
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to a new class of synthetic building blocks via trifluoromethoxylation of pyridines and pyrimidines†
Pengju
Feng‡
ab,
Katarzyna N.
Lee‡
 ab,
Johnny W.
Lee
ab,
Chengbo
Zhan
ab and
Ming-Yu
Ngai
*ab
ab,
Johnny W.
Lee
ab,
Chengbo
Zhan
ab and
Ming-Yu
Ngai
*ab
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA
bInstitute of Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: ming-yu.ngai@stonybrook.edu
First published on 7th October 2015
Abstract
Since the first synthesis of trifluoromethyl ethers in 1935, the trifluoromethoxy (OCF3) group has made a remarkable impact in medicinal, agrochemical, and materials science research. However, our inability to facilely incorporate the OCF3 group into molecules, especially heteroaromatics, has limited its potential across a broad spectrum of technological applications. Herein, we report a scalable and operationally simple protocol for regioselective trifluoromethoxylation of a wide range of functionalized pyridines and pyrimidines under mild reaction conditions. The trifluoromethoxylated products are useful scaffolds that can be further elaborated by amidation and palladium-catalysed cross coupling reactions. Mechanistic studies suggest that a radical O-trifluoromethylation followed by the OCF3-migration reaction pathway is operable. Given the unique properties of the OCF3 group and the ubiquity of pyridine and pyrimidine in biologically active molecules and functional materials, trifluoromethoxylated pyridines and pyrimidines could serve as valuable building blocks for the discovery and development of new drugs, agrochemicals, and materials.
Introduction
The trifluoromethoxy (OCF3) group has made a significant impact in medicinal, agrochemical, life- and materials science research1–5 since Booth and Burchfield reported the first synthesis of trifluoromethyl ethers in 1935.6 The increasing importance of the OCF3 group can be attributed to its unique structural and electronic properties. First of all, in aryl trifluoromethyl ethers the OCF3 moiety lies in the plane orthogonal to arene ring (Fig. 1a)7 and studies have shown that this unusual orientation may be beneficial for providing additional binding affinity in drug–target complexes.8 In addition, the OCF3 group is among the most electronegative groups (χ(F) = 4.0, χ(OCF3) = 3.7).9 Molecules bearing an electron-withdrawing group have better metabolic stability. Moreover, the OCF3 group has an excellent lipophilicity (πx(SCF3) = +1.44, πx(SF5) = +1.23, πx(OCF3) = +1.04, πx(CF3) = +0.88, πx(OCH3) = −0.02);10 compounds with higher lipophilicity show enhancement in their in vivo uptake and transport in biological systems. Therefore, the OCF3 group is introduced into biologically active molecules to improve their efficacy and minimize their side effects (Fig. 1b).1,2,5 Furthermore, incorporation of the OCF3 group into organic molecules can increase their melting point and boiling point difference under ambient pressure, and lower their surface tension, dielectric constant, and pour point.1,11,12 These properties are particularly useful in designing electronic devices and materials; as a result, the OCF3-containing molecules can be found in electro-optical materials used for the development of liquid crystal displays,13 soluble organic semi-conductor,14 and melt-processable fluoropolymers such as perfluoroalkoxy alkanes.12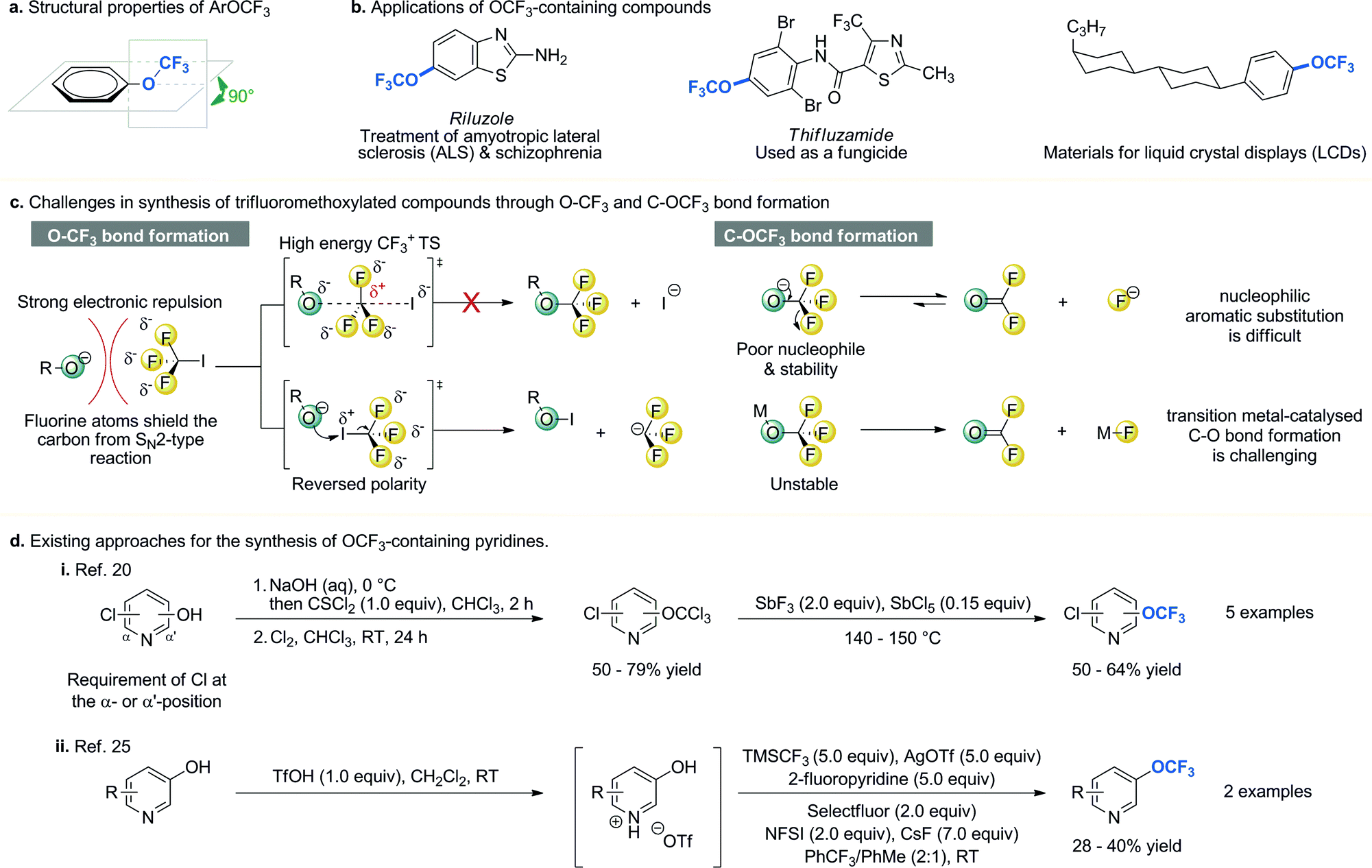 | ||
| Fig. 1 Properties, applications, synthetic challenges and methods of synthesis of OCF3-containing compounds. NFSI = N-fluorobenzenesulfonimide, TMSCF3 = trifluoromethyltrimethylsilane. | ||
Given the unique properties of the OCF3 group and the ubiquity of pyridines and pyrimidines in biologically active molecules and functional materials, trifluoromethoxylated pyridines and pyrimidines could serve as valuable synthetic building blocks for the discovery and development of new drugs, agrochemicals, and functional materials. However, synthesis of OCF3 containing heteroarenes through either O–CF3 or C–OCF3 bond formation remains a formidable challenge in organic synthesis (Fig. 1c).1–5,15 Unlike its analogous methoxy (OCH3) group, the OCF3 group cannot be formed via trifluoromethylation of hard nucleophiles such as phenoxides with CF3I through SN2 type mechanism.11,16,17 This is due to (i) strong electron repulsion between three fluorine atoms and an incoming nucleophile; (ii) formation of energetically disfavoured CF3 carbocation transition state structure (TS); and (iii) competing iodination of nucleophiles due to the reversed electron density. In addition, the thermal instability of transition metal–OCF3 complexes (they readily decompose to form fluorophosgene and metal fluoride)18 and the poor nucleophilicity of the OCF3 anion (a reactive electrophile is needed for the C–OCF3 bond formation)19 have hampered the development of the C–OCF3 bond formation through either transition metal-catalysed C–O bond formation or nucleophilic substitution. Strategies for the synthesis of trifluoromethoxylated heteroaromatic compounds are very rare.20–25 Leroux and co-workers reported a detailed examination of several different approaches and concluded that the presence of a chlorine atom at the α-, and/or α′- position of hydroxy-pyridines is critical (Fig. 1d).20 Without it, little or no desired product was isolated. This requirement greatly limited its application. Recently, Qing and co-workers reported a novel, direct synthesis of pyridyl trifluoromethyl ethers from unprotected hydroxypyridines.25 However, excess amounts of reagents and oxidants were required. In addition, only two examples with moderate yield were reported. Due to the lack of a general synthetic method for the synthesis of trifluoromethoxylated pyridines and pyrimidines, their full potential has not been fully exploited in pharmaceutical, agrochemical, and materials applications.
Herein, we report a scalable and operationally simple protocol for regioselective synthesis of trifluoromethoxylated functionalized pyridines and pyrimidines. Several unique features distinguish our strategy from the existing approaches: (i) many substrates with complex skeletons are trifluoromethoxylated at or below room temperature (17 out of the 30 examples); (ii) a wide range of functional groups and substitution patterns are tolerated; (iii) this transformation is amenable to gram-scale synthesis; (iv) halogen or amino group is used as synthetic handles for further elaborations, and (v) the operational simplicity of our protocol would render trifluoromethoxylation available to broader synthetic community. More importantly, this strategy allows access to a new class of synthetic building blocks to aid the discovery and development of new functional molecules.
Results and discussion
It is known that the N–O bond is relatively weak (bond dissociation energy = ∼57 kcal mol−1) due to the lone-pair electron repulsion between the nitrogen and the oxygen atoms. In addition, electron withdrawing O-substituent and/or electron donating N-substituent could promote heterolytic cleavage of the N–O bond to form nitrenium ion and oxy-anion.26 Recently, we took advantage of these properties and successfully synthesized trifluoromethoxylated aromatic compounds through O-trifluoromethylation of N-aryl-N-hydroxylamine derivatives to form N-OCF3 compounds followed by thermally induced OCF3-migration.27 However, to apply this strategy to the synthesis of trifluoromethoxylated heteroaromatic compounds such as pyridine and pyrimidine, two challenges had to be addressed.First of all, reaction conditions for the synthesis of N-acetyl/methoxycarbonyl-N-pyridinylhydroxylamine precursors are very limited,28–30 because the presence of heteroarenes complicates their synthesis. For instance, reduction of nitro-pyridines to the corresponding N-pyridinyl-N-hydroxylamines often accompanied by over reduction side-products (i.e. formation of aminopyridines). In addition, formation of undesired bis-protected side products and pyridinium salts during the protection of N-pyridinyl-N-hydroxylamines lowered the yields of precursors. After extensive optimization, we were able to obtain pure precursors in good to excellent yields. We have identified a robust catalytic hydrazine reduction protocol (using 5% rhodium on carbon as a catalyst) for converting nitropyridines and pyrimidines to N-heteroaryl-N-hydroxylamines. This method is general and high yielding; most of the reduction products can be used directly without further purification (only filtration through celite to remove Rh/C followed by the removal of the solvent is required). We have also observed that isolation of the intermediate hydroxylamines is not necessary; subsequent N-protection can be performed one-pot and the selectivity can be controlled by the rate of addition of acyl chloride or methylchloroformate at appropriate temperature (see ESI† for detail experimental procedures).
Another challenge was that the O-trifluoromethylation and OCF3-migration processes for the protected N-heteroaryl-N-hydroxylamine derivatives were inefficient under our previously developed reaction conditions. Exposure of methyl (5-bromo-6-methoxypyridin-3-yl)(hydroxyl)carbamate (1a) to 1-trifluoromethyl-1,2-benziodoxol-3(1H)-one (Togni reagent II, B)31,32 in the presence of cesium carbonate (Cs2CO3, 0.1 equiv.) in chloroform (CHCl3) at room temperature for 15 hours afforded the desired product 2a in only 29% yield (Table 1, entry 1).27 Examination of different trifluoromethylation reagents such as 3,3-dimethyl-1-(trifluoromethyl)-1,2-benziodoxole (Togni reagent I, A),31 5-(trifluoromethyl)dibenzothiophenium trifluoromethanesulfonate (Umemoto reagent, C),33 and [(oxido)phenyl(trifluoromethyl)-λ4-sulfanylidene] dimethylammonium tetrafluoroborate (Shibata–Johnson reagent, D)34 revealed that Togni reagent I was superior to other reagents and delivered the desired product 2a in 42% yield (entries 2–4). Higher yield was obtained in the absence of Cs2CO3 (entry 5). Screening of solvents showed that CH2Cl2 gave the best result, providing the desired product 2a in 87% NMR yield (entries 6–10).
|
|
||||
|---|---|---|---|---|
| Entry | Reagent | Base (0.1 equiv.) | Solvent | Yielda |
| a Yields were determined by 1H-NMR analysis using benzotrifluoride as the internal standard. | ||||
| 1 | B | Cs2CO3 | CHCl3 | 29% |
| 2 | A | Cs2CO3 | CHCl3 | 42% |
