
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pluripotent stem cell derived hepatocytes: using materials to define cellular differentiation and tissue engineering
B.
Lucendo-Villarin
,
H.
Rashidi
,
K.
Cameron
 and
D. C.
Hay
*
and
D. C.
Hay
*
Medical Research Council Centre for Regenerative Medicine, University of Edinburgh, 5 Little France Drive, Edinburgh, EH16 4UU, Scotland, UK. E-mail: davehay@talktalk.net; Tel: +44(0)1316519500
First published on 15th April 2016
Abstract
Pluripotent stem cell derived liver cells (hepatocytes) represent a promising alternative to primary tissue for biological and clinical applications. To date, most hepatocyte maintenance and differentiation systems have relied upon the use of animal derived components. This serves as a significant barrier to large scale production and application of stem cell derived hepatocytes. Recently, the use of defined biologics has overcome those limitations in two-dimensional monolayer culture. In order to improve the cell phenotype further, three-dimensional culture systems have been employed to better mimic the in vivo situation, drawing upon materials chemistry, engineering and biology. In this review we discuss efforts in the field, to differentiate pluripotent stem cells towards hepatocytes under defined conditions.
 B. Lucendo-Villarin | Dr Baltasar Lucendo-Villarin completed his PhD under the supervision of Dr David Hay at the MRC Centre for Regenerative Medicine, University of Edinburgh, using synthetic materials to stabilise the hepatocellular phenotype. Currently he is a post-doctoral fellow in Hay's lab where he is modelling the impact of maternal drug use on liver development. His research interest includes tissue engineering, regenerative medicine, and PSC-based disease modelling. |
 H. Rashidi | Dr Hassan Rashidi is a Postdoctoral Research Fellow at University of Edinburgh. Dr Rashidi joined the University of Edinburgh after completing his PhD in Stem Cell Biology and a postdoctoral study in Bone Tissue Engineering, both at University of Nottingham. His research focuses on development of 3D liver models. This work is funded by the UK Regenerative Medicine Platform (UKRMP). |
 K. Cameron | Dr Kate Cameron carried out her PhD at the University of Edinburgh, studying the effects of calcium phosphate based substrates on mesenchymal stem cell differentiation and new bone formation. She is currently involved in a UK wide project on regenerative medicine (UKRMP) developing stem cell derived hepatocytes for liver repair. Her focus is on utilising pluripotent stem cells and optimising their differentiation, under defined culture conditions, using novel techniques and biomaterials to deliver cells for clinical use. Since completing her PhD, Dr Cameron continues to work in Edinburgh at the Scottish Centre for Regenerative Medicine. |
 D. C. Hay | Dr David Hay is a Principal Investigator at the University of Edinburgh's MRC Centre for Regenerative Medicine. David has worked in the field of stem cell biology and differentiation over the last fifteen years. David and his team have highlighted the important role that pluripotent stem cells have to play in modelling human liver biology ‘in a dish’. The impact of this work has led to a number of publications and regular appearances at high profile international conferences. |
1. Introduction
The advent of human pluripotent stem cells (hPSCs) and their efficient differentiation allows users to custom-make human tissue ‘in a dish’. This has major implication in biomedicine and will likely lead to personalised regenerative medicines of the future. Our particular interest is in the liver, and the generation of functional tissue from human pluripotent stem cells. The major cell type of the liver is the hepatocyte and we, and others,1 have been working to produce these cells at the scale for basic research and therapeutic purposes. While freshly isolated human hepatocytes represent the current gold standard,2,3 they are a scarce and expensive resource with variable performance. The isolation of primary hepatocytes commences with collagenase digestion of the liver followed by density-gradient centrifugation.4 Post-isolation, hepatocyte phenotype is lost and cells begin to senesce, limiting their widespread use.5–9 In an effort to preserve the cell phenotype, a number of approaches have been developed, including the modification of culture media, the use of different extracellular matrices, and the development of co-culture formats.5,7,10,11 Despite the advantages of these approaches, phenotypic instability still hinders the routine use of primary human hepatocytes.12 As a consequence, alternative models have been developed to study human liver biology and model cell based therapy. Those include the use of human cancer cell lines, and animal derived hepatocytes.1 While these cell types are promising, they also suffer from limitations which limit their routine deployment. These include genomic instability,13 incomplete gene expression,14–17 scale-up limitation,18 heterogeneous culture and species differences.19While the field faces major challenges, progress is being made. Recent studies provide hope that some of the previous limitations associated with hepatic progenitor cell isolation and expansion have been addressed. Hepatic progenitor cells (HPCs), possess the capacity to regenerate liver epithelia. Although HPCs are extremely rare in healthy liver, their scalability and plasticity makes them an attractive cell source of hepatocytes for application. Recently, Lu et al. isolated a defined population of HPCs from the mouse liver. The resulting cells were expandable and displayed stability following long term maintenance in vitro and in vivo.20
Recently, the limited proliferative capacity of adult human hepatocytes has been studied by Levy et al. The authors created an oncostatin M dependent expansion system for primary hepatocytes using human papilloma virus oncoproteins.21 We have also studied hepatocyte expansion, differentiation and stabilisation using hPSC-derived hepatocyte-like cells (HLCs). In these experiments HLC stability was maintained for over twenty days, revealing a novel gene signature associated with a stable hepatocyte phenotype. Importantly, these findings were successfully translated to GMP grade hESC lines promising therapeutic application in the future.22 Most recently, we have employed recombinant laminins to drive hepatocyte differentiation and self-organisation of HLCs from hESC lines available at GMP grade.23
We believe that the development of defined culture systems, and novel tissue engineering processes are essential for the delivery of stable, scalable and functional human liver tissue and this is discussed in the review.
2. Pluripotent stem cells
Pluripotent stem cells (PSCs) are defined as cells which give rise to all somatic cell types found in the body. Human embryonic stem cells (hESCs) and the more recently described induced pluripotent stem cells (iPSCs) represent the two major sources of pluripotent stem cells (Fig. 1).24–26 Human embryonic stem cells are derived from the inner cell mass of blastocyst stage embryos which are not suitable for human implantation.27 Pioneering studies of mouse ESCs28,29 and of culturing techniques developed in non-human ESC lines30,31 and EC (embryonal carcinoma) lines32 led to the isolation and propagation of hESC lines for the first time.27 While hESCs are highly promising for the field, they have raised ethical issues. In 2006 and 2007 Shinya Yamanaka's laboratory, inspired by the successes in mammalian nuclear transfer,33 delivered a PSC population from somatic cells, in a process called reprogramming.34,35 In these studies the authors used a core set of transcription factors (Oct 4, Sox2, Klf4 and c-Myc) to reprogram somatic cells to a pluripotent state. Today, PSCs serve as an important resource to generate human tissues. | ||
| Fig. 1 Sources and properties of human pluripotent stem cells (hPSCs). hPSCs can be derived either from somatic cells isolated from the human body or from cells isolated from the inner cell mass (ICM) of human embryos. Pluripotent stem cells possess two important attributes: the ability to self-renew and the capacity to differentiate into any cell type in the human body. Somatic cells can be generated from pluripotent stem cells using multistep differentition procedures or through the use of transcription factor combinations. | ||
3. Transdifferentiation
In addition to reprogramming, other groups have developed a process called transdifferentiation. This is the direct reprogramming of somatic cells into another type of somatic cell, by-passing the requirement for pluripotency.36 Traditional methods of transdifferentiation rely on the expression of a single or a cocktail of tissue specific transcription factors to specify somatic cell types that are representative of all three germ layers.37–474. Hepatocyte differentiation
During liver development various cell types and signalling pathways orchestrate the formation of a highly organised, functional and regenerative organ. The liver acinus represents the basic functional unit of the liver. During acinar formation, the extracellular matrix plays an important role in cell organisation and function and its composition varies depending on the zone of the liver.48–51 Therefore, many factors must be considered if one wishes to generate HLCs from human PSCs or from other stem cell populations. To date, hepatocyte differentiation from PSCs has been achieved using two different formats, either three-dimensional cellular aggregation or two-dimensional monolayer culture.4.1 Hepatocyte differentiation in three dimensions
Embryoid bodies (EBs) are three-dimensional (3D) aggregates that permit spontaneous differentiation of hPSCs in suspension, aiming to mimic the 3D tissue niche. Seminal studies reported by Lavon et al. demonstrated the ability to isolate and purify HLCs using GFP under control of the albumin promoter.52 EB formation, tissue specific reporting and FACS were also employed by Duan et al. to differentiate, purify and implant stem cell derived hepatocytes in vivo.53 Following on from this, Basma et al. used EB based systems to derive and enrich HLCs, prior to their characterisation in vitro and in vivo.54 The effect of different ECM components on hepatic differentiation has also been studied by several groups. Schwartz et al. observed improved endoderm gene expression in hESC-derived EBs on type I collagen coated plates in the presence of fibroblast growth factor 4 (FGF4) and HGF.55 Sirahashi et al. highlighted the importance of cell maintenance media, ECM substrates, foetal bovine serum (FBS) and growth factors in hepatocyte differentiation.56 More recently, Vosough et al. developed a scalable 3D differentiation approach in stirred flasks.57 While providing a proof of concept, the presence of undefined components in these experiments have complicated large scale cell production and therefore application. We believe that this highlights the need to truly define and simplify HLC differentiation methodology for use with PSCs.4.2 Directed hepatocyte differentiation in two dimensions
To address the issue of heterogeneous cell differentiation in EBs, research has focused on two dimensional differentiation systems. This requires the stepwise addition of key drivers which move the cells from pluripotency, through definitive endoderm to hepatic progenitors and finally functional hepatocytes. Various procedures exist between labs (Table 1), but most draw upon the seminal observations of D'Amour et al. In these studies, hESCs were driven efficiently to definitive endoderm using activin A.58 Endoderm differentiation has also been achieved in the presence of multiple factors including Activin A, HGF, bone morphogenic protein 4 (BMP4), FGF2, FGF4, glucocorticoids, insulin and all-trans-retinoic acid (ATRA) on different extracellular matrices.59 The WNT signalling pathway has also been shown to be important in human endoderm specification.60–62 Following endoderm specification, hepatic differentiation has been achieved using a number of approaches. Cai and coworkers developed a differentiation protocol mimicking in vivo development using, FGF4 and BMP2 followed by HGF, OSM and dexomethasone.63 Agarwal et al. employed FGF4 and HGF to promote hepatic specification of hESC-derived endoderm on type I collagen. Following this, hepatocyte differentiation and maturation of the endoderm-derived cells were induced using a combination of BSA, FGF4, HGF, OSM and dexamethasone.4 Hay et al. induced hepatic progenitor specification from hESC-derived endodermal cells by supplementing the media with 1% DMSO prior to induction of hepatocyte differentiation in media supplemented with HGF and OSM.65–67 Brolen et al. demonstrated hepatic specification using combinations of BMP2 and -4 with FGF1, -2 and -4, followed by a cocktail of different factors including the epithelial growth factor (EGF), insulin, transferrin, ascorbic acid, FGF4, HGF, dexamethasone, dimethyl sulfoxide (DMSO) and OSM.68| Substrate | Definitive endoderm induction | Hepatic specification | Hepatic differentiation | Ref. |
|---|---|---|---|---|
| MEFs | AA (3d) | FGF4, BMP2 (5d) | HGF (5d) + OSM (5d) | Cai et al., 200724 |
| MEFs/collagen I | AA, FBS, KOSR (5d) | FGF4, HGF, KOSR (3d) | BSA, FGF4, HGF, OSM, Dex (2d) + FGF4, HGF, OSM, Dex (9d) | Argawal et al., 200864 |
| Matrigel | AA, Wnt3a (3d) | 1% DMSO, 20% KOSR (5d) | HGF, OSM (9d) | Hay et al., 200865 |
| MEFs | AA, FGF2 (5d) | BMP 2/4, FGF1/2 (11d) | EGF, insulin, hydrocortisone, transferrin (28d) | Brolen et al., 200968 |
| MEFs | AA, bFGF (3d) | HGF, DMSO (8d) | Dex (3d) | Basma et al., 200954 |
| Matrigel | AA (5d) | FGF4, BMP2 (5d) | HGF (5d) + OSM (5d) | Si-Tayeb et al., 201071 |
| Matrigel | AA, Wnt3a, (3d); +AA (2d) | 1% DMSO, 20% KOSR (5d) | OSM, HGF (5d) | Sullivan et al., 201072 |
| Matrigel | AA, BMP4,FGF2 (3d) | FGF10 (3d) | FGF4, HGF, EGF (8d) | Toboul et al., 201073 |
| Fibronectin | AA, FGF2, BMP4, Ly294002, CHIR99021 (1d) + AA, FGF2, Ly294003 (1d)++AA, FGF2 (1d) | AA (5d) | HGF, OSM (17d) | Rashid et al., 201074 |
| MEFs | AA, Wnt3a, HFG (3d) | 1% DMSO, 20% KOSR (5d) | OSM/HGF (7d) | Chen et al., 201275 |
| Matrigel | AA, Wnt3a (3d) | 1% DMSO, 20% KOSR (5d) | HGF, OSM (9d) | Szkolnicka et al., 2014;76 Rashidi et al., 201677 |
| Matrigel | AA, Wnt3a, HFG (3d) | 1% DMSO, 20% KOSR (5d) | OSM, FGF2, insulin, Dex (7d) + OSM, FGF2, insulin, Dex, LCA, MK4 (4d) | Avior et al., 201578 |
| Laminin | AA, Wnt3a (3d) | 1% DMSO, 20% KOSR (5d) | HGF, OSM (9d) | Cameron et al., 2015 et al.23 |
Importantly, the hepatocyte differentiation procedures developed in hESCs have been successfully translated to iPSCs.69 Song et al. reported that iPSC derived HLCs expressed liver cell markers and related functions, including urea production, albumin secretion and cytochrome P450 activity, which were comparable to hESC-derived HLCs.70 Si-Tayeb et al. described a four step differentiation protocol in low oxygen to obtain functional HLCs with similarities to hESC-derived HLCs.71 The differentiation protocol developed by Hay et al. was successfully translated to iPSC technologies, with HLCs displaying metabolic activity and secreting liver proteins.72 This was followed by Touboul et al. who obtained hepatic progenitor cells from hESC-derived definitive endoderm cells upon treatment with FGF10, retinoic acid and SB431542, a pharmacological inhibitor of TGFβ signaling. Hepatocyte differentiation was induced using FGF4, HGF and EGF.73 Following on from this, Rashid et al. developed a three step differentiation protocol containing Activin A, BMP4 and FGF2 in the presence of glycogen synthase kinase 3β (GSK3β) and phosphoinositide 3 kinase (PI3K) pathway inhibitors. Notably, iPSC derived HLCs using this method were found to recapitulate key pathological features observed in human monogenic liver disease.74
4.3 Somatic cell transdifferentiation
As described previously, differentiation can be achieved without the need for pluripotency. Sekiya et al. transfected murine embryonic and adult fibroblasts with HNF4α plus FoxA1, -A2 or -A3, successfully obtaining induced hepatocytes (iHeps).79 Another seminal study, performed by Huang et al., employed mouse tail fibroblasts as their starting cell population. Using lentivirus over-expression of GATA4, HNF1α and Fox3 and inactivated p19Arf they successfully generated iHeps.37 The same group recently reported successful reprogramming of human fibroblasts into iHeps by lentivirus expression of FoxA1, HNF1α and HNF4α. The resulting iHeps displayed hepatic function80 and excitingly fueled a bio-artificial liver device that improved animal survival followed by acute liver failure.81 Therefore, iHeps and HLCs represent a reliable alternative to primary human hepatocytes, holding great potential for biological and clinical applications. These cells have been employed to accurately predict human drug metabolism and drug responses,82–84 as a model to study hepatitis C and B viral life cycle,85–89 to study the mechanisms behind the drug-induced liver injury90,91 and to modulate drug overdose by using non-coding RNAs.67 However, improvements in cell fidelity are required.92 One key void in current approaches is the niche for driving cell differentiation and maintaining the cell phenotype. This is an important area of focus which requires interdisciplinary research.225. Biomaterials in stem cell biology
Traditionally, hPSCs were cultured on murine embryonic fibroblast layers or extracellular matrices derived from mice, such as Matrigel. However, the undefined nature of these environments, along with the potential pathogenic and immunogenic issues, is known to be problematic for clinical application and serves as a barrier to reproducible cell based modelling. In an attempt to overcome these limitations, research studies have focused on developing xeno-free and defined substrates that can efficiently replace the use of animal-derived culture components.23,93–96 In an attempt to reduce the costs associated with cellular differentiation, attention has also focussed on developing cheap synthetic substrates for PSC maintenance and differentiation (Fig. 2).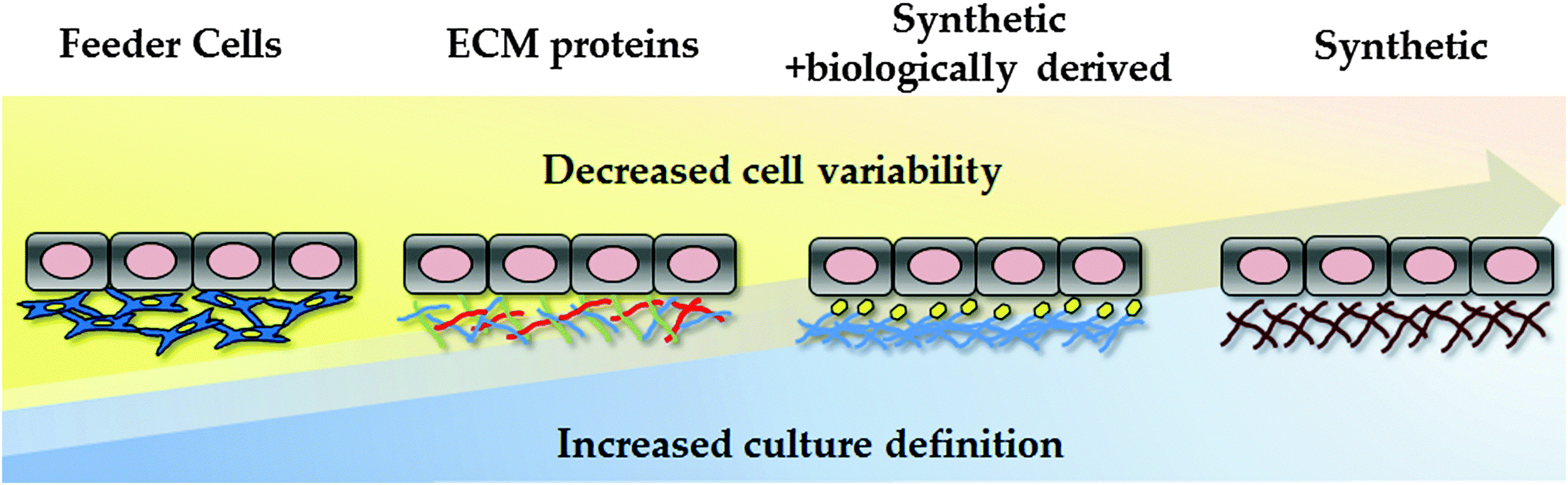 | ||
| Fig. 2 Evolution of human pluripotent stem cell culture and differentiation. Culture conditions are becoming increasingly defined, reducing cell variability and scale-up costs. The field has moved away from animal feeder layers, using undefined ECM formulations, such as Matrigel. As Matrgiel is an undefined mixture of murine proteins and growth factors, the field has now moved toward recombinant proteins and/or synthetic substrates to support hPSC culture and differentiation. | ||
5.1 Synthetic polymers
Biomaterials can be defined as a material or a combination of materials that can be used to support cells, tissues or organs in vitro and in vivo. According to their make-up, biomaterials can be classified as polymers, metals, ceramics and composites. Natural and synthetic polymers have gained the attention of researchers in the stem cell field due to their inert nature, diverse composition, capacity to interact with other synthetic or natural substrates, and reliable cost-effective scale-up.Combinatorial approaches involving the use of synthetic polymers and biologics have been employed in the maintenance and differentiation of hPSCs with promising results. For example, polymers with high acrylate content combined with vitronectin have been reported to successfully maintain the pluripotency properties and colony formation of PSCs for a prolonged period of time.97 In addition, to support hPSC maintenance, combinatorial polymer screens have led to the development of hepatic differentiation or co-culture strategies.98–100 Examples of successful application of biomaterials in the formation of organoid cultures, include the improved expression of hepatocyte growth factor mRNA with faster formation of viable 3D spheroids following co-culture of hepatocytes with stellate cells on a PLGA substrate.101 Increased albumin secretion has also been achieved following the manufacture of endothelialized hepatic spheroids in PDMS honeycomb microwells and poly-L-lactide fibers.102 However, batch-to-batch variation, the risk of pathogens from undefined media components and the high cost associated with the production of some biologics compromise technology scale up and application.105 Therefore, to deliver somatic cells under defined conditions research has focussed on cheap and scalable synthetic materials and renewable cell sources.
Poly [2-(methacryloyloxy) ethyl dimethyl-(3-sulfopropyl) ammonium hydroxide] (PMEDSAH) is a well-characterised synthetic substrate employed in the maintenance of PSCs. It is thought that the sulfonic group mimics heparin sulphate proteoglycans, which are important extracellular components in hPSC culture systems99,103–106 and mesenchymal stem cells derived from hPSCs.107 Aminopropylmethacrylamide (APMAAm) represents another example of a synthetic substrate successfully employed in the long-term maintenance of undifferentiated PSCs, by promoting the adsorption of different proteins present in the culture media.108 The importance of media protein adsorption in the maintenance of PSCs was also revealed by Mei et al., by identifying a polymer generated from monomers with high acrylate content, displaying the capacity of fixing vitronectin from the culture media and promoting colony formation. This system also revealed the importance of physical parameters of the polymers including wettability, surface topography and surface chemistry.97
5.2 3D culture systems
Synthetic polymers have been proven to be economical and effective for the long-term self-renewal, large-scale expansion and direct differentiation of hPSCs. However, these platforms are traditionally applied in two-dimensional (2D) systems. Although 2D systems have proved to be invaluable for studying basic cell biology, cells in these systems are forced to change their cytoskeleton towards flattened shapes, affecting cell-to-cell and cell-to-extracellular environment contacts. These forces lead to reduced polarisation and modification of important signalling pathways, affecting stem cell pluripotency and differentiation.109 As a consequence different approaches have been employed to emulate 3D tissue structure. These approaches can be broadly divided into scaffold-based and scaffold-free culture systems (Fig. 3).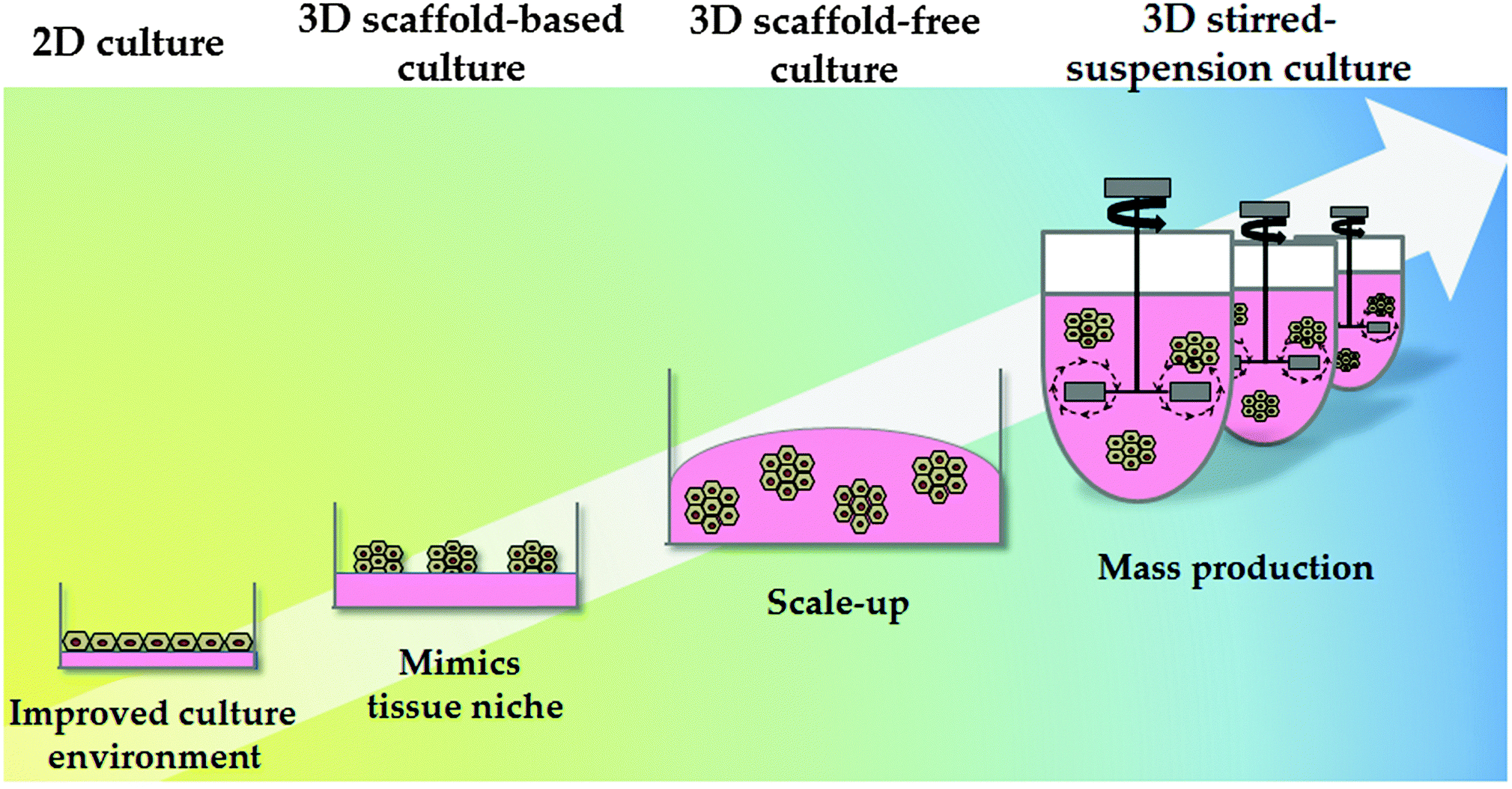 | ||
| Fig. 3 Culture system physical properties. The physical environment of pluripotent and differentiated cells has also evolved; from static 2D culture to 3D culture systems. Static 2D cultures do not accurately mimic the in vivo environment and frequently result in alterations in cell biology. 3D culture systems can be divided into scaffold-based and scaffold-free systems. 3D stirred-suspension cultures represent a more advanced culture system, with cells homogeneously exposed to the different factors in the medium. Further research and development is required in this space to permit cost effective technology scale up. | ||
Natural hydrogels have also been employed for cell culture. Alginate, a polysaccharide derived from the cell wall of marine algae, is one example and has been used to successfully culture hepatocytes in 3D. Interestingly, cell aggregation, essential to ensure correct cell function, was mediated by E-cadherin.114 In another study, Fang et al. demonstrated that exogenous growth factors were required for hepatocyte differentiation via EBs in alginate microbeads.115 More recently, Jitraruch et al. described an intraperitoneal transplantation of alginate-microencapsulated human hepatocytes in a murine model that reduced the severity of liver damage.116
There are several examples of the scaffold-free culture system applied in hPSC technology. Amit et al. developed serum free suspension culture methodology using an IL6R-IL6 (interleukin-6 receptor interleukin-6) fusion protein supporting to support cell expansion and pluripotency.117 Steiner employed an alternative approach supporting suspension cultures of hPSCs using neurobasal media supplemented with serum replacement and different ECM components.118 More recently, Vosough et al. described a stirred-suspension bioreactor culture to obtain iPSC-derived HLCs that post-transplantation supported mice with acute liver damage.57
6. Current challenges and future directions
Although current differentiation approaches from pluripotent stem cells generate HLCs displaying hepatocyte markers and function, they still express features of foetal hepatocytes. In support of this, Godoy et al. compared gene regulatory networks between pluripotent stem cell derived HLCs and adult human hepatocytes. In comparison to PHHs, HLCs expressed the majority of hepatic genes (∼70%). However, there were many genes which did not approach the levels of primary hepatocytes (∼30%). They also identified the expression of colon, fibroblasts and stem cell-associated transcription factors, indicating mixed HLC cultures in vitro.92 These studies concluded that in vitro culture conditions for primary hepatocytes and HLCs were responsible for their instability and/or not reaching a fully differentiated status. Importantly, those studies provided a ‘blue-print’ to help eliminate non-desired cell traits.Liver development is a process that extends beyond birth, when there is a switch from placental to enteral nutrition. Fatty acids from breastfeeding become the main energy source, and those are further metabolised by the gut microbiome to secondary metabolites, which lead to liver development in the neonate. In line with this, Avior et al. recently demonstrated that a secondary metabolite, lithocholic acid (LCA), or the use of Vitamin K may drive maturation of HLCs. They demonstrated that these additives synergise, regulating the activity of a key nuclear factor, PXR, improving cytochrome P450 enzymes 2C9 and 3A4 expression and function.127 While promising, stem cell derived HLC cultures generated using this method are short lived, limiting technology scale up and application.
Searching for defined matrices compatible with maintenance of pluripotency and differentiation capacities of pluripotent stem cells has been traditionally slow and has shown low throughput. However, in recent years, high-throughput (HTP) approaches such as microarraying have allowed rapid screening of chemically diverse substrates that modulate or control cell biology, thus representing an important tool for discovering new materials. HTP technologies have identified polymers with trapping and release properties119 for cell isolation, proliferation and differentiation.120 Recently, Celiz et al. demonstrated that polymer high-throughput screening technology could be used to identify polymer(s) that support stem cell self-renewal and stem cell differentiation to cells representative of the three germ layers. They identified a polymer (HPhMA-co-HEMA), which resulted from the polymerization of 5 (N-(4-hydroxyphenyl)methacrylamide) and poly (2-hydroxyethyl methacrylate) (polyHEMA), supporting both hPSC culture and pluripotency in a defined environment.121 In a separate study a synthetic polyurethane, PU134, was identified employing HTP technology100 and facilitated the maturation and maintenance of HLCs from research and GMP grade hESC lines.22
Concerning the cell niche, most current differentiation approaches lack many aspects of the micro-environment. Therefore the development of strategies employing relevant cell types from the organ of interest in 3D is required. In this vein, Takebe et al. developed an innovative approach. Endothelial and mesenchymal cells, in combination with PSC-HLCs, self-organised into 3D liver-like tissue structures which were functional in vitro and in vivo. Moreover, the authors demonstrated liver function could be supplied from the mesentery, representing an ideal target site for future cell based therapies.122
The emergence of bioprinting in the recent years has also provided new opportunities for the production of printed human liver tissue. Recently Organovo's exVive3D bioprinted liver tissue (Organovo®; USA) has been shown to secrete fibrinogen, albumin and transferrin in proportion to levels observed in vivo.123 Regarding PSCs, valve-based cell printers have been developed to print viable PSC and HLC populations, and offer the promise of automated tissue manufacture for clinical and research applications.124
A common problem associated with most differentiation systems is their static nature. These approaches lack fluid circulation resulting in poor cell perfusion. Recently, Giobbe et al. reported a microfluidic system on a chip to differentiate PSCs into HLCs which could predict drug toxicity.125 Additionally, Berger et al. described a successful microfluidic system employing co-cultures of PSC-derived HLCs and stroma cells, reporting enhanced maturity and polarity.126 More recently, we have shown that pluripotent stem cell-derived HLCs can be further improved by exposing them to fluid shear stress.77
In conclusion, the ability to derive somatic cell types from hPSCs represents an attractive strategy for human tissue engineering. Despite promising advances in the field, further sophistication is necessary to improve cell type fidelity and stability. To date, differentiation protocols are mainly performed using 2D culture systems lacking the relevant cell types. Recent progress in the field, including; combinatorial approaches and three-dimensional and co-culture strategies provide the promise of more sophisticated systems in the future.
Acknowledgements
Dr Lucendo-Villarin was supported by a grant MR/L010011/1. Dr Cameron, Dr Rashidi and Dr Hay were supported by the UK Regenerative medicine platform (MR/K026666/1 and MR/L022974).Notes and references
- D. Szkolnicka, W. Zhou, B. Lucendo-Villarin and D. C. Hay, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 147–159 CrossRef CAS PubMed.
- R. S. Obach, Curr. Opin. Drug Discovery Dev., 2009, 12, 81–89 CAS.
- N. J. Hewitt, E. L. LeCluyse and S. S. Ferguson, Xenobiotica, 2007, 37, 1196–1224 CrossRef CAS PubMed.
- E. L. LeCluyse, E. Alexandre, G. A. Hamilton, C. Viollon-Abadie, J. Coon, S. Jolley and L. Richert, Basic Cell Cult., 2005, 290, 207–229 Search PubMed.
- E. L. LeCluyse, P. L. Bullock and A. Parkinson, Adv. Drug Delivery Rev., 1996, 22, 133–136 CrossRef CAS.
- K. F. Nelson, D. Acosta and J. V. Bruckner, Biochem. Pharmacol., 1982, 31, 2211–2214 CrossRef CAS PubMed.
- E. L. LeCluyse, Eur. J. Pharm. Sci., 2001, 13, 343–368 CrossRef CAS PubMed.
- A. Bader, I. Rinkes, E. I. Closs and C. M. Ryan, Biotechnol. Prog., 1992, 8, 219–225 CrossRef CAS PubMed.
- J. C. Dunn, M. L. Yarmush, H. G. Koebe and R. G. Tompkins, FASEB J., 1989, 3, 147–177 Search PubMed.
- Y. Meng, S. Eshghi, Y. J. Li, R. Schmidt, D. V. Schaffer and K. E. Healy, FASEB J., 2010, 24, 1056–1065 CrossRef CAS PubMed.
- L. G. Griffith and M. A. Swartz, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS PubMed.
- I. J. Fox and S. C. Strom, Gastroenterology, 2008, 134, 878–881 CrossRef PubMed.
- N. Wong, P. Lai, E. Pang, T. Leung and J. Lau, Hepatology, 2000, 121, 9–16 Search PubMed.
- K. Takahashi, T. Ichisaka and S. Yamanaka, Methods Mol. Biol., 2006, 329, 449–458 Search PubMed.
- C. Rodríguez-Antona, M. T. Donato, A. Boobis, R. J. Edwards, P. S. Watts, J. V. Castell and M.-J. Gómez-Lechón, Xenobiotica, 2002, 32, 505–520 CrossRef PubMed.
- E. F. A. Brandon, C. D. Raap, I. Meijerman, J. H. Beijnen and J. H. M. Schellens, Toxicol. Appl. Pharmacol., 2003, 189, 233–246 CrossRef CAS PubMed.
- L. Richert, M. J. Liguori, C. Abadie, B. Heyd, G. Mantion, N. Halkic and J. F. Waring, Drug Metab. Dispos., 2006, 34, 870–879 CrossRef CAS PubMed.
- P. Chao, T. Maguire, E. Novik, K. C. Cheng and M. L. Yarmush, Biochem. Pharmacol., 2009, 78, 625–632 CrossRef CAS PubMed.
- S. Greenhough, C. N. Medine and D. C. Hay, Toxicology, 2010, 278, 250–255 CrossRef CAS PubMed.
- W. Y. Lu, T. G. Bird, L. Boulter, A. Tsuchiya, A. M. Cole, T. Hay, R. V. Guest, D. Wojtacha, T. Y. Man, A. Mackinnon, R. A. Ridqway, T. Kendall, M. J. Williams, T. Jamienson, A. Raven, D. C. Hay, J. P. Iredale, A. R. Clarke, O. J. Sansom and S. J. Forbes, Nat. Cell Biol., 2015, 17, 971–983 CrossRef CAS PubMed.
- G. Levy, D. Bomze, S. Heinz, S. D. Ramachandran, A. Noerenberg, M. Cohen, O. Shibolet, E. Sklan, J. Braspenning and Y. Nahmias, Nat. Biotechnol., 2015, 33, 1264–1271 CrossRef CAS PubMed.
- B. Lucendo-Villarin, K. Cameron, D. Szkolnicka, H. Rashidi, N. Bates, S. J. Kimber, O. Flint, S. J. Forbes, J. P. Irelade, M. Bradley and D. C. Hay, Adv. Healthcare Mater., 2015, 4, 1820–1825 CrossRef PubMed.
- K. Cameron, R. Tan, W. Schmidt-Heck, G. Campos, M. J. Lyall, Y. Wang, B. Lucendo-Villarin, D. Szkolnicka, N. Bates, S. J. Kimber, J. G. Hengstler, P. Godoy, S. J. Forbes and D. C. Hay, Stem Cell Rep., 2015, 5, 1250–1262 CrossRef CAS PubMed.
- J. Cai, J. Chen, Y. Liu, T. Miura, Y. Luo, J. F. Loring, W. J. Freed, M. S. Rao and X. Zeng, Stem Cells, 2006, 24, 516–530 CrossRef CAS PubMed.
- L. M. Hoffman and M. K. Carpenter, Nat. Biotechnol., 2005, 23, 699–708 CrossRef CAS PubMed.
- T. Vazin and W. J. Freed, Restor. Neurol. Neurosci., 2010, 28, 589–603 CAS.
- J. A. Thomson, Science, 1998, 282, 1145–1147 CrossRef CAS PubMed.
- G. R. Martin, Proc. Natl. Acad. Sci U. S. A., 1981, 78, 7634–7638 CrossRef CAS.
- M. J. Evans and M. H. Kaufman, Nature, 1981, 292, 154–156 CrossRef CAS PubMed.
- R. Josephson, G. Sykes, Y. Liu, C. Ording, W. Xu, X. Zeng, S. Shin, J. Loring, A. Maitra, M. S. Rao and J. M. Auerbach, BMC Biol., 2006, 4, 28 CrossRef PubMed.
- J. A. Thomson, J. Kalishman, T. G. Golos, M. Durning, C. P. Harris and J. P. Hearn, Biol. Reprod., 1996, 55, 254–259 CrossRef CAS PubMed.
- J. K. Henderson, J. S. Draper, H. S. Baillie, S. Fishel, J. A. Thomson, H. Moore and P. W. Andrews, Stem Cells, 2002, 20, 329–337 CrossRef CAS PubMed.
- K. H. Campbell, J. McWhir, W. A. Ritchie and I. Wilmut, Nature, 1996, 380, 64–66 CrossRef CAS PubMed.
- K. Takahashi and S. Yamanaka, Cell, 2006, 126, 663–676 CrossRef CAS PubMed.
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, Cell, 2007, 131, 861–872 CrossRef CAS PubMed.
- T. Graf and T. Enver, Nature, 2009, 462, 587–594 CrossRef CAS PubMed.
- P. Huang, Z. He, S. Ji, H. Sun, D. Xiang, C. Liu, Y. Hu, X. Wang and L. Hui, Nature, 2011, 475, 386–389 CrossRef CAS PubMed.
- R. L. Davis, H. Weintraub and A. B. Lassar, Cell, 1987, 51, 987–1000 CrossRef CAS PubMed.
- C. N. Shen, J. Slack and D. Tosh, Nat. Cell Biol., 2000, 2, 879–887 CrossRef CAS PubMed.
- Z. P. Pang, N. Yang, T. Vierbuchen, A. Ostermeier, D. R. Fuentes, T. Q. Yang, A. Citri, V. Sebastiano, S. Marro, T. C. Sudhof and M. Wernig, Nature, 2011, 476, 220–223 CAS.
- M. Caiazzo, M. T. Dell'Anno, E. Dvoretskova, D. Lazarevic, S. Taverna, D. Leo, T. D. Sotnikova, A. Menegon, P. Roncaglia, G. Colciago, G. Russo, P. Carninci, G. Pezzoli, R. R. Cainetdinov, S. Gustincich, A. Dityatev and V. Broccoli, Nature, 2011, 476, 224–227 CrossRef CAS PubMed.
- T. Vierbuchen, A. Ostermeier, Z. P. Pang, Y. Kokubu, T. C. T. C. Sudhof and M. Wernig, Nature, 2010, 463, 1035–1041 CrossRef CAS PubMed.
- U. Pfisterer, A. Kirkeby, O. Torper, J. Wood, J. Nelander, A. Dufour, A. Bjorklund, O. Lindvall, J. Jakobsson and M. Parmar, Proc. Natl. Acad. Sci U. S. A., 2011, 108, 10343–10348 CrossRef CAS PubMed.
- K. Batta, M. Florkowska, V. Kouskoff and G. Lacaud, Cell Rep., 2014, 9, 1871–1884 CrossRef CAS PubMed.
- M. Ieda, J. D. Fu, P. Delgado-Oliquin, V. Vedantham, Y. Hayashi, B. G. Bruneau and D. Srivastava, Cell, 2010, 142, 375–386 CrossRef CAS PubMed.
- K. S. Velloven, M. Malinen, E. Mannermaa, A. Subrizi, E. Toropainen, Y. R. Lou, H. Kidron, M. Yliperttula and A. Urtti, J. Controlled Release, 2014, 190, 94–114 CrossRef PubMed.
- J. K. Takeuchi and B. G. Bruneau, Nature, 2009, 459, 708–711 CrossRef CAS PubMed.
- A. Martinez and P. S. Amenta, FASEB J., 1995, 9, 1401–1410 Search PubMed.
- A. Martinez, Lab. Invest., 1984, 51, 57–74 Search PubMed.
- D. M. Bissell, J. M. Caron, L. E. Babiss and J. M. Friedman, Mol. Biol. Med., 1990, 7, 187–197 CAS.
- D. M. Bissell, D. M. Arenson, J. J. Maher and F. J. Roll, J. Clin. Invest., 1987, 79, 801–812 CrossRef CAS PubMed.
- N. Lavon, O. Yanuka and N. Benvenisty, Differentiation, 2004, 72, 230–238 CrossRef CAS PubMed.
- Y. Duan, A. Catana, Y. Meng, N. Yamamoto, S. He, S. Gupta, S. S. Gambhir and M. A. Zern, Stem Cells, 2007, 25, 3058–3068 CrossRef CAS PubMed.
- H. Basma, A. S. Gutiérrez, G. R. Yannam, L. Liu, R. Ito, T. Yamamoto, E. Ellis, S. D. Carson, S. Sato, Y. Chen, D. Muirhead, N. N. Álvarez, R. J. Wong, J. R. Chowdhury, J. L. Platt, D. F. Mercer, J. D. Miller, S. C. Strom, N. Kobayashi and I. J. Fox, Gastroenterology, 2009, 136, 990–999 CrossRef CAS PubMed.
- R. E. Schwartz, J. L. Linehan, M. S. Painschab, W. S. Hu, C. M. Verfaillie and D. S. Kaufman, Stem Cells Dev., 2005, 14, 643–655 CrossRef CAS PubMed.
- H. Shirahashi, J. Wu, N. Yamamoto, A. Catana, H. Wege, B. Wager, K. Okita and M. A. Zern, Cell Transplant., 2004, 13, 197–211 Search PubMed.
- M. Vosough, E. Omidinia, M. Kadivar, M.-A. Shokrgozar, B. Pournasr, N. Aghdami and H. Baharvand, Stem Cells Dev., 2013, 22, 2693–2705 CrossRef CAS PubMed.
- K. A. D'Amour, A. D. Agulnick, S. Eliazer, O. G. Kelly, E. Kroon and E. E. Baetge, Nat. Biotechnol., 2005, 23, 1534–1541 CrossRef PubMed.
- T. Ishii, K. Fukumitsu, K. Yasuchika, K. Adachi, E. Kawase, H. Suemori, N. Nakatsuji, I. Ikai and S. Uemoto, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 295, G313–G321 CrossRef CAS PubMed.
- Z. D. Burke, S. Thowfeequ and D. Tosh, Curr. Biol., 2006, 16, R688–R690 CrossRef CAS PubMed.
- V. A. McLin, S. A. Rankin and A. M. Zorn, Development, 2007, 134, 2207–2217 CrossRef CAS PubMed.
- J. Fletcher, W. Cui, K. Samuel, J. R. Black, Z. Hannoun, I. S. Currie, J. D. Terrace, C. Payne, C. Filippi, P. Newsome, S. J. Forbes, J. A. Ross, J. P. Iredale and D. C. Hay, Cloning Stem Cells, 2008, 10, 331–340 CrossRef CAS PubMed.
- J. Cai, Y. Zhao, Y. Liu, F. Ye, Z. Song, H. Qin, S. Meng, Y. Chen, R. Zhou, X. Song, Y. Guo, M. Ding and H. Deng, Hepatology, 2007, 45, 1229–1239 CrossRef CAS PubMed.
- S. Agarwal, K. L. Holton and R. Lanza, Stem Cells, 2008, 26, 1117–1127 CrossRef CAS PubMed.
- D. C. Hay, J. Fletcher, C. Payne, J. D. Terrace, R. C. J. Gallagher, J. Snoeys, J. R. Black, D. Wojtacha, K. Samuel, Z. Hannoun, A. Pryde, C. Filippi, I. S. Currie, S. J. Forbes, J. A. Ross, P. N. Newsome and J. P. Iredale, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12301–12306 CrossRef CAS PubMed.
- D. C. Hay, D. Zhao, J. Fletcher, Z. A. Hewitt, D. McLean, A. Urruticoechea-Uriguen, J. R. Black, C. Elcombe, J. A. Ross, R. Wolf and W. Cui, Stem Cells, 2008, 26, 894–902 CrossRef CAS PubMed.
- D. Szkolnicka, B. Lucendo-Villarin, J. Moore, K. Simpson, S. J. Forbes and D. C. Hay, Stem Cells Transl. Med., 2016 DOI:10.5966/sctm.2015-0117.
- G. Brolén, L. Sivertsson, P. Björquist, G. Eriksson, M. Ek, H. Semb, I. Johansson, T. B. Andersson, M. Ingelman-Sundberg and N. Heins, J. Biotechnol., 2009, 145, 284–294 CrossRef PubMed.
- D. Szkolnicka and D. C. Hay, Stem Cells, 2016 DOI:10.1002/stem.2368.
- Z. Song, J. Cai, Y. Liu, D. Zhao, J. Yong, S. Duo, X. Song, Y. Guo, Y. Zhao, H. Qin, X. Yin, C. Wu, J. Che, S. Lu, M. Ding and H. Deng, Cell Res., 2009, 19, 1233–1242 CrossRef PubMed.
- K. Si-Tayeb, F. K. Noto, M. Nagaoka, J. Li, M. A. Battle, C. Duris, P. E. North, S. Dalton and S. A. Duncan, Hepatology, 2010, 51, 297–305 CrossRef CAS PubMed.
- G. J. Sullivan, D. C. Hay, I.-H. Park, J. Fletcher, Z. Hannoun, C. M. Payne, D. Dalgetty, J. R. Black, J. A. Ross, K. Samuel, G. Wang, G. Q. Daley, J.-H. Lee, G. M. Church, S. J. Forbes, J. P. Iredale and I. Wilmut, Hepatology, 2009, 51, 329–335 CrossRef PubMed.
- T. Touboul, L. Vallier and A. Weber, Med. Sci., 2010, 26, 10611066 Search PubMed.
- S. T. Rashid, S. Corbineau, N. Hannan, S. J. Marciniak, E. Miranda, G. Alexander, I. Huang-Doran, J. Griffin, L. Ahrlund-Ritcher, J. Skepper, R. Semple, A. Webber, D. A. Lomas and L. Vallier, J. Clin. Invest., 2010, 120, 127–136 Search PubMed.
- Y. F. Chen, C. Y. Tseng, H. W. Wang, H. C. Kuo, V. W. Yang and O. K. Lee, Hepatology, 2012, 55, 1193–1203 CrossRef CAS PubMed.
- D. Szkolnicka, S. L. Farnworth, B. Lucendo-Villarin B and D. C. Hay, Curr. Protoc. Stem Cell Biol., 2014, 30, G.5.1–12 Search PubMed.
- H. Rashidi, S. Alhaque, D. Szkolnicka, O. Flint and D. C. Hay, Arch. Toxicol., 2016 DOI:1510.1007/s00204-016-1689-8.
- Y. Avior, G. Levy, M. Zimerman, D. Kitsberg, R. Schwartz, R. Sadeh, A. Moussaieff, M. Cohen, J. Itskovitz-Eldor and Y. Nahmias, Hepatology, 2015, 62, 265–278 CrossRef CAS PubMed.
- S. Sekiya and A. Suzuki, Nature, 2011, 475, 390–393 CrossRef CAS PubMed.
- P. Huang, L. Zhang, Y. Gao, Z. He, D. Yao, Z. Wu, J. Cen, X. Chen, C. Liu, Y. Hu, D. Lai, Z. Hu, L. Chen, Y. Zhang, X. Cheng, X. Ma, G. Pan, X. Wang and L. Hui, Cell Stem Cell, 2014, 14, 370–384 CrossRef CAS PubMed.
- X. L. Shi, Y. Gao, Y. Yan, H. Ma, L. Sun, P. Huang, X. Ni, L. Zhang, X. Zhao, H. Ren, D. Hu, Y. Shou, F. Tian, Y. Ji, X. Cheng, G. Pan, Y. T. Ding and L. Hui, Cell Res., 2016 DOI:10.1038/cr.2016.6.
- C. N. Medine, B. Lucendo-Villarin B, C. Storck, F. Wang, D. Szkolnicka, F. Khan, S. Pernagallo, J. R. Black, H. M. Marriage, J. A. Ross, M. Bradley, J. P. Iredale, O. Flint and D. C. Hay, Stem Cells Transl. Med., 2013, 2, 505–509 CrossRef CAS PubMed.
- G. Holmgren, A. K. Sjögren, I. Barragan, A. Sabirsh, P. Sartipy, J. Synnergren, P. Björquist, M. Ingelman-Sundberg, T. B. Andersson and J. Edsbagge, Drug Metab. Dispos., 2014, 42, 1401–1406 CrossRef PubMed.
- K. Takayama, Y. Morisaki, S. Kuno, Y. Nagamoto, K. Harada, N. Furukawa, M. Ohtaka, K. Nishimura, K. Imagawa, F. Sakurai, M. Tachibana, R. Sumazaki, E. Noguchi, M. Nakanishi, K. Hirata, K. Kawabata and H. Mizuguchi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16772–16777 CrossRef CAS PubMed.
- A. Shlomai, Y. P. de Jong and C. M. Rice, Semin. Cancer Biol., 2014, 26, 78–88 CrossRef CAS PubMed.
- X. Zhou, P. Sun, B. Lucendo-Villarin, A. G. Angus, D. Szkolnicka, K. Cameron, S. L. Farnworth, A. H. Patel and D. C. Hay, Stem Cell Rep., 2014, 3, 204–214 CrossRef CAS PubMed.
- P. Roelandt, S. Obeid, J. Paeshuyse, J. Vanhove, A. Van Lommel, Y. Nahmias, F. Nevens, J. Neyts and C. M. Verfaillie, J. Hepatol., 2012, 57, 246–251 CrossRef CAS PubMed.
- R. E. Schwartz, K. Trehan, L. Andrus, T. P. Sheahan, A. Ploss, S. A. Duncan, C. M. Rice and S. N. Bhatia, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2544–2548 CrossRef CAS PubMed.
- C. Y. Wu, Y. J. Chen, H. J. Ho, Y. C. Hsu, K. N. Kuo, M. S. Wu and L. T. Lin, JAMA, 2012, 308, 1906–1914 CrossRef CAS PubMed.
- K. Takayama, M. Inamura, K. Kawabata, K. Katayama, M. Higuchi, K. Tashiro, A. Nonaka, F. Sakurai, T. Hayakawa, M. K. Furue and H. Mizuguchi, Mol. Ther., 2012, 20, 127–137 CrossRef CAS PubMed.
- D. Szkolnicka, S. L. Farnworth, B. Lucendo-Villarin, C. Storck, W. Zhou, J. P. Iredale, O. Flint and D. C. Hay, Stem Cells Transl. Med., 2014, 3, 141–148 CrossRef CAS PubMed.
- P. Godoy, W. Schmidt-Heck, K. Natarajan, B. Lucendo-Villarin, D. Szkolnicka, A. Asplund, P. Bjorquist, A. Widera, R. Stoeber, G. Campos, S. Hammad, A. Sachinidi, U. Chaudhari, G. Damm, T. S. Weiss, A. Nussler, J. Synnergren, K. Edlund, B. Küppers-Munther, D. C. Hay and J. Hengstler, J. Hepatol., 2015, 63, 934–942 CrossRef CAS PubMed.
- D. A. Brafman, C. W. Chang, A. Fernandez, K. Willert, S. Varghese and S. Chien, Biomaterials, 2010, 31, 9135–9144 CrossRef CAS PubMed.
- S. Rodin, L. Antonsson, C. Niaudet, O. E. Simonson, E. Salmela, E. M. Hansson, A. Domogatskaya, Z. Xiao, P. Damdimopoulou, M. Sheikhi, J. Inzunza, A.-S. Nilsson, D. Baker, R. Kuiper, Y. Sun, E. Blennow, M. Nordenskjöld, K.-H. Grinnemo, J. Kere, C. Betsholtz, O. Hovatta and K. Tryggvason, Nat. Commun., 2014, 5, 3195, DOI:10.1038/ncomms4195.
- S. Rodin, A. Domogatskaya, S. Ström, E. M. Hansson, K. R. Chien, J. Inzunza, O. Hovatta and K. Tryggvason, Nat. Biotechnol., 2010, 28, 611–615 CrossRef CAS PubMed.
- S. R. Braam, C. Denning, E. Matsa, L. E. Young, R. Passier and C. L. Mummery, Nat. Protoc., 2008, 3, 1435–1443 CrossRef CAS PubMed.
- Y. Mei, K. Saha, S. R. Bogatyrev, J. Yang, A. L. Hook, Z. I. Kalcioglu, S. W. Cho, M. Mitalipova, N. Pyzocha, F. Rojas, K. J. Van Vliet, M. C. Davies, M. R. Alexander, R. Langer, R. Jaenisch and D. G. Anderson, Nat. Mater., 2010, 9, 768–778 CrossRef CAS PubMed.
- C. Du, K. Narayanan, M. F. Leong and A. Wan, Biomaterials, 2014, 35, 6006–6014 CrossRef CAS PubMed.
- S. Musah, S. A. Morin, P. J. Wrighton, D. B. Zwick, S. Jin and L. L. Kiessling, ACS Nano, 2012, 6, 10168–10177 CrossRef CAS PubMed.
- D. C. Hay, S. Pernagallo, J. J. Diaz-Mochon, C. N. Medine, S. Greenhough, Z. Hannoun, J. Schrader, J. R. Black, J. Fletcher, D. Dalgetty, A. I. Thompson, P. N. Newsome, S. J. Forbes, J. A. Ross, M. Bradley and J. P. Iredale, Stem Cell Res., 2011, 6, 92–102 CrossRef CAS PubMed.
- R. J. Thomas, R. Bhandari, D. A. Barrett, A. J. Bennett, J. R. Fry, D. Powe, B. J. Thomson and K. M. Shakesheff, Cells Tissues Organs, 2005, 181, 67–79 CrossRef PubMed.
- Y. Pang, K. Montagne, M. Shinohara, K. Komori and Y. Sakai, Biofabrication, 2012, 4, 045004 CrossRef CAS PubMed.
- J. R. Klim, L. Li, P. J. Wrighton, M. S. Piekarczyk and L. L. Kiessling, Nature, 2010, 7, 989–994 CAS.
- L. Gasimli, R. J. Linhardt and J. S. Dordick, Biotechnol. Appl. Biochem., 2012, 59, 65–76 CrossRef CAS PubMed.
- L. G. Villa-Diaz, H. Nandivada, J. Ding, N. C. Nogueira-de-Souza, P. H. Krebsbach, K. S. O'Shea, J. Lahann and G. D. Smith, Nat. Biotechnol., 2010, 28, 581–583 CrossRef CAS PubMed.
- H. Nandivada, L. G. Villa, K. S. O'Shea, G. D. Smith, P. H. Krebsbach and J. Lahann, Nat. Protoc., 2011, 23, 1037–1043 CrossRef PubMed.
- L. G. Villa-Diaz, S. E. Brown, Y. Liu, A. M. Ross, J. Lahann, J. M. Parent and P. H. Krebsbach, Stem Cells, 2012, 30, 1174–1181 CrossRef CAS PubMed.
- E. F. Irwin, R. Gupta, D. C. Dashti and K. E. Healy, Biomaterials, 2011, 32, 6912–6919 CrossRef CAS PubMed.
- K. G. Chen, B. S. Mallon, R. D> G. McKay and P. G. Robey, Cell Stem Cell, 2014, 14, 13–26 CrossRef CAS PubMed.
- S. Liu, M. Jin, Y. Quan, F. Kamiyama, H. Katsumi, T. Saskane and A. Yamamoto, J. Controlled Release, 2012, 161, 933–941 CrossRef CAS PubMed.
- D. A. Brafman, K. D. Shah, T. Fellner, S. Chien and K. Willert, Stem Cells Dev., 2009, 18, 1141–1154 CrossRef PubMed.
- R. Zhang, H. K. Mjoseng, M. A. Hoeve, N. G. Bauer, S. Pells, R. Besseling, S. Velugotla, G. Tourniaire, R. E. B. Kishen, Y. Tsenkina, C. Armit, C. R. E. Duffy, M. Helfen, F. Edenhofer, P. A. de Sousa and M. Bradley, Nat. Commun., 2013, 4, 1335 CrossRef PubMed.
- T. P. Kraehenbuehl, R. Langer and L. S. Ferreira, Nat. Methods, 2011, 8, 731–736 CrossRef CAS PubMed.
- T. Maguire, A. E. Davidovich, E. J. Wallenstein, E. Novik, N. Sharma, H. Perdersen, I. P. Androulakis, R. Schloss and M. Yarmush, Biotechnol. Bioeng., 2007, 15, 631–644 CrossRef PubMed.
- S. Fang, Y. D. Qiu, L. Mao, X. L. Shi, D. C. Yu and Y. T. Ding, Acta Pharmacol. Sin., 2007, 28, 1924–1930 CrossRef CAS PubMed.
- S. Jitraruch, A. Dhawan, R. D. Hughes, C. Filippi, D. Soong, C. Philippeos, S. C. Lehec, N. D. Heaton, M. S. Longhi and R. R. Mitry, PLoS One, 2014, 9, e113609 Search PubMed.
- M. Amit, J. Chebath, V. Margulets, I. Laevsky, Y. Miropolsky, K. Shariki, M. Peri, I. Blais, G. Slutsky, M. Revel and J. Itskovitz-Eldor, Stem Cell Rev., 2010, 6, 248–259 CrossRef PubMed.
- D. Steiner, H. Khaner, M. Cohen, S. Even-Ram, Y. Gil, P. Itsykson, M. Idelson, E. Aizenman, R. Ram, Y. Berma-Zaken and B. Reubinoff, Nature, 2010, 28, 361–364 CAS.
- R. S. Tare, F. Khan, G. Tourniaire, S. M. Morgan, M. Bradley and R. O. Oreffo, Biomaterials, 2009, 30, 1045–1055 CrossRef CAS PubMed.
- R. Zhang, A. Liberski, R. Sanchez-Martin and M. Bradley, Biomaterials, 2009, 30, 6193–6201 CrossRef CAS PubMed.
- A. D. Celiz, J. G. Smith, A. K. Patel, A. L. Hook, D. Rajamohan, V. T. George, L. Flatt, M. J. Patel, V. C. Epa, T. Singh, R. Langer, D. G. Anderson, N. D. Allen, D. C. Hay, D. A. Winkler, D. A. Barrett, M. C. Davies, L. E. Young, C. Denning and M. R. Alexander, Adv. Mater., 2015, 27, 4006–4012 CrossRef CAS PubMed.
- T. Takebe, K. Sekine, M. Enomura, H. Koike, M. Kimura, T. Ogaeri, R.-R. Zhang, Y. Ueno, Y.-W. Zheng, N. Koike, S. Aoyama, Y. Adachi and H. Taniguchi, Nature, 2013, 499, 481–484 CrossRef CAS PubMed.
- D. A. Visk, In Vitro Toxicol., 2015, 1, 79–82 CrossRef.
- A. Faulkner-Jones, C. Fyfe, D. J. Cornelissen, J. Gardner, J. King, A. Courtney and W. Shu, Biofabrication, 2015, 7, 044102 CrossRef PubMed.
- G. G. Giobbe, F. Michielin, C. Luni, S. Giulitti, S. Martewicz, S. Dupont, A. Floreani and N. Evassore, Nat. Methods, 2015, 12, 637 CrossRef CAS PubMed.
- D. R. Berger, B. R. Ware, M. D. Davidson, S. R. Allsup and S. R. Khetani, Hepatology, 2015, 61, 1370–1381 CrossRef CAS PubMed.
- Y. Avior, G. Levy, M. Zimerman, D. Kitsberg, R. Schwartz, R. Sadeh, A. Moussaieff, M. Cohen, J. Itskovitz-Eldor and Y. Nahmias, Hepatology, 2015, 62, 265–278 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |