DOI:
10.1039/C6RA04531F
(Paper)
RSC Adv., 2016,
6, 28947-28955
Safe and efficient membrane permeabilizing polymers based on PLLA for antibacterial applications†
Received
20th February 2016
, Accepted 14th March 2016
First published on 15th March 2016
Abstract
Poly(N,N-dimethylaminoethyl methacrylate)-block-poly(L-lactic acid)-block-poly(N,N-dimethylaminoethyl methacrylate) conjugated with poly(ethylene glycol) (D-PLLA-D@PEG) copolymers were synthesized. These non-aggregating polymers showed low MIC values against Gram-negative and Gram-positive, including methicillin-resistant Staphylococcus aureus (MRSA), bacteria. The polymers exhibited minimal toxicity and are promising antibacterial agents for biomedical applications.
1. Introduction
Antimicrobial agents are commonly used in consumer products to inhibit microbial growth for preventing infections and product decomposition. Most antimicrobial agents used in consumer products are small molecules. Among these, triclosan is one of the most extensively used antimicrobial agents. However, the use of triclosan has caused many concerns, and will be banned in consumer products in Europe and the USA within 2 years. Many strains of microbes have intrinsic resistance to existing antimicrobial agents in consumer products. Moreover, they are not effective against biofilms. The overuse of these small antimicrobial agents has also led to drug resistance in microbes. With the growing resistance to conventional antimicrobial agents, it is imperative to discover materials that are resistant to colonization by harmful microbes. Developing new agents to defeat bacterial and other infections is undoubtedly one of the most important scientific challenges of our age. Britain's Chief Medical Officer, Professor Dame Sally Davies recently said, “If we don't take action, then we may all be back in an almost 19th century environment where infections kill us as a result of routine operations. We won't be able to do a lot of our cancer treatments or organ transplants.” A recent US Center for Disease Control and Prevention report highlighted one particular example: “The bacteria, Carbapenem-Resistant Enterobacteriaceae (CRE), kill up to half of patients who get bloodstream infections from them… CRE are nightmare bacteria. Our strongest antibiotics don't work and patients are left with potentially untreatable infections”. Antibiotic-resistant infections in the healthcare field have evolved rapidly and have become a significant concern within the medical industry. This has sparked the increase in interest in development of antibacterial materials.1 Synthetic polymers and host defense peptides are widely studied as antibacterial materials.2,3 These materials possess cationic and amphiphilic structures to selectively target and disintegrate bacterial membranes via electrostatic interaction and insertion into the membrane lipid domains and reduces potential bacterial resistance.4,5 There are limitations associated with both peptides and synthetic polymers. Peptides suffer from scale up issues, enzyme susceptibility and unknown pharmacokinetics.6–10 Synthetic polymers face challenges in their biocompatibility and/or biodegradability. This matter can be addressed through the construction of biodegradable and biocompatible amphiphilic polymers based on polyesters such as poly(L-lactic acid) (PLLA), which are FDA approved. To date, studies on antibacterial poly(lactic acid) has been limited to polymer blends and insoluble polymers.11–13 Here, we describe the synthesis of PEG conjugation of PLLA based polyelectrolytes for use as antibacterial compounds. In this work, PLLA is the biodegradable segment which hydrolyses into lactic acid after use. Poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) functions as the quarternizable amine for effective antibacterial function. PEG was selected as the haemocompatible and biocompatible component to minimize toxicity to red blood cells and the body. In this work, we develop novel antimicrobial polymers which can be used as coatings for use in consumer products, hospital gowns, and surfaces in hospitals, gyms, aircrafts, trains, buses and bathrooms, to prevent multidrug-resistant infections. The unique properties of these materials include non-toxicity and broad spectrum activities against multidrug-resistant microbes and biofilms. We report the antibacterial properties of these polymers and explore the structure property relationship in the development of effective antibacterial polymers.
2. Experimental
2.1. Materials
2-Bromoethanol (95%), sodium azide (NaN3, >99.5%), 2-bromopropionyl bromide (97%), poly(ethylene glycol) methyl ether (MPEG, Mn = 550), propargyl bromide solution (80 wt% in toluene), sodium hydride (NaH, 60% dispersion in mineral oil), α-bromoisobutyryl bromide (98%), triethylamine (>99%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), copper(I) bromide (CuBr, 99%), stannous octoate [Sn(Oct)2] (95%), anhydrous 1,4-dioxane (99.8%), anhydrous toluene (99.8%), and anhydrous N,N-dimethylformamide (99.8%) were obtained from Sigma-Aldrich. 2-(Dimethylamino)ethyl methacrylate (DMAEMA) stabilized with hydroquinone monomethyl ether was obtained from Merck and used as received. Ethylene glycol (99.8%, Sigma-Aldrich) was distilled over CaH2 before use. L-Lactide (L-LA) (Purac Biochem, The Netherlands) were used without further purification. Alkyne-ended MPEG was prepared according to the previous procedures.14
2.2. Synthesis of PLLA-diBr macroinitiators
PLLA-diBr macroinitiators were synthesized using ring opening polymerization (ROP), and followed by terminal groups modification. The process was based on the previously described method with some modification (Scheme S1†).15 Typically, L-LA monomers were weighed into a dry and nitrogen purged flask together with ethylene glycol as initiator. The molar ratio of initiator and monomer was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 for both reactions. The polymerization was performed in concentrated solution (ca. 3.0 M in anhydrous toluene) at 130 °C for 24 h in the presence of Sn(Oct)2 as catalyst. Purified PLLA-diol was obtained by precipitation of the reaction mixture into excess cold methanol twice, followed by overnight vacuum drying at 80 °C. Subsequently, PLLA-diol were modified into ATRP macroinitiator by esterification of the hydroxyl end groups with α-bromoisobutyryl bromide in anhydrous THF. A 20 times excess of α-bromoisobutyryl bromide with respect to –OH end groups was added and triethylamine was used to trap hydrobromic acid generated during the reaction. Insoluble ammonium salt produced was first removed by centrifugation and the reaction mixture was further purified by passing through a short Al2O3 column using THF as eluent. The dilute solution was concentrated and precipitated into excess cold methanol twice. PLLA-diBr macroinitiators were obtained after drying under vacuum at 50 °C overnight.
:25 for both reactions. The polymerization was performed in concentrated solution (ca. 3.0 M in anhydrous toluene) at 130 °C for 24 h in the presence of Sn(Oct)2 as catalyst. Purified PLLA-diol was obtained by precipitation of the reaction mixture into excess cold methanol twice, followed by overnight vacuum drying at 80 °C. Subsequently, PLLA-diol were modified into ATRP macroinitiator by esterification of the hydroxyl end groups with α-bromoisobutyryl bromide in anhydrous THF. A 20 times excess of α-bromoisobutyryl bromide with respect to –OH end groups was added and triethylamine was used to trap hydrobromic acid generated during the reaction. Insoluble ammonium salt produced was first removed by centrifugation and the reaction mixture was further purified by passing through a short Al2O3 column using THF as eluent. The dilute solution was concentrated and precipitated into excess cold methanol twice. PLLA-diBr macroinitiators were obtained after drying under vacuum at 50 °C overnight.
2.3. Synthesis of PDMAEMA-b-PLLA-b-PDMAEMA (D-PLLA-D) triblock copolymers
PDMAEMA-b-PLLA-b-PDMAEMA triblock copolymers were prepared by controlled ATRP. Molar feed ratio of [PLLA-diBr]:[DMAEMA]:[CuBr]:[HMTETA] = 1:1000:1:2 was applied for all polymer synthesis. As a typical example, PLLA-diBr was first introduced into a nitrogen filled round bottom flask (RBF) followed by successive addition of 1,4-dioxane and DMAEMA monomer through syringe injection. Afterwards, the RBF was purged and refilled with nitrogen using vacuum-nitrogen-cycling system three times. HMTETA and CuBr were added quickly under nitrogen atmosphere. Polymerization was allowed to proceed under continuous stirring at 60 °C for a desired reaction time. The molecular weight was monitored by gel permeation chromatography (GPC) analysis. After polymerization, the reaction was stopped by diluting the reaction mixture with THF and exposing it to ambient atmosphere for 1 h. Catalyst complex was removed by passing the reaction mixture through a short neutral Al2O3 column. After concentrating the filtrates, the solutions were precipitated into excess ether and the final product PDMAEMA-b-PLLA-b-PDMAEMA (D-PLLA-D) was obtained through centrifugation.
2.4. Synthesis of 2-azidoethyl-2-bromopropanoate (AEBP)
AEBP was prepared from the synthesis of 2-azidoethanol, followed by esterification with 2-bromopropionyl bromide. First, 2-azidoethanol was prepared according to a reported procedure.16 In a typical reaction, 2-bromoethanol (8.8 g, 0.07 mol) and sodium azide (8.7 g, 0.14 mol) were placed into the reaction flask together with 100 mL of water. The mixture was stirred at 50 °C for 24 h and then cooled to room temperature. The solution was extracted with 100 mL ether and repeated for three cycles, followed by drying with magnesium sulphate overnight and filtered. Purified 2-azidoethanol was obtained as a colorless liquid after vacuum dry. For the synthesis of AEBP, 2-azidoethanol (6.0 g, 0.07 mol) and anhydrous Et3N (10.6 mL, 0.08 mol) were dissolved in 50 mL anhydrous THF in a 150 mL RBF. A 1.1 times excess of 2-bromopropionyl bromide (7.9 mL, 0.08 mol) with respect to the hydroxyl groups was added dropwise into the flask at 4 °C over a period of 1 h. After the addition was completed, the reaction mixture was continually stirred for another 24 h at room temperature. The undissolved solid was removed by centrifuge and the concentrated solution was further purified by a silica gel column chromatograph using THF/hexane (1:4 v/v) as an eluent. The solvent was removed by vacuum drying and purified AEBP was obtained as a colourless oil.17
2.5. Synthesis of alkyne-terminated PEG
Alkyne-terminated PEG was synthesized according to previous method.14 Typically, NaH (60% w/w in mineral oil, 0.77 g, 32.01 mmol, 1.1 equiv.) was added into 80 mL of anhydrous THF solution dissolved with 16 g of MPEG (29.1 mmol). Traces of water in PEG were removed by azeotropic distillation with toluene. The reaction was kept at 0 °C for 15 min with frequent venting. Then, propargyl bromide (80% in toluene, 4.74 mL, 32.01 mmol, 1.1 equiv.) was added dropwise through the dropping funnel, after which the mixture was stirred at 25 °C for 24 h. For purification, the solution was concentrated and extracted with 400 mL H2O/CHCl3 (1/3, v/v) and repeated for three cycles, followed by drying with magnesium sulphate and filtered. The concentrated solution was precipitated in excess hexane and vacuum dried to obtain the final purified product which is colourless liquid (yield, 87.5%).
2.6. Synthesis of PEG conjugated D-PLLA-D (D-PLLA-D@PEG) copolymers
D-PLLA-D@PEG was synthesized through one-pot approach using AEBP as coupling agent. In a typical procedure, D-PLLA-D (1.0 g) prepared as described above was dissolved in DMF (10 mL) and AEBP (0.072 mL) was then introduced into the mixture. The [AEBP]/[DMAEMA] molar ratio was fixed at 1:10, targeting at a 10% quaternization degree of D-PLLA-D@N3. After stirring at 50 °C for 48 h, the mixture was cooled to room temperature. Next, 0.55 g of propargyl-terminated PEG and 0.09 g of PMDETA were added and the flask was purged with N2 for 30 min. CuBr (0.07 g) were added quickly under nitrogen atmosphere. After stirring for 24 h at ambient temperature, the solution was dialyzed against deionized water using a dialysis membrane (spectrum dialysis membrane, MWCO 1000) for 48 h to remove the excess PEG, and the final product (D-PLLA-D@PEG) was subsequently collected through lyophilization.
2.7. Molecular characterization
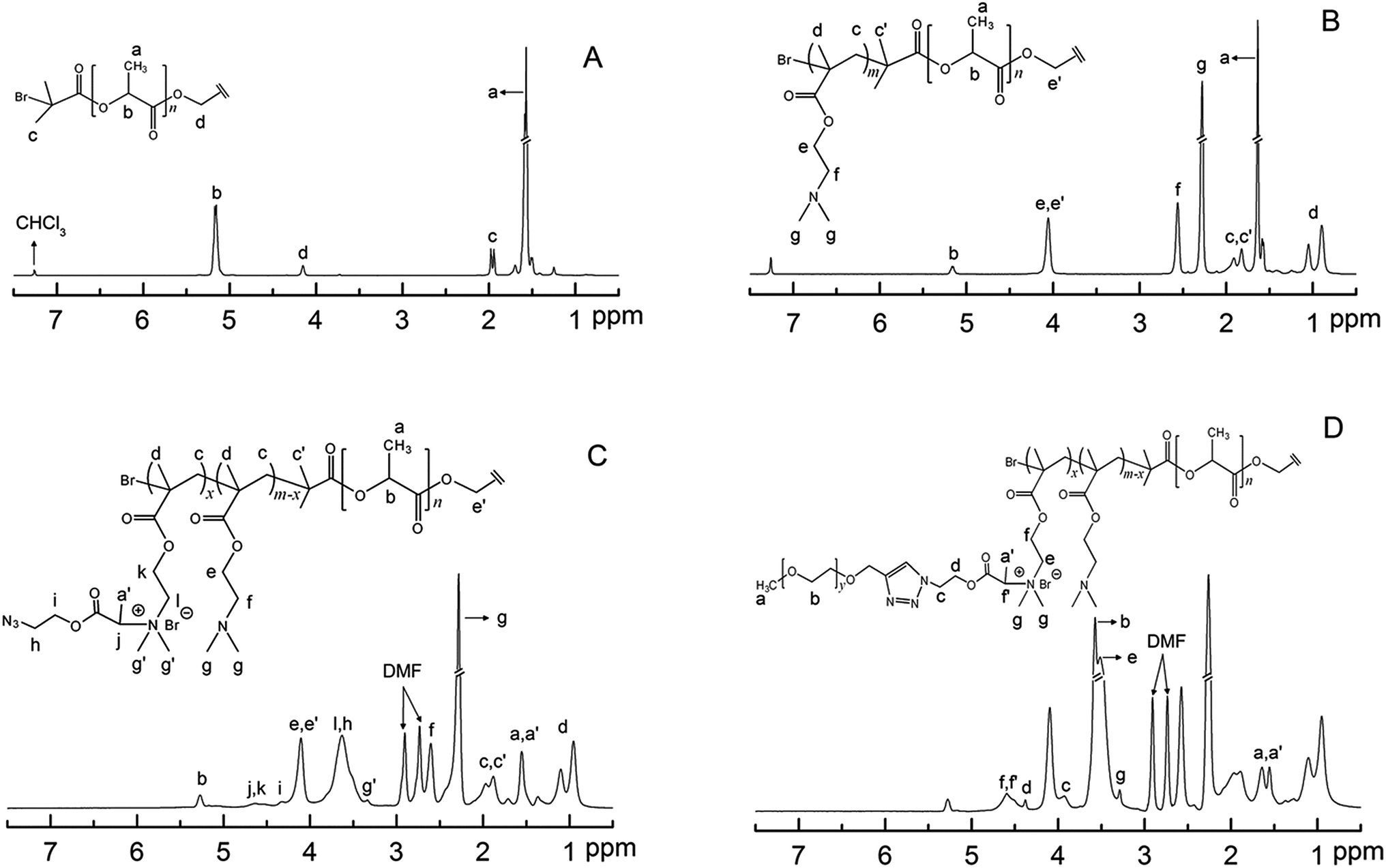
Nuclear magnetic resonance (1H-NMR). 1H-NMR spectra were recorded on a Bruker AV-400 NMR spectrometer at room temperature. Chemical peaks are reported in ppm with reference to solvent peaks (DMF: δ 8.03, 2.92 and 2.75 ppm; CHCl3: δ 7.3 ppm; and H2O: δ 4.8 ppm). Chemical compositions of the copolymers were evaluated from the proton integral regions as assigned in Fig. 1.
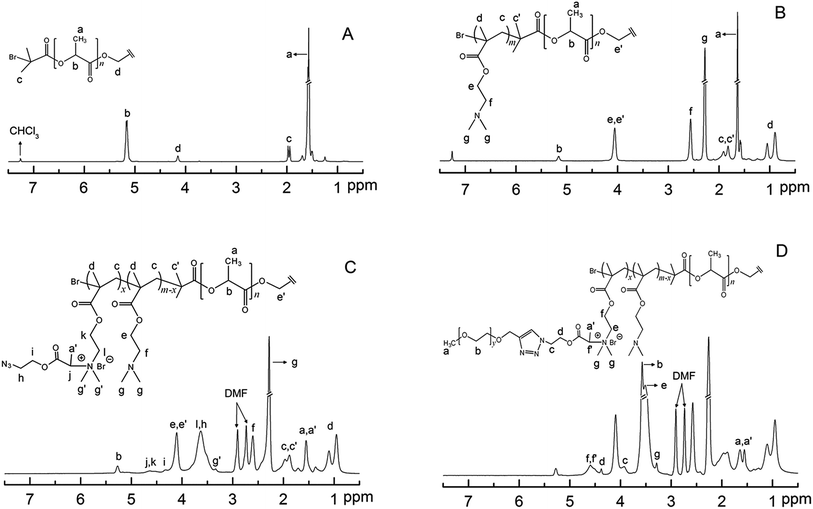 |
| | Fig. 1 1H NMR spectra of (A) PLLA-diBr and (B) D-PLLA-D in CDCl3, (C) D-PLLA-D@Q and (D) D-PLLA-D@QPEG-2 in d-DMF. | |
Gel permeation chromatography (GPC). The molecular weight and polydispersity of the as-synthesized copolymers were determined by GPC (Shimadzu SCL-10A and LC-8A system) equipped with a Shimadzu RID-10A refractive index detector. DMF (0.1 M LiBr) was used as the eluent at a flow rate of 1.0 mL min−1 at 40 °C. Monodispersed poly(methylmethacrylate) (PMMA) standards were used to obtain a calibration curve. The PEG conjugation percentage was calculated from the molecular weight difference before and after the conjugated reaction.
2.8. Cell viability assay
The cytotoxicity of the polymers was evaluated using the MTT assay in HEK 293T and Hela cell lines. They were cultured in DMEM, supplemented with 10% FBS, 100 units per mL of penicillin/streptomycin at 37 °C under 5% CO2, and 95% relative humidity atmosphere. The cells were seeded in a 96-well plate at a density of 1 × 104 cells per well and incubated in 100 μL of DMEM per well for 24 h. The culture media were replaced with fresh culture media containing serial dilution of polymers (15.6 to 500 μg mL−1), and the cells were incubated for 24 h. Then, 10 μL of sterile-filtered MTT stock solution in PBS (5 mg mL−1) was added to each well. After 4 h, the unreacted dye was removed by aspiration and the produced formazan crystals were dissolved in 100 μL of DMSO per well. The absorbance was measured using a microplate reader (Infinite M200, Tecan) at wavelength of 570 nm. The cell viability (%) of polymers was the absorbance of polymer treated cells divided by the absorbance of control cells.
2.9. Determination of minimum inhibitory concentration (MIC)
Stock solutions were prepared by dissolving the polymers in deionized water and subsequently diluted to 256 μg mL−1 in Mueller Hinton broth (MHB). 100 μL of the polymer solution was added to two wells of the 96 well plates for duplicates and another 200 μL was added to one well for zero reference. Serial dilution was performed to obtain the following concentrations: 256 μg mL−1, 128 μg mL−1, 64 μg mL−1, 32 μg mL−1, 16 μg mL−1, 8 μg mL−1, 4 μg mL−1 and 2 μg mL−1. The inoculum suspension was prepared by adding a few colonies extracted from the Tryptic Soy Agar (TSA) plate into the MHB. The concentration of the inoculum suspension was adjusted according to the 0.5 McFarland standard and the final concentration of each well was ∼105 colony forming units (CFU) mL−1. Upon the addition of 100 μL of the inoculum suspension into the polymers solutions, the plates were incubated at 37 °C for 24 hours. Absorbance was then read at 600 nm. The positive and the negative controls used in the experiment was inoculum with MHB and MHB only. Vancomycin solution was also used as a control drug.
2.10. Hemolysis experiments
Hemolysis was performed using fresh rabbit red blood cells which were isolated from the whole blood of the New Zealand white rabbits. The retrieval process follows the standards of the Association for Research in Vision and Ophthalmology and approval was obtained from the IACUC of SingHealth. The whole blood was centrifuged at 3000 rpm at 4 °C for 10 min. The supernatant was discarded while the red blood cells were subjected to two times of washing with 2 mL sterile PBS. The purified red blood cells were then diluted with 10 mM sterile PBS (pH 7) to make 8% (v/v) red blood cell stock solution. Polymer solutions were prepared and mixed with the red blood cells solutions to obtain the desired concentrations with 4% (v/v) red blood cells. Negative and positive controls used in the experiment were PBS with red blood cells and Triton X with red blood cells respectively. Following the addition of the red blood cells to the polymer solutions, the solutions were incubated at 37 °C for 1 h. After the incubation, the solutions were centrifuged at 3000 rpm for 3 min and 100 μL of the supernatant was transferred to a 96 well plate. The absorbance of the supernatants were measured using TECAN infinite 200 microplate reader at 576 nm and the values were subsequently input into the following equation to calculate the % hemolysis.
| % hemolysis = ([Abs] − [Neg])/([Pos] − [Neg]) × 100% |
where [Abs] is the absorbance reading of the supernatant, [Neg] is the absorbance reading of the negative control and [Pos] is the absorbance reading of the positive control.
2.11. Cytoplasmic membrane depolarization assay
The effect of the polymers on the membrane potential of Pseudomonas aeruginosa (ATCC 9027) was probed by membrane sensitive DiSC35 fluorescent assay. Briefly, Pseudomonas aeruginosa (ATCC 9027) was harvested at an early exponential growth phase and washed with buffer solution (5 mM HEPES at pH 7) and resuspended in the same buffer until an optical density of 0.09 at 620 nm [OD620] was obtained. The cell suspension was incubated with 0.4 μM DiSC35 (Invitrogen) and 0.1 M potassium chloride (KCl) solution at 37 °C until DiSC35 uptake was maximal (when the reduction of fluorescence intensity was stable due to self-quenching of DiSC35 in the untreated bacteria). The desired concentration of polymer was added into a stirred cuvette. The fluorescence reading was monitored for 500 s with a Photon Technology International Model 814 fluorescence spectrophotometer, at an excitation wavelength of 660 nm and an emission wavelength of 675 nm. DMF alone had no effect on depolarization. Experiments were repeated at least three times and were reproducible.
2.12. Nitrocefin assay
Nitrocefin was used as the probe to characterize the outer membrane permeabilization. 10 mg mL−1 stock of nitrocefin was prepared with DMSO as the solvent. This stock solution was then diluted to 2 mg mL−1 using sterile PBS. A stock solution of bacteria and nitrocefin was prepared in PBS. Colonies of bacteria were added to a 100 mM PBS solution to achieve an optical density of 0.4. Nitrocefin was then added to the prepared solution in the ratio of 1 μL of nitrocefin to 200 μL of solution. 200 μL of the stock solution was added to each well in a 96 well plate. Reading was taken for 5 min which accounts for the baseline. Stock solutions of the various polymers were then added to attain final concentrations of: 1 μg mL−1, 2 μg mL−1, 4 μg mL−1, 8 μg mL−1, 16 μg mL−1, 32 μg mL−1, 64 μg mL−1, 128 μg mL−1, 256 μg mL−1, 512 μg mL−1 and 1024 μg mL−1. Absorbance of the wells was recorded for 40 min using Perkin-Elmer Multimode plate reader at an excitation wavelength of 500 nm while a reading was collected every minute.
The typical method employed in determining the PC50 values of the polymers with regards to their potential in outer membrane permeabilization involved determining the difference in absorbance between the baseline and the final plateau of the curve. The average of the first five values read before polymer addition was subtracted from the average of the final five values after the addition of the polymer. The same analysis was also run on positive control PMB, and the computed results from the polymers were taken as percentage values of the obtained value from PMB. The percentage values of each concentration per polymer were determined, and plotted against the polymer concentration. This allowed for the generation of a curve that could be fitted to the one phase association model. Through the equation of the fitted model, interpolation could be conducted to determine the PC50 values of the polymers.
2.13. Antibacterial activity in chemically defined media
Bacteria were diluted to ∼5 × 105 CFU mL−1 in M9 minimal media (as per ATCC medium: 2511 M9 minimal broth containing inorganic salts, glucose and thiamine, pH = 7.4) for E. coli and minimum essential medium (MEM) for S. aureus. 50 μL of the polymers were added to a 96 well plate containing 150 μL bacterial solutions. The plate was then incubated at 37C for a period of 18–24 h. 20 μL of bacterial suspension was spot-plated on agar plates. The viable colonies (<100) were counted after 48 h incubation at 37 °C.
3. Results and discussion
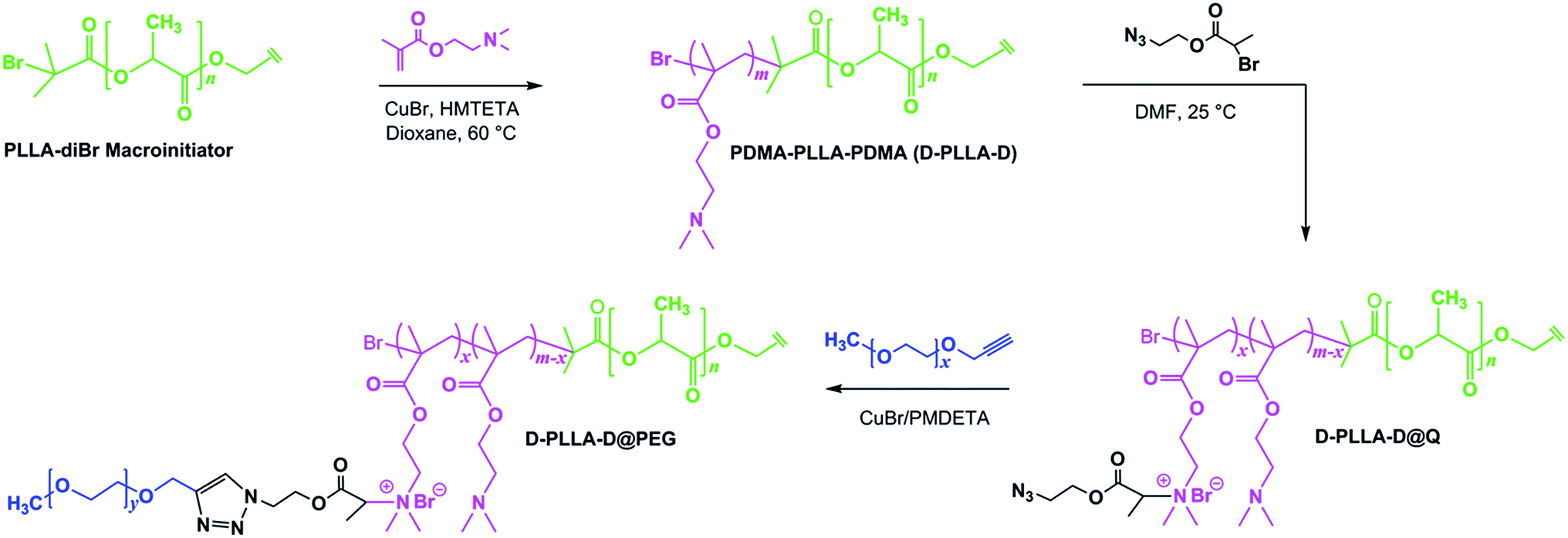
The structure of D-PLLA-D@PEG copolymers is shown (Scheme 1). The complete synthetic procedure is described in the ESI† and was previously reported by us.18 First, the starting PLLA-diBr macroinitiator for ATRP was prepared from the reaction of PLLA-diol with 2-bromoisobutyryl bromide according to our previous report.15 The Mn (NMR) of the obtained starting macroinitiator was 3.3 kDa (Fig. 1A). The degree of Br substitution in PLLA-diBr was 95%. D-PLLA-D triblock copolymers were synthesized from PLLA-diBr macroinitiators (Scheme 1). Finally, D-PLLA-D@PEG copolymers (Fig. 1D) were synthesized using a sequential reactions consisting of quaternization of PDMAEMA chains in D-PLLA-D triblock copolymers, and subsequent azide–alkyne cycloadditions with alkyne-end PEG through a one-pot approach (Scheme 1). To facilitate these reactions, a bifunctional AEBP linker with bromopropionyl and azide group at opposite chain termini was designed. AEBP was prepared according to the reaction sequence shown in Scheme S1.† The quaternized D-PLLA-D@Q copolymers with two different chemical compositions (D-PLLA-D@Q 1:12.3%; D-PLLA-D@Q 2:26.2%) were synthesized by varying the AEBP feed. These two copolymers were used as intermediate products for subsequent PEG conjugation. PEG with molecular weight of 550 Da was conjugated via Cu(I)-catalyzed azide–alkyne click reaction between alkyne-end PEG and azide groups in the freshly prepared D-PLLA-D@N3 copolymer solutions (Scheme S1†).
 |
| | Scheme 1 Synthesis of PEG conjugated PDMA-b-PLLA-b-PDMA@PEG (D-PLLA-D@PEG) copolymers by the combination of ATRP. | |
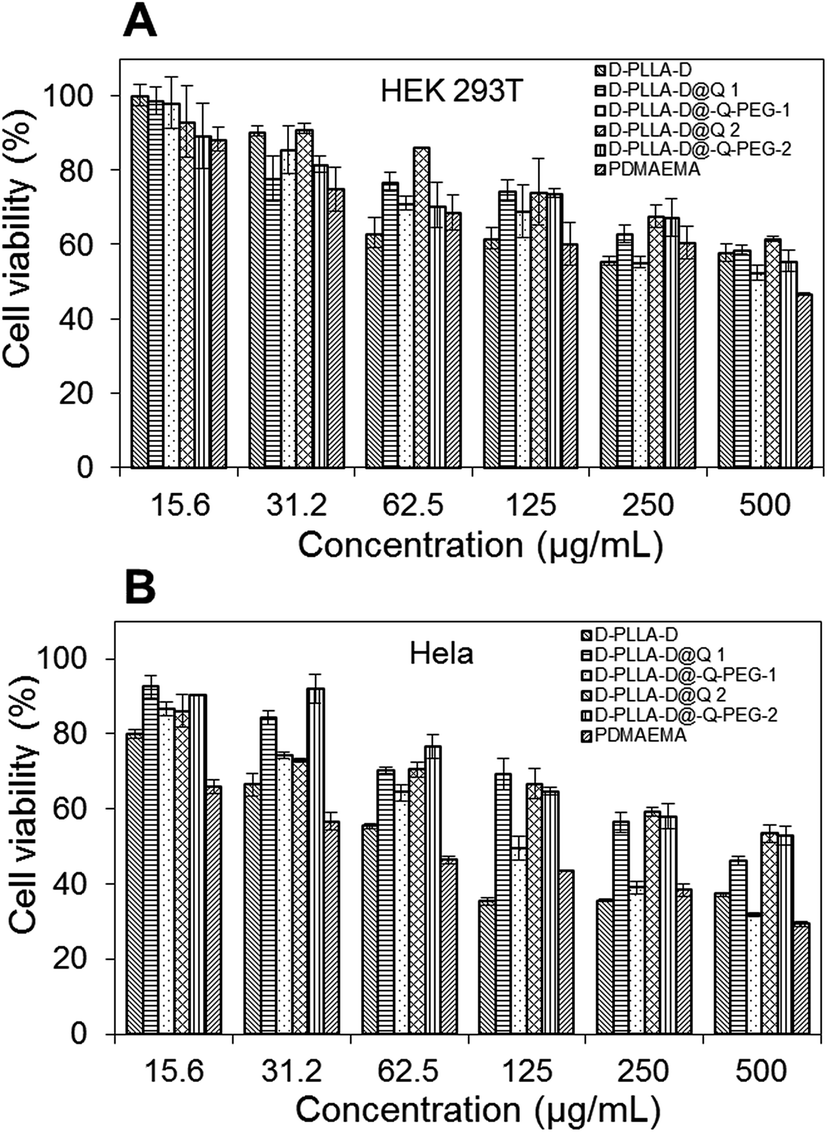
The terminal hydroxyl group of MPEG was esterified to introduce alkyne (Scheme S1(B)†). Alkyne-end PEG was then conjugated with D-PLLA-D@N3 copolymers to give D-PLLA-D@Q-PEG-1 and D-PLLA-D@Q-PEG-2. The relative molecular weight and polydispersity of the as-synthesized copolymers are presented in Table 1. The in vitro cytotoxicity of these copolymers in HEK 293T and Hela cells was studied in comparison with PDMAEMA polymers. Based on the MTT results in Fig. 2, the IC50 of these polymers were estimated. In HEK 293T cells, the IC50 for D-PLLA-D, D-PLLA-D@Q 1, D-PLLA-D@Q-PEG-1, D-PLLA-D@Q 2, and D-PLLA-D@Q-PEG-2 copolymers were 452 μg mL−1, 473 μg mL−1, >500 μg mL−1, >500 μg mL−1, and >500 μg mL−1 respectively. Their toxicity in Hela cells was relatively higher, with IC50 of 63 μg mL−1, 100 μg mL−1, 141 μg mL−1, 406 μg mL−1, 536 μg mL−1, and >500 μg mL−1, respectively. Generally, the cytotoxicity of these copolymers increased as the DMAEMA block length increased in all the cell lines.19 The toxicity decreased in both cell lines following this trend D-PLLA-D > D-PLLA-D@Q 1 > D-PLLA-D@Q-PEG-1 > D-PLLA-D@Q 2 > D-PLLA-D@Q-PEG-2. Additionally, the toxicity of these copolymers was lower than polymer PDMAEMA with highest DMAEMA content (IC50 of 414 μg mL−1 in HEK 293T and IC50 of 63.5 μg mL−1 in Hela cells).
Table 1 Molecular characteristics of D-PLLA-D@Q-PEG copolymers and their prepolymers
| Samplesa |
Block length (kDa) |
Qd (%) |
PEG conjugatione (%) |
[Mn]f/kDa |
| PLAb |
PDMAEMAc |
| PEG conjugated PDMAEMA-b-PLLA-b-PDMAEMA triblock copolymers are denoted as D-PLLA-D@PEG, where D represents PDMAEMA. Estimated from 1H NMR spectroscopy based on intensity ratio of PLA methine proton (δ 5.16 ppm) and the methylene proton (δ 4.15 ppm) in the ROP initiator of ethylene glycol. Calculated based on intensity ratio of PLA methine proton (δ 5.16 ppm) and PDMAEMA methylene proton (δ 2.56 ppm). Quaternizatino ratio as obtained from the residual intensities of –N–CH2– peak at 2.56 ppm in PDMAEMA. Evaluated from GPC, by calculating the molecular weight difference before and after PEG conjugation. Calculated based on GPC results. |
| PLLA-diBr |
3.3 |
— |
— |
— |
3.0 |
| D-PLLA-D |
3.3 |
8.5 |
— |
— |
11.4 |
| D-PLLA-D@Q 1 |
3.3 |
7.4 |
12.3 |
— |
13.1 |
| D-PLLA-D@Q 2 |
3.3 |
6.3 |
26.2 |
— |
14.6 |
| D-PLLA-D@Q-PEG-1 |
3.3 |
7.4 |
12.3 |
11.3 |
16.2 |
| D-PLLA-D@Q-PEG-2 |
3.3 |
6.3 |
26.2 |
22.1 |
20.9 |
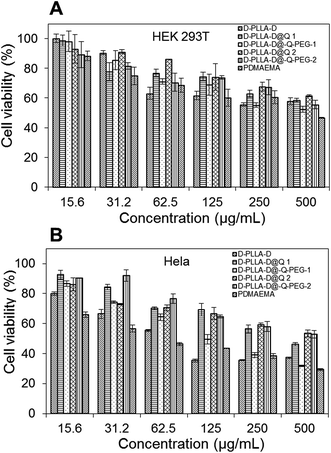 |
| | Fig. 2 Toxicity of polymers against HEK 293T and Hela cells. | |
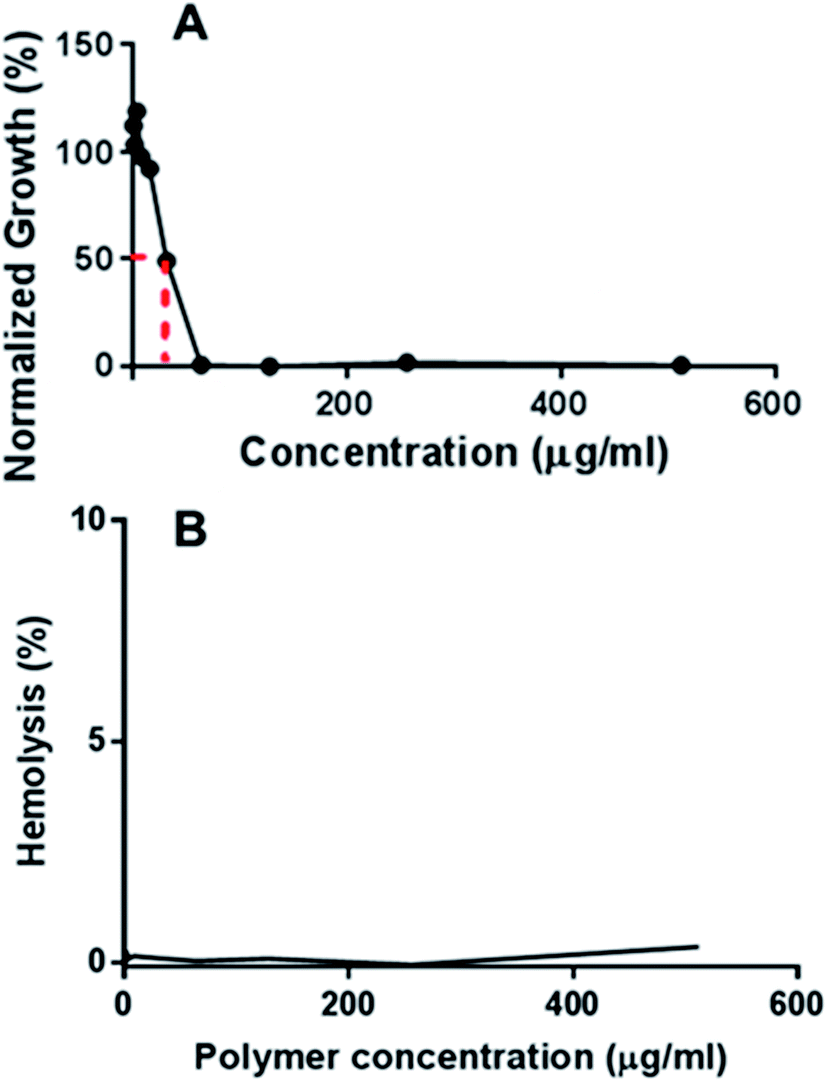
MRSA, which is the leading cause of nosocomial infection globally, has acquired resistance against various antibiotic classes, such as β-lactams, fluoroquinolones, tetracyclines, macrolides, lincosamides, aminoglycosides as well as the newest drugs licensed to treat MRSA infections such as linezolid and daptomycin.20 MRSA infections have also been linked with greater hospital death rates than even methicillin-susceptible S. aureus (MSSA) infections.21 The in vitro antimicrobial activities of the polymers were screened against various strains of MRSA. According to Table 2, all polymers showed good activities against MRSA, with the minimum inhibitory concentration (MIC) values ranging from 14 to 520 mg L−1. Fig. 3A shows a typical plot of the growth of bacteria as a function of polymer concentration from which MIC50 was determined. The other plots for the other polymers are shown in Fig. S1–S5.† In this example, the growth of MRSA 42412 in the presence of different concentrations of D-PLLA-D@Q-PEG-2 is shown. The polymers showed relatively high MIC values against MRSA 57964. D-PLLA-D performed the best against MRSA in general, however, it has to be noted that this polymer was also the most cytotoxic amongst the polymers surveyed. From the table, the conjugation of PEG does not significantly decrease the antibacterial activity of the polymer while significantly enhancing the compatibility of the polymer against mammalian cell lines. We have also conducted a screening of the antimicrobial effect of the polymers against a panel of Gram-positive and Gram-negative bacteria as well as yeast pathogens (Tables S1–S4†). The MIC values were between 16 and 64 μg mL−1, showing its potential activity against a wide spectrum of bacteria and pathogens. Some of the origins of the bacteria and pathogens are clinical in nature showing that these polymers could reasonably be extended for biomedical applications. The MIC values reported in these tables are for 100% growth inhibition.
Table 2 Minimum inhibitory concentrations (MICs) of antibacterial polymers against methicillin-resistant Staphylococcus aureus (MRSA)
| Bacteria strains/MIC (μg mL−1) |
D-PLLA-D |
D-PLLA-D@Q 1 |
D-PLLA-D@Q 2 |
D-PLLA-D@Q-PEG-1 |
D-PLLA-D@Q-PEG-2 |
| MRSA 57964 |
133 ± 7 |
224 ± 9 |
222 ± 8 |
84 ± 7 |
520 ± 22 |
| MRSA 42412 |
15 ± 1 |
62 ± 3 |
62 ± 4 |
16 ± 1 |
63 ± 4 |
| MRSA 21595 |
16 ± 6 |
60 ± 4 |
126 ± 3 |
30 ± 3 |
248 ± 6 |
| MRSA 9808 |
15 ± 2 |
258 ± 4 |
127 ± 7 |
14 ± 1 |
256 ± 10 |
| MRSA 6506 |
25 ± 3 |
30 ± 3 |
231 ± 6 |
43 ± 6 |
118 ± 6 |
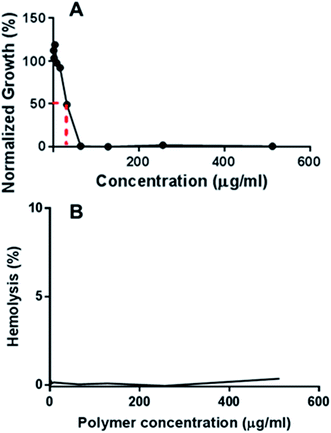 |
| | Fig. 3 (A) Growth of MRSA 42412 in the presence of different concentrations of D-PLLA-D@Q-PEG-2 (B) hemolysis profile of D-PLLA-D@Q-PEG-2 at different polymer concentrations. | |
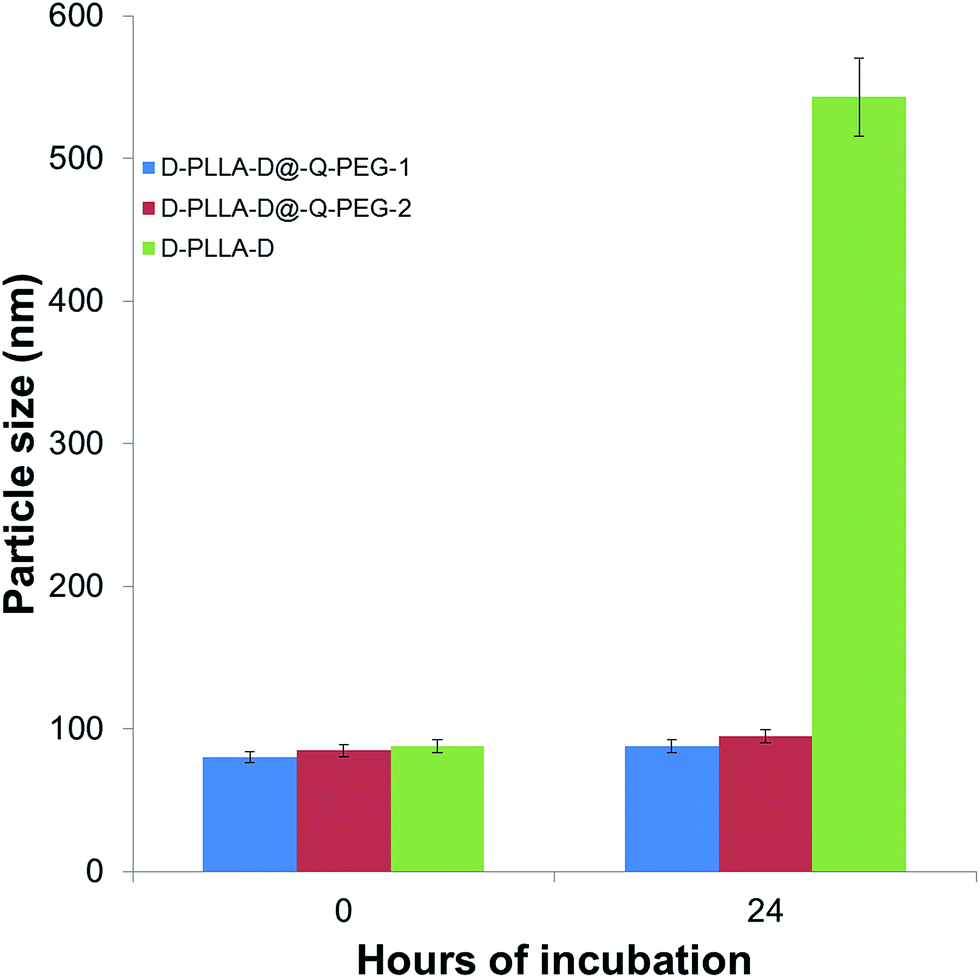
Our PEG-containing quarternised polymers showed MIC values of 14–520 μg mL−1 against MRSAs. Previous reports on chitosan based antimicrobial compounds exhibited MIC values of 98 μg mL−1 against Gram-negative bacteria and 49 μg mL−1 against Gram-positive bacteria.22 A main factor for consideration in this case would be that the bacteria were all obtained from commercial sources and not clinical isolates. Another report also reported MIC values of 31 to 98 μg mL−1 for various bacteria.23 In light of these, the performance of our polymers compare favourably against these reports. Chemical media has been reported to influence the activities of cationic polymers.24 The polymers were tested in chemically defined media against E. coli and S. aureus as previously reported (Tables S5 and S6†).25 It appears that there is not much change in the MIC values for the PEG conjugated polymers showing that the incorporation of PEG minimises the variation of the MIC in different media. Furthermore, the PEGylated particles did not aggregate after incubation for 24 h in medium whereas the D-PLLA-D polymers showed significant aggregation (Fig. 4). The aggregation has an effect on the activity of the polymers. Upon a high degree of aggregation, the MIC values increase to above 512 μg mL−1 for D-PLLA-D whereas the non-aggregating particles, D-PLLA-D@Q-PEG, maintained their MIC values (Tables S7 and S8†).
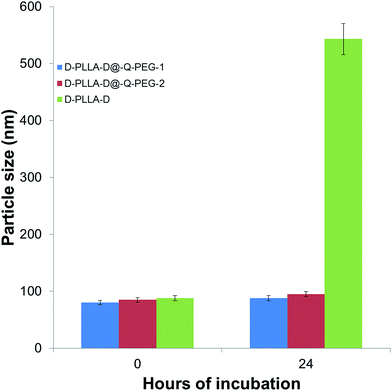 |
| | Fig. 4 Effect on PEGylation on the aggregation potential of the micelles. | |
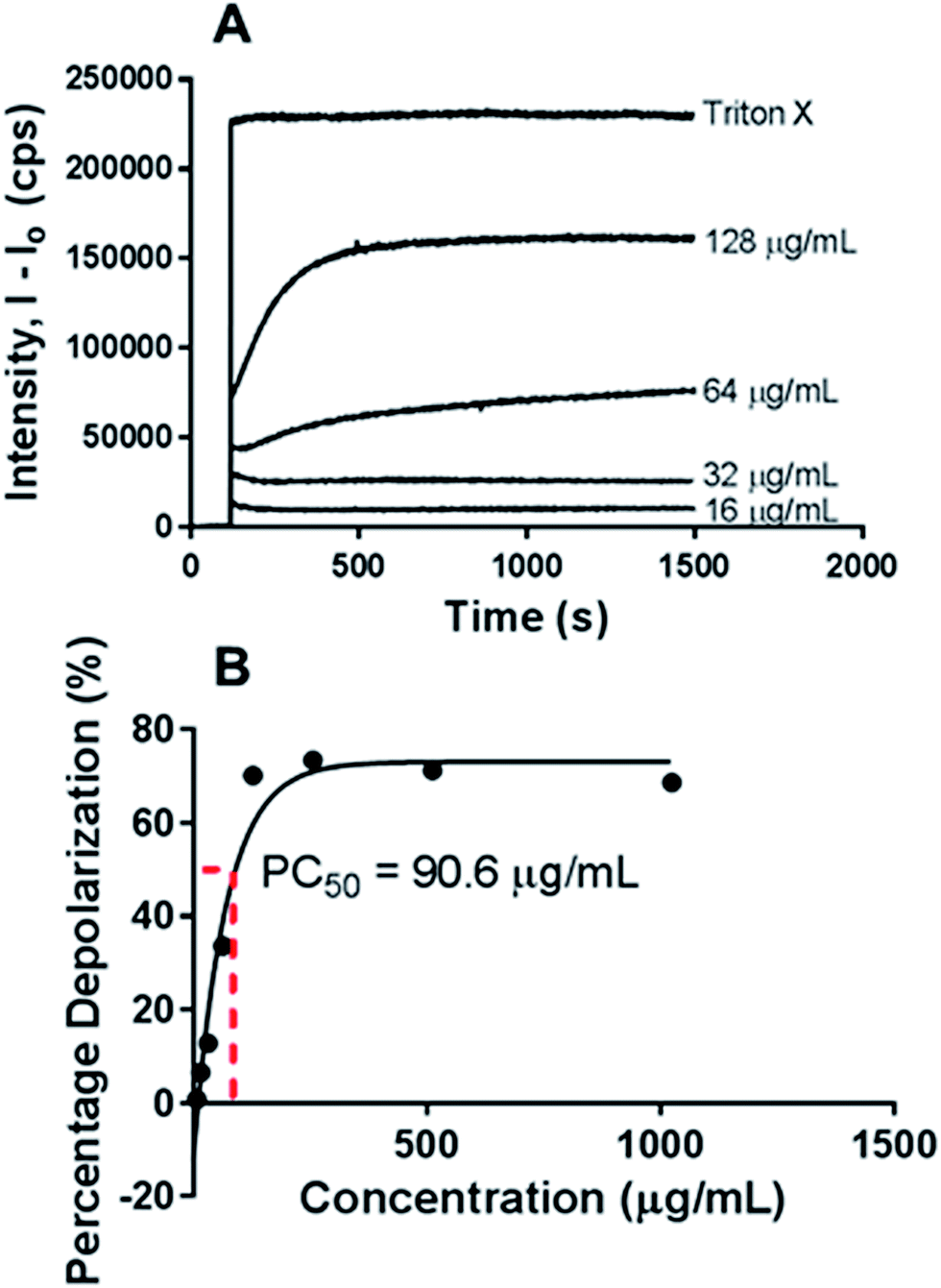
Next, the haemolytic activities of the polymers were evaluated using 4% (v/v) rabbit blood as a measure of their toxicity against mammalian cells. Fig. 3B & S6† showed that the percentage values of haemolysis were less than 5% for all the polymers and even at high concentrations of 5× MIC, less than 5% haemolysis observed. These results suggest that the polymers have good selectivity in targeting the bacteria membranes and can be used at high concentrations without having adverse impact on the mammalian membrane. PEGylation has been previously shown to improve the hemocompatibility of polymers.26,27 In our case, the PEGylation of the copolymers improved the biocompatibility as well as the hemocompatibility of the polymers. Quantification of the bacterial membrane depolarization was conducted using the membrane sensitive probe, DiSC35.1 (Fig. 5A & S7†) DiSC35 possesses membrane potential sensitivities due to the self-quenching of its fluorescence upon its partitioning onto the surface of a polarized bacteria cell. As this dye partitioning happens on a polarized cell, it can be prevented upon the depolarization of the cell, which results in the release of the dye into the media and causes a consequential increase in fluorescence intensity. It can hence be expected that the fluorescence intensity of DiSC35 detected is directly proportional to the degree of membrane depolarization, allowing for its quantification. Using the aforementioned procedure, the assay showed an almost immediate increase of detected fluorescence intensity upon the addition of the polymer, which was indicative of rapid membrane depolarization. The degree and speed of membrane depolarization were concentration-dependent until a maximal depolarization was reached, with higher concentrations of the polymers showing increased and faster depolarization effects. The calculation of PC50 values, based on Triton X, as a positive control demonstrated that relatively low concentrations of the polymers were required to achieve significant depolarization.
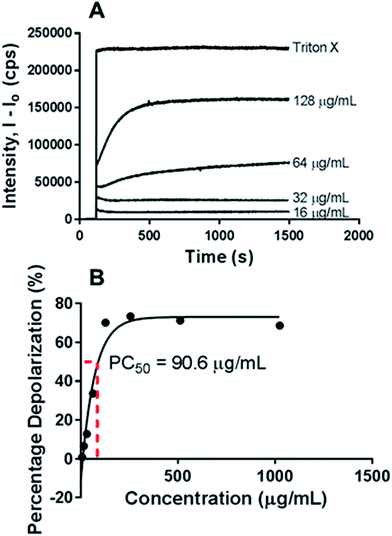 |
| | Fig. 5 D-PLLA-D@Q-PEG-1 as a cytoplasmic membrane disruption agent. (A) Effects of different concentrations of D-PLLA-D@Q-PEG-1 on the depolarization of inner membrane monitored by a membrane-sensitive DiSC35 assay, incubated with Pseudomonas aeruginosa (ATCC 9027) (B) membrane permeabilization monitored by DiSC35 assay. Increase in fluorescence intensity of DiSC35 is plotted against the concentration of D-PLLA-D@Q-PEG-1 and PC50 was estimated from the fitted graph. | |
Nitrocefin was used as the probe to characterize the outer membrane permeabilization. Nitrocefin interacts with β-lactamase, which is an enzyme inherently present within certain bacterium strains. When encountering bacteria with an intact outer cell membrane, nitrocefin is excluded from the inner periplasmic space, where β-lactamase is localized. However, upon the compromising of the outer membrane, nitrocefin is able to reach β-lactamase, which results in the cleavage of nitrocefin, causing a change in colour from yellow to red. This change thus allows for a method of monitoring the permeabilization of the outer membrane. All the tested samples are promising in bactericidal action through outer membrane permeabilization. A concentration-dependent degree of permeabilization was observed in all samples until a maximum value was reached (Table 3, Fig. 5B & S8†). PC50 values obtained through the comparison of these values to positive control polymyxin B suggested that relatively low concentrations of polymer (10 to 800 μg mL−1) were required to achieve significant effects in outer membrane disruption.
Table 3 PC50 values of nitrocefin assay tests of antibacterial polymers against Pseudomonas aeruginosa (ATCC 9027)
| Polymer |
D-PLLA-D |
D-PLLA-D@Q 1 |
D-PLLA-D@Q 2 |
D-PLLA-D@Q-PEG-1 |
D-PLLA-D@Q-PEG-2 |
| PC50 (μg mL−1) |
7 ± 1 |
13 ± 2 |
55 ± 5 |
38 ± 4 |
780 ± 20 |
4. Conclusion
To overcome problems of increasing bacterial infections, we have developed biodegradable, potent and cost-effective antimicrobial polymers for healthcare applications. These materials act via membrane-lytic antimicrobial mechanism, will have broad spectrum antimicrobial activities against multidrug-resistant bacteria. The novelty of our approach include: (1) excellent control over molecular composition and structure, which is important for producing materials with reproducible antimicrobial properties; (2) ease of incorporating functionalities essential to achieve high potency and selectivity towards microbes; (3) facile and scalable synthesis. The PEG conjugated polymers could be used as antibacterials as they are able to achieve quick depolarization to a high degree at low concentrations. The versatile synthetic platform has been exemplified by the synthesis of hemo-compatible biodegradable polymers. The copolymers showed improved biocompatibility upon PEGylation while retaining good antibacterial effect against both Gram-positive and Gram-negative bacteria. Membrane permeabilization is the probable mechanism behind the activity against the wide spectrum of bacteria.
Acknowledgements
The authors would like to thank Ms Hang Xinran and Ms Yew Pek Yin Michelle for their assistance in performing the experiments, Dr Aung Thet Tun for the blood collection, Ms Eunice Goh Tze Leng for her guidance in the experiments and the members of SERI for their useful comments and suggestions. RL thanks the funding support from the National Medical Research Council's Co-operative Basic Research Grant (NMRC/CBRG/0048/2013).
References
- J. Li, S. Liu, J.-J. Koh, H. Zou, R. Lakshminarayanan, Y. Bai, K. Pervushin, L. Zhou, C. Verma and R. W. Beuerman, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1023–1031 CrossRef CAS PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef.
- S. R. Deka, A. K. Sharma and P. Kumar, Curr. Top. Med. Chem., 2015, 15, 1179–1195 CrossRef CAS PubMed.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- G. N. Tew, D. H. Liu, B. Chen, R. J. Doerksen, J. Kaplan, P. J. Carroll, M. L. Klein and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5110–5114 CrossRef CAS PubMed.
- A. C. Engler, N. Wiradharma, Z. Y. Ong, D. J. Coady, J. L. Hedrick and Y.-Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y.-Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- S. Venkataraman, Y. Zhang, L. Liu and Y.-Y. Yang, Biomaterials, 2010, 31, 1751–1756 CrossRef CAS PubMed.
- H. Wang, K. Xu, L. Liu, J. P. K. Tan, Y. Chen, Y. Li, W. Fan, Z. Wei, J. Sheng, Y.-Y. Yang and L. Li, Biomaterials, 2010, 31, 2874–2881 CrossRef CAS PubMed.
- N. Wiradharma, U. Khoe, C. A. E. Hauser, S. V. Seow, S. Zhang and Y.-Y. Yang, Biomaterials, 2011, 32, 2204–2212 CrossRef CAS PubMed.
- J. Bonilla, E. Fortunati, M. Vargas, A. Chiralt and J. M. Kenny, J. Food Eng., 2013, 119, 236–243 CrossRef CAS.
- T. Jin and H. Zhang, J. Food Sci., 2008, 73, M127–M134 CrossRef CAS PubMed.
- J.-W. Rhim, H.-M. Park and C.-S. Ha, Prog. Polym. Sci., 2013, 38, 1629–1652 CrossRef CAS.
- F. Liu, J. Hu, G. Liu, S. Lin, Y. Tu, C. Hou, H. Zou, Y. Yang, Y. Wu and Y. Mo, Polym. Chem., 2014, 5, 1381–1392 RSC.
- X. Zhang, B. H. Tan and C. He, Macromol. Rapid Commun., 2013, 34, 1761–1766 CrossRef CAS PubMed.
- Y. Li, J. Yang and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4300–4308 CrossRef CAS.
- X. Wu, X. He, L. Zhong, S. Lin, D. Wang, X. Zhu and D. Yan, J. Mater. Chem., 2011, 21, 13611–13620 RSC.
- Z. Li, D. Yuan, X. Fan, B. H. Tan and C. He, Langmuir, 2015, 31, 2321–2333 CrossRef CAS PubMed.
- X. Liu, H. Yin, Z. Zhang, B. Diao and J. Li, Colloids Surf., B, 2015, 125, 230–237 CrossRef CAS PubMed.
- K. M. G. O'Connell, J. T. Hodgkinson, H. F. Sore, M. Welch, G. P. C. Salmond and D. R. Spring, Angew. Chem., Int. Ed., 2013, 52, 10706–10733 CrossRef PubMed.
- H. Hanberger, S. Walther, M. Leone, P. S. Barie, J. Rello, J. Lipman, J. C. Marshall, A. Anzueto, Y. Sakr, P. Pickkers, P. Felleiter, M. Engoren and J.-L. Vincent, Int. J. Antimicrob. Agents, 2011, 38, 331–335 CrossRef CAS PubMed.
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani, R. W. Beuerman, E.-T. Kang, Y. Mu, C. M. Li, M. W. Chang, S. S. Jan Leong and M. B. Chan-Park, Nat. Mater., 2011, 10, 149–156 CrossRef CAS PubMed.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y.-Y. Yang, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- H. Choi, S. Chakraborty, R. Liu, S. H. Gellman and J. C. Weisshaar, PLoS One, 2014, 9, e104500 Search PubMed.
- D. S. S. M. Uppu, S. Samaddar, C. Ghosh, K. Paramanandham, B. R. Shome and J. Haldar, Biomaterials, 2016, 74, 131–143 CrossRef CAS PubMed.
- J. Guo, Y. Feng, Y. Ye and H. Zhao, J. Appl. Polym. Sci., 2011, 122, 1084–1091 CrossRef CAS.
- W.-H. Kuo, M.-J. Wang, C.-W. Chang, T.-C. Wei, J.-Y. Lai, W.-B. Tsai and C. Lee, J. Mater. Chem., 2012, 22, 9991–9999 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra04531f |
|
| This journal is © The Royal Society of Chemistry 2016 |