| 3 | C | Cs2CO3 | CHCl3 | 10% |
| 4 | D | Cs2CO3 | CHCl3 | 4% |
| 5 | A | — | CHCl3 | 72% |
| 6 | A | — | DMF | 24% |
| 7 | A | — | THF | 48% |
| 8 | A | — | CH3CN | 69% |
| 9 | A | — | CH3NO2 | 74% |
| 10 | A | — | CH2Cl2 | 87% |
|
||||

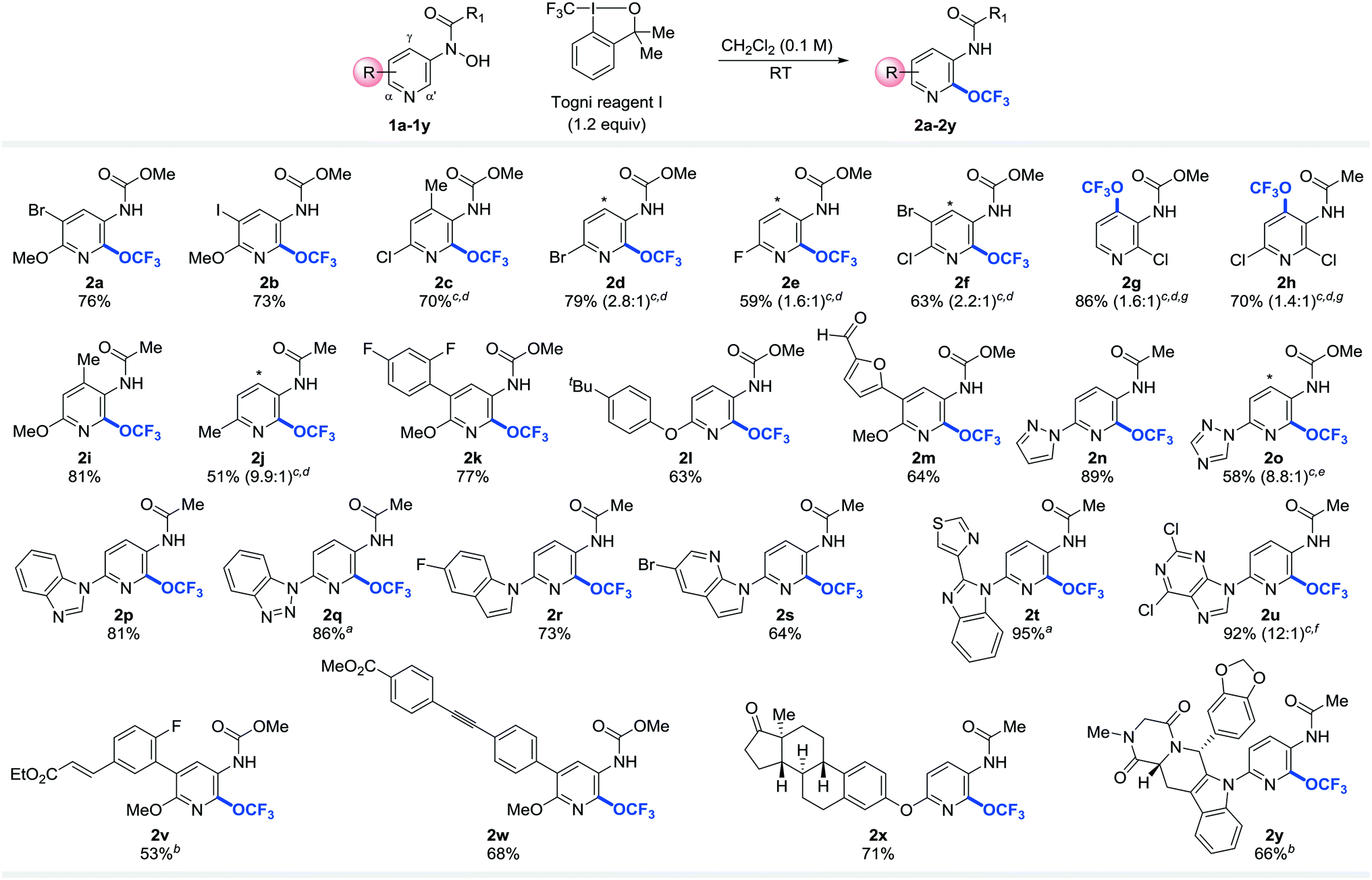
With the optimized reaction conditions in hand, we next directed our attention to exploring the scope and generality of the trifluoromethoxylation reaction. Gratifyingly, a wide range of functional groups and substitution patterns were tolerated (Table 2). Halogen functionalities remain intact after the reaction, providing useful synthetic handles for further functionalization (2a–h, 2k, 2r, 2s, 2u, 2v). Other functional groups such as alkyl- and aryl-ethers (2a, 2b, 2i, 2k–m, 2v–x), aldehydes (2m), ketone (2x), alkene (2v), alkyne (2w), amide (2y), esters (2v–w), and benzo[1,3]dioxole (2y) proved compatible under the reaction conditions. In addition, this methodology was successfully applied to pyridines bearing a wide array of heteroaryl substituents such as furan (2m), pyrazole (2n), 1,2,4-triazole (2o), benzimidazole (2p), benzotriazole (2q), indole (2r, 2y), 7-azaindole (2s), thiazole (2t), and 2,6-dichloropurine (2u). These products could be useful building blocks for drug and agrochemical discovery and development because it is estimated that over 70% of all pharmaceutical products bear heterocyclic moieties.35 More excitingly, our mild reaction conditions allow late-stage trifluoromethoxylation of complex organic molecules. For examples, estrone and Tadalafil (Cialis, sales in 2013: US$2.159 billion)36 conjugated pyridines (1x, 1y) were trifluoromethoxylated to afford desired products 2x and 2y in 71% and 66% yield, respectively. Remarkably, no epimerization was observed under our reaction conditions. These results further demonstrated the synthetic utility of our strategy.
| a Cited yields and isomeric ratios are of isolated material by column chromatography. RT then 50 °C in CH2Cl2. b 4 °C in CH2Cl2 (0.01 M). c Following the O-trifluoromethylation reaction in CH2Cl2 at RT, the reaction mixture was concentrated, the residue was dissolved in MeNO2, and the resulting mixture was heated. d 120 °C. e 80 °C. f 60 °C. g Atropisomeric ratio. |
|---|
|
Several features of the reaction are noteworthy. First of all, the reaction is sensitive to the electronic properties of substituents on pyridine. Substrates with an electron donating substituent para to the protected N-hydroxylamine readily undergo rearrangement to yield the desired products of trifluoromethoxylation at or below room temperature (2a–b, 2i, 2k–n, 2p, 2r–2s, 2v–y). In the absence of such substituents, higher reaction temperatures are required for the OCF3-migration step (2c–h, 2o, 2q, 2t–u). These observations are consistent with the formation of nitrenium ion through heterolytic cleavage of N–O bond (vide infra).26 Secondly, for the reactions that take place at or below room temperature, the OCF3 group is introduced exclusively to the α′-position.37–39 Since α- and α′-carbon of pyridines are metabolically labile sites, incorporation of an electron withdrawing OCF3 group to the α′-position could improve their metabolic stability.40,41 If the α′-position is blocked, product of γ-OCF3 pyridine is formed instead (2g and 2h). Interestingly, atropisomers are obtained in these cases. This is because the OCF3 group lies in the plane orthogonal to the pyridine ring (Fig. 1a), which prevents the free rotation of the adjacent amide or carbamate group (see ESI†).7,42–45 Finally, the regioselectivity erodes as the reaction temperature increases (2d–f, 2o, 2u).
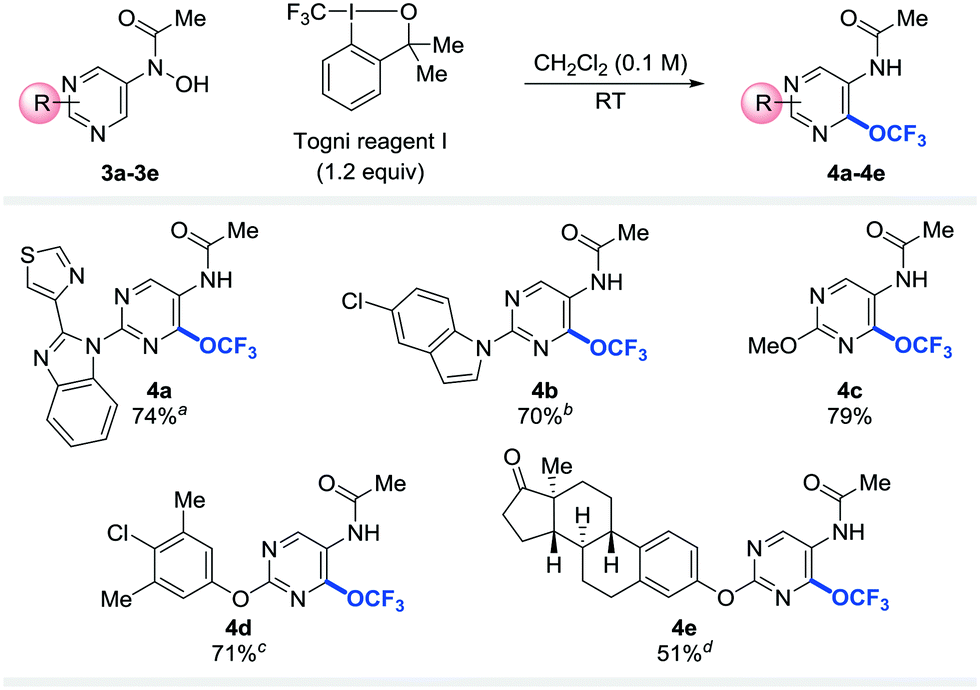
To probe the applicability of the trifluoromethoxylation reaction to other heteroarenes, pyrimidines substituted with benzimidazolyl (3a), indolyl (3b), methoxy (3c), phenoxy (3d), or estronyl (3e) groups were examined (Table 3). To our delight, these substrates were trifluoromethoxylated to afford the corresponding desired products (4a–4e) in good yields. Notably, none of the trifluoromethoxylated pyridines and pyrimidines reported here has ever been prepared before.
| a Cited yields are of isolated material by column chromatography. Following the O-trifluoromethylation reaction in CH2Cl2 at RT, the reaction mixture was concentrated, the residue was dissolved in MeNO2, and the resulting mixture was heated at 80 °C. b CH2Cl2 (0.01 M). c RT then 50 °C in CH2Cl2 (0.03 M). d RT then 50 °C in CH2Cl2. |
|---|
|
To ensure that our products can serve as useful building blocks for molecular screening, our protocol must be scalable and further functionalization of the trifluoromethoxylated products must be possible. To evaluate the reaction efficacy on preparative scale, a gram-scale reaction of 1a (1.39 g, 5.00 mmol) was performed (Scheme 1a) and the efficiency of the small-scale reaction was retained upon scale-up. Our trifluoromethoxylated products also proved to be versatile (Scheme 1b). For instance, 2a could be further elaborated through palladium-catalysed Suzuki and Sonogashira couplings to afford the desired products (6a, 8a) in good yields. In addition, deprotected amino-pyridine (2a′) could be efficiently incorporated into other molecules through amidation and palladium-catalysed Buchwald–Hartwig coupling (5a, 7a).
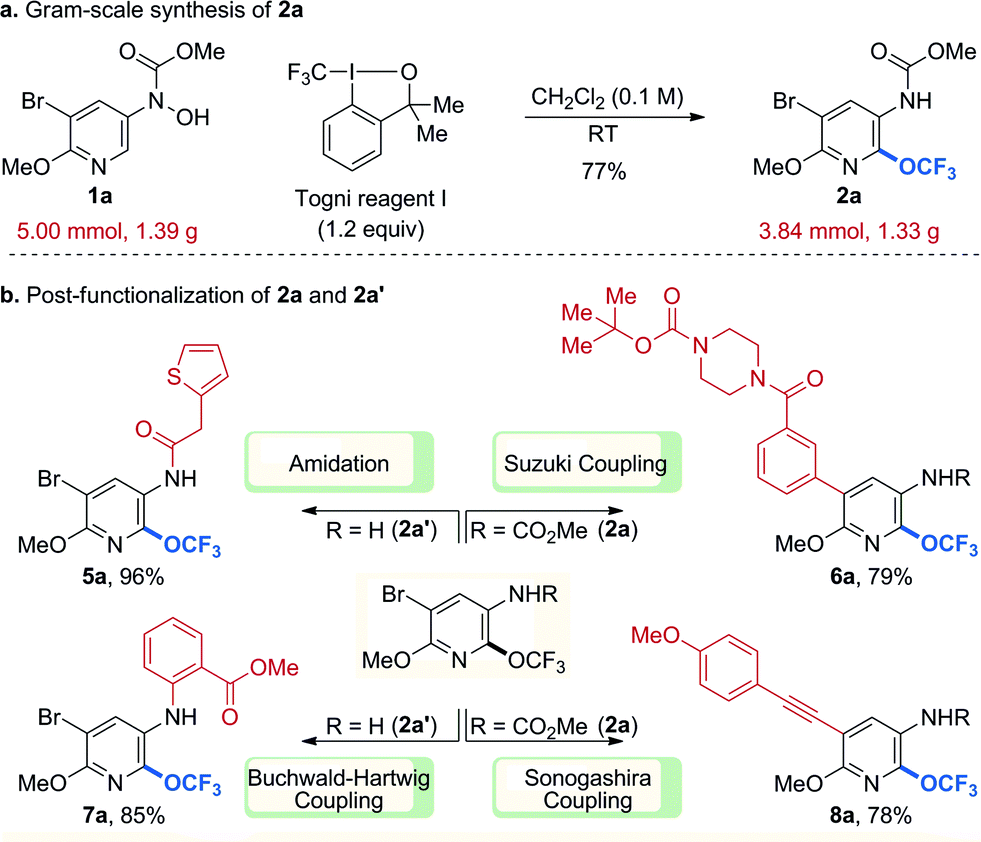 | ||
| Scheme 1 Gram-scale synthesis and post-functionalization. Cited yields are of isolated material by column chromatography. | ||
Although our strategy is operationally simple and scalable, has broad substrate scope, and tolerates a wide range of functional groups, this procedure, much like other methods, is not without limitations. First of all, substrates with the protected N-hydroxylamino-group at α-, γ-, or α′-position do not give the product of trifluoromethoxylation. Presumably, the formation of nitrenium ion is energetically disfavoured in these cases, because it involves placing the positive charge on the endocyclic nitrogen atom.46 In addition, preparation of N-heteroaryl-N-hydroxylamine precursors is required for this transformation. Furthermore, Togni reagent I is relatively expensive, and thus large-scale synthesis would be costly. Therefore, more improvements are needed for the development of a truly general and industrially practical trifluoromethoxylation reaction. Nevertheless, with the method accessing unprecedented and versatile synthetic building blocks in hand, the discovery and development of new pharmaceuticals, agrochemicals, and functional materials can be expected.
To gain some insight into the reaction mechanism, we performed reactions in the presence of radical trap butylated hydroxytoluene (BHT) (Scheme 2a). We chose to use substrate 1d because we could isolate the O-trifluoromethylated intermediate 1d′ and study each step (i.e. O-trifluoromethylation and OCF3-migration) separately. Addition of BHT (1 equiv.) to a reaction mixture of 1d and Togni reagent I had detrimental effect to the formation of O-trifluoromethylated N-hydroxylamine intermediate 1d′. This result strongly suggests the involvement of radical species in the reaction pathway, which is in agreement with literature precedents.47,48 On the other hand, BHT did not affect the reaction yield for the OCF3-migration process (step 2, Scheme 2a). These experiments argue against the presence of long-lived radical species in the OCF3-migration process and are consistent with our previous finding.27 Moreover, introduction of electron rich substituent para to the N-OCF3 group facilitates the OCF3-migration process. These observations support the formation of nitrenium ion and trifluoromethoxide.26,27 On the basis of these results, a plausible mechanism for the trifluoromethoxylation reaction is illustrated in Scheme 2b. Deprotonation of 1d forms N-hydroxyl anion 9, which undergoes single-electron transfer (SET) with Togni reagent I to generate N-hydroxyl radical 10, trifluoromethyl radical, and alkoxide 11.48 Reaction of N-hydroxyl radical and trifluoromethyl radical affords the O-trifluoromethylated hydroxylamine 1d′, which could be isolated and characterized. This intermediate will then undergo thermally induced heterolytic cleavage of the N–O bond to form a tight ion pair of nitrenium ion 12 and trifluoromethoxide. Rapid recombination of this ion pair gives 13, which upon tautomerization (i.e. migration of proton) yields the desired product 2d.
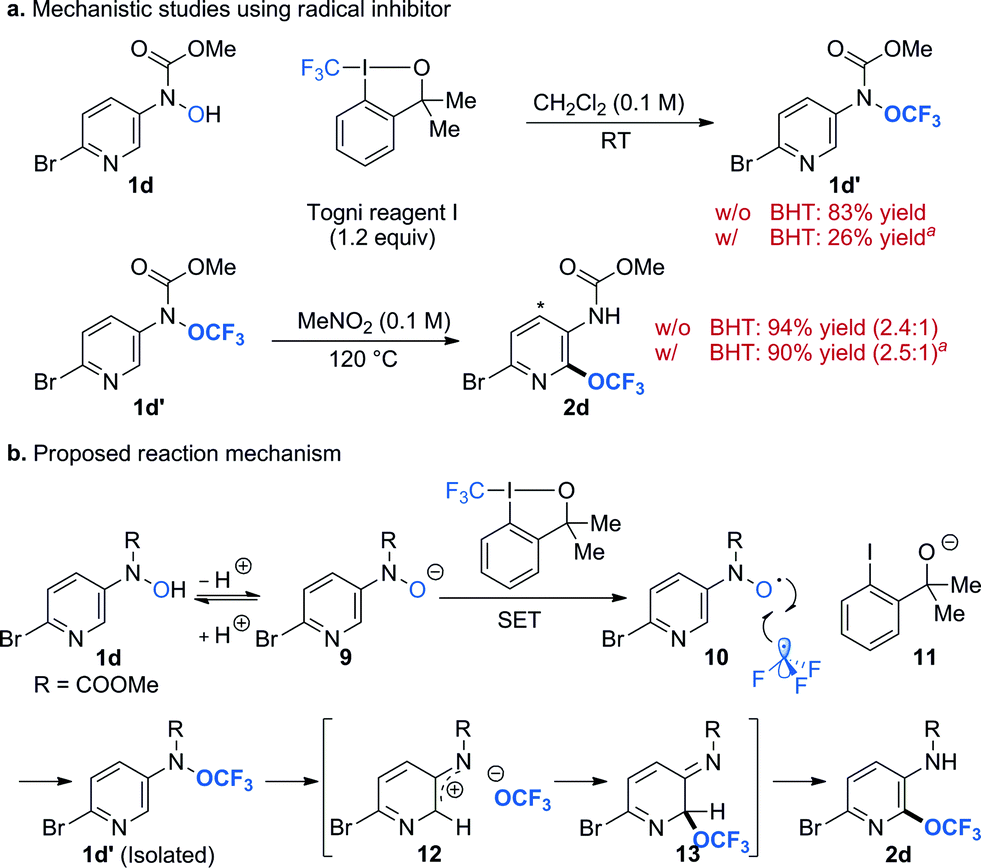 | ||
| Scheme 2 Mechanistic studies and proposed reaction mechanism. a1 equiv. of BHT was used. Yields were determined by 1H-NMR analysis using benzotrifluoride as the internal standard. BHT = butylated hydroxytoluene. w/ = with. w/o = without. | ||
Conclusions
In summary, we reported an operationally simple protocol for the regioselective trifluoromethoxylation of functionalized pyridines and pyrimidines. The strategy uses commercially available and bench stable Togni reagent I, features a broad substrate scope, tolerates a wide range of functional groups, and is amenable to gram scale synthesis. With this procedure, a variety of highly functionalized pyridines and pyrimidines could be trifluoromethoxylated under mild reaction conditions. Since heteroarenes are ubiquitous in biologically active natural products, pharmaceuticals, and agrochemicals, we expect that our work will provide valuable OCF3-containing heteroaromatic building blocks for the discovery and development of new drugs, agrochemicals, and functional materials.Acknowledgements
This paper is dedicated to Prof. Iwao Ojima on the occasion of his 70th birthday. We acknowledge generous start-up funds from Stony Brook University in support of this work. K.N.L. is grateful for the Chemistry Graduate Research Fellowship provided by the Department of Chemistry. We thank TOSOH F-Tech, Inc. for their gift of TMSCF3 for the preparation of Togni reagent I. Fuhua Zhao is acknowledged for his contribution to the preparation of 2-substituted nitropyridine precursors. We express our special thanks to the American Chemical Society Division of Organic Chemistry for providing summer undergraduate research fellowship (SURF) for Fuhua Zhao.Notes and references
- F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef CAS PubMed.
- P. Jeschke, E. Baston and F. R. Leroux, Mini-Rev. Med. Chem., 2007, 7, 1027–1034 CrossRef CAS PubMed.
- F. R. Leroux, B. Manteau, J. P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4 DOI:10.3762/bjoc.4.13.
- B. Manteau, S. Pazenok, J. P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef CAS.
- G. Landelle, A. Panossian and F. R. Leroux, Curr. Top. Med. Chem., 2014, 14, 941–951 CrossRef CAS PubMed.
- H. S. Booth and P. E. Burchfield, J. Am. Chem. Soc., 1935, 57, 2070–2070 CrossRef CAS.
- D. Federsel, A. Herrmann, D. Christen, S. Sander, H. Willner and H. Oberhammer, J. Mol. Struct., 2001, 567, 127–136 CrossRef.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- M. A. Mcclinton and D. A. Mcclinton, Tetrahedron, 1992, 48, 6555–6666 CrossRef CAS.
- C. Hansch and A. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York, 1979 Search PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry : Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- G. Siegemund, W. Schwertfeger, A. Feiring, B. Smart, F. Behr, H. Vogel and B. McKusick, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a11_349.
- P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4217–4235 CrossRef.
- M. Mamada, H. Shima, Y. Yoneda, T. Shimano, N. Yamada, K. Kakita, T. Machida, Y. Tanaka, S. Aotsuka, D. Kumaki and S. Tokito, Chem. Mater., 2015, 27, 141–147 CrossRef CAS.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214–231 CrossRef CAS PubMed.
- T. Umemoto, K. Adachi and S. Ishihara, J. Org. Chem., 2007, 72, 6905–6917 CrossRef CAS PubMed.
- C. P. Zhang and D. A. Vicic, Organometallics, 2012, 31, 7812–7815 CrossRef CAS.
- A. A. Kolomeitsev, M. Vorobyev and H. Gillandt, Tetrahedron Lett., 2008, 49, 449–454 CrossRef CAS.
- B. Manteau, P. Genix, L. Brelot, J. P. Vors, S. Pazenok, F. Giornal, C. Leuenberger and F. R. Leroux, Eur. J. Org. Chem., 2010, 2010, 6043–6066 CrossRef.
- A. Fuss and V. Koch, Synthesis, 1990, 1990, 604–608 CrossRef.
- K. Morimoto, K. Makino and G. Sakata, J. Fluorine Chem., 1992, 59, 417–422 CrossRef CAS.
- D. Guiadeen, S. Kothandaraman, L. H. Yang, S. G. Mills and M. MacCoss, Tetrahedron Lett., 2008, 49, 6368–6370 CrossRef CAS.
- T. M. Sokolenko, Y. A. Davydova and Y. L. Yagupolskii, J. Fluorine Chem., 2012, 136, 20–25 CrossRef CAS.
- J.-B. Liu, C. Chen, L. Chu, Z.-H. Chen, X.-H. Xu and F.-L. Qing, Angew. Chem., Int. Ed., 2015, 54, 11839–11842 CrossRef CAS PubMed.
- A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
- K. N. Hojczyk, P. Feng, C. Zhan and M.-Y. Ngai, Angew. Chem., Int. Ed., 2014, 53, 14559–14563 CrossRef CAS PubMed.
- M. Novak and T. M. Nguyen, J. Org. Chem., 2003, 68, 9875–9881 CrossRef CAS PubMed.
- S. Rajagopal, M. E. Brooks, T. M. Nguyen and M. Novak, Tetrahedron, 2003, 59, 8003–8010 CrossRef CAS.
- M. Novak, K. Toth, S. Rajagopal, M. Brooks, L. L. Hott and M. Moslener, J. Am. Chem. Soc., 2002, 124, 7972–7981 CrossRef CAS PubMed.
- P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579–2586 CrossRef CAS PubMed.
- V. Matousek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763–6768 CrossRef CAS PubMed.
- T. Umemoto, Chem. Rev., 1996, 96, 1757–1777 CrossRef CAS PubMed.
- S. Noritake, N. Shibata, S. Nakamura, T. Toru and M. Shiro, Eur. J. Org. Chem., 2008, 2008, 3465–3468 CrossRef.
- G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728–4750 RSC.
- F. Weber and G. Sedelmeier, Nachr. Chem., 2014, 62, 997–997 CrossRef.
- V. N. Novikov, A. F. Pozharskii and V. N. Doronkin, Khim. Geterotsikl. Soedin., 1976, 12, 244–252 Search PubMed.
- M. Hirota, H. Masuda, Y. Hamada and I. Takeuchi, Bull. Chem. Soc. Jpn., 1979, 52, 1498–1505 CrossRef CAS.
- C. K. Mcgill and A. Rappa, Adv. Heterocycl. Chem., 1988, 44, 1–79 CrossRef CAS.
- H. Ishida, S. Isami, T. Matsumura, H. Umehara, Y. Yamashita, J. Kajita, E. Fuse, H. Kiyoi, T. Naoe, S. Akinaga, Y. Shiotsu and H. Arai, Bioorg. Med. Chem. Lett., 2008, 18, 5472–5477 CrossRef CAS PubMed.
- D. J. St Jean and C. Fotsch, J. Med. Chem., 2012, 55, 6002–6020 CrossRef CAS PubMed.
- I. F. Shishkov, H. J. Geise, C. van Alsenoy, L. V. Khristenko, L. V. Vilkov, V. M. Senyavian, B. van der Veken, W. Herrebout, B. V. Lokshin and O. G. Garkusha, J. Mol. Struct., 2001, 567–568, 339–360 CrossRef CAS.
- E. G. Kapustin, V. M. Bzhezovsky and L. M. Yagupolskii, J. Fluorine Chem., 2002, 113, 227–237 CrossRef CAS.
- J. Klocker, A. Karpfen and P. Wolschann, Chem. Phys. Lett., 2003, 367, 566–575 CrossRef CAS.
- H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- M. B. Sullivan and C. J. Cramer, J. Am. Chem. Soc., 2000, 122, 5588–5596 CrossRef CAS.
- Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221–8224 CrossRef CAS PubMed.
- V. Matoušek, E. Pietrasiak, L. Sigrist, B. Czarniecki and A. Togni, Eur. J. Org. Chem., 2014, 2014, 3087–3092 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for new compounds. See DOI: 10.1039/c5sc02983j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |