controlling the dynamic assemblies (Fig. 1); then we highlight recent progress towards designing and controlling the assembly and disassembly of small molecules, both under cell free conditions and in the cellular environment; finally, we suggest several future directions for research on supramolecular catalysis and dynamic assemblies for medicine.
 | ||
| Fig. 1 Schematic illustration of regulating dynamic assemblies via self-assembly and disassembly processes for therapeutics and diagnostics. | ||
Rational design
The past two decades have witnessed significant progress in the development of functional nanostructures resulting from simple and basic building blocks (i.e., small molecules) that are held together via supramolecular (i.e., noncovalent) interactions. Recently, the research of supramolecular chemistry is shifting from constructing static nanostructures under thermodynamic control to bioinspired, dynamic nanosystems that are kinetic.11 Such dynamic nanosystems, with a high degree of complexity, are expected to exhibit rich and complex functions. The extensive examples of dynamic assemblies to perform sophisticate biological functions of the cells, which are an unlimited source of inspiration for scientists, provide a rationale for designing controlled dynamic assemblies via supramolecular catalysis. One of the most fundamental kinetic controls would be biological catalysts (e.g., enzymes), which play a central role in all biochemical processes, and are possibly the key elements of the origin of life. The integration of enzymatic reactions with the assembly/disassembly process enables control over the rate of assembly/disassembly by the reaction rate. Inspired by these fundamental facts, we have developed enzyme-instructed self-assembly (EISA),12 as a form of supramolecular catalysis. EISA is a powerful concept to generate functions. For example, EISA selectively generates supramolecular assemblies of small molecules13–15in situ for potential cancer therapy16,17 and molecular imaging.18 Moreover, the combination of enzyme-instructed assembly and disassembly affords a reaction cycle to switch between monomers and assemblies,19 as well as an approach to target downregulation (or loss of functions) in cancer cells.20 Hamachi and coworkers demonstrated the coupling of enzymatic reactions with stimuli responsive hydrogels for logic-gate sensing of disease-related biomarkers.21 Besides the enzymes, photocatalysts22 serve as simple tools to control the rates of the formation and breaking of assemblies via the incorporation of catalytically controlled steps in self-assembly processes.The aforementioned studies converge to the concept/design illustrated in Fig. 1. In essence, the formation of assemblies from monomers and the dissipation of the assemblies are dynamic, reversible processes, which promise applications in molecular imaging, analyte detection, anticancer therapy, and treatment of neurodegenerative diseases. Supramolecular catalysis, which utilizes enzymes10 or synthetic catalysts (i.e., acids or bases)23 to control the kinetics of assembly/disassembly, affords spatiotemporal regulation over the system. In addition, the ligand–receptor24 or protein–protein interactions25 could provide another level of precision to design controllable dynamic assemblies. In the following sections, we discuss recent studies on controlling the dynamic assembly, disassembly, or both for a variety of applications in biomedicine.
Assembly
The inverse association between cancer and neurodegenerative diseases26 suggests the feasibility of using molecular assemblies for cancer inhibition, but, due to the cytotoxicity of the assemblies to normal cells, the challenge is to inhibit cancer cells selectively. Providing spatiotemporal control, the dynamic generation of assemblies via EISA enables selective inhibition of cancer cells that exhibit high enzyme activity, which is always positively correlated with enzyme expression levels. This notion has led to the first case of designing EISA precursors according to cancer genetics. As the example illustrated in Fig. 2A, a peptidic precursor (1) turns into a hydrogelator (2) upon dephosphorylation catalyzed by overexpressed alkaline phosphatase (ALP)16 on certain cancer cells, the assemblies of peptides form selectively and in situ on cancer cells. The assemblies present autocrine proapoptotic ligands to their cognate receptors in a juxtacrine manner and directly cluster the death receptors (i.e., TRAIL, TNF-α, CD95), thus activating the extrinsic death signalling of cancer cells (Fig. 2B) and selectively inhibiting cancer cells over normal cells in a co-culture. Besides peptide derivatives, other phosphorylated small molecules (e.g., carbohydrates,15 lipids,14 or sterols13) act as substrates of EISA. For example, enzymatic dephosphorylation converts a phosphotyrosine–cholesterol conjugate (3) to a tyrosine–cholesterol conjugate (4, Fig. 2C), resulting in nanoparticles (Fig. 2D) that confirm an increased degree of noncovalent oligomerization.13 Generated by EISA, the assemblies of 4 interact with actin filaments, microtubules, and lipid rafts for selectively inducing the death of platinum-resistant ovarian cancer cells, with higher efficacy than the clinic drug cisplatin (Fig. 2E). In addition to enzymatic reactions, ligand–receptor interactions also provide precise control over the production of assemblies.24 The interaction of vancomycin with a D-Ala–D-Ala-containing peptide derivative 5 (Fig. 2F) catalyzes the aggregation of 5 (Fig. 2G), which likely induces cancer cell death via necroptosis.24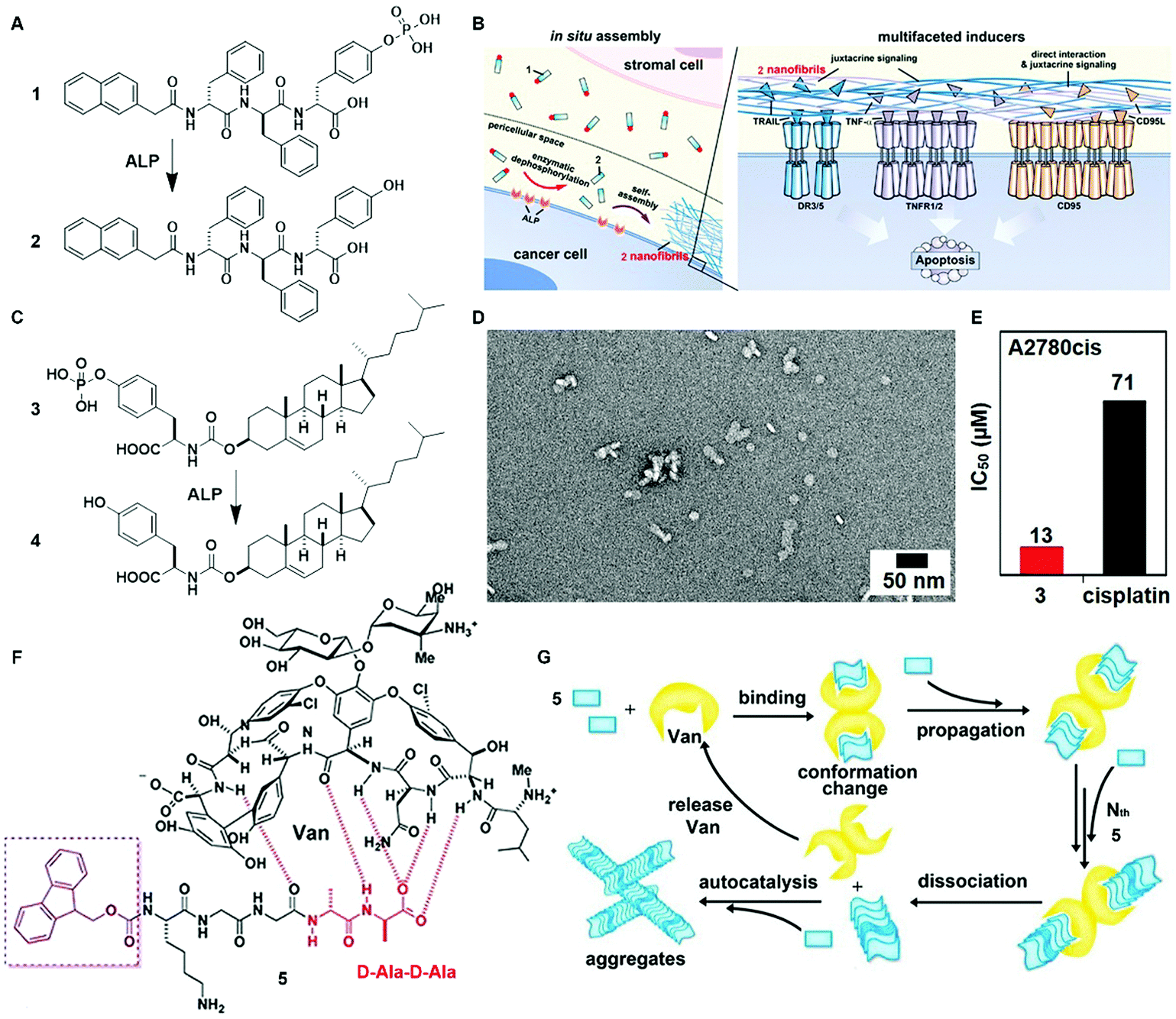 | ||
| Fig. 2 (A) Molecular structures of EISA precursor 1 and the self-assembling molecule 2 after dephosphorylation. (B) Illustration of the pericellular nanofibrils of 2 formed via EISA to selectively inhibit cancer cells in a co-culture by activating cell death signaling. (C) Molecular structure of tyrosine–cholesterol conjugate 3 which converts into 4 upon ALP treatment. (D) TEM image of the nanoparticles formed by adding ALP into 3. (E) The IC50 (48 h) values of 3 against platinum-resistant ovarian cancer cells (A2780cis) compared with the clinic drug cisplatin. (F) Chemical structures of the ligand (Van) and receptor 5 containing D-Ala–D-Ala. (G) The ligand–receptor interaction-catalyzed molecular aggregation. Adapted with permission from ref. 13, 16 and 24. Copyright 2016, American Chemical Society (ref. 13). Copyright 2017, Nature Publishing Group (ref. 16). Copyright 2015, American Chemical Society (ref. 24). | ||
The most exciting aspect of the assemblies of small molecules is that the assemblies are able to interact with multiple cellular targets or subcellular organelles, which may reduce the odds of acquired drug resistance. Being generated from precursor 6via EISA and targeting mitochondria, the assemblies of 7 selectively inhibit cancer cells (i.e., Sasos2) without inducing acquired drug resistance (Fig. 3B).17 Designed for both cell and subcellular targeting, precursor 6 consists of an environment-sensitive fluorophore 4-nitro-2,1,3-benzoxadiazole (NBD) for visualizing the assemblies, a self-assembling module (e.g., Phe–Phe–Tyr–Lys (FFYK)), an enzyme substrate (e.g., phosphotyrosine), and a mitochondria targeting motif (e.g., triphenylphosphinium (TPP)) (Fig. 3A). Being generated on Saos2 cells, the assemblies of 7 not only disrupt the dynamics of cytoskeletons, but also enter the cells via endocytosis and then partially escape from lysosomes (Fig. 3E). After internalization, the assemblies co-localize with mitochondria (Fig. 3F) and disrupt the homeostasis of mitochondria, thus triggering the release of cyt c (Fig. 3C) to activate the caspase cascade of cell death pathways. Most importantly, repeated stimulation of Saos2 cells with 6 results in similar cell viability to the unstimulated cells (Fig. 3D), suggesting that Saos-2 cells are unable to develop acquired drug resistance to 6, or more precisely, the assemblies of 7. This result illustrated the promise of multiple targeting by EISA in minimizing the acquired drug resistance.
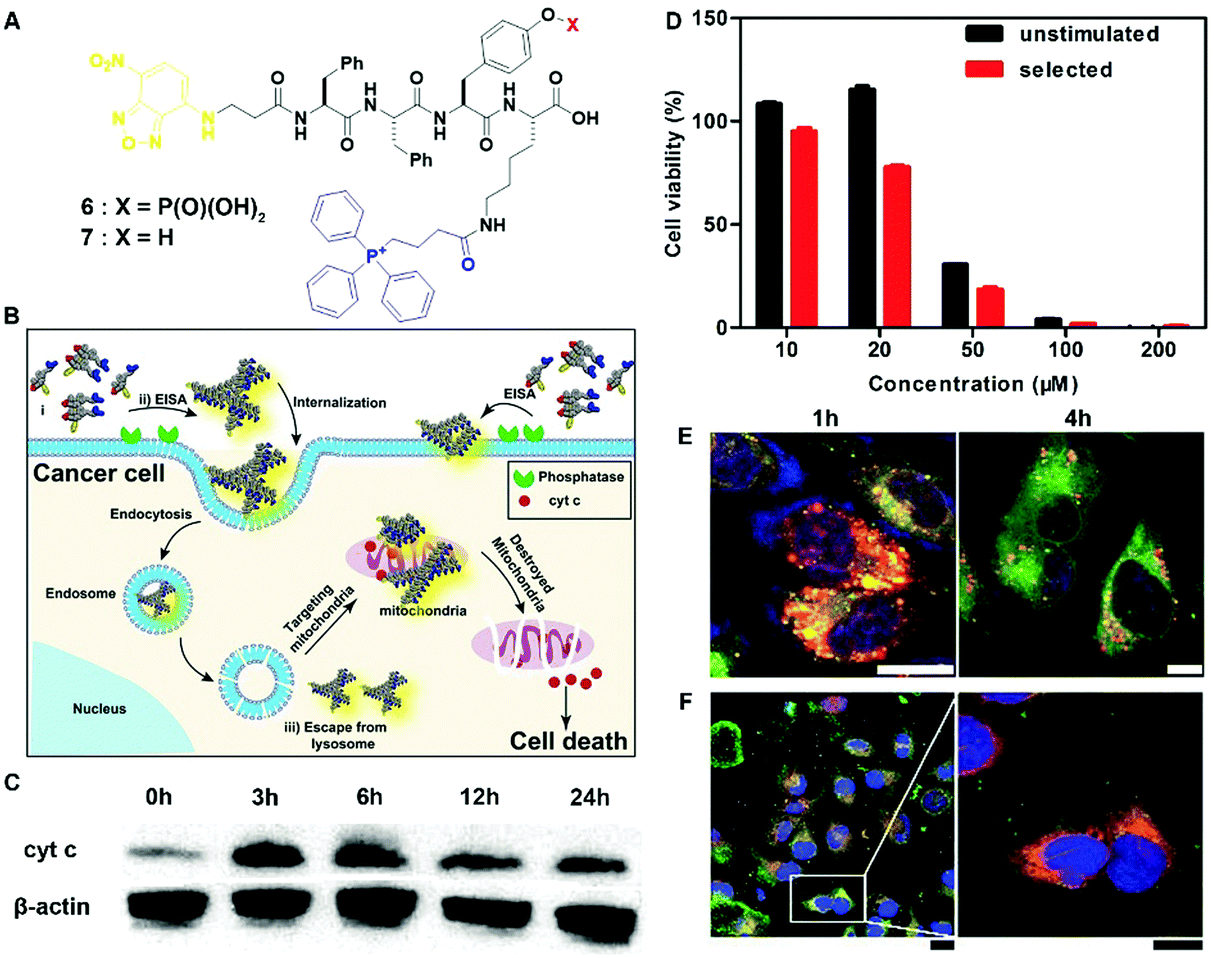 | ||
| Fig. 3 (A) Molecular structures of EISA precursor 6 and hydrogelator 7. (B) Illustration of EISA for targeting mitochondria and inducing cancer cell death. (C) Time-dependent western blot analysis of cytochrome c (cyt c) from the cytosolic fraction of Saos2 cell treated with 6 (50 μM). (D) Cell viability of the unstimulated Saos2 cell line or the selected Saos2 cell line (after five weeks treatment of 6 with gradually increasing concentrations) incubated with 6 for 48 h. (E) CLSM images of Saos2 cells treated with 6 (50 μM) for 1 or 4 h with Lyso-Tracker co-staining. The scale bar is 10 μm. (F) CLSM images of Saos2 cells treated with 6 (50 μM) for 4 h, with Mito-tracker co-staining. The scale bar for low magnification is 25 μm and for higher is 15 μm. Adapted with permission from ref. 17. Copyright 2016, American Chemical Society. | ||
Besides the above examples of utilizing supramolecular catalysis to regulate the self-assembly, another promising strategy is to control the conversion of assemblies. Ulijn et al. reported a peptide based enzyme-responsive system which undergoes a morphological switch from micellar aggregates to fibers via the catalysis of matrix metalloproteinases (MMPs).27 The enzyme-triggered micelle to fiber transition allows the localized encapsulation and controlled release of doxorubicin, serving as a potential carrier for drug delivery. More recently, Ulijn et al. demonstrated a biocatalytic self-assembly cascade regulated by two kinetically competing enzymatic reactions,28 where the structure, composition and morphology of the nanostructures formed are dynamically controlled by the sequence and ratio of two biocatalysts (e.g., phosphatase and thermolysin).
Disassembly
While the above examples report forming assemblies, the reverse process, disassembly, is underexplored, despite its importance. Hamachi et al. reported the combination of enzymatic reactions with a H2O2-responsive hydrogel for the detection of variety of biomarkers through the naked eye.21 As shown in Fig. 4A, peptide precursor 8, with a p-boronophenylmethoxycarbonyl (BPmoc) group at the N-terminus, reacts with H2O2 to generate unmodified peptide 9 and p-quinonemethide. The decomposition of the BPmoc group diminishes the intermolecular interactions and disassembles the nanofiber network of 8, resulting in a gel–sol transition that is capable of sensing analytes. Encapsulating oxidases (e.g., a glucose oxidase (GOx)), the hydrogel matrix of 8 degrades upon the generation of H2O2 resulting from the addition of the substrate (e.g., glucose) of GOx (Fig. 4B). Such a design allows the selective degradation of hydrogel–enzyme hybrids when there are specific biomolecules. For example, the GOx containing hydrogel only responds to glucose. The selective responsiveness of the hydrogel–enzyme hybrids suggests that it is feasible to incorporate other enzymes for sensing a variety of biomarkers.29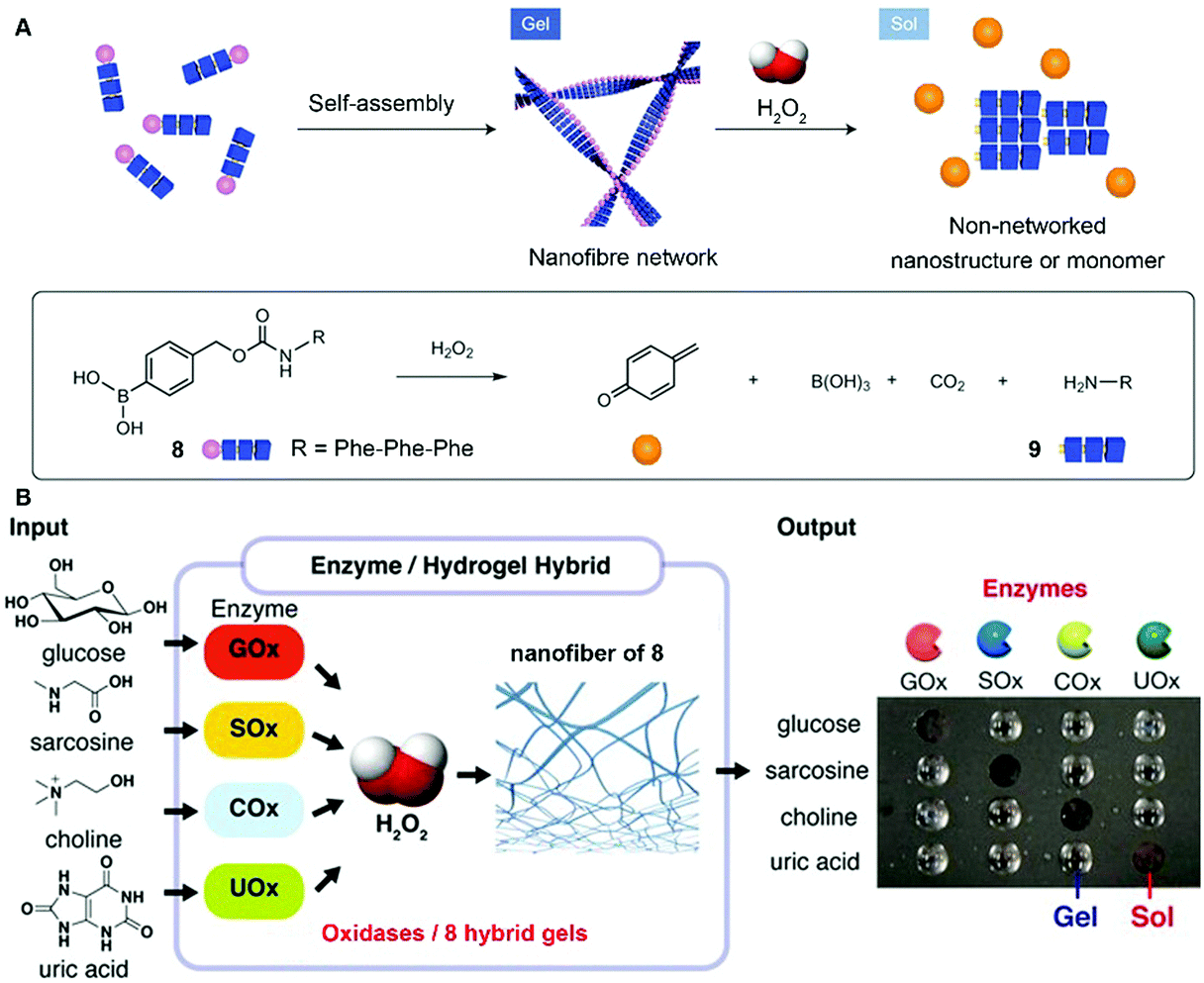 | ||
| Fig. 4 (A) Schematic illustration of the self-assembly of 8 to form a nanofiber network (gel) and its degradation to 9 upon H2O2 oxidation. (B) Biomolecule-responsive supramolecular hydrogels consisting of H2O2-responsive gel fibers of 8 and oxidases. Oxidases receive the inputs and convert the corresponding substrates generating H2O2 as a byproduct, which eventually gives rise to a gel–sol change as output through the degradation of gel fibers. Adapted with permission from ref. 21 and 29. Copyright 2014, Nature Publishing Group (ref. 21). Copyright 2017, American Chemical Society (ref. 29). | ||
Besides the disassembling based on enzymatic actions, molecular assemblies that respond to nonenzymatic proteins significantly expand the scope of the disassembly approach.30 Thayumanavan et al. demonstrated that the nanoassemblies of a ligand-decorated polypeptide P10 were programmed to disassemble in response to specific proteins via ligand–protein interactions (Fig. 5A).31 Driven by hydrophobic forces, the benzenesulfonamide containing polypeptide P10 self-assembles to form spherical nanoparticles as shown in Fig. 5B. The addition of bovine carbonic anhydrase II (bCA-II), which specifically binds to benzenesulfonamide, significantly decreases the size of the assemblies due to the change in the hydrophilic–lipophilic balance (Fig. 5C). Such a design allows protein-specific delivery, like the example shown in Fig. 5D in which the fluorescent dye DiI is released from P10 assemblies due to the disassembly resulting from the specific ligand–protein interaction.
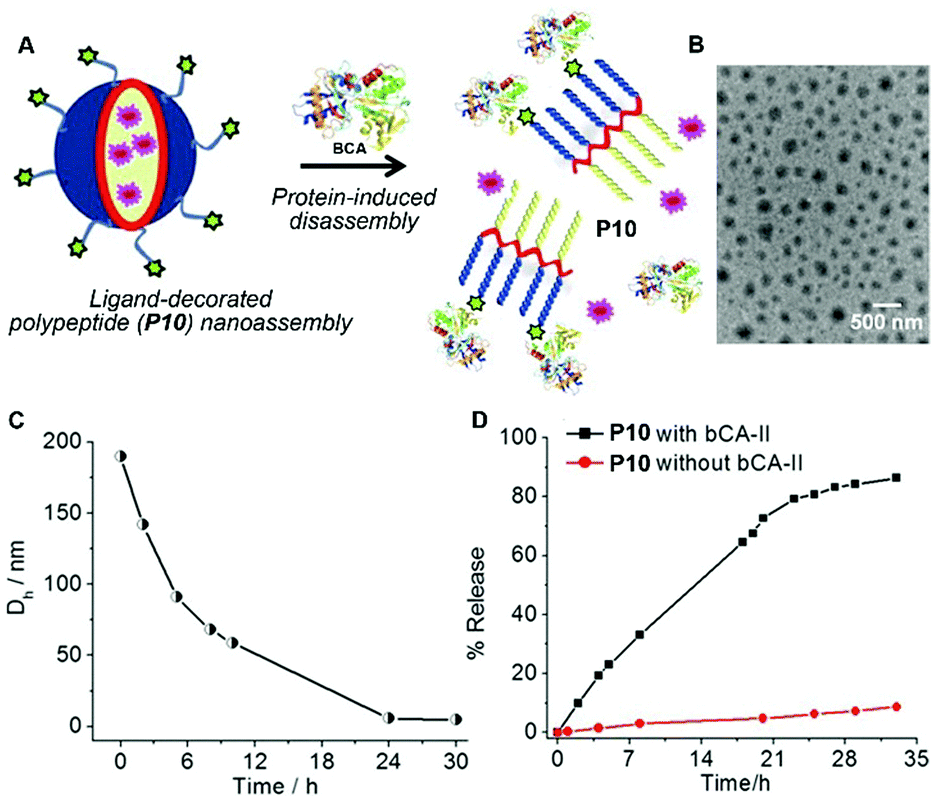 | ||
| Fig. 5 (A) Schematic representation of the protein binding induced disassembly of the supramolecular assemblies of polypeptide P10. (B) TEM image of spherical assemblies of P10. (C) Time-dependent size variation profile of the particle of P10 in the presence of the bovine carbonic anhydrase II (bCA-II) protein. (D) Percentage of guest molecules released from assemblies of P10 in the presence and absence of bCA-II. Adapted with permission from ref. 31. Copyright 2015, American Chemical Society. | ||
The recent progress in the development of synthetic peptides to mimic the protein interactions32 indicates the usefulness of small peptides in controlling the dynamic assemblies via non-covalent interactions. For example, the binding between glycoconjugate 12 and peptide 11 disrupts the self-assembly of 11 to generate monomers of 11 for proteolysis, thus accelerating the degradation of molecular nanofibrils (Fig. 6A).33 Serving as a model system of amyloids of pathogenic peptides or proteins, peptide 11 self-assembles in aqueous solution to form a hydrogel that consists of nanofibrils with a width of 9 ± 2 nm (Fig. 6B). The addition of four equivalent glycopeptides 12 into the hydrogel of 11 promotes a gel–sol transition to yield a solution with thinner and less-entangled nanofibrils (Fig. 6C), indicating that the binding between 11 and 12 disrupts the nanofibrils of 11. The analysis of the proteolytic stability (Fig. 6D) shows that the binding of 12 dramatically increases the degradation of the hydrogel of 11, which bears proteolysis resistance. This work illustrates a new method of using glycopeptides to assist in the elimination of cytotoxic oligomers of peptides or aberrant proteins for potential applications such as the treatment neurodegenerative diseases.
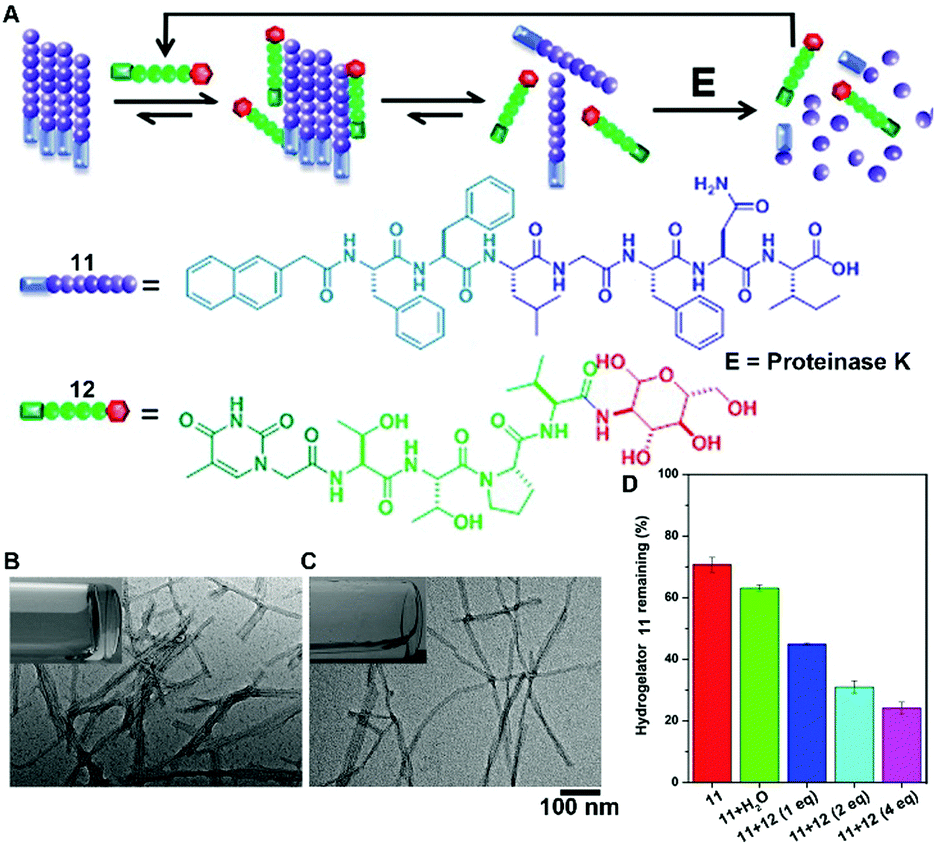 | ||
| Fig. 6 (A) Illustration of supramolecular glycosylation to assist the dissociation of molecular nanofibrils and acceleration of the peptide nanofibril degradation via supramolecular interactions. Molecular structures of 11 and 12. TEM images of (B) the hydrogel of 11 and (C) solution of 11 with 4 equiv. of 12 (insets: corresponding optical images). (D) Percentage of hydrogelator 11 remaining in the mixtures of 11 and 12 after treatment with proteinase K (3.2 U mL−1) at 37 °C for 24 h. Adapted with permission from ref. 33. Copyright 2015, American Chemical Society. | ||
Assembly and disassembly
The dynamic consequences of the interplay between assembly and disassembly play crucial roles in many complex physiological responses via precise spatiotemporal control. The regulations of such systems often rely on the activity of a pair of counteractive enzymes (i.e., enzyme switch). For example, reversible phosphorylation and dephosphorylation control the disassembly and reassembly of nuclear lamins during mitosis.34 The first synthetic example, inspired by reversible phosphorylation catalyzed by kinase/phosphatase, is the enzymatic switch between hydrogelator 13 and precursor 14 to regulate supramolecular hydrogels.19 As shown in Fig. 7A, a kinase phosphorylates 13 to yield 14, and the dephosphorylation of 14 by a phosphatase results in 13. The hydrogelator (13) self-assembles in aqueous solution to form a hydrogel (Fig. 7C), and the kinase converts 13 to 14, leading to a gel–sol transition (Fig. 7D). The addition of the phosphatase into the solution in Fig. 7D restores the hydrogel (Fig. 7E). This result confirms the use of an enzymatic switch to control the phase transition of supramolecular hydrogels. Moreover, the subcutaneous injection of 14 results in the formation of a supramolecular hydrogel in vivo (Fig. 7B). These results promise a new approach for designing dynamic assemblies via regulating the assembly and disassembly processes, which are controlled by a pair of counteractive enzymes. Using a similar approach, Stupp et al. employed a pair of counteractive enzymes for switching the self-assembly in peptide amphiphile nanostructures.35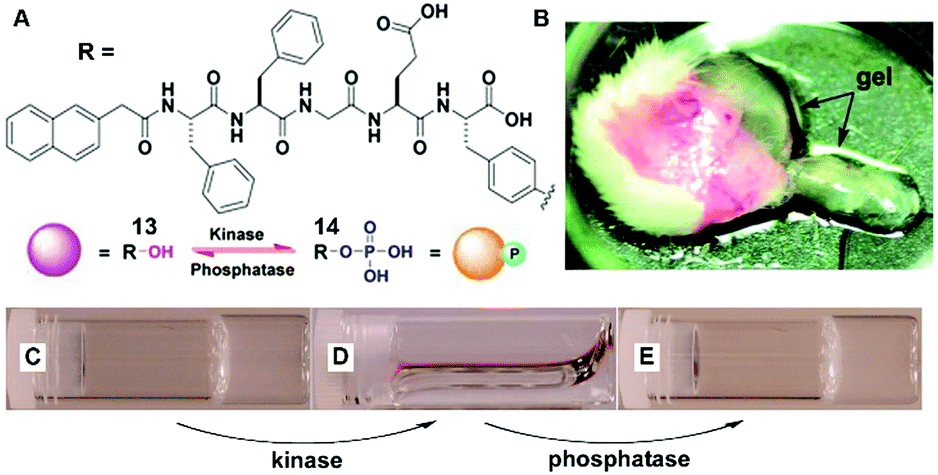 | ||
| Fig. 7 (A) Molecular structures of kinase substrate 13 and phosphatase substrate 14, this pair of counteractive enzymes affords an enzymatic switch between 13 and 14; (B) optical image of the hydrogel formed subcutaneously after injecting 14 for 1 h into the mice. Optical images of (C) the gel of 13; (D) the solution formed by adding kinase to (C); and (E) the gel restored by adding phosphatase into solution of (D). Adapted with permission from ref. 19. Copyright 2006, American Chemical Society. | ||
The combination of enzyme-instructed assembly and disassembly regulates the assembling states of molecular assemblies, providing a way to target the downregulation (or loss-of-function) in cancer cells, which remains a challenge in translational medicine.20 As the substrate of both carboxylesterases (CESs) and ALP, precursor 15 turns into 16 upon the treatment of ALP, converts into 17 by the ester bond cleavage catalyzed by CES, and generates 18via the actions of ALP and CESs (Fig. 8A). The measurement of the critical micelle concentration (CMC) shows that the self-assembling ability of above four compounds follows the order of 16 > 15 > 18 > 17 (Fig. 8D). Such a design allows ALP to convert precursor 15 to self-assembling molecule 16 to form nanofibrils, and CESs to convert 16 to 18 to result in the dissociation of the nanofibrils (Fig. 8C). Regulated by the ALP and CESs on cancer cells, precursor 15 selectively generates the assemblies of 16 on cancer cells expressing ALP and downregulating CESs (e.g., OVSAHO), thus inhibiting the OVSAHO cells while being innocuous to the cells that upregulate CESs (e.g., HepG2) (Fig. 8B). Moreover, the addition of CES inhibitors significantly increases the cytotoxicity of 15 against HepG2 cells, but hardly shows any effect on the viability of OVSAHO, suggesting the critical role of the action of CESs in selectively inhibiting the cancer cells (Fig. 8E).
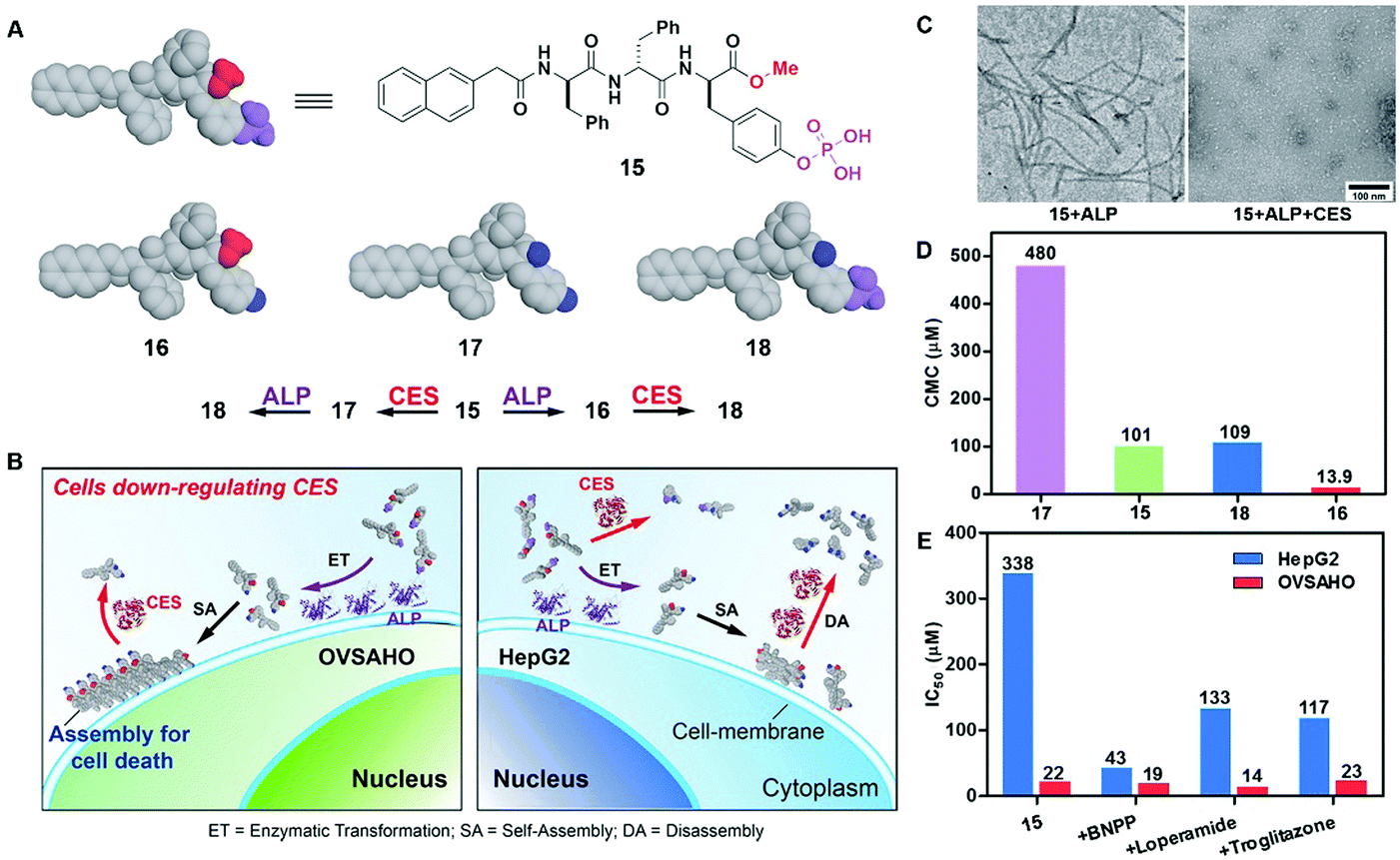 | ||
| Fig. 8 (A) Structures of precursor 15 and its hydrolysis products 16, 17 and 18. (B) Illustration of the concept of targeting the cells that downregulate carboxylesterases (CESs). (C) TEM of the nanostructures formed by 15 (100 μM) after the addition of ALP or both ALP and CESs in PBS (pH 7.4) (D) CMCs for 15, 16, 17 and 18. (E) IC50 values (at 72 h) of 15 against HepG2 or OVSAHO cells without/with addition of the inhibitors of esterases: BNPP (nonspecific), loperamide (CES2), and troglitazone (CES1). Adapted with permission from ref. 20. Copyright 2017, American Chemical Society. | ||
In addition to enzyme-switched reversible self-assembly and disassembly processes of supramolecular assemblies, a self-assembling nanoprobe, developed by Hamachi et al., is able to sense intracellular protein dynamics based on specific ligand–protein interaction.36 As shown in Fig. 9C, probe 19 consists of a ligand (i.e., methotrexate) for targeting dihydrofolate reductase (eDHFR), a fluorophore (i.e., tetramethylrhodamine) for imaging, a hydrophobic module (i.e., phenylalanine) for tuning the self-assembling ability, and a linker. In aqueous solution, probe 19 spontaneously forms spherical or oval aggregates (Fig. 9B), suppressing its fluorescence. The specific ligand–protein recognition causes the disassembly of the aggregates to light up the fluorescence, while a competitive inhibitor exchanges the probe to result in aggregates and a decrease of the fluorescence (Fig. 9A). Incubating with HeLa cells overexpressing GFP-fused eDHFR (HeLa-DG cell), strong intracellular fluorescence co-localizes with GFP fluorescence, suggesting that the binding between eDHFR and the ligand induces the disassembly of aggregates (Fig. 9D). Moreover, the release of probe 19, driven by the ligand exchange by a competitive inhibitor, significantly decreases the fluorescence inside the cells (Fig. 9D).
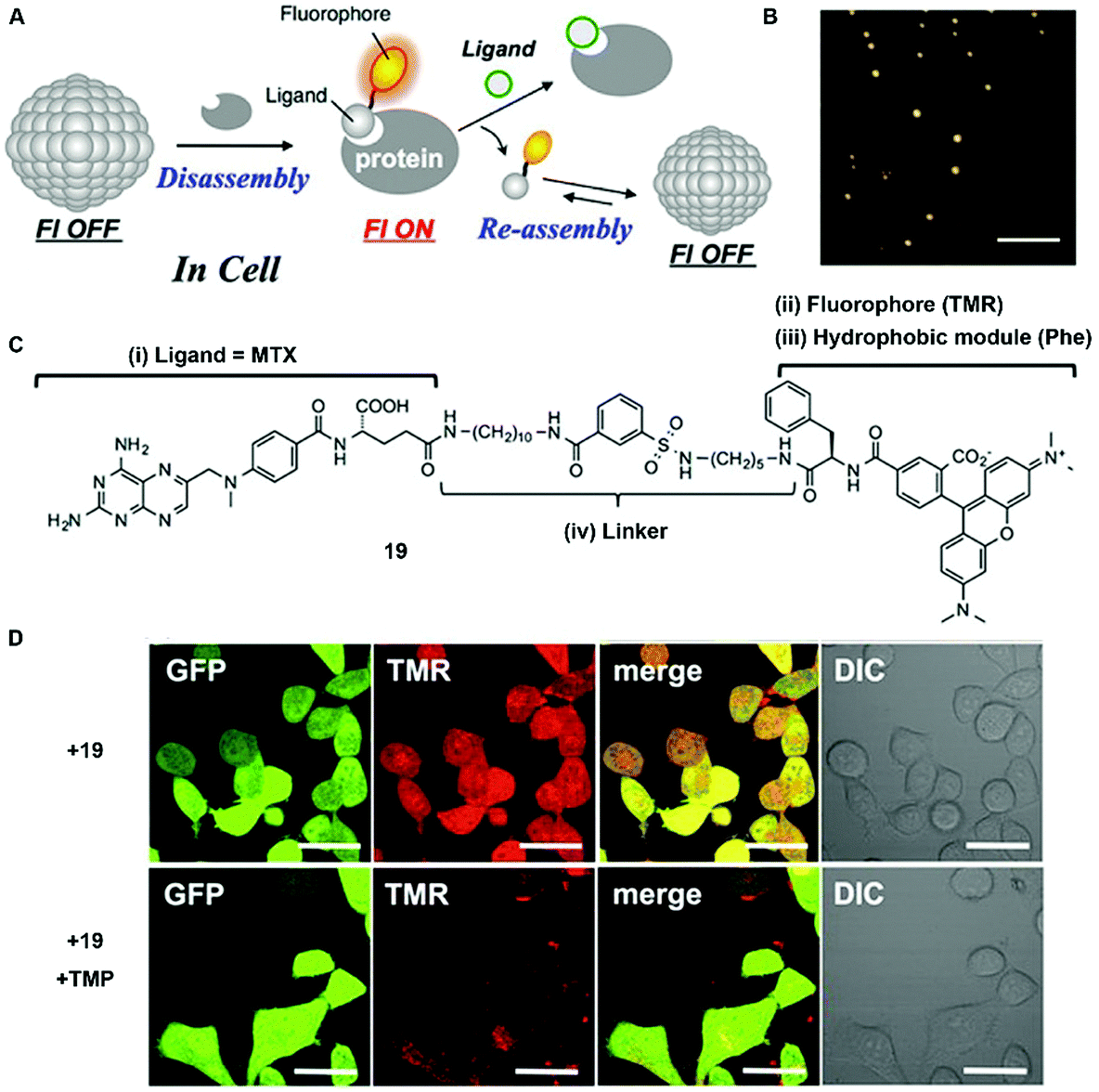 | ||
| Fig. 9 (A) Schematic illustration of protein-responsive nanoaggregates for specific and reversible intracellular protein sensing. (B) Atomic force microscopy (AFM) image of the self-assembled probe 19 (10 μM) (scale bar 500 nm). (C) Chemical structure of ligand-tethered probe 19 for intracellular eDHFR imaging. (D) CLSM images of the HeLa cell line stably overexpressing GFP-fused eDHFR (HeLa-DG cell) treated with probe 19 (2 μM) for 12 h at 37 °C or with probe 19 (2 μM) for 12 h at 37 °C, and then treated with TMP (10 μM) (scale bars, 20 μm). Adapted with permission from ref. 36. Copyright 2015, American Chemical Society. | ||
Future and outlook
The rapid development in structural biology has shed light on the structure and dynamics of the higher order assemblies, which serve as signalling complexes and directly control their biological functions via the switch of assembling states in living cells.37 Moreover, disrupting the dynamics of these assemblies often associates with pathological conditions, for example, the mutations of heterogeneous nuclear ribonucleoproteins potentiate the conversion to amyloid states in degenerative diseases (e.g., amyotrophic lateral sclerosis).38 These new insights into biology provide a scientific premise not only for studying dynamic assemblies and supramolecular catalysis, but also for developing novel molecular assemblies, based on dynamic assemblies and supramolecular catalysis, as new medicine, particularly for cancer therapy and neurodegenerative diseases.Cancer remains the major public health problem worldwide due to the ubiquitous genomic instability and complex microenvironment of tumors.39 In the clinic, the invasion and metastasis found at the diagnosis, off-target effects, side-effects from the damages to normal cells, the quickly developed drug resistance, and persisted activation of alternative signalling pathways all limit the benefits of traditional chemotherapy and molecular therapy for cancer patients. Because inefficient drug delivery and non-specific distribution is the first barrier for cancer treatment, hydrogels, as the direct consequence of supramolecular catalysis, can serve as promising carriers for chemotherapy regimen delivery, either encapsulating drugs in hydrogels or conjugating drugs covalently with the hydrogelators.40 This approach promises high selectivity, sustainable release, lower therapeutic dosage, boosted efficiency, and minimized adverse effects. Moreover, supramolecular catalysis and dynamic assemblies are uniquely suitable for actively targeting cancer cells by taking the advantages of the endogenic enzyme activities of cancer cells. One area that deserves more attention is to integrate metabolic reprogramming of cancer cells, an important cancer hallmark,39 with supramolecular catalysis and dynamic assemblies. In addition, it is worthwhile to explore supramolecular catalysis and dynamic assemblies in the context of personalized medicine because the trigger-responsive delivery of active regimens is essential for targeting the real-time genetics and disease status and maintaining safety dosage. As a promising frontier of supramolecular chemistry, more sophisticated supramolecular assembling systems harboring endogenic anticancer effects in vivo may ultimately lead to another “breakthrough” in cancer treatment.
Another promising and challenging area for applying supramolecular catalysis and dynamic assemblies in clinical cancer therapy would be cancer immunotherapy. Cancer immunotherapy, aiming to harness patients' own immune system to fight cancer, is emerging as the forth treatment for cancer after surgery, radiation, and chemotherapy.41 Because immunotherapies depend highly on the activity of the immune system itself, developing supramolecular catalysis and dynamic assemblies for increasing the response rates of cancer patients to cancer immunotherapies would lead to great clinical benefit. But this area remains exploration. The cross talk from the microenvironment is critical for carcinogenesis. Thus, the complicated and dynamic environment may alter both the pathology in situ and supramolecular assemblies in vivo. Therefore, how to translate them to cancer patients will be extremely challenging. The controllable and reproducible drug synthesis, pharmacokinetics, pharmacodynamics, potential adverse effects, and patient protocols of dynamic assemblies still need more studies. Nevertheless, we expect that supramolecular catalysis and dynamic assemblies would bring creative innovation for cancer therapy.
Besides the developments in cancer therapy based on the in situ generation of assemblies,42 manipulating dynamic assemblies may help to understand neurodegenerative diseases33 and to treat bacterial infections.43 Associating with the accumulation of protein aggregates in a variety of forms, neurodegenerative diseases remain incurable due to the difficulties in understanding the pathogenic assemblies, especially their dynamic process.44 While the functional assemblies have extremely tight regulation of the assembly/disassembly process,45 the accumulation of pathological assemblies may result from dysfunction in regulation mechanisms. Therefore, the precise control of dynamic assemblies is necessary and critical in developing novel therapies, especially in the controlled disassembly process.
Utilizing dynamic assemblies for therapeutics requires solving several challenges, including how to achieve spatial control and precise regulation of assembly/disassembly in living organisms. For functional assemblies, their synthesis, degradation and post-translational modification are tightly regulated in cellular systems,46 suggesting the rationale to employ supramolecular catalysis for controlling the dynamic assemblies. Based on enzymatic transformation, EISA provides a methodology to instruct assemblies of small molecules in complex and dynamic biological environments such as cells.47 Despite the recent progress in the instruction of dynamic assemblies, regulation of their disassembly remains rather unexplored except in the field of controlled drug release.48 The dynamic switch between assembling and dissociating states is essential to allow sensitive responses to small changes in the system.49 Coupling catalysis reactions enables the construction of hierarchical networks that mimic the cellular signal transduction cascades with feedback loops.49 In addition to enzymatic control, allosteric regulation is another universal mechanism to modulate the assembling states of proteins. Moreover, the concurrence of enzymatic reactions and ligand–receptor interactions provides a new strategy to design sophisticated molecular systems under dual control.50 With more understanding of the self-assembling process and crystal structures of biomacromolecules in different intermediate states at the atomic level, dynamic assemblies provide a platform technology for developing next generation biomaterials with precisely controlled functions. In addition, addressing these challenges requires the interdisciplinary collaboration of chemists, molecular biologists, physicists, physiologists, structural biologists, clinicians as well as computer scientists.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the NIH (R01CA142746) and NSF (MRSEC-1420382).Notes and references
- T. D. Pollard and J. A. Cooper, Science, 2009, 326, 1208–1212 CrossRef CAS PubMed.
- B. Lemon and R. Tjian, Genes Dev., 2000, 14, 2551–2569 CrossRef CAS PubMed.
- E. Latz, T. S. Xiao and A. Stutz, Nat. Rev. Immunol., 2013, 13, 397–411 CrossRef CAS PubMed.
- Y. G. Shi, Curr. Opin. Cell Biol., 2006, 18, 677–684 CrossRef CAS PubMed.
- C. G. Dos Remedios, D. Chhabra, M. Kekic, I. V. Dedova, M. Tsubakihara, D. A. Berry and N. J. Nosworthy, Physiol. Rev., 2003, 83, 433–473 CrossRef CAS PubMed.
- R. J. Hung, C. W. Pak and J. R. Terman, Science, 2011, 334, 1710–1713 CrossRef CAS PubMed.
- A. Akhmanova and M. O. Steinmetz, Nat. Rev. Mol. Cell Biol., 2015, 16, 711–726 CrossRef CAS PubMed.
- A. Lu and H. Wu, FEBS J., 2015, 282, 435–444 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- J. Zhou and B. Xu, Bioconjugate Chem., 2015, 26, 987–999 CrossRef CAS PubMed.
- H. M. Wang, Z. Q. Q. Feng, D. D. Wu, K. J. Fritzsching, M. Rigney, J. Zhou, Y. J. Jiang, K. Schmidt-Rohr and B. Xu, J. Am. Chem. Soc., 2016, 138, 10758–10761 CrossRef CAS PubMed.
- A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS PubMed.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- X. Du, J. Zhou, H. Wang, J. Shi, Y. Kuang, W. Zeng, Z. Yang and B. Xu, Cell Death Dis., 2017, 8, e2614 CrossRef CAS PubMed.
- H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046–16055 CrossRef CAS PubMed.
- Y. Gao, J. Shi, D. Yuan and B. Xu, Nat. Commun., 2012, 3, 1033 CrossRef PubMed.
- Z. M. Yang, G. L. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS PubMed.
- Z. Feng, H. Wang, R. Zhou, J. Li and B. Xu, J. Am. Chem. Soc., 2017, 139, 3950–3953 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- J. Shi, X. Du, Y. Huang, J. Zhou, D. Yuan, D. Wu, Y. Zhang, R. Haburcak, I. R. Epstein and B. Xu, J. Am. Chem. Soc., 2015, 137, 26–29 CrossRef CAS PubMed.
- Y. S. Bai, Q. Luo and J. Q. Liu, Chem. Soc. Rev., 2016, 45, 2756–2767 RSC.
- J. A. Driver, A. Beiser, R. Au, B. E. Kreger, G. L. Splansky, T. Kurth, D. P. Kiel, K. P. Lu, S. Seshadri and P. A. Wolf, Br. Med. J., 2012, 344, e1442 CrossRef PubMed.
- D. Kalafatovic, M. Nobis, N. Javid, P. W. J. M. Frederix, K. I. Anderson, B. R. Saunders and R. V. Ulijn, Biomater. Sci., 2015, 3, 246–249 RSC.
- J. K. Sahoo, C. G. Pappas, I. R. Sasselli, Y. M. Abul-Haija and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 6828–6832 CrossRef CAS PubMed.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740–750 CrossRef CAS PubMed.
- M. A. Azagarsamy, V. Yesilyurt and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 4550–4551 CrossRef CAS PubMed.
- M. R. Molla, P. Prasad and S. Thayumanavan, J. Am. Chem. Soc., 2015, 137, 7286–7289 CrossRef CAS PubMed.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2493 CrossRef CAS PubMed.
- D. Yuan, J. F. Shi, X. W. Du, N. Zhou and B. Xu, J. Am. Chem. Soc., 2015, 137, 10092–10095 CrossRef CAS PubMed.
- S. Guttinger, E. Laurell and U. Kutay, Nat. Rev. Mol. Cell Biol., 2009, 10, 178–191 CrossRef PubMed.
- M. J. Webber, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2011, 7, 9665–9672 RSC.
- T. Yoshii, K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2014, 136, 16635–16642 CrossRef CAS PubMed.
- H. Wu and M. Fuxreiter, Cell, 2016, 165, 1055–1066 CrossRef CAS PubMed.
- A. Molliex, J. Temirov, J. Lee, M. Coughlin, A. P. Kanagaraj, H. J. Kim, T. Mittag and J. P. Taylor, Cell, 2015, 163, 123–133 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS PubMed.
- D. N. Soft MatterCellKhalil, E. L. Smith, R. J. Brentjens and J. D. Wolchok, Nat. Rev. Clin. Oncol., 2016, 13, 273–290 CrossRef PubMed.
- R. Gallardo, M. Ramakers, F. De Smet, F. Claes, L. Khodaparast, L. Khodaparast, J. R. Couceiro, T. Langenberg, M. Siemons, S. Nystrom, L. J. Young, R. F. Laine, L. Young, E. Radaelli, I. Benilova, M. Kumar, A. Staes, M. Desager, M. Beerens, P. Vandervoort, A. Luttun, K. Gevaert, G. Bormans, M. Dewerchin, J. Van Eldere, P. Carmeliet, G. Vande Velde, C. Verfaillie, C. F. Kaminski, B. De Strooper, P. Hammarstrom, K. P. R. Nilsson, L. Serpell, J. Schymkowitz and F. Rousseau, Science, 2016, 354, aah4949 CrossRef PubMed.
- Z. Yang, G. Liang, Z. Guo, Z. Guo and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 8216–8219 CrossRef CAS PubMed.
- D. W. Sanders, S. K. Kaufman, B. B. Holmes and M. I. Diamond, Neuron, 2016, 89, 433–448 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annual Review of Biochemistry, 2006, vol. 75, pp. 333–366 Search PubMed.
- J. Gsponer and M. M. Babu, Cell Rep., 2012, 2, 1425–1437 CrossRef CAS PubMed.
- H. M. Wang, Z. Q. Q. Feng and B. Xu, Chem. Soc. Rev., 2017, 46, 2421–2436 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- B. N. Kholodenko, Nat. Rev. Mol. Cell Biol., 2006, 7, 165–176 CrossRef CAS PubMed.
- R. Haburcak, J. Shi, X. Du, D. Yuan and B. Xu, J. Am. Chem. Soc., 2016, 138, 15397–15404 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |