
Synthesis of the 1,2,4-thiadiazole alkaloids polycarpathiamines A and B†
Emma K.
Davison
and
Jonathan
Sperry
*
School of Chemical Sciences, University of Auckland, 23 Symonds Street, Auckland, New Zealand. E-mail: j.sperry@auckland.ac.nz
First published on 30th November 2015
Abstract
In the presence of 1 mol% of a copper(II) catalyst and air, a readily available N1-acetyl-N3-thioacylguanidine undergoes a one-pot benzylic oxidation–oxidative heterocyclization sequence to give the 3-amino-5-acyl-1,2,4-thiadiazole core of polycarpathiamines A (2) and B (3) and thus facilitating the first synthesis of both natural products. This methodology offers a straightforward alternative to the low yielding dipolar cycloaddition used to access 3-amino-5-acyl-1,2,4-thiadiazoles.
 Jonathan Sperry | Jonathan Sperry obtained his BSc (Hons) (2002) and Ph.D. (2006, Professor Chris Moody) from the University of Exeter, UK. He spent 3.5 years as a postdoctoral researcher with distinguished Professor Margaret Brimble at the University of Auckland, before taking up a lectureship at the same institution in 2009, where he is currently a Senior Lecturer and a Rutherford Discovery Fellow. |
Introduction
The 1,2,4-thiadiazole heterocycle is present in a small family of natural products (Fig. 1), of which dendrodoine (1), isolated from the murine tunicate Dendroba grossularia (Styélidés),1 was the sole member for over thirty years. This statistic changed in 2013 upon the isolation of polycarpathiamines A (2) and B (3),2 from the ascidian Polycarpa aurata and the unnamed enantiomeric pair of alkaloids (+)-4 and (−)-4,3 from the herbaceous plant Isatis indigotica.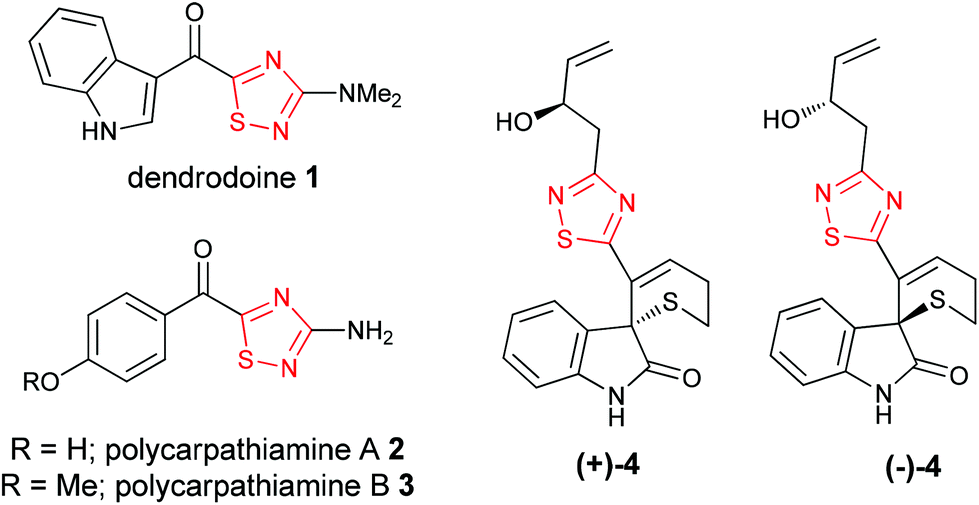 | ||
| Fig. 1 Natural products containing a 1,2,4-thiadiazole (red). | ||
The 3-amino-5-acyl-1,2,4-thiadiazole subunit of natural products 1–3 is a rare heterocyclic motif that to the best of our knowledge has only been the target for synthesis in the context of these three natural products (Scheme 1).2,4,5 The synthetic method of choice to access this motif is the dipolar cycloaddition between an acyl cyanide and N,N-dimethylaminonitrile sulfide 5 (generated in situ from 5-dimethylamino-1,3,4-oxathiazol-2-one) (Scheme 1), an approach that was employed in the synthesis of dendrodoine A (1)4 and the 1,2,4-thiadiazole 6, the latter of which was used to aid the structural determination of the polycarpathiamines.2 The harsh conditions and poor yields associated with this reaction prompted us to develop an alternative synthetic route to this heterocyclic subunit en route to the natural products polycarpathiamines A (2) and B (3).
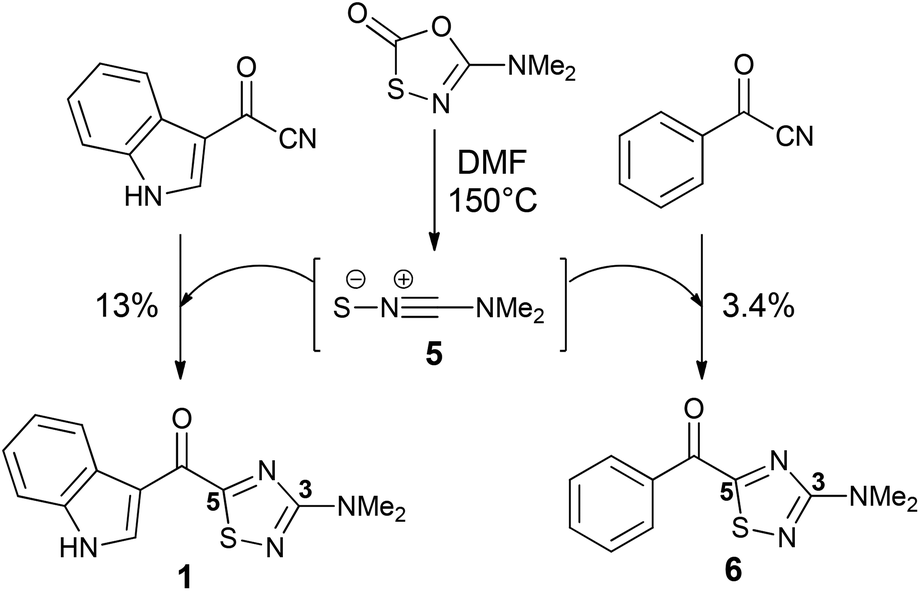 | ||
| Scheme 1 Synthesis of 3-amino-5-acyl-1,2,4-thiadiazoles by dipolar cycloaddition.2,4 | ||
We envisioned that the 3-amino-5-acyl-1,2,4-thiadiazole core (8) of the polycarpathiamines could be obtained from N1-acetyl-N3-thioacylguanidine 7 by a one-pot benzylic oxidation–oxidative heterocyclization6–8 sequence, a challenging transformation that requires uncovering oxidation conditions that effect the overall conversion of 7 to 8 but do not promote oxidative desulfurization9 or intermolecular dehydrogenative coupling of the electron rich aromatic ring (Scheme 2). Once 8 is secured, deprotection steps would deliver polycarpathiamines A (2) and B (3).
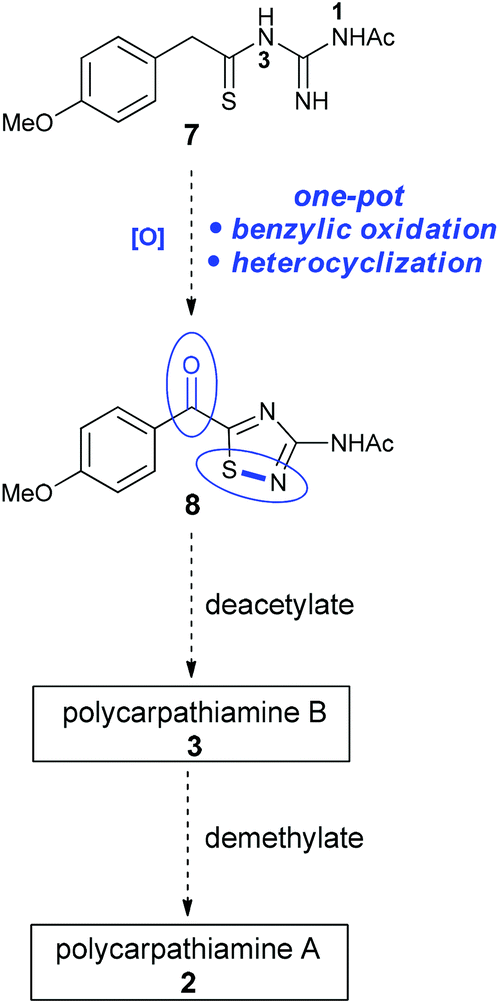 | ||
| Scheme 2 Proposed route to 3-amino-5-acyl-1,2,4-thiadiazole 8en route to polycarpathiamines A and B. | ||
The N1-acetyl-N3-thioacylguanidine 7 was readily prepared by the base-mediated condensation of N-acetylguanidine with thionoester 10 obtained from treating the commercially available ester 9 with Lawesson's reagent (Scheme 3).
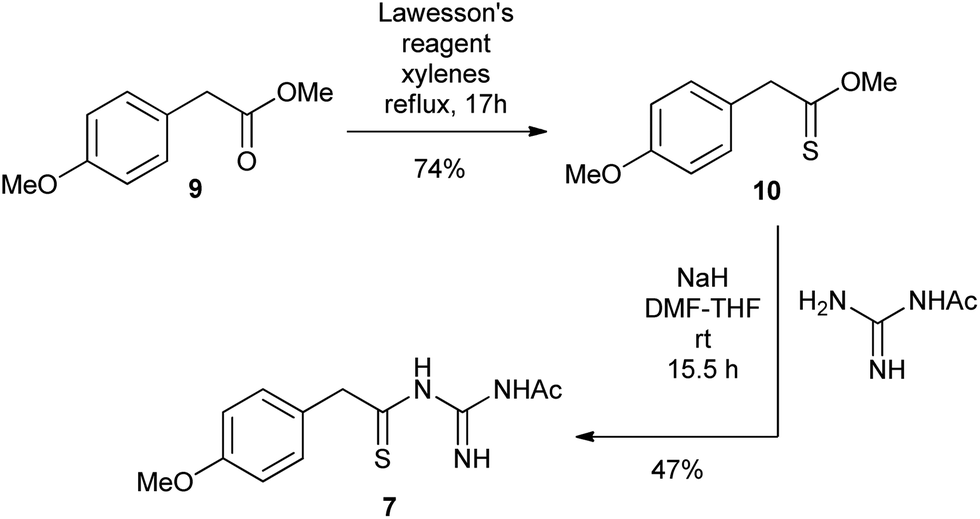 | ||
| Scheme 3 Synthesis of N1-acetyl-N3-thioacylguanidine 7. | ||
When considering the proposed overall oxidative transformation of 7 to 8, aerobic copper-catalyzed conditions were deemed an appropriate starting point, given their ability to effect both benzylic oxidation and intramolecular N–S bond formation under mild conditions,10 but need to be used forcefully to cause desulfurization reactions.10 The reported copper(II)-chloride-catalyzed α-oxygenation of arylthioacetamides to their corresponding α-keto arylthioamides11 was initially attempted, affording the desired 3-amino-5-acyl-1,2,4-thiadiazole (8) in a moderate yield (Table 1, entry 1) that provided an excellent starting point for further optimization. In the presence of air instead of neat oxygen, the yield increased to 67% and simplified the procedure so it could be carried out in an open-flask (entry 2). When caesium carbonate was employed as base, a slight drop in yield was noted, but increasing the amount of base to two equivalents remedied the yield and shortened the reaction time (entries 3 and 4) compared to potassium carbonate (entry 2). A screen of copper(II)-salts revealed that copper(II)-acetate was the best yielding (87%, entries 5–8), and there was a decrease in yield when changing the solvent from DMF to acetonitrile and toluene (entries 9 and 10). It is of note that in all of the reactions described in Table 1, the product 8 is isolated essentially pure after work-up, with no flash chromatography required. Upon tracking the reaction of 7 to 8 by TLC analysis, it is readily apparent that the reaction proceeds through a polar intermediate (assumed to be the benzoyl thioacylguanidine) that undergoes oxidative heterocyclization (N–S bond formation) to give 8. All attempts to isolate this intermediate or characterise it by NMR failed as it is quickly converted to the 1,2,4-thiadiazole 8 during workup/chromatography.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (1 mol%) | Oxidant | Base (eq.) | Solvent | Time (h) | Yield 8 (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a All reactions above carried out at 80 °C on 0.04–0.05 mmol scale except entries 1 (0.13 mmol) and 2 (0.11 mmol). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | CuCl2·2H2O | O2 | K2CO3 (1) | DMF | 1.75 | 56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | CuCl2·2H2O | Air | K2CO3 (1) | DMF | 2.75 | 67 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | CuCl2·2H2O | Air | Cs2CO3 (1) | DMF | 1.0 | 59 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | CuCl2·2H2O | Air | Cs2CO3 (2) | DMF | 1.5 | 69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | CuBr2 | Air | Cs2CO3 (2) | DMF | 0.5 | 56 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Cu(OTf)2 | Air | Cs2CO3 (2) | DMF | 0.5 | 70 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | CuSO4 | Air | Cs2CO3 (2) | DMF | 0.5 | 57 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Cu(OAc) 2 ·H2O | Air | Cs 2 CO 3 (2) | DMF | 0.5 | 87 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Cu(OAc)2·H2O | Air | Cs2CO3 (2) | MeCN | 0.5 | 77 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Cu(OAc)2·H2O | Air | Cs2CO3 (2) | PhMe | 3.0 | 38 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
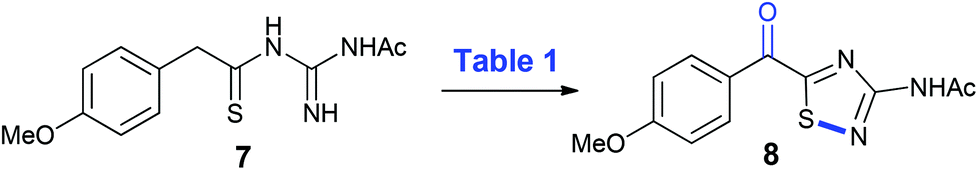
With 8 in hand, its conversion to the polycarpathiamine natural products was undertaken (Scheme 4). Deacetylation of 8 gave a synthetic sample of polycarpathiamine B (3), which itself underwent demethylation12 to give polycarpathiamine A (2). The spectroscopic data for synthetic 2 and 3 were in near perfect agreement with those in the isolation report (Table 2).2
 | ||
| Scheme 4 Synthesis of polycarpathiamines A (2) and B (3). | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
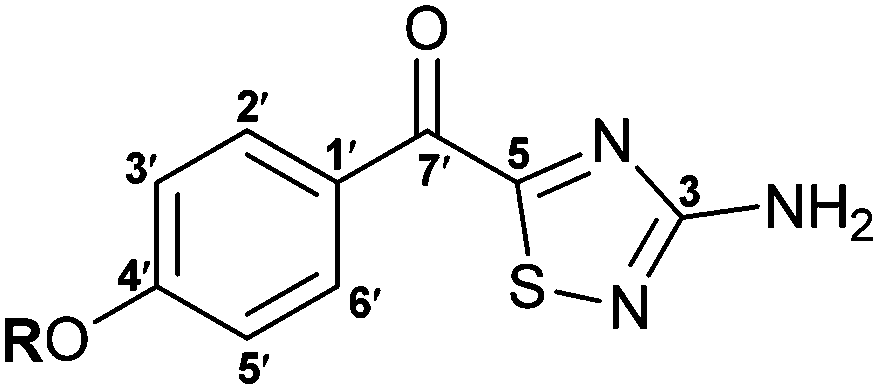
| Atom no. | R = H; polycarpathiamine A (2) (DMSO-d6) | R = Me; polycarpathiamine B (3) (DMSO-d6) | ||||||
| Synthetic | Natural product | Synthetic | Natural product | |||||
| 1H (400 MHz) | 13C (100 MHz) | 1H (400 MHz) | 13C (100 MHz) | 1H (400 MHz) | 13C (100 MHz) | 1H (300 MHz) | 13C (75 MHz) | |
| C (3) | 171.7 | 171.7 | 171.8 | 171.8 | ||||
| NH2 (3) | 7.09 (br s) | 7.08 (s) | 7.12 (br s) | 7.12 (s) | ||||
| C (5) | 185.7 | 185.7 | 185.4 | 185.4 | ||||
| C (1′) | 125.2 | 125.1 | 126.6 | 126.6 | ||||
| CH (2′/6′) | 8.37 (d, J 8.8) | 133.6 | 8.37 (d, J 8.8) | 133.6 | 8.44 (d, J 9.2) | 133.3 | 8.44 (d, J 9.0) | 133.3 |
| CH (3′/5′) | 6.94 (d, J 8.8) | 115.7 | 6.94 (d, J 8.8) | 115.6 | 7.15 (d, J 9.2) | 114.3 | 7.15 (d, J 9.0) | 114.3 |
| C (4′) | 163.9 | 163.8 | 164.6 | 164.6 | ||||
| CO (7′) | 180.6 | 180.6 | 180.9 | 180.9 | ||||
| OR (4′) | 10.80 (br s) | 10.79 (br s) | 3.90 (s) | 55.8 | 3.90 (s) | 55.8 | ||
Conclusions
In summary, the N1-acetyl-N3-thioacylguanidine 7 undergoes a copper(II)-air mediated one-pot benzylic oxidation–oxidative heterocyclization process to provide the 3-amino-5-acyl-1,2,4-thiadiazole core of polycarpathiamines A (2) and B (3), facilitating the first synthesis of the two natural products. This methodology provides a straightforward alternative to the low yielding dipolar cycloaddition used to access 3-amino-5-acyl-1,2,4-thiadiazoles and should find utility in the construction of this heterocyclic motif, for example in the synthesis of analogues of polycarpathiamine A given it displays significant cytotoxic effects against L5178Y murine lymphoma cells (IC50 0.41 μM).2Experimental procedures
General
Commercially available reagents were used throughout without purification unless otherwise stated. Anhydrous solvents were used as supplied. Tetrahydrofuran was distilled from sodium benzophenone ketyl under nitrogen atmosphere. Dichloromethane was distilled from calcium hydride under a nitrogen atmosphere. Ether refers to diethyl ether. All reactions were routinely carried out in oven-dried glassware under a nitrogen or argon atmosphere unless otherwise stated. Analytical thin layer chromatography was performed using silica plates and compounds were visualized at 254 and/or 360 nm ultraviolet irradiation followed by staining with either alkaline permanganate or ethanolic vanillin solution. Infrared spectra were obtained using a Perkin Elmer spectrum One Fourier Transform Infrared spectrometer as thin films between sodium chloride plates. Absorption maxima are expressed in wavenumbers (cm−1). Melting points were recorded on an electrothermal melting point apparatus and are uncorrected. NMR spectra were recorded as indicated on either a spectrometer operating at 400 MHz for 1H nuclei and 100 MHz for 13C nuclei or on a spectrometer operating at 500 MHz and 125 MHz for 1H and 13C nuclei, respectively. Chemical shifts are reported in parts per million (ppm) relative to the tetramethylsilane peak recorded as δ 0.00 ppm in CDCl3/TMS solvent, or the residual chloroform (δ 7.26 ppm), DMSO (δ 2.50 ppm) or methanol (δ 3.31 ppm) peaks. The 13C NMR values were referenced to the residual chloroform (δ 77.1 ppm), DMSO (δ 39.5 ppm) or methanol (δ 49.0 ppm) peaks. 13C NMR values are reported as chemical shift δ, multiplicity and assignment. 1H NMR shift values are reported as chemical shift δ, relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), coupling constant (J in Hz) and assignment. Assignments are made with the aid of DEPT 135, COSY, NOESY, HMBC and HSQC experiments. High resolution mass spectra were obtained by electrospray ionization in positive ion mode at a nominal accelerating voltage of 70 eV on a microTOF mass spectrometer.O-Methyl 2-(4-methoxyphenyl)ethanethioate (10)
To a stirred solution of methyl-(4-methoxy)phenylacetate 9 (500 mg, 2.78 mmol) in xylenes (40 mL) was added Lawesson's reagent (1.55 g, 3.82 mmol) and the mixture was heated at reflux for 17 h. The solution was cooled and concentrated in vacuo and the crude material purified by flash chromatography on silica gel eluting with ethyl acetate–hexanes (1:19) to afford the title compound (402 mg, 2.05 mmol, 74%) as a yellow oil. HRMS Found: [M + Na]+, 219.0446, [C10H12O2S + Na+] requires 219.0450; IR (neat): 2942, 1610, 1510, 1244, 1175, 1120, 1032, 924, 820, 766 cm−1; 1H NMR (400 MHz, CDCl3) 7.23 (2H, d, J 8.5, 2 × ArH), 6.86 (2H, d, J 8.5, 2 × ArH), 4.06 (3H, s, OMe), 4.00 (2H, s, CH2), 3.80 (3H, s, OMe); 13C NMR (100 MHz, CDCl3) 222.2, 158.9, 130.3 (2 × CH), 128.3, 114.1 (2 × CH), 59.5 (OMe), 55.4 (OMe), 52.6 (CH2).
N-(N′-(2-(4-Methoxyphenyl)ethanethioyl)carbamimidoyl)acetamide (7)
To a stirred solution of N-acetylguanidine (114 mg, 1.13 mmol) in THF (3.8 mL) and DMF (2.0 mL) cooled to 0 °C, was added sodium hydride (60% in mineral oil, 64 mg, 1.60 mmol). The mixture was stirred for 10 min before adding thioester 10 (200 mg, 1.02 mmol) in THF (2.0 mL) dropwise. The resulting solution was allowed to warm to room temperature and stirring continued for 15.5 h, before being cooled back to 0 °C prior to the addition of water (7 mL). The aqueous layer was then separated, and extracted with ethyl acetate (3 × 10 mL) until the aqueous layer became colourless. The combined organic extracts were dried using Na2SO4, filtered, then concentrated in vacuo. The crude product was purified by flash chromatography on silica gel eluting with dichloromethane–methanol (49:1) to afford the title compound (126 mg, 0.475 mmol, 47%) as a yellow oil. HRMS Found: [M + H]+, 266.0963, [C12H15N3O2S + H+] requires 266.0958; IR (neat): 3295, 2932, 1697, 1611, 1509, 1449, 1242, 1176, 1029, 820, 734 cm−1; 1H NMR (400 MHz, DMSO-d6) 11.61 (1H, br s, NH), 11.14 (1H, br s, NH), 9.71 (1H, br s, NH), 7.23 (2H, d, J 8.7, 2 × ArH), 6.84 (2H, d, J 8.7, 2 × ArH), 3.87 (2H, s, CH2), 3.72 (3H, s, OMe), 2.15 (3H, s, COMe); 13C NMR (100 MHz, DMSO-d6) 218.4, 174.2, 159.0, 157.8, 130.5, 129.8 (2 × CH), 113.5 (2 × CH), 58.5 (CH2), 55.0 (OMe), 24.2 (CO ).
).
N-Acetamide-5-(4-methoxybenzoyl)-1,2,4-thiadiazole (8)
To a stirred solution of Cu(OAc)2·H2O (0.10 mg, 0.0005 mmol, 1 mol%) in DMF (0.1 mL) was added Cs2CO3 (28 mg, 0.086 mmol) and a solution of 7 (13 mg, 0.049 mmol) in DMF (0.3 mL). The resulting solution was heated to 80 °C in an open flask for 30 min. Upon completion of the reaction, the solution was cooled to room temperature, and concentrated in vacuo. The crude material was then redissolved in ethyl acetate (5 mL) and washed with water (2 × 5 mL). The aqueous layers were then combined, and extracted with ethyl acetate (2 × 5 mL). The combined organic extracts were dried using Na2SO4, filtered, then concentrated in vacuo to afford the title compound (11.8 mg, 0.0426 mmol, 87%) as a dark yellow solid, which was used directly in the next step without further purification. HRMS Found: [M + Na]+, 300.0409, [C12H11N3O3S + Na+] requires 300.0413; IR (neat): 3217, 3099, 1704, 1593, 1562, 1493, 1451, 1262, 1231, 1174, 1121, 1023, 983, 876, 854, 700 cm−1; 1H NMR (400 MHz, DMSO-d6) 11.50 (1H, br s, NH), 8.49 (2H, d, J 9.1, 2 × ArH), 7.16 (2H, d, J 9.1, 2 × ArH), 3.91 (3H, s, OMe), 2.17 (3H, s, COMe); 13C NMR (100 MHz, DMSO-d6) 186.3, 180.6, 168.1, 164.9, 163.2, 133.5 (2 × CH), 126.3, 114.4 (2 × CH), 55.9 (OMe), 23.8 (CO ).
).
Polycarpathiamine B (3)
To a stirred solution of 8 (51 mg, 0.184 mmol) in ethanol (4 mL) was added conc. hydrochloric acid (0.06 mL). The resulting solution was heated at reflux for 30 min, then was cooled and concentrated in vacuo. The crude material was redissolved in water (3 mL) and 1 M NaOH was added to obtain a solution of ∼pH 10. The aqueous solution was extracted with ethyl acetate (3 × 5 mL) and the combined organic extracts were dried using Na2SO4, filtered, then concentrated in vacuo to afford the title compound (40 mg, 0.170 mmol, 92%) as a dark yellow solid, which did not require further purification. Mp: 171.5–174.3 °C {lit.2 mp: not given}; HRMS Found: [M + Na]+, 258.0311, [C10H9N3O2S + Na+] requires 258.0308; IR (neat): 3450, 3340, 3232, 2925, 1633, 1599, 1537, 1511, 1480, 1297, 1260, 1127, 1024, 856, 842, 758 cm−1; 1H and 13C NMR data, see Table 2.Polycarpathiamine A (2)
To a stirred solution of polycarpathiamine B (3) (30 mg, 0.128 mmol) in dichloromethane (1.5 mL) cooled to −78 °C, was added boron tribromide (1 M solution in dichloromethane, 0.54 mL, 0.536 mmol) dropwise. The resulting solution was stirred for 5 minutes, before warming to room temperature, then heating at reflux. The reaction mixture was heated at reflux for 2 h, before being cooled to room temperature, diluted with dichloromethane (5 mL) and quenched with sodium hydrogen carbonate solution (10 mL). The layers were separated, and the organic layer washed again with sat. sodium hydrogen carbonate (10 mL). The organic layer was dried using Na2SO4, filtered, then concentrated in vacuo. The crude product was purified by flash chromatography on silica gel eluting with dichloromethane–methanol (19:1) to afford the title compound (4.2 mg, 0.0190 mmol, 15%) as a dark yellow solid. Mp: 170.9–176.8 °C {lit.2 mp: not given}; HRMS Found: [M + Na]+, 244.0156, [C9H7N3O2S + Na+] requires 244.0151; IR (neat): 3430, 3218, 2918, 1595, 1536, 1510, 1465, 1292, 1245, 1174, 1123, 1015, 841 cm−1; 1H and 13C NMR data, see Table 2.
Acknowledgements
We thank the University of Auckland for a Doctoral Scholarship (E. K. D.) and the Royal Society of New Zealand for the award of a Rutherford Discovery Fellowship (J. S.).References
- S. Heitz, M. Durgeat, M. Guyot, C. Brassy and B. Bachet, Tetrahedron Lett., 1980, 21, 1457 CrossRef CAS.
- C.-D. Pham, H. Weber, R. Hartmann, V. Wray, W. Lin, D. Lai and P. Proksch, Org. Lett., 2013, 15, 2230 CrossRef CAS PubMed.
- M. Chen, S. Lin, L. Li, C. Zhu, X. Wang, Y. Wang, B. Jiang, S. Wang, Y. Li, J. Jiang and J. Shi, Org. Lett., 2012, 14, 5668 CrossRef CAS PubMed.
- For the synthesis of a 3-(imidazol-1-yl)-5-acyl-1,2,4-thiadiazole by palladium-catalyzed cross-coupling, see: T. C. Gahman, H. Lang, M. R. Herbert, A. M. Thayer, C. A. Hassig, S. A. Noble, R. D. Cousins, H. Zhuang, C. R. Santos and X. Chen, U.S. Patent544182000, 2006 Search PubMed; Chem. Abstr. 2006, 145, 8185 Search PubMed.
- I. T. Hogan and M. Sainsbury, Tetrahedron, 1984, 40, 681 CrossRef CAS.
- Although the formation of 1,2,4-thiadiazoles by oxidative intramolecular N–S bond formation is well established, it has never been used to construct 3-amino-5-acyl-1,2,4-thiadiazoles, see: (a) F. Kurzer and S. A. Taylor, J. Chem. Soc., 1958, 379 RSC; (b) F. Kurzer and F. S. A. Taylor, J. Chem. Soc., 1959, 1064 RSC; (c) F. Kurzer and S. A. Taylor, J. Chem. Soc., 1962, 4191 RSC; (d) B. Junge, Liebigs Ann. Chem., 1975, 1961 CrossRef CAS; (e) C. G. Newton, W. D. Ollis and D. E. Wright, J. Chem. Soc., Perkin Trans. 1, 1984, 75 RSC; (f) F. Kurzer, J. Chem. Soc., Perkin Trans. 1, 1985, 311 RSC.
- For some further references on the formation of 1,2,4-thiadiazoles by intramolecular oxidative N–S bond formation, see: (a) H.-Y. Kim, S. H. Kwak, G.-H. Lee and Y.-D. Gong, Tetrahedron, 2014, 70, 8737 CrossRef CAS and references therein; (b) Y. Kihara, S. Kabashima, K. Uno, T. Okawara, T. Yamasaki and M. Furukawa, Synthesis, 1990, 1020 CrossRef CAS.
- For the synthesis of 3-amino-5-substituted-1,2,4-thiadiazoles that do not rely on an intramolecular oxidative heterocyclization, see: (a) K. Gewald, O. Calderon and U. Hain, J. Prakt. Chem., 1986, 328, 741 CrossRef CAS; (b) S. Buscemi and N. Vivona, Heterocycles, 1994, 38, 2423 CrossRef CAS; (c) A. H. M. Al-Shaar, D. W. Gilmour, D. J. Lythgoe, I. McClenaghan and C. A. Ramsden, J. Chem. Soc., Perkin Trans. 1, 1988, 3019 RSC; (d) P. M. When, P. E. Harrington and J. E. Eksterowicz, Org. Lett., 2009, 11, 5666 CrossRef PubMed; (e) M. C. McKie and R. M. Paton, ARKIVOC, 2002, vi, 15 Search PubMed.
- For a review on the oxidation of thiocarbonyls to carbonyls, see: A. Corsaro and V. Pistará, Tetrahedron, 1998, 54, 15027 CrossRef CAS.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- F. M. Moghaddam, Z. Mirjafary, H. Saeidian and M. J. Javan, Synlett, 2008, 892 CrossRef CAS.
- A comprehensive screen of alternative demethylation conditions was performed in an effort to increase the yield for this step, to no avail.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all novel compounds. See DOI: 10.1039/c5qo00367a |
| This journal is © the Partner Organisations 2016 |