
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the heteroatom on the optoelectronic properties and transistor performance of soluble thiophene-, selenophene- and tellurophene–vinylene copolymers†
Mohammed
Al-Hashimi
 *ab,
Yang
Han
ac,
Jeremy
Smith
c,
Hassan S.
Bazzi
b,
Siham Yousuf A.
Alqaradawi
d,
Scott E.
Watkins
e,
Thomas D.
Anthopoulos
c and
Martin
Heeney
*a
*ab,
Yang
Han
ac,
Jeremy
Smith
c,
Hassan S.
Bazzi
b,
Siham Yousuf A.
Alqaradawi
d,
Scott E.
Watkins
e,
Thomas D.
Anthopoulos
c and
Martin
Heeney
*a
aDept. Chemistry and Centre for Plastic Electronics, Imperial College London, Exhibition Rd, London, SW7 2AZ, UK. E-mail: m.heeney@imperial.ac.uk
bDept. Chemistry, Texas A&M University at Qatar, P.O. Box 23874, Doha, Qatar. E-mail: mohammed.al-hashimi@qatar.tamu.edu
cDept. Physics and Centre for Plastic Electronics, Imperial College London, Exhibition Rd, London, SW7 2AZ, UK
dDept. of Chemistry & Earth Sciences, Qatar University, P.O. Box 110003, Doha, Qatar
eCSIRO, Molecular and Health Technologies, VIC 3169, Australia
First published on 2nd November 2015
Abstract
We report the first soluble poly(3-dodecyl tellurophenylene-vinylene) polymer (P3TeV) by Stille copolymerization and compare its properties to the analogous thiophene and selenophene containing polymers. The optical band gap of the polymers is shown to systematically decrease as the size of the heteroatom is increased, mainly as a result of a stabilization of the LUMO energy, resulting in a small band gap of 1.4 eV for P3TeV. Field effect transistors measurements in variety of architectures demonstrate that the selenophene polymer exhibits the highest mobility, highlighting that increasing the size of the heteroatom is not always beneficial for charge transport.
Introduction
Conjugated organic polymers continue to attract widespread interest for a wide range of applications due to their potential for large area, low-cost fabrication.1,2 The ability of synthetic chemistry to manipulate the structure and properties of the conjugated aromatic monomers, as well as the almost limitless possibilities to co-polymerize such building blocks leads to a huge scope to tune the polymer performance for each application.3 Despite the large tool-box of organic chemistry, many of the polymers reported to date are based upon thiophene, or its fused ring analogues.4,5 One fascinating opportunity to manipulate the polymer properties is by changing the heteroatom in the five membered heterocycle from sulfur to other elements within group 16.6,7 Switching to heavier elements with larger polarizability such as selenium or tellurium is of potential interest since the larger atomic size may facilitate solid state intermolecular interactions, as well as allowing a route to control the HOMO and LUMO levels and optical band gap.8,9Reports of tellurophene containing polymers are considerably less numerous than selenophene containing materials, probably in part due to the synthetic challenges of tellurophene chemistry.10,11 In addition many of the examples reported to date are co-polymers, with relatively low loadings of tellurophene in which there is a potential that the intrinsic influence of the heteroatom is masked.12–20 Homo-polymers of tellurophene are rare, with the first substituted tellurophene polymer, poly(3,4-dimethoxytellurophene),21 prepared as an insoluble film by electrochemical polymerization. A significant step was the recent report of the first soluble tellurophene polymer, poly(3-alkyltellurophene), P3ATe which was prepared by both electrochemical and Kumada catalyst transfer polymerization methods.22 Regioregular P3ATe exhibited a red shifted absorption compared to the thiophene or selenophene analogues, although the preliminary performance reported in solar cell was modest (PCE 1.1%).23
In this paper we report the synthesis and characterization of an analogous series of vinylene copolymers containing 3-dodecylthiophene, selenophene or tellurophene to allow the systematic investigation of the role of the heteroatom. 3-Alkylthiophene and 3-alkylselenophene–vinylene co-polymers have previously shown promise as the active semiconducting component in both FETs and OPVs,24,25 as well as in singlet fission applications.26 The tellurophene analogues have never been reported to the best of our knowledge. Promisingly the regioregularity of the alkyl chains has less influence on crystallinity in these systems than the non-vinylene co-polymers, due to the reduction of torsional disorder with the vinylene spacer.27 Herein, we report the synthesis of a novel 2,5-dibrominated 3-dodecyltellurophene monomer and its copolymerization with (E)1,2-bis(tributylstannyl)ethylene to afford poly(3-dodecyl-2,5-tellurophenylenevinylene) (P3TeV). We compare the optoelectronic properties to the analogous thiophene (P3TV) and selenophene (P3SV) containing polymers, as well as report the properties of all three polymer organic field-effect transistors (OFETs).
Experimental
General procedures
All chemicals were purchased from Aldrich or Fisher. All reactions were carried out under argon using solvents and reagents as commercially supplied, unless otherwise stated. 1H and 13C NMR spectra were recorded on a Bruker AV-400 (400 MHz), using the residual solvent resonance of CDCl3 or TMS as an internal reference and are given in ppm. Microwave reactions were run in a Biotage Initiator at constant temperature. Number-average (Mn) and weight-average (Mw) were determined by Agilent Technologies 1200 series GPC running in chlorobenzene at 80 °C. The system used two PL mixed B columns in series, and were calibrated against narrow polydispersity polystyrene standards. Electrospray mass spectrometry was performed with a Thermo Electron Corporation DSQII mass spectrometer. UV-Vis spectra were recorded on a Perkin Elmer Lambda 900 UV-Vis spectrometer. Flash chromatography (FC) was performed on silica gel (Merck Kieselgel 60 F254 230–400 mesh) unless otherwise indicated. Thin Layer Chromatography (TLC) was performed on Merck aluminium-backed plates pre-coated with silica (0.2 mm, 60 F254). Photo Electron Spectroscopy in Air (PESA) measurements were recorded with a Riken Keiki AC-2 PESA spectrometer with a power setting of 5 nW and a power number of 0.5. Samples for PESA were prepared on glass substrates as drop cast films. 3-Dodecylselenophene was prepared according to the published procedure.28General polymerisation procedure
To the appropriate 2,5-dibromo-3-dodecyl monomer in a microwave vial was added Pd(PPh3)4 (1 mol%), toluene or chlorobenzene (0.6 mL) and equal molar amount of (E)-1,2-bis-(tributylstannyl)ethylene. The resultant mixture was degassed for 30 min with argon and securely sealed. The glass vial was either placed into an oil bath and was heated at 110 °C for 24 h when using toluene or placed into a microwave at 120 °C for 5 min, 140 °C for 5 min and 180 °C for 30 min when using chlorobenzene.All the polymers where purified as follows: the polymerisation was cooled to room temperature, and added dropwise into a rapidly stirring mixture of methanol (200 mL)/concentrated hydrochloric acid (2 mL) and allowed to stir for 2 h. The polymeric material was then filtered under reduced pressure into a cellulose thimble and was purified by Soxhlet extraction with methanol (12 h) acetone (12 h) and hexane (12 h) in that order. The remaining, polymer was dissolved in chloroform and precipitated into methanol, filtered and dried under vacuum to achieve the desired polymer.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mn = 10100 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 566 nm, λmax (film): 572 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 6.88–7.03 (br, 3H), 2.72 (br, 2H), 1.74 (br, 2H), 1.38 (br, 18H), 0.97 (br, 3H).
000, Mn = 12000 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 609 nm, λmax (film): 613 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 6.87–7.05 (br, 3H), 2.68 (br, 2H), 1.72 (br, 2H), 1.38 (br, 18H), 0.97 (br, 3H).
000, Mn = 10000 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 637 nm, λmax (film): 652 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 7.41–6.68 (br, 3H), 2.67 (br, 2H), 1.70 (br, 2H), 1.37 (br, 18H), 0.97 (br, 3H).
000, Mn = 10100 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 566 nm, λmax (film): 572 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 6.88–7.03 (br, 3H), 2.72 (br, 2H), 1.74 (br, 2H), 1.38 (br, 18H), 0.97 (br, 3H).
000, Mn = 12000 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 609 nm, λmax (film): 613 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 6.87–7.05 (br, 3H), 2.68 (br, 2H), 1.72 (br, 2H), 1.38 (br, 18H), 0.97 (br, 3H).
000, Mn = 10000 g mol−1. UV-Vis λmax (dilute chlorobenzene solution): 637 nm, λmax (film): 652 nm. 1H NMR (500 MHz, C2D2Cl4 at 130 °C) δ 7.41–6.68 (br, 3H), 2.67 (br, 2H), 1.70 (br, 2H), 1.37 (br, 18H), 0.97 (br, 3H).
Transistor fabrication and testing
Results and discussion
There are limited reports for the preparation of 3-alkyltellurophenes, and here we based our synthesis upon slight modifications of the previous reports.22 In order to enable ready comparison to the thiophene and selenophene–vinylene co-polymers, which were both synthesized form the respective 2,5-dibromo-3-alkylchalcogenophene monomers by polymerization with (E)1,2-bis(tributylstannyl)ethylene, the obvious starting material was 2,5-dibromo-3-dodecyltellurophene. However all of our attempts at electrophilic bromination of 3-dodecyltellurophene with either NBS or elemental bromine proved problematic. Complex inseparable mixtures were obtained, which we attributed to bromination of the Te atom,29 followed by decomposition of the resulting heterocycle. This could be avoided by di-lithiation of 3-dodecyl tellurophene with excess n-BuLi at room temperature followed by the low temperature addition of 1,2-dibromotetrachlorethane to afford 2,5-dibromo-3-dodecyltellurophene in 55% yield as a yellow oil. The structure of the product was confirmed by 1H and 13C NMR and high-resolution mass-spectrometry. The product was stable, with no signs of degradation seen upon storage at 5 °C over several months.With all three dibrominated monomers in hand, the three polymers P3TV, P3SV and P3TeV were synthesized by Stille coupling between the appropriate 2,5-dibromo-3-dodecyl monomers and (E)1,2-bis(tributylstannyl)ethylene (Scheme 1). Polymers P3TV and P3SV where synthesized using microwave assisted heating in chlorobenzene, whereas polymer P3TeV was synthesized using conventional heating in toluene. Attempted microwave polymerization of P3TeV resulted in insoluble polymers, which we attributed to the formation of insoluble high molecular weight material. The crude polymers were all purified by precipitation and Soxhlet extraction with methanol and acetone to remove catalyst residues and low molecular weight oligomers, followed by precipitation of a chloroform solution of the polymer into methanol to afford the polymers in good yield. Clear differences were observed in the qualitative solubility of the polymers, which decreased in the trend P3TV > P3SV > P3TeV. The P3TeV polymer was only slightly soluble at room temperature in chlorinated solvents like chlorobenzene or chloroform, but could be dissolved upon prolonged heating. Nevertheless it had a high tendency to precipitate upon cooling, resulting in difficulties in the formation of smooth films.
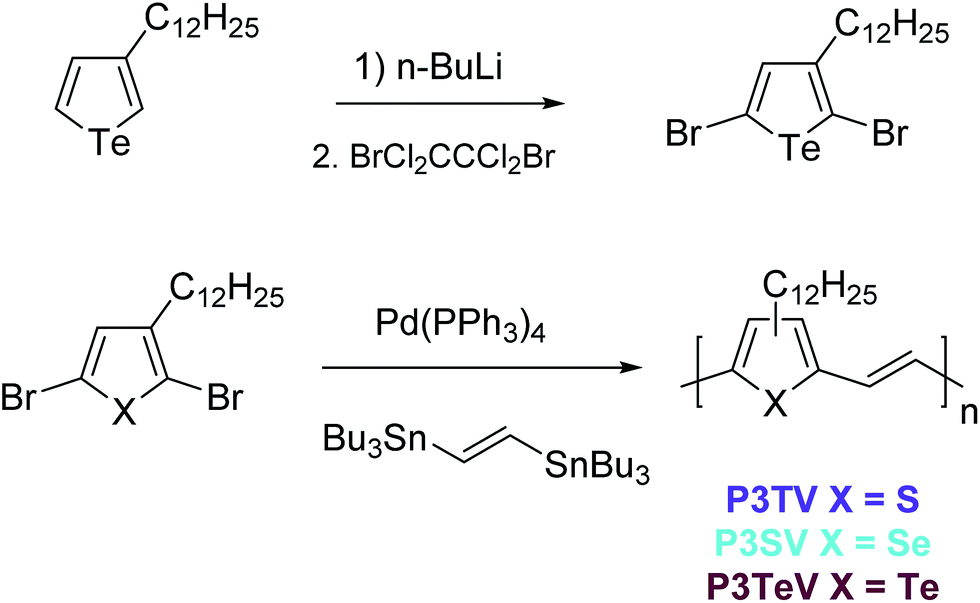 | ||
| Scheme 1 Synthesis of 2,5-dibromo-3-dodecyltellurophene and polymerization to afford P3TV, P3SV and P3TeV. | ||
The molecular weight (Mn and Mw) and dispersity index (Ð) of the copolymers were determined by gel permeation chromatography (GPC) relative to polystyrene standards in hot chlorobenzene at 80 °C. The three polymers exhibit similar average molecular weights (Mw) in the range of 16–24 kDa, although there is some variation in the dispersity between the polymers (Table 1). It is worth highlighting that GPC calibrated against flexible polystyrene standards often significantly overestimates the molecular weight of semi-flexible conjugated polymers, suggesting these are likely upper estimates of the actual values.30,31
| Polymers | M n (kDa) | M w (kDa) | Ð | λ max soln [nm] | λ max film [nm] | λ onset film [nm] | HOMO (PESA)b [eV] | LUMOc [eV] | HOMO (CV)d (Eox, onset V) | E optg [eV] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC and reported as their polystyrene equivalents. b Determined as a thin film by UV-PESA error (±0.05 eV). c Estimated using HOMOb − Eoptg. d CV measurements performed on as cast polymer films on a Pt working electrode in CH3CN with 0.1 M TBAPF6. The scan rate was 50 mV s−1. EHOMO = −(φonset ox − φFc/Fc+ + 4.8). e Determined by onset of optical absorption of thin film deduced from the equation, Eoptg = 1240/λonset. | ||||||||||
| P3TV | 10.1 | 16 | 1.58 | 566 | 572 | 725 | −4.93 | −3.22 | −5.21 (0.70) | −1.71 |
| P3SV | 12 | 24 | 2 | 609 | 613 | 800 | −4.95 | −3.40 | −5.17 (0.66) | −1.55 |
| P3TeV | 10 | 24 | 2.4 | 637 | 652 | 875 | −4.89 | −3.50 | −5.08 (0.57) | −1.42 |
The chemical structure of the three polymers were investigated using 1H NMR spectroscopy in 1,1,2,2-tetrachloroethane-d2 at 130 °C (Fig. 1). Loss of resolution and broadening of the spectra can be seen due to the high degree of polymer aggregation in solution even at 130 °C. Nevertheless, the signals corresponding to the aromatic and vinyl protons of P3TV, P3SV and P3TeV can be resolved in the range of δ 6.6 to 7.5 ppm. Interestingly, P3TV and P3SV polymers show similar and overlapping chemical shifts at δ 6.87 to 7.05 ppm for the aromatic and vinylene protons as previously observed,25,32 whereas P3TeV shows significant differences, with the aromatic proton shifted downfield to a broad multiplet around 7.41 ppm, and the vinylene protons are shifted upfield to a doublet 6.78–6.68 ppm. The broadness of the tellurophene signal is probably related to end group effects due to the low molecular weight. The methylene protons for all polymers all fall as a broad peak in a similar region around δ 2.72–2.65 ppm. Studies on P3TV's of well-defined regularity show that the methylene region is not very sensitive to the regiochemistry of the coupling, and that the aromatic region is more informative.33 However it is difficult to assess the regioregularity of the polymers due to the broadness of the aromatic peaks in the current case, but we can say all polymers are similar. Although early reports of PTV's by Stille polymerization suggested a regioregularity around 85–90%,32 later reports33 suggest the polymers are regiorandom. As noted earlier, low regioregularity is not expected to prevent backbone planarization in this case.
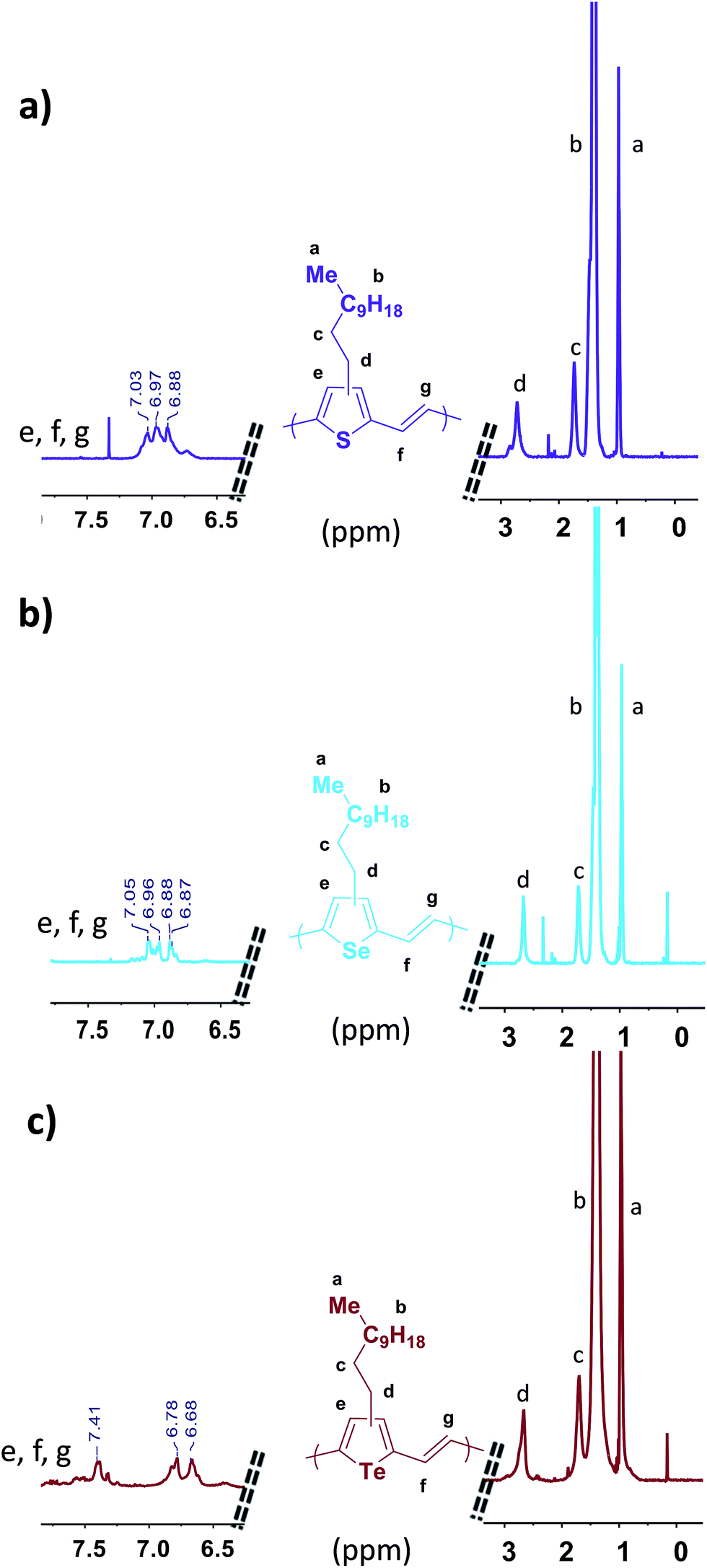 | ||
| Fig. 1 1H NMR spectra of the three polymers; (a) P3TV, (b) P3SV and (c) P3TeV (500 MHz, C2D2Cl4, 130 °C). | ||
The absorption spectra of P3TV, P3SV and P3TeV in solution and as drop cast films are shown in Fig. 2 and the data is summarized in Table 1. There is a clear trend across the series, with the spectra in both solution and thin film red-shifting as the heteroatom is changed, hence moving from sulfur (P3TV) to tellurium (P3TeV) results in a red shift of 71 nm in solution and 80 nm in film, whereby the polymer containing the tellurium atom shows the largest red-shift absorption in both solution and film. In solution the spectra all display similar shape, with an absorption maxima (λmax) at 566, 609 and 637 nm for P3TV, P3SV and P3TeV respectively. There is also a pronounced shoulder at longer wavelengths for all polymers, this is typically associated with vibronic coupling and suggests some degree of order for the polymers, even in solution. Upon film formation the λmax of all polymers red-shifts slightly (6–15 nm) and broaden significantly. The vibronic shoulder appears to become more pronounced on moving down the periodic table, becoming especially prominent for P3TeV. The optical band gaps of the three polymers as estimated from the onset absorption of the thin film are P3TV = 1.71 eV, P3SV = 1.55 eV and P3TeV = 1.42 eV, and follow a similar trend to the shift in λmax. Although the vibronic shoulders are indicative of some short range aggregation in the solid state, investigation of the polymers by differential scanning calorimetry (DSC) was uninformative (ESI†), with either no peaks observed (P3SV, P3TeV) or only very broad transitions (P3TV) upon cycling to 200 °C. Heating above 200 °C was previously noted to result in irreversible changes in phase behavior.25 The lack of peaks in the current examples versus those previously observed for P3TV and P3SV25 is likely due to the lower molecular weight and regioregularity of the current materials versus previous examples.
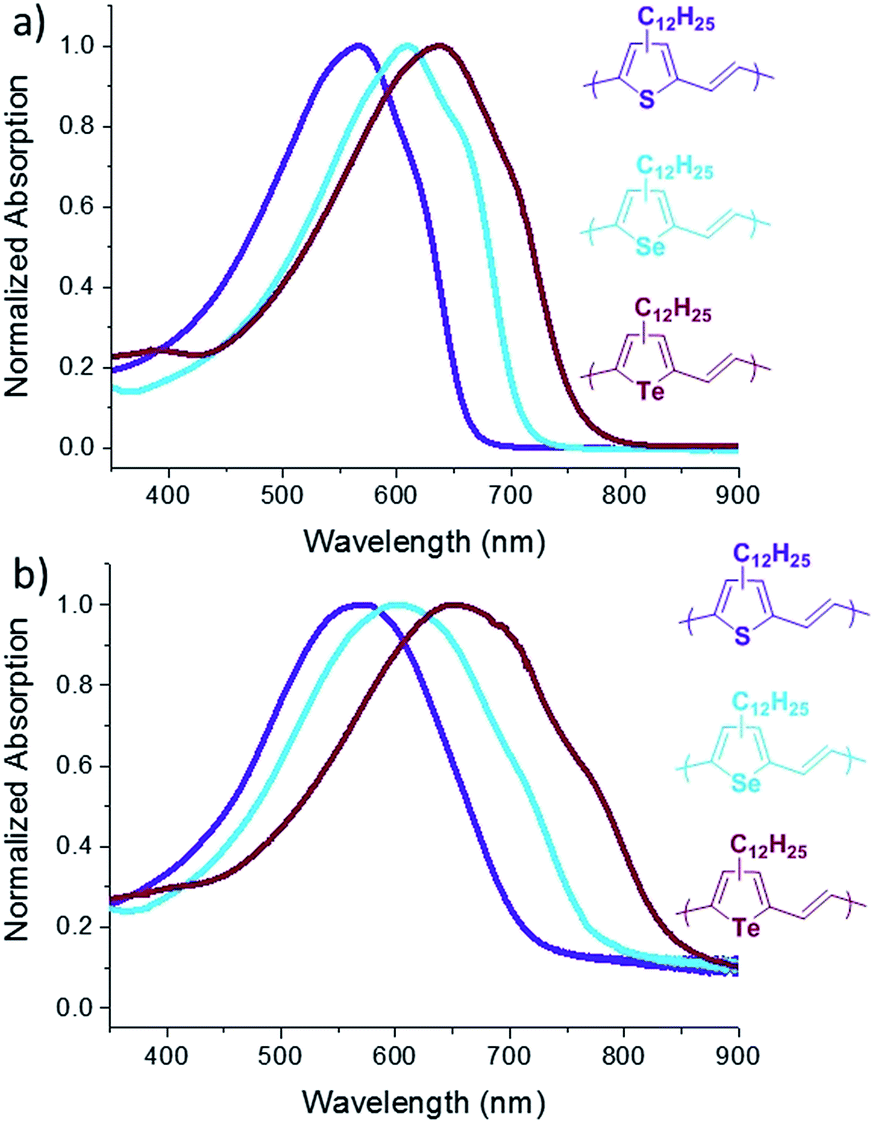 | ||
| Fig. 2 UV-Vis spectra of P3TV, P3SV and P3TeV in dilute chlorobenzene (a) and in thin films (b). | ||
The oxidation potentials of the polymers were obtained using cyclic voltammetry (CV) measurements of drop-cast polymers films on a platinum working electrodes. The cyclic voltagrams are shown in Fig. 3 and show quasi-reversible oxidation waves in the case of P3TV and P3SV. In the case of P3TeV a fully reversible oxidation is observed, with a second reversible oxidation observed at higher voltages (Eox2 onset 1.04 V). We were not able to observe reproducible reduction for any polymer under our test conditions. The HOMO energies for P3TV, P3SV and P3TeV were estimated by the onset of oxidation (Eox onset, see Table 1) against Ag/AgCl reference, assuming the ferrocene/ferrocenium reference redox system is 4.8 eV below the vacuum level. The ionization potentials were also measured as thin films using photoelectron spectroscopy (PESA, Fig. S1–S3†). The absolute values measured by either technique are different, but there is reasonable agreement in the trends observed. Thus both CV and PESA find similar HOMO values for the P3TV and P3SV, with the differences within the error of either technique, suggesting that the substitution of sulfur for selenium has a relatively minor influence of the oxidation potential. The clear difference in optical band gap therefore suggests a greater perturbation to the LUMO, with the decrease in band gap suggesting a stabilizing influence on the LUMO on moving to the larger heteroatom.34,35 On moving down the periodic table, both PESA and CV suggest the HOMO of P3TeV moves closer to the vacuum level compared to the sulfur or selenium analogues. The increase in the HOMO level (ca. 0.05–0.1 eV depending on technique) is substantially less than the reduction in optical band gap, suggesting that the perturbation to the LUMO is greater than the HOMO. The increase in the HOMO level observed is in agreement with other reports on tellurophene containing polymers,10 and with the observation that the aromaticity of tellurophene is low in comparison to thiophene and selenophene as a result of the reduced overlap between the larger heteroatom and the adjacent carbon based π orbitals.36
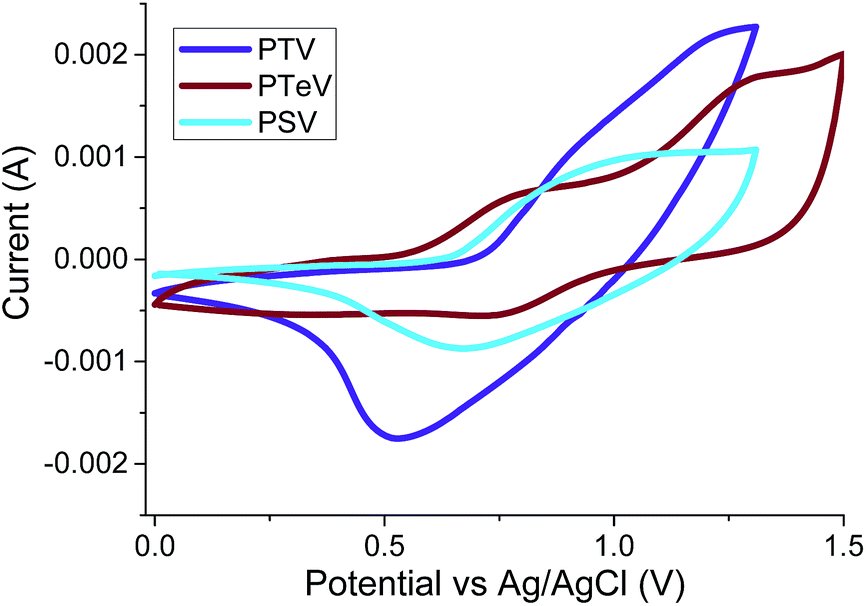 | ||
| Fig. 3 Thin-film cyclic voltammograms of P3TV, P3SV and P3TeV on Pt electrodes in CH3CN/0.1 M [nBu4N]+[PF6]− at 50 mV s−1. | ||
Finally the charge carrier mobilities of the polymers were investigated in field effect transistors, using a variety of device architectures including bottom gate, bottom contact (BG/BC), bottom gate, top contact (BG/TC) and top gate, bottom contact (TG/BC). In all cases the processing of the films was problematic due to the moderate solubility and this was a particular problem for P3TeV which rapidly precipitated from solution on cooling. Although heating of the substrate prior to spin-casting helped, the device yield remaining low. P3TV was also problematic due to wetting issues, particularly for BG/BC devices. Nevertheless working devices could be obtained for all polymers in at least two of the device architectures, and the data is summarized in Table 2, with individual transfer characteristics in the ESI.† All polymers were p-type semiconductors with no evidence of electron transport observed in these configurations. The devices showed some hysteresis between the forward and reverse scans in most cases, which may relate to the difficulty in film formation. In terms of mobility, relatively modest saturated mobilities were observed for all polymers, in agreement with the previous reports of P3TV and P3SV prepared by Stille polymerization.25,27 Direct comparisons between the polymers are complicated by the film forming issues, but it is clear that the selenophene polymer consistently displays the highest transistor mobility across all devices. The incorporation of tellurium does not seem to confer any beneficial increase in mobility, despite the large size of Te and the possibility of interchain Te⋯Te interactions.37
| Polymer | Field effect mobility (cm2 V−1 s−1) | ||
|---|---|---|---|
| TG/BC | BG/BC | BG/TC | |
| P3TV | 5 × 10−4 | — | 1.8 × 10−3 |
| P3SV | 0.05 | 0.02 | 3.6 × 10−3 |
| P3TeV | 1 × 10−4 | 0.001 | — |
To the best of our knowledge there is only one report of the transistor properties of a tellurophene homo polymer, in which the performance of regioregular P3ATe with varying sidechain lengths is reported with moderate mobility.38 There are even fewer examples comparing the performance of polymers with different chalcogenophenes and all of these examples have been co-polymers in which the tellurophene comprises a relatively minor fraction of the backbone. In these limited reports, the inclusion of Te has been shown to improve mobility over S and Se.13,15,17 In the case of vacuum deposited small molecules, Te inclusion has also resulted in a reduction in charge carrier mobility compared to S or Se.39 In our case any beneficial influence of the Te on mobility in terms of enhanced molecular contacts seems countered by the reduced solubility resulting in poor film forming ability. These results suggest attempts to fully harness the potential of all tellurophene containing polymers need to pay close attention to sidechain engineering to enhance solubility.
Conclusions
In conclusion we have reported the synthesis of 2,5-dibromo-3-dodecyltellurophene and co-polymerized it with (E)1,2-bis(tributylstannyl)ethylene to afford the first tellurophene vinylene co-polymer. Analogous thiophene and selenophene containing polymers were prepared by the same methodology to produce polymers of similar molecular weight and regioregularity. Increasing the size of the heteroatom results in a reduction in solubility as well as a reduction in the optical band gap, mainly as a result of the stabilization of the polymer LUMO level. Field effect transistors in a variety of device architectures show that the selenophene containing polymers consistently exhibit the highest mobility, whereas the low solubility of the tellurophene containing polymer inhibited device performance, highlighting the requirement to carefully tune sidechains for solubility in the case of heavy atom containing polymers.Acknowledgements
This report was made possible by a NPRP award [NPRP 6-452-1-089] from the Qatar National Research Fund (a member of The Qatar Foundation). The statements made herein are solely the responsibility of the authors.Notes and references
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef CAS PubMed.
- Y. He, W. Hong and Y. Li, J. Mater. Chem. C, 2014, 2, 8651 RSC.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef CAS PubMed.
- C. B. Nielsen and I. McCulloch, Prog. Polym. Sci., 2013, 38, 2053 CrossRef CAS.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253 CrossRef CAS.
- X. He and T. Baumgartner, RSC Adv., 2013, 3, 11334 RSC.
- A. A. Jahnke and D. S. Seferos, Macromol. Rapid Commun., 2011, 32, 943 CrossRef CAS PubMed.
- L. Zhang, S. M. Fakhouri, F. Liu, J. C. Timmons, N. A. Ran and A. L. Briseno, J. Mater. Chem., 2011, 21, 1329 RSC.
- E. I. Carrera and D. S. Seferos, Macromolecules, 2015, 48, 297 CrossRef CAS.
- E. Rivard, Chem. Lett., 2015, 44, 730 CrossRef CAS , advpub.
- A. A. Jahnke, G. W. Howe and D. S. Seferos, Angew. Chem., Int. Ed., 2010, 49, 10140 CrossRef CAS PubMed.
- M. Kaur, D. S. Yang, J. Shin, T. W. Lee, K. Choi, M. J. Cho and D. H. Choi, Chem. Commun., 2013, 49, 5495 RSC.
- G. He, L. Kang, W. Torres Delgado, O. Shynkaruk, M. J. Ferguson, R. McDonald and E. Rivard, J. Am. Chem. Soc., 2013, 135, 5360 CrossRef CAS PubMed.
- E. H. Jung, S. Bae, T. W. Yoo and W. H. Jo, Polym. Chem., 2014, 5, 6545 RSC.
- M. Kaur, D. H. Lee, D. S. Yang, H. A. Um, M. J. Cho, J. S. Kang and D. H. Choi, Chem. Commun., 2014, 50, 14394 RSC.
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2014, 137, 1314 CrossRef PubMed.
- M. Planells, B. C. Schroeder and I. McCulloch, Macromolecules, 2014, 47, 5889 CrossRef CAS.
- Y. S. Park, Q. Wu, C. Y. Nam and R. B. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 10691 CrossRef CAS PubMed.
- A. K. Mahrok, E. I. Carrera, A. J. Tilley, S. Ye and D. S. Seferos, Chem. Commun., 2015, 51, 5475 RSC.
- A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Org. Lett., 2009, 11, 1487 CrossRef CAS PubMed.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. B. Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951 CrossRef CAS PubMed.
- W.-H. Lee, S. Kyu Lee, W. Suk Shin, S.-J. Moon and I.-N. Kang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2753 CrossRef CAS.
- J. Sun, C. Zhang, S. Venkatesan, R. Li, S. S. Sun and Q. Qiao, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 917 CrossRef CAS.
- M. Al-Hashimi, M. A. Baklar, F. Colleaux, S. E. Watkins, T. D. Anthopoulos, N. Stingelin and M. Heeney, Macromolecules, 2011, 44, 5194 CrossRef CAS.
- A. J. Musser, M. Al-Hashimi, M. Maiuri, D. Brida, M. Heeney, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2013, 135, 12747 CrossRef CAS PubMed.
- J. C. Speros, H. Martinez, B. D. Paulsen, S. P. White, A. D. Bonifas, P. C. Goff, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2013, 46, 5184 CrossRef CAS.
- C. Mahatsekake, J. M. Catel, C. G. Andrieu, M. Ebel and Y. Mollier, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 35 CrossRef CAS.
- T. M. McCormick, A. A. Jahnke, A. J. Lough and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 3542 CrossRef CAS PubMed.
- F. P. V. Koch, P. Smith and M. Heeney, J. Am. Chem. Soc., 2013, 135, 13695 CrossRef CAS PubMed.
- J. Liu, R. S. Loewe and R. D. McCullough, Macromolecules, 1999, 32, 5777 CrossRef CAS.
- R. S. Loewe and R. D. McCullough, Chem. Mater., 2000, 12, 3214 CrossRef CAS.
- C. Zhang, J. Sun, R. Li, S.-S. Sun, E. Lafalce and X. Jiang, Macromolecules, 2011, 44, 6389 CrossRef CAS.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007, 5061 RSC.
- S. S. Zade, N. Zamoshchik and M. Bendikov, Chem.–Eur. J., 2009, 15, 8613 CrossRef CAS PubMed.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777 CrossRef CAS PubMed.
- K. Kobayashi, H. Masu, A. Shuto and K. Yamaguchi, Chem. Mater., 2005, 17, 6666 CrossRef CAS.
- A. A. Jahnke, L. Yu, N. Coombs, A. D. Scaccabarozzi, A. J. Tilley, P. M. DiCarmine, A. Amassian, N. Stingelin and D. S. Seferos, J. Mater. Chem. C, 2015, 3, 3767 RSC.
- K. Takimiya, Y. Kunugi, Y. Konda, N. Niihara and T. Otsubo, J. Am. Chem. Soc., 2004, 126, 5084 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Transistor transfer and output plots and PESA plots. See DOI: 10.1039/c5sc03501e |
| This journal is © The Royal Society of Chemistry 2016 |