
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis†
Zheng
Zhao
ab,
Yang
Yang
b,
Yong
Zeng‡
*bc and
Mei
He‡
*ad
aDepartment of Biological and Agricultural Engineering, Kansas State University, Manhattan, KS 66506, USA. E-mail: meih@ksu.edu
bDepartment of Chemistry, University of Kansas, Lawrence, KS 66045, USA. E-mail: yongz@ku.edu
cUniversity of Kansas Cancer Center, Kansas City, KS 66160, USA
dTerry C. Johnson Cancer Research Center, Kansas State University, Manhattan, KS 66506, USA
First published on 20th November 2015
Abstract
Tumor-derived circulating exosomes, enriched with a group of tumor antigens, have been recognized as a promising biomarker source for cancer diagnosis via a less invasive procedure. Quantitatively pinpointing exosome tumor markers is appealing, yet challenging. In this study, we developed a simple microfluidic approach (ExoSearch) which provides enriched preparation of blood plasma exosomes for in situ, multiplexed detection using immunomagnetic beads. The ExoSearch chip offers a robust, continuous-flow design for quantitative isolation and release of blood plasma exosomes in a wide range of preparation volumes (10 μL to 10 mL). We employed the ExoSearch chip for blood-based diagnosis of ovarian cancer by multiplexed measurement of three exosomal tumor markers (CA-125, EpCAM, CD24) using a training set of ovarian cancer patient plasma, which showed significant diagnostic power (a.u.c. = 1.0, p = 0.001) and was comparable with the standard Bradford assay. This work provides an essentially needed platform for utilization of exosomes in clinical cancer diagnosis, as well as fundamental exosome research.
1. Introduction
Extracellular vesicles, particularly exosomes, have become essential for intercellular communications involved in many pathophysiological conditions, such as cancer progression and metastasis.1–6 Exosomes are a distinct population of small microvesicles (50–150 nm) that are released from multivesicular bodies (MVBs) through an endolysosomal pathway, as opposed to other subcellular membrane derived vesicles.7,8 Studies have shown that exosomes are abundant in cancer patient blood.9–11 Probing of tumor-derived circulating exosomes has been emerging to better aid in non-invasive cancer diagnosis and monitoring of treatment response.12 However, exosome biogenesis at the molecular level is still not well understood, as well as clinical utilization of exosomes lags, due to current technical challenges in rapid isolation and molecular identification of exosomes.8,13The most common procedure for purifying exosomes involves a series of high-speed ultracentrifugation steps in order to remove cell debris and pellet exosomes. However, this procedure does not discriminate exosomes from other vesicular structures or large protein aggregates.14–16 Moreover, the isolation protocols are extremely tedious, time-consuming (>10 h), and inefficient especially for blood samples, making clinical application difficult.17–20 Although physical size is employed to define exosomes, this property has not completely distinguished exosomes as a specific population apart from other vesicles that originate from different cellular locations, such as apoptotic vesicles, exosome-like vesicles, membrane particles, and ectosomes.5 Exosomes carry a group of specific proteins, RNAs, and mitochondrial DNA, that represents their cells of origin.21,22 The molecular signature of exosomes is essential for defining exosome populations and origins.23,24 However, conventional flow cytometry for molecular marker identification is limited by detectable size (>200 nm), thereby excluding the majority of exosomes.25 Standard benchtop ultracentrifugation, western blotting and enzyme-linked immunosorbent assay (ELISA) require lengthy processes and large amounts of purified, concentrated exosomes from blood (~2 mL) or cell culture media (~300 mL).15,26
Herein, we developed a simple and robust microfluidic continuous-flow platform (ExoSearch chip) for rapid exosome isolation streamlined with in situ, multiplexed detection of exosomes. Several microfluidic approaches have been previously developed for exosome study,27,28 such as isolation,29,30 quantification,31,32 and molecular profiling.33–35 However, these platforms require either complicated fabrication or sophisticated sensing methods. We previously developed a microfluidic system for integrated exosome lysis and detection of intravesicular protein markers that exosomes carry.28 However, on-chip isolation and enrichment of exosomes streamlined with multiplexed detection of marker combinations have not been established yet. In addition, the previous approach involves off-chip exosome capture using a small amount of magnetic beads and thus lacks the ability to prepare large-scale enriched exosomes for variable downstream molecular characterizations. Therefore, we developed the ExoSearch chip which combines on-chip continuous-flow mixing and immunomagnetic isolation with an in situ, multiplexed exosome immunoassay. Compared to other existing microfluidic methods, the ExoSearch chip possesses distinct features: first, continuous-flow operation affords dynamic scalability in processing sample volumes from microliter for on-chip analysis to millilitre preparation for variable downstream measurements; second, it enables multiplexed quantification of marker combinations in one sample with much improved speed (~40 min); lastly, because of simplicity, cost-effectiveness and robustness, the ExoSearch chip holds the potential to be developed into a viable technology in point-of-care and clinical settings. The one-step exosome assay enabled by the ExoSearch chip has been applied for ovarian cancer diagnosis via quantifying a panel of tumor markers from exosomes in a small-volume of blood plasma (20 μL), which showed significant diagnostic accuracy and was comparable with the standard Bradford assay.
2. Experimental
2.1 ExoSearch chip fabrication and operation
The microfluidic chip was fabricated using a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PDMS base and curing agent over a master wafer, and then bound with a microscope glass slide. The master was the pattern of SU8 photoresist on a 4-inch silicon wafer and was silanized to facilitate generation of many replicas as needed. A 2 mm magnet disk was molded into a PDMS layer during the curing process at the desired location; the magnet is removable for switching off magnetic force. A surface treatment for the PDMS chip was applied to avoid non-specific adsorption and generation of bubbles in the microchannel, using a blocking buffer (2.5 w/w% BSA and 0.01 w/w% Tween-20 in 1× PBS) with 30 min flushing at a flow rate of 1 μL min−1. A programmable syringe pump (picoliter precision) with two 20 μL micro-syringes was used to provide the optimized flow rate for continuous, on-line mixing of the plasma sample and immunomagnetic beads. The magnetic beads (2.8 μm, 0.1 mg mL−1) were conjugated with capture antibodies for isolating intact plasma exosomes. A washing buffer (1 w/w% BSA in 1× PBS) was applied for 5 min after exosome capturing. A mixture of three probing antibodies (anti CA-125/A488, anti EpCAM/A550, anti CD24/A633) labeled with distinct fluorescent dyes was introduced afterwards for 10 min incubation at a slow flow rate of 100 nL min−1, followed by 5 min washing. The non-specific adsorption, specificity of probing antibodies, and incubation were well characterized in the ESI.†
:1 mixture of PDMS base and curing agent over a master wafer, and then bound with a microscope glass slide. The master was the pattern of SU8 photoresist on a 4-inch silicon wafer and was silanized to facilitate generation of many replicas as needed. A 2 mm magnet disk was molded into a PDMS layer during the curing process at the desired location; the magnet is removable for switching off magnetic force. A surface treatment for the PDMS chip was applied to avoid non-specific adsorption and generation of bubbles in the microchannel, using a blocking buffer (2.5 w/w% BSA and 0.01 w/w% Tween-20 in 1× PBS) with 30 min flushing at a flow rate of 1 μL min−1. A programmable syringe pump (picoliter precision) with two 20 μL micro-syringes was used to provide the optimized flow rate for continuous, on-line mixing of the plasma sample and immunomagnetic beads. The magnetic beads (2.8 μm, 0.1 mg mL−1) were conjugated with capture antibodies for isolating intact plasma exosomes. A washing buffer (1 w/w% BSA in 1× PBS) was applied for 5 min after exosome capturing. A mixture of three probing antibodies (anti CA-125/A488, anti EpCAM/A550, anti CD24/A633) labeled with distinct fluorescent dyes was introduced afterwards for 10 min incubation at a slow flow rate of 100 nL min−1, followed by 5 min washing. The non-specific adsorption, specificity of probing antibodies, and incubation were well characterized in the ESI.†
For comparison with standard benchtop approaches, differential centrifugations were carried out on the collected fresh frozen blood plasma (2 mL) to obtain exosomes. The amount of protein recovered from pellets was measured by the Bradford assay (BioRad). The exosome vesicles were conserved at −80 °C until use. Nanoparticle tracking analysis (NTA) was performed using NanoSight V2.3 following the standard protocols. By monitoring the trajectory of microvesicle movement, the particle numbers within the size range of 0–500 nm were estimated in serial dilutions. The concentrations were calibrated back to the human plasma concentration. For consistent reading, the measurement settings were optimized and five replicas were performed to obtain the average measurements. Transmission electron microscopy and image analysis were performed for characterizing the morphology and size of the exosomes captured on the bead surface. The agar and resin embedding protocols were employed to ensure that the exosome morphology was maximally maintained under TEM imaging. Ultra-thin sections (80 nm) were cut on a Leica Ultracut-S Ultramicrotome and viewed after counterstaining in a JEOL JEM-1400 Transmission Electron Microscope operating at 80 kV. Micrographs were prepared to a known scale, and exosome sizes were measured and calculated using TEM imaging software with a ruler function at 20 K magnification and normalized to the scale bar.
2.2 Data collection and analysis
Fluorescence images were collected by an inverted epifluorescence microscope with a 20× (N.A. = 0.35) Zeiss objective lens and a scientific CMOS camera (OptiMOS, QImaging). The camera exposure time was set to 2000 ms with a 10 MHz frequency controlled by the open source software Micro-Manager 1.4. The filter sets of FITC, Rhodamine and Cy-5 were used for multiplexed three-color fluorescence detection with a LED light source for excitation. Fluorescence image analysis was performed using ImageJ with an in-house written Macro to determine 1000 points randomly across consistent regions of bead aggregates for obtaining averaged fluorescence intensity. Two fluorescence images were collected right before and after antibody detection in three fluorescence channels respectively, for calculating the difference of fluorescence signals. The measured fluorescence signal was then normalized to the background.3. Results & discussion
3.1 Working principle of the ExoSearch chip
Exosomes contain a variety of surface markers originating from their host cells.36,37 Selective isolation and specific analysis of disease-responsive exosome subpopulations is essential to evaluate the clinical relevance of circulating exosomes.25,38–40 To this end, the ExoSearch chip is designed to specifically isolate exosome subpopulations and simultaneously measure a panel of tumor markers for better defining disease, compared to single-marker detection. As shown in Fig. 1a, the ExoSearch chip consists of a Y-shaped injector, a serpentine fluidic mixer for bead-based exosome capture (~25.5 cm in length), and a microchamber (4 mm in diameter) with a replaceable magnet for collection and detection of exosomes. The microchannel is 300 μm wide and 50 μm deep. Such a microfluidic geometry was adapted from our previous studies on on-chip mixing and magnetic bead capture.28 The operation was simply driven by a programmable microsyringe pump with picoliter resolution. Briefly, a plasma sample and immunomagnetic beads were introduced at the same flow rate from the injection channels (Fig. 1b) through the long serpentine channel where they are uniformly mixed to facilitate exosome binding with the beads (Fig. 1c). No significant aggregation of beads by interactions with exosomes or other plasma components was observed during flow mixing at the bead concentrations and flow rates used here (Fig. 1b and c). Magnetic beads with bound exosomes can be retained as a tight aggregate in the downstream microchamber by magnetic force (Fig. 1d). The amount of beads retained in the chamber was found to be proportional to the injection volume, allowing for quantitative isolation and detection of exosomes.28 A mixture of antibodies labeled with unique fluorescent dyes was injected into the chamber to stain the exosomes for multi-color fluorescence imaging. Total analysis is completed with as low as 20 μL plasma samples in ~40 min. Alternatively, the beads can be released by removing the magnet and collected off the chip to yield purified and enriched exosome samples for variable benchtop measurements, such as morphological studies by transmission electron microscopy (TEM, Fig. 1e and Fig. S1†). While a 20 μL sample volume was used throughout this study, the smallest sample volume that can be reliably handled was estimated to be 10 μL, given the dead volume of the system (i.e., syringes, tubing and the chip). Our previous results showed that the magnetic bead aggregate formed in the chamber increased linearly in size by a factor of 8 with a 50-fold increase in the total injected bead number.28 Note that ~106 beads formed an aggregate of ~1 mm size. Based on the chamber size (4 mm in diameter) and the bead concentration used (~106 per mL), it is reasonable to estimate that this device can readily process 10 mL of plasma in a single continuous run. The processing capacity can be increased by operating in a repetitive capture-and-release manner (Fig. S1†). The single-channel device is readily scaled up to multi-channel systems for high-throughput exosome immuno-isolation and analysis.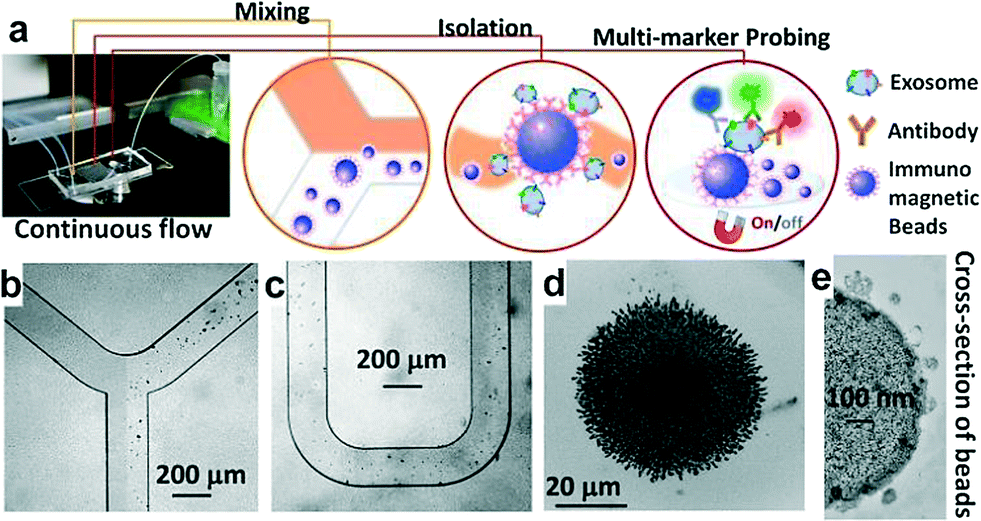 | ||
| Fig. 1 (a) Workflow of the ExoSearch chip for continuous mixing, isolation and in situ, multiplexed detection of circulating exosomes. (b)–(c) Bright-field microscope images of immunomagnetic beads manipulated in the microfluidic channel for mixing and isolation of exosomes. (d) Exosome-bound immunomagnetic beads aggregated in the microchamber with an on/off switchable magnet for continuous collection and release of exosomes. (e) TEM image of an exosome-bound immunomagnetic bead in a cross-sectional view. | ||
3.2 Characterization of microfluidic continuous-flow mixing for exosome isolation
We systematically characterized the on-chip mixing behaviour of particles in various sizes for efficient exosome isolation. First, fluorescently labeled nanoparticles (50 nm) and micro-sized magnetic beads (2.8 μm) were flowed through the ExoSearch chip, respectively, in order to mimic the mixing process for exosome isolation (Fig. 2a). In both cases, two streams were well mixed passively by the serpentine channel, showing uniform distribution of particles across the channel width. Mixing of fluorescently labelled exosomes with antibody-conjugated microbeads was then studied. We observed uniform distributions of both exosome stream and microbeads that emitted bright fluorescence due to the binding of exosomes on the bead surface (Fig. 2a). The microbeads were dominant for effective mixing which leads to much faster mixing. The minimum flow travel distance required for complete mixing in the microchannel was measured for each case, which exhibited a linear semi-log response to the flow rates applied (50 to 104 nL min−1) (Fig. 2b). Higher mixing efficiency was observed at relatively low flow rates for all three cases. Low-Reynolds-number conditions allow the exosomes and magnetic-bead suspension to flow side by side. Thus, complete mixing is critical and determines the effective residence time (incubation time), which in turn determines the effective capture. In the serpentine microchannel, mixing is promoted by the Dean flow and inertial lift.41 For larger particles, the lift force increases rapidly and positions the particles across the channel.42 Therefore, the micro-sized magnetic beads showed faster mixing, compared to the smaller exosomes and nanoparticles (Fig. 2a & b). In addition, in such a mixing system, the shear stress is low and particularly suitable for isolating and collecting intact exosomes.41,42 For all flow rates we studied (50 to 104 nL min−1), effective mixing was completely achieved, which can significantly reduce the incubation time for efficient immunomagnetic capture of exosomes.43 We also investigated the exosome capture efficiency by comparing the fluorescence intensity of flows at the inlet and outlet of the capture chamber. The capture efficiency of 42–97.3% was achieved at flow rates from 50 to 104 nL min−1 (Fig. 2c). Subsequent studies were performed at the flow rate of 1 μL min−1 which result in a fairly good capture efficiency of 72%. This flow speed allows exosome isolation from a 20 μL plasma sample in 20 min. For preparing enriched exosomes from large-volume samples, the throughput can be increased by using a relatively faster flow rate or expanding the single-channel device to a multi-channel system. For instance, a 2 mL blood plasma can be processed within 3 hours (10 μL min−1) without the need of manual intervention, which is at least 3 times faster than standard ultracentrifugation for processing the same amount of plasma with only 25% exosome recovery rate.44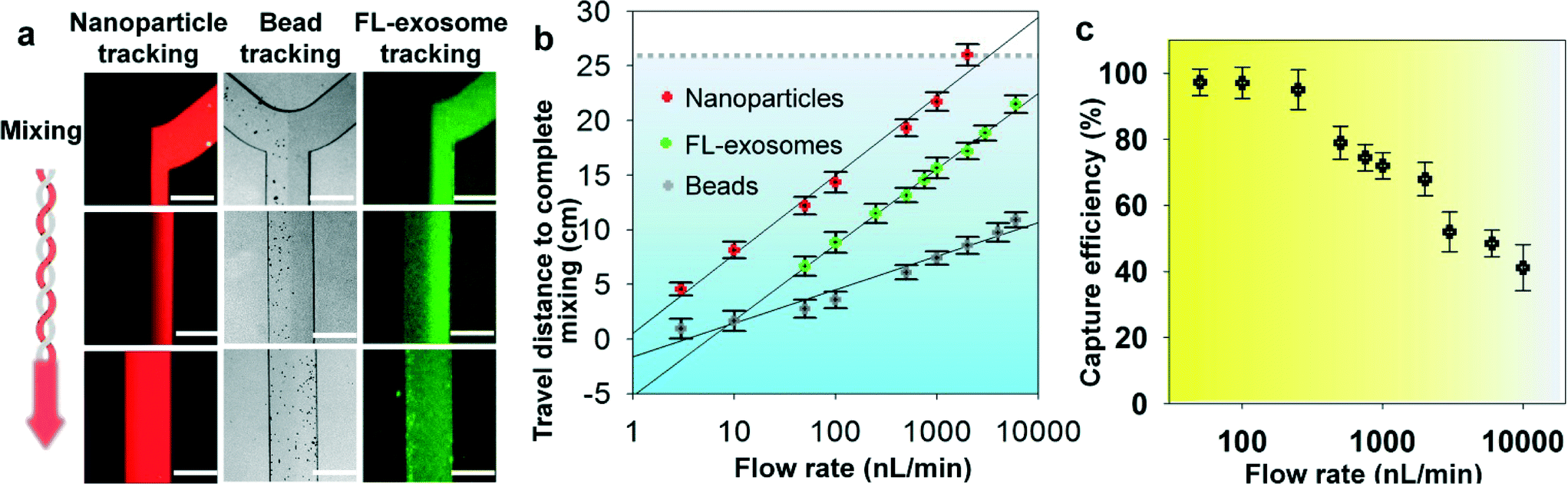 | ||
| Fig. 2 Microfluidic continuous-flow mixing for efficient exosome isolation. (a) Two-stream particle mixing in the microchannel. Left: Fluorescence CCD images of the mixing process for a stream of Texas Red labeled nanoparticles (50 nm) co-flowed with a bead solution. Middle: Immunomagnetic beads (2.8 μm) tracked under bright field for mixing with human blood plasma. Right: Mixing of fluorescently labeled exosomes with antibody beads. Exosomes were purified from ovarian cancer patient plasma by ultracentrifugation. Scale bars: 300 μm. (b) Plots of the minimum travel length required for uniform mixing over a flow rate range. The grey dashed line indicates the mixing channel length in the ExoSearch chip. (c) Exosome capture efficiency as a function of the mixing flow rate measured using purified, fluorescently labeled exosomes and capture beads. RSD is ∼5% from replicate measurements. | ||
3.3 Specificity for isolating tumor-derived exosomes
Recent studies have suggested that both tumor cells and normal cells secrete exosomes, although significantly higher amounts of exosomes have been observed from tumor cells.45 Therefore, specifically isolating, purifying and characterizing tumor cell derived exosomes are essential.46 We characterized the specificity for on-chip immunomagnetic isolation of exosomes from ovarian cancer patient blood plasma. On-chip isolation of variable exosome subpopulations was conducted by targeting both ovarian tumor-associated markers (EpCAM and CA-125) and common exosomal markers (CD9, CD81, and CD63). EpCAM is a cargo protein in exosomes and is highly overexpressed in multiple types of carcinomas, including ovarian tumor. CA-125 antigen is the most commonly measured biomarker for epithelial ovarian tumors, which accounts for 85–90% of ovarian cancer. The exosome-bound beads were washed on the chip and then released and concentrated for morphology evaluation and counting of intact exosomes using TEM, as presented in Fig. 3a. Significantly higher amounts of round membrane vesicles (smaller than 150 nm) were observed for EpCAM+, CA-125+, and CD9+ subpopulations from ovarian cancer plasma, compared to healthy controls. Negative control beads with IgG conjugation showed negative capture of vesicles, demonstrating a good specificity of immunomagnetic isolation. The relative expression levels of the five surface markers were measured by counting the number of intact exosomes bound to the beads (n = 25). The results showed a ~3–5 fold increase in expression levels of the five markers from the ovarian cancer patient, compared to the healthy control (Fig. 3b, p = 0.001). | ||
| Fig. 3 Microfluidic ExoSearch chip for specific isolation of ovarian cancer plasma derived exosomes. (a) TEM images of on-chip immunomagnetically isolated exosomes from ovarian cancer plasma, compared to healthy control. Scale bar is 100 nm. IgG-conjugated immunomagnetic beads were negative control beads. (b) Exosome counts analyzed from surfaces of variable capture beads (EpCAM+, CA-125+, CD9+, CD81+, CD63+) using TEM particle analysis (n = 25, CV = 2.8–10%). Single bead diameter was 2.8 μm and sliced bead layer was 80 nm thick. (c) Size distribution of on-chip isolated exosomes (CD9+) using TEM particle analysis, compared to standard NTA analysis of ultracentrifugation-purified exosomes. Dashed lines were log-normal fit (R2 > 0.98). | ||
To verify the results of on-chip isolation, we conducted nanoparticle tracking analysis (NTA) of ultracentrifugation-isolated exosomes to measure their size distribution and concentrations. In Fig. 3c, on-chip isolated exosomes (CD9+) exhibited a notably narrower range with the log-normal fitted size distribution (R2 > 0.98). The smaller size than 150 nm is a commonly used criterion to differentiate exosomes from larger microvesicles.5 Compared to ultracentrifugation approaches, microfluidic immunoaffinity isolation yields a higher percentage of vesicles smaller than 150 nm (~79.7% vs. 60.7%), suggesting that the developed ExoSearch chip offers high specificity in isolation of circulating exosomes.
3.4 Quantitative and multiplexed exosomal marker detection
We first characterized the ExoSearch chip for quantitative isolation and detection of exosomes. Fig. 4a shows the fluorescence images of exosomes isolated from serial dilutions of purified, fluorescently labeled plasma exosomes. The concentrations of purified plasma exosomes were determined by NTA measurements. Employing the same mixing and isolating conditions, increased fluorescence signals (ΔFL) were observed and found to be proportional to exosome concentrations. Using fluorescently labeled anti-EpCAM as the detection antibody, exosome titration curves were obtained for a healthy plasma sample and ovarian cancer plasma, which exhibited good linear response as seen in Fig. 4b (R2 > 0.98, CV = ~5%). The small variation of measurements indicates the good robustness of the method. Moreover, a much higher ΔFL signal (~30-fold increase) was observed for the ovarian cancer sample, compared to the healthy control under the same concentration. These results demonstrated the ability of the ExoSearch chip to quantitatively measure exosome surface markers for differentiating changes associated with disease. The results were consistent with other recent reports that EpCAM is highly overexpressed in ovarian tumor exosomes.47 The quantitative detection of intact exosomes was achieved with a limit of detection of 7.5 × 105 particles per mL (LOD, S/N = 3), which is 1000-fold sensitive than Western blotting.34 While such sensitivity is comparable with that of the previously reported microfluidic method,34 our method features simple fabrication, easy operation and low cost.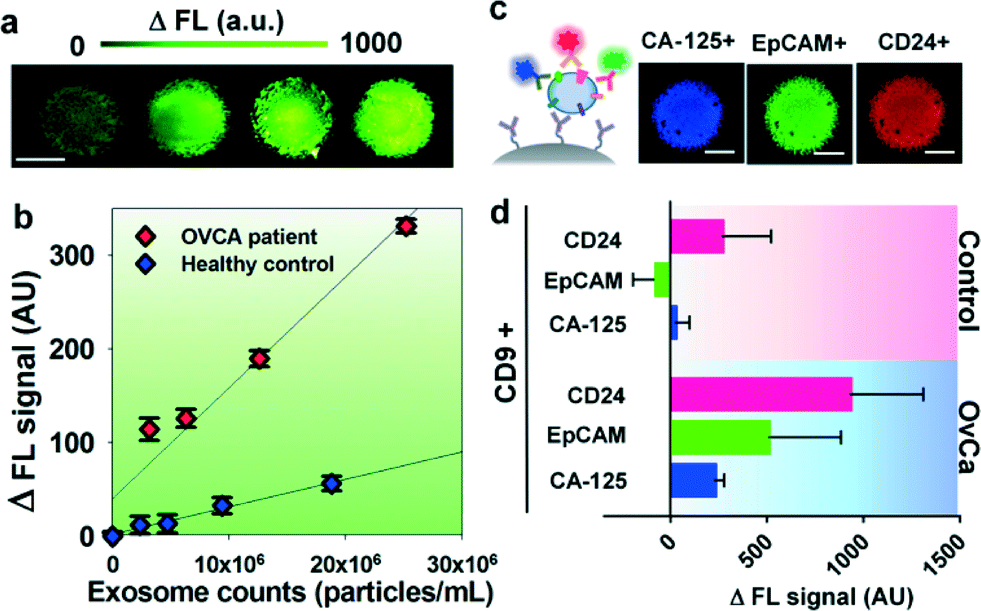 | ||
| Fig. 4 (a) CCD images of bead aggregates in the ExoSearch chip captured with fluorescence-labeled plasma exosomes in serial dilutions (from left to right: 5 × 105, 1 × 106, 5 × 106, 1 × 107 particles per mL). Scale bar was 100 μm. (b) Calibration curves for quantitative detection of intact exosomes (R2 > 0.98, CV = ~5%). Exosomes were purified from one healthy control plasma and one ovarian cancer patient plasma using ultracentrifugation. Concentrations were measured by NTA. (c) CCD images of multiplexed three-color fluorescence detection of tumor markers (CA-125, EpCAM, CD24) from captured exosome subpopulation (CD9+). Scale bar was 50 μm, indicating the bead aggregate size. (d) Average expression levels of three ovarian tumor markers measured by the ExoSearch chip from 20 human subjects (nOvCa = 15, nhealthy = 5). Error bars indicate standard deviations. | ||
In situ, multiplexed biomarker detection was then developed for rapid and quantitative microfluidic analysis of ovarian tumor derived plasma exosomes. We chose the common exosome marker CD9 as the capture antibody for selective isolation of exosomes, because of the consistently high expression of CD9 we observed from human plasma derived exosomes (Fig. S3†). In addition to the established ovarian cancer biomarker CA-125, human epididymis protein 4 (HE4) has been recognized for improving the diagnostic specificity of CA-125 in pathological tests. We did not observe substantial expression of HE4 from the exosome samples (Fig. S2†), which could be due to the different secretion pathway of HE4.22 This observation was consistent with other recent reports.34,48 Previous observations have indicated that CD24 could be a significant marker in ovarian tumor prognosis and diagnosis.49 Therefore, we developed a multiplexed sandwich immunofluorescence assay to quantify isolated exosomes by targeting three markers, CA-125, EpCAM, and CD24 from the same population of exosomes, as exemplified in Fig. 4c. Quantitative tests of raw human plasma collected from 20 subjects (nOvCa = 15, nhealthy = 5) were conducted for three-marker classification of ovarian tumor derived exosomes, and a distinctive three-marker expression pattern was observed for ovarian cancer patients (Fig. 4d). The average expression levels of individual exosomal markers from ovarian cancer patients were statistically higher as compared to healthy controls (CD24: 3-fold increase, p = 0.003; EpCAM: 6.5-fold increase, p = 0.0009; CA-125: 12.4-fold increase, p < 0.0001).
Non-specific adsorption of exosomes and antibody cross-reactivity were characterized in Fig. S2.† The negative and positive control experiments were designed and conducted in parallel for testing the four antibodies we used in this study (CA-125, EpCAM, CD24, and HE4). Slight auto-fluorescence of capture beads and negligible non-specific adsorption fluorescence were observed, and no cross-reaction was observed between antibodies. The positive control (ovarian cancer patient plasma exosomes) showed strong fluorescence signals after antibody probing (CA-125, EpCAM, and CD24). However, we did not observe an acceptable positive response from HE4 antibody probing, as HE4 is not expressed on the exosome surface which demonstrates the negligible non-specific adsorption onto captured exosomes (Fig. S2†). In addition, Fig. 4d shows low signal intensities for these three markers when their expression levels are low in healthy exosomes. This result also indicates negligible non-specific interference from non-specific antibody adsorption or cross-reactivity.
3.5 ExoSearch chip for blood-based ovarian cancer diagnosis
Currently, there is no single marker that can detect early-stage ovarian cancer with desired sensitivity and specificity (>98%).50 A large number of combinations of biomarkers have been investigated to improve diagnostic sensitivity and specificity.51 Circulating exosomes, enriched with a group of tumor antigens, provide a unique opportunity for cancer diagnosis using multi-marker combination. To this end, we employed the ExoSearch chip for blood-based diagnosis of ovarian cancer by simultaneously detecting three tumor antigens present in the same exosome subpopulation. Standard Bradford assay of total protein levels in ultracentrifugation-purified exosomes from matched human subjects was performed for parallel comparison. A total of 20 human subjects (nOvCa = 15, nhealthy = 5) were chosen for evaluating diagnostic accuracy, based on receiver operator characteristic analysis of adequate sample size (Table S1†). Both ExoSearch and Bradford assay showed significantly increased level of exosome proteins from ovarian cancer patients, compared to healthy controls (Fig. 5a, Bradford assay p = 0.001; ExoSearch chip p < 0.001). Particularly, the ExoSearch chip gave individual exosomal protein expression levels and the levels of CA-125 and EpCAM showed extremely significant differences between ovarian cancer patients and healthy controls (EpCAM, p = 0.0009; CA-125, p < 10−4). The area under the receiver operator characteristic curve (a.u.c.) represents the overall accuracy of a test (Table S2†). To determine the diagnostic accuracy of the ExoSearch chip assay, we analyzed the true positives (sensitivity) and false positives (one-specificity) using receiver operating characteristic (ROC) curves. The areas under the curves (a.u.c.) obtained for CA-125, EpCAM, and CD24 were 1.0, 1.0 and 0.91, respectively, which were comparable with the standard Bradford assay (a.u.c. = 1.0, 95% CI) (Fig. 5b and c). However, the diagnostic accuracy of using exosomal particle concentrations measured by NTA was relatively poor with an a.u.c. of only 0.67 (Fig. 5c, Fig. S4,† 95% CI). It could be attributed to the variation of NTA measurement which gives a relatively large uncertainty in size and concentration.51,52 In addition, the results were consistent with recent reports showing that counting exosomes alone was insufficient for cancer diagnosis and targeting specific exosome phenotypes could markedly improve the diagnostic accuracy.53 By ROC analysis (Table S3†), the ExoSearch chip assay was highly accurate in discriminating plasma exosomes from ovarian cancer patients versus healthy individuals. The above results suggested that the ExoSearch chip enables sensitive multiplexed exosomal marker detection for blood-based diagnosis of ovarian cancer with significant predictive power. The combination of plasma exosomal markers CA-125, EpCAM, and CD24 provided desirable diagnostic accuracy for non-invasive, early detection of ovarian cancer (Table S3†).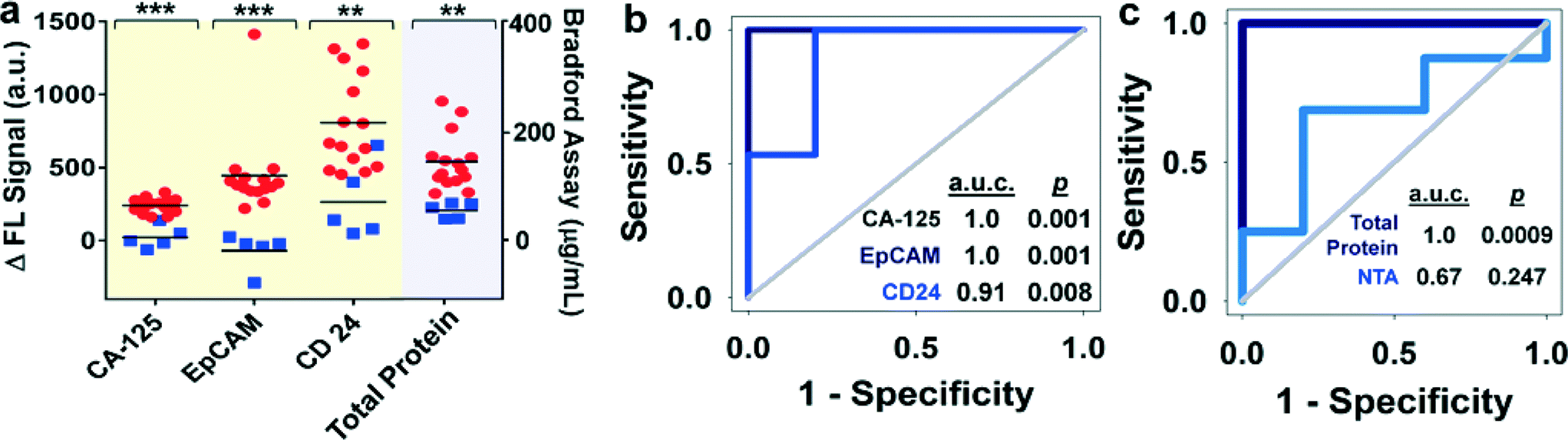 | ||
| Fig. 5 (a) Scattering plots of expression levels of three tumor markers (CA-125, p < 10−4; EpCAM, p = 0.0009; CD24, p = 0.003) from blood plasma derived exosomes (nOvCa = 15, nhealthy = 5), compared to the standard Bradford assay of total proteins (p = 0.0013) in ultracentrifugation-purified exosomes from matched human subjects. Black lines indicate the average expression levels of each group. Ovarian cancer patients were represented by red dots, and healthy controls were represented by blue dots. (b) ROC analysis of the ExoSearch chip assay for in situ, multiplexed detection of three ovarian tumor exosomal markers (CA-125 a.u.c. = 1.0, p = 0.001; EpCAM a.u.c. = 1.0, p = 0.001; CD24 a.u.c. = 0.91, p = 0.008). Confidence interval (CI) is 95%. (c) ROC analysis of standard benchtop measurements (Bradford assay of total exosome protein, and NTA of exosome concentration) of blood plasma exosomes from matched patients in Fig. 5b. | ||
4. Conclusions
Because exosomes differ 5-fold in size and 104-fold in concentration in biological samples, and can contain other membrane derived subcellular structures,8 accurate measurement of exosome concentration in biofluids is challenging. For conventional approaches, such as NTA and flow cytometry, exosome quantitation is limited primarily by minimum detectable vesicle sizes, resulting in relatively large variation (CV = ~20%).52,54 The ExoSearch chip enables simultaneous, quantitative evaluation of multiple markers from the same exosome subpopulation with much improved measurement reproducibility (CV < 10%), indicating the good robustness of this method. Such robustness is essential for precision medicine and diagnostics involving exosomes. In addition, the continuous-flow design affords capability for obtaining distinct populations of exosomes from a wide range of preparation volumes (10 μL to 10 mL), which is useful for downstream comparative molecular profiling or therapeutic use.As surrogates of tumor cells, exosomes hold great promise for precise and personalized cancer diagnosis. Combinations of exosomal protein markers may constitute a “cancer signature” and provide improved detection as the first step in multimodal screening.54 However, to our best knowledge, multiplexed assay of exosomes has not been well established yet. We demonstrated the feasibility of ExoSearch chip for non-invasive diagnosis of ovarian cancer using a combination of three exosomal tumor markers (CA-125, EpCAM, CD24), which showed comparable accuracy and diagnostic power (a.u.c. = 1.0, p = 0.001) with the standard Bradford assay (a.u.c. = 1.0, p = 0.0009). However, the ExoSearch chip requires only 20 μL of human plasma for multiplexed detection of the three tumor proteins within 40 min, as compared to ~1 mL of plasma and ~12 hours required by the Bradford assay.
To date, conventional tissue biopsy for pathological diagnosis of ovarian cancer is extremely invasive, as a difficult surgery. General imaging screenings, including tomography (CT) scans and magnetic resonance imaging (MRI) scans, are costly and unavailable in a majority of clinics. Therefore, blood-based assay for pre-screening is highly valuable and can dramatically decrease healthcare costs. The ExoSearch chip provides a cost-effective, accessible approach for specific, rapid isolation of blood diagnostic exosomes, paving the way for clinical utilization of exosomes. We will further validate the diagnostic effectiveness of the ExoSearch chip in various sample cohorts and enhance the disease discrimination power, including the use of large-scale sample size and benign tumor group as a positive control. This work, as a preliminary proof-of-concept in discovery phase, is an essential step and could serve as a basic platform for developing clinical tests in other diseases, as well as fundamental laboratory research.
Acknowledgements
We would like to acknowledge the University of Kansas Cancer Center's Biospecimen Repository Core Facility for providing the human specimens and the Microfabrication and Microfluidics Core at the KU COBRE Center for device fabrication. This study was supported by the K-INBRE Developmental Research Project Award from NIH/NIGMS (P20GM103418) and the Innovative Research Award from the Terry C. Johnson Cancer Research Center to M. H., and the J. R. and Inez Jay Award from KU, the COBRE Research Project Award under P20GM103638 (NIGMS) and the NIH/NCI grant R21CA186846 to Y. Z.References
- F. Wendler, N. Bota-Rabassedas and X. Franch-Marro, J. Extracell. Vesicles, 2013, 2 Search PubMed.
- S. A. Melo, H. Sugimoto, J. T. O'Connell, N. Kato, A. Villanueva, A. Vidal, L. Qiu, E. Vitkin, L. T. Perelman, C. A. Melo, A. Lucci, C. Ivan, G. A. Calin and R. Kalluri, Cancer Cell, 2014, 26, 707–721 CrossRef CAS PubMed.
- G. K. Alderton, Nat. Rev. Cancer, 2012, 12, 447 CrossRef PubMed.
- G. Camussi, M. C. Deregibus, S. Bruno, V. Cantaluppi and L. Biancone, Kidney Int., 2010, 78, 838–848 CrossRef CAS PubMed.
- C. Thery, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CAS.
- M. Iero, R. Valenti, V. Huber, P. Filipazzi, G. Parmiani, S. Fais and L. Rivoltini, Cell Death Differ., 2008, 15, 80–88 CrossRef CAS PubMed.
- S. A. Melo, H. Sugimoto, J. T. O'Connell, N. Kato, A. Villanueva, A. Vidal, L. Qiu, E. Vitkin, L. T. Perelman, C. A. Melo, A. Lucci, C. Ivan, G. A. Calin and R. Kalluri, Cancer Cell, 2014, 26, 707–721 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- L. Muller, C. S. Hong, D. B. Stolz, S. C. Watkins and T. L. Whiteside, J. Immunol. Methods, 2014, 411, 55–65 CrossRef CAS PubMed.
- V. S. Alikhani, A. Malmer, K. Ekstrom, A. Bossios, M. Sjostrand and J. Lotvall, Allergy, 2007, 62, 452 CrossRef PubMed.
- A. S. V. Linan, G. S. de las Heras, J. C. Tebar, J. A. G. Masia, J. I. M. de Llama, J. L. R. Martinez, A. P. Calero, M. A. Martinez, B. M. Flores, G. B. Blesa, A. A. O. Manyari, C. C. Dorado, P. C. Sanchez and P. G. Masegosa, Br. J. Surg., 2015, 102, 8 CrossRef PubMed.
- G. K. Alderton, Nat. Rev. Cancer, 2015, 15, 453 CrossRef CAS PubMed.
- F. Momen-Heravi, L. Balaj, S. Alian, P.-Y. Mantel, A. E. Halleck, A. J. Trachtenberg, C. E. Soria, S. Oquin, C. M. Bonebreak, E. Saracoglu, J. Skog and W. P. Kuo, Biol. Chem., 2013, 394, 1253–1262 CrossRef CAS PubMed.
- D. W. Greening, R. Xu, H. Ji, B. J. Tauro and R. J. Simpson, Methods Mol. Biol., 2015, 1295, 179–209 Search PubMed.
- D. D. Taylor, W. Zacharias and C. Gercel-Taylor, Methods Mol. Biol., 2011, 728, 235–246 CAS.
- D. K. Jeppesen, M. L. Hvam, B. Primdahl-Bengtson, A. T. Boysen, B. Whitehead, L. Dyrskjot, T. F. Orntoft, K. A. Howard and M. S. Ostenfeld, J. Extracell. Vesicles, 2014, 3, 25011 CrossRef PubMed.
- M. Li, A. J. Rai, G. Joel DeCastro, E. Zeringer, T. Barta, S. Magdaleno, R. Setterquist and A. V. Vlassov, Methods, 2015, 87, 26–30 CrossRef CAS.
- J. Schageman, E. Zeringer, M. Li, T. Barta, K. Lea, J. Gu, S. Magdaleno, R. Setterquist and A. V. Vlassov, BioMed Res. Int., 2013, 2013, 253957 Search PubMed.
- K. Rekker, M. Saare, A. M. Roost, A. L. Kubo, N. Zarovni, A. Chiesi, A. Salumets and M. Peters, Clin. Biochem., 2014, 47, 135–138 CrossRef CAS PubMed.
- R. J. Lobb, M. Becker, S. W. Wen, C. S. Wong, A. P. Wiegmans, A. Leimgruber and A. Moller, J. Extracell. Vesicles, 2015, 4, 27031 Search PubMed.
- P. Peng, Y. Yan and S. Keng, Oncol. Rep., 2011, 25, 749–762 CAS.
- A. Beach, H. G. Zhang, M. Z. Ratajczak and S. S. Kakar, J. Ovarian Res., 2014, 7 Search PubMed.
- S. Fontana, L. Saieva, S. Taverna and R. Alessandro, Proteomics, 2013, 13, 1581–1594 CrossRef CAS.
- J. M. Aliotta, J. Gastrointest. Oncol., 2011, 2, 203–205 Search PubMed.
- H. Kalra, C. G. Adda, M. Liem, C. S. Ang, A. Mechler, R. J. Simpson, M. D. Hulett and S. Mathivanan, Proteomics, 2013, 13, 3354–3364 CrossRef CAS PubMed.
- K. Koga, K. Matsumoto, T. Akiyoshi, M. Kubo, N. Yamanaka, A. Tasaki, H. Nakashima, M. Nakamura, S. Kuroki, M. Tanaka and M. Katano, Anticancer Res., 2005, 25, 3703–3707 CAS.
- A. Liga, A. D. B. Vliegenthart, W. Oosthuyzen, J. W. Dear and M. Kersaudy-Kerhoas, Lab Chip, 2015, 15, 2388–2394 RSC.
- M. He, J. Crow, M. Roth, Y. Zeng and A. K. Godwin, Lab Chip, 2014, 14, 3773–3780 RSC.
- R. T. Davies, J. Kim, S. C. Jang, E. J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- S. M. Santana, M. A. Antonyak, R. A. Cerione and B. J. Kirby, Biomed. Microdevices, 2014, 16, 869–877 CrossRef CAS PubMed.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC.
- L. Zhu, K. Wang, J. Cui, H. Liu, X. L. Bu, H. L. Ma, W. Z. Wang, H. Gong, C. Lausted, L. Hood, G. Yang and Z. Y. Hu, Anal. Chem., 2014, 86, 8857–8864 CrossRef CAS PubMed.
- R. Vaidyanathan, M. Naghibosadat, S. Rauf, D. Korbie, L. G. Carrascosa, M. J. A. Shiddiky and M. Trau, Anal. Chem., 2014, 86, 11125–11132 CrossRef CAS.
- H. Im, H. L. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- H. L. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS PubMed.
- S. N. Thomas, Z. Liao, D. Clark, Y. Chen, R. Samadani, L. Mao, D. K. Ann, J. E. Baulch, P. Shapiro and A. J. Yang, Proteomes, 2013, 1, 87–108 CrossRef CAS PubMed.
- H. Ji, D. W. Greening, T. W. Barnes, J. W. Lim, B. J. Tauro, A. Rai, R. Xu, C. Adda, S. Mathivanan, W. Zhao, Y. Xue, T. Xu, H. J. Zhu and R. J. Simpson, Proteomics, 2013, 13, 1672–1686 CrossRef CAS PubMed.
- R. Crescitelli, C. Lasser, T. G. Szabo, A. Kittel, M. Eldh, I. Dianzani, E. I. Buzas and J. Lotvall, J. Extracell. Vesicles, 2013, 2 Search PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ji, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56, 293–304 CrossRef CAS PubMed.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Thery, J. Extracell. Vesicles, 2012, 1 Search PubMed.
- F. Jiang, K. S. Drese, S. Hardt, M. Kupper and F. Schonfeld, AIChE J., 2004, 50, 2297–2305 CrossRef CAS.
- J. M. Martel and M. Toner, Sci. Rep., 2013, 3 Search PubMed.
- A. H. C. Ng, U. Uddayasankar and A. R. Wheeler, Anal. Bioanal. Chem., 2010, 397, 991–1007 CrossRef CAS.
- H. G. Lamparski, A. Metha-Damani, J. Y. Yao, S. Patel, D. H. Hsu, C. Ruegg and J. B. Le Pecq, J. Immunol. Methods, 2002, 270, 211–226 CrossRef CAS PubMed.
- P. Kharaziha, S. Ceder, Q. Li and T. Panaretakis, Biochim. Biophys. Acta, 1826, 2012, 103–111 Search PubMed.
- N. M. Mahaweni, M. E. H. Kaijen-Lambers, J. Dekkers, J. G. J. V. Aerts and J. P. J. J. Hegmans, J. Extracell. Vesicles, 2013, 2 Search PubMed.
- S. Runz, S. Keller, C. Rupp, A. Stoeck, Y. Issa, D. Koensgen, A. Mustea, J. Sehouli, G. Kristiansen and P. Altevogt, Gynecol. Oncol., 2007, 107, 563–571 CrossRef CAS PubMed.
- B. Liang, P. Peng, S. Chen, L. Li, M. J. Zhang, D. Y. Cao, J. X. Yang, H. X. Li, T. Gui, X. L. Li and K. Shen, J. Proteomics, 2013, 80, 171–182 CrossRef CAS PubMed.
- G. Kristiansen, C. Denkert, K. Schluns, E. Dahl, C. Pilarsky and S. Hauptmann, Am. J. Pathol., 2002, 161, 1215–1221 CrossRef CAS PubMed.
- B. M. Nolen and A. E. Lokshin, Mol. Diagn. Ther., 2013, 17, 139–146 CrossRef CAS PubMed.
- V. Sokolova, A. K. Ludwig, S. Hornung, O. Rotan, P. A. Horn, M. Epple and B. Glebel, Colloids Surf., B, 2011, 87, 146–150 CrossRef CAS PubMed.
- E. van der Pol, F. A. W. Coumans, A. E. Grootemaat, C. Gardiner, I. L. Sargent, P. Harrison, A. Sturk, T. G. van Leeuwen and R. Nieuwland, J. Thromb. Haemostasis, 2014, 12, 1182–1192 CrossRef CAS PubMed.
- S. A. Melo, L. B. Luecke, C. Kahlert, A. F. Fernandez, S. T. Gammon, J. Kaye, V. S. LeBleu, E. A. Mittendorf, J. Weitz, N. Rahbari, C. Reissfelder, C. Pilarsky, M. F. Fraga, D. Piwnica-Worms and R. Kalluri, Nature, 2015, 523, 177–U182 CrossRef CAS.
- A. Srivastava, J. Filant, K. M. Moxley, A. Sood, S. McMeekin and R. Ramesh, Curr. Gene Ther., 2015, 15, 182–192 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The ovarian cancer patient sample information, cross-reactivity and non-specific characterization, sample size justification, as well as the ROC analysis were detailed. See DOI: 10.1039/c5lc01117e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |