
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive mapping of O-GlcNAc modification sites using a chemically cleavable tag†
Matthew E.
Griffin
a,
Elizabeth H.
Jensen
a,
Daniel E.
Mason
b,
Courtney L.
Jenkins
a,
Shannon E.
Stone
a,
Eric C.
Peters
b and
Linda C.
Hsieh-Wilson
*a
aDepartment of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: lhw@caltech.edu
bGenomics Institute of the Novartis Research Foundation, San Diego, CA 92121, USA
First published on 30th March 2016
Abstract
The post-translational modification of serine or threonine residues of proteins with a single N-acetylglucosamine monosaccharide (O-GlcNAcylation) is essential for cell survival and function. However, relatively few O-GlcNAc modification sites have been mapped due to the difficulty of enriching and detecting O-GlcNAcylated peptides from complex samples. Here we describe an improved approach to quantitatively label and enrich O-GlcNAcylated proteins for site identification. Chemoenzymatic labelling followed by copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) installs a new mass spectrometry (MS)-compatible linker designed for facile purification of O-GlcNAcylated proteins from cell lysates. The linker also allows subsequent quantitative release of O-GlcNAcylated proteins for downstream MS analysis. We validate the approach by unambiguously identifying several established O-GlcNAc sites on the proteins α-crystallin and O-GlcNAc transferase (OGT), as well as discovering new, previously unreported sites on OGT. Notably, these novel sites on OGT lie in key functional domains of the protein, underscoring how this site identification method may reveal important biological insights into protein activity and regulation.
The dynamic and reversible post-translational modification of intracellular proteins by β-linked O-GlcNAc, known as O-GlcNAcylation, is necessary for the regulation of numerous cellular processes, including transcription, translation, protein homeostasis, and metabolism.1–6 Alterations in O-GlcNAcylation are associated with human diseases such as cancer, diabetes, and neurodegeneration.6–8 However, understanding the roles of O-GlcNAcylation in specific physiological contexts will require a more comprehensive characterization of the O-GlcNAc proteome and the modification sites on proteins. Notably, although thousands of proteins have been putatively shown to be O-GlcNAcylated,9–15 relatively few glycosylation sites have been mapped. Mass spectrometric (MS) identification of O-GlcNAcylated peptides from complex mixtures has been challenging due to the substoichiometric nature of O-GlcNAcylation and is further exacerbated by suppression of O-GlcNAc peptide ionization in the presence of the unmodified peptide.16 Thus, improved methods to enrich O-GlcNAcylated peptides or proteins are much needed, particularly approaches that can be directly used in conjunction with MS/MS sequencing to achieve a more comprehensive understanding of O-GlcNAc modification sites.
Robust enrichment of O-GlcNAcylated proteins can be accomplished using a two-step chemoenzymatic approach.11,17 First, the O-GlcNAc moiety is tagged with a non-natural azide group by treatment of cell lysates with UDP-GalNAz 1 and a mutant galactosyltransferase (Y289L GalT)18 that specifically recognizes terminal GlcNAc moieties (Fig. 1). Next, a biotin group is attached via copper(I)-catalysed azide–alkyne cycloaddition (CuAAC),19 which allows for affinity purification. Although a limited set of alkyne-biotin linkers are commercially available, many existing linkers are not ideal for mapping O-GlcNAc modification sites. In particular, harsh conditions are usually required to disrupt the femtomolar biotin–streptavidin interaction,20 which may hydrolyse the labile O-GlcNAc moiety. Additionally, many linkers contain a large spacer between the biotin group and the alkyne functionality, which appends a relatively large mass to the glycopeptide and can preclude its sequencing by mass spectrometry.20 Therefore, a facile method to release the labelled peptides and proteins with minimal added mass would greatly facilitate downstream analysis.
Several cleavable linkers have been previously developed for the enrichment of O-GlcNAcylated proteins.13,15,16,21 However, each suffers from significant drawbacks for site identification. For example, a photocleavable linker was employed in conjunction with UDP-GalNAz 1 and Y289L GalT to sequence modified peptides from mouse brain lysate.15,16 Importantly, the moiety retained after cleavage provided a positively-charged amine group, which increased the overall peptide charge and facilitated ionization by electron-transfer dissociation (ETD), the most successful MS/MS method for O-GlcNAc peptide sequencing.12,22 Unfortunately, cleavage of the linker was found to be incomplete.16 In a recent report, a dibromine-containing, acid-cleavable linker was employed to identify various glycan modifications including O-GlcNAc.21 However, cleavage of the linker revealed only a neutral hydroxyl group, and the halogenated glycopeptides demonstrated poor fragmentation efficiency using ETD. Therefore, we aimed to develop a tag that would be quantitatively appended and released, as well as incorporate a positive charge upon cleavage to facilitate ETD-MS detection.
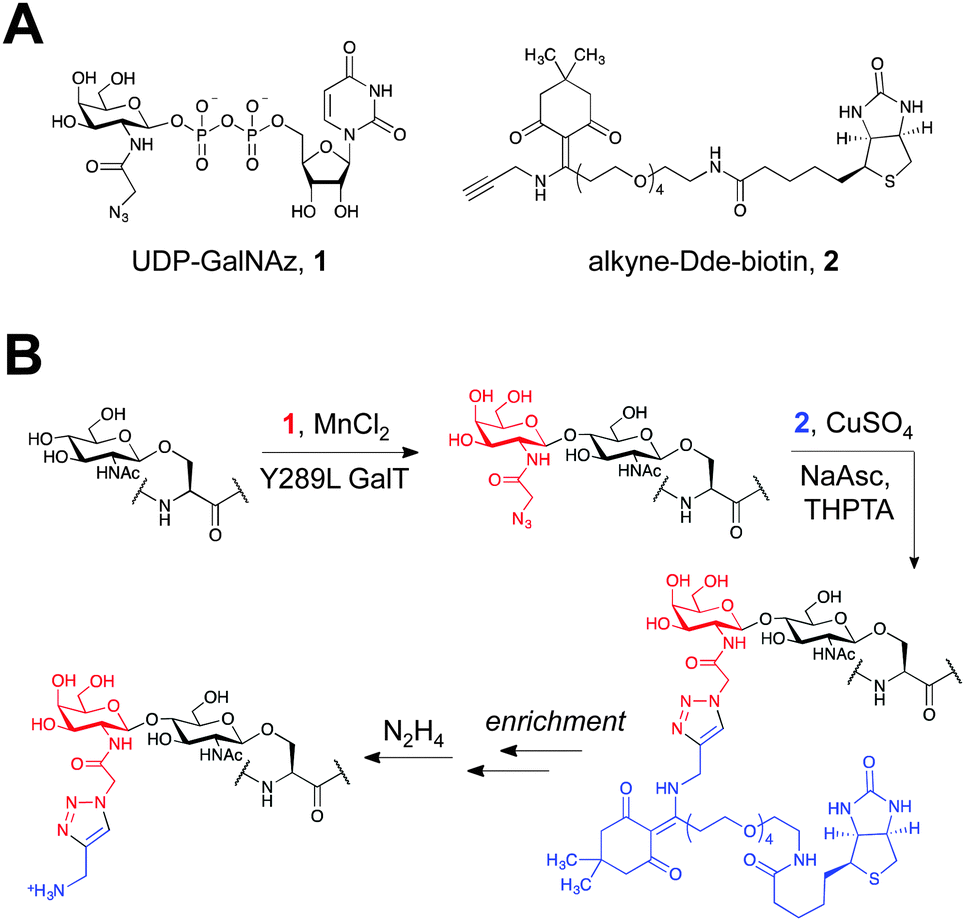 | ||
| Fig. 1 (A) Chemicals used in the labelling protocol. (B) Schematic of O-GlcNAc protein enrichment and elution using the two-step chemoenzymatic/CuAAC labelling protocol. | ||
To achieve these dual goals, we chose to examine the 1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (Dde) functional group (Fig. 1A). The Dde moiety has been used extensively as a protecting group for lysine in peptide synthesis,23 demonstrating its compatibility with biomolecules. The group is stable to both acid and base and can be quantitatively removed by hydrazine.24 However, it was reported that the Dde group is incompatible with sodium dodecyl sulphate (SDS) and amine-containing buffers, common additives to protein labelling protocols.25
We first investigated the labelling of a model O-GlcNAcylated peptide with commercially available alkyne-Dde-biotin 2 followed by cleavage of the linker using liquid chromatography (LC)-MS (Fig. 1B and 2). Commercially available peptide TAPT(gS)TIAPG (Fig. 2A), where gS is the O-GlcNAcylated residue, was incubated with 100 ng μL−1 of Y289L GalT and 1 mM of UDP-GalNAz 1 in 10 mM HEPES pH 7.9, 5.5 mM MnCl2 overnight at 4 °C. LC-MS analysis revealed quantitative conversion to the desired GalNAz-labeled product (Fig. 2B). Next, the azide-containing peptide was reacted with 100 μM of 2 in 10 mM sodium phosphate pH 7.6 containing 2 mM sodium ascorbate (NaAsc), 100 μM THPTA, and 1 mM CuSO4. After 1 h, stoichiometric biotinylation of the peptide was observed (Fig. 2C). Treatment with 2% aqueous hydrazine for 1 h at RT resulted in quantitative cleavage of the linker to afford a minimal, positively-charged aminomethyltriazolyl group (Fig. 1B and 2D). To test whether the linker would be stable under stringent wash conditions, we incubated the labelled peptide with 1% RapiGest, a MS-compatible analogue of SDS, or 6 M urea for 1 h at RT (Fig. S1, ESI†). In both cases, the linker remained intact, highlighting the compatibility of the linker with rigorous washing steps.
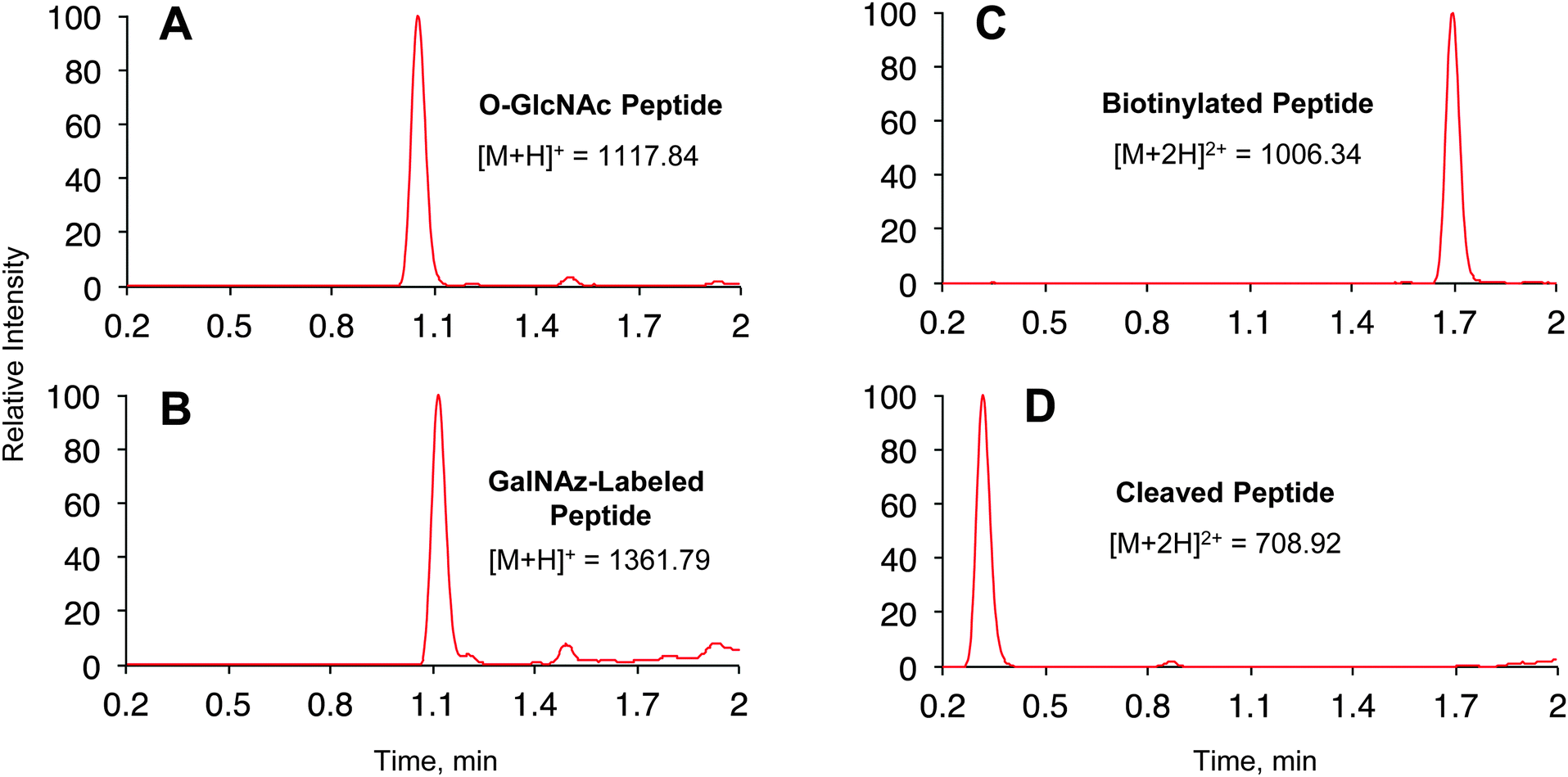 | ||
| Fig. 2 Labelling and cleavage reactions proceed quantitatively. Reverse phase LC-MS analysis of O-GlcNAc peptide labelling reactions at (A) time 0, (B) 16 h after addition of 1 and Y289L GalT, (C) 1 h after CuAAC with 2, and (D) 1 h after cleavage with 2% aqueous hydrazine. See ESI† for experimental details. (A) and (B) show base peak chromatograms. (C) and (D) show extracted ion chromatograms of the starting material and product within ±1 m/z of calculated values. | ||
We next tested the performance of our linker in comparison to a previously described, widely utilized photocleavable linker (alkyne-PC-biotin).16 Briefly, HEK-293T cell lysate was subjected to chemoenzymatic labelling with 1 using Y289L GalT as described above. The azide-labelled protein was then split into two equal fractions and reacted with either 2 or alkyne-PC-biotin by CuAAC. An aliquot of each sample was reserved for analysis, and the remainder of each sample was subjected to cleavage using 2% hydrazine monohydrate or by UV irradiation at 365 nm. The samples were resolved by SDS-PAGE and probed for biotin using streptavidin conjugated to AlexaFluor 680 dye (Fig. 3). Notably, a stronger biotin signal was observed for lysate labelled with 2 compared to alkyne-PC-biotin, suggesting higher labelling efficiency with the Dde linker. Furthermore, although both linkers cleaved well, the photocleavable linker showed slightly higher residual signal compared to 2, suggesting that the Dde moiety was also released more efficiently than the photocleavable group. These results demonstrate that our new approach provides an improvement in both labelling efficiency and recovery of O-GlcNAcylated proteins compared to the most widely used method.
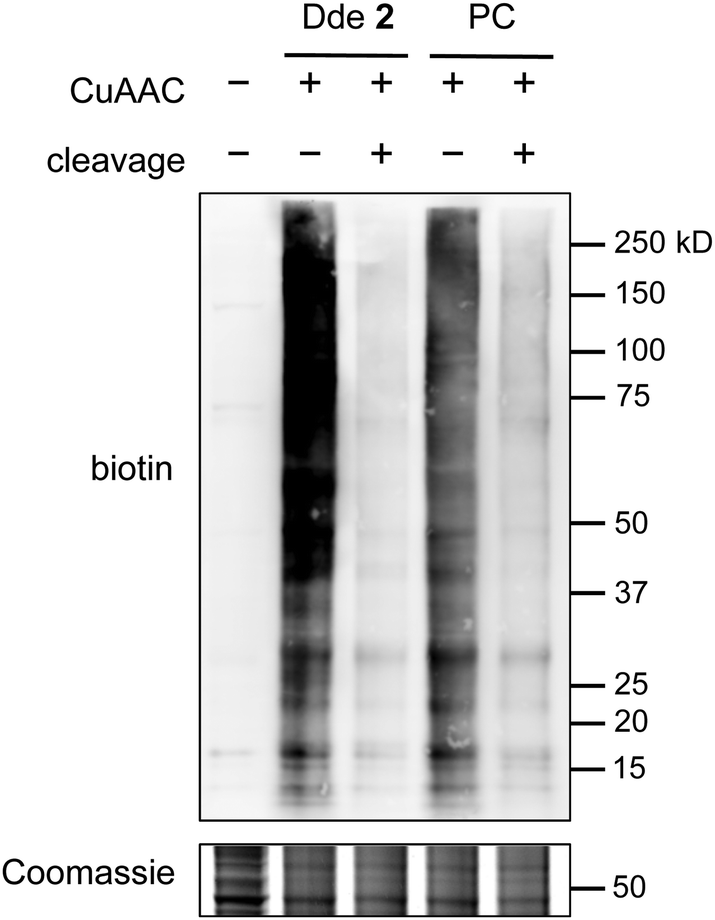 | ||
| Fig. 3 Alkyne-Dde-biotin linker 2 outperforms a widely used alkyne-photocleavable-biotin linker (PC). Protein lysates labelled with 2 show higher biotin signal after labelling (lane 2 vs. lane 4) and lower residual signal after cleavage (lane 3 vs. lane 5) compared to the PC linker. See Fig. S2 (ESI†) for full Coomassie gel. | ||
We then evaluated the potential of the approach to pull down known O-GlcNAcylated proteins and identify sites of modification. The well-characterized O-GlcNAcylated protein α-crystallin was selected to assess the sensitivity of the method because it has a relatively low glycosylation stoichiometry (<10%).26 Short-form OGT (sOGT)27,28 from Sf9 cells has multiple sites of O-GlcNAcylation12 and was thus used to determine whether comprehensive site mapping could be achieved. To test the robustness of our method in a complex mixture, each protein was added to 200 μg of adult mouse cortical lysate and subjected to chemoenzymatic labelling and CuAAC using 2. The labelled proteins were applied to high-capacity Neutravidin resin and washed with 1% SDS, 6 M urea, and phosphate buffered saline (PBS). The resin was then incubated for 1 h with 2% aqueous hydrazine to cleave the O-GlcNAcylated proteins from the resin. Eluted samples were precipitated, re-dissolved in denaturing buffer, and subjected to reduction, alkylation, and proteolytic digestion. Digested peptides were separated by nanoLC-MS and analysed on an LTQ-Velos by a combination of collision-induced dissociation (CID) and ETD-MS.
Impressively, a large number of O-GlcNAcylation sites were identified on α-crystallin and sOGT (Table 1). The known O-GlcNAc site on α-crystallin A (Ser-162)29 was readily recognized despite the low abundance of the O-GlcNAc modification at this site. Importantly, we observed both known and novel sites on sOGT.12,30 For example, we identified the previously reported Thr-662 site, which is found in the catalytic domain of sOGT.15 The new linker design also revealed a number of new O-GlcNAcylation sites within the N-terminal tetratricopeptide repeat-containing (TPR) domains (Ser-10/Thr-12, Ser-20, Ser-52, and Ser-56) of sOGT, and we observed a doubly modified peptide at both Ser-10/Thr-12 and Ser-20, highlighting the sensitivity of the approach to identify novel glycosylation sites and multiply occupied states. As the TPR domains of OGT are thought to mediate protein–protein interactions,31–33 such modifications could play an integral role in OGT regulation and may provide a mechanism to selectively modulate its activity toward specific substrates.
| Protein | Peptide sequence | Site(s) | Mascot ion score | Mascot delta ion score | Method |
|---|---|---|---|---|---|
| α-Crystallin A |

|
Ser-162 | 24.9 | 23.5 | ETD |
| sOGT |
|
Ser-10*/Thr-12* | 46.5 | — | ETD |
| sOGT |

|
Ser-20* | 21.6 | 13.6 | ETD |
| sOGT |

|
Ser-10*/Thr-12*, Ser-20* | 38.4 | — | ETD |
| sOGT |
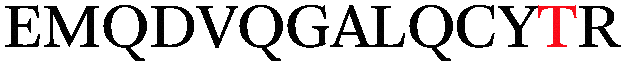
|
Thr-38 | 41.8 | 35.0 | CID |
| sOGT |
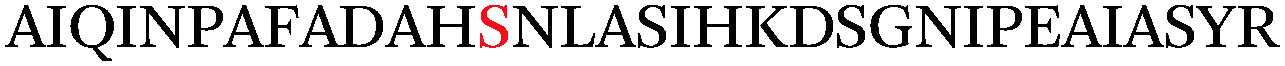
|
Ser-52* | 53.5 | 7.9 | ETD |
| sOGT |

|
Ser-56* | 56.8 | 15.7 | ETD |
| sOGT |
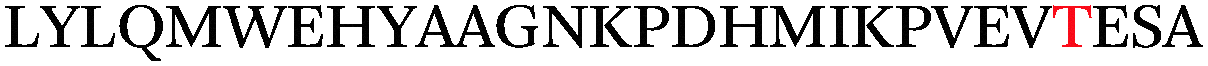
|
Thr-662 | 33.1 | 8.0 | ETD |
Conclusions
Herein we describe an improved method to facilitate the comprehensive mapping of O-GlcNAc modification sites. Chemoenzymatic attachment of an azide-containing monosaccharide onto O-GlcNAc sugars provides a stoichiometric, bioorthogonal handle, which is further functionalized with an alkyne-Dde-biotin linker to isolate and enrich O-GlcNAcylated proteins. The cleavable Dde linker provides numerous benefits over other reported structures. First, the linker is commercially available and inexpensive. Second, it is stable to rigorous, denaturing wash conditions and can be quantitatively cleaved under mild chemical conditions. Finally, the cleaved moiety that remains on the modified peptide minimally changes the peptide mass and generates an additional positive charge, which facilitates peptide sequencing by ETD. Together, in combination with the commercially available chemoenzymatic labelling kit,11 the method provides an accessible and practical system for the broader community. Using this approach, we identified established sites on both α-crystallin and sOGT and new modification sites on sOGT, including novel sites for OGT regulation. Our results showcase the method's potential to comprehensively profile protein O-GlcNAcylation. The stoichiometric nature of the chemoenzymatic labelling, CuAAC reaction, and elution steps provides an ideal platform for future quantitative MS analyses to profile global O-GlcNAcylation and will enable the discovery of novel functional roles for O-GlcNAcylation in diverse biological settings.Acknowledgements
We thank Dr. M. Shahgholi for assistance with peptide analysis by LC-MS. This research was supported by the National Institutes of Health (R01-GM084724), the National Science Foundation (GRFP DGE-1144469, M. E. G.), and the Department of Defense (NDSEG, E. H. J.).Notes and references
- K. Sakabe, Z. Wang and G. W. Hart, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19915–19920 CrossRef CAS PubMed.
- J. E. Rexach, P. M. Clark, D. E. Mason, R. L. Neve, E. C. Peters and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2012, 8, 253–261 CrossRef CAS PubMed.
- Y. Zhu, T.-W. Liu, S. Cecioni, R. Eskandari, W. F. Zandberg and D. J. Vocadlo, Nat. Chem. Biol., 2015, 11, 319–325 CrossRef CAS PubMed.
- W. H. Yang, J. E. Kim, H. W. Nam, J. W. Ju, H. S. Kim, Y. S. Kim and J. W. Cho, Nat. Cell Biol., 2006, 8, 1074–1083 CrossRef CAS PubMed.
- H.-B. Ruan, X. Han, M.-D. Li, J. P. Singh, K. Qian, S. Azarhoush, L. Zhao, A. M. Bennett, V. T. Samuel, J. Wu, J. R. Yates III and X. Yang, Cell Metab., 2012, 16, 226–237 CrossRef CAS PubMed.
- W. Yi, P. M. Clark, D. E. Mason, M. C. Keenan, C. Hill, W. A. Goddard III, E. C. Peters, E. M. Driggers and L. C. Hsieh-Wilson, Science, 2012, 337, 975–980 CrossRef CAS PubMed.
- J. R. Erickson, L. Pereira, L. Wang, G. Han, A. Ferguson, K. Dao, R. J. Copeland, F. Despa, G. W. Hart, C. M. Ripplinger and D. M. Bers, Nature, 2013, 502, 372–376 CrossRef CAS PubMed.
- S. A. Yuzwa, X. Shan, M. S. Macauley, T. Clark, Y. Skorobogatko, K. Vosseller and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 393–399 CrossRef CAS PubMed.
- J. Ma and G. W. Hart, Clin. Proteomics, 2014, 11, 8 CrossRef PubMed.
- N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13132–13137 CrossRef CAS PubMed.
- P. M. Clark, J. F. Dweck, D. E. Mason, C. R. Hart, S. B. Buck, E. C. Peters, B. J. Agnew and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 11576–11577 CrossRef CAS PubMed.
- N. Khidekel, S. B. Ficarro, P. M. Clark, M. C. Bryan, D. L. Swaney, J. E. Rexach, Y. E. Sun, J. J. Coon, E. C. Peters and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2007, 3, 339–348 CrossRef CAS PubMed.
- B. W. Zaro, Y.-Y. Yang, H. C. Hang and M. R. Pratt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8146–8151 CrossRef CAS PubMed.
- J. C. Trinidad, D. T. Barkan, B. F. Gulledge, A. Thalhammer, A. Sali, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2012, 11, 215–229 Search PubMed.
- J. F. Alfaro, C.-X. Gong, M. E. Monroe, J. T. Aldrich, T. R. W. Clauss, S. O. Purvine, Z. Wang, D. G. Camp II, J. Shabanowitz, P. Stanley, G. W. Hart, D. F. Hunt, F. Yang and R. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7280–7285 CrossRef CAS PubMed.
- Z. Wang, N. D. Udeshi, M. O'Malley, J. Shabanowitz, D. F. Hunt and G. W. Hart, Mol. Cell. Proteomics, 2010, 9, 153–160 CAS.
- N. Khidekel, S. Arndt, N. Lamarre-Vincent, A. Lippert, K. G. Poulin-Kerstien, B. Ramakrishnan, P. K. Qasba and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2003, 125, 16162–16163 CrossRef CAS PubMed.
- B. Ramakrishnan and P. K. Qasba, J. Biol. Chem., 2002, 277, 20833–20839 CrossRef CAS PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- J. Szychowski, A. Mahdavi, J. J. L. Hodas, J. D. Bagert, J. T. Ngo, P. Landgraf, D. C. Dieterich, E. M. Schuman and D. A. Tirrell, J. Am. Chem. Soc., 2010, 132, 18351–18360 CrossRef CAS PubMed.
- C. M. Woo, A. T. Iavarone, D. R. Spiciarich, K. K. Palaniappan and C. R. Bertozzi, Nat. Methods, 2015, 12, 561–567 CrossRef CAS PubMed.
- S. A. Myers, S. Daou, E. B. Affar and A. L. Burlingame, Proteomics, 2013, 982–991 CrossRef CAS PubMed.
- B. W. Bycroft, W. C. Chan, S. R. Chhabra and N. D. Hone, J. Chem. Soc., Chem. Commun., 1993, 778–779 RSC.
- S. R. Chhabra, H. Parekh, A. N. Khan, B. W. Bycroft and B. Kellam, Tetrahedron Lett., 2001, 42, 2189–2192 CrossRef CAS.
- Y. Yang and S. H. L. Verhelst, Chem. Commun., 2013, 49, 5366–5368 RSC.
- R. J. Chalkley and A. L. Burlingame, J. Am. Soc. Mass Spectrom., 2001, 12, 1106–1113 CrossRef CAS PubMed.
- J. A. Hanover, S. Yu, W. B. Lubas, S.-H. Shin, M. Ragano-Caracciola, J. Kochran and D. C. Love, Arch. Biochem. Biophys., 2003, 409, 287–297 CrossRef CAS PubMed.
- B. D. Lazarus, D. C. Love and J. A. Hanover, Glycobiology, 2006, 16, 415–421 CrossRef CAS PubMed.
- E. P. Roquemore, A. Dell, H. R. Morris, M. Panico, A. J. Reason, L. A. Savoy, G. J. Wistow, J. S. Zigler Jr., B. J. Earles and G. W. Hart, J. Biol. Chem., 1992, 267, 555–563 CAS.
- H.-C. Tai, N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2004, 126, 10500–10501 CrossRef CAS PubMed.
- Q. Chen, Y. Chen, C. Bian, R. Fujiki and X. Yu, Nature, 2012, 493, 561–564 CrossRef PubMed.
- N. Zeytuni and R. Zarivach, Structure, 2012, 20, 397–405 CrossRef CAS PubMed.
- S. P. N. Iyer and G. W. Hart, J. Biol. Chem., 2003, 278, 24608–24616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6mb00138f |
| This journal is © The Royal Society of Chemistry 2016 |