
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: recent advances
Felix E.
Held
and
Svetlana B.
Tsogoeva
*
Department of Chemistry and Pharmacy, Organic Chemistry Chair I and Interdisciplinary Center for Molecular Materials, University of Erlangen-Nürnberg, Henkestraße 42, 91054 Erlangen, Germany. E-mail: Svetlana.Tsogoeva@fau.de; Fax: +49 (0)9131 85 26865
First published on 7th December 2015
Abstract
High on the list of challenges in organic chemistry is the development of new efficient chiral catalysts for enantioselective cycloaddition reactions, which are among the most useful processes in chemical synthesis. In the past few decades, various highly enantioselective bifunctional organocatalysts for different versatile cycloaddition reactions have been developed. In most cases, these organocatalytic cycloadditions (e.g. [4 + 2], [3 + 2], formal [3 + 2], formal [3 + 3], formal [5 + 1], [5 + 2], 1,3-dipolar cycloadditions and Tamura cycloaddition) provide the most convenient and economical routes to nitrogen- and oxygen-containing heterocyclic bioactive molecules. This minireview summarizes the recent developments in this field using chiral bifunctional amine-thiourea and amine-squaramide organocatalysts.
 Felix E. Held | Felix E. Held was born in Fürth (Germany) in 1986. He received his Diploma degree in Chemistry from the University of Erlangen-Nuremberg in 2011. The same year he began his graduate studies under the supervision of Prof. S. B. Tsogoeva. His research interests include synthesis and application of organocatalysts for domino reactions and one-pot processes. |
 Svetlana B. Tsogoeva | Svetlana B. Tsogoeva was born in 1973, studied chemistry at St. Petersburg State University, where she completed her doctoral thesis in 1998. Then, she moved to the Johann Wolfgang Goethe-University, Frankfurt am Main, for a postdoctoral research. In July 2000 she joined the Degussa AG Fine Chemicals Division as a research scientist. In January 2002 she was appointed a first junior professor in Germany at the Georg-August-University of Göttingen. Since February 2007, she is professor of organic chemistry at the Friedrich-Alexander-University of Erlangen-Nuremberg. Her research is currently focused on organocatalytic multi-component domino reactions, asymmetric oxidations with non-heme iron complexes and synthesis of natural product hybrids with anticancer, antiviral and antimalarial activities. |
1. Introduction
The year 2000 saw the rebirth of catalysis by small organic molecules and marked the beginning of the explosive growth of the exciting field now known as organocatalysis.1,2 In particular, the rediscovery of the versatile catalytic nature of L-proline by List, Lerner and Barbas III3 occurring via enamine intermediates and the disclosure of the highly enantioselective imidazolidinone catalyzed Diels–Alder reaction by MacMillan,4 which is promoted by an iminium intermediate, have been the biggest breakthroughs in this field of research.The asymmetric (hetero) Diels–Alder reaction is the most frequently used catalytic method for the synthesis of natural products, drugs, and agrochemicals.5,6 Over the last several years considerable effort has been devoted to the development of this [4 + 2] and related cycloaddition reactions (e.g. [3 + 2], formal [3 + 2], formal [3 + 3], [5 + 2], 1,3-dipolar cycloadditions and Tamura cycloaddition) employing chiral organocatalysts.7,8
In their pioneering investigations Rawal and Yamamoto developed chiral H-bonding diols as efficient hydrogen bond donor organocatalysts for asymmetric hetero Diels–Alder reactions.9 Simultaneously, new catalyst scaffolds which possess both a H-bonding group (urea, thiourea or squaramide) and basic/nucleophilic moiety (primary, secondary or tertiary amine), and which act cooperatively, have been developed by several research groups for a broad range of enantioselective transformations. The ability of these bifunctional compounds to activate both nucleophilic and electrophilic reaction components synergistically and highly stereoselectively by basic/nucleophilic moiety and H-bonding group, respectively, is the key to the success of this type of organocatalysts. Particularly, the dual-activation by bifunctional thioureas/squaramides has been shown to be very powerful to a variety of asymmetric cycloaddition reactions. This minireview focuses on the remarkable progress since 2010 in enantioselective cycloaddition transformations using bifunctional thiourea and squaramide organocatalysts with the general structures presented in Fig. 1.
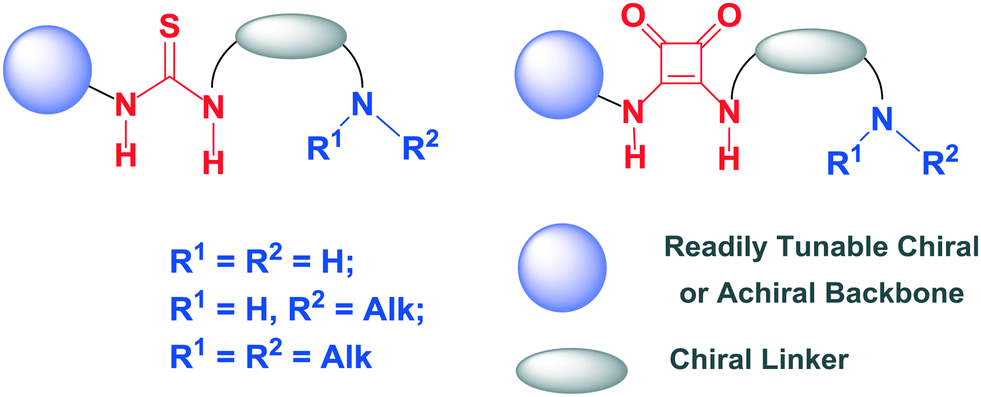 | ||
| Fig. 1 General presentation of bifunctional thiourea and squaramide organocatalysts discussed in this article. | ||
After giving a short overview of the development of the corresponding bifunctional organocatalysts, a variety of cycloaddition reactions and their formal versions ([4 + 2], [3 + 2], formal [3 + 2], formal [3 + 3], formal [5 + 1], [5 + 2], 1,3-dipolar cycloadditions and Tamura cycloaddition), catalyzed by these bifunctional compounds, will be highlighted.
2. Development of primary amine-thiourea organocatalysts and their applications to cycloaddition reactions
Thiourea derivatives, as potent hydrogen bond donors, became very attractive for organocatalyst design in recent years.10,11 Pioneering examples include the work of Curran,12 Jacobsen,13 Schreiner14 and Takemoto15 (Fig. 2).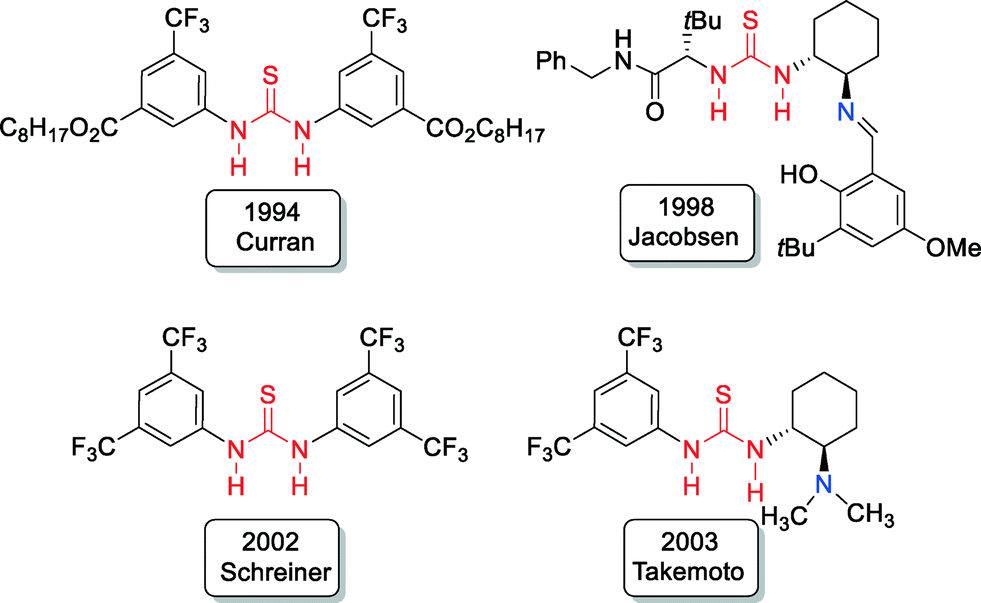 | ||
| Fig. 2 Pioneering examples of thiourea organocatalysts. | ||
Developed in 1998 by Jacobsen, bifunctional Schiff base-thioureas13 were found to be excellent asymmetric catalysts for different organic transformations.11 Chiral bifunctional organic catalysts that contain a thiourea structural unit became of growing importance in the development of asymmetric catalysis since that time.
In comparison to the pioneering investigations with bifunctional Schiff base-thioureas by Jacobsen in 199813 and tertiary amine-thioureas by Takemoto in 2003,15 the potential of primary amine-thioureas in asymmetric bifunctional organocatalysis was completely overlooked at that time, probably because of their known lower basicity and/or nucleophilicity in comparison to tertiary and secondary amines, as well as Schiff bases.
This is particularly surprising taking into consideration the fact that primary amine catalysis is of enormous importance in enzyme catalysis. For example, primary amines occur in the catalytic sites of several enzymes, such as type I aldolases, dehydratases, and decarboxylases.16 Therefore, primary amines as organocatalysts possess of particular appeal.
The investigations of bifunctional primary amine-thioureas were based on the initial studies of N-terminal primary amino dipeptides as chiral catalysts.17 In 2004 the Tsogoeva group reported the first example of an N-terminal primary amine based unmodified dipeptide being used in organocatalysis,17 which stimulated further work on primary amine based dipeptide catalysts.18–21 The bifunctional character of the dipeptide is evident (Fig. 3), since the donor can be activated via enamine formation, and the acceptor – via hydrogen bonding with the NH-group and the C-terminal carboxyl group of dipeptide. Tsogoeva and co-workers envisioned that a better hydrogen-bond donor incorporated into the N-terminal primary amino dipeptide-like structure might be advantageous for the development of more powerful primary amine-derived bifunctional catalysts. Therefore, they have chosen the thiourea moiety as a known excellent hydrogen-bond donor10,11,22,23 and designed the first chiral bifunctional organic catalysts that possess of both a thiourea moiety and a primary amine group as a base (Fig. 3 and 4), and which they applied for nitro-Michael reactions.24,25
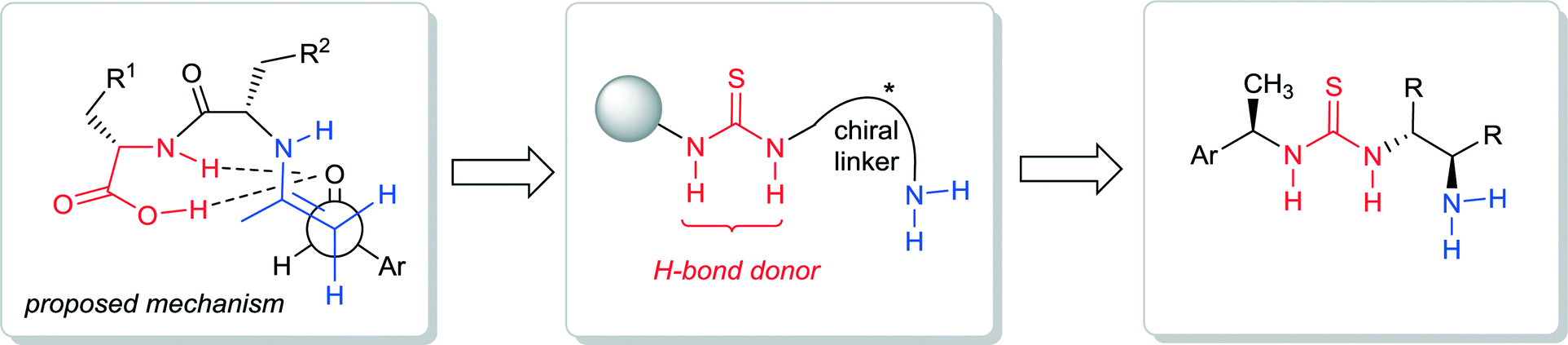 | ||
| Fig. 3 Design of primary amine-thiourea organocatalysts based on N-terminal primary amino dipeptide catalysis. | ||
 | ||
| Fig. 4 Selected examples of primary amine-thiourea organocatalysts. | ||
Meanwhile, the Jacobsen group reported another new primary amine-thiourea catalyst for the same nitro-Michael reaction.26 These findings on primary amine based organocatalysts have created new impulses to this field and also motivated several other scientists to develop new primary amine based organocatalysts. In general, over 30 new primary amine-thiourea catalysts have been developed since 2006.27 Just some selected representatives are shown in Fig. 4.
Thus, in the area of organocatalysis, chiral primary amines have emerged as new and powerful catalysts for many important organic transformations, including [4 + 2] and [5 + 2] cycloaddition reactions. The primary amine-thiourea catalyzed [4 + 2] cycloadditions represent facile methods for the generation of structurally complex spirocyclic skeletons28 and indolo- and benzoquinolizidine frameworks,29 which have a widespread occurrence in pharmaceutically relevant compounds and, hence, are of significant interest for the biological and medicinal chemistry. In particular, the spirocyclic scaffold, containing a quaternary carbon stereogenic center, is a versatile structural element, being also a crucial part of a large number of naturally occurring compounds.30
Also the primary amine-thiourea catalyzed [5 + 2] cycloaddition is a very useful reaction in organic synthesis for the construction of several complex natural product derivatives like chiral 8-oxabicyclo[3.2.1]octanes. Being on the one hand a moiety occurring in several natural compounds,31 8-oxabicyclo[3.2.1]octane architectures can additionally act as highly convenient precursors for further transformations.32
In the following we outline these useful primary amine-thiourea catalyzed [4 + 2] and [5 + 2] cycloadditions along with the discussion of the mechanistic proposals.
2.1. [4 + 2] cycloadditions
Wang and co-workers introduced in 2012 an efficient enantioselective organocatalytic synthesis of spirocyclic compounds via an inverse-electron-demand Diels–Alder reaction that is promoted by bifunctional rosin-derived primary amine-thiourea 1 in combination with a Brønsted acid additive such as acetic acid (Scheme 1).33 | ||
| Scheme 1 [4 + 2] cycloaddition reaction towards azaspirocyclic skeletons. | ||
In fact, the results showed that the asymmetric reaction was promoted by the acetic acid salt of the bifunctional primary amine-thiourea 1. Mechanistically, the LUMO energy of the N-tosyl-2-methylenebut-3-enoate, which serve as a diene, is lowered via hydrogen-bonding to the thiourea moiety, whereas the HOMO energy of cyclic keto/enolate salt is raised by the simultaneous formation of ketiminium cation and protonation by the acetic acid additive (Fig. 5).
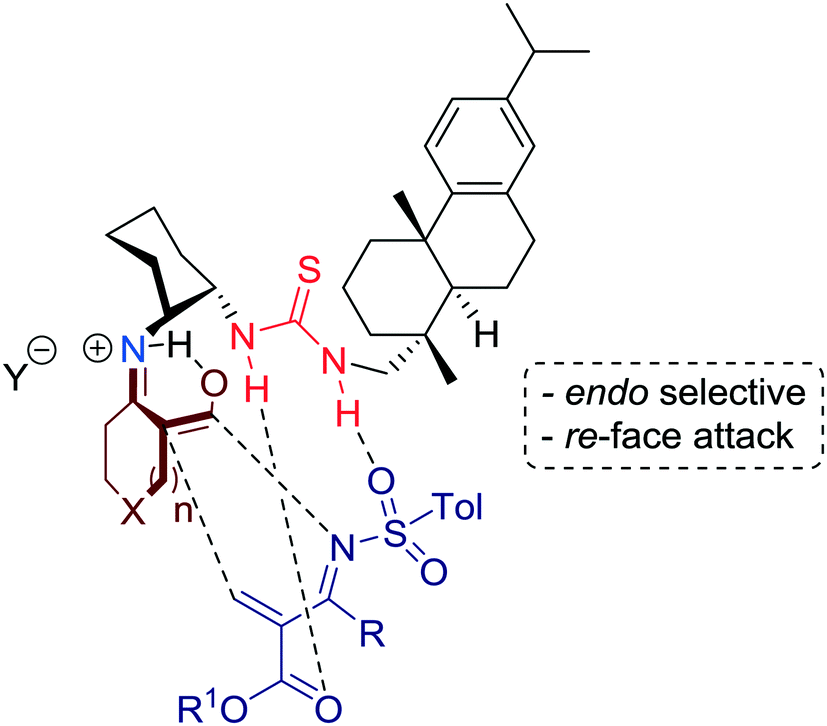 | ||
| Fig. 5 Proposed activation model for the re-face attack. | ||
In 2013, the group of Jacobson reported a sophisticated highly enantio- and diastereoselective method for generation of chiral indolo- and benzoquinolizidine frameworks through the formal aza-Diels–Alder reactions between enones and cyclic imines catalyzed by bifunctional primary amine-thiourea 2 (Scheme 2).34
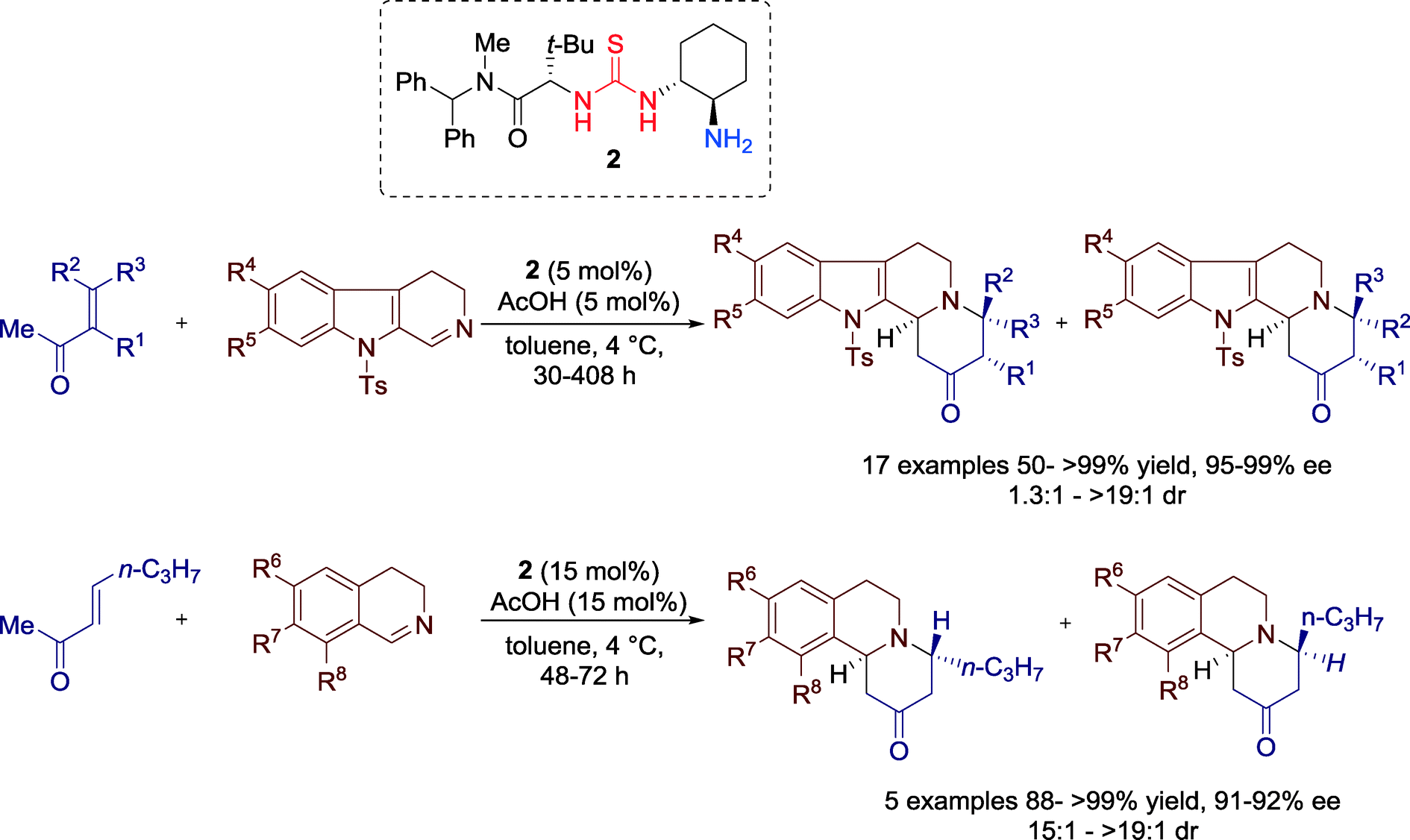 | ||
| Scheme 2 Primary amine-thiourea-catalyzed formal aza-Diels–Alder reactions. | ||
The broad scope of these reactions is demonstrated by the use of different substituted enones, 9-tosyl-3,4-dihydro-β-carboline imines and 3,4-dihydroisoquinolines.
The authors presented a plausible cooperative mechanism wherein the enone is activated by the catalyst through generation of the covalently bound dienamine, while the imine is simultaneously converted to a thiourea-bound iminium ion, which promotes the subsequent irreversible concerted [4 + 2] cycloaddition reaction (Fig. 6). Alternatively, the cyclization step can also be described by a stepwise Mannich-conjugate addition.
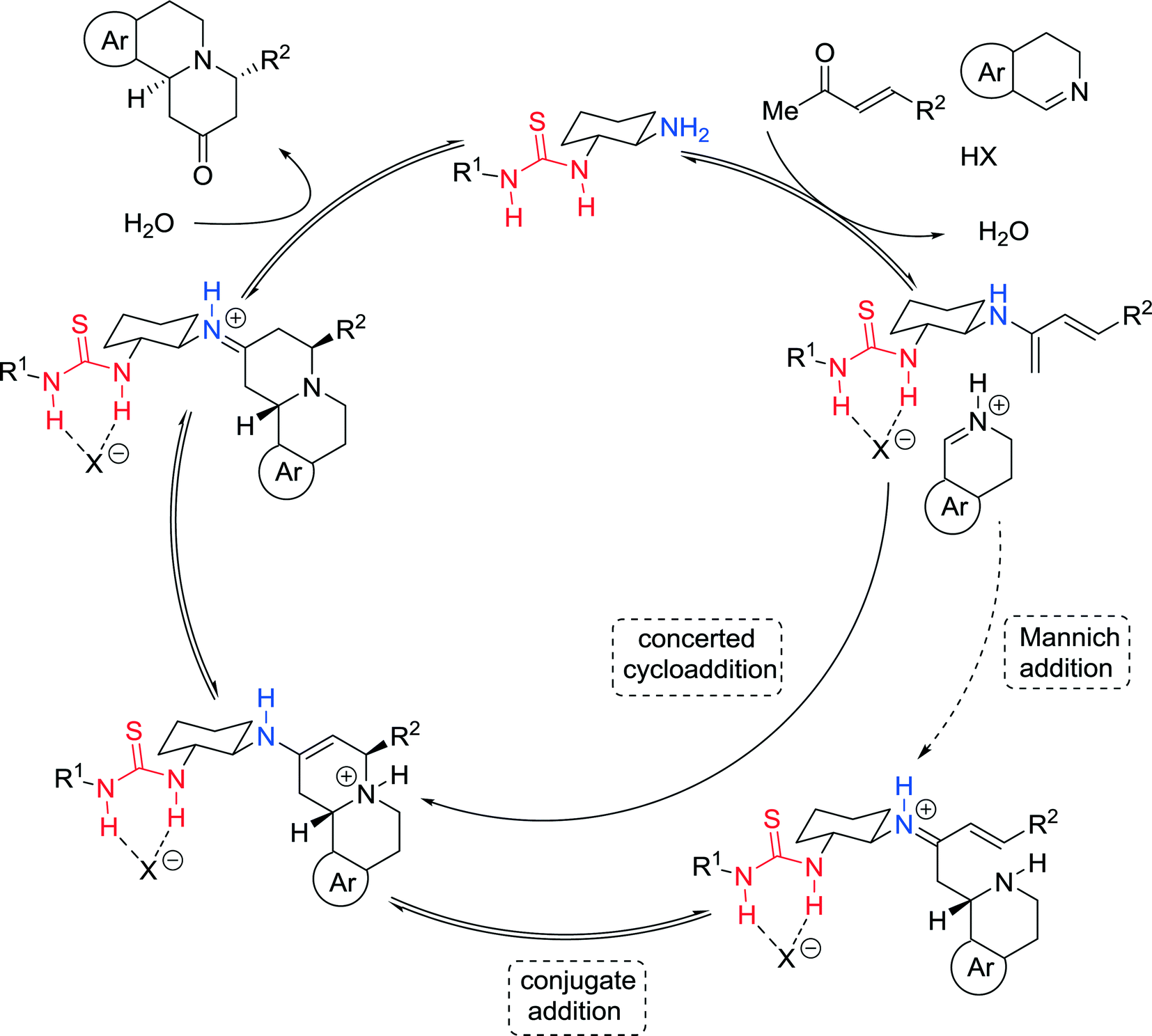 | ||
| Fig. 6 Proposed catalytic cycle for the formal aza-Diels–Alder reaction. | ||
2.2. [5 + 2] cycloadditions
An effective dual catalyst system combining a chiral primary amine-thiourea 3 with achiral thiourea 4 (Schreiner's catalyst) was demonstrated in 2011 by Jacobsen and co-workers for enantioselective synthesis of useful tricyclic structures (8-oxabicyclo[3.2.1]octanes) via intramolecular oxidopyrylium [5 + 2] cycloadditions (Scheme 3).35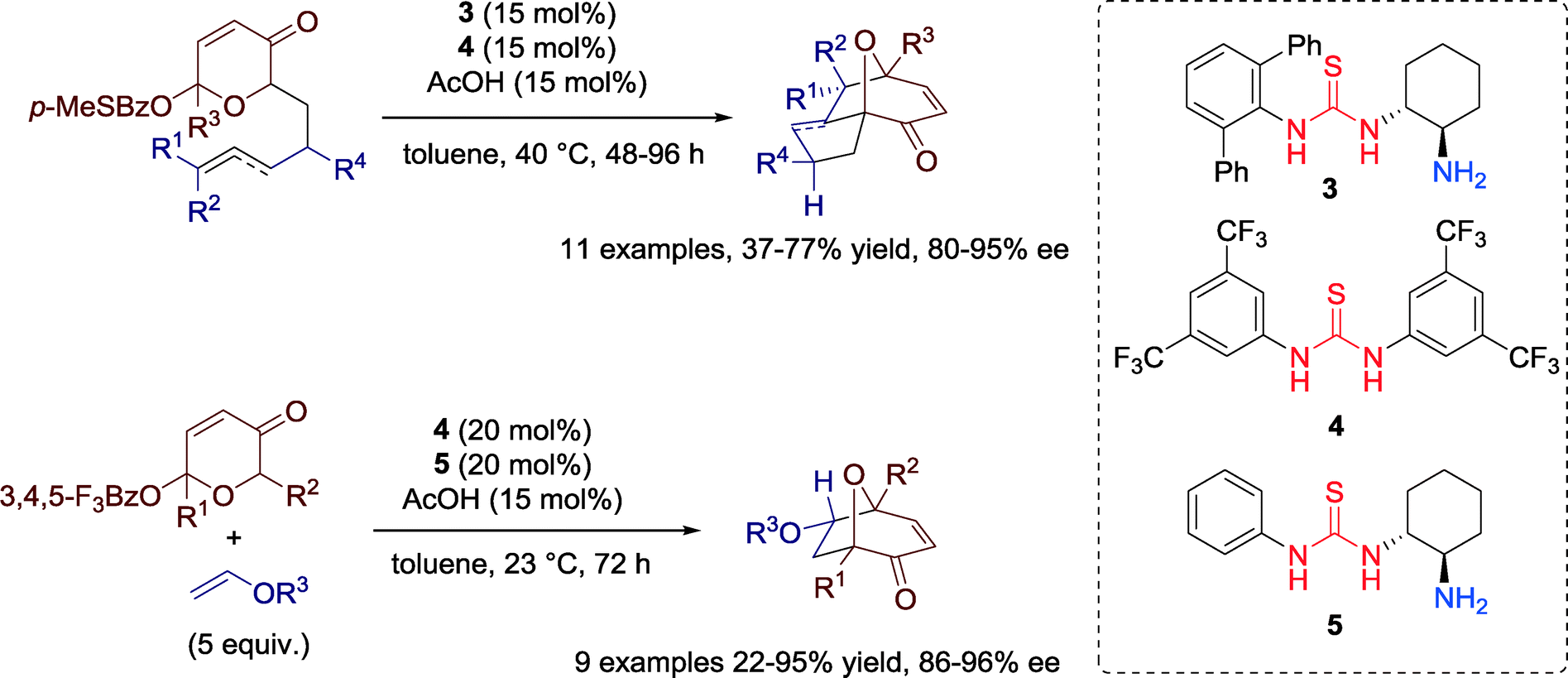 | ||
| Scheme 3 Cooperative catalysis of a chiral primary amine-thiourea and an achiral thiourea in oxidopyrylium cycloadditins. | ||
Computational frontier molecular orbital (FMO) studies were carried out to gain insights into the role of both catalysts 3 and 4. The achiral thiourea 4 acts as a carboxylate-binding agent in the pyrylium cycloaddition reaction, cooperating with chiral primary-amine thiourea 3 to generate a reactive ion pair A poised to undergo the cycloaddition step (Fig. 7).
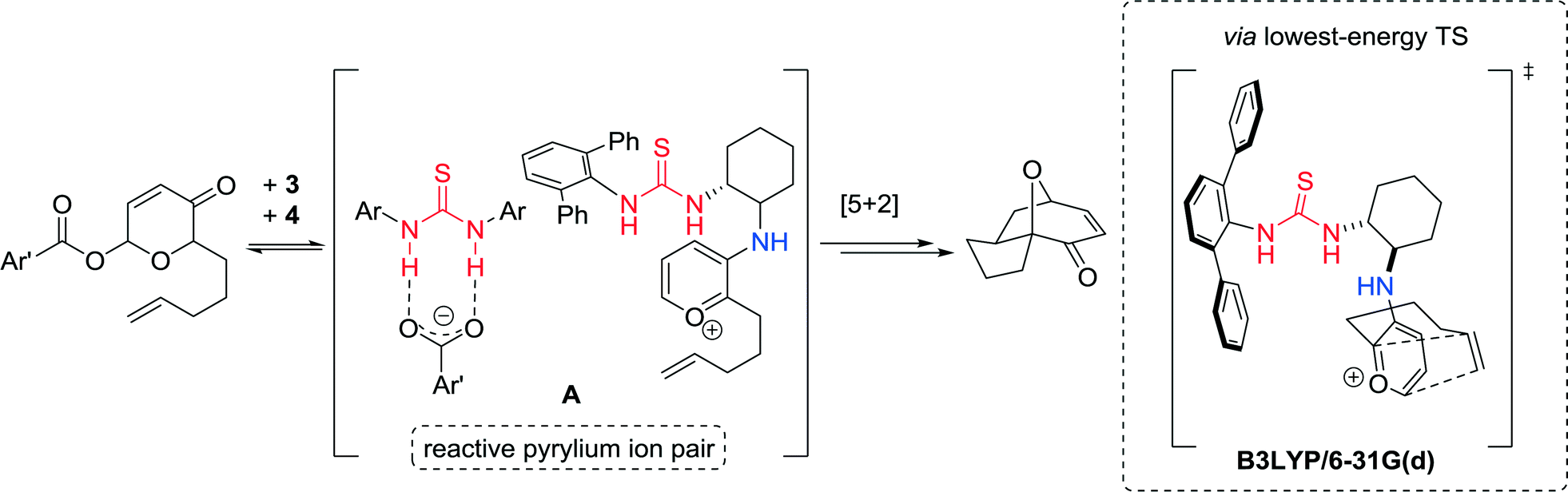 | ||
| Fig. 7 Proposed mode of catalysis and transition state structure calculated at the B3LYP/6-31G(d) level of theory. | ||
Although initial efforts to carry out an intermolecular version of this [5 + 2] cycloaddition proved unsuccessful,35 in 2014 the same research group developed the highly enantioselective intermolecular variant of the [5 + 2] pyrylium cycloaddition reaction using a dual catalyst system composed of a chiral primary-amine thiourea 5 and an achiral thiourea 4 (Scheme 3).36
These reactions provide multifunctional compounds, which can serve as versatile building blocks for further stereoselective complexity-generating transformations.
Looking to the future, we expect that a sufficiently large scope of other new exciting pericyclic reactions catalyzed by chiral primary amine-thioureas will be developed and detailed insights into the factors controlling chemo-, regio-, and stereoselectivities may be provided.
3. Cycloadditions catalyzed by tertiary amine-thioureas
In 2003, Takemoto pioneered a first bifunctional tertiary amine-thiourea organocatalyst 6,15 which has found a broad application in asymmetric synthesis and has been established as an efficient chiral catalyst for different organic transformations,22,23 including cycloaddition reactions such as [3 + 2], 1,3-dipolar cycloadditions and formal cycloadditions such as formal [3 + 2] and [5 + 1]. These facile tertiary amine-thiourea catalyzed cycloadditions are able to generate multifunctional chiral chroman derivatives with four vicinal stereogenic carbon centers,37 functionalized cyclopentanes,38 highly functionalized spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic skeletons,39 azlactones and methyleneindolinones,40 4-nitropyrazolidines,41 functionalized pyrolidines as well as thiopyrano-indole annulated heterocycles42 and spirooxindole δ-lactones.43 All these structural motifs are common to numerous bioactive compounds, natural products, pharmaceuticals and are also valuable chiral precursors for further organic transformations.3.1. [3 + 2] cycloadditions
In 2010, Xie and co-workers established a kinetic resolution of racemic 3-nitro-2H-chromene derivatives via [3 + 2] cycloaddition reaction with α-amino malonate imine catalyzed by Takemoto's chiral thiourea 6 (Scheme 4).37 This resolution process leads to multifunctional chroman derivatives with four vicinal carbon centers which can be of interest for pharmaceutical chemistry.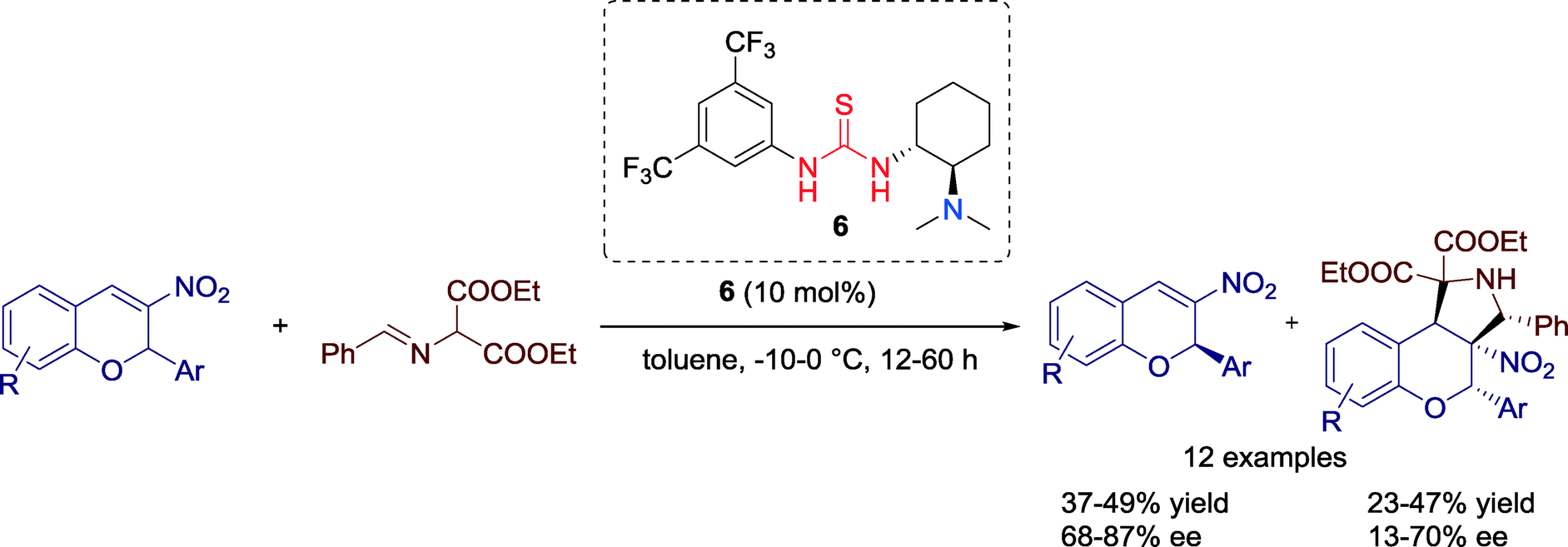 | ||
| Scheme 4 Asymmetric kinetic resolution of racemic 3-nitro-2H-chromene derivatives via enantioselective [3 + 2] cycloaddition reaction. | ||
The authors suggest a dual activation in a bifunctional manner of both substrates by the thiourea and the tertiary amine units (Fig. 8).
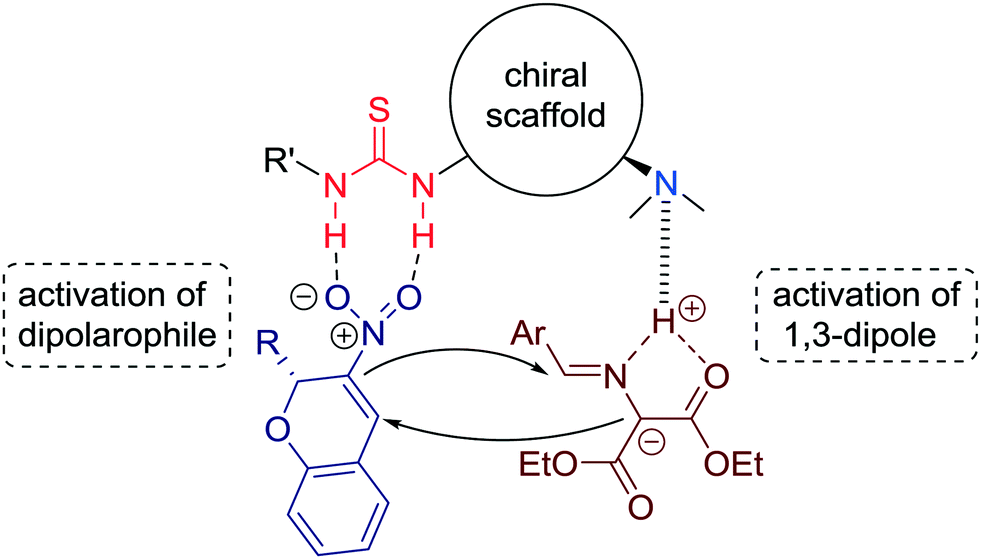 | ||
| Fig. 8 Proposed mechanistic model for the [3 + 2] cycloaddition reaction catalyzed by tertiary amine-thiourea. | ||
Bonne, Rodriguez and co-workers demonstrated in 2010 an interesting route towards highly functionalized cyclopentanes with an additionally oxime moiety, involving an enantioselective organocatalytic Michael addition followed by a highly diastereoselective [3 + 2] cycloaddition-fragmentation step.38
Within this one-pot sequence catalyzed by Takemoto's tertiary amine-thiourea 6 or by a cinchona alkaloid catalyst 7, respectively, up to three stereocenters with excellent enantioselectivity were created (Scheme 5). In addition, the tolerance for different β-nitrostyrenes is proof of the versatility in practical utility.
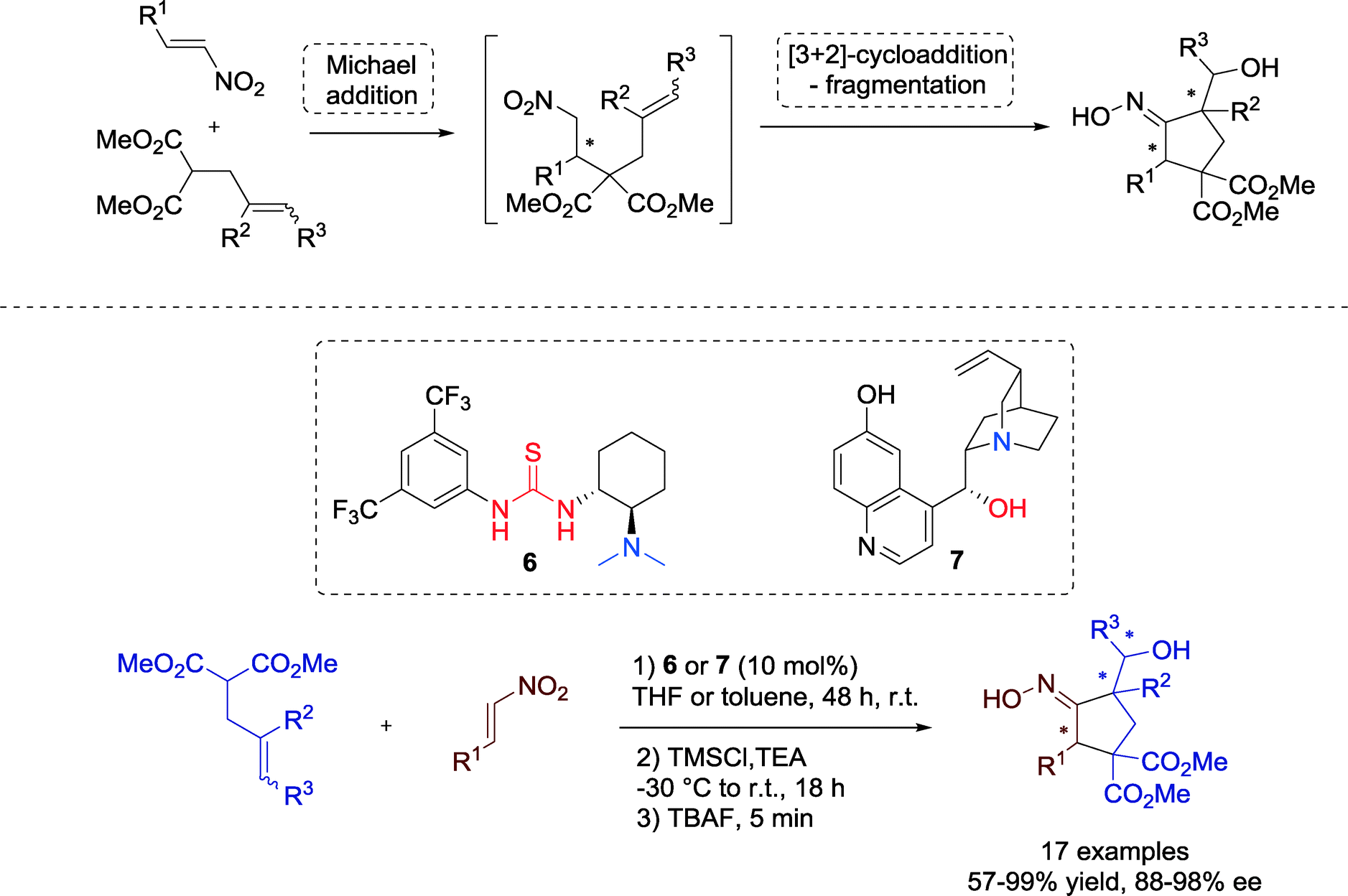 | ||
| Scheme 5 One-pot process for the enantioselective cyclocarbohydroxylation sequence. | ||
3.2. 1,3-Dipolar cycloadditions
In 2011, Wang, Xu and co-workers furnished optically active functionalized pyrrolidines in excellent yields and enantioselectivities via a metal-free 1,3-dipolar cycloaddition reaction between azomethine ylides and N-arylmaleimides catalyzed by a chiral tertiary amine-thiourea 8 (Scheme 6).44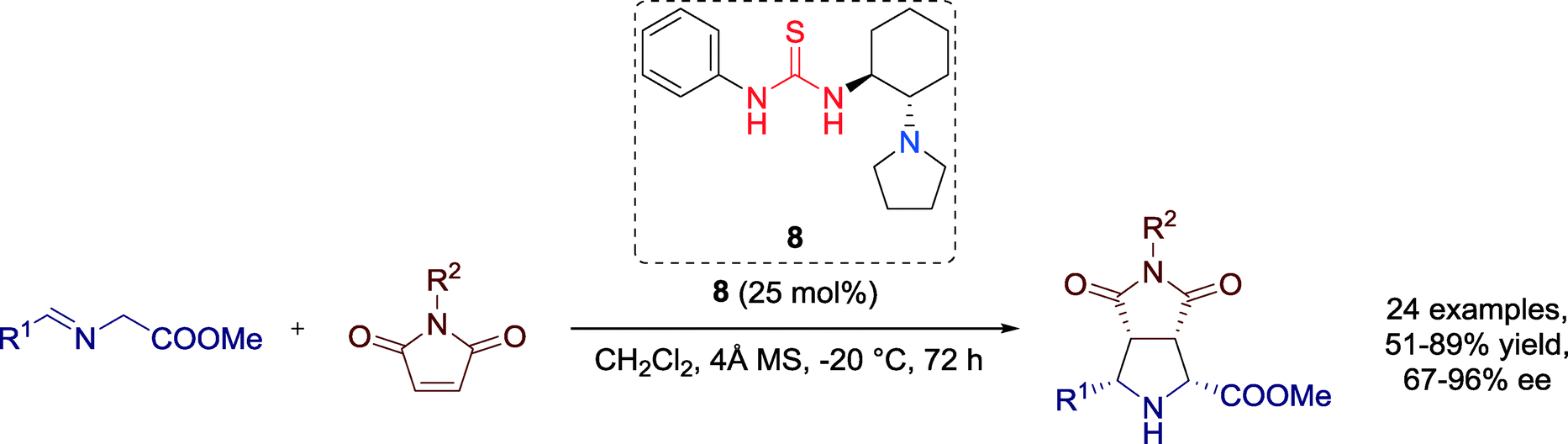 | ||
| Scheme 6 1,3-Dipolar cycloaddition towards functionalized pyrrolidines. | ||
All products bearing aromatic substituents could be obtained with moderate to good yields and good enantioselectivities, the performance of the catalyst, however, dropped significantly for aliphatic N-substituted maleimide, respecting the enantioselectivity (30% ee). In general, strong electron-withdrawing groups on the phenyl ring of the azomethine ylides had a negative influence both on the yield and the stereoselectivity of the reaction, whereas electron-rich substituents affected the enantioselectivity in a positive way.
The authors, moreover, proposed a plausible transition state structure for the reaction wherein the α-amino esters are deprotonated and activated by the tertiary amine moiety of the catalyst to create azomethine ylides and, simultaneously, the N-arylmaleimide is activated by the thiourea group of the catalyst (Fig. 9). As a result of these synergistic interactions, the high stereoselectivities within the cycloaddition reaction can be explained.
 | ||
| Fig. 9 Postulated transition state with synergistic interactions of the thiourea catalyst. | ||
In 2013 Wang and co-workers developed a highly enantioselective 1,3-dipolar cycloaddition reaction of homoserine lactone derived imino esters with methyleneindolinones to provide spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic scaffolds with four stereocenters, two of which are spiro quaternary stereocenters (Scheme 7).39
 | ||
| Scheme 7 1,3-Dipolar cycloaddition reaction of homoserine lactone derived imino ester with methyleneindolinones. | ||
The excellent performance of the catalyst 9 doesn't rely on the position or electronic nature of substituents in the starting compounds and, hence, desired products were generated with high yields and excellent enantioselectivities.
The suggested mechanism, which involves simultaneous activation of 1,3-dipole and of dipolarophile through bifunctional tertiary amine-thiourea is presented in Fig. 10.
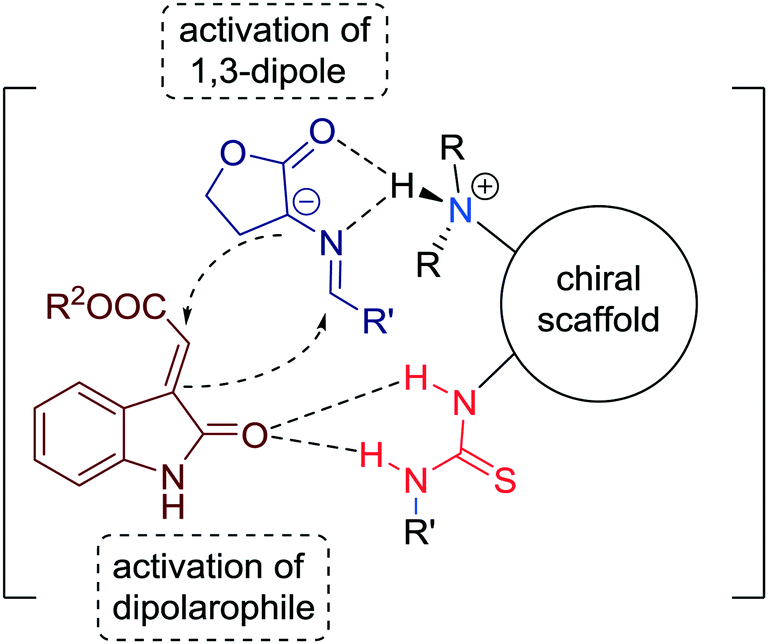 | ||
| Fig. 10 Bifunctional organocatalytic strategy for direct construction of spirocyclic skeletons. | ||
Azlactones, bearing three reactive sites at the C2, C4, and C5 atoms are highly versatile and convenient for the transformation to amino acids and heterocycles, generally via 1,2 or 1,4-additions and [4 + 2] cycloaddition reactions. Hong, Wang and co-workers successfully employed these starting compounds in 2013 for a 1,3-dipolar cycloaddition reaction with methyleneindolinones towards highly functionalized spirocyclic oxindole derivatives with high levels of enantioselectivity (Scheme 8).40
 | ||
| Scheme 8 (a) Stereochemical control by dipole formation through interaction with a chiral base; (b) enantioselective 1,3-dipolar cycloaddition between azlactones and methyleneindolinones. | ||
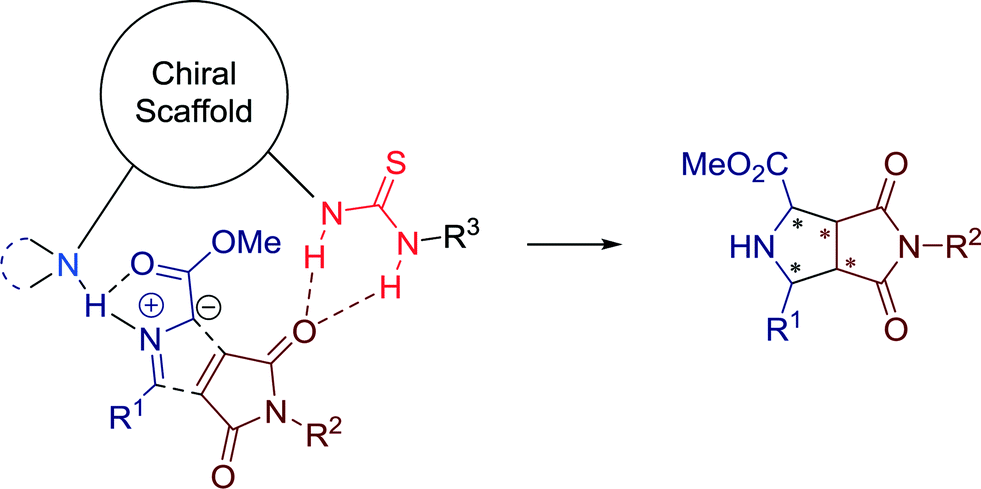
The usefulness of this asymmetric bifunctional catalysis is demonstrated by the broad substrate scope. Mechanistically, a deprotonation of the azlactones by the basic group of the chiral amine-thiourea catalyst 10, generating a dipole intermediate, suitable for an enantioselective 1,3-dipolar cycloaddition with dipolarophiles, could be proposed.
In 2014, Jørgensen and co-workers presented the first catalytic enantio- and diastereoselective synthesis of 4-nitropyrazolidines. The highly enantioselective 1,3-dipolar cycloaddition reaction with hydrazones is catalyzed by thiourea catalyst 11 (Scheme 9).41
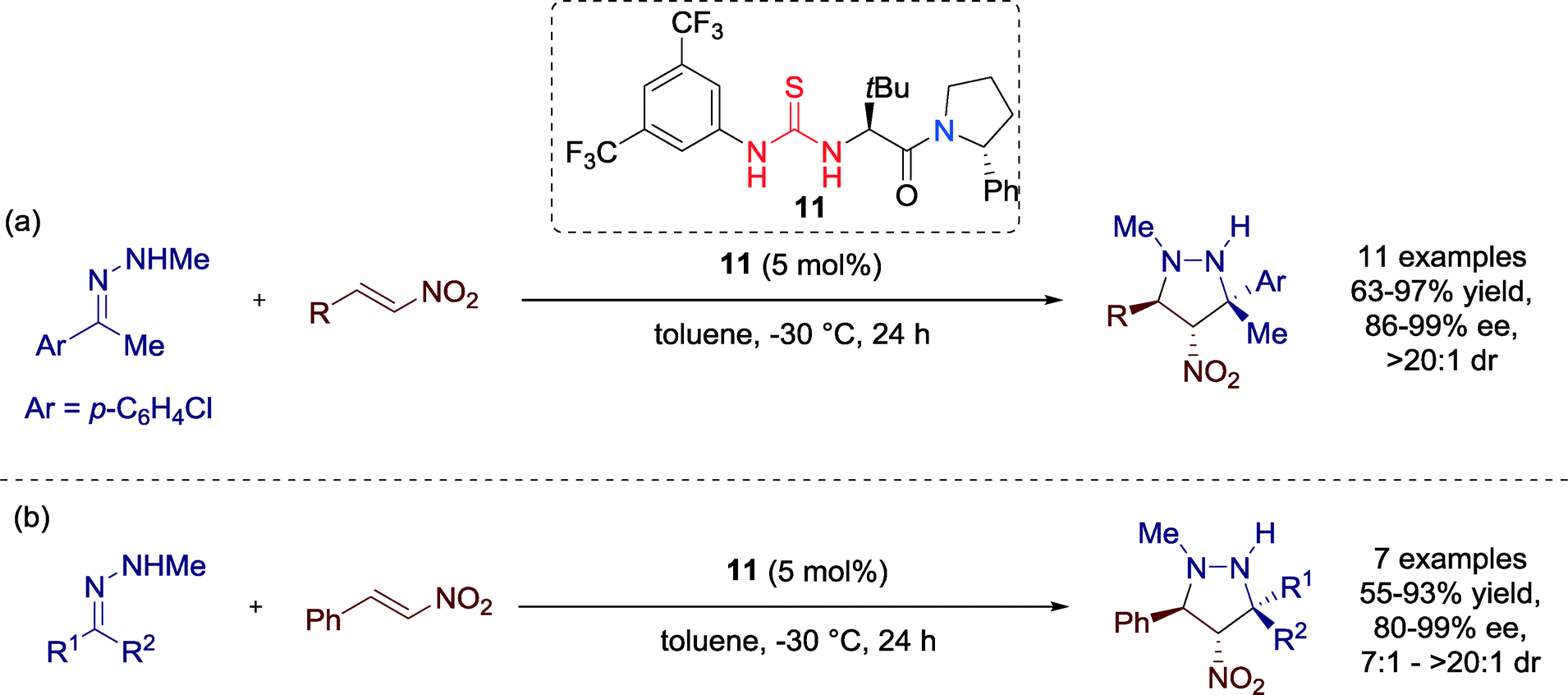 | ||
| Scheme 9 Organocatalytic asymmetric synthesis of 4-nitropyrazolidines, applying (a) different nitro-olefins and (b) various hydrazones. | ||
Having the optimized conditions with catalyst 11 in hands, the authors focused on extending the substrate scope by employing a range of aromatic and aliphatic nitro-olefins and hydrazones. All substrates were applicable for the reaction without limitations and furnished the 4-nitropyrazolidines in high to excellent yields as single diastereomers with good to excellent enantioselectivities.
Moreover, to further demonstrate the potential of 4-nitropyrazolidines as useful precursors, the transformations towards 1,2,3-triamines, subunits of several biologically active natural products and pharmaceutical compounds, were examined (Scheme 10).
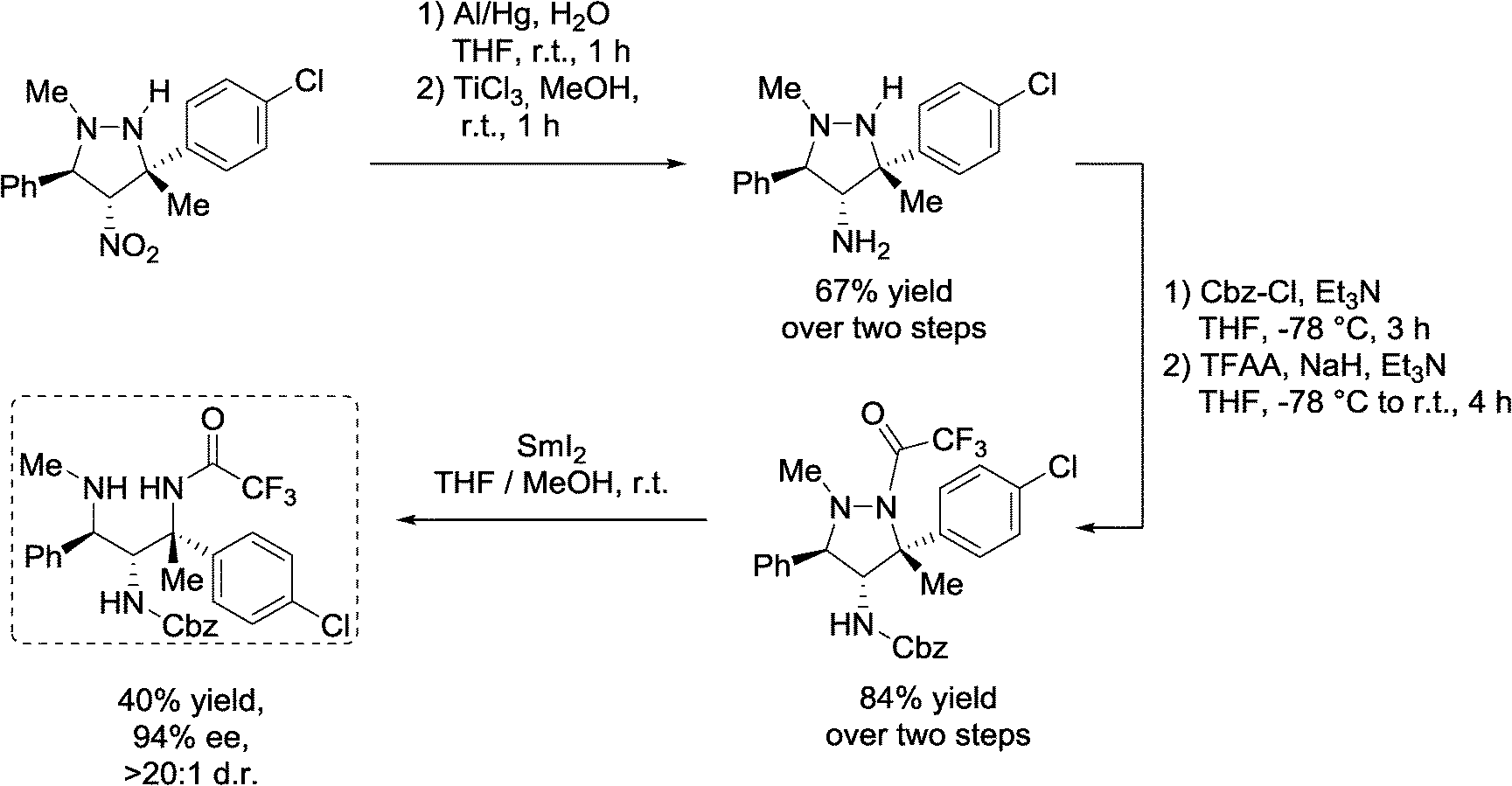 | ||
| Scheme 10 Exemplary synthesis of 1,2,3-triamine from 4-nitropyrazolidine. | ||
3.3. Formal [3 + 3] and [5 + 1] cycloadditions
In 2014, the group of Wang reported the first catalytic asymmetric method towards optically active thiopyrano-indole annulated heterocycles in a highly enantioselective way and with good to excellent yields via formal [3 + 3] cycloaddition, employing bifunctional thiourea–tertiary amine catalyst 12 (Scheme 11).42 Organosulfur compounds have great significance for the biological and medicinal chemistry.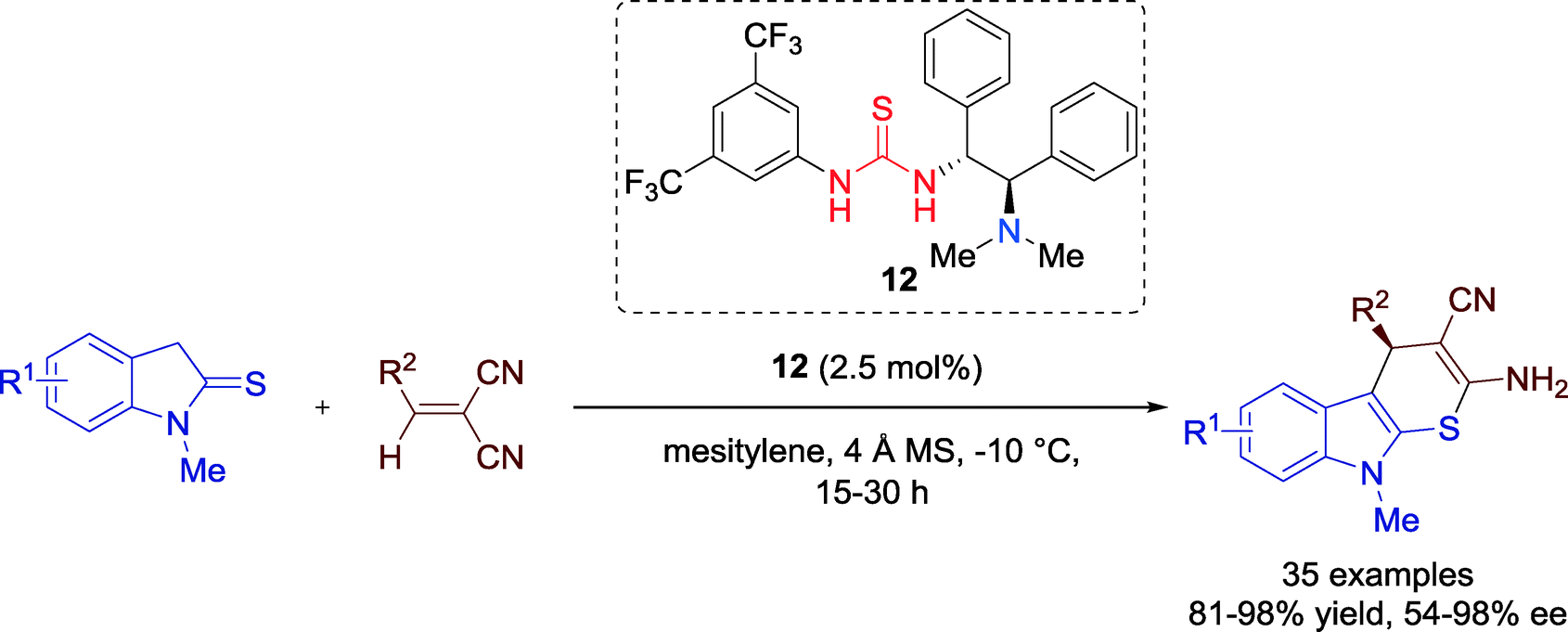 | ||
| Scheme 11 Formal enantioselective thio [3 + 3] cycloaddition towards thiopyrano-indole heterocycles. | ||
A variety of several different indoline-2-thione and 2-benzylmalononitriles could be employed for the reaction and both electron-withdrawing and -donating groups were well tolerated, producing the product with a high efficiency with respect to the yield and enantioselectivity.
Thus, the enormous substrate scope of the organocatalytic cascade thio-Michael-cyclization reaction reveals the generality of this transformation. The only limitation was observed for indoline-2-thione, bearing a bromo substituent at the 4-position, which gave just a moderate performance (81 yield, 54% ee).
Furthermore, all mechanistic aspects of this reaction were supported by computational calculations. The authors reached important conclusions by DFT (density functional theory) calculations. The rather easy enolization of indoline-2-thione (9.85 kcal mol−1) as compared to indoline-2-one (16.61 kcal mol−1) confirm the high reactivity of keto-S. The simultaneous activation of both reactants, forming a stable multiple H-bonded complex (S-M1, 3.75 kcal mol−1) with the bifunctional thiourea catalyst leads to S-TS–1 and, thus, the C–C bond formation step takes place in a highly chiral environment (Fig. 11).
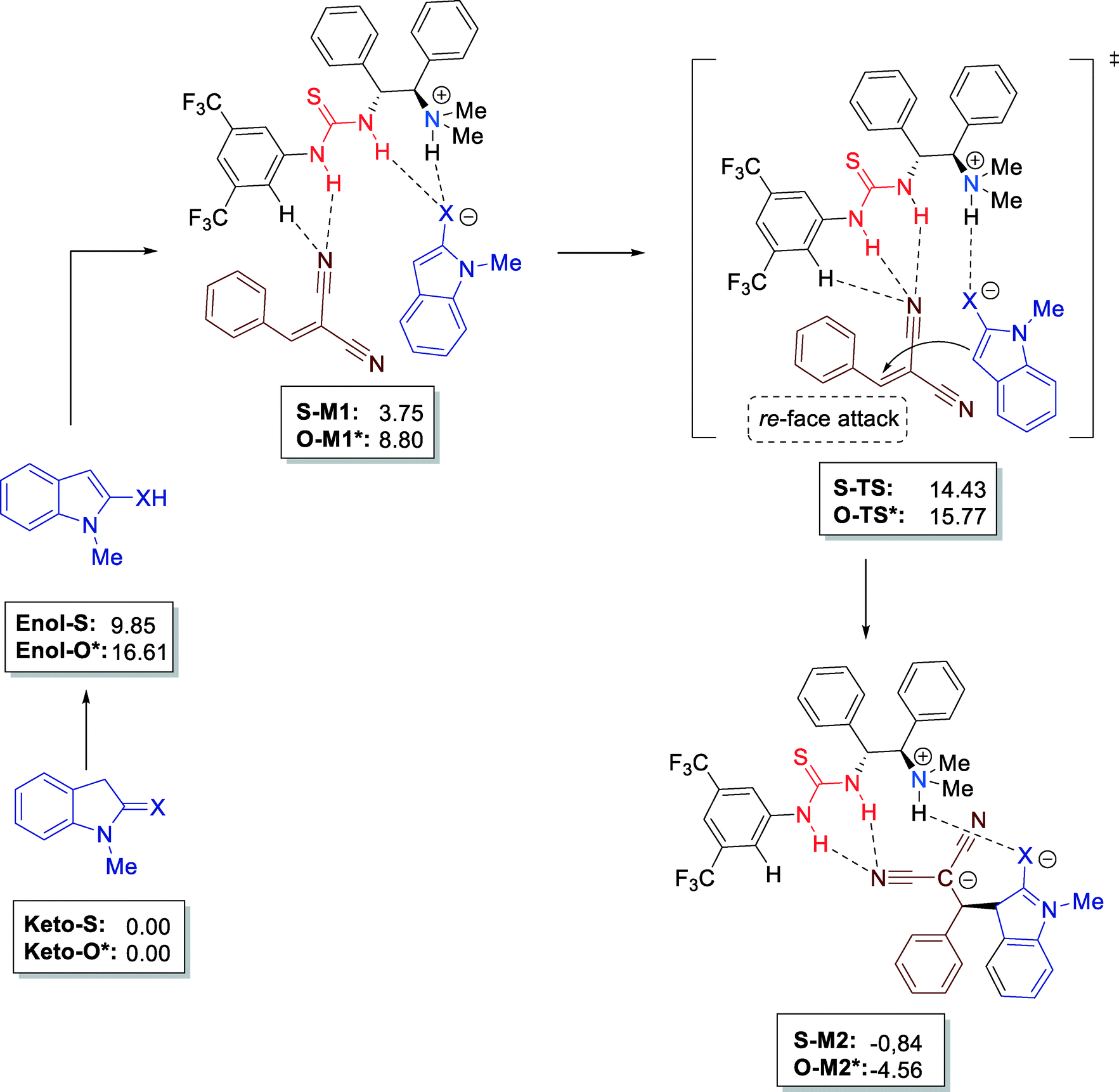 | ||
| Fig. 11 Mechanistic studies at DFT level B3LYP/6-311++G(d,p) (energies in frames in kcal mol−1) using the CPCM solvent model; * values for indoline-2-one (X = O). | ||
Trisubstituted spirooxindole δ-lactones with three contiguous stereocenters are common in many naturally occurring substances and pharmaceuticals. In 2014, Xu and co-workers established a highly diastereo- and enantioselective novel formal [5 + 1] cycloaddition reaction towards these compounds, catalyzed by bifunctional thiourea catalyst 13 (Scheme 12).43
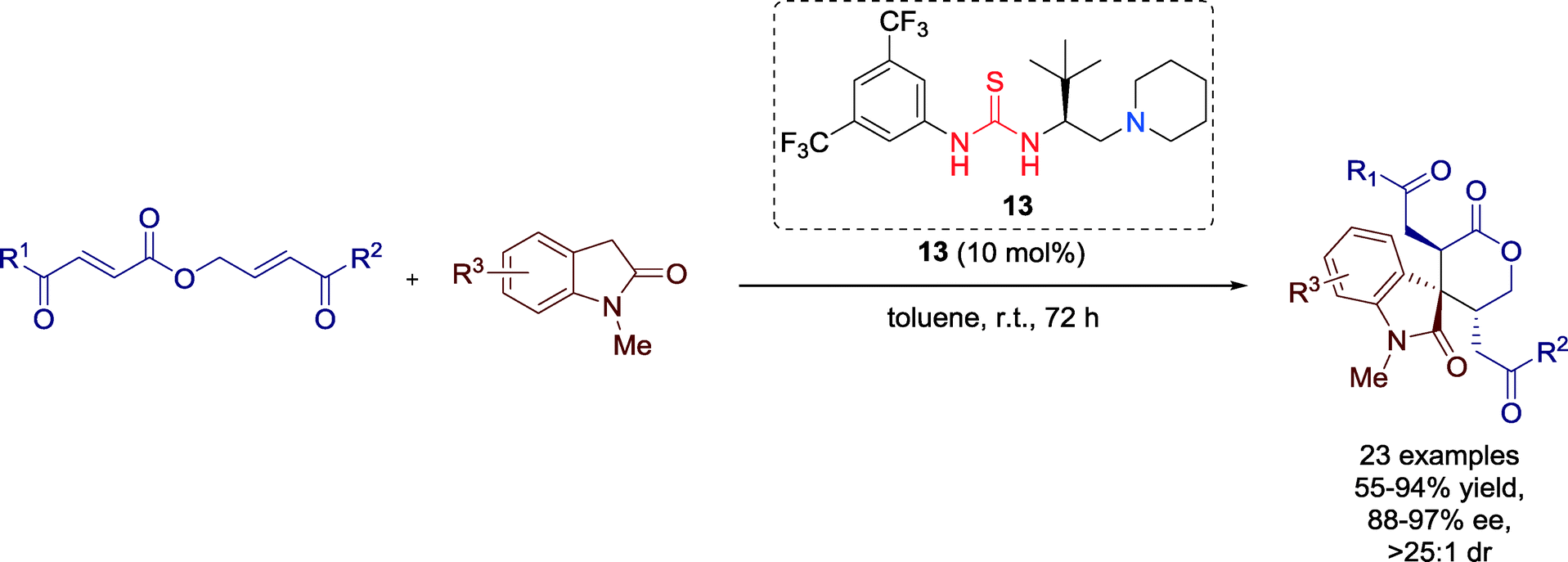 | ||
| Scheme 12 Formal [5 + 1] cycloaddition towards chiral spirooxindole δ-lactones. | ||
During investigations of the substrate scope the authors revealed that the performance of the reaction depended more on the electronic properties of the substituents rather than their positions. Electron-withdrawing substituents furnished products with higher yields but lower enantioselectivities, electron-donating groups, however, had positive influence on the ee values, albeit the yields dropped.
Furthermore, a plausible reaction mechanism was proposed, that is, initially the concerted activation of the oxindole and the enone by the bifunctional thiourea catalyst via the formation of TS-1. After an intramolecular Michael addition, a stable six-membered transition state TS-2 is generated, creating a chiral environment for the subsequent intermolecular Michael reaction of the oxindole with the si-face of the bound enone (Fig. 12).
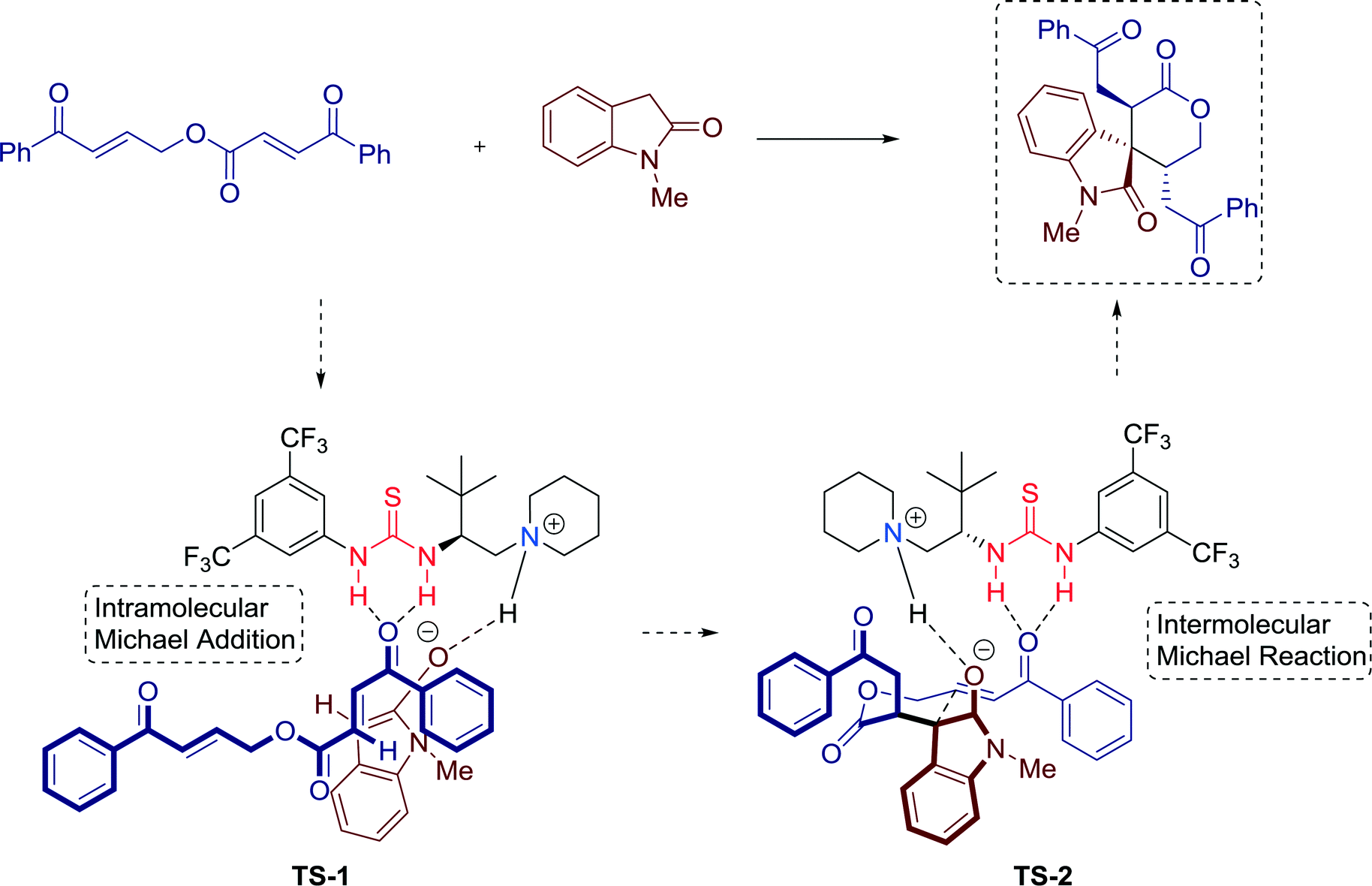 | ||
| Fig. 12 Proposed mechanism of the spirooxindole δ-lactone-forming tandem reaction. | ||
These examples of cycloaddition reactions demonstrate the power of bifunctional tertiary amine-thiourea catalysis, and suggest that this important mode of catalysis may be broadly applicable to the wide range of other pericyclic reactions, which are expected to be discovered in the near future.
4. Cycloadditions catalyzed by cinchona alkaloid derived thioureas
Bifunctional Cinchona-alkaloid derived thiourea catalysts are an important subgroup of tertiary amine–thioureas. This family of bifunctional organocatalysts was introduced for the first time in 2005 by Chen and co-workers.45 Meanwhile, the research groups of Soós, Connon and Dixon reported another new Cinchona-alkaloid derived thiourea catalysts, which were employed for enantioselective conjugate additions,46–48 gaining further attention in the following years as catalysts for several sorts of transformations.49–51 Furthermore, Cinchona-derived thiourea catalysts successfully promoted cycloaddition reactions such as [4 + 2], [3 + 2] and their formal versions, being well suited, for instance for the synthesis of 2H-pyran-2,5-diones,52 3,3′-pyrrolidinyl spirooxindoles,53,54 1,3-dioxolanes,55 α-aryl isocyanoacetates and isatins,56 as well as 2-oxazolidinones.574.1. [4 + 2] cycloadditions
In 2011, Lee and co-workers established a highly enantioselective Diels–Alder reaction of 2H-pyran-2,5-diones promoted by Cinchona-derived thiourea catalyst 14 (Scheme 13).52 The highly functionalized bridged bicyclic lactones and α-hydroxycyclohexanones, obtained in this reaction, might be useful building blocks for natural product synthesis.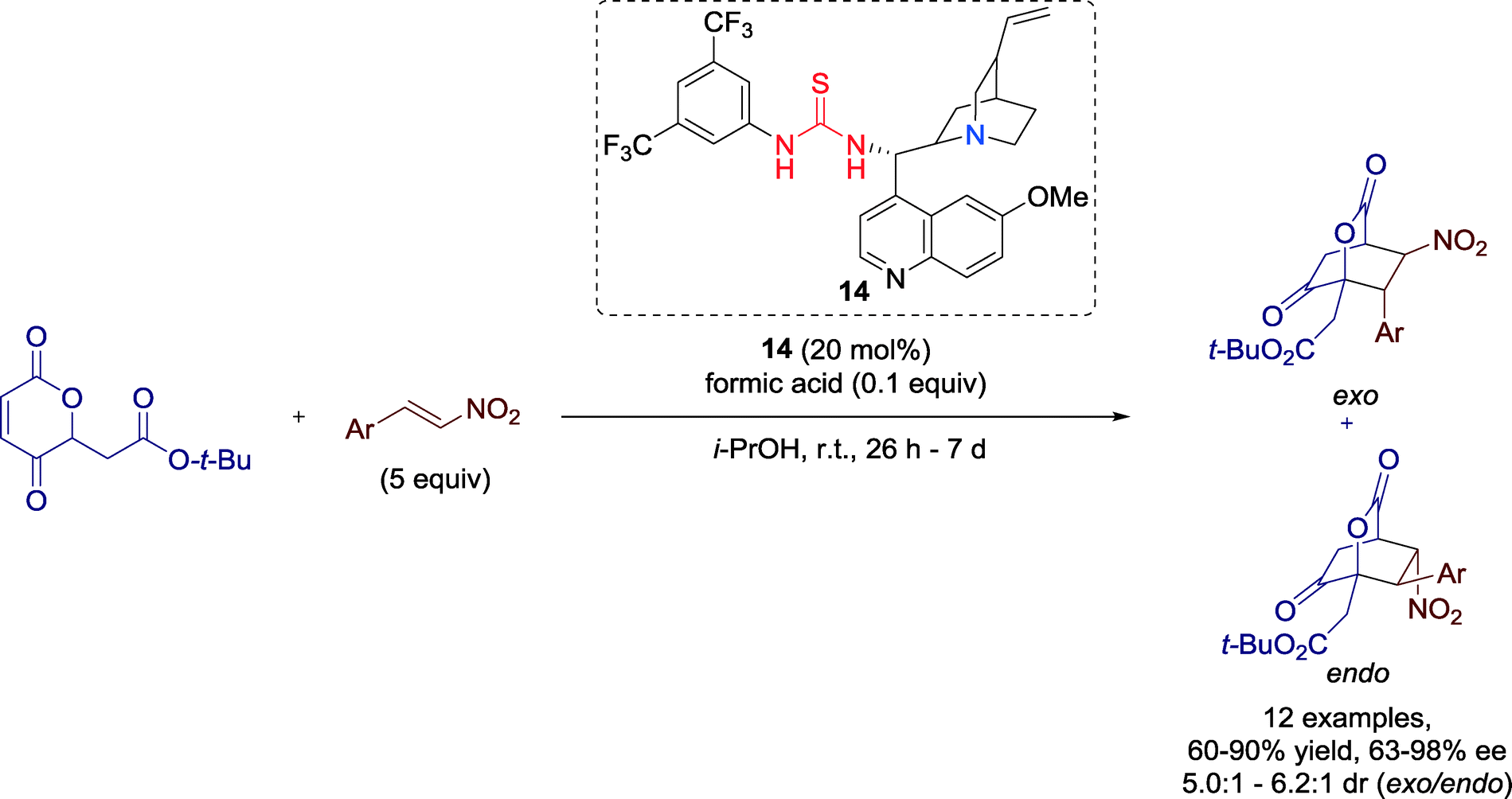 | ||
| Scheme 13 [4 + 2] cycloaddition promoted by a Cinchona-derived thiourea catalyst. | ||
The yields and enantioselectivities of the transformation were significantly affected by the substituents of the aromatic ring of the nitrostyrene, whereas the exo/endo ratio, in general, remained constant.
The mechanism may be described as a concerted dual activation of both components: The enolization of the 2H-pyran-2,5-dione to 5-hydroxy-2-pyrone might be enabled by the Brønsted base of the Cinchona moiety of the catalyst. Simultaneously, the nitro group of the dienophile is proposed to be activated via hydrogen bonds with the acidic thiourea motif of the catalyst (Fig. 13). Thus, enclosed by the chiral environment of the catalyst, the cycloaddition proceeds with high enantioselectivity and products can be furnished in high optical purity.
 | ||
| Fig. 13 Proposed transition state structure for the Diels–Alder reaction. | ||
4.2. [3 + 2] and formal [3 + 2] cycloadditions
Wang, Xu and co-workers presented in 2012 a highly stereoselective [3 + 2] cycloaddition of isocyanoesters and methyleneindolinones catalyzed by Cinchona alkaloid derived thiourea catalyst 15 to produce 3,3′-pyrrolidinyl spirooxindoles (Scheme 14).53 These structural motifs are found in many natural alkaloids and bioactive molecules and, thus, they are of great interest for the pharmaceutical industry.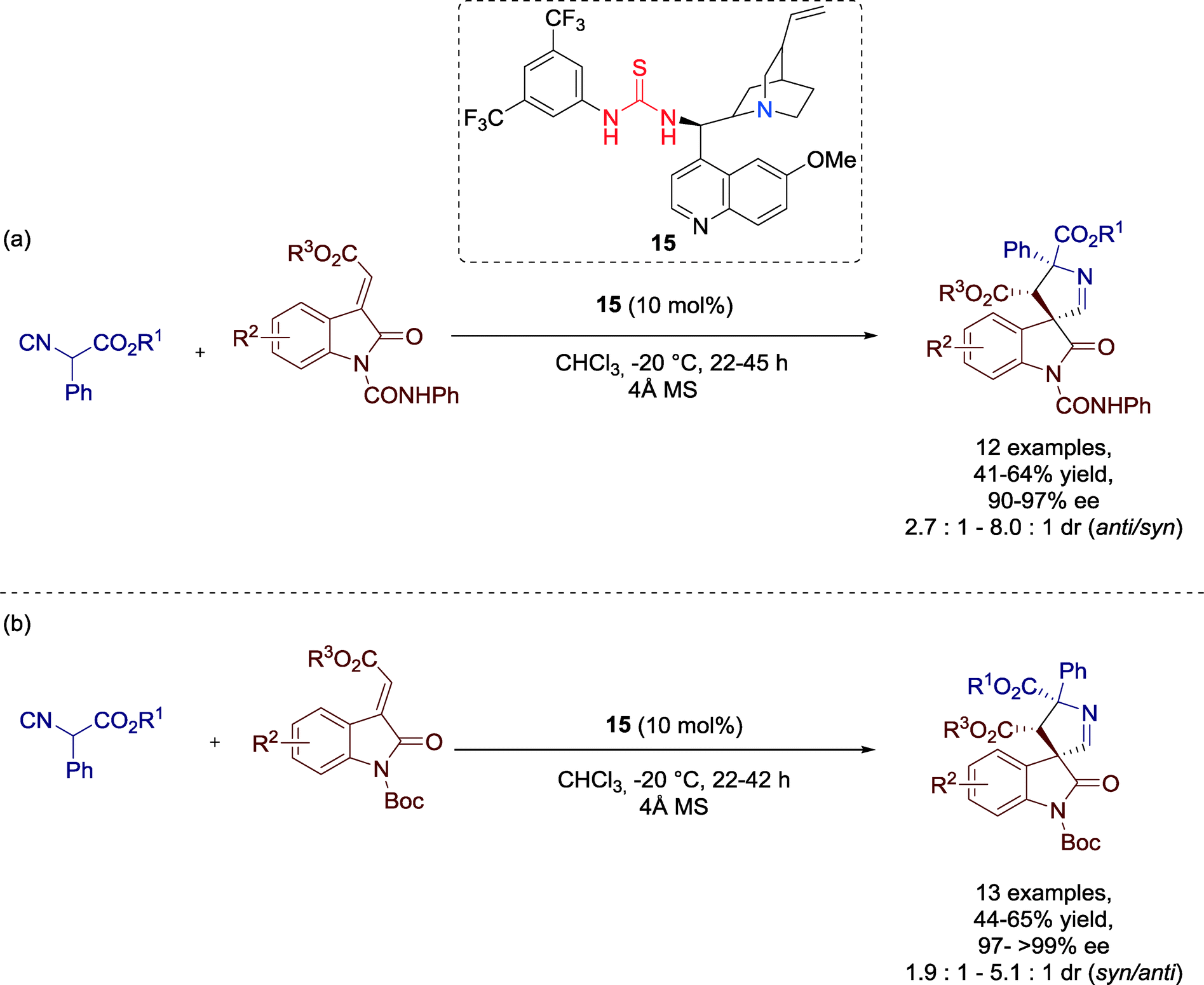 | ||
| Scheme 14 [3 + 2] cycloaddition reaction with methyleneindolinones protected (a) by N-phenyl amide or (b) by N-tert-butoxycarbonyl. | ||
The broad substrate scope gives prove of the generality of the cycloaddition reaction for a broad range of methyleneindolinones gave good performance for the transformation. All spirooxindoles were generated with moderate to good yields and excellent enantioselectivities independent from the electronic nature and position of the substituent on the aromatic ring of the methyleneindolinones (Scheme 14a and b).
Furthermore, the authors recognized the reliance of the diastereoselectivity on the N-protecting group: methyleneindolines protected by phenylamide (–CONHPh) furnished the anti-diastereomer (Scheme 14a), whereas the tert-butoxycarbonyl (boc) protected compound favored the generation of syn-product, which could be, moreover, isolated optically pure in most reactions (Scheme 14b).
In the same year, Zhong and Barbas III described an alternative route towards 3,3′-pyrrolidonyl spirooxindoles, employing simple starting materials via [3 + 2] cycloaddition promoted by Cinchona-alkaloid derived thiourea catalyst 16 (Scheme 15).54
 | ||
| Scheme 15 Organocatalytic asymmetric [3 + 2] cycloaddition towards 3,3′-pyrrolidinyl spirooxindoles. | ||
A variety of diversely substituted methyleneindolinone derivatives could be applied for this straightforward process, regardless of the electronic and steric properties of the substituents and all products were generated with good to excellent yields and enantioselectivities. Additionally, the diastereoselectivity of the reaction was on a very high level.
Considering the benefits of this cycloaddition, which makes use of rather simple starting material, this reaction might be an attractive candidate for the medicinal chemistry.
A mechanistic explanation was proposed to account for the stereoselectivity of the cycloaddition reaction, wherein a concerted activation of both substrates by the bifunctional catalyst was proposed by the authors (Fig. 14). A multiple H-bonded transition state is formed, wherein the reactants are constrained to a well-defined orientation, required for asymmetric induction.
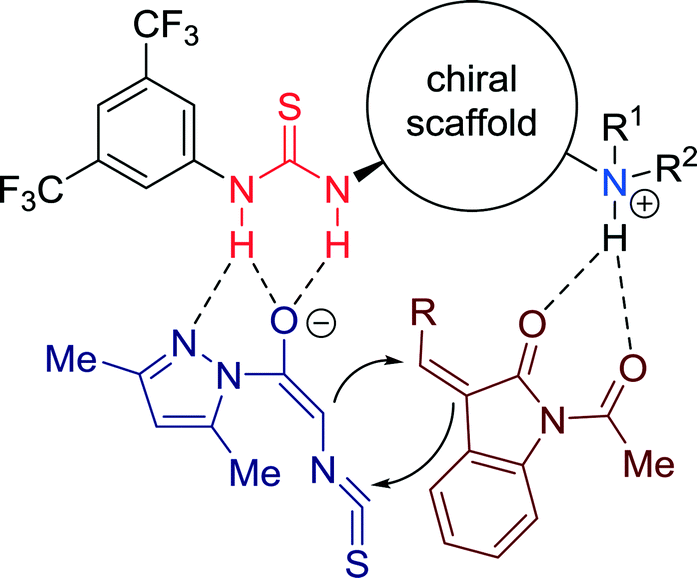 | ||
| Fig. 14 Proposed dual activation model of substrates. | ||
In 2012, Matsubara and Asano developed a highly enantioselective formal [3 + 2] cycloaddition reaction promoted by Cinchona alkaloid-based bifunctional organocatalyst 15 (Scheme 16).55 The optically active 1,3-dioxolanes are structural motifs in several biologically active products.
 | ||
| Scheme 16 Organocatalytic formal [3 + 2] cycloaddition towards optically active 1,3-dioxolanes; CPME = cyclopentyl methyl ether. | ||
Etheral solvents like cyclopentyl methyl ether (CPME) gave the best performance for the reaction regarding the yield and enantioselectivity. Independent from the electronic nature of the substituents, a variety of different aldehydes and electron-deficient ketones could be successfully employed.
The reaction is predicted to proceed by a stepwise rather than a concerted mechanism via the formation of hemiacetal intermediates between γ-hydroxy-α,β-unsaturated ketones and aldehydes (Fig. 15).
 | ||
| Fig. 15 Reaction pathway via formation of a hemiacetal intermediate. | ||
The mechanism is not completely investigated yet, further experiments by the authors, however, indicate that the oxa-Michael addition from the hemiacetal intermediates B might be the enantioselective step of this reaction.
Moreover, the versatility of the 1,3-dioxolanes was demonstrated on the basis of two examples: (a) the transformation to chiral triols via a reduction/deacetalization pathway and (b) a stereospecific ring cleavage could be successfully carried out without any loss of enantioselectivity (Scheme 17).
 | ||
| Scheme 17 Transformation of 1,3-cioxolanes (a) towards chiral triazoles and (b) via ring cleavage. | ||
In 2013, Shi, Zhao and co-workers developed a highly enantioselective [3 + 2] cycloaddition reaction of α-aryl isocyanoacetates and isatins towards optically active spirooxindole oxazolines catalyzed by Cinchona alkaloid derived bifunctional amine-thiourea-bearing sulfonamide 17 (Scheme 18).56
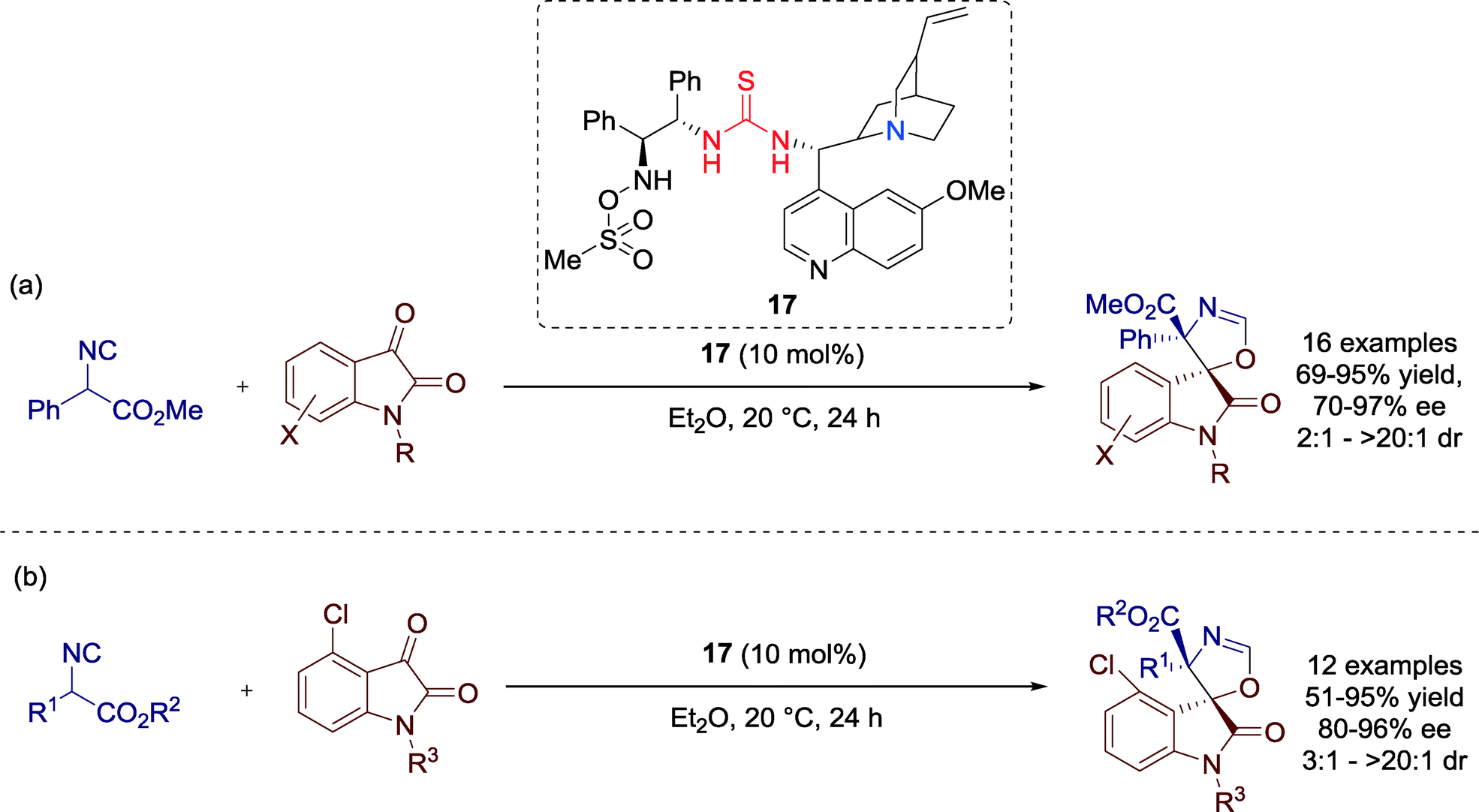 | ||
| Scheme 18 Enantioselective [3 + 2] cycloaddition, employing (a) different isatin derivatives and (b) various α-substituted isocyanoacetates. | ||
Although, the electronic and steric properties of the isatin derivatives had significant influence on the reaction, the authors could furnish all products with moderate to excellent yields and enantioselectivities. A limitation of the cycloaddition reaction was observed in attempts employing an ortho-substituted aryl group (R2 = 2-MeC6H4) or an α-isopropyl isocyanoacetate (R2 = i-Pr), respectively, which showed no conversion.
Additionally, a plausible transition-state model was proposed by the group (Fig. 16). Firstly, the catalyst deprotonates the isocyanate, building concerted hydrogen bonds with the hydroxy group and the two carbonyl groups of the isatin and, thus, directing the orientation of the attack. Simultaneously, π–π stacking interactions, formed between the phenyl ring of isocyanoacetate and the isatin moiety, facilitate the re-face attack of the enolized isocyanoacetate. Finally, the spirocyclic products are created via an intramolecular reaction between the hydroxy group of the formed aldol intermediate and the isocyano group.
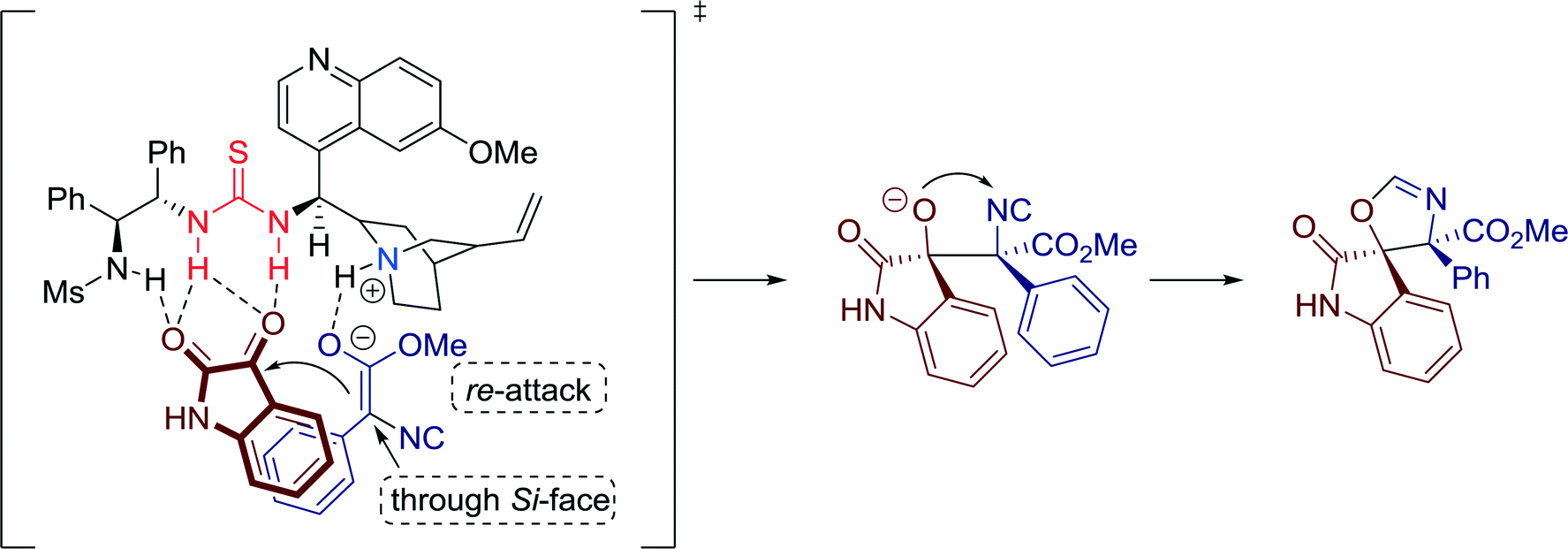 | ||
| Fig. 16 Proposed transition state model; Ms = mesyl (methanesulfonyl). | ||
Matsubara, Asano and co-workers presented in 2013 a formal [3 + 2] cycloaddition of γ-hydroxy-α,β-unsaturated carbonyls and an isocyanate towards 2-oxazolidinones catalyzed by Cinchona-alkaloid-derived amine-thiourea catalyst 15. Additionally, they observed an extraordinary procedure-controlled enantioselective switch and, thus, the two enantiomers could be furnished selectively without varying the reaction conditions (Scheme 19).57
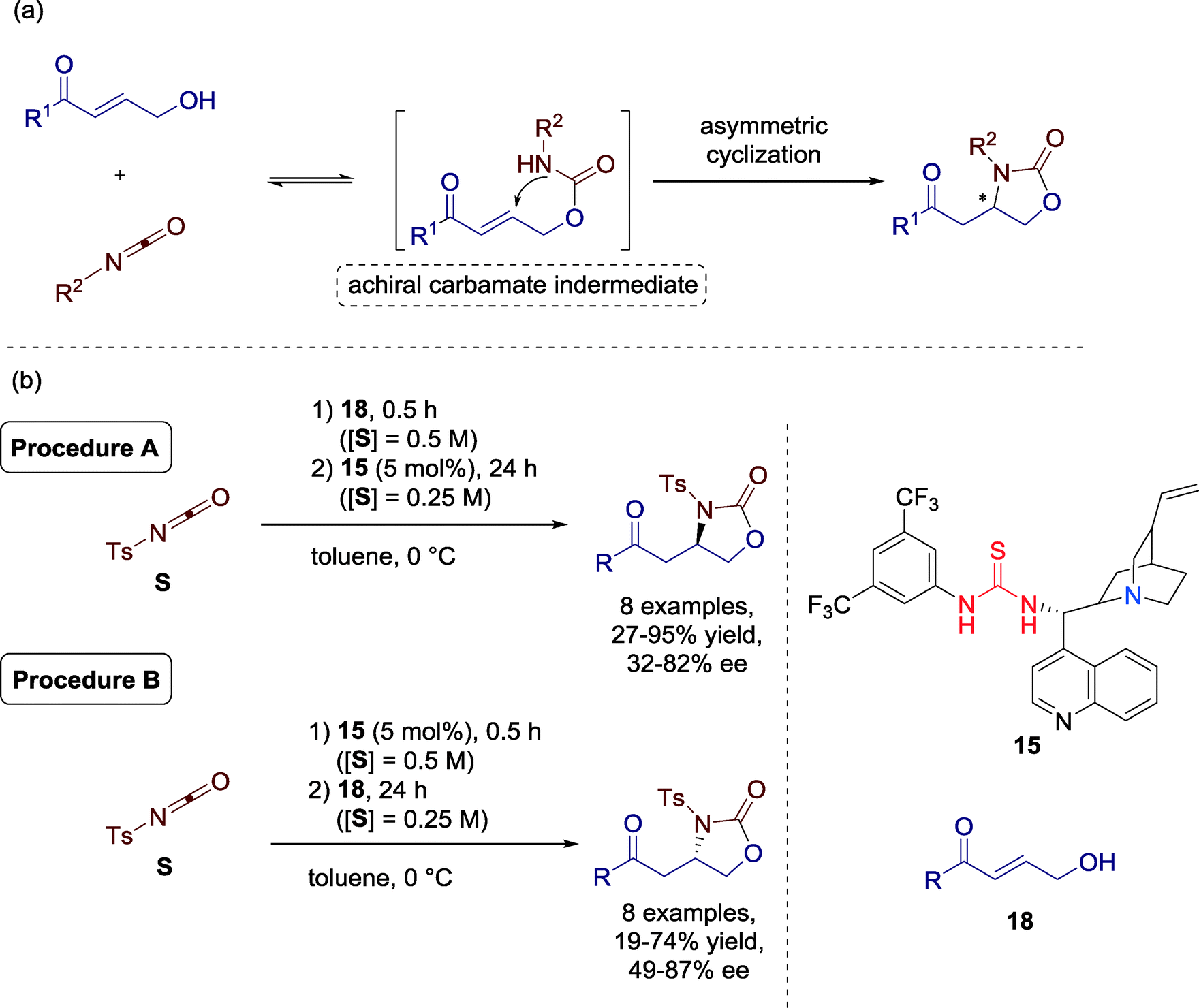 | ||
| Scheme 19 (a) Formal [3 + 2] cycladdition via asymmetric intramolecular hetero-Michael addition; (b) organocatalytic procedure-controlled cycloaddition. | ||
The versatility of the current protocol was demonstrated using a range of γ-hydroxy-α,β-unsaturated ketones. Both electron donating and withdrawing substituents were well tolerated for the reaction. Varying the addition sequence of the reactants generally resulted in a switch or, in one single cycloaddition, an increased stereoselectivity, albeit with the same configuration (10% ee (R) to 76% (R)). Additionally, enones with bulky biphenyl and naphthyl groups gave good performance under the established reaction conditions.
Based on 1H and 13C NMR studies and high-resolution mass spectrometry analysis the authors proposed that the Cinchona-alkaloid-derived amine-thiourea catalyst might be transformed in the presence of the isocyanate (Fig. 17), leading to the opposite enantiomer. Therefore, this zwitterionic adduct significantly affects the selectivity of the reaction and, thus, switch of diastereoselectivity can be achieved by a slight variation of the reaction parameters.
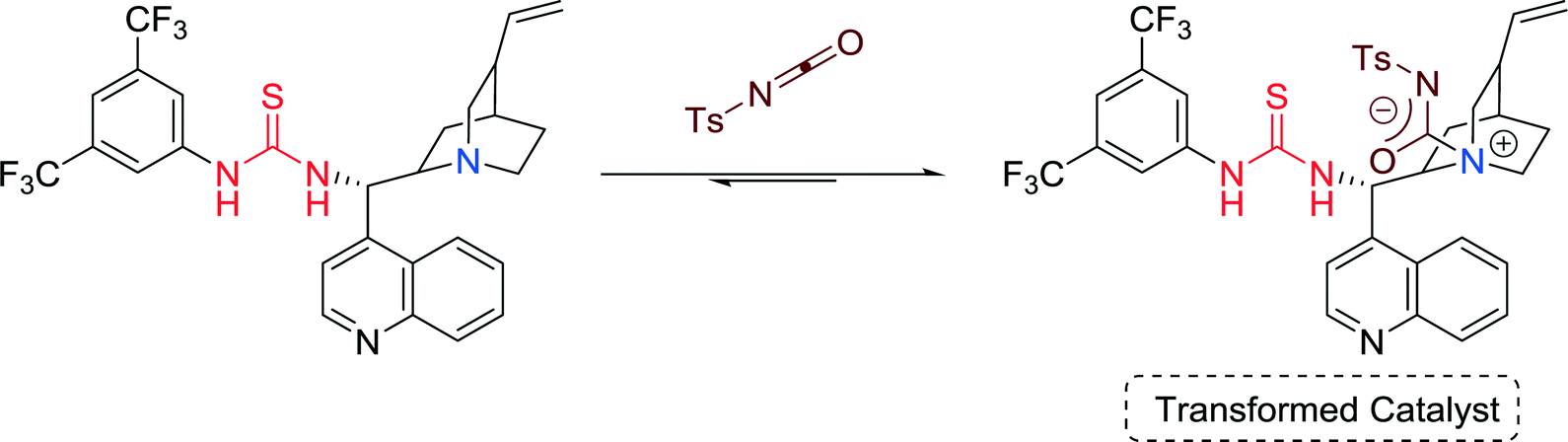 | ||
| Fig. 17 Proposed structure of the transformed catalyst. | ||
5. Cycloadditions catalyzed by squaramide derived catalysts
Beside thiourea catalysts, squaramide derivatives represent an alternative type of H-bond donor catalysts, developed and employed for the first time in 2008 by Rawal and co-workers for a highly enantioselective Michael addition of 1,3-dicarbonyl compounds to nitroolefins.58Due to a larger distance between the two N–H groups and some further structural alterations such as an enhanced delocalization through the cyclobutenedione system of the nitrogen lone pair, the pKa value of the squaramide moiety is lower compared to their thiourea analogues (Fig. 18). Hence, activity of the corresponding squaramide catalyst is raised.59
 | ||
| Fig. 18 Calculated H-bond distances in the thiourea- and squaramide moiety. | ||
This unique characteristic and the capability to effectively act as H-bond donor catalysts are useful attributes that make them an effective alternative to thiourea catalysts.
Thus, it is no surprising that squaramide derived catalysts can also be employed to successfully catalyze cycloaddition reactions including [4 + 2] and [5 + 2] in order to generate valuable compounds such as tetrahydroxanthone derivatives,60 3,3-spirooxindoles,61 as well as 8-oxabicyclo[3.2.1]octanes.62 These products might be of pharmacological and medicinal interest.63,64
5.1. [4 + 2] cycloadditions
In 2012, Jørgensen and co-workers developed the first H-bond-directed trienamine-mediated [4 + 2] cycloaddition. Herein, they employed different 2,4-dienals and 3-cyanochromones in the presence of squaramide-containing amino-catalyst 19 towards optically active tetrahydroxanthone derivatives,60 structural motifs common in several pharmacologically compounds (Scheme 20).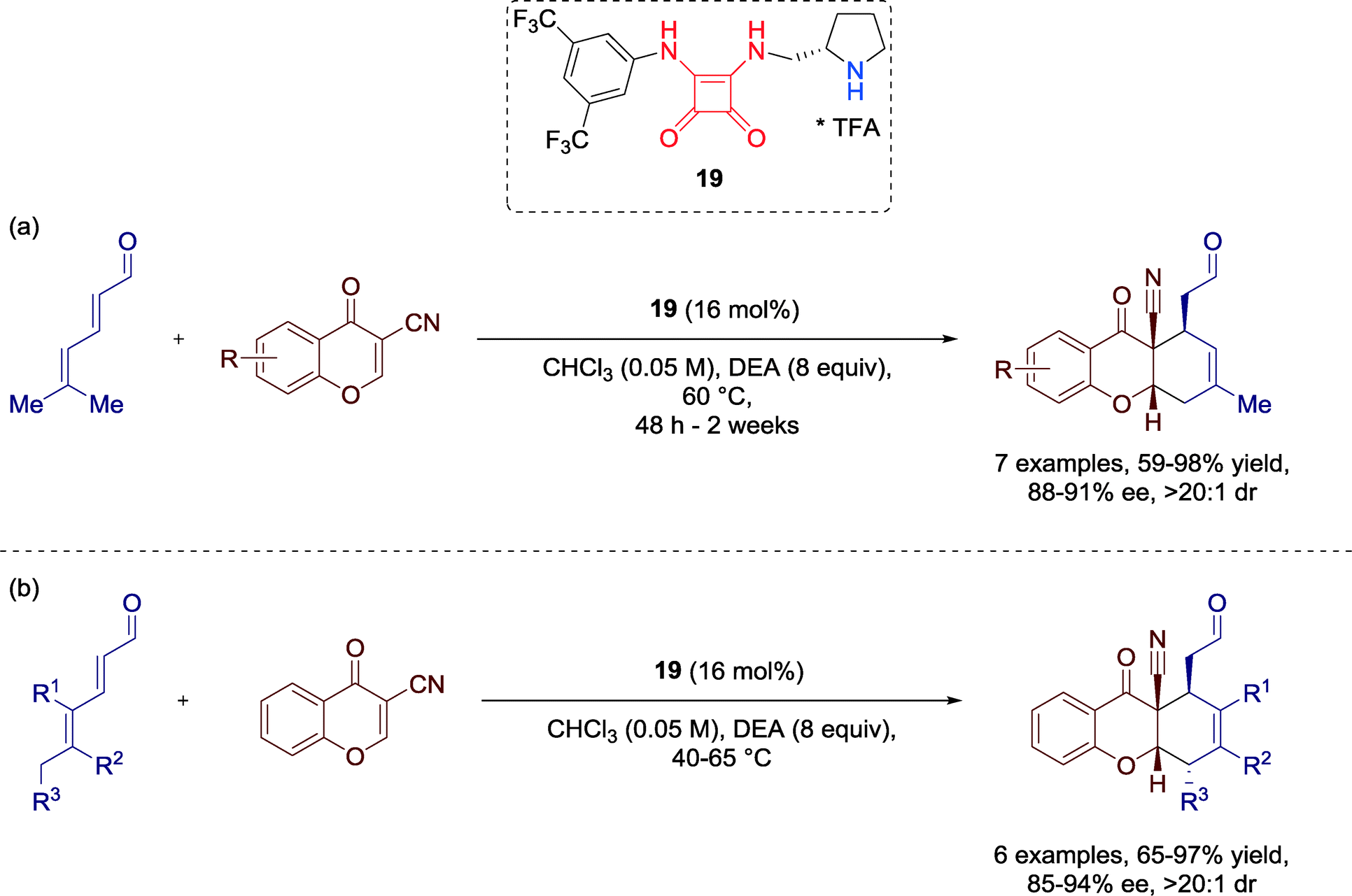 | ||
| Scheme 20 Enantioselective [4 + 2] cycloaddition towards optically active tetrahydroxanthones; DEA = diethanolamine. | ||
Moreover, the scope of this methodology could be successfully extended to other chromones and diversely substituted 2,4-dienals to furnish the tetrahydroxanthones in good yields and with high enantioselectivities.
In addition, the authors predicted two plausible transition state models for the cycloaddition reaction, depending on the residues at the 4-position of the aldehydes.
Aldehydes without substituent on the 4-position are proposed to be activated via transition state TS-1. Herein, the trienamine intermediate reacts in the s-cis (C3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4–C5C6) and s-cis (C1C2–C3C4) conformation, whereas transition state TS-2 is, due to steric repulsion, favored for aldehydes with residues at the 4-position, generating the trans relationship between the C1 and C4 stereogenic centers (Fig. 19).
C4–C5C6) and s-cis (C1C2–C3C4) conformation, whereas transition state TS-2 is, due to steric repulsion, favored for aldehydes with residues at the 4-position, generating the trans relationship between the C1 and C4 stereogenic centers (Fig. 19).
 | ||
| Fig. 19 Two transition state models for the cycloaddition with the corresponding products: for (a) unsubstituted and (b) substituted aldehydes in 4-position. | ||
In both cases, H-bonds are involved, generating an interaction between the electron-rich cyano group of the cyanochromone and the squaramide moiety of the catalyst. Additionally, secondary orbital overlap of the chromone carbonyl group and π–π-stacking interactions between the aromatic ring of the chromone and the conjugated π-system of the trienamine might further stabilize the transition states.
Moreover, the versatility of tetrahydroxanthones was demonstrated by several highly practical transformations towards polycyclic structures with moderate to good yields (Fig. 20).
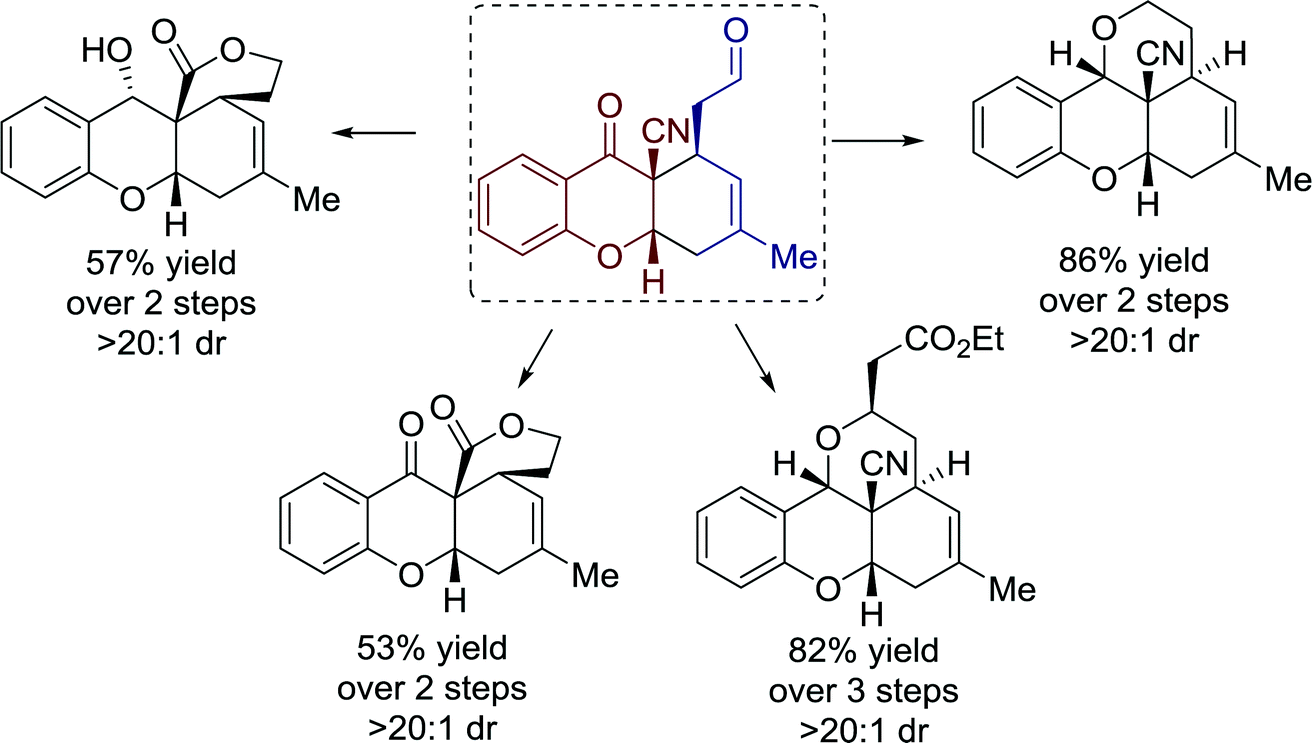 | ||
| Fig. 20 Synthetic utilization of tetrahydroxanthones. | ||
5.2. Tamura cycloaddition
In 2014, Connon and co-workers developed the first catalytic asymmetric Tamura cycloaddition reaction. By the aid of Cinchona-alkaloid squaramide-based catalyst 20 the authors generated densely functionalized 3,3-spirooxindoles, potential bioactive natural compounds of medicinal interest (Scheme 21).61,64 | ||
| Scheme 21 Organocatalytic asymmetric Tamura cycloaddition involving a squaramide catalyst. | ||
An assortment of enolizable anhydrides gave good performance for the cycloaddition reaction and all products could be isolated with good to excellent yields and high levels of enantioselectivity. Moreover, only one single diastereomer was observed in the crude reaction mixture. The great merit compared to earlier attempts of the Tamura cycloaddition is that this methodology is neither limited to homophthalic anhydride derivatives nor to α-succinic analogues. Furthermore, a tricyclic anhydride could be successfully employed for the reaction and the complex cycloaddition product was generated nearly enantiopure and in high yields.
Additionally, the authors observed a significant influence of the reaction temperature on the diastereocontrol of the reaction. A stepwise reduction from 30 to −50 °C resulted in a shift of the diastereomeric ratio and, furthermore, benefits the enantioselectivity of the reaction. At higher temperatures, diastereomer C is favored, whereas at 0 °C diastereoselectivity switches and kinetic product D is furnished as the major diastereomer (Scheme 22), an effect which further intensifies at lower temperatures. The authors suggested that a previously E to Z isomerization of the alkylidene oxindole might be responsible for this unique outcome, offering a more accurate control over the stereochemical outcome of the reaction.
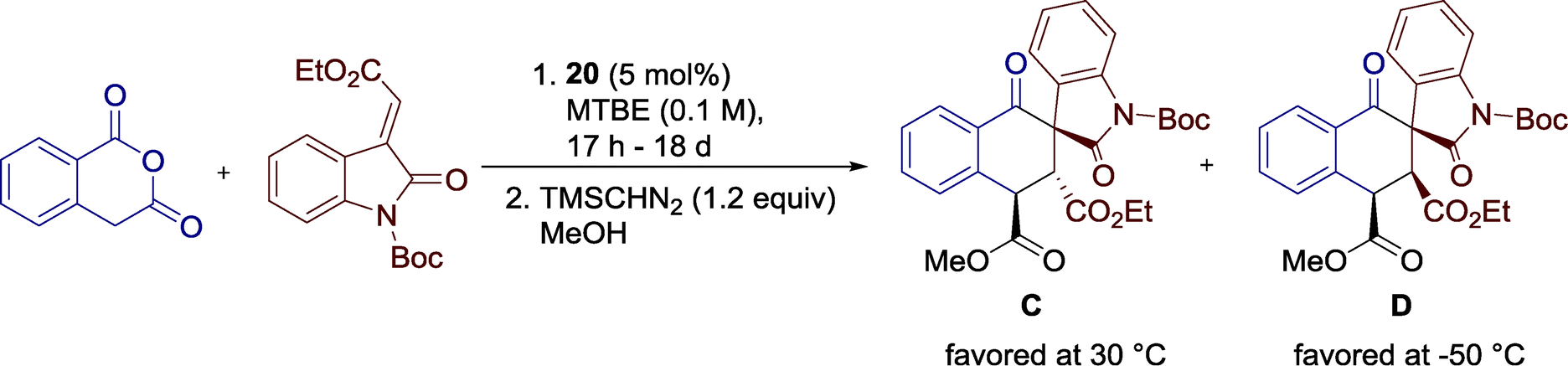 | ||
| Scheme 22 The effect of the temperature on diastereocontrol. | ||
5.3. [5 + 2] cycloadditions
Very recently, Vicario and Reyes successfully employed bifunctional secondary amine-squaramide catalyst 21 to establish a highly diastereo- and enantioselective [5 + 2] cycloaddition with oxidopyrylium ylides and enals towards compounds, bearing a 8-oxabicyclo[3.2.1]octane moiety (Scheme 23).62 | ||
| Scheme 23 (a) General reaction pathway for the [5 + 2] cycloaddition reaction employing (b) different α,β-unsaturated aldehydes and (c) various pyranones. | ||
After the optimization of reaction conditions with catalyst 21, the authors focused on extending the substrate scope. The performance of the reaction was satisfactory respecting both the yields and the enantioselectivities and different aliphatic and aromatic α,β-unsaturated aldehydes were well tolerated for the reaction (Scheme 23b). Furthermore, several pyranones could be successfully employed for the transformation (Scheme 23c).
Mechanistic studies indicated that a dienamine intermediate is formed after condensation of the enal with the secondary amine-squaramide catalyst, raising the HOMO energy of the dipolarophile and, thus, creating an exclusive β,γ-reactivity (Fig. 21).
 | ||
| Fig. 21 Catalytic enantioselective [5 + 2] cycloaddition via dienamine activation. | ||
6. Conclusions
Cycloaddition reactions are powerful synthetic tools for the generation of complex nitrogen- and oxygen-containing heterocyclic molecules in organic chemistry. This minireview has summarized the recent efforts and developments in different asymmetric cycloaddition reactions using bifunctional amine-thiourea and amine-squaramide organocatalysts. Whereas majority of the efforts to use bifunctional thiourea-based catalysts were directed on tertiary amine-thiourea/squaramide organocatalysts, since recently primary amine-thiourea and secondary amine-squaramide organocatalysts certainly delighted chemists by most unexpected activities in a variety of cycloaddition reactions.The scope of amine-thiourea and amine-squaramide organocatalysts is increasing constantly, and even versatile cycloaddition methods ([4 + 2], [3 + 2], formal [3 + 2], formal [3 + 3], formal [5 + 1], [5 + 2], 1,3-dipolar cycloadditions and Tamura cycloaddition) have been successfully developed with this type of bifunctional catalysis.
Despite the large number of excellent results obtained with different cycloaddition reactions, the versatility of amine-thiourea and amine-squaramide organocatalysts is still far from being fully explored and, thus, the entire reaction scope of different pericyclic transformations still remains to be uncovered in the near future.
Acknowledgements
S. B. T. gratefully acknowledges the financial support by grants from Deutsche Forschungsgemeinschaft (DFG, Projects TS 87/15-1 and TS 87/17-1) and thanks the Interdisciplinary Center for Molecular Materials (ICMM) & Erlangen Catalysis Resource Center (ECRC) for the generous research support.References
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS PubMed.
- B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- D. A. Evans and J. S. Johnson, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed.
- M. Trost and C. Jiang, Synthesis, 2006, 3, 369–396 CrossRef.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- F. E. Held, D. Grau and S. B. Tsogoeva, Molecules, 2015, 20, 16103–16126 CrossRef CAS PubMed.
- A. K. Unni, N. Takenaka, H. Yamamoto and V. H. Rawal, J. Am. Chem. Soc., 2005, 127, 1336–1337 CrossRef CAS PubMed.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- D. P. Curran and L. H. Kuo, J. Org. Chem., 1994, 59, 3259–3261 CrossRef CAS.
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS.
- P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217–220 CrossRef CAS PubMed.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- D. J. Hupe, The Chemistry of Enzyme Action, Elsevier, Amsterdam, 1984 Search PubMed.
- S. B. Tsogoeva and S. B. Jagtap, Synlett, 2004, 14, 2624–2626 CrossRef.
- S. B. Tsogoeva and S. Wei, Tetrahedron: Asymmetry, 2005, 16, 1947–1951 CrossRef CAS.
- A. L. Weber and S. Pizzarello, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12713–12717 CrossRef CAS PubMed.
- A. Córdova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes and Y. Xu, Chem. – Eur. J., 2006, 12, 5383–5397 CrossRef PubMed.
- M. Freund and S. B. Tsogoeva, Peptides for asymmetric catalysis, John WILEY & Sons, 2011 Search PubMed.
- Y. Takemoto, Org. Biomol. Chem., 2005, 4299–4306 CAS.
- S. J. Connon, Chem. – Eur. J., 2006, 12, 5418–5427 CrossRef PubMed.
- D. A. Yalalov, S. B. Tsogoeva and S. Schmatz, Adv. Synth. Catal., 2006, 348, 826–832 CrossRef CAS.
- S. B. Tsogoeva and S. Wei, Chem. Commun., 2006, 1451–1453 RSC.
- H. Huang and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 7170–7171 CrossRef CAS PubMed.
- O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051–7071 CAS.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- B. E. Maryanoff, D. F. McComsey, J. Russell, J. Taylor and J. F. Gardocki, J. Med. Chem., 1981, 24, 79–88 CrossRef CAS PubMed.
- K. Takahashi, B. Witkop, A. Brossi, M. A. Maleque and E. X. Albuquerque, Helv. Chim. Acta, 1982, 65, 252–261 CrossRef CAS.
- R. Ratnayake, D. Covell, T. T. Ransom, K. R. Gustafson and J. A. Beutler, Org. Lett., 2008, 11, 57–60 CrossRef PubMed.
- U. Murali Krishna, Tetrahedron Lett., 2010, 51, 2148–2150 CrossRef CAS.
- X. Jiang, X. Shi, S. Wang, T. Sun, Y. Cao and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 2084–2087 CrossRef CAS PubMed.
- M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891–1894 CrossRef CAS PubMed.
- N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed.
- M. R. Witten and E. N. Jacobsen, Angew. Chem., Int. Ed., 2014, 53, 5912–5916 CrossRef CAS PubMed.
- J.-W. Xie, L.-P. Fan, H. Su, X.-S. Li and D.-C. Xu, Org. Biomol. Chem., 2010, 8, 2117–2122 CAS.
- W. Raimondi, G. Lettieri, J.-P. Dulcère, D. Bonne and J. Rodriguez, Chem. Commun., 2010, 7247–7249 RSC.
- L. Wang, X.-M. Shi, W.-P. Dong, L.-P. Zhua and R. Wang, Chem. Commun., 2013, 3458–3460 RSC.
- W. Sun, G. Zhu, C. Wu, G. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 8633–8637 CrossRef CAS PubMed.
- L. Lykke, B. D. Carlsen, R. S. Rambo and K. A. Jørgensen, J. Am. Chem. Soc., 2014, 136, 11296–11299 CrossRef CAS PubMed.
- X. Chen, Z.-H. Qi, S.-Y. Zhang, L.-P. Kong, Y. Wang and X.-W. Wang, Org. Lett., 2014, 17, 42–45 CrossRef CAS PubMed.
- S. Zhao, J.-B. Lin, Y.-Y. Zhao, Y.-M. Liang and P.-F. Xu, Org. Lett., 2014, 16, 1802–1805 CrossRef CAS PubMed.
- J.-F. Bai, L.-L. Wang, L. Peng, Y.-L. Guo, J.-N. Ming, F.-Y. Wang, X.-Y. Xu and L.-X. Wang, Eur. J. Org. Chem., 2011, 2011, 4472–4478 CrossRef CAS.
- B.-J. Li, L. Jiang, M. Liu, Y.-C. Chen, L.-S. Ding and Y. Wu, Synlett, 2005, 4, 603–606 Search PubMed.
- B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS PubMed.
- S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS PubMed.
- J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC.
- J. Wang, H. Li, L. Zu, W. Jiang, H. Xie, W. Duan and W. Wang, J. Am. Chem. Soc., 2006, 128, 12652–12653 CrossRef CAS PubMed.
- Y.-Q. Wang, J. Song, R. Hong, H. Li and L. Deng, J. Am. Chem. Soc., 2006, 128, 8156–8157 CrossRef CAS PubMed.
- Y. Wang, R. G. Han, Y. L. Zhao, S. Yang, P. F. Xu and D. J. Dixon, Angew. Chem., Int. Ed., 2009, 48, 9834–9838 CrossRef CAS PubMed.
- W. Wu, L. Min, L. Zhu and C.-S. Lee, Adv. Synth. Catal., 2011, 353, 1135–1145 CrossRef CAS.
- L.-L. Wang, J.-F. Bai, L. Peng, L.-W. Qi, L.-N. Jia, Y.-L. Guo, X.-Y. Luo, X.-Y. Xu and L.-X. Wang, Chem. Commun., 2012, 48, 5175–5177 RSC.
- B. Tan, X. Zeng, W. W. Y. Leong, Z. Shi, C. F. Barbas and G. Zhong, Chem. – Eur. J., 2012, 18, 63–67 CrossRef CAS PubMed.
- K. Asano and S. Matsubara, Org. Lett., 2012, 14, 1620–1623 CrossRef CAS PubMed.
- M.-X. Zhao, H. Zhou, W.-H. Tang, W.-S. Qu and M. Shi, Adv. Synth. Catal., 2013, 355, 1277–1283 CrossRef CAS.
- Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2013, 135, 12160–12163 CrossRef CAS PubMed.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed.
- X. Ni, X. Li, Z. Wang and J. P. Cheng, Org. Lett., 2014, 16, 1786–1789 CrossRef CAS PubMed.
- Ł. Albrecht, F. Cruz Acosta, A. Fraile, A. Albrecht, J. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 9088–9092 CrossRef PubMed.
- F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 2628–2632 CrossRef CAS PubMed.
- A. Orue, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Angew. Chem., Int. Ed., 2015, 54, 3043–3046 CrossRef CAS PubMed.
- R. N. Kharwar, A. Mishra, S. K. Gond, A. Stierle and D. Stierle, Nat. Prod. Rep., 2011, 28, 1208–1228 RSC.
- L. Hong and R. Wang, Adv. Synth. Catal., 2013, 355, 1023–1052 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |