
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective binding in different adsorption sites of a 2D covalent organic framework†
D.
Cui
 a,
J. M.
MacLeod
*ab,
M.
Ebrahimi
a and
F.
Rosei
*ac
a,
J. M.
MacLeod
*ab,
M.
Ebrahimi
a and
F.
Rosei
*ac
aCentre Énergie, Matériaux et Télécommunications, Institut National de la Recherche Scientifique, 1650 Boulevard Lionel-Boulet, Varennes, Québec J3X 1S2, Canada. E-mail: rosei@emt.inrs.ca
bSchool of Chemistry, Physics, and Mechanical Engineering, Queensland University of Technology, Brisbane, 4000 QLD, Australia. E-mail: jennifer.macleod@qut.edu.au
cInstitute of Fundamental and Frontier Science, University of Electronic Science and Technology of China, Chengdu 610054, PR China
First published on 24th March 2017
Abstract
This study shows that surface-supported two-dimensional (2D) porous covalent organic frameworks (COFs) can selectively bind different molecules at specific sites via different types of interactions. Scanning tunneling microscopy (STM) images collected at the liquid/solid interface reveal the adsorption of 1,2,4-trichlorobenzene (TCB) in the hexagonal pore of a COF-1 template. A well-defined loop boundary formed by a chain of pentagonal and heptagonal pores allowed the investigation of the effect of pore shape and size on TCB adsorption, suggesting that both geometrical and size effects are important in binding TCB. When both C60 and TCB are present at the solution/solid interface, the TCB molecules are selectively trapped in the pore-site, whereas fullerenes are adsorbed on the top-site of COF-1. While the former structure is stabilized by Cl⋯H hydrogen bonds, the latter is controlled by van der Waals interactions. These results suggest that COFs may offer a powerful platform for the recognition and patterning of guest molecules.
Introduction
Host/guest (H/G) chemistry in two-dimensional (2D) supramolecular networks has been extensively studied as a means to immobilize a variety of guest molecules, with potential applications in separation technology, molecular recognition, sensing, catalysis and nanoscale patterning.1,2 In general, H/G networks rely on hierarchical interactions: the interactions binding the host together are stronger than the interactions binding the guest to the host. The host networks are typically sustained by hydrogen bonding,3–6 van der Waals (vdW) forces,7,8 or metal–ligand interactions,9 whereas the adsorption of guest molecules mainly occurs via weaker interactions, typically dispersive interactions with the host as well as with the underlying substrate. However, moving away from self-assembled templates may offer some new opportunities for the stabilization of guest molecules. 2D covalent organic frameworks (COFs) have recently been explored as host systems,10–12 opening the possibility of using stronger interactions to stabilize molecules into pores. COF-1, a well ordered 2D porous material that can be synthesized through the on-surface polycondensation of benzenediboronic acid (BDBA),13 is a promising candidate for this application.The mutually specific recognition between the host network and the guest molecules, i.e., a form of molecular recognition, is at the core of host–guest chemistry.14,15 Design strategies for selectivity in guest bonding have often relied on tuning the host geometry, such as pore size,16,17 shape18 and chirality.19 Much less is known about design based on the interaction between a host and a guest.20 This is mainly due to limitations of hierarchical interaction strength imposed by self-assembled host templates. This aspect can be addressed by using a covalently-bonded template, such as COF-1, which enables a route to molecular recognition through a range of different interactions, including hydrogen bonding, halogen bonding or vdW interactions.
Here, we show that a template of COF-1 can host 1,2,4-trichlorobenzene (TCB) guest molecules that are stabilized through X⋯H hydrogen bonding. The adsorption is enabled through a combination of factors, including the host pore size and shape, and the specific stereochemical arrangement of binding sites. By introducing a solution of C60 in TCB onto the COF-1 template, we observe that selective adsorption of C60 on top of the COF-1 template is obtained simultaneously with trapping of TCB within the pores. This work demonstrates a strategy to separate and recognize molecules through different interactions within a covalent porous 2D template.
Experimental section
Monolayer COF-1 on highly oriented pyrolytic graphite (HOPG) was formed through a modification of procedures described previously, where heptanoic acid was used as a solvent.13,21 In the present experiments, 1.8 mg of 1,4-benzenediboronic acid (Tokyo Chemical Industry Co. Ltd) was added to 1.2 ml of 1,2,4-trichlorobezene (99%, Sigma-Aldrich), and then sonicated for approximately 30 min. This produced a whitish suspension. 15 μl of this BDBA suspension was dropped onto freshly cleaved HOPG (Structure Probe International, grade SPI-2) and placed into a reactor with a volume of ∼16 ml. 130 μl of deionized water was added to the bottom of the reactor, and a valve to atmosphere was left slightly open to maintain an open system. The entire reactor was placed in an oven preheated to 125 °C and left for 60 min. After the thermal treatment, the reactor was taken out of the oven and allowed to cool down for at least 20 min before the samples were removed.Following the confirmation of monolayer COF-1 on the HOPG surface using scanning tunneling microscopy (STM), 15 μl of 1,2,4-TCB (99%, Sigma-Aldrich) was applied onto the substrate. Subsequently, the samples were characterized by STM at the solvent/solid interface. To investigate the adsorption of fullerenes, 15 μl of a supernatant solution of C60 (99.5%, Sigma-Aldrich) in 1,2,4-TCB was applied onto a pre-prepared COF-1 template.
STM was performed at room temperature at the liquid–solid interface, using a Digital Instruments STM equipped with a Nanoscope IIIa controller. Tips were cut from a Pt0.8Ir0.2 wire (Nanoscience Instruments). Bias voltages are reported with respect to the STM tip. STM images were calibrated with the COF-1 lattice parameter (1.476 nm) using free WSxM software.22
Gas-phase density functional theory (DFT) calculations were carried out using Gaussian09.23 In previous work, we compared the performance of a number of functionals and basis sets for calculations of self-assembled systems comprising X⋯H and X⋯X interactions,24,25 and found that the combination of M06-2X26/LANL2DZ provides an accurate description of energies for halogen-bonded systems. To simulate surface adsorption, all geometries were constrained to remain planar.
Results and discussion
Using TCB as a solvent, the synthesis of COF-1 on HOPG produces a high density of small domains across the surface, as shown in Fig. S1a.† The obtained surface-confined porous network is qualitatively identical to the one formed using heptanoic acid as a solvent.10 In both cases, large COF sheets appear to comprise multiple domains that have grown together, leaving disordered regions evident at the domain boundaries. However, when using TCB, we also observed ordered domain boundaries, as described below.As shown in Fig. 1a, the COF-1 polycrystalline film synthesized in TCB is composed of two different domain orientations tilted at an angle α = 30 ± 1° to each other and separated by a grain boundary (GB). In our analysis of the epitaxial relationship of COF-1 and HOPG (Fig. 1b), we attribute the dominant domain as phase I, i.e., the COF-1 lattice aligned with HOPG with a 6 × 6 epitaxial unit cell, which corresponds to a lattice parameter of a1 = 1.476 nm, as reported in previous work.10 The lattice of phase I is represented by small blue hexagons in Fig. 1b. The smaller domain is denoted as phase II. The orientation of this domain, along the HOPG armchair direction, is nearly commensurate to a 10 × 10 superstructure (corresponding to 3 × 3 unit cells). We previously calculated that the phase II epitaxial orientation of COF-1 should be energetically disfavoured,10 consistent with our infrequent experimental observation of the phase. Phase II has also occasionally been observed for COF-1 synthesized from heptanoic acid. (See Fig. S1†).
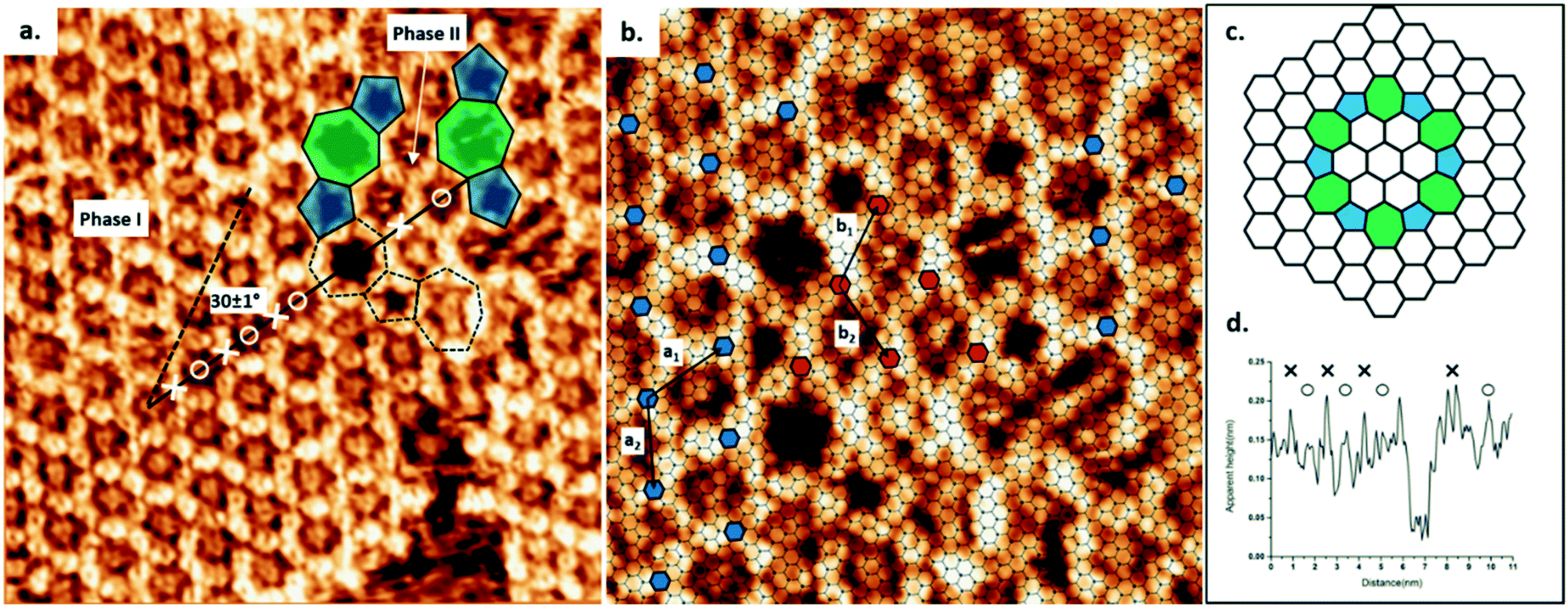 | ||
| Fig. 1 (a) COF-1 STM image showing the grain boundary consisting of pentagon and heptagon rings, connecting two domains rotated by 30 ± 1° with respect to one another, as shown by the dashed and solid black lines. The image was collected at the interface of TCB and HOPG. Image width: 15 nm. Scanning conditions: V = −1000 mV, I = 100 pA. (b) The epitaxy of COF-1 and HOPG at the grain boundary region. Phase I: a1 = a2 = 1.476 nm; phase II: b1 = b2 = 1.42 nm. Image width: 9.6 nm. (c) Schematic structure of the rotational grain boundary identified in (b), i.e., a loop defect with C6 symmetry. Five- and sevenfold rings are shaded by blue and green, respectively. (d) Line profile of the measured height along the black solid line in (a). The positions of benzene rings in COF-1 are marked as × and the positions of adsorbed TCB molecules are marked as ○. | ||
The GB region in Fig. 1 consists of elementary topological defects, pentagons (shaded blue) and heptagons (shaded green). These defects represent some of the basic building blocks of the disordered COF-1 network that have been observed on oriented noble metal surfaces under ultra-high vacuum conditions (UHV),27,28 where the random distribution of these topological defects can be attributed to kinetic trapping during synthesis, since the water-driven self-correction mechanism is not available under UHV conditions. However, the ordered pentagonal/heptagonal defects that we observe in this work are directly analogous to defects formed in another single layer covalent system, i.e., single layer graphene.29–31 According to the atomic model proposed by Cockayne et al.,32 the chain of alternating pentagon and heptagon defects is consistent with a loop GB of graphene, as shown in Fig. 1c. Compared with the linear GB, reported by Xu et al. in COFs formed by benzene-1,3,5-tricarbaldehyde (BTA) and p-phenylenediamine (PDA),33 the loop defect boundary has the lowest energy per dislocation core, suggesting that this defect is likely to form under conditions where mobile dislocations exist. This is consistent with our COF-1 synthesis method, since defect correction within the COF lattice leads to improved structural order under humid synthesis conditions.13 In our case this translates to the minimization of the energy at domain boundaries through the inclusion of well-defined Stone–Wales type defects.29
The presence of the loop boundary defect enables the identification of the adsorption of TCB in the COF-1 pore. Fig. 1d shows the apparent height profile along the zigzag direction of the phase I COF-1 lattice (the black solid line in Fig. 1a). The adsorption of TCB in hexagonal rings can be clearly distinguished by comparison with the apparent height of the heptagonal ring.
High resolution STM images allow the elucidation of the host–guest structure formed by the hexagonal pore of COF-1 and a TCB molecule, as shown in Fig. 2a. The six phenyl rings in the backbone of COF-1 can be easily distinguished, as are submolecular features associated with the adsorbed TCB molecule. These features are consistent with the benzene ring and the chlorine atoms, and their presence suggests the stable adsorption of the TCB molecule (i.e. that the molecule is not rotating).34 Our gas-phase DFT calculations suggest that the TCB adsorption is stabilized through Cl⋯H hydrogen bonding interactions, as indicated in Fig. 2b; consistent with previous work, we identify these interactions through Cl⋯H distances that are shorter than the sum of the Cl and H vdW radii, as shown in Table S1 in the ESI.24,35† The electron density associated with the chlorine atoms in the TCB molecule is anisotropically distributed, as shown in Fig. S2.†36 The nucleophilic belt orthogonal to the covalent bond, with higher electron density and showing negative electrostatic potential, forms X⋯H hydrogen bonds with neighboring hydrogen atoms on the COF-1 backbone (dashed blue line).37 Although the σ-hole of the chlorine atoms points toward the nucleophilic oxygen atom of the COF-1 (dotted black line in Fig. 2b), the interaction between the chlorine and the oxygen is screened by the hydrogens on the phenyl rings, as shown in Fig. S2.† Our DFT calculation confirms that the chlorine–oxygen distance is too large (∼4.38 Å) to allow a significant interaction.
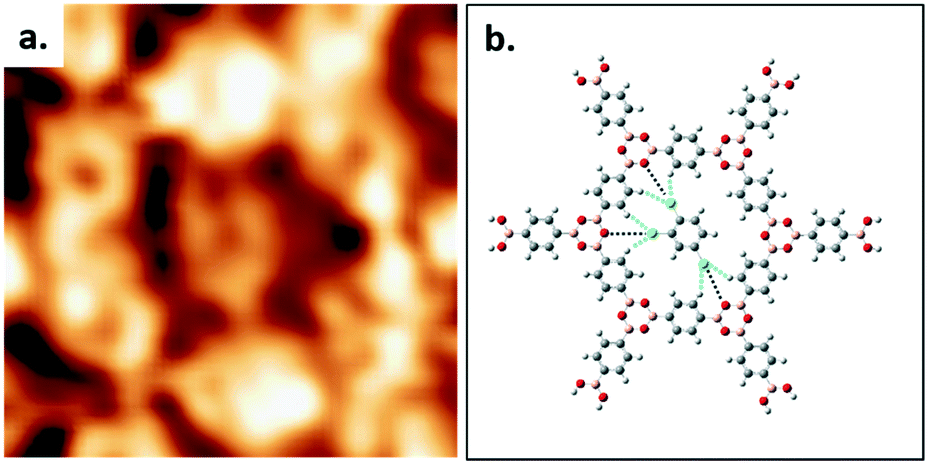 | ||
| Fig. 2 (a) The detailed STM image of one TCB molecule adsorbing into the hexagonal ring formed by COF-1. Image width: 2.1 nm. Scanning conditions: V = −1000 mV, I = 100 pA. (b) DFT-calculated structure of one TCB molecule in the hexagonal pore (M06-2X/LANL2DZ). Dashed blue and black lines represent the interactions of Cl⋯H and Cl⋯O, respectively. | ||
The presence of the pentagonal/heptagonal defects at the GB allows the exploration of the topological and pore size dependence of the guest molecule adsorption. In previous work, fullerene guest molecules were shown to preferentially adsorb in the irregularly-shaped pores present at domain boundaries in a nanoporous hydrogen-bonded network of trimesic acid (TMA).38 However, in contrast to the TMA network where the domain boundary dynamically evolves,39 the loop defects in COF-1 are stable during STM scanning and present a well-defined molecular template, as shown in Fig. 3. The diameters of pentagonal rings (blue dashed line) and heptagonal rings (green dashed line) are 1.381 nm and 1.959 nm respectively, compared to the hexagonal pore size (red dashed line) of 1.758 nm.27 Pores of different sizes can adsorb different numbers of guest molecules, as shown in Fig. 3a–c. There is no adsorption of TCB in pentagonal rings; basic geometric considerations reveal that a TCB molecule cannot be included in this pore without creating interference between hydrogen atoms on the TCB and the pentagonal ring (multiple H⋯H contacts with distances smaller than twice the hydrogen vdW radius), as shown in Fig. S3.†
 | ||
| Fig. 3 (a–c) The sequential collection of STM images showing the evolution of adsorption of TCB in the grain boundary defects. Different geometries are marked by different color dashed circles: heptagonal ring (green circle); hexagonal ring (red circle); and pentagonal ring (blue circle). Image width: 5.8 nm. Scanning conditions: V = −1000 mV, I = 100 pA. | ||
Two TCB molecules can be adsorbed simultaneously in the heptagonal pore, as shown in Fig. 3. The adsorption of a different number of guest molecules within different pore sizes is driven by the commensurability of host and guest molecules. Networks with sufficiently large cavities can host clusters of molecules.4,9,40 The adsorption of TCB in porous self-assembled networks has been reported previously for large-pore self-assembled templates,17,41 including instances where TCB was trapped in close-packed domains within the pore.41,42 In the present study, DFT calculations suggest that both inter-TCB Cl⋯Cl interactions and TCB–COF Cl⋯H interactions stabilize the adsorption of the TCB pair within the heptagonal pore (see the ESI†).
The occupation states of the pentagonal and hexagonal pores appear unchanged with time under STM scanning: the pentagonal pores remained empty, whereas the hexagonal pores retain a single TCB molecule. Conversely, the adsorption of TCB molecules in heptagonal rings exhibits dynamical change. The adsorption–desorption–readsorption process of two TCB molecules can be clearly distinguished in the STM images, as marked by green circles in Fig. 3a–c. Although both empty pores and double-occupied pores were observed, we did not detect the intermediate state, i.e., single TCB adsorbed in a heptagonal ring. The most likely explanation for this is the relatively weak stabilization for an individual TCB molecule in a heptagonal pore. Unlike the matched symmetry between the TCB and the hexagonal pore, the reduced symmetry of the heptagonal pore is a poor match for the TCB molecule, reducing the number of possible Cl⋯H contacts for a single molecule, and rendering single-molecule adsorption unfavourable.
Previous work showed that C60 introduced from a solution containing heptanoic acid as a solvent is adsorbed on the COF-1 template in two different sites, the top-site and pore-site.10 The top-site geometry corresponds to the adsorption of a fullerene molecule on the boroxine ring and the pore-site corresponds to the adsorption of a fullerene molecule in the hexagonal pore. Our present experiments reveal that C60 introduced from a solution in TCB exhibits a different behaviour, as shown in Fig. 4a. Our interpretation of the STM data is shown in Fig. 4b, which is consistent with the results obtained in the absence of C60. Therefore, we attribute the tightly-packed hexagonal pattern to TCB molecules adsorbed in the COF-1 pores (indicated as shaded blue circles in Fig. 4a and b), and the high-contrast, larger-periodicity hexagonal domain to C60 molecules adsorbed on top-sites. The top-site adsorption of C60 is consistent with the calculated geometry, where the architecture is stabilized by vdW interactions.10
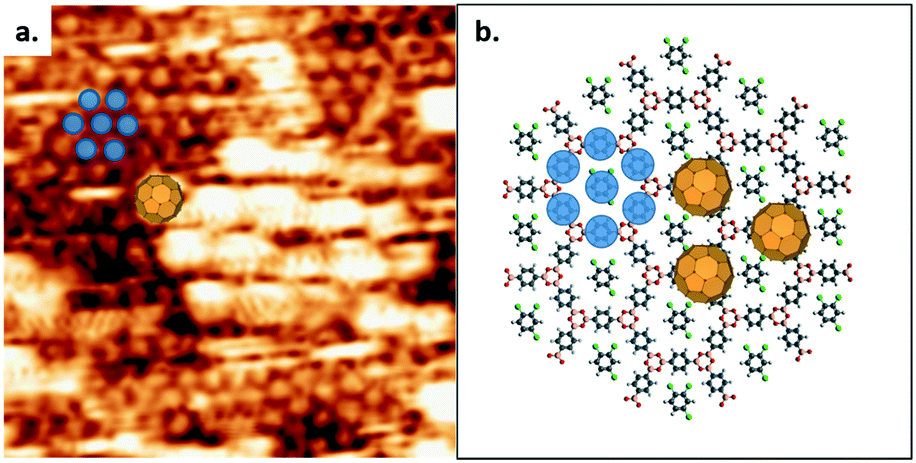 | ||
| Fig. 4 (a) The top-site selective formation in the C60/COF-1 system. Image width: 12 nm. Scanning conditions: V = −800 mV, I = 100 pA. (b) Schematic image of top-site selective formation in the C60/COF-1 system. The shaded blue circles over benzene rings represent the closed packed background in STM image. Top-site C60 molecules are placed over boroxine rings, corresponding to the large bright spots in (a). | ||
The growing body of literature on H/G architectures based on COF-1 suggests that the solvent used for C60 deposition plays an important role in the adsorption of the guest molecules. Plas' work shows that C60 molecules grow layer-by-layer sequentially from a COF-1 template dispersed in 1-phenyloctane, suggesting that the solvent does not compete with C60 for adsorption in the pore site.11 Using heptanoic acid as a solvent, we anecdotally observed a preference for top-site adsorption of fullerenes, suggesting that heptanoic acid may compete for adsorption in the pore-site. Combined with the present results, this suggests that the solvent can be used to select the initial adsorption site for fullerenes, that is TCB and 1-phenyloctane will favor top-site and pore-site adsorption, respectively, whereas heptanoic acid cannot predictably favor either adsorption site.
Conclusions and perspectives
A 2D porous COF-1 layer was synthesized on HOPG from solution in TCB. Two different epitaxial orientations of COF-1 were observed, with a well-defined loop boundary defect formed by a chain of pentagonal rings and heptagonal rings separating the two domain orientations. STM images collected at the TCB/solid interface reveal the adsorption of TCB within the hexagonal pores of the COF-1 template. Our DFT calculations show that this host/guest structure is stabilized by Cl⋯H hydrogen bonding. The presence of the loop boundary defect permitted the investigation of the effect of pore shape and size on TCB adsorption: no TCB was adsorbed in the pentagonal pores, but the larger heptagonal pores accommodated two TCB molecules, stabilized through a combination of Cl⋯H and Cl⋯Cl bonding. Including C60 in the TCB solution allowed the investigation of the molecular recognition of the template. When both C60 and TCB are present at the solution/solid interface, TCB molecules are selectively trapped in the pores of the COF-1 template, whereas fullerenes are adsorbed on top of the COF-1 in sites identified as top sites. The selective adsorption of TCB and C60 in COF-1 suggests that the different adsorption sites in the COF-1 lattice can be used to separate guest molecules by different interactions, opening opportunities for applications in molecular patterning and recognition.Acknowledgements
F. R. acknowledges the support from NSERC (Discovery Grant) as well as the Canada Research Chair program for funding and partial salary support. J. M. M., acknowledges financial support from Queensland University of Technology (QUT) and from an Australian Research Council (ARC) Discovery Early Career Researcher Award (DE170101170). Computational resources and services used in this work were provided by the HPC and the Research Support Group, QUT. F. R. is also grateful to the government of China for a short term Chang Jiang Scholar Award and to the Sichuan province for a short term 1000 Talents Award.References
- J. Teyssandier, S. De Feyter and K. S. Mali, Chem. Commun., 2016, 52, 11465–11487 RSC.
- F. Cicoira, C. Santato and F. Rosei, in Stm and Afm Studies on (bio) molecular systems: Unravelling the nanoworld, Springer, 2008, pp. 203–267 Search PubMed.
- S. J. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 11556–11560 CrossRef CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS PubMed.
- J. M. MacLeod, O. Ivasenko, C. Fu, T. Taerum, F. Rosei and D. F. Perepichka, J. Am. Chem. Soc., 2009, 131, 16844–16850 CrossRef CAS PubMed.
- O. Ivasenko, J. M. MacLeod, K. Y. Chernichenko, E. S. Balenkova, R. V. Shpanchenko, V. G. Nenajdenko, F. Rosei and D. F. Perepichka, Chem. Commun., 2009, 1192–1194 RSC.
- E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2006, 18, 447–451 CrossRef CAS.
- S. Lei, K. Tahara, X. Feng, S. Furukawa, F. C. De Schryver, K. Müllen, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2008, 130, 7119–7129 CrossRef CAS PubMed.
- S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E. Delvigne, N. Lin, X. Deng, C. Cai, J. V. Barth and K. Kern, Nat. Mater., 2004, 3, 229–233 CrossRef CAS PubMed.
- D. Cui, J. MacLeod, M. Ebrahimi, D. Perepichka and F. Rosei, Chem. Commun., 2015, 51, 16510–16513 RSC.
- J. Plas, O. Ivasenko, N. Martsinovich, M. Lackinger and S. De Feyter, Chem. Commun., 2016, 52, 68–71 RSC.
- J. Sun, X. Zhou and S. Lei, Chem. Commun., 2016, 52, 8691–8694 RSC.
- J. R. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed.
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem – Eur. J., 2009, 15, 7004–7025 CrossRef CAS PubMed.
- M. Li, Q. Zeng and C. Wang, Sci. China: Phys., Mech. Astron., 2011, 54, 1739–1748 CrossRef CAS.
- M. Li, K. Deng, S. B. Lei, Y. L. Yang, T. S. Wang, Y. T. Shen, C. R. Wang, Q. D. Zeng and C. Wang, Angew. Chem., 2008, 120, 6819–6823 CrossRef.
- K. Tahara, K. Nakatani, K. Iritani, S. De Feyter and Y. Tobe, ACS Nano, 2016, 10, 2113–2120 CrossRef CAS PubMed.
- J. Adisoejoso, K. Tahara, S. Okuhata, S. Lei, Y. Tobe and S. De Feyter, Angew. Chem., 2009, 121, 7489–7493 CrossRef.
- E. Ghijsens, H. Cao, A. Noguchi, O. Ivasenko, Y. Fang, K. Tahara, Y. Tobe and S. De Feyter, Chem. Commun., 2015, 51, 4766–4769 RSC.
- K. Tahara, K. Katayama, M. O. Blunt, K. Iritani, S. De Feyter and Y. Tobe, ACS Nano, 2014, 8(8), 8683–8694 CrossRef CAS PubMed.
- J. R. F. Dienstmaier, A. M. Gigler, A. J. Goetz, P. Knochel, T. Bein, A. Lyapin, S. Reichlmaier, W. M. Heckl and M. Lackinger, ACS Nano, 2011, 5, 9737–9745 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. Gomez-Rodriguez, J. Colchero, J. Gómez-Herrero and A. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Gatti, J. M. MacLeod, J. A. Lipton-Duffin, A. G. Moiseev, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2014, 118, 25505–25516 CAS.
- R. Gutzler, C. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Nanoscale, 2012, 4, 5965–5971 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- O. Ourdjini, R. Pawlak, M. Abel, S. Clair, L. Chen, N. Bergeon, M. Sassi, V. Oison, J.-M. Debierre and R. Coratger, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 125421 CrossRef.
- N. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- J. Ma, D. Alfe, A. Michaelides and E. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 033407 CrossRef.
- S. Kurasch, J. Kotakoski, O. Lehtinen, V. Skákalová, J. Smet, C. E. Krill III, A. V. Krasheninnikov and U. Kaiser, Nano Lett., 2012, 12, 3168–3173 CrossRef CAS PubMed.
- J. MacLeod and F. Rosei, Small, 2014, 10, 1038–1049 CrossRef CAS PubMed.
- E. Cockayne, G. M. Rutter, N. P. Guisinger, J. N. Crain, P. N. First and J. A. Stroscio, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195425 CrossRef.
- L. Xu, X. Zhou, W. Q. Tian, T. Gao, Y. F. Zhang, S. Lei and Z. F. Liu, Angew. Chem., Int. Ed., 2014, 53, 9564–9568 CrossRef CAS PubMed.
- S. J. Griessl, M. Lackinger, F. Jamitzky, T. Markert, M. Hietschold and W. M. Heckl, Langmuir, 2004, 20, 9403–9407 CrossRef CAS PubMed.
- R. Gutzler, O. Ivasenko, C. Fu, J. L. Brusso, F. Rosei and D. F. Perepichka, Chem. Commun., 2011, 47, 9453–9455 RSC.
- I. S. Gutiérrez, F.-Y. Lin, K. Vanommeslaeghe, J. A. Lemkul, K. A. Armacost, C. L. Brooks and A. D. MacKerell, Med. Chem., 2016, 24, 4812–4825 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- D. den Boer, G. D. Han and T. M. Swager, Langmuir, 2014, 30, 762–767 CrossRef CAS PubMed.
- M. Lackinger, S. Griessl, L. Kampschulte, F. Jamitzky and W. M. Heckl, Small, 2005, 1, 532–539 CrossRef CAS PubMed.
- M. O. Blunt, J. C. Russell, N. R. Champness and P. H. Beton, Chem. Commun., 2010, 46, 7157–7159 RSC.
- M. O. Blunt, J. Adisoejoso, K. Tahara, K. Katayama, M. Van der Auweraer, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2013, 135, 12068–12075 CrossRef CAS PubMed.
- R. Gutzler, S. Lappe, K. Mahata, M. Schmittel, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 680–682 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, details of DFT calculations, and additional STM images. See DOI: 10.1039/c7ce00263g |
| This journal is © The Royal Society of Chemistry 2017 |