
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Potential and challenges of zeolite chemistry in the catalytic conversion of biomass
Thijs
Ennaert
,
Joost
Van Aelst
,
Jan
Dijkmans
,
Rik
De Clercq
,
Wouter
Schutyser
,
Michiel
Dusselier
,
Danny
Verboekend
* and
Bert F.
Sels
*
Centre for Surface Chemistry and Catalysis, Faculty of Bioscience Engineering, KU Leuven, Celestijnenlaan 200f, B-3001, Heverlee, Belgium. E-mail: bert.sels@biw.kuleuven.be; danny.verboekend@biw.kuleuven.be; Fax: +32 16 321 998; Tel: +32 16 321 610
First published on 21st December 2015
Abstract
Increasing demand for sustainable chemicals and fuels has pushed academia and industry to search for alternative feedstocks replacing crude oil in traditional refineries. As a result, an immense academic attention has focused on the valorisation of biomass (components) and derived intermediates to generate valuable platform chemicals and fuels. Zeolite catalysis plays a distinct role in many of these biomass conversion routes. This contribution emphasizes the progress and potential in zeolite catalysed biomass conversions and relates these to concepts established in existing petrochemical processes. The application of zeolites, equipped with a variety of active sites, in Brønsted acid, Lewis acid, or multifunctional catalysed reactions is discussed and generalised to provide a comprehensive overview. In addition, the feedstock shift from crude oil to biomass involves new challenges in developing fields, like mesoporosity and pore interconnectivity of zeolites and stability of zeolites in liquid phase. Finally, the future challenges and perspectives of zeolites in the processing of biomass conversion are discussed.
 Thijs Ennaert | Thijs Ennaert obtained his MSc in Catalytic Technology (Bioscience Engineering) at KU Leuven (Belgium) in 2012. He did his master thesis at the Center for Surface Chemistry and Catalysis, where he explored the stability of USY zeolites in hot liquid water. He is currently doing a PhD as a IWT-funded fellow, also under the guidance of Prof. Bert F. Sels. His research focuses on the stability and application of zeolite catalysts in biomass transformations. |
 Danny Verboekend | Danny Verboekend obtained his PhD at ETH Zurich (Switzerland, 2012) on the preparation and exploitation of hierarchical zeolite catalysts. Afterwards, he continued to work at the same university in the context of a post-doctoral stay (2012–2014). He is currently working at the KU Leuven (Belgium) as a FWO-funded post-doctoral fellow on the topic of heterogeneous catalysis. His general interest focuses on combining applied and sustainable research with the highest degree of fundamental understanding. |
 Bert F. Sels | Bert F. Sels, currently full professor at the KU Leuven, obtained his PhD in 2000 in the field of heterogeneous oxidation catalysis. He was awarded the DSM Chemistry Award in 2000, the Incentive Award by the Societè Chimique Belge in 2005 and the Green Chemistry Award in 2015. He was recently elected co-chair of the IZA Catalysis Commission and is international board member of ChemSusChem. He heads a research group in the Centre for Surface Chemistry and Catalysis, designing heterogeneous catalysis for future challenges in the industrial organic and environmental catalysis. His expertise includes heterogeneous catalysis in biorefineries and the spectroscopic and kinetic study of active sites for small molecules activation. |
Introduction
The limited supply of cheap petroleum has renewed the interest towards more sustainable energy sources like wind, solar and hydro technology. While these renewable energy sources have the potential to constitute an important part of the energetic matrix, the production of chemicals and fuels requires a renewable carbon source, such as biomass (Fig. 1). Accordingly, these possibilities have sprouted a widely-varied research attention on the valorisation of various biomass resources (Fig. 2).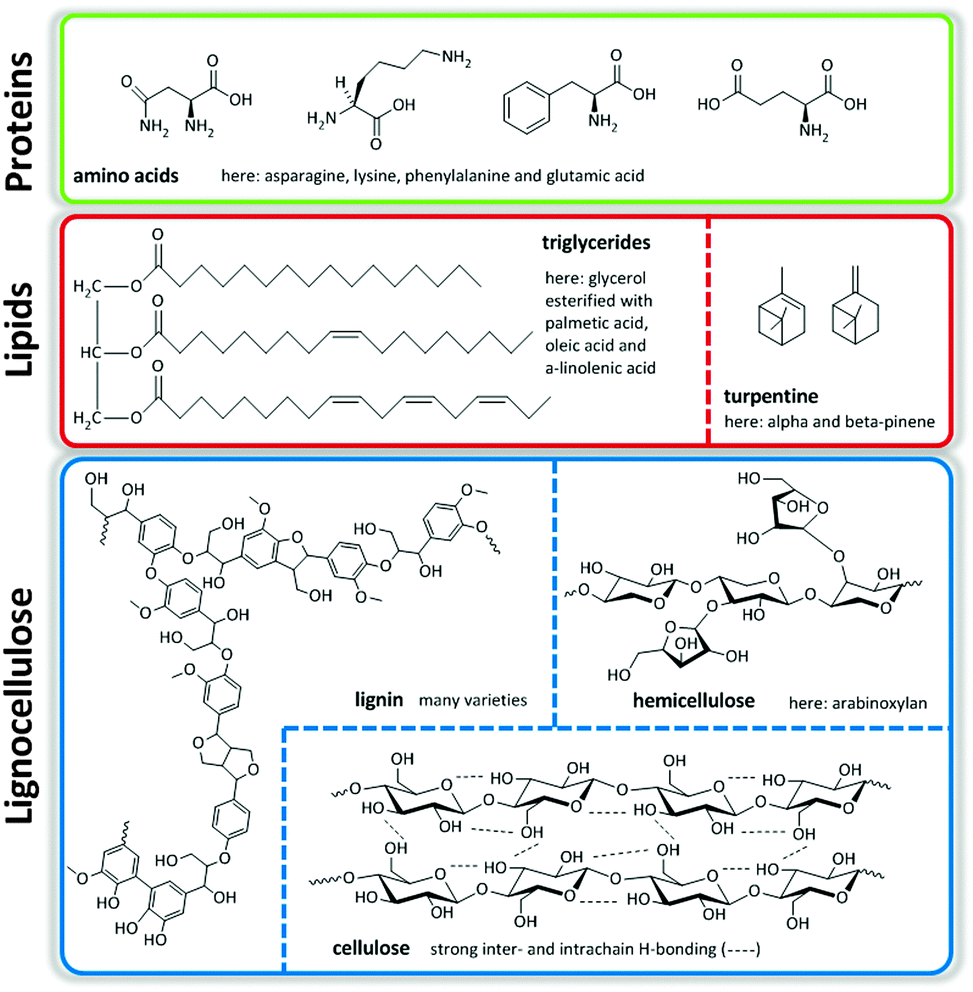 | ||
| Fig. 1 Overview of the prominent fractions of the biomass feedstock. | ||
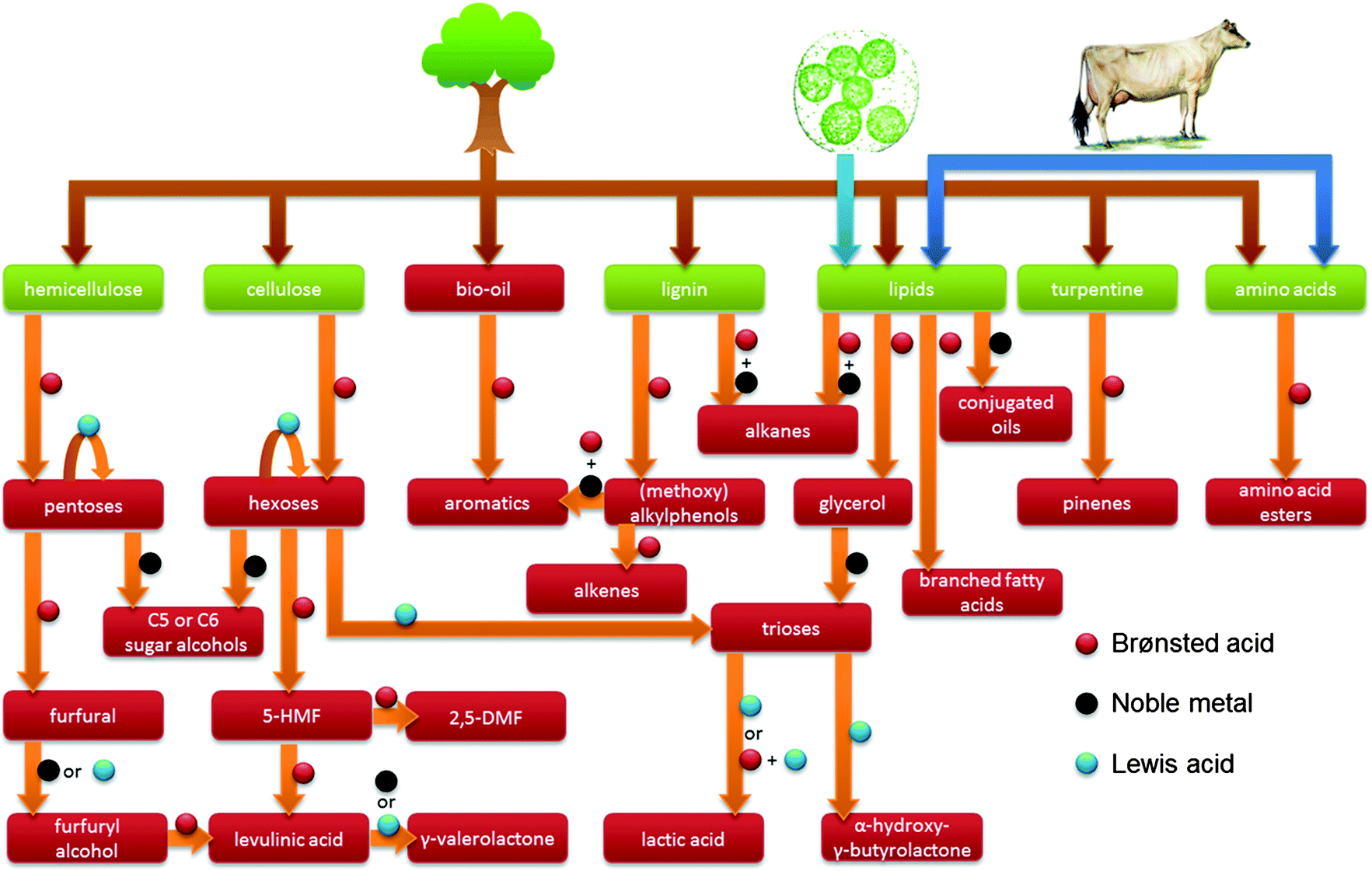 | ||
| Fig. 2 Overview of the use of zeolites and the respective catalytic active sites in a selection of biomass transformations discussed in this review. | ||
Major streams of biomass are lignocellulose, lipids containing triglycerides from animal fats, vegetable origin and microalgae, and turpentine streams. Of these, lignocellulose, composed of cellulose, hemicellulose and lignin, represents the most abundant one. Finally, there is a smaller group of protein-based fractions of animal or vegetable origin (Fig. 1).1,2 The structural nature of these molecules determines the ultimate transformation routes needed to convert them to useful fuels, chemicals and materials.
Three distinct modes of valorising the carbon content of biomass may be distinguished (Fig. 3). The first mode represents biological transformations, exemplified by microbial or yeast-based fermentation processes, such as the commercial conversion of carbohydrate biomass into ethanol or lactic acid.3–8 The second mode is thermal, ranging from burning biomass for heat generation over gasification to biogas or liquefaction and pyrolysis to bio-oils.9 The third mode encompasses all specific chemical and catalytic processes, which target a single chemical or narrow range of products, perhaps with the exception of catalytic fast pyrolysis (CFP).
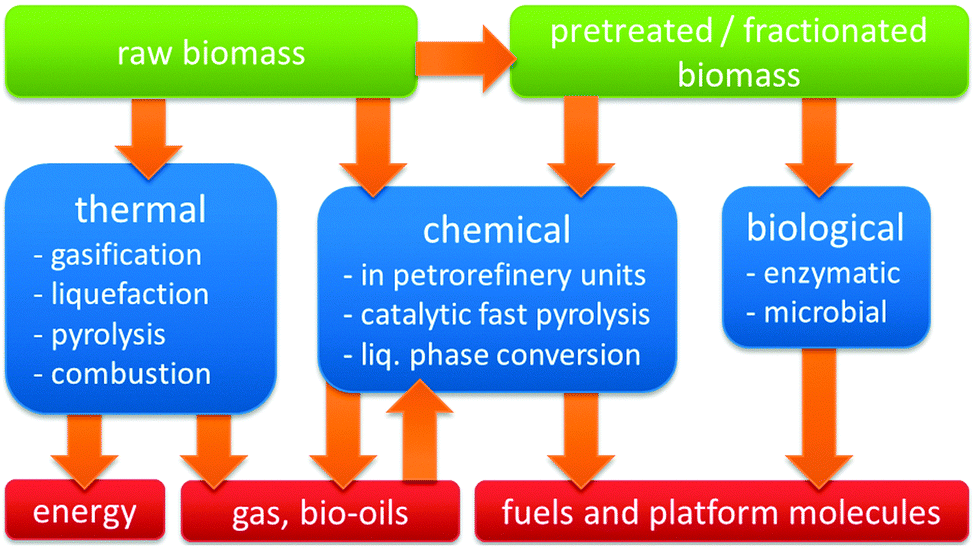 | ||
| Fig. 3 Schematic representation of the different modes of biomass conversion. | ||
Zeolite catalysis plays a significant role in many of the chemical biomass conversion routes (Fig. 2 and 3). First, there is the concept of blending-in biomass fractions, pyrolysis oils or platform molecules into existing petroleum refinery operations.10–12 Secondly, in CFP, either on platform compounds, such as glucose or furans, or raw biomass feedstock, zeolites have proven promising efficiency.13–16 Thirdly, specific liquid phase catalytic processes have been proposed, where zeolites contribute in converting raw biomass feedstock. The last, and most documented use of zeolites in biomass valorisation, is found one-step down from the raw or pretreated feedstock in Fig. 1, namely in the upgrading of bio-derived platform molecules to fuels, chemicals and materials by specific transformations. In general, these processes target the on-purpose synthesis of one final product or intermediate from a biomass-derived (platform) molecule.10,12,17–25
The wide-scale application of zeolites in biomass conversion can be explained by their numerous positive attributes. Zeolites are aluminosilicates with a crystalline, microporous framework built from oxide tetrahedra.26 Accordingly, zeolite crystals are highly porous, with precisely-defined micropores (0.4–1 nm). Combined with the ability to load them with exchangeable cations makes them useful as adsorbents, molecular sieves, ion exchangers and catalysts. For example, since breakthrough work in the early 1960s, synthetic zeolites have become the most prominent heterogeneous catalysts in the refining and petrochemical industries.27–31
In fossil fuel-based refineries, zeolite-based catalysts play a key role.32 Of the different applications, fluid catalytic cracking (FCC), where the atmospheric crude oil distillation residue is transformed into automotive (gasoline) fuel components, represents in volume terms by far the largest zeolite consumption. Typical FCC catalysts are mainly comprised of ultra-stable zeolite Y (USY) combined with amorphous aluminosilicates as catalytically active matrix. The key catalytic parameters are the strength and density of the Brønsted acidic sites, which arise when protons neutralise the negative charge introduced by Al substitution in the framework.
The presence of extra-framework Al (EFAl) species in USY zeolites, generated during the steaming process, was shown to positively affect the cracking performance. Next to improving the Brønsted acidic function, EFAl is also a source of Lewis acidity, a second prominent zeolite function. Besides EFAl, also the incorporation of heteroatoms in the zeolite structure creates Lewis acidic sites in the so-called zeotypes. TS-1, a titanosilicate of the MFI topology, is probably the most familiar example. These high-silica molecular sieves serve in the functionalisation of hydrocarbon derived base-chemicals, such as oxidation reactions. Newer examples for instance contain incorporated Sn, like in Sn-beta.33–39
In the catalytic processes that follow FCC, the different fractionated oil and gas streams are further upgraded. Therein, zeolites play a crucial role, among others, in removal of functional groups via hydrotreating and hydroisomerisation processes, alkylation, chlorination and oxidation. Hereby, besides monofunctional Brønsted acidic sites, also bifunctional or multifunctional catalytic properties are employed, usually by the addition of metallic nanoparticles. These particles can easily be introduced in zeolites with high metal dispersions, due to the ion-exchange capacity generated by the exchangeable nature of the charge-compensating cations, which afford a hydrogenation component after reduction. A fourth less-studied functionality is caused by the introduction of alkali metals providing the zeolite with basic properties.
Zeolites excel in catalytic processes starting from petroleum feedstock based on their strong and tuneable acidity, their microporous dimensionality (<2 nm, either in 1, 2 or 3D) allowing molecular traffic control and high reaction surface areas, their robustness and high thermal stability leading to easy regeneration at elevated temperatures, and finally, their adjustable nature so that they match the need of the desired chemical transformation. The latter is exemplified by the ease of engineering the pores, adjusting composition and crystal size or shaping them into a catalyst pellet.40 Literature on the use of zeolites in petrochemical and refining industries is vast and many synthesis–property–function (spf) relations have been established.41,42 This stands in great contrast with the field of biomass, where, although zeolites are routinely applied,32,43,44 tailor-made zeolite design remains at its infancy.
In this contribution, we highlight the progress made in zeolite catalysed conversion of biomass and consider herein the most recognised concepts extrapolated from established petrochemical processes. This rationale structures the review into four prominent zeolite catalysis concepts with parallels to petrochemistry and two new challenges arising from the transition of fossils towards renewable feedstocks (Fig. 4). An insightful comparison between zeolite-catalysis in petrochemistry (and refining) and biomass conversion is presented, with details on how key zeolite concepts like strong Brønsted and Lewis acidity, multifunctional catalysis and shape-selectivity are transferred. One of the challenges is the intrinsic microporosity of the zeolites. As pores are too small to allow diffusion of large biopolymers, creation of a secondary level of porosity (mesopores) is profitable, enhancing number of accessible active sites. A second challenge regards the stability of zeolites in aqueous solutions. In contrast to the importance of a zeolite's steam-stability in the gas phase, the behaviour of zeolites in the condensed phase, such as hot liquid water (HLW), is quite distinct. Finally, the work is concluded and perspectives are discussed.
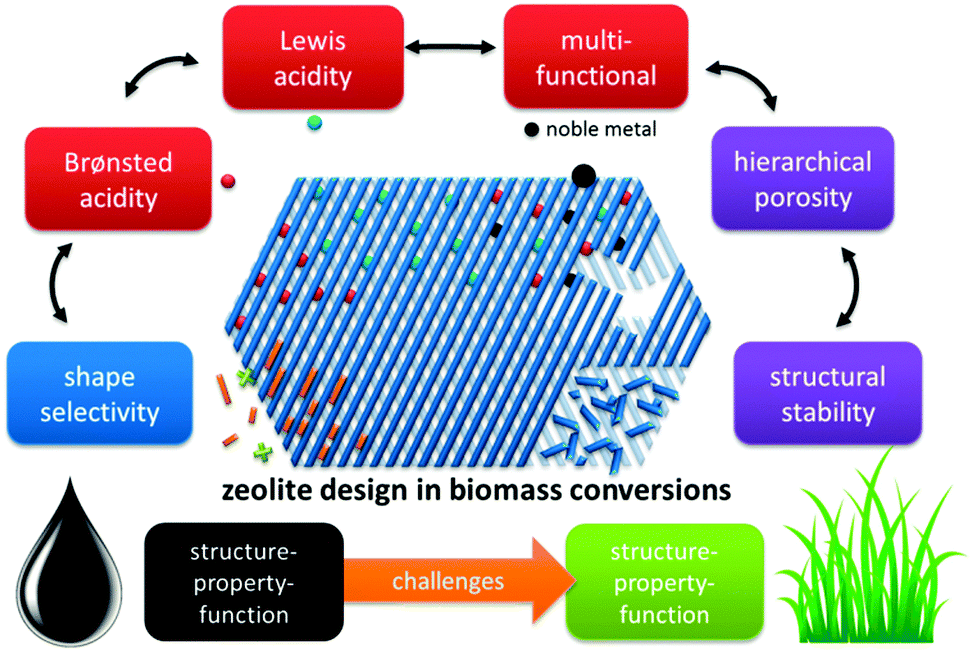 | ||
| Fig. 4 Concepts of zeolite catalysis found in biomass conversion, paralleled with petroleum-based processes. | ||
Concept 1: Brønsted acidity
Biomass, alike crude oil, constitutes of high-molecular weight components, viz. (hemi)cellulose, lignin and triglycerides. In accordance with crude oil refining, the first objective of biomass valorisation consists of decreasing the molecular mass. Therefore, in an extension to their widespread use in oil refinery and petrochemistry, Brønsted acidic zeolites should also be able to play a pivotal role in cracking and upgrading the biomass components. However, one can expect that the Brønsted acidic activation mechanism of large and bulky biomass substrates containing high oxygen content will be more challenging than Brønsted acidic activation of hydrocarbons.Conversion of fats and oils
Fats and oils, having long linear hydrocarbon chains, are structurally most related to petrochemical hydrocarbons. Their isomerisation and cracking chemistry is therefore closely associated to the well-known acid catalysed carbocation chemistry. The natural presence of double bonds in fatty molecules makes them very sensitive to acid catalysed isomerisation (Scheme 1), with for example the formation of mono-branched-chain unsaturated fatty acids (MoBUFA), but also to less-desired oligomerisation. However, isomerisation does not form a pure MoBUFA stream, but rather a mixture of a variety of branched and straight-chain C10–C20 components.45–47 As MoBUFA are interesting compounds as lubricants and for their use in cosmetics,48 there exists a need for more selective processes. While some clays give moderate yields,49–52 zeolites are used with more success53–58 due to their relative small channels and pores.47 As the zeolite pores are already blocked at the beginning of the reaction due to cokes formation,47 the isomerisation takes place solely at the entrance of the micropores (Fig. 5), making it a classic example of pore mouth catalysis.47 Based on the pore geometry, ferrierite-based catalysts were established as superior catalysts,53 especially for the formation of methyl MoBUFA,59 while zeolites with larger pores (Y, USY and beta) mainly produce ethyl and propyl MoBUFA.55 Deeper study showed that MoBUFA are only selectively produced when the Brønsted acid site speciation and density is optimized59 (up to 85% selectivity).53 For example, to suppress the formation of polymeric products on the acid sites at the outer surface of the zeolite, a small amount of bulky triphenylphosphine (TPP) base is added to the reaction mixture (Fig. 5).60 Although very elegant, it remains unclear whether the less-selective surface sites cannot be removed prior to reaction by acid-treatment of the zeolites.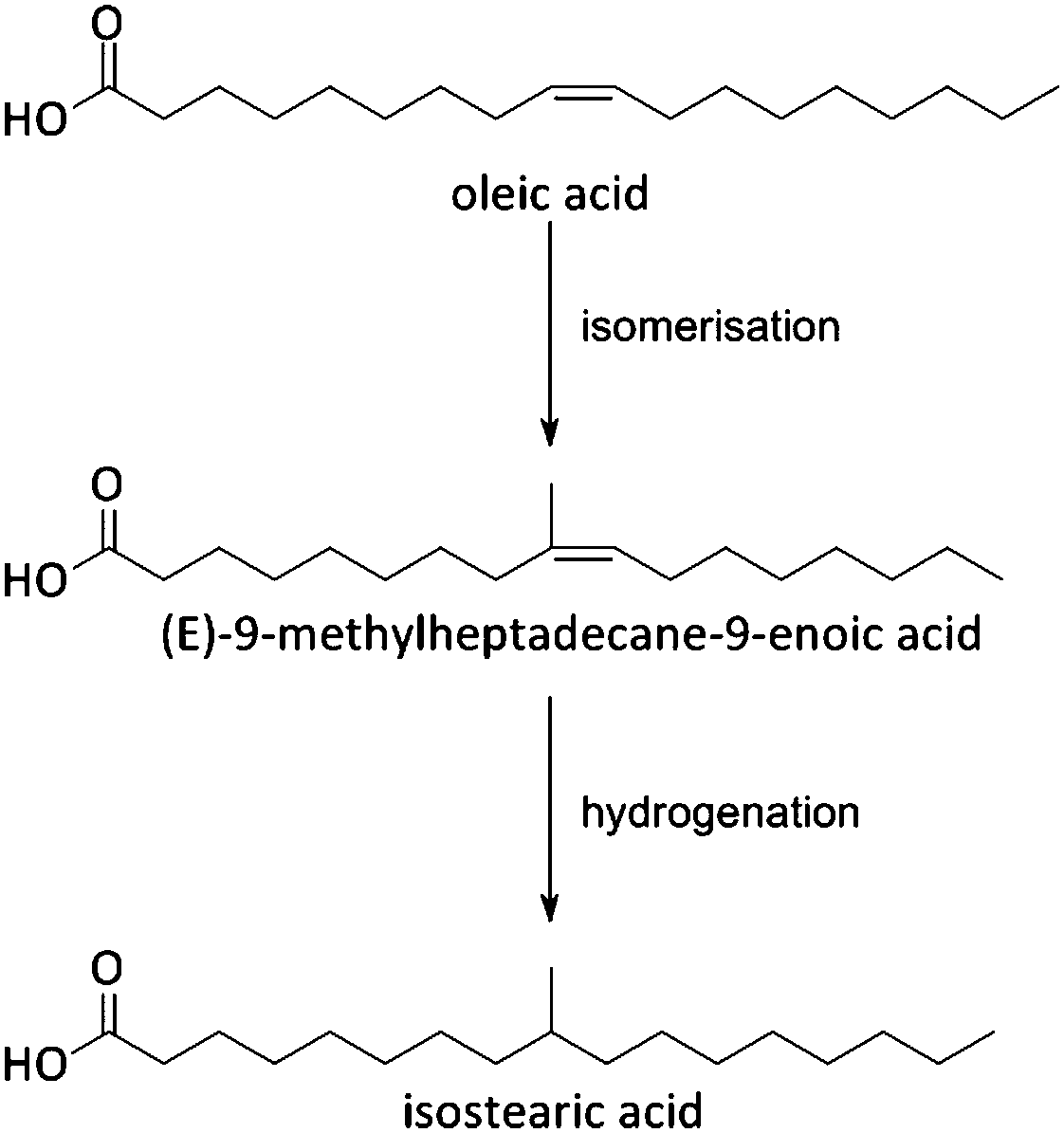 | ||
| Scheme 1 Isomerisation of oleic acid to isostearic acid. | ||
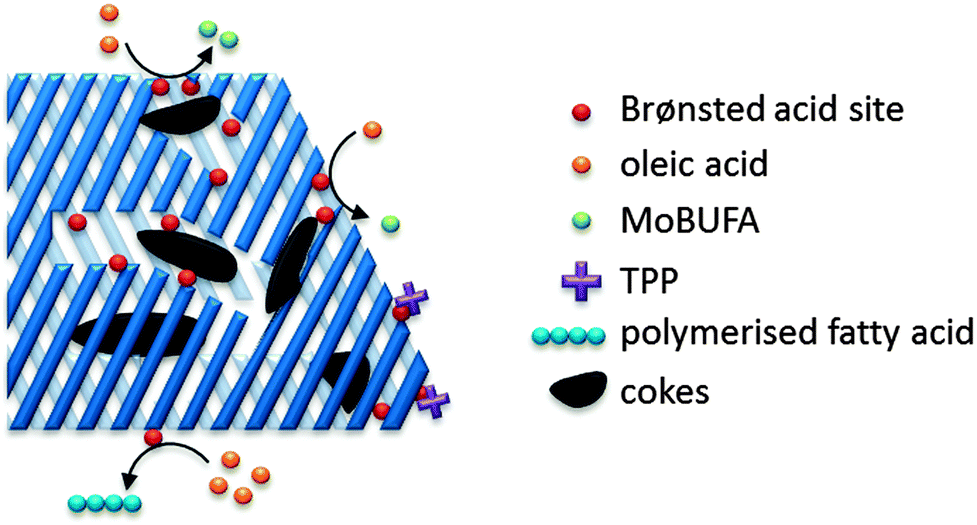 | ||
| Fig. 5 Schematic presentation of the pore mouth catalytic conversion of oleic acid over Brønsted acidic zeolites towards MoBUFA in the presence of TPP. | ||
Higher temperatures with protonated zeolites allow C–C bond cracking61–66 forming gasoline and light naphtha fractions (with especially ZSM-5), diesel (with USY and beta), hydrocarbon gases (like C3–C5 paraffins and olefins) and cokes,64,67,68 the latter being particularly pronounced for the less selective large-pore zeolites. Obviously, not only the zeolite structure but also the type and origin of the substrate influences the product distribution. Fats with only an alcohol function (fatty alcohols) produce more valuable products than fatty acids.69 The presence of unsaturations in the alkyl chain affords more aromatic products than in case of saturated fats.69,70 Yet, it is preferable to prevent the extensive formation of aromatics as these have the tendency to undergo polymerisation reactions towards cokes.10 Finally, triglycerides, being too bulky to easily access the micropores, display lower conversions and product selectivities than fatty acids or alcohols.69 Accordingly, they first require thermal or catalytic decomposition prior to efficiently entering the zeolite micropores. However, these decomposition products have higher tendencies to form condensation products, resulting in higher amounts of aromatics, heavy products and cokes. As an alternative to direct cracking, fats and oils can firstly undergo deoxygenation towards pure hydrocarbons which can consequently be cracked in classic refinery units.12,71–79
(Catalytic) fast pyrolysis of lignocellulose and upgrading of pyrolysis vapours
As lignocellulose molecules are much bulkier than fatty acids, zeolite cracking of lignocellulose is always preceded by a pretreatment step, mostly a thermal degradation. One of the economically most interesting thermochemical pathways for lignocellulose degradation, besides liquefaction, is a catalyst-free fast pyrolysis of dry biomass.80–82 During fast pyrolysis, the biomass substrate is rapidly heated up to 400–600 °C for short residence times (1–2 s).9 Afterwards, it is quickly cooled down whereby a thermally unstable black liquor, called bio-oil, is formed, containing up to 300 different compounds.83 However, the obtained bio-oil is not compatible with conventional liquid transportation fuels like gasoline and diesel due to the high oxygen content. Therefore, zeolite-based processes play an important role to extensively adjust the characteristics of bio-oils to become compatible with conventional transportation fuels.84The upgrading of pyrolysis vapours is pioneered in the late 1980's and early 1990's.85–89 It was demonstrated that the pyrolysis vapours are very reactive with Brønsted acidic ZSM-5 zeolites forming aromatics, both mono- and poly-aromatics.84,86–93 Especially the group of Huber extensively studied the potential of Brønsted acidic zeolites in upgrading pyrolysis products. They integrated the zeolite catalysed upgrading step in the pyrolysis process (i.e. CFP). During this process, the pyrolysis is carried out in the direct presence of the zeolite catalyst, which leads to a more intense contact between substrate and zeolite catalyst, resulting in a much higher zeolite impact compared to catalytic upgrading of pyrolysis vapours. Yet, a major disadvantage of CFP is the higher cokes deposition on the catalyst, leading to faster catalyst deactivation. Therefore, proper catalyst selection by optimal tuning of the catalyst pore structure and active sites, adjustments where zeolite catalysts are well suited for, is necessary to reduce the cokes and increase the aromatics yield.
It was shown that both the Brønsted acidic sites as well as the presence of a crystalline pore structure are prerequisites for aromatics production from carbohydrate feedstocks.94,95 Whereas ZSM-5 yields over 30% aromatics, USY and beta mainly produce cokes, indicating the importance of the specific pore structure of ZSM-5.15,90 These observations are supported by the fact that only medium pore zeolites, with pore sizes between 5.2 and 5.9 Å, selectively produce aromatics, due to an appropriate combination of pore size and cavity dimensions.91 In contrast, small pore zeolites produce mainly oxygenates and cokes as the diffusion of reactants and products is strongly hindered. Despite of the faster reactant diffusion in large pore zeolites, these zeolites are also not well suited as the large pores allow formation of poly-aromatics due to the lack of reactant/transition state confinement (see Concept 4 for more details about shape-selectivity).
Compared to cracking of fossil fuels, biomass cracking requires not only breaking of C–C bonds but also a considerable amount of C–O bonds. This change in substrate composition also translates into a switch of the optimal Si/Al needed, with higher Si/Al ratios for biomass cracking. C–O bonds have lower dissociation energies, so they do not need zeolites with high acid density or acid strength. Where typical fossil fuel cracking catalysts show the highest activity for Si/Al in the range of 5–8,92 the highest activity in biomass cracking can be found around 15.93 Such medium Si/Al zeolites show the best balance between availability of Brønsted acid sites on the one hand and maintaining enough space between the Brønsted acid sites to prevent side reactions on the other hand.93
A major obstacle during pyrolysis is catalyst deactivation due to cokes deposition induced by the high oxygen content of the substrate.85 Moreover, it has been suggested that the cokes formed during pyrolysis are mainly attributed to the presence of lignin.94,95 Therefore, removal of lignin prior to pyrolysis or more selective pyrolysis approaches with lignin will reduce the cokes formation. Yet, specific research about CFP of lignin is limited,94–105 therefore, deeper insight in pyrolysis of lignin towards gasoline range products or petrochemicals like toluene, phenol or benzene will be a capital gain for CFP of biomass in general. As cokes are also an issue in zeolite catalysis in petrochemistry, reactor set-ups used in commercial refinery plants can provide ideas to enhance the regeneration capacity of zeolites in pyrolysis of biomass. The optimal manner to study the regeneration capacity is operating at continuous mode or in line connected set-ups. Only a few studies already addressed this problem. For example, for zeolites ZSM-5 and USY, a 3 h steady state operation in a circulating fluid bed reactor, consisting of a riser reactor, fluid bed regenerator and a stripper was proposed.106 Another idea is a bubbling fluidised bed reactor with on-stream addition and removal of spray-dried zeolite catalyst. When the CFP of pine wood with ZSM-5 was studied in this set-up, it was possible to maintain a constant aromatics yield of 16% over an extended reaction period of 6 h.107
Liquid phase catalytic conversion of sugar-derived compounds
Besides the thermochemical conversion of biomass, chemocatalytic conversion is a promising strategy as it enables, in contrast to pyrolysis, targeting one single product or a narrow range of products. Chemocatalytic conversions of biomass are mostly liquid phase processes which is a major difference with the gas phase processes in crude oil refining and upgrading of pyrolysis products. This leads to several new challenges, including the (until recently) unknown behaviour of zeolites in hot liquid water (HLW), tackling the low stability of some targeted oxygen-rich products, as well as gaining more insight on the exact nature of the active sites of zeolites in aqueous conditions. Due to the manifold use of zeolites in gas phase processes, the dynamics and stability of the catalytic sites under such conditions are well-known.108–111 The nature of the sites directly determines the intrinsic strength of the sites. Brønsted acid sites are highly polarised hydroxyl groups, while Lewis acid sites are coordinatively unsaturated cationic sites accepting electron pairs of guest molecules.112 Yet, when these active sites are surrounded by polar molecules (e.g. condensed water) complex equilibria between the surface groups of the zeolite and the solvent molecules occur. Solvent molecules may react with Lewis acid sites generating Brønsted acidic sites.113 Besides, when surface hydroxyl groups ionize as Brønsted acids or bases, altered reactivity of surface sites can be observed.113Zeolites have already shown potential in various Brønsted acidic reactions in condensed phase. A well-known example hereof is the dehydration of sugars towards platform molecules like 5-hydroxymethylfurfural (5-HMF) (Fig. 2 and Scheme 2).314,315 Sugar dehydration has already been studied for nearly two centuries. The first reports date from 1840,114 but the major breakthrough came at the end of the 19th century, when the synthesis of 5-HMF, the dehydration product of glucose and fructose, was firstly described. Dehydration of pentoses, like xylose, into furfural was first described half a century later.115 In the subsequent decades, there was an ever increasing interest in the dehydration of sugars.18,116–118 Only since 1990, zeolites were implemented in dehydration reactions of sugars. However, the obtained 5-HMF yields with zeolites, mainly zeolite Y and ZSM-5, are low, generally between 10 and 25%.119–121 The low yields are mainly rooted in the instability of the highly oxygenated molecules (both sugars, intermediates as end-products) at high temperatures and in acidic conditions, especially in aqueous media. For example, 5-HMF is prone to consecutive transformations into levulinic acid and formic acid, 2,5-dimethylfuran (2,5-DMF) (Fig. 2 and Scheme 2) or to cross-polarizations towards coloured soluble polymers and insoluble humins.122–124
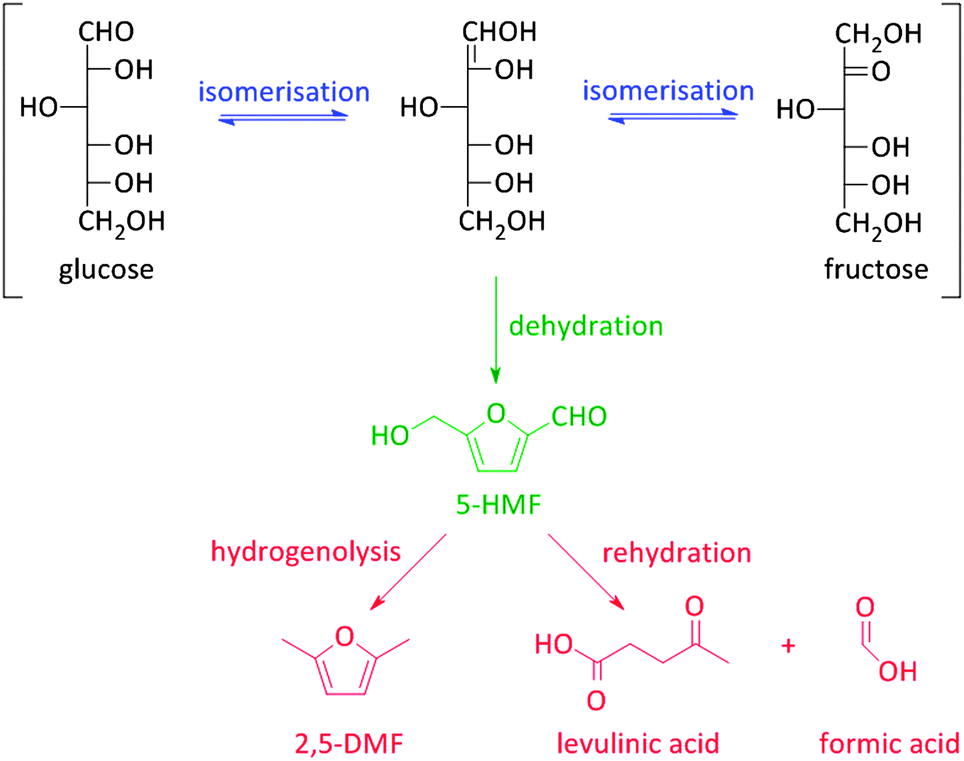 | ||
| Scheme 2 Formation and degradation reactions of 5-HMF. | ||
Alternatively to aqueous media, non-aqueous systems were proposed. However, also the use of these latter solvents has several drawbacks such as tough process conditions (in the case of high boiling solvents like DMSO123,125) or consecutive reactions when low-molecular weight alcohols are used; for example, in ethanol,126 5-ethoxymethulfurfural, an alternative biofuel,129,130 is formed. An elegant solution is the use of mixed reaction systems, especially biphasic liquid systems. In this case, the unstable dehydration products formed in the reactive water phase are continuously extracted into the non-polar solvent phase, where they are protected from subsequent reactions and related selectivity drops. Numerous reports involving biphasic reactions are known, like water–MIBK,127–131 water–toluene,119,130–148 water–THF,149 water-1-butanol,149,150 water-p-xylene134 and water–DMSO–acetone.151 Low 5-HMF yields are also rooted in the moderate glucose dehydration reactivity. As a result, some researchers have focused on the conversion of fructose or fructans towards 5-HMF. Due to the lower stability of fructose ring structures compared to glucose rings,122 higher activities can be obtained.119,127–129 Yet, poly-fructans are much more expensive than cellulose or starch. As a solution, the Brønsted acidic dehydration can be preceded by the Lewis acidic isomerisation of glucose to fructose (the latter will be discussed in more detail in Concept 2). In this process, glucose or cellulose can be used as feedstock but thanks to the Lewis acid catalysed isomerisation step, the more difficult dehydration of glucose is evaded. For the dehydration step, mostly mineral acids, e.g. HCl, are used.149 Later, fully heterogeneous systems, with a solid Brønsted acid catalyst, were described.126,152
As stated before, 5-HMF is very prone to undergo side reactions. One of these side reactions is the rehydration and rearrangement towards levulinic acid (Fig. 2 and Scheme 2). However, this rehydration step is not necessarily undesirable as levulinic acid153–155 and its hydrogenated product gamma-valerolactone156–172 can serve as platform molecules. Soluble acids and commercial resins have already been applied for the selective production of levulinic acid, however, with little success.125 Recently, zeolites were proposed as 5-HMF can be trapped in the pores and cages where it can be further converted into levulinic acid by the strong Brønsted acidity of the zeolite.173–175 Yet, starting from C6 sugars entails the production of one molecule of formic acid with every formed molecule of levulinic acid. Therefore, using C5 sugars like xylose, or consequently furfural, can serve as a valuable alternative as it represents a higher atom economy due to the preservation of all carbon atoms, although a hydrogenation step is needed (Scheme 3). This route was recently explored with furfuryl alcohol, the hydrogenated product of furfural, towards ethyl levulinate over a series of sulphonated polystyrene resins and zeolites. While the ion-exchange resins show the highest performances, zeolites demonstrate to have much better regeneration capacities176 which may indicate the higher potential of zeolites for this reaction provided that their performances increase.176
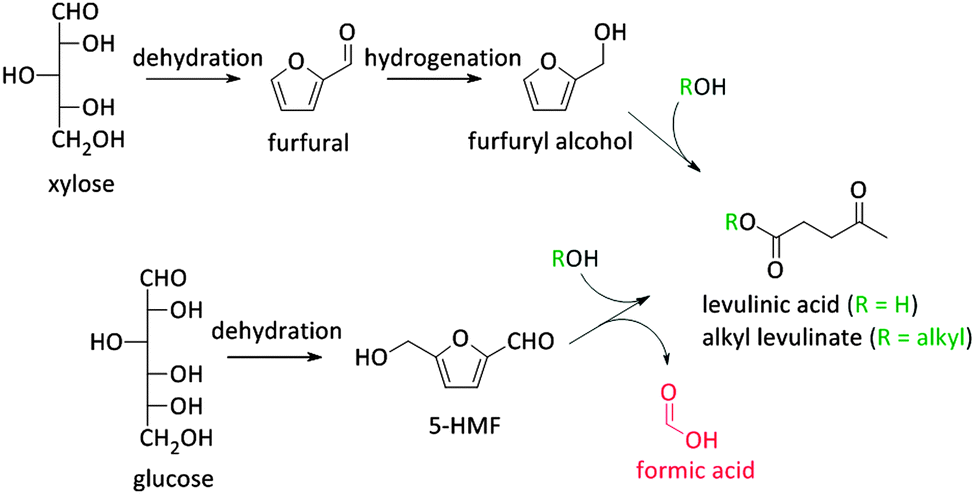 | ||
| Scheme 3 Formation of levulinic acid starting from both C6- and C5-sugars. | ||
5-HMF can also be transformed into 2,5-dimethyl furan (2,5-DMF; Fig. 2), a possible feedstock for the production of p-xylene, a precursor for terephthalic acid and consequently PET. Via a [4+2] cycloaddition of ethylene and 2,5-DMF followed by subsequent dehydration, p-xylene can be formed over zeolite Y or beta (Scheme 4).177–182 Hereof, zeolite Y exhibits the best performance.180 The major disadvantage of this process is the requirement of ethylene. Recently, an alternative was proposed whereby acrolein is used instead of ethylene. However, the researchers only obtained moderate yields, indicating that this reaction is not industrially practical on the short term. Nevertheless, further research is desirable as this reaction can be a very valuable and sustainable route to produce p-xylene and derivatives. Acrolein can, for instance, easily be produced from the dehydration of glycerol,183–187 a readily available by-product of the soap and biodiesel industry.188–190
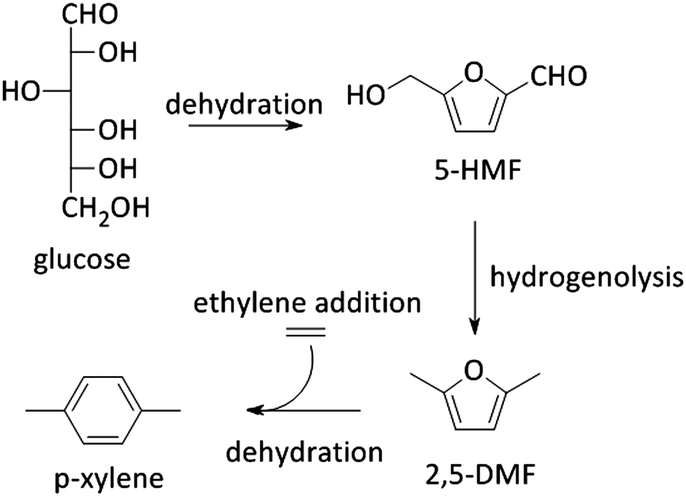 | ||
| Scheme 4 Reaction pathway from glucose towards p-xylene. | ||
Liquid phase catalytic conversion of lignin
Brønsted acidic zeolites find also gradual entrance in aqueous phase lignin processing. For instance, USY has recently been used in a one-pot depolymerisation and conversion of lignin into value-added aromatic monomers (Fig. 2). Herewith, aromatic monomer yields up to 60% could be obtained from dealkalined lignin.191 Similar yields were obtained with ZSM-5. Unfortunately, the reusability of the USY zeolite catalyst is questionable, as the yield dropped from 60% during a first run to 22% after a third run. This activity drop could be related to the poisoning of the Brønsted acidic sites by Na+ present in the lignin substrates. However, even when Na+-free lignin substrates, as for example organosolv lignin were used, significant activation loss was observed. After a characterisation of the fresh and spent catalyst, the researchers observed also a decreased catalyst crystallinity and changed pore structure induced by the applied reaction conditions,191 pointing to the importance of zeolite reusability and structural stability in condensed phase reaction conditions (see Concept 6).Concept 2: Lewis acidity
Zeolites are interesting scaffold structures for hosting Lewis acid properties. In 1983, the discovery of Ti-silicalite-1, a Ti-containing molecular sieve with the MFI topology, opened a new field of catalytic opportunities for zeotypes, as up to this point, the use of zeolites in catalysis has been limited to Brønsted acid reactions.192 It was found that presence of isolated tetrahedral Ti-species, substituting Si-atoms in the zeolite framework, resulted in Lewis acid sites. Since then, aside from Ti-silicalite-1, several other topologies and metal substitutions have been reported to result in Lewis acidity (Sn,192–194 Zr,195–197 Hf,198 V,199 Nb,200,201 Ta201), though the genuine T site substitution of some of them is debated. Furthermore, some extra-framework species (Ga,202 Al,203 Zn204) have also been found to result in Lewis acid properties. Whereas use of Lewis acid zeolites in petrochemical applications is somewhat limited by the restricted presence of electron-rich substituents (like carbonyl and alcohol groups) in fossil oils or natural gas, such groups are plentiful in biomolecules. The development and use of Lewis acid zeolite catalysts will therefore be expected to take a bigger role in the bio-based chemistry.The exact nature of Lewis acid sites in zeolites is a subject of debate. For example in Al-containing zeolites, it is suggested that, next to the EFAL species, some tri-coordinated framework species are a major source of Lewis acidity,205 and in Sn-zeolites two separate framework species, a fully framework connected (SiO)4Sn and a partially hydrated (SiO)3SnOH species, are assumed. A high catalytic activity has been attributed to the latter Sn-species, as Sn–OH species or proximate Si–OH groups are assumed to interfere in the reaction mechanism resulting in transition state stabilisation and therefore higher reaction rates.196,206–208 Keeping this in mind, the idea of the most active Lewis acid metal site might therefore be shifting from perfectly incorporated hetero-elements to lower-coordinated metal species with higher site accessibility and higher numbers of possible interaction sites.209 In this aspect, the emergence of post-synthetic procedures is interesting to mention, as they likely introduce Lewis acid metals into preformed zeolite structures different than the classic hydrothermal synthesis procedures.36,37,210–215 In comparison with hydrothermal synthesis, lower coordinated metal sites are formed more easily resulting in highly active catalyst materials.209 But also, there is a relationship between the reaction type and the activity of different sites, isolated Sn sites being required for reactions like sugar isomerisation, while clusters are also very active in reactions like Baeyer–Villiger.36,215
In Lewis acid catalysed reactions, apolar and aprotic solvents are often used, as the combination of Lewis acids and protic solvents, like water, result in a significant decrease in catalyst activity. Electron-rich oxygen atoms of water molecules tend to coordinate the Lewis acid sites leading to suppression of the catalytic activity by hindering substrate coordination or even decomposition of the sites.216,217 Hydrophobic zeolite structures, which limit the presence of water molecules within its structure, are an elegant solution for performing Lewis acid catalysis in presence of water. One such example is Sn-beta, a silicious beta framework containing small amounts of tetrahedral framework incorporated Sn. The large pore diameter of BEA makes this topology suitable for use in biomass transformations.
Sugar isomerisation
In 2010, the Davis group demonstrated a good activity for Sn-beta in aqueous hexose isomerisation reactions (Fig. 2 and Scheme 5), an important sub-reaction in various biomass-to-chemicals reaction schemes and industrially relevant in the case of fructose production, where the zeolite (Table 1, entry 1 and Scheme 5) could replace an enzyme-based process.218,219 Detailed characterisation of the Sn-zeolite showed that the active site of the catalytic material is similar to the active cleft of the used xylose isomerase.220 Besides that, the more robust nature of zeolites, in comparison to enzymes, allows its combination with other catalytic reactions in harsh conditions. The Lewis acidic isomerisation activity, as shown with isotope experiments to proceed via hydride shift, was for instance combined with Brønsted acid catalysts in the direct conversion of glucose to 5-HMF or derivatives (as already discussed in Concept 1).126,149,221 Later, Sn-beta has also been used in the isomerisation and epimerisation of other monosaccharides like galactose, pentoses and trioses, and even disaccharides.36,222–224 It has been recently demonstrated that isolated Sn sites, present in beta zeolites with low Sn content, are the most active sites.36,215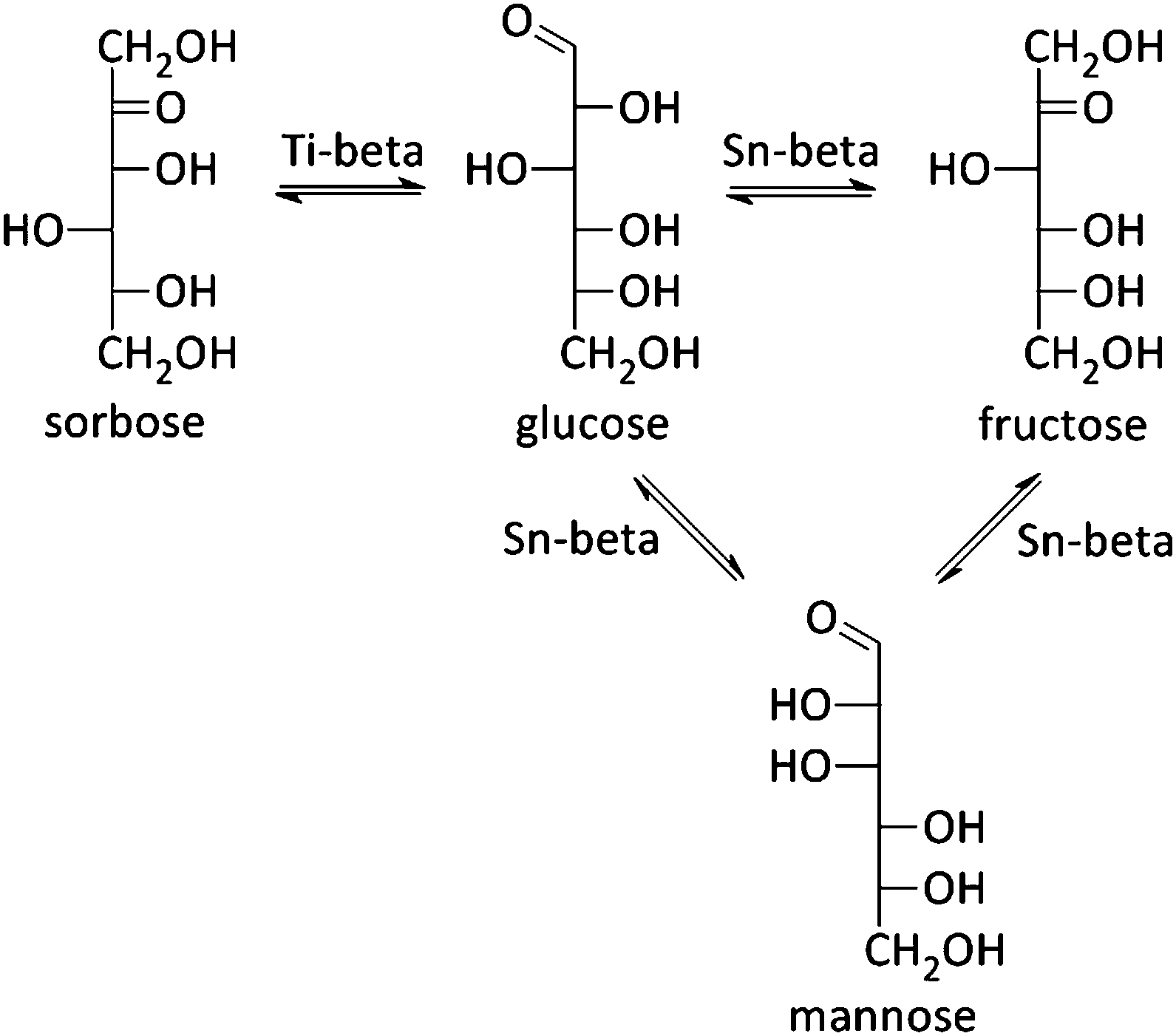 | ||
| Scheme 5 Isomerisation of C6 sugars with Lewis acidic beta zeolites. | ||
| Entry | Catalyst | Solvent | Conversion/% | Monosaccharide distribution/% | Ref. | ||
|---|---|---|---|---|---|---|---|
| Fructose | Mannose | Sorbose | |||||
| 1 | Sn-beta | H2O | 6.4 | 93 | 7 | n.d. | 218–220 and 227 |
| 2 | Sn-beta + SB | H2O | 16 | 6 | 94 | n.d. | 223 |
| 3 | Sn-beta | CH3OH | 23.2 | 73 | 27 | n.d. | 226 and 227 |
| 4 | Sn-beta (Na+) | H2O | 6.8 | 41 | 59 | n.d. | 227 |
| 5 | Sn-beta (Na+) | CH3OH | 12.4 | 0 | 100 | n.d. | 227 |
| 6 | Ti-beta | H2O | 19 | 66 | <1 | 33 | 228 |
| 7 | Ti-beta | CH3OH | 21 | 31 | 6 | 63 | 228 |
Mannose, the epimer of glucose, is a typical by-product in the isomerisation reaction of glucose. But combining a Sn-beta zeolite with sodium borate salts, shifts the selectivity of the isomerisation reaction predominantly to the epimer formation (Table 1, entry 2 and Scheme 5).223,225 Where Sn is responsible for an intramolecular hydride shift in the isomerisation reaction, presence of the borate–monosaccharide complexes shifts the catalytic activity of the metal toward an intramolecular carbon shift in the epimerisation reaction. It was furthermore shown that the epimerisation of sugars can also be achieved by changing the reaction solvent from water to methanol (Table 1, entry 3) or by exchanging a silanol group, adjacent to the Sn-atom, with Na+ cations (Table 1, entries 4 and 5).226,227 In both cases, a carbon shift mechanism is responsible for the monosaccharide transformation. Finally, use of the Lewis acid Ti-beta zeolite in the isomerisation of glucose does not result into fructose or mannose, but sorbose is formed by a C5–C1 intramolecular hydride shift (Table 1, entries 6 and 7 and Scheme 5).228
Transformations of 5-HMF and alkyl lactate/lactic acid formation
Other uses of Lewis acid zeolites in biomass conversions make use of sugar derived 5-HMF or trioses (glyceraldehyde (GLY) and 1,3-dihydroxyacetone (DHA), the two retro-aldol components of fructose). Mixtures of trioses may also be obtained from glycerol oxidation (Fig. 2).229–231 5-HMF was converted in presence of Hf-, Zr- and Sn-beta into the high energy density fuel additive 2,5-bis(alkoxymethyl)furans (Fig. 2) by a transfer hydrogenation and etherification with alcohol.221DHA and GLY were converted into lactic acid or alkyl lactates (Scheme 6), potential biomass derived platform molecules,232 when performed in aqueous or alcoholic media, respectively (Fig. 2). This reaction scheme has been described for various catalysts, both homogeneous as heterogeneous.233–237 The importance of Lewis acid catalysis in lactic acid chemistry was recently analysed and reviewed in depth.2,232,238 The multistep mechanism comprises a dehydration of the substrate C3-sugar to methyl glyoxal, followed by an addition of a solvent molecule and a hydride shift (Scheme 6). The dehydration reaction is catalysed by Lewis acidity, but it is more efficiently done by Brønsted acid sites, as will be discussed later in the part on multifunctional catalysis. Here, we limit the discussion to purely Lewis acid catalysts. The Lewis acidic Sn zeolites have been described as catalytically active moieties that efficiently catalyses the DHA to lactate transformation. Use of Sn-MFI,224 Sn-beta,224,239,240 and Sn-MWW241 has been reported. For Sn-MFI, a high activity was found for reaction in water, yielding lactic acid, but when performing the reaction in methanol, a significantly lower activity was observed due to pore-size restrictions in the MFI structure.224 For Sn-beta, which contains larger pores, such restriction was found in methanol nor ethanol and near-quantitative methyl lactate yields could be obtained.239,240 Similar results were obtained with Sn-MWW.241 Next to Sn, various Ga-containing zeolites, synthesised by post-synthetic alkaline-assisted galliation, have been described as efficient catalysts for this conversion whereby FAU-type zeolites were found to show the highest activity.242
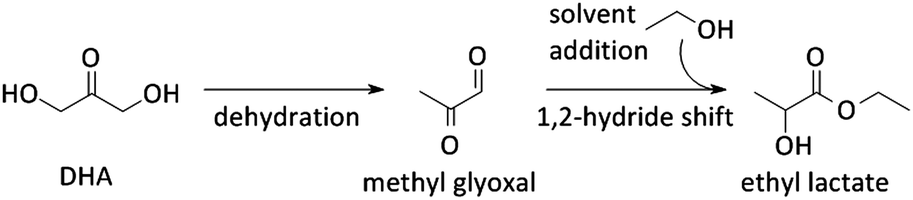 | ||
| Scheme 6 Pathway of dihydroxyacetone towards ethyl lactate. | ||
Other Lewis acid catalysed reactions
DHA, amongst others, has also been used in Sn-beta catalysed C–C-coupling reactions with formaldehyde. The formed product, α-hydroxy-γ-butyrolactone (Fig. 2), is an important intermediate for the production of pharmaceuticals and herbicides.243 Other Lewis acid catalysed C–C coupling examples include the conversion of terpenes. For instance, the transformation of β-pinene to nopol, a precursor to pesticides and perfumes, by an intramolecular Prins reaction with formaldehyde is reported in presence of Sn-beta, as well as the intramolecular cyclisation of citronellal to isopulegol.244,245 Terpenes can also be used in other Lewis acid catalysed reactions such as Baeyer–Villiger oxidation of dihydrocarvone with hydrogen peroxide. The nature of the substituted heteroatom governs the chemoselectivity, while Sn-beta produces the corresponding lactone, use of Ti-beta favours the epoxide product.246 Zr-beta is the Lewis acid zeolite of choice to rearrange β-pinene oxide into myrtanal, which has pharmaceutical uses.247 These examples nicely illustrate the favourite role of Lewis acid zeolites in the production of various chemicals from natural resources.As most work on Lewis acid catalysed conversions focuses on converting cellulose(-derived) substrates, examples of transformations of lignin-derived molecules into useful chemicals using Lewis acid catalysis are extremely scarce. Recently, Sn-containing Lewis acid zeolites were used to convert cyclohexanone compounds, obtained in high yields from lignin-derived syringols and guaiacols, into their corresponding caprolactone derivatives by Baeyer–Villiger oxidation in presence of Sn-beta with aqueous H2O2 as oxidant. Such lactones may be useful in the synthesis of various novel polymers.248
Concept 3: multifunctional zeolite designs
Reaction schemes for transforming raw biomass and biomass derived platform molecules into chemicals and fuels often exist of a cascade of reactions. If the different reaction steps require identical sites, one catalyst type can be used.198,221,243,249,250 One example is the Sn catalysed conversion of sucrose into methyl lactate. A cascade of isomerisation, retro-aldol, dehydration and H-shift sub-reactions are needed to complete this transformation, all catalysed by the same catalytic site.251 Other examples are the Lewis acid catalysed sequential transfer hydrogenation and etherification of 5-HMF to 2,5-bis(alkoxymethyl)furan, a useful diesel additive221 and the one-step synthesis of butadiene from ethanol with Zr-incorporated zeolites.252 As the latter allows the production of green polybutadiene, many researchers are looking for novel catalysts.253–256 Critical reaction steps in the cascade to butadiene are the C–C bond formation and a Meerwein–Pondorf–Verley step, both being catalysed by the Lewis acid Zr site.252However, usually different active sites are required in the sequence of reactions. Basically three approaches can be discerned: the use of multiple catalytic systems in series, multiple catalysts in one-pot or a one-pot reaction with different active sites on one material (Fig. 6). The former can be considered when the intermediates are appropriately stable and have potential value themselves.152,257 Moreover, it could be the preferred technique when the different reaction steps require reaction conditions which are not compatible. With this approach, the temperature and pressure can be optimally tuned for each active site individually. On the other hand, when the reaction conditions of the different steps are more or less compatible, the use of different catalysts in one-pot (Fig. 6b) could be the approach of choice, as it does not require separation of the catalysts and intermediates after every reaction step. This is for example illustrated for the one-pot conversion of hemicellulose-derived furfural in gamma-valerolactone (Fig. 2 and Scheme 7) in the absence of H2. While the Lewis acidity of Zr-beta catalyses a transfer hydrogenation of furfural with 2-butanol solvent to form furfuryl alcohol, it is the Brønsted acidity of an Al-MFI nanosheet zeolite that converts it further to levulinic acid. Next, the latter undergoes a second Lewis acid catalysed transfer hydrogenation into 4-hydroxypentanoate which is finally followed by a cyclisation to gamma-valerolactone (Scheme 7).258 As the used mild reaction conditions (120 °C) are compatible with those used in hemicellulose hydrolysis and dehydration towards furfural, this approach can potentially be extended towards the raw feedstock leading to an ultimate example of the elegancy of multifunctional catalysed biomass processing.
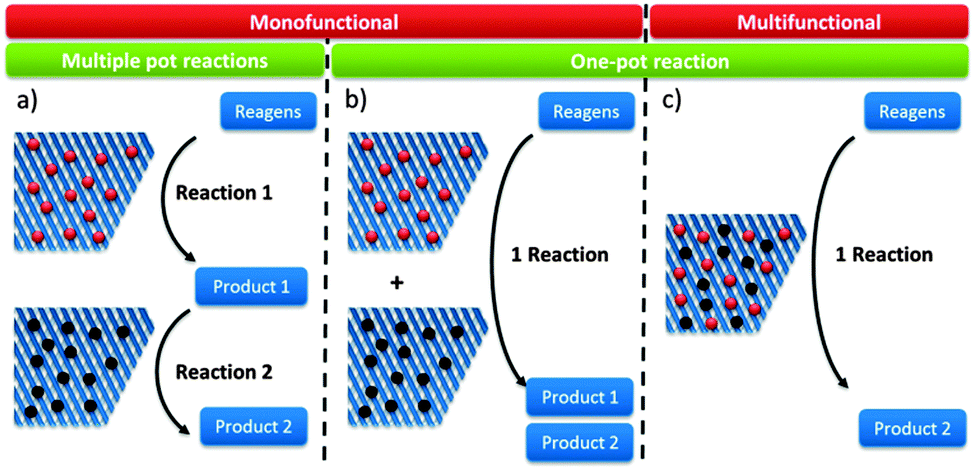 | ||
| Fig. 6 Schematic representation of the modes to use multiple different catalytic sites. | ||
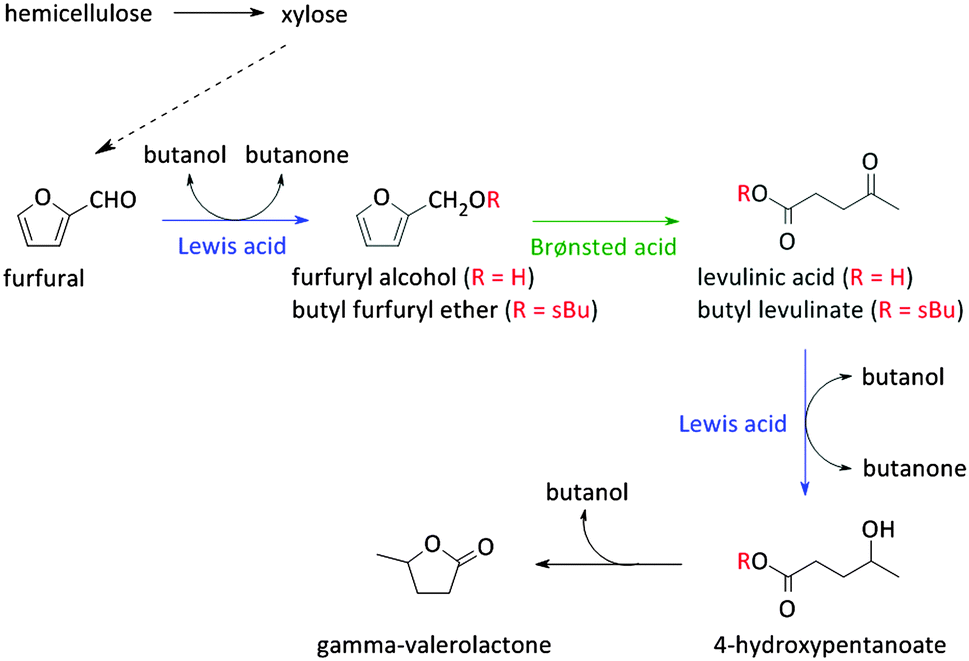 | ||
| Scheme 7 Reaction scheme for the conversion of hemicellulose/furfural into gamma-valerolactone. | ||
Multifunctional catalysis with sugar(-derived) compounds
Systems with multiple catalyst materials in one-pot are only successful when the intermediates are relatively stable molecules. When the intermediates are very reactive molecules, which is often the case with biomass, catalysis by multifunctional materials, i.e. different active sites on one material (Fig. 6c), might be a better choice. Zeolites are interesting materials to organise such a close vicinity of different catalytically active sites. Proceeding the entire cascade in the pores of the zeolite, therefore, usually leads to higher product yields. Synergistic effects between the different sites have recently been identified in such multifunctional designs.259 Site-ratio balancing though is the major challenge and requires sufficient insight in the kinetics of the underlying reaction network.Several examples were reported using the multifunctional catalysis concept. A first example combines the two types of acidity in a single material for the direct conversion of trioses, GLY and DHA, into alkyl lactates (Fig. 2). Though the reaction was successfully catalysed with the Lewis acid Sn-beta (see Concept 2),240,251 it was demonstrated that dehydration of the triose molecule to methyl glyoxal, the first step in the cascade mechanism (Scheme 6), is rate determining. As such dehydration is more accelerated by Brønsted acid sites, a Sn-beta containing tetrahedral Brønsted acid Al3+ sites was synthesised by a two-step post-synthetic procedure.260 Increasing Al contents indeed fastened the overall reaction. A clear contribution of the two sites was observed: the Sn active site catalyses the hydride shift of methyl glyoxal to lactate, while the Al3+ active site efficiently accelerates the formation of methyl glyoxal from DHA. A good balance of both sites, in this example corresponding to a molar Sn to Al ratio of 2, is important to combine a high reaction rate with excellent selectivity. In contrast to the multifunctional Al/Sn-beta zeolite, use of a physical mixture of Sn-beta and Al-beta catalysts showed modest lactate yield. Another example favouring multifunctional catalysis with Al/Sn-beta describes the conversion of cortalcerone, enzymatically produced from glucose, over furyl glyoxal hydrate into furyl glycolic acid. Similarly, Brønsted acid catalysed dehydration is nicely combined with Lewis acid catalysed intra-molecular hydride shift.261
Incorporation of heteroatoms is not always required to create multifunctional acid zeolites. Lewis–Brønsted acid zeolites may for instance be produced by classic steaming of the parent Al-containing zeolite. USY zeolites for instance have been found to be very active in the conversion of trioses to lactate, especially the ones with a high content of extra-framework Lewis acid Al sites,236,237 though their stability in hot liquid water is questionable (see Concept 6).262,263 Steam-treated Al-beta also outperforms the parent material when used in sugar cascade reactions for producing 5-HMF, the introduced Lewis acidity being responsible for fastening glucose to fructose reaction.264
Besides the one-pot Lewis–Brønsted catalysis, other elegant multifunctional designs combine redox and Lewis acid catalysis. Recently, Pt catalysed oxidation of biodiesel waste glycerol to DHA, followed by a Sn Lewis acid catalysed conversion to lactic acid was described (Fig. 2 and Scheme 8).265 The highest substrate conversion rates were found when the two sites were residing on the same material (TOF 0.07 vs. 1.95 for Pt/AC with Sn-MFI and Pt/Sn-MFI respectively). A substantially faster conversion of trioses to lactic acid after adsorption of the Pt nanoparticles was invoked to explain the observed difference between both zeolite topologies.
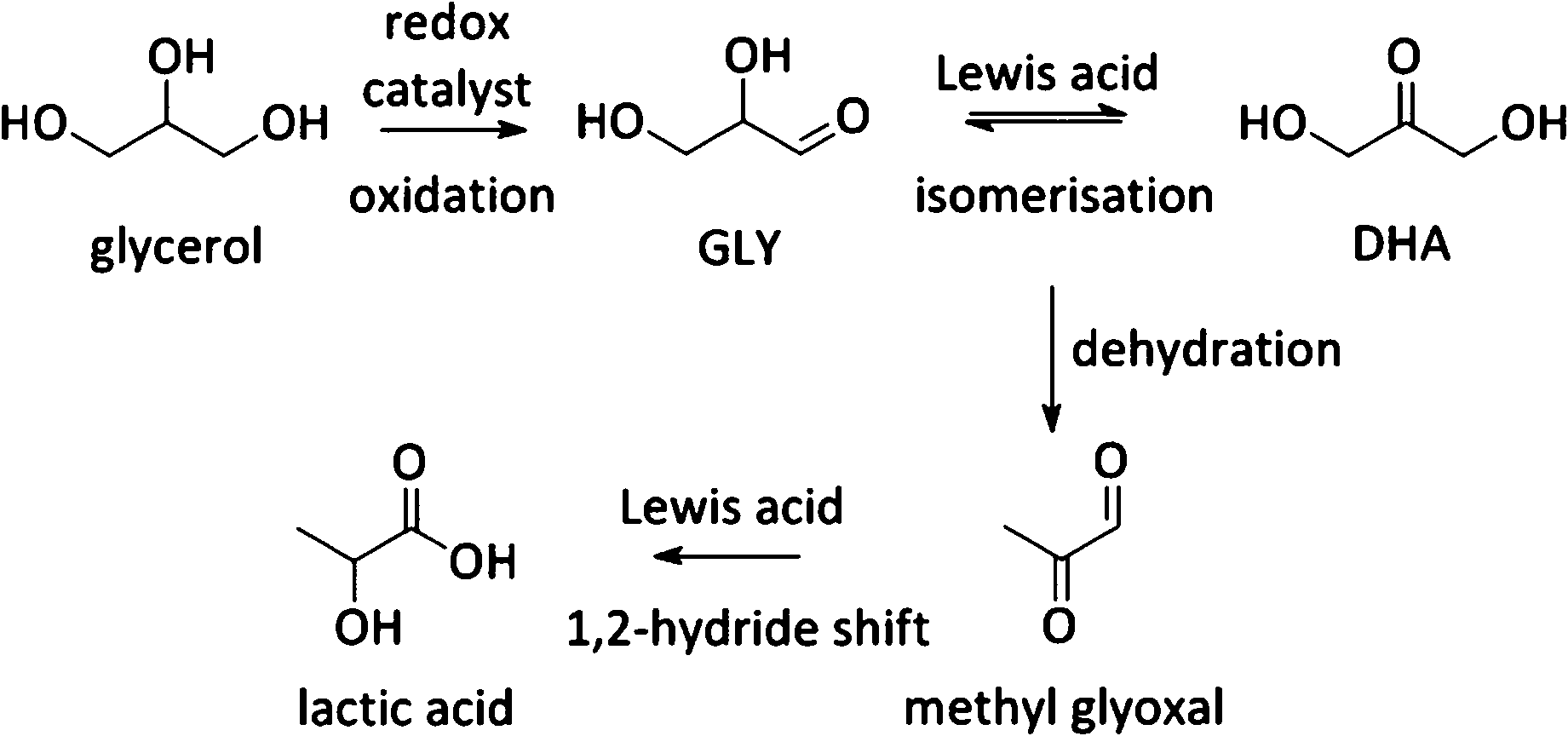 | ||
| Scheme 8 Formation of lactic acid starting from glycerol. | ||
As already mentioned before, the choice of a multifunctional catalyst, rather than performing reactions with multiple catalysts, is most advised when the intermediate products are too reactive. A good example is the reductive splitting of carbohydrate polymers into sugar alcohols (Fig. 2 and Scheme 9). Since biopolymers like (hemi)cellulose are resistant to chemocatalytic conversion, fast hydrolysis is only possible when applying harsh reaction conditions with temperatures above 150 °C and/or in presence of substantial acid quantities. As the hydrolysate compounds, like glucose, are too reactive under these severe conditions312,313 and degrade into caramel and tars, a second catalytic function is introduced to reductively stabilize the sugars into hexitols like sorbitol and sorbitans.
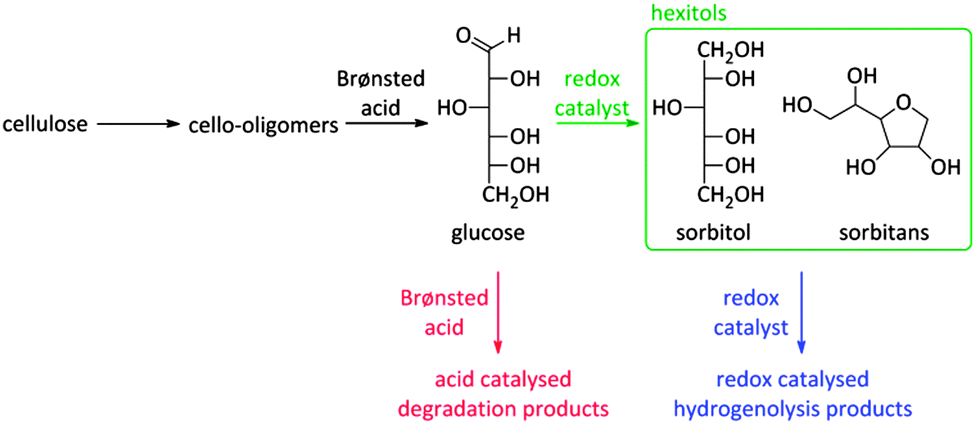 | ||
| Scheme 9 Reaction scheme for the reductive splitting of cellulose. | ||
The first systematic studies about this process were done by Russian researchers more than 50 years ago.266 With noble metals on charcoal in acidic media, high hexitol yields were obtained.266,267 A few decades later, this catalytic system was translated into a completely heterogeneous system by replacing the mineral acid by a heterogeneous acid catalyst loaded with ruthenium particles.268 However, due to sintering of the metal particles, the lifetime of the catalyst was limited. This problem was solved when a patent appeared about Ru/H-USY catalysts, showing high hexitol selectivities from starch on the long-term.269 This system was later adopted for the conversion of the more recalcitrant cellulose. Herewith, hexitols could be obtained with nearly quantitative yields for multiple consecutive runs.262,270 Ball-milling, to create reactive cellulose, as well as the addition of minute amounts of HCl, in order to facilitate cellulose solubilisation, seemed to be important for high activities.
Scheme 9 represents the reaction network. Unsoluble cellulose is firstly hydrolysed into soluble cello-oligomers by the hydrolysing action of hot water and the minute amounts of mineral acid. Remarkably, it was found that also the zeolite can assist during this first step, indicating the appearance of solid–solid interactions between Brønsted acid sites of the zeolite and large cellulose polymers.262 Once small enough, the cello-oligomers are rapidly hydrolysed to glucose in the pores of the zeolite by the Brønsted acidic sites and hydrogenated to sorbitol by Ru nanoparticles (Scheme 9). A major obstacle for long-term catalysis seems the loss of Ru dispersion and not the zeolite stability.17,262,271–274 The latter will be discussed into more depth in Concept 6. A few research groups now have developed a similar bifunctional catalytic system for converting cellulose into sugar alcohols such as Pt promoted Ni-beta,275 Ni on ZSM-5276,277 and Ru or Ir on beta,278 but so far the hexitol yield of Ru/H-USY remained unsurpassed.
Critical to obtain high sugar alcohol selectivities is working at an optimal balance between the two catalytic sites (Fig. 7). When the Brønsted acidity is too dominant, selectivity loss through thermal and acid degradation of sugars will occur (Scheme 9 and Fig. 7).279–281 However, when the amount of active metal species is too high, typical metal catalysed hydrogenolysis products will be formed, like ethylene glycol, 1,2-propanediol, glycerol and other C4 and C5 polyol isomers (Scheme 9 and Fig. 7).279,281,282 So, highly selective formation of platform chemicals directly from sugar polymers requires working on a thin balance between both catalytic functions.
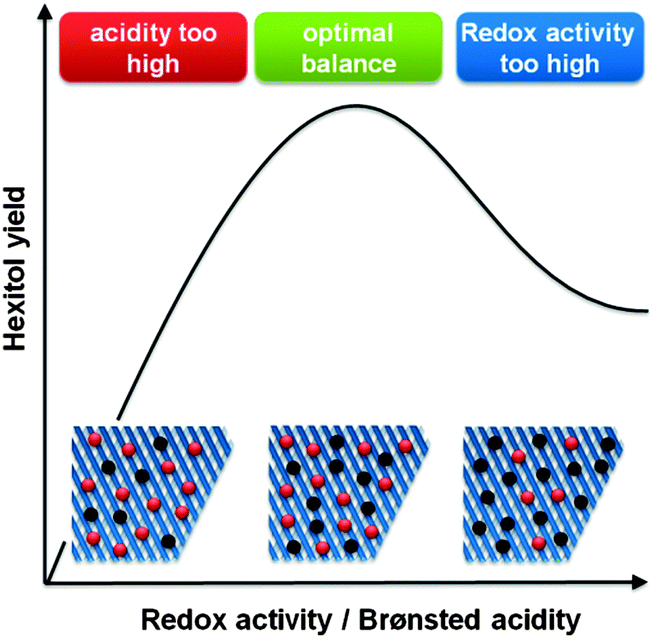 | ||
| Fig. 7 Influence of the balance between redox (black sites) and Brønsted (red) sites on the hexitol selectivity during hydrolytic hydrogenation of cellulose. | ||
Such balance is, due to the very reactive nature of many biomass-derived chemicals, of utmost importance in biomass sequences. Note that balancing of active sites is not a new phenomenon resulting from biomass processing as it can also be retrieved in traditional oil refineries. During hydrocracking the balance between the amount and strength of hydrogenation (noble metals) and cracking activity (acid sites) of the catalysts determines the product distribution. For instance, strong acidic zeolites in combination with moderate hydrogenation activity yields especially gasoline and lighter products, while weak acidic zeolites with a strong hydrogenation catalyst leads to the formation of high middle-distillates.283
Basically the same exercise can be done with other sugar polymers like hemicellulose. However, up to now, the reductive splitting of hemicellulose fractions from lignocellulosic feedstock is somewhat overlooked. Yet, the valorisation of the hemicellulose fraction is important in view of the economics of a commercial biorefinery. The few reports in literature focus mainly on arabinogalactan.284–288 In principle, it seems reasonable to expect that the hydrolytic hydrogenation of hemicellulose will be much easier, thanks to the less recalcitrant nature of the feedstock, though literature only reports sugar alcohol yields as high as 20 to 25% with Ru loaded beta and USY zeolites.284–286
Multifunctional catalysis with lignin-derived compounds
Bifunctional metal-loaded zeolites are also reported as catalysts for the conversion of biomass to high-quality drop-in biofuels, for instance for the conversion of carbohydrate-derived compounds to alkanes.289–294 As mentioned earlier, pyrolysis of biomass is negatively affected by the presence of lignin (see Concept 1). The removal of lignin prior to pyrolysis could therefore be necessary to improve the CFP selectivity. Besides, lignin is a current waste product in the pulp and paper industry and in second generation bioethanol biorefineries where it is mainly burned for energy recuperation. The search for valuable lignin valorisation routes has recently been considered as highly important in view of the economics of the biorefinery.25,295–297 Recently, a cost profit of about 30%, providing that lignin is converted to value-added chemicals like phenolic monomers and oligomers, was showed.298In order to produce fuels from lignin, the phenolic monomers can be converted to alkanes (mono- and bicycloalkanes) or aromatics over bifunctional metal-loaded zeolites, like Pt on H-Y, H-beta or H-ZSM-5,299–301 Pd/H-beta,302 Ni/H-ZSM-5303–306 and Ru/H-ZSM-5,307 or over a combination of a redox catalyst and a zeolite, like Pd/C and H-ZSM-5308 or La/H-beta.309 These transformations involve a series of reactions like hydrolysis, dehydration, hydrogenation, cracking, alkylation and dealkylation. Bicycloalkanes can be produced by bifunctional catalysts comprising zeolites with large pores, like H-beta and H-Y,299,302,309 as they enable the coupling of phenolic monomers through alkylation reactions inside the zeolite pores. For selective bicycloalkane formation, an optimal balance of the rates of metal catalysed hydrogenation and acid catalysed dehydration and alkylation, thus pointing to an optimal acid site/metal site balance, is critical.302,309 In this way, the phenolic monomers can thus be converted to both light (monocycloalkanes, C6–C9) as well as heavier (bicycloalkanes, C12–C18) hydrocarbon fuels. Lignin can also directly be converted to hydrocarbons over bifunctional metal-loaded zeolites like Ni/H-ZSM-5 and Ni/H-beta.310 Hydrocarbon yields up to 70% can be obtained, with monocycloalkanes being the main products. Also in this case, Ni/H-beta shows a higher selectivity for bicycloalkanes than Ni–H-ZSM-5.
Multifunctional catalysis with triglyceride-based compounds
Another nice example of the use of biomass for biofuels production using bifunctional zeolites comprises the conversion of triglycerides and/or derivatives towards products in the range of diesel and aviation fuels.78,79,311–323 Hydrogenolysis and deoxygenation are carried out on the highly dispersed metal function, whereas the combined action with acid sites of the zeolite enables the hydro-isomerisation and hydro-cracking of the formed long-chain alkanes. Both for diesel and aviation fuel, isomerisation to branched alkanes is necessary to meet demanded cold-flow properties. Cracking to shorter-chain alkanes on the other hand is only desired when aviation fuel is aimed for. The carbenium ion intermediate formed upon dehydrogenation (metal site) and protonation (acid site) either can be isomerised and again hydrogenated or cracked. Therefore, similar to the earlier described reductive splitting of cellulose, the metal/acid site balance is a crucial parameter of these bifunctional zeolite-based catalysts. In general, moderate acidic supports combined with an optimised, sufficiently high metal/acid balance favours isomerisation, whereas more acidic supports or non-optimal metal loadings enhance the degree of cracking towards the aviation fuel range.78,79,311,315,318,321Concept 4: shape-selectivity
The concept of shape-selectivity was introduced in 1960 by researchers from Mobil, who observed exceptional micropore size-dependent catalytic performance in the cracking of decane and the dehydration of butanols.31,324,325 Ever since, this concept has had a tremendous impact on the design of new catalytic processes in petrochemistry and refining. In its most basic form, shape-selectivity can be defined as a deviation, induced by constraints on molecular motion or product formation, from the product distribution obtained in absence of spatial constraint.326,327 Generally, the three most basic types of shape-selectivity, i.e. reactant type, transition-state and product type shape selectivity, visually explained in Fig. 8b, can be described by a well-defined shape-selective factor S, being larger than 1 when shape-selective effects are present (Fig. 8a).326 Over the years, other shape-selective effects have been documented, but these are often case-specific (such as the window and nest effects328). A related, somewhat more general concept is that of molecular traffic control,329 based on the fact that reactant molecules may enter through one type of pore, while products diffuse out of the other, intersecting pores.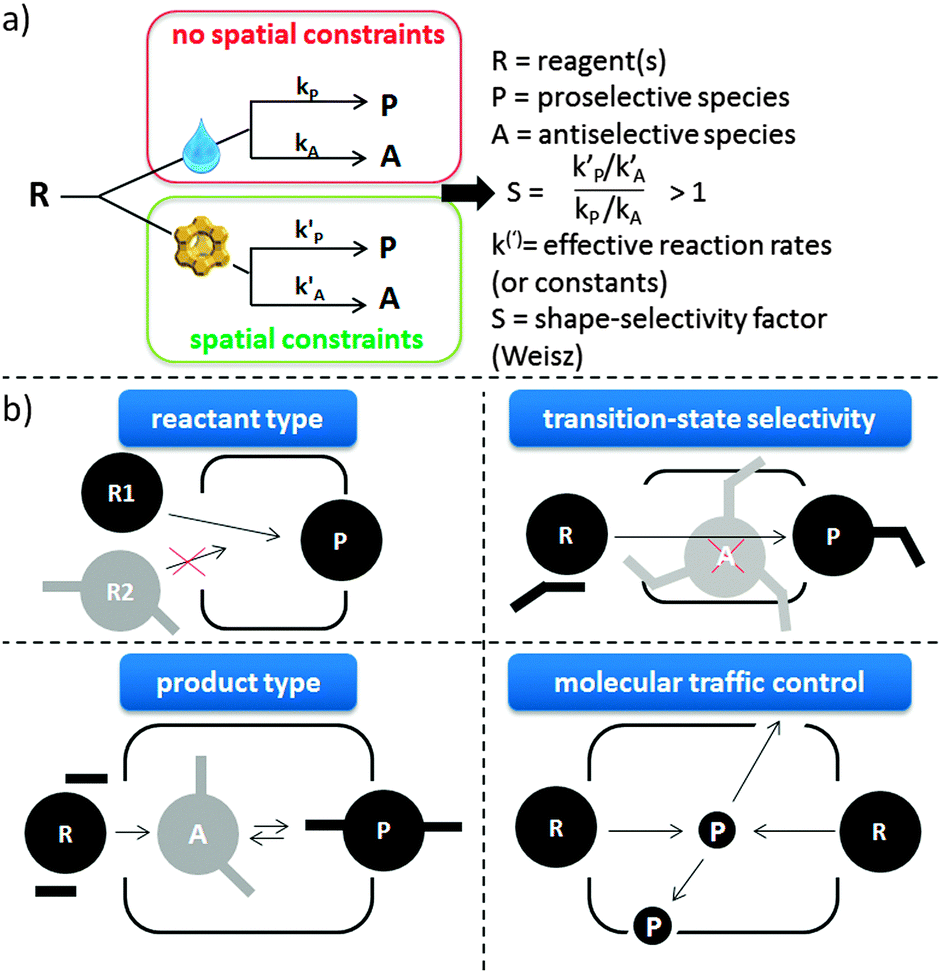 | ||
| Fig. 8 (a) The most basic definition of shape-selectivity, defined when S > 1. The drop represents reaction in absence of structural constraints on molecular motion or formation. (b) The 3 foremost types of shape-selectivity along with molecular traffic control. | ||
The oldest mention of shape-selectivity in biomass conversion is the direct conversion of rubber latex, corn oil and other oils over H-ZSM-5.330 The latter famous example of product-type shape-selectivity is based on the fact that the largest hydrocarbon pool intermediates are not able to diffuse out of the crystal while the gasoline-type (boiling point ca. 70–140 °C) compounds can.331 In recent years, this process is being exploited in the petrochemical industry to convert alcohols into olefins by using, instead of the 10 membered-ring (MR) ZSM-5 micropores, smaller 8 MR molecular sieves, such as SAPO-34 or SSZ-13.332,333 Nowadays, shape-selective effects can to some extent be found in all areas of biomass conversion where zeolites have been put forward, but they are most present and highly important in CFP (see Concept 1 for more details). In other areas, such as the upgrading of bio-derived platform molecules via specific processes, examples are limited to the observation of diffusion limitations for larger reactants, that can be used to steer the selectivity in favour of smaller reactants with zeolite catalysis.
A reactant-type of shape selectivity was recently reported in the form of the hydrogenation of triacylglycerols (TAG) over Pt/Na-ZSM-5.334 In this case, the preferred reactant was the central fatty acid chain of the triglyceride (sn-2), as explained by a pore mouth adsorption in tuning fork configuration. In addition, the enhanced affinity of more polar chains for the polar MFI pore mouths leads to a selective hydrogenation to the mono-unsaturated level and reduced amounts of saturated products. Therefore, an intermediately melting TAG product with desirable physical properties for applications was obtained.335 Furthermore, the shape-selective properties of MFI induce a preferred hydrogenation of the slimmer trans-configured unsaturated fatty acids over cis-isomers, allowing the selective removal of undesired trans-isomers.334 As a result, the obtained high-oleic and low-trans hydrogenated product is beneficial from a nutritional point of view, but also for bio-lubricant applications. Interestingly, the selective hydrogenation of trans-isomers on Pt/Na-ZSM-5 was proven by sorption experiments with fatty acid methyl esters (FAMEs).336
Other examples of shape-selectivity in biomass conversions can be found in the upgrading of bio-derived platform molecules. Shape-selective zeolite based conversions in this field are somewhat different than what is known from petrochemistry due to the often different reaction phase. Reactions discriminating between substrates with large size differences can be found in condensed phase resulting in reactant-type shape selectivity. The latter is exemplified by the absence of hexose isomerisation over Sn-MFI337 or Ti-MFI338 as compared to its active isomerisation to fructose over Sn-beta338 (or mannose220,226). Trioses (and in an intermediate case pentoses) however, can be efficiently converted over 10 MR frameworks, such as Sn-MFI, or even desilicated MFI zeolites.211,212 To exploit this concept, recently Sn-MFI was combined with MoO3 in a unique tandem catalytic approach to produce lactates from hexose sugars. The MoO3 catalyst enables retro-aldol scission of fructose, remarkably already at 90 °C, while the Sn-MFI zeolite converts the resulting small trioses in lactates. The larger fructose can hardly access the Lewis acid Sn sites and their hydride-shift activity. The result is a selective (up to 75%), low-temperature conversion of common sugars into ethyl lactate, an important building block for polyesters and sustainable solvents.339
Recently a novel process for facilitating the production of the biodegradable plastic polylactic acid (PLA) itself, also based on shape-selective zeolite catalysis,340 was presented. A large bottleneck in PLA production is the time-, energy-, and carbon-inefficient synthesis of lactide, the actual building block for the ring-opening polymerisation. The current route for making lactide (Scheme 10) involves a two-step procedure, with formation of an intermediate pre-polymer, due to the absence of selectivity control for lactide during condensation from concentrated lactic acid solutions. The two-step route also induces racemisation due to the severe conditions and the use of metals.232 Using concentrated lactic acid (9–18 wt% with respect to the solvent) in a refluxing aromatic solvent with phase-settler for water removal, the use of a H-beta zeolite (Si/Al 12.5) allows lactide yields of over 80% in less than an hour, with short lactyl oligomers as only side-products. Key here is the restricted formation of larger oligomers in the pores of the zeolite, resembling the characteristics of transition-state shape-selectivity. Mesoporous, macroporous or soluble acid catalysts mainly produce (long) oligomers instead of lactide, while H-beta forms lactide with 98% purity without racemisation. Since this zeolite could be reused 6 times without structural changes, this pioneering example of shape-selectivity in bioplastics production offers the promise of a zero-waste process for lactide synthesis.340 Another example of transition-state selectivity control was recently given for the conversion of tetrose sugars to C4-α-hydroxy esters, similar to lactic acid. A direct relationship was found between the catalyst pore sizes and product distribution. Here, the mesoporous Sn–MCM-41, Sn–SBA-15 and soluble SnCl4·5H2O strongly prefer the formation of the more bulky methyl-4-methoxy-2-hydroxybutanoate, whereas the microporous Sn-beta significantly favours the production of the smaller methyl vinyl glycolate.341
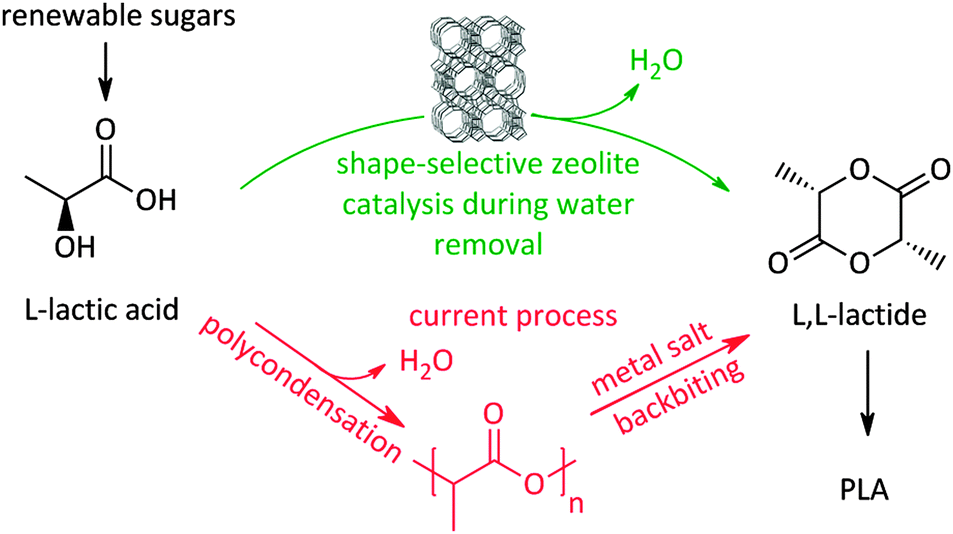 | ||
| Scheme 10 Shape-selective zeolite catalysed process340 for lactide (and PLA) production compared to the classic route. | ||
The shape-selective properties of a specific zeolite can be altered by changing the accessibility of the active site. This was for example demonstrated by narrowing the pore mouth of ZSM-5 by chemical liquid deposition of tetraethyl-orthosilicate.16 The obtained catalyst was applied in the Diels–Alder conversion of 2-methylfuran with propylene into xylenes16 (a variation on the reaction presented in Scheme 4), where a selectivity increase of the para-form from 32 to 96% could be realised.
In the conversion of lignin-derived phenolic monomers to alkanes over metal-loaded zeolites, the shape-selective properties of zeolites are also used to tune the product selectivity (see Concept 3). When using a zeolite with small pores, like H-ZSM-5, mainly monocycloalkanes are obtained,299,303,304,310 while zeolites with larger pores, like H-beta or H-Y,299,302,309,310 can selectively yield bicycloalkanes (see Concept 3).
Concept 5: hierarchical zeolites
The narrow micropores of zeolites enable several key properties as described in previous parts of this review. However, they also imply access and diffusion limitations (Fig. 9).342–351 As a result, the class of hierarchical (mesoporous) zeolites was conceived, coupling a secondary level of porosity to the intrinsic zeolitic micropores. The hierarchically structured porosity facilitates diffusion inside the zeolite crystal and enlarges the number of closely accessible active sites. This relatively novel type of zeolites has attained promising results in a vast variety of petrochemical reactions.352 Since biomass related reactions often comprise substantially larger molecules than those derived from fossil fuels, the potential of hierarchical zeolites may be even larger.32,44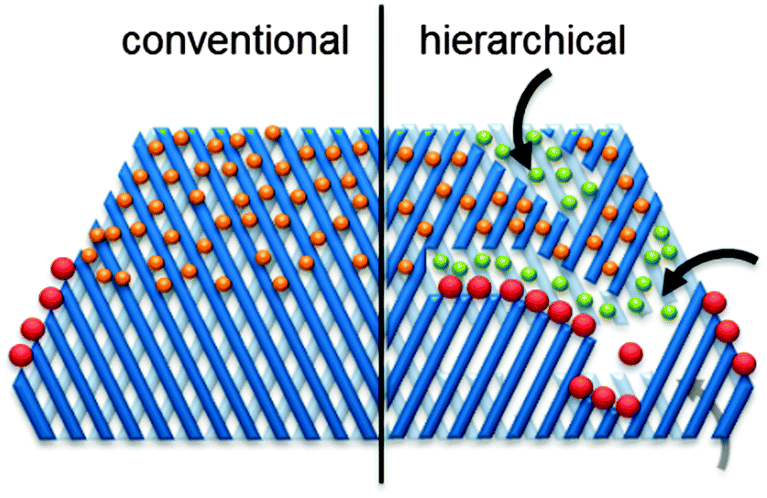 | ||
| Fig. 9 Schematic representation of access and transport/diffusion limitations in conventional and hierarchical zeolites. The orange spheres represent molecules that suffer from single-file diffusion. The secondary porosity enhances the number of pore mouths to diffuse in and out of, as well as the diffusion properties within the pores (indicated by the green spheres). In the latter case, the introduction of external surface often leads to enhanced selectivity (in for example cracking365 or isomerisation366). The red dots represent bulky molecules which can only react on pore mouths. | ||
Nowadays, a large variety of bottom-up and top-down strategies have been developed to enable the formation of hierarchical zeolites.346,353 Especially, top-down post-synthetic modifications have shown promise as they are highly efficient, tuneable, scalable, and enable to prepare any zeolite to its hierarchical form.349,351 Moreover, it was recently demonstrated that these methods also enable to control the economic and environmental footprint of the synthetic protocol, increase reactor productivity, recycle waste streams, prevent the combustion of organic compounds, and minimize separation efforts.354 These synthetic aspects are often overlooked, but play a critical role, particularly regarding the importance of maintaining a green fingerprint in the valorisation of biomass.
The most abundant zeolite catalyst used today (primarily in FCC) comprises the faujasite family, and more specifically the siliceous USY variants. Commercial USY zeolites comprise a 3D network of 12 MR micropores (0.74 nm). Although the micropores are relatively large, it was demonstrated that many petrochemical conversions occur predominately on the external surface,342,343,345,347,348,351 emphasizing the potential of their hierarchical analogues. Taking into account the abundance of USY zeolites and their organic-free hydrothermal synthesis, their hierarchical form can be considered as an ideal sustainable catalyst for biomass related conversions.354 Since conventional USYs are prepared by steaming and acid leaching of Y zeolites, bottom-up strategies cannot be used to prepare their hierarchical analogues. Moreover, the faujasite family has proven to be a highly illustrative case study,262,355 due to the counterintuitive stability of faujasites in the liquid phase compared to the well-known gas phase (see Concept 6).
The variety of post-synthetic modifications protocols that have been reported is quite substantial,262,342,343,345,347,348,351,354–356 and are recently summarised.357 Although each has a certain advantage, it is important that both the individual steps as the synthesis sequence of steps should be optimised. A good example hereof is the synthesis of hierarchical Y zeolites, requiring an optimised sequence of acid–base–acid treatments.355 In addition, to enable a true rational design of zeolite catalysts, solid synthesis–property–function relationships should be established (Fig. 10). Therefore, systematic synthetic efforts must be combined with in-depth characterisation of all zeolitic and non-zeolite properties, and subsequently be correlated to the catalytic performance.
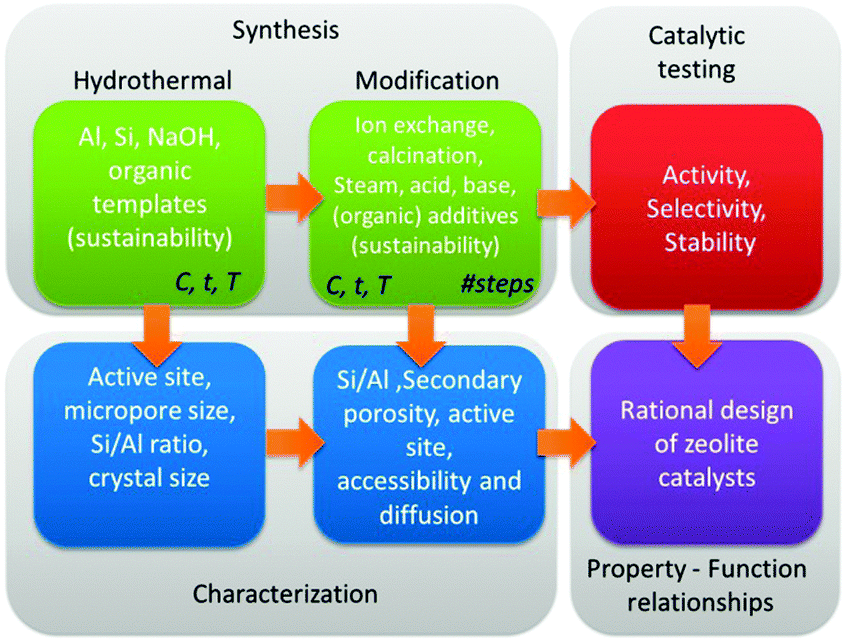 | ||
| Fig. 10 Rational design of catalysts by synthesis, characterisation and catalytic testing of hierarchical zeolites. | ||
A large part of biomass conversion relies on the breaking down of bulky polymers into (mixtures of) basic molecules and/or processing the latter in order to obtain high-value end-products. In both cases, access limitations could be dominant, particularly when dealing with bulky substrates. Accordingly, the enhanced external surface and/or the improved diffusion rate in the zeolite crystal should yield strong activity benefits, as observed for hydrolysis of (hemi)cellulose,262,358 pyrolysis of lignocellulose,359 upgrading of bio-oil,360–362 alkylation with HMF,363 and isomerisation of DHA,212 α-pinene356,364 and safflower oil.356
A higher accessibility to the active sites may also lead to a better diffusion of products out of the crystal, resulting in a decreased contact time of products with the active sites. Therefore, due to the lower susceptibility towards secondary reactions, selectivity enhancements can be obtained, as observed in the isomerisation of n-alkanes.366 Also for biomass conversions, this advantage has led to higher yields of the preferred products, i.e. reducing sugars after hydrolysis of hemicellulose358 or primary isomers after conjugation of safflower oil and α-pinene.356
Particularly, in reactions that can involve acid catalysed breaking of bonds, the hierarchical porosity should prove a powerful tool to steer the degree of cracking to the desired fraction and to control the amount of unwanted by-products. These advantages were already observed for vacuum gasoil cracking using hierarchical USY faujasites365,367 and hierarchical mordenites,368 but also apply to biomass conversion. During the upgrading of bio-oils by catalytic cracking with desilicated Y zeolite (Fig. 2), the selectivity to primary cracking and deoxygenation products is increased, while the selectivity to aromatic hydrocarbons is decreased due to a lower probability for secondary oligomerisation, cyclisation and hydrogen transfer reactions.369 Also in catalytic pyrolysis of biomass (model) compounds, hierarchical zeolites have already proven their value, resulting in high-quality bio-oils and/or lower coke yields.359,361,370 Furthermore, conversion of the resulting biomass pyrolysis vapours to C8 and C9 mono-aromatics was enhanced by mesopore introduction in ZSM-5.371
A particular important factor in the conversion of biomass is the catalyst lifetime, which could be limited due to the formation of cokes, possibly blocking zeolite pores. Hierarchisation of zeolites can offer a solution to improve this lifetime. Whereas the larger external surface prone to coking gives a slower deactivation of the active sites, the lower retention time of products in the micropores could also lead to less secondary coke product formation. A better catalytic stability is reported for bio-oil upgrading with hierarchical zeolites,361,372 whereas a lower amount of formed and/or deposited coke was reported in the isomerisation of α-pinene356 and CFP of lignocellulose.359
Concept 6: zeolite stability
A shift from gas to liquid phase reactions has important consequences for the stability of the used catalysts. Common industrial catalysts are typically developed for gas phase processes. When such catalysts are used in liquid phase reactions, several issues emerge to the stability of these heterogeneous catalysts.373–375 The liquid solvent can irreversibly deactivate the catalyst by hydrolysing the catalyst or its support, inducing leaching of catalyst elements or sintering of metal particles. A second possible issue is deactivation by reaction products or reactants, for example when acids or bases are formed or used. Therefore, besides activity and selectivity also a thorough assessment of catalyst stability is needed under relevant biomass processing conditions.When the stability of zeolites is taken into account, it is important to define the exact nature of the treatment conditions. Traditionally, the stability of zeolites is based on their stability in the gas phase (steam stability). However, with the increasing importance for liquid phase processes and the use of zeolites at different pH, the traditional concept of zeolite stability becomes extremely relative (vide infra), and needs to be refined. Fig. 11 represents a flow chart of the different conditions where zeolites can be used. The first distinction is the phase of the medium (gas vs. liquid phase). Zeolites are commonly used in gas phase processes containing H2O (hydrothermal gas phase, also known as steaming) as well as in the absence of H2O (thermal gas phase). Condensed conditions however, can be divided into atmospheric (P = 1 atm) and super atmospheric (P > 1 atm) pressure. Both cases are dealing with liquid water but super atmospheric conditions (also known as HLW) implies processes at temperatures above the boiling point of water (under elevated pressure), while atmospheric conditions limits the processes under 100 °C (atmospheric liquid water, ALW). Finally, both can also be further specified based on the pH (alkaline, neutral or acidic).
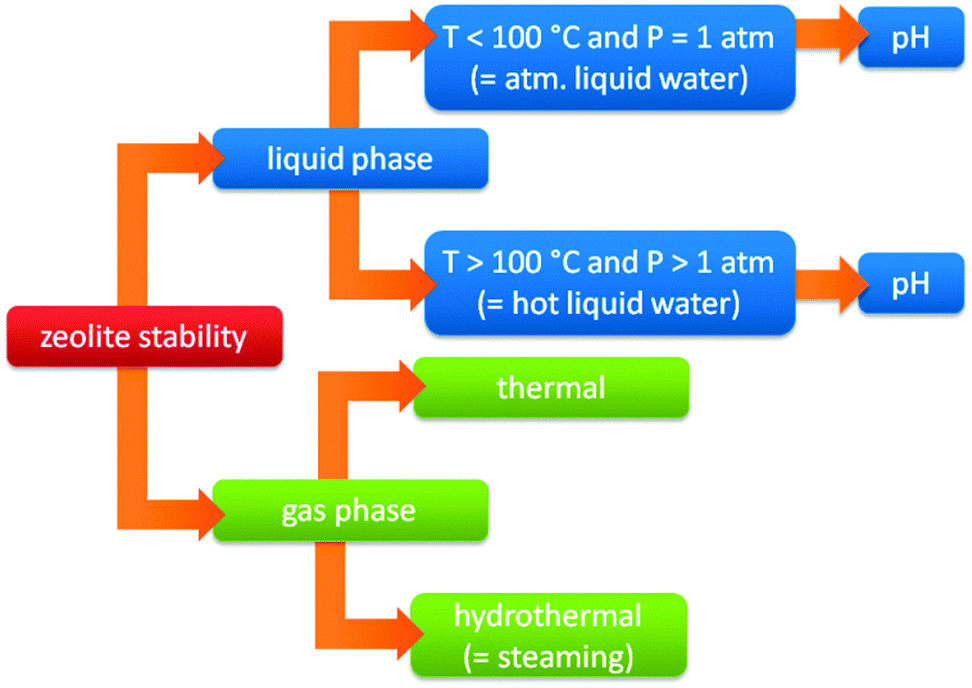 | ||
| Fig. 11 Flow chart with the division of the different conditions where zeolites are used. | ||
Extensive knowledge of the stability of zeolites is mostly restricted to their synthesis conditions and post-synthetic modifications. For example, comprehensive knowledge of zeolite stability in basic HLW during zeolite synthesis is extremely important as the synthesis of the desired metastable zeolite should be interrupted at the appropriate time to prevent the formation of undesirable more dense phases.110,376–379 The stability of zeolites in ALW (at alkaline or acidic conditions) is well-known from post-synthetic modification strategies, and were recently generalised based on the relative abundance of charge-balancing counter cations (CBCC).380,381 As shown in Fig. 12, high-silica zeolites containing low amounts of CBCC, are unstable at basic conditions due to the high sensitivity of Si–O–Si bonds to base catalysed hydrolysis.380,381 On the other hand, zeolites with a high amount of CBCC, offer better resistance to alkaline conditions but are relatively unstable at low pH as Si–O–Al bonds are very sensitive to acid catalysed hydrolysis (Fig. 12).380,381 However, the behaviour of zeolites in neutral or acidic HLW is relatively unknown.
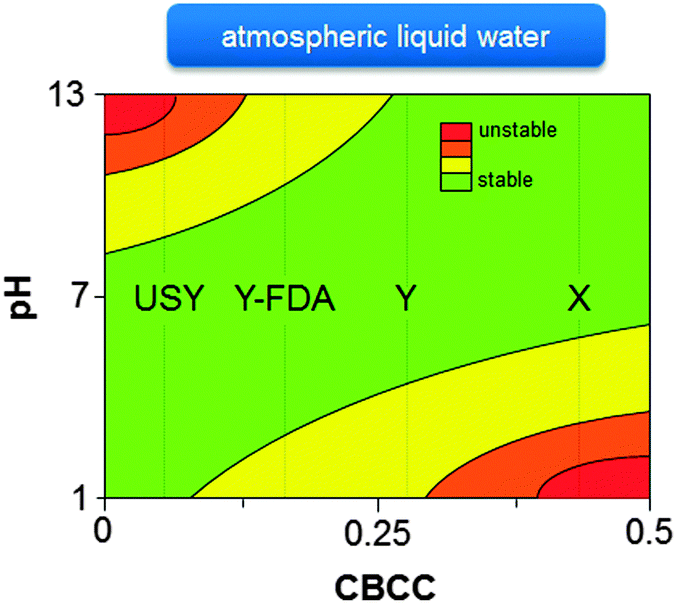 | ||
| Fig. 12 Stability diagram of faujasite zeolites with different charge-balancing counter cations (CBCC). The stability of zeolites in HLW resembles the trend at high pH, whereas the stability of zeolites under steaming conditions resembles the trend at low pH. Y-FDA: framework dealuminated Y zeolite. | ||
Stability of zeolites in acidic HLW
Especially in the case of sugar transformations, the use of zeolites in acidic HLW gains importance as some interesting sugar derived chemicals, like lactic acid and levulinic acid, are (weak) organic acids. When these products are targeted or used as platform chemical, the reaction medium is, dependent on the concentration, slightly to strongly acidified which may have implications on the stability of the used catalysts. For example, irreversible framework damage could be observed during the USY catalysed conversion of DHA into alkyl lactates and lactic acid (Scheme 6).236 Especially in water, with formation of lactic acid, the USY zeolite is strongly affected by severe loss of pore volume, surface area and acidity. In contrast, when the reaction is performed in methanol (with methyl lactate as end-product) only minor zeolite deterioration could be observed.236 Further research demonstrated that the formed (lactic) acid was the main cause of the zeolite degradation with selective dealumination, in line with Fig. 12, leading to a decreased acidity.212,236The same could be observed when zeolite-supported metal-catalysts were used for the hydrogenation of levulinic acid. In this case, ZSM-5 and especially beta show gradual deactivation caused by structure degradation in 2-ethylhexanoic acid.382 The organic acid induces the conversion of Brønsted acid sites into Lewis acid sites, besides an overall decrease in the amount of Al.
Similar behaviour was observed when the reaction is performed in neat levulinic acid but, as the corrosive agent (levulinic acid) disappears upon reaction, the degradation is less pronounced.382 However, when the solvent is changed from organic acids to dioxane, no structural changes could be observed.382,383 These studies show that Al-rich zeolites have potential for reactions with organic acids in aqueous phase when low acid concentrations and temperatures are applied. Otherwise, working in alcoholic media (with the consequent formation of the alkyl esters) or in other non-corrosive solvents shows little problems.
Stability of zeolites in neutral HLW
Besides acidified HLW at 100–150 °C, zeolites are more and more used in neutral HLW at higher temperatures (150–250 °C). Due to the absence of a corrosive agent (like an acid), these conditions seem less stringent. However, in practice, the stability of zeolites under these conditions appears to be less evident than supposed. This is rooted in the dissociation equilibrium of H2O (eqn (1)):| Keq = aH3O+ × aOH− | (1) |
The HLW stability is strongly dependent on the zeolite topology. For example, there is a dependency between the framework stability of a zeolite and the framework density.389 Zeolites with high framework densities, like MOR and MFI, are relatively stable up to 250 °C, while topologies with low density (BEA and especially FAU) undergo already extensive transformations at 150 °C and are largely transformed into an amorphous silica–alumina after a treatment at 200 °C.263,389 Within one topology, the nature of the CBCC greatly influences the stability. Up to now, this is only systematically studied for high CBCC zeolites, such as X and A, showing an increasing HLW stability by increasing base strength of the metal hydroxide.390–392 This was rationalised by the assumption that a stronger base counteracts more efficiently the structure degradation.391 This explanation assumed that, just like in steam, the main degradation mechanism of zeolites in HLW is Si–O–Al hydrolysis.391 However, as mentioned above, the main degradation method in HLW resembles more the degradation pathway in alkaline ALW, namely Si–O–Si hydrolysis, than dealumination under steaming conditions.262 Further investigations elucidating the exact influence of the nature of the CBCC on the HLW stability can clarify this issue.
Yet, when the purpose is to use zeolites in Brønsted acid catalysed reactions, one is restricted to the use of zeolites in their protonic form or at least in a mixed form with protons as one of the CBCC. Therefore, several research groups studied in more detail the potential of H-USY in HLW. All investigated H-USY zeolites degraded to an extent depending on the Si/Al ratio.262 USY zeolites with a high Si/Al ratio transform within 2 to 6 hours into completely amorphous materials which have lost nearly all micropores and Brønsted acid sites. On the other hand, USY zeolites with intermediate Si/Al ratios degrade at a slower rate due to the presence of the Al centres which counteract the hydrolysis of framework bonds.262,263 Another indication for the stabilizing nature of aluminium was observed when salts, like NaCl, were added to the treatment medium. As the presence of these salts enhances the hydrolysis of Si–O–Al bonds, the zeolite stability was significantly affected in a negative sense.393 These observations show that the most stable zeolites during hydrothermal/steam treatment, extensively dealuminated USY zeolites, are the least stable in HLW. From this point of view, the name ‘ultra-stable Y zeolite’ (USY) is an unsuitable name as it is not the most stable Y zeolite under all conditions. Yet, throughout this paper we have used ‘USY’ for clarity.
Recently, it was demonstrated that not only the amount of aluminium but also its location and nature is of particular interest.262 By comparing two FAU zeolites with the same overall Si/Al ratio, e.g. one Y zeolite without EFAl and a slightly steamed USY zeolite with EFAl, it could be concluded that the USY zeolite with EFAl shows a higher resistance to HLW than the Y zeolite without EFAl.262 These conclusions are in line with earlier observations, which have shown that steamed Y zeolites, containing EFAl, are more stable than Y zeolites dealuminated by substitution, consequently containing no EFAl.381,394,395 The higher stability of slightly steamed zeolites was related to the presence of the low water-soluble amorphous EFAl species, in line with the low water solubility of alumina gel.262 Besides, by interacting with terminal Si–OH groups, EFAl protects such hydrolysis sensitive groups against the attack of OH− anions.386,396 It could be demonstrated that not only steaming, but also treatment in HLW induces the formation of Al-rich species at the outer surface, forming a poorly soluble, protective layer around the zeolite. Therefore, slightly dealuminated USY zeolites with Si/Al ratio lower than 3 self-stabilize over time during treatment in HLW.262
Improving the HLW stability of zeolites
The stabilizing role of EFAl provides a suitable tool to tailor the stability of zeolites under HLW conditions by, for example, realuminating the surface of (extensively) dealuminated USY zeolites.395–397 An alkaline alumination method was suggested whereby two consecutive steps are involved to form aluminosilicate species at the outer surface. First, the alkaline conditions lead to a partial dissolution of the zeolite, afterward these dissolved silicon species precipitate with the externally added aluminate on the zeolite surface.395,396 The success rate is very sensitive to the extent of alkalinity during the modification process. If the solution is too alkaline, the zeolite dissolves completely, while a too low alkaline medium leads to insufficient aluminate concentrations for total coverage of the zeolite surface.395,396 During this assessment only the zeolite structure after treatment at intermediate temperatures (150–160 °C) was studied, while the catalytic active sites were not taken into account. Further research is needed to investigate the potential of such realuminated zeolites as Brønsted acid catalysts in liquid water at high(er) temperatures. Strikingly, the (re)alumination process is also crucial in the synthesis of hierarchical zeolites by base treatment in aqueous solutions.398,399 Therefore, it is likely that stabilisation and mesopore formation can be executed simultaneously.Another option to prevent extensive degradation of high silica zeolites, is to limit the contact of the zeolite with water. A common strategy therefore is increasing the hydrophobicity of the material.400 Increasing the Si/Al ratio, so decreasing the Al content, is an effective method to increase the zeolite hydrophobicity. Yet, when Al is eliminated from the framework, Brønsted acid sites are immolated, which is detrimental for the use of these catalysts in Brønsted acid catalysed reactions. Besides, it was demonstrated that high silica USY zeolites are unstable in HLW, despite the higher hydrophobicity.262,263 A better way to improve the hydrophobicity of a zeolite is by functionalizing the surface with hydrophobic species to increase the water repulsion.400 For example, by modifying high silica H-USY zeolites with organosilanes, USY zeolites with hydrophobic surfaces are created.401,402 The chloro ligands in the alkyltrichlorosilanes react with the surface OH groups of the zeolite.401 Due to the higher hydrophobicity, these modified zeolites retain a high fraction of their micropore volume and crystallinity after recycling, showing an enhanced stability compared to non-functionalised high silica USY zeolites.401,402 The economic feasibility of the method is likely the next challenge before such modified catalyst can be used in industrial applications.
Conclusions and perspectives
Besides their pivotal role in crude oil refining, zeolites can potentially also play an important role in biomass conversions based on the high similarity between these two. Both conversions require the degradation and transformation of a complex feedstock towards useful components. However, based on the higher oxygen content of biomass compared to crude oil, there are also some major differences affecting the valorisation (Fig. 13). | ||
| Fig. 13 Schematic overview of the challenges arising from the shift from crude oil towards biomass feedstock. | ||
First, where crude oil refining is mostly a gas phase process, biomass conversions take mainly place in condensed phase, mostly aqueous media, which has major implications on the stability of the catalysts. Common industrial zeolite catalysts are mainly designed to withstand gas phase processes, which does not necessarily mean that they also endure in aqueous conditions. The latter becomes particularly relevant when the stability of metal particles in zeolites is considered.17,262,271–274 By further establishing the stability mechanisms of the zeolite catalysts in aqueous phase conditions, stable zeolites may be designed with simple procedures, which are essential for industrial processes.
Despite the success of Brønsted acidic zeolites in biomass valorisation, the exact nature of the active site in polar aqueous solutions is still vague. Due to the polar solvent molecules the intrinsic nature of the acid sites can be altered by solvation effects. Deeper insight in this matter may be accompanied by progress of the development of Brønsted acidic catalysts used in aqueous conditions. In comparison with petrorefinery, Lewis acid catalysis will play a much more important role in biorefineries. The numerous examples of Lewis acid catalysed biomass conversions indicate the potential of these pathways for the valorisation of biomass. Moreover, the limited knowledge on the nature of these Lewis acid sites indicates the room for improvements.
Due to the reactive nature of biomass derivatives, it is in some specific cases preferred to use basic sites instead of acidic ones. For example, basic catalysts have much lower deactivation tendencies and consequently higher yields in the aldol condensation of aldehydes.403 Traditionally, basicity was introduced in zeolites by ion-exchange of CBCC with alkali metals, such as caesium.404–406 As the basicity of these zeolites correlates with the aluminium content of the zeolite and the size of the alkali metal cations, mostly high aluminium zeolites were loaded with consequently high amounts of alkali metals.407 Due to diffusion constraints, a particularly large part of the introduced metal cations are not used resulting in modest activities while having high cost per actually used basic site. New approaches to synthesize active but less expensive basic zeolites are explored. For example, by a mild base treatment of silicon-rich zeolites, Na+-stabilised deprotonated silanol groups are created as base centers.360,408 However, the susceptibility of silicon-rich zeolites towards dissolution and amorphisation under basic conditions, as used during the synthesis procedure, poses severe challenges to the aforementioned method. Alternatively, alcoholic solvents can be used for the generation of basic sites, avoiding the alkaline degradation of the zeolites during synthesis.403 Also, an acidic method was reported to produce alkaline-earth loaded (Mg, Ca, Sr or Ba) silicalite-1 showing basic properties.409 Recently, acid–base pairs in Lewis acidic zeolites (Hf-, Sn- and Zr-beta) were described.209,410 Such materials are suggested as highly effective catalysts for the cross-aldol condensation of aromatic aldehydes with acetone.410 The zeolitic metal center polarizes the carbonyl group of the aldehyde allowing the framework oxygen, bound to the metal atom, to act as a base and abstracting the α-proton. Hereby, a silanol group and a metal enolate are created, whereby the latter rapidly undergoes C–C coupling with other aldehydes. Such type of catalysts are extremely elegant as they remain active in the presence of water and in acidic solutions. Especially the latter is important as minute amounts of acids, such as acetic acid, are ubiquitous in biomass processing.
Due to the low stability of some high-oxygenated components, it may be necessary to convert these products immediately to more stable ones. Therefore, strategic combinations of the active sites is essential, in which the amount and strength of the present functions must be optimally balanced. Moreover, as this balance is in some cases very thin, it is recommended that all active sites are easily accessible for the reactants. When the active sites of one of the catalytic functions are, for example, significant less accessible, the ratio between both catalytic functions is drifted away from the optimal point resulting in an excess of the other catalytic function(s) leading to unwanted side reactions.
Although hierarchical zeolites haven been firmly established in classical petrochemical conversions, enhancing the accessibility of active sites by increasing the external surface may not always lead to an enhanced catalytic performance in biomass-related conversions. This should be largely due to the more polar feedstock and solvents. For example, the high polarity may hamper the desorption of a formed product. The latter, particularly in combination with relative mild reaction conditions, may lead to a saturation of the zeolite's micropores with products, ultimately ceasing all catalytic activity. In this case, the use of sweeping agents may be of use to ‘free’ the zeolite's porosity and maintain catalytic activity. Only then, with the catalytic cycle optimised, can the full potential of the zeolite be addressed using secondary porosity. This principle was recently demonstrated in the zeolite catalysed dealkylation of alkylphenols derived from lignin.411 In this case, co-feeding of steam enabled to remove phenol from the ZSM-5 catalyst enabling highly selective and stable formation of phenol and olefins for several days with time on stream.
The use of biomass instead of fossil fuels is seen as a more sustainable approach to produce chemicals and fuels. However, it is also important to study the sustainability of synthetic aspects in zeolite-based catalysts as this is often overlooked. For example, application of Sn-beta zeolites is hampered by a time-consuming (up to 40 days) and environmentally unfriendly (use of HF) synthesis procedure. The last years however, new protocols are described to synthesize Sn-beta in shorter synthesis times33–35 or by using post-processing procedures on commercially available beta.36–38
The concept of shape-selectivity could be exploited more thoroughly in biomass conversion. Especially in the conversion of platform molecules to fuels and chemicals via specific processes, zeolite confinement effects could be beneficial. Actually, less examples are identified in biomass conversion compared to petrochemistry. This may be due to the more divergent nature of the biomass conversion products, rooted in the more functional nature of the starting feedstock. In CFP, shape-selective effects have been exploited in more depth, and have in fact proven crucial. Exciting progress based on transferring this original petrochemical concept has been achieved recently.16 Not surprisingly, the targeted products in these processes are less functionalised, often aromatic, petrochemical(-like) molecules.
It should be stressed that, when doubt arises on the recognition of shape-selective effects, the universal shape-selectivity factor, as defined by Weisz, can offer solution (Fig. 8).326 In contrast to the early work on shape-selectivity, which was developed for gas phase catalysis, the conversion of biomass compounds in liquid phase might lead to the recognition of different types of or induce unknown complexities in shape-selectivity. It is important to note that the origin of common shape-selective patterns are sometimes not yet fully understood. Whether they originate from thermodynamic reasons of adsorption327 or rather from pure steric constraints on molecular motion or product formation326,412 often remains an open question.
Biorefineries may only be competitive with crude oil refineries when the entire entity of the feedstock is used. In other words, it is important to develop valorisation paths for every incoming stream (cellulose, hemicellulose, lignin, triglycerides, turpentine, amino acids etc.) to minimise the amount of feedstock that will end as waste or is burned off. In addition, the role of the pretreatment is often overlooked (or avoided by using model compounds) in biomass research.413 Nevertheless, the use of industrial feedstocks often requires some specific changes to the used catalysts. Therefore, integrating industrial feedstocks in the designed lab-scale processes is, from an industrial point of view, at least as important as the catalytic process itself.
The catalytic applicability of zeolites in various biomass processes is, in the academic context, often performed in batch reaction systems. Thereby catalyst deactivation by cokes deposition is not critically addressed. Here, testing in line linked set-ups or even better in continuous mode is necessary and will give a better insight in the process design or in the feasibility to scale-up for industrial use. For example, due to the high oxygen content of biomass and the lower stability of the intermediates, severe coking may occur. Inventive reactor set-ups, where inspiration can be derived from petrochemistry, could give a solution.
Biomass components are often solid substrates due to their high oxygen content, which hampers the interaction with solid catalysts. This may lead to low space-time yields, by, for example, relatively high catalyst-substrate ratios, occasionally up to 1 or 2. Finding alternatives to increase the interaction between the catalyst and the substrate may be crucial in decreasing this ratio. To this end, it is important, instead of focusing merely on the catalytic process, to pay more attention to the engineering part of the process.
Research and development of catalysts does not end when active and selective powder catalysts are developed. Besides the upscaling of the process itself, another important hurdle should be taken, namely the transformation of a research catalyst to an industrial catalyst. Such industrial catalysts contain, besides the active phase, multiple additives to provide desired physical, chemical and mechanical properties. This is exemplified by FCC catalysts, where around 10 wt% of the industrial catalyst may constitute the active phase (USY zeolite).414–420 The other 90% comprises binders, catalyst matrix, fillers and other additives.10 Some of these components, like silica–alumina materials, also exercise a catalytic role by pre-cracking the large oil molecules which facilitates the diffusion towards the active phase. As biomolecules are also bulky molecules, the use of an active pre-cracking matrix, whether or not preceded by a thermal/chemical pretreatment step, may be an essential element for an industrial biorefinery catalyst. However, literature about the composition of industrial (biorefinery) catalysts remains rare.421 Also here, the abovementioned differences between both feedstocks will lead to important alterations pertaining to FCC catalysts. There is, for example, less need to design catalysts with active components for the removal of traces of N and S, while hydrogenation of double bonds and/or deoxygenation will be largely in focus.
Acknowledgements
T. E., J. V. A. and R. D. C. thank IWT-Vlaanderen for their doctoral fellowship. W. S., M. D. and D. V. acknowledge Research Foundation-Flanders (FWO) for financial support. B. F. S. thanks the Research Council of KU Leuven and the Flemish government (FISCH ARBOREF biomass program). The Belgian Government is acknowledged for financial support through IAP funding (Belspo).References
- G. Centi and R. A. van Santen, Catalysis for Renewables: From Feedstock to Energy Production, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- M. Dusselier, M. Mascal and B. F. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, Topics in Current Chemistry, ed. K. M. Nicholas, Springer International Publishing, 2014, vol. 353, ch. 544, pp. 1–40 Search PubMed.
- N. Qureshi and T. C. Ezeji, Biofuels, Bioprod. Biorefin., 2008, 2, 319–330 CrossRef CAS.
- B. D. Solomon, J. R. Barnes and K. E. Halvorsen, Biomass Bioenergy, 2007, 31, 416–425 CrossRef.
- M. A. Abdel-Rahman, Y. Tashiro and K. Sonomoto, Biotechnol. Adv., 2013, 31, 877–902 CrossRef CAS PubMed.
- S. Nanda, A. K. Dalai and J. A. Kozinski, Energy Sci. Eng., 2014, 2, 138–148 CrossRef CAS.
- N. Qureshi, B. C. Saha, B. Dien, R. E. Hector and M. A. Cotta, Biomass Bioenergy, 2010, 34, 559–565 CrossRef CAS.
- Y.-N. Zheng, L.-Z. Li, M. Xian, Y.-J. Ma, J.-M. Yang, X. Xu and D.-Z. He, J. Ind. Microbiol. Biotechnol., 2009, 36, 1127–1138 CrossRef CAS PubMed.
- A. V. Bridgwater, Chem. Eng. J., 2003, 91, 87–102 CrossRef CAS.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- A. Corma, G. W. Huber, L. Sauvanaud and P. O'Connor, J. Catal., 2007, 247, 307–327 CrossRef CAS.
- G. W. Huber, P. O'Connor and A. Corma, Appl. Catal., A, 2007, 329, 120–129 CrossRef CAS.
- T. R. Carlson, Y.-T. Cheng, J. Jae and G. W. Huber, Energy Environ. Sci., 2011, 4, 145–161 CAS.
- T. R. Carlson, J. Jae, Y.-C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS.
- T. R. Carlson, T. R. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397–400 CrossRef CAS PubMed.
- Y.-T. Cheng, Z. Wang, C. J. Gilbert, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 11097–11100 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2013, 114, 1827–1870 CrossRef PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- IZA-Structure-Commission, Database of Zeolite Structures, http://www.iza-online.org/.
- E. M. Flanigen, Pure Appl. Chem., 1980, 52, 2191–2211 CrossRef CAS.
- R. A. Meyers, Handbook of Petroleum Refining Processes, McGraw-Hill Education, New York, 3 edn, 2004 Search PubMed.
- H. Van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Introduction to Zeolite Science and Practice, Elsevier, 2001 Search PubMed.
- W. Vermeiren and J. P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- P. B. Weisz and V. J. Frilette, J. Phys. Chem., 1960, 64, 382 CrossRef CAS.
- P. A. Jacobs, M. Dusselier and B. F. Sels, Angew. Chem., Int. Ed., 2014, 53, 8621–8626 CrossRef CAS PubMed.
- C.-C. Chang, Z. Wang, P. Dornath, H. Je Cho and W. Fan, RSC Adv., 2012, 2, 10475–10477 RSC.
- Z. Kang, X. Zhang, H. Liu, J. Qiu, W. Han and K. L. Yeung, Mater. Chem. Phys., 2013, 141, 519–529 CrossRef CAS.
- Z. Kang, X. Zhang, H. Liu, J. Qiu and K. L. Yeung, Chem. Eng. J., 2013, 218, 425–432 CrossRef CAS.
- J. Dijkmans, D. Gabriels, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- C. Hammond, S. Conrad and I. Hermans, Angew. Chem., Int. Ed., 2012, 51, 11736–11739 CrossRef CAS PubMed.
- P. Li, G. Liu, H. Wu, Y. Liu, J.-G. Jiang and P. Wu, J. Phys. Chem. C, 2011, 115, 3663–3670 CAS.
- S. Tolborg, A. Katerinopoulou, D. D. Falcone, I. Sadaba, C. M. Osmundsen, R. J. Davis, E. Taarning, P. Fristrup and M. S. Holm, J. Mater. Chem. A, 2014, 2, 20252–20262 CAS.
- W. Vermeiren and J. P. Gilson, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co, 2 edn, 2008 Search PubMed.
- D. Kubička, I. Kubičková and J. Čejka, Catal. Rev., 2013, 55, 1–78 Search PubMed.
- D. Kubička and O. Kikhtyanin, Catal. Today, 2015, 243, 10–22 CrossRef.
- I. M. J. A. M. Den Otter, Fette, Seifen, Anstrichm., 1970, 72, 667–673 CrossRef.
- M. J. A. M. Den Otter, Fette, Seifen, Anstrichm., 1970, 72, 875–883 CrossRef CAS.
- S. C. C. Wiedemann, J. A. Stewart, F. Soulimani, T. van Bergen-Brenkman, S. Langelaar, B. Wels, P. de Peinder, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2014, 316, 24–35 CrossRef CAS.
- U. Biermann and J. O. Metzger, Eur. J. Lipid Sci. Technol., 2008, 110, 805–811 CrossRef CAS.
- T. A. Foglia, T. Perlstein, Y. Nakano and G. Maerker, US4371469A, 1983.
- Y. Nakano, T. A. Foglia, H. Kohashi, T. Perlstein and S. Serota, J. Am. Oil Chem. Soc., 1985, 62, 888–891 CrossRef CAS.
- M. Neuss and H. Eierdanz, US5364949A, 1994.
- R. M. Koster, M. Bogert, B. de Leeuw, E. K. Poels and A. Bliek, J. Mol. Catal. A: Chem., 1998, 134, 159–169 CrossRef CAS.
- H. L. Ngo, A. Nuñez, W. Lin and T. A. Foglia, Eur. J. Lipid Sci. Technol., 2007, 109, 214–224 CrossRef CAS.
- S. Zhang and Z. C. Zhang, Catal. Lett., 2007, 115, 114–121 CrossRef CAS.
- Z. Zhang, M. Dery, S. Zhang and D. Steichen, J. Surfactants Deterg., 2004, 7, 211–215 CrossRef CAS.
- T. Tomifuji, H. Abe, Y. Matsumura and Y. Sakuma, US5677473A, 1997.
- W. R. Hodgson, W. T. Koetsier, C. M. Lok and G. Roberts, EP0774451A1, 1996.
- W. R. Hodgson, C. M. Lok, G. Roberts and W. T. Koetsier, US5856539A, 1999.
- S. C. C. Wiedemann, A. Muñoz-Murillo, R. Oord, T. van Bergen-Brenkman, B. Wels, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2015, 329, 195–205 CrossRef CAS.
- H. L. Ngo, E. Hoh and T. A. Foglia, Eur. J. Lipid Sci. Technol., 2012, 114, 213–221 CrossRef CAS.
- L. Dandik, H. A. Aksoy and A. Erdem-Senatalar, Energy Fuels, 1998, 12, 1148–1152 CrossRef CAS.
- S. P. R. Katikaneni, J. D. Adjaye and N. N. Bakhshi, Energy Fuels, 1995, 9, 599–609 CrossRef CAS.
- T. A. Milne, R. J. Evans and N. Nagle, Biomass, 1990, 21, 219–232 CrossRef CAS.
- F. A. Twaiq, N. A. M. Zabidi and S. Bhatia, Ind. Eng. Chem. Res., 1999, 38, 3230–3237 CrossRef CAS.
- F. A. A. Twaiq, A. R. Mohamad and S. Bhatia, Fuel Process. Technol., 2004, 85, 1283–1300 CrossRef CAS.
- P. S. Yarlagadda, Y. Hu and N. N. Bakhshi, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25, 251–257 CrossRef CAS.
- S. P. R. Katikaneni, J. D. Adjaye and N. N. Bakhshi, Can. J. Chem. Eng., 1995, 73, 484–497 CrossRef CAS.
- T. Y. Leng, A. R. Mohamed and S. Bhatia, Can. J. Chem. Eng., 1999, 77, 156–162 CrossRef CAS.
- R. Černý, M. Kubů and D. Kubička, Catal. Today, 2013, 204, 46–53 CrossRef.
- T. J. Benson, R. Hernandez, W. T. French, E. G. Alley and W. E. Holmes, J. Mol. Catal. A: Chem., 2009, 303, 117–123 CrossRef CAS.
- R. W. Gosselink, S. A. W. Hollak, S.-W. Chang, J. van Haveren, K. P. de Jong, J. H. Bitter and D. S. van Es, ChemSusChem, 2013, 6, 1576–1594 CrossRef CAS PubMed.
- D. Kubička, P. Šimáček and N. Žilková, Top. Catal., 2009, 52, 161–168 CrossRef.
- S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, ChemSusChem, 2009, 2, 1109–1119 CrossRef CAS PubMed.
- M. Mohammad, T. Kandaramath Hari, Z. Yaakob, Y. Chandra Sharma and K. Sopian, Renewable Sustainable Energy Rev., 2013, 22, 121–132 CrossRef CAS.
- E. Santillan-Jimenez and M. Crocker, J. Chem. Technol. Biotechnol., 2012, 87, 1041–1050 CrossRef CAS.
- A. K. Sinha, M. Anand, B. S. Rana, R. Kumar, S. A. Farooqui, M. G. Sibi, R. Kumar and R. K. Joshi, Catal. Surv. Asia, 2013, 17, 1–13 CrossRef CAS.
- M. Toba, Y. Abe, H. Kuramochi, M. Osako, T. Mochizuki and Y. Yoshimura, Catal. Today, 2011, 164, 533–537 CrossRef CAS.
- C. Wang, Q. Liu, J. Song, W. Li, P. Li, R. Xu, H. Ma and Z. Tian, Catal. Today, 2014, 234, 153–160 CrossRef CAS.
- C. Wang, Z. Tian, L. Wang, R. Xu, Q. Liu, W. Qu, H. Ma and B. Wang, ChemSusChem, 2012, 5, 1974–1983 CrossRef CAS PubMed.
- M. M. Wright, D. E. Daugaard, J. A. Satrio and R. C. Brown, Fuel, 2010, 89, S2–S10 CrossRef CAS.
- M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard and D. D. Hsu, Techno-Economic Analysis of Biomass Fast Pyrolysis to Transportation Fuels, Report NREL/TP-6A20-46586, National Renewable Energy Laboratory, 2010 Search PubMed.
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89, S29–S35 CrossRef CAS.
- D. Mohan, C. U. Pittman, Jr. and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- J. Diebold and J. Scahill, ACS Symp. Ser., 1988, 376, 264–276 CrossRef CAS.
- P. A. Horne, N. Nugranad and P. T. Williams, J. Anal. Appl. Pyrolysis, 1995, 34, 87–108 CrossRef CAS.
- P. A. Horne and P. T. Williams, J. Anal. Appl. Pyrolysis, 1995, 34, 65–85 CrossRef CAS.
- P. T. Williams and P. A. Horne, Fuel, 1995, 74, 1839–1851 CrossRef CAS.
- P. T. Williams and P. A. Horne, J. Anal. Appl. Pyrolysis, 1995, 31, 39–61 CrossRef CAS.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Top. Catal., 2009, 52, 241–252 CrossRef CAS.
- J. Jae, G. A. Tompsett, A. J. Foster, K. D. Hammond, S. M. Auerbach, R. F. Lobo and G. W. Huber, J. Catal., 2011, 279, 257–268 CrossRef CAS.
- A. Corma and A. Martínez, in Stud. Surf. Sci. Catal., ed. J. Čejka and H. V. Bekkum, Elsevier, 2005, vol. 157, pp. 337–366 Search PubMed.
- A. J. Foster, J. Jae, Y.-T. Cheng, G. W. Huber and R. F. Lobo, Appl. Catal., A, 2012, 423, 154–161 CrossRef.
- K. Wang, K. H. Kim and R. C. Brown, Green Chem., 2014, 16, 727–735 RSC.
- J. Q. Bond, A. A. Upadhye, H. Olcay, G. A. Tompsett, J. Jae, R. Xing, D. M. Alonso, D. Wang, T. Zhang, R. Kumar, A. Foster, S. M. Sen, C. T. Maravelias, R. Malina, S. R. H. Barrett, R. Lobo, C. E. Wyman, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2014, 7, 1500–1523 CAS.
- H. Ben and A. J. Ragauskas, RSC Adv., 2012, 2, 12892–12898 RSC.
- M. A. Jackson, D. L. Compton and A. A. Boateng, J. Anal. Appl. Pyrolysis, 2009, 85, 226–230 CrossRef CAS.
- X. Li, L. Su, Y. Wang, Y. Yu, C. Wang, X. Li and Z. Wang, Front. Environ. Sci. Eng., 2012, 6, 295–303 CrossRef CAS.
- C. A. Mullen and A. A. Boateng, Fuel Process. Technol., 2010, 91, 1446–1458 CrossRef CAS.
- R. W. Thring, S. P. R. Katikaneni and N. N. Bakhshi, Fuel Process. Technol., 2000, 62, 17–30 CrossRef CAS.
- Y. Zhao, L. Deng, B. Liao, Y. Fu and Q.-X. Guo, Energy Fuels, 2010, 24, 5735–5740 CrossRef CAS.
- H. Ben and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2013, 1, 316–324 CrossRef CAS.
- J. Jae, G. A. Tompsett, Y.-C. Lin, T. R. Carlson, J. Shen, T. Zhang, B. Yang, C. E. Wyman, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2010, 3, 358–365 CAS.
- Z. Ma, E. Troussard and J. A. van Bokhoven, Appl. Catal., A, 2012, 423, 130–136 CrossRef.
- Y. Yu, X. Li, L. Su, Y. Zhang, Y. Wang and H. Zhang, Appl. Catal., A, 2012, 447, 115–123 CrossRef.
- A. A. Lappas, M. C. Samolada, D. K. Iatridis, S. S. Voutetakis and I. A. Vasalos, Fuel, 2002, 81, 2087–2095 CrossRef CAS.
- J. Jae, R. Coolman, T. J. Mountziaris and G. W. Huber, Chem. Eng. Sci., 2014, 108, 33–46 CrossRef CAS.
- R. A. Beyerlein, C. Choi-Feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4, 27–42 CrossRef CAS.
- G. T. Kerr, J. Phys. Chem., 1967, 71, 4155–4156 CrossRef CAS.
- G. T. Kerr, Appl. Catal., 1969, 15, 200–204 CrossRef CAS.
- M. Briend-Faure, O. Cornu, D. Delafosse, R. Monque and M. J. Peltre, Appl. Catal., 1988, 38, 71–87 CrossRef CAS.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- B. Kasprzyk-Hordern, Adv. Colloid Interface Sci., 2004, 110, 19–48 CrossRef CAS PubMed.
- G. J. Mulder, J. Prakt. Chem., 1840, 21, 203–240 CrossRef.
- F. H. Newth, in Adv. Carbohydr. Chem., ed. S. H. Claude and M. C. Sidney, Academic Press, 1951, vol. 6, pp. 83–106 Search PubMed.
- R. Karinen, K. Vilonen and M. Niemelä, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Energy Environ. Sci., 2011, 4, 793–804 CAS.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Molecules, 2010, 15, 3863–3877 CrossRef CAS PubMed.
- K. Lourvanij and G. L. Rorrer, Ind. Eng. Chem. Res., 1993, 32, 11–19 CrossRef CAS.
- K. Lourvanij and G. L. Rorrer, J. Chem. Technol. Biotechnol., 1997, 69, 35–44 CrossRef CAS.
- B. F. M. Kuster, Starch-Stärke, 1990, 42, 314–321 CrossRef CAS.
- K.-i. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849–1853 CrossRef CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- Y. Nakamura and S. Morikawa, Bull. Chem. Soc. Jpn., 1980, 53, 3705–3706 CrossRef CAS.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS.
- C. Moreau, R. Durand, C. Pourcheron and S. Razigade, Ind. Crops Prod., 1994, 3, 85–90 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- C. Moreau, R. Durand, D. Peyron, J. Duhamet and P. Rivalier, Ind. Crops Prod., 1998, 7, 95–99 CrossRef CAS.
- A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, Carbohydr. Res., 2006, 341, 2946–2953 CrossRef CAS PubMed.
- S. Kim, S. You, Y. Kim, S. Lee, H. Lee, K. Park and E. Park, Korean J. Chem. Eng., 2011, 28, 710–716 CrossRef CAS.
- R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751–761 CrossRef CAS PubMed.
- S. J. You and E. D. Park, Microporous Mesoporous Mater., 2014, 186, 121–129 CrossRef CAS.
- S. Lima, A. Fernandes, M. Antunes, M. Pillinger, F. Ribeiro and A. Valente, Catal. Lett., 2010, 135, 41–47 CrossRef CAS.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141–148 CrossRef CAS.
- J. Lessard, J.-F. Morin, J.-F. Wehrung, D. Magnin and E. Chornet, Top. Catal., 2010, 53, 1231–1234 CrossRef CAS.
- X. Shi, Y. Wu, H. Yi, G. Rui, P. Li, M. Yang and G. Wang, Energies, 2011, 4, 669–684 CrossRef CAS.
- M. M. Antunes, S. Lima, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2012, 417–148, 243–252 CrossRef.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Güemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS PubMed.
- A. S. Dias, S. Lima, D. Carriazo, V. Rives, M. Pillinger and A. A. Valente, J. Catal., 2006, 244, 230–237 CrossRef CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, Microporous Mesoporous Mater., 2006, 94, 214–225 CrossRef CAS.
- C. García-Sancho, I. Agirrezabal-Telleria, M. B. Güemez and P. Maireles-Torres, Appl. Catal., B, 2014, 152–1153, 1–10 CrossRef.
- S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144–2148 CrossRef CAS.
- B. Pholjaroen, N. Li, Z. Wang, A. Wang and T. Zhang, J. Energy Chem., 2013, 22, 826–832 CrossRef CAS.
- I. Sádaba, S. Lima, A. A. Valente and M. López Granados, Carbohydr. Res., 2011, 346, 2785–2791 CrossRef PubMed.
- T. Suzuki, T. Yokoi, R. Otomo, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2011, 408, 117–124 CrossRef CAS.
- E. Nikolla, Y. Romàn-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- J. Zhang, J. Zhuang, L. Lin, S. Liu and Z. Zhang, Biomass Bioenergy, 2012, 39, 73–77 CrossRef CAS.
- W. Daengprasert, P. Boonnoun, N. Laosiripojana, M. Goto and A. Shotipruk, Ind. Eng. Chem. Res., 2011, 50, 7903–7910 CrossRef CAS.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- R. H. Leonard, Ind. Eng. Chem., 1956, 48, 1330–1341 CrossRef.
- V. Ghorpade and M. Hanna, in Cereals, ed. G. Campbell, C. Webb and S. McKee, Springer US, 1997, ch. 7, pp. 49–55 Search PubMed.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2013, 6, 76–80 CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- D. Fegyverneki, L. Orha, G. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078–1081 CrossRef CAS.
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef PubMed.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed.
- I. T. Horvath, Green Chem., 2008, 10, 1024–1028 RSC.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 122, 4581–4585 CrossRef.
- J.-P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488–3490 RSC.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- R. Palkovits, Angew. Chem., Int. Ed., 2010, 49, 4336–4338 CrossRef CAS PubMed.
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- I. van der Meulen, E. Gubbels, S. Huijser, R. Sablong, C. E. Koning, A. Heise and R. Duchateau, Macromolecules, 2011, 44, 4301–4305 CrossRef CAS.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Curr. Opin. Chem. Eng., 2012, 1, 218–224 CrossRef CAS.
- J. Jow, G. L. Rorrer, M. C. Hawley and D. T. A. Lamport, Biomass, 1987, 14, 185–194 CrossRef CAS.
- K. Lourvanij and G. L. Rorrer, Appl. Catal., A, 1994, 109, 147–165 CrossRef CAS.
- W. Zeng, D.-g. Cheng, H. Zhang, F. Chen and X. Zhan, React. Kinet., Mech. Catal., 2010, 100, 377–384 CAS.
- J.-P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437–441 CrossRef CAS PubMed.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- N. Nikbin, P. T. Do, S. Caratzoulas, R. F. Lobo, P. J. Dauenhauer and D. G. Vlachos, J. Catal., 2013, 297, 35–43 CrossRef CAS.
- R. E. Patet, N. Nikbin, C. L. Williams, S. K. Green, C.-C. Chang, W. Fan, S. Caratzoulas, P. J. Dauenhauer and D. G. Vlachos, ACS Catal., 2015, 5, 2367–2375 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- R. Xiong, S. I. Sandler, D. G. Vlachos and P. J. Dauenhauer, Green Chem., 2014, 16, 4086–4091 RSC.
- A. S. Nagpure, N. Lucas and S. V. Chilukuri, ACS Sustainable Chem. Eng., 2015, 3, 2909–2916 CrossRef CAS.
- B. O. Dalla Costa, M. A. Peralta and C. A. Querini, Appl. Catal., A, 2014, 472, 53–63 CrossRef CAS.
- J. L. Dubois, C. Duquenne and W. Hoelderlich, WO2006087083, 2006.
- C.-J. Jia, Y. Liu, W. Schmidt, A.-H. Lu and F. Schüth, J. Catal., 2010, 269, 71–79 CrossRef CAS.
- H. Kasuga, M. Kirishiki, E. Matsunami, M. Okada, M. Okuno and T. Takahashi, WO2007132926, 2007.
- H. Kasuga and M. Okada, JP2008137950, 2008.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass – Volume I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Report DOE/GO-102004-1992 Pacific Northwest National Laboratory, Richland, Washington, 2004.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- B. Katryniok, S. Paul, V. Belliere-Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079–2098 RSC.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- M. Taramasso, G. Perego and B. Notari, US4410501A, 1983.
- N. K. Mal and A. V. Ramaswamy, Chem. Commun., 1997, 425–426 RSC.
- N. K. Mal, V. Ramaswamy, S. Ganapathy and A. V. Ramaswamy, J. Chem. Soc., Chem. Commun., 1994, 1933–1934 RSC.
- M. Moliner, Dalton Trans., 2014, 43, 4197–4208 RSC.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Phys. Chem. C, 2013, 117, 3976–3986 CAS.
- Y. Ko and W. Ahn, Korean J. Chem. Eng., 1998, 15, 423–428 CrossRef CAS.
- J. D. Lewis, S. Van de Vyver, A. J. Crisci, W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, ChemSusChem, 2014, 7, 2255–2265 CrossRef CAS PubMed.
- S.-H. Chien, J.-C. Ho and S.-S. Mon, Zeolites, 1997, 18, 182–187 CrossRef CAS.
- S. Dzwigaj, Y. Millot, C. Méthivier and M. Che, Microporous Mesoporous Mater., 2010, 130, 162–166 CrossRef CAS.
- A. Corma, F. X. Llabrés, I. Xamena, C. Prestipino, M. Renz and S. Valencia, J. Phys. Chem. C, 2009, 113, 11306–11315 CAS.
- R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2406 CrossRef CAS PubMed.
- E. A. Pidko, E. J. M. Hensen and R. A. van Santen, Proc. R. Soc. London, Ser. A, 2012, 468, 2070–2086 CrossRef CAS.
- V. S. Marakatti and A. B. Halgeri, RSC Adv., 2015, 5, 14286–14293 RSC.
- J. Brus, L. Kobera, W. Schoefberger, M. Urbanová, P. Klein, P. Sazama, E. Tabor, S. Sklenak, A. V. Fishchuk and J. Dědeček, Angew. Chem., Int. Ed., 2014, 54, 541–545 Search PubMed.
- M. Boronat, P. Concepción, A. Corma, M. Renz and S. Valencia, J. Catal., 2005, 234, 111–118 CrossRef CAS.
- M. Boronat, A. Corma, M. Renz, G. Sastre and P. M. Viruela, Chem. – Eur. J., 2005, 11, 6905–6915 CrossRef CAS PubMed.
- G. Li, E. A. Pidko and E. J. M. Hensen, Catal. Sci. Technol., 2014, 4, 2241–2250 CAS.
- J. Dijkmans, M. Dusselier, W. Janssens, M. Trekels, A. Vantomme, E. Breynaert, C. E. A. Kirschhock and B. F. Sels, ACS Catal., 2016, 6, 31–46 Search PubMed.
- P. Y. Dapsens, C. Mondelli, B. T. Kusema, R. Verel and J. Perez-Ramirez, Green Chem., 2014, 16, 1176–1186 RSC.
- P. Y. Dapsens, C. Mondelli and J. Perez-Ramirez, Chem. Soc. Rev., 2015, 44, 7025–7043 RSC.
- P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ChemSusChem, 2013, 6, 831–839 CrossRef CAS PubMed.
- J. Jin, X. Ye, Y. Li, Y. Wang, L. Li, J. Gu, W. Zhao and J. Shi, Dalton Trans., 2014, 43, 8196–8204 RSC.
- J. Wang, K. Okumura, S. Jaenicke and G.-K. Chuah, Appl. Catal., A, 2015, 493, 112–120 CrossRef CAS.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- Y. Román-Leshkov and M. E. Davis, ACS Catal., 2011, 1, 1566–1580 CrossRef.
- M. Saito, H. Ikeda, Y. Horiuchi and M. Matsuoka, Res. Chem. Intermed., 2014, 40, 87–96 CrossRef CAS.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef PubMed.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Román-Leshkov, S.-J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS PubMed.
- J. Jae, E. Mahmoud, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 508–513 CrossRef CAS.
- R. S. Assary and L. A. Curtiss, J. Phys. Chem. A, 2011, 115, 8754–8760 CrossRef CAS PubMed.
- W. R. Gunther, Y. Wang, Y. Ji, V. K. Michaelis, S. T. Hunt, R. G. Griffin and Y. Román-Leshkov, Nat. Commun., 2012, 3, 1109–1116 CrossRef PubMed.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Microporous Mesoporous Mater., 2012, 153, 55–58 CrossRef CAS.
- W. R. Gunther, Q. Duong and Y. Román-Leshkov, J. Mol. Catal. A: Chem., 2013, 379, 294–302 CrossRef CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- R. Gounder and M. E. Davis, ACS Catal., 2013, 3, 1469–1476 CrossRef CAS.
- S. Hirasawa, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2012, 2, 1150–1152 CAS.
- Y. Kwon, Y. Birdja, I. Spanos, P. Rodriguez and M. T. M. Koper, ACS Catal., 2012, 2, 759–764 CrossRef CAS.
- R. M. Painter, D. M. Pearson and R. M. Waymouth, Angew. Chem., Int. Ed., 2010, 49, 9456–9459 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 CAS.
- Y. Hayashi and Y. Sasaki, Chem. Commun., 2005, 2716–2718 RSC.
- K. P. F. Janssen, J. S. Paul, B. F. Sels and P. A. Jacobs, Stud. Surf. Sci. Catal., 2007, 170B, 1222–1227 CrossRef CAS.
- C. B. Rasrendra, B. A. Fachri, I. G. B. N. Makertihartha, S. Adisasmito and H. J. Heeres, ChemSusChem, 2011, 4, 768–777 CrossRef CAS PubMed.
- R. M. West, M. S. Holm, S. Saravanamurugan, J. Xiong, Z. Beversdorf, E. Taarning and C. H. Christensen, J. Catal., 2010, 269, 122–130 CrossRef CAS.
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1083–1089 RSC.
- P. Mäki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef PubMed.
- C. M. Osmundsen, M. S. Holm, S. Dahl and E. Taarning, Proc. R. Soc. A, 2012, 468, 2000–2016 CrossRef CAS.
- E. Taarning, S. Saravanamurugan, S. M. Holm, J. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS PubMed.
- Q. Guo, F. Fan, E. A. Pidko, W. N. P. van der Graaff, Z. Feng, C. Li and E. J. M. Hensen, ChemSusChem, 2013, 6, 1352–1356 CrossRef CAS PubMed.
- P. Y. Dapsens, B. T. Kusema, C. Mondelli and J. Pérez-Ramírez, J. Mol. Catal. A: Chem., 2014, 388–189, 141–147 CrossRef.
- S. Van de Vyver, C. Odermatt, K. Romero, T. Prasomsri and Y. Román-Leshkov, ACS Catal., 2015, 5, 972–977 CrossRef CAS.
- A. Corma and M. Renz, Chem. Commun., 2004, 550–551 RSC.
- A. Corma and M. Renz, ARKIVOC, 2007, 40–48 CAS.
- A. Corma, M. T. Navarro and M. Renz, J. Catal., 2003, 219, 242–246 CrossRef CAS.
- O. de la Torre, M. Renz and A. Corma, Appl. Catal., A, 2010, 380, 165–171 CrossRef CAS.
- W. Schutyser, S. Van den Bosch, J. Dijkmans, S. Turner, M. Meledina, G. Van Tendeloo, D. P. Debecker and B. F. Sels, ChemSusChem, 2015, 8, 1805–1818 CrossRef CAS PubMed.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS PubMed.
- M. S. Holm, Y. J. Pagan-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin and I. I. Ivanova, ACS Catal., 2015, 5, 4833–4836 CrossRef CAS.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degreve, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- E. V. Makshina, W. Janssens, B. F. Sels and P. A. Jacobs, Catal. Today, 2012, 198, 338–344 CrossRef CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, ChemSusChem, 2015, 8, 994–1008 CrossRef CAS PubMed.
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2013, 6, 1595–1614 CrossRef CAS PubMed.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Ind. Eng. Chem. Res., 2012, 51, 5364–5366 CrossRef CAS.
- L. Bui, H. Luo, W. R. Gunther and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025 CrossRef CAS PubMed.
- W. Song, Y. Liu, E. Barath, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC.
- J. Dijkmans, M. Dusselier, D. Gabriëls, K. Houthoofd, P. C. M. M. Magusin, S. Huang, Y. Pontikes, M. Trekels, A. Vantomme, L. Giebeler, S. Oswald and B. F. Sels, ACS Catal., 2015, 5, 928–940 CrossRef CAS.
- T. J. Schwartz, S. M. Goodman, C. M. Osmundsen, E. Taarning, M. D. Mozuch, J. Gaskell, D. Cullen, P. J. Kersten and J. A. Dumesic, ACS Catal., 2013, 3, 2689–2693 CrossRef CAS.
- T. Ennaert, J. Geboers, E. Gobechiya, C. M. Courtin, M. Kurttepeli, K. Houthoofd, C. E. A. Kirschhock, P. C. M. M. Magusin, S. Bals, P. A. Jacobs and B. F. Sels, ACS Catal., 2015, 5, 754–768 CrossRef CAS.
- R. M. Ravenelle, F. Schüßler, A. D'Amico, N. Danilina, J. A. van Bokhoven, J. A. Lercher, C. W. Jones and C. Sievers, J. Phys. Chem. C, 2010, 114, 19582–19595 CAS.
- R. Otomo, T. Yokoi, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2014, 470, 318–326 CrossRef CAS.
- H. J. Cho, C.-C. Chang and W. Fan, Green Chem., 2014, 16, 3428–3433 RSC.
- A. Balandin, N. Vasyunina, S. Chepigo and G. Barysheva, Dokl. Akad. Nauk SSSR, 1959, 128, 941–944 CAS.
- V. I. Sharkov, Angew. Chem., Int. Ed., 1963, 2, 405–409 CrossRef.
- J. Sabadie, D. Barthomeuf, H. Charcosset and G. Descotes, Bull. Soc. Chim. Fr., 1981, 288–292 CAS.
- P. A. Jacobs and H. Hinnekens, EP0329923, 1989.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- O. A. Abdelrahman, A. Heyden and J. Q. Bond, ACS Catal., 2014, 4, 1171–1181 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2003, 81, 547–559 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, V. Oleshko and R. J. Davis, J. Phys. Chem. B, 2006, 110, 7869–7876 CrossRef CAS PubMed.
- Y. Schuurman, B. F. M. Kuster, K. van der Wiele and G. B. Marin, Appl. Catal., A, 1992, 89, 47–68 CrossRef CAS.
- A. Shrotri, A. Tanksale, J. N. Beltramini, H. Gurav and S. V. Chilukuri, Catal. Sci. Technol., 2012, 2, 1852–1858 CAS.
- G. Liang, H. Cheng, W. Li, L. He, Y. Yu and F. Zhao, Green Chem., 2012, 14, 2146–2149 RSC.
- G. Liang, L. He, H. Cheng, W. Li, X. Li, C. Zhang, Y. Yu and F. Zhao, J. Catal., 2014, 309, 468–476 CrossRef CAS.
- A. Negoi, K. Triantafyllidis, V. I. Parvulescu and S. M. Coman, Catal. Today, 2014, 223, 122–128 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. A. Jacobs and B. F. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS PubMed.
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef CAS PubMed.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- J. Weitkamp, ChemCatChem, 2012, 4, 292–306 CrossRef CAS.
- L. Faba, B. T. Kusema, E. V. Murzina, A. Tokarev, N. Kumar, A. Smeds, E. Díaz, S. Ordóñez, P. Mäki-Arvela, S. Willför, T. Salmi and D. Y. Murzin, Microporous Mesoporous Mater., 2014, 189, 189–199 CrossRef CAS.
- B. T. Kusema, L. Faba, N. Kumar, P. Mäki-Arvela, E. Díaz, S. Ordóñez, T. Salmi and D. Y. Murzin, Catal. Today, 2012, 196, 26–33 CrossRef CAS.
- D. Y. Murzin, B. Kusema, E. V. Murzina, A. Aho, A. Tokarev, A. S. Boymirzaev, J. Wärnå, P. Y. Dapsens, C. Mondelli, J. Pérez-Ramírez and T. Salmi, J. Catal., 2015, 330, 93–105 CrossRef CAS.
- D. Y. Murzin, E. V. Murzina, A. Tokarev, N. D. Shcherban, J. Wärnå and T. Salmi, Catal. Today, 2015, 257(part 2), 169–176 CAS.
- M. Käldström, N. Kumar, M. Tenho, M. V. Mokeev, Y. E. Moskalenko and D. Y. Murzin, ACS Catal., 2012, 2, 1381–1393 CrossRef.
- Y. Nakagawa, S. Liu, M. Tamura and K. Tomishige, ChemSusChem, 2015, 8, 1114–1132 CrossRef CAS PubMed.
- S.-i. Oya, D. Kanno, H. Watanabe, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2015, 8, 2472–2475 CrossRef CAS PubMed.
- B. L. Wegenhart, L. Yang, S. C. Kwan, R. Harris, H. I. Kenttämaa and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2742–2747 CrossRef CAS PubMed.
- J. Yang, N. Li, S. Li, W. Wang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 4879–4884 RSC.
- G. Li, N. Li, J. Yang, L. Li, A. Wang, X. Wang, Y. Cong and T. Zhang, Green Chem., 2014, 16, 594–599 RSC.
- A. Deneyer, T. Renders, J. Van Aelst, S. Van den Bosch, D. Gabriëls and B. F. Sels, Curr. Opin. Chem. Biol., 2015, 29, 40–48 CrossRef CAS PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass – Volume II: Results of Screening for Potential Candidates from Biorefinery Lignin, Report PNNL-16983, Pacific Northwest National Laboratory, Richland, Washington, 2007.
- R. Davis, L. Tao, E. C. D. Tan, M. J. Biddy, G. T. Beckham, C. Scarlata, J. Jacobson, K. Cafferty, J. Ross, J. Lukas, D. Knorr and P. Schoen, Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbons: Dilute-Acid and Enzymatic Deconstruction of Biomass to Sugars and Biological Conversion of Sugars to Hydrocarbons, Report NREL/TP-5100-60223, National Renewable Energy Laboratory, 2013.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 6185 CrossRef PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 CAS.
- D.-Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2011, 281, 21–29 CrossRef CAS.
- X. Zhu, L. Nie, L. L. Lobban, R. G. Mallinson and D. E. Resasco, Energy Fuels, 2014, 28, 4104–4111 CrossRef CAS.
- C. Zhao, D. M. Camaioni and J. A. Lercher, J. Catal., 2012, 288, 92–103 CrossRef CAS.
- C. Zhao, S. Kasakov, J. He and J. A. Lercher, J. Catal., 2012, 296, 12–23 CrossRef CAS.
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS PubMed.
- C. Zhao, Y. Yu, A. Jentys and J. A. Lercher, Appl. Catal., B, 2013, 132, 282–292 CrossRef.
- W. Song, Y. Liu, E. Barath, C. Zhao and J. A. Lercher, Green Chem., 2015, 17, 1204–1218 RSC.
- W. Zhang, J. Chen, R. Liu, S. Wang, L. Chen and K. Li, ACS Sustainable Chem. Eng., 2014, 2, 683–691 CrossRef CAS.
- C. Zhao and J. A. Lercher, ChemCatChem, 2012, 4, 64–68 CrossRef CAS.
- C. Zhao, W. Song and J. A. Lercher, ACS Catal., 2012, 2, 2714–2723 CrossRef CAS.
- S. Kasakov, H. Shi, D. M. Camaioni, C. Zhao, E. Barath, A. Jentys and J. A. Lercher, Green Chem., 2015, 17, 5079–5090 RSC.
- H. Chen, Q. Wang, X. Zhang and L. Wang, Appl. Catal., B, 2015, 166–167, 327–334 CrossRef CAS.
- N. Chen, S. Gong and E. W. Qian, Appl. Catal., B, 2015, 174–175, 253–263 CrossRef CAS.
- J. Cheng, T. Li, R. Huang, J. Zhou and K. Cen, Bioresour. Technol., 2014, 158, 378–382 CrossRef CAS PubMed.
- M. Herskowitz, M. V. Landau, Y. Reizner and D. Berger, Fuel, 2013, 111, 157–164 CrossRef CAS.
- Q. Liu, H. Zuo, T. Wang, L. Ma and Q. Zhang, Appl. Catal., A, 2013, 468, 68–74 CrossRef CAS.
- Y. Liu, L. Yao, H. Xin, G. Wang, D. Li and C. Hu, Appl. Catal., B, 2015, 174–175, 504–514 CrossRef CAS.
- B. Peng, Y. Yao, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 2072–2075 CrossRef CAS PubMed.
- E. W. Qian, N. Chen and S. Gong, J. Mol. Catal. A: Chem., 2014, 387, 76–85 CrossRef CAS.
- N. Shi, Q.-y. Liu, T. Jiang, T.-j. Wang, L.-l. Ma, Q. Zhang and X.-h. Zhang, Catal. Commun., 2012, 20, 80–84 CrossRef CAS.
- D. Verma, R. Kumar, B. S. Rana and A. K. Sinha, Energy Environ. Sci., 2011, 4, 1667–1671 CAS.
- D. Verma, B. S. Rana, R. Kumar, M. G. Sibi and A. K. Sinha, Appl. Catal., A, 2015, 490, 108–116 CrossRef CAS.
- C. Wang, Q. Liu, X. Liu, L. Yan, C. Luo, L. Wang, B. Wang and Z. Tian, Cuihua Xuebao, 2013, 34, 1128–1138 CAS.
- S. Zhao, M. Li, Y. Chu and J. Chen, Energy Fuels, 2014, 28, 7122–7132 CrossRef CAS.
- V. J. Frilette, P. B. Weisz and R. L. Golden, J. Catal., 1962, 1, 301–306 CrossRef CAS.
- P. B. Weisz, V. J. Frilette, R. W. Maatman and E. B. Mower, J. Catal., 1962, 1, 307–312 CrossRef CAS.
- P. B. Weisz, Pure Appl. Chem., 1980, 52, 2091–2103 CrossRef CAS.
- B. Smit and T. L. M. Maesen, Nature, 2008, 451, 671–678 CrossRef CAS PubMed.
- T. F. Degnan Jr, J. Catal., 2003, 216, 32–46 CrossRef.
- E. G. Derouane and Z. Gabelica, J. Catal., 1980, 65, 486–489 CrossRef CAS.
- P. B. Weisz, W. O. Haag and P. G. Rodewald, Science, 1979, 206, 57–58 CAS.
- P. Dejaifve, J. C. Védrine, V. Bolis and E. G. Derouane, J. Catal., 1980, 63, 331–345 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- Y. Bhawe, M. Moliner-Marin, J. D. Lunn, Y. Liu, A. Malek and M. Davis, ACS Catal., 2012, 2, 2490–2495 CrossRef CAS.
- A. Philippaerts, S. Paulussen, A. Breesch, S. Turner, O. I. Lebedev, G. Van Tendeloo, B. Sels and P. Jacobs, Angew. Chem., Int. Ed., 2011, 50, 3947–3949 CrossRef CAS PubMed.
- A. Philippaerts, A. Breesch, G. Cremer, P. Kayaert, J. Hofkens, G. Mooter, P. A. Jacobs and B. F. Sels, J. Am. Oil Chem. Soc., 2011, 88, 2023–2034 CrossRef CAS.
- A. Philippaerts, S. Paulussen, S. Turner, O. I. Lebedev, G. Van Tendeloo, H. Poelman, M. Bulut, F. De Clippel, P. Smeets, B. Sels and P. Jacobs, J. Catal., 2010, 270, 172–184 CrossRef CAS.
- C. M. Lew, N. Rajabbeigi and M. Tsapatsis, Microporous Mesoporous Mater., 2012, 153, 55–58 CrossRef CAS.
- Y. Roman-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef CAS PubMed.
- M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11777–11782 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- R. De Clercq, M. Dusselier, C. Christiaens, J. Dijkmans, R. I. Iacobescu, Y. Pontikes and B. F. Sels, ACS Catal., 2015, 5, 5803–5811 CrossRef CAS.
- R. Chal, C. Gérardin, M. Bulut and S. van Donk, ChemCatChem, 2011, 3, 67–81 CrossRef CAS.
- S. Lopez-Orozco, A. Inayat, A. Schwab, T. Selvam and W. Schwieger, Adv. Mater., 2011, 23, 2602–2615 CrossRef CAS PubMed.
- M. Milina, S. Mitchell, Z. D. Trinidad, D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2012, 2, 759–766 CAS.
- K. Moller and T. Bein, Chem. Soc. Rev., 2013, 42, 3689–3707 RSC.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- W. J. Roth and J. Cejka, Catal. Sci. Technol., 2011, 1, 43–53 CAS.
- D. P. Serrano, J. M. Escola and P. Pizarro, Chem. Soc. Rev., 2013, 42, 4004–4035 RSC.
- D. Verboekend, T. C. Keller, M. Milina, R. Hauert and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 1947–1959 CrossRef CAS.
- D. Verboekend, T. C. Keller, S. Mitchell and J. Pérez-Ramírez, Adv. Funct. Mater., 2013, 23, 1923–1934 CrossRef CAS.
- D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1, 879–890 CAS.
- M. S. Holm, E. Taarning, K. Egeblad and C. H. Christensen, Catal. Today, 2011, 168, 3–16 CrossRef CAS.
- Z. L. Hua, J. Zhou and J. L. Shi, Chem. Commun., 2011, 47, 10536–10547 RSC.
- D. Verboekend and J. Pérez-Ramírez, ChemSusChem, 2014, 7, 753–764 CrossRef CAS PubMed.
- D. Verboekend, G. Vilé and J. Pérez-Ramírez, Adv. Funct. Mater., 2012, 22, 916–928 CrossRef CAS.
- J. Van Aelst, A. Philippaerts, N. Nuttens, D. Verboekend, M. Kurttepeli, E. Gobechiya, M. Haouas, S. P. Sree, J. F. M. Denayer, J. A. Martens, C. E. A. Kirschhock, F. Taulelle, S. Bals, G. V. Baron, P. A. Jacobs and B. F. Sels, Adv. Funct. Mater., 2015 DOI:10.1002/adfm.201502772.
- D. Verboekend, N. Nuttens, R. Locus, J. Van Aelst, P. Verolme, J. C. Groen, J. Perez-Ramirez and B. F. Sels, Chem. Soc. Rev., 2015 10.1039/C5CS00520E.
- L. Zhou, M. Shi, Q. Cai, L. Wu, X. Hu, X. Yang, C. Chen and J. Xu, Microporous Mesoporous Mater., 2013, 169, 54–59 CrossRef CAS.
- J. Li, X. Li, G. Zhou, W. Wang, C. Wang, S. Komarneni and Y. Wang, Appl. Catal., A, 2014, 470, 115–122 CrossRef CAS.
- T. C. Keller, S. Isabettini, D. Verboekend, E. G. Rodrigues and J. Perez-Ramirez, Chem. Sci., 2014, 5, 677–684 RSC.
- H. I. Lee, H. J. Park, Y.-K. Park, J. Y. Hur, J.-K. Jeon and J. M. Kim, Catal. Today, 2008, 132, 68–74 CrossRef CAS.
- M. Milina, S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2014, 235, 176–183 CrossRef CAS.
- K. S. Arias, M. J. Climent, A. Corma and S. Iborra, Energy Environ. Sci., 2015, 8, 317–331 CAS.
- N. Nuttens, D. Verboekend, A. Deneyer, J. Van Aelst and B. F. Sels, ChemSusChem, 2015, 8, 1197–1205 CrossRef CAS PubMed.
- C. Martinez, D. Verboekend, J. Perez-Ramirez and A. Corma, Catal. Sci. Technol., 2013, 3, 972–981 CAS.
- J. A. Martens, D. Verboekend, K. Thomas, G. Vanbutsele, J. Pérez-Ramírez and J.-P. Gilson, Catal. Today, 2013, 218–219, 135–142 CrossRef CAS.
- K. P. de Jong, J. Zečević, H. Friedrich, P. E. de Jongh, M. Bulut, S. van Donk, R. Kenmogne, A. Finiels, V. Hulea and F. Fajula, Angew. Chem., Int. Ed., 2010, 122, 10272–10276 CrossRef.
- S. Stefanidis, K. Kalogiannis, E. F. Iliopoulou, A. A. Lappas, J. M. Triguero, M. T. Navarro, A. Chica and F. Rey, Green Chem., 2013, 15, 1647–1658 RSC.
- J. R. García, M. Bertero, M. Falco and U. Sedran, Appl. Catal., A, 2015, 503, 1–8 CrossRef.
- G. T. Neumann, B. R. Pimentel, D. J. Rensel and J. C. Hicks, Catal. Sci. Technol., 2014, 4, 3953–3963 CAS.
- S. Kelkar, C. M. Saffron, Z. Li, S.-S. Kim, T. J. Pinnavaia, D. J. Miller and R. Kriegel, Green Chem., 2014, 16, 803–812 RSC.
- I. Graça, A. M. Carmo, J. M. Lopes and M. F. Ribeiro, Fuel, 2015, 140, 484–494 CrossRef.
- I. Sadaba, M. Lopez Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- H. Xiong, H. N. Pham and A. K. Datye, Green Chem., 2014, 16, 4627–4643 RSC.
- J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186–13197 CrossRef CAS PubMed.
- K. G. Strohmaier, Zeolites and Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 57–86 Search PubMed.
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1–78 CrossRef CAS.
- C. E. A. Kirschhock, E. J. P. Feijen, P. A. Jacobs and J. A. Martens, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- J. C. Jansen, in Stud. Surf. Sci. Catal., ed. H. van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Elsevier, 2001, vol. 137, ch. 5A, pp. 175–227 Search PubMed.
- D. Verboekend, M. Milina and J. Pérez-Ramírez, Chem. Mater., 2014, 26, 4552–4562 CrossRef CAS.
- K. Ehrhardt, M. Suckow and W. Lutz, in Stud. Surf. Sci. Catal., ed. H. G. K. I. K. H. K. Beyer and J. B. Nagy, Elsevier, 1995, vol. 94, pp. 179–186 Search PubMed.
- W. Luo, U. Deka, A. M. Beale, E. R. H. van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- W. Luo, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2014, 320, 33–41 CrossRef CAS.
- A. V. Bandura and S. N. Lvov, J. Phys. Chem. Ref. Data, 2006, 35, 15–30 CrossRef CAS.
- A. Vjunov, M. A. Derewinski, J. L. Fulton, D. M. Camaioni and J. A. Lercher, J. Am. Chem. Soc., 2015, 137, 10374–10382 CrossRef CAS PubMed.
- A. Vjunov, J. L. Fulton, D. M. Camaioni, J. Z. Hu, S. D. Burton, I. Arslan and J. A. Lercher, Chem. Mater., 2015, 27, 3533–3545 CrossRef CAS.
- R. K. Iler, The chemistry of silica: solubility, polymerization, colloid and surface properties, and biochemistry, John Wiley & Sons, USA, 1978 Search PubMed.
- L. Zhang, K. Chen, B. Chen, J. L. White and D. E. Resasco, J. Am. Chem. Soc., 2015, 137, 11810–11819 CrossRef CAS PubMed.
- W. Lutz, H. Toufar, R. Kurzhals and M. Suckow, Adsorption, 2005, 11, 405–413 CrossRef CAS.
- H. Fichtner-Schmittler, W. Lutz, S. Amin, A. Dyer and M. Wark, Zeolites, 1992, 12, 750–755 CrossRef CAS.
- W. Lutz, M. Bülow, N. N. Feoktistova, L. B. Vtjurina, S. P. Shdanov and H. Siegel, Cryst. Res. Technol., 1989, 24, 161–165 CrossRef CAS.
- J. C. Buhl, M. Gerstmann, W. Lutz and A. Ritzmann, Z. Anorg. Allg. Chem., 2004, 630, 604–608 CrossRef CAS.
- D. W. Gardner, J. Huo, T. C. Hoff, R. L. Johnson, B. H. Shanks and J.-P. Tessonnier, ACS Catal., 2015, 5, 4418–4422 CrossRef CAS.
- W. Lutz, E. Löffler and B. Zibrowius, in Stud. Surf. Sci. Catal., ed. B. Laurent and K. Serge, Elsevier, 1995, vol. 97, pp. 327–334 Search PubMed.
- W. Lutz, B. Zibrowius and E. Löffler, in Stud. Surf. Sci. Catal., ed. J. Weitkamp, H. G. Karge, H. Pfeifer and W. Hölderich, Elsevier, 1994, vol. 84, pp. 1005–1012 Search PubMed.
- W. Lutz, W. Gessner, R. Bertram, I. Pitsch and R. Fricke, Microporous Mater., 1997, 12, 131–139 CrossRef CAS.
- W. Lutz, E. Löffler and B. Zibrowius, Zeolites, 1993, 13, 685–686 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérez-Ramírez, Chem. – Eur. J., 2005, 11, 4983–4994 CrossRef CAS PubMed.
- D. Verboekend and J. Pérez-Ramírez, Chem. – Eur. J., 2011, 17, 1137–1147 CrossRef CAS PubMed.
- R. Gounder, Catal. Sci. Technol., 2014, 4, 2877–2886 CAS.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570–8578 CrossRef CAS PubMed.
- P. A. Zapata, Y. Huang, M. A. Gonzalez-Borja and D. E. Resasco, J. Catal., 2013, 308, 82–87 CrossRef CAS.
- T. C. Keller, K. Desai, S. Mitchell and J. Perez-Ramirez, ACS Catal., 2015, 5, 5388–5396 CrossRef CAS.
- Y. Ono, J. Catal., 2003, 216, 406–415 CrossRef CAS.
- R. A. Sheldon and H. van Bekkum, Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH Verlag GmbH, 2007, ch. 7, pp. 309–349 Search PubMed.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- D. Barthomeuf, Catal. Rev., 1996, 38, 521–612 Search PubMed.
- T. C. Keller, E. G. Rodrigues and J. Pérez-Ramírez, ChemSusChem, 2014, 7, 1729–1738 CrossRef CAS PubMed.
- Y. Zhou, Y. Jin, M. Wang, W. Zhang, J. Xie, J. Gu, H. Wen, J. Wang and L. Peng, Chem. – Eur. J., 2015, 21, 15412–15420 CrossRef CAS PubMed.
- J. D. Lewis, S. Van de Vyver and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2015, 54, 9835–9838 CrossRef CAS PubMed.
- D. Verboekend, Y. Liao, W. Schutyser and B. F. Sels, Green Chem., 2015 10.1039/C5GC01868D.
- S. M. Csicsery, Pure Appl. Chem., 1986, 58, 841–856 CrossRef CAS.
- T. Ennaert, B. Op de Beeck, J. Vanneste, A. T. Smit, W. J. J. Huijgen, A. Vanhulsel, P. A. Jacobs and B. F. Sels, Green Chem., 2015 10.1039/C5GC02346G.
- J. M. Maselli and A. W. Peters, Catal. Rev., 1984, 26, 525–554 CAS.
- H. W. Beck, J. D. Carruthers, E. B. Cornelius, R. A. Kmecak, S. M. Kovach and W. P. Hettinger, US4480047A, 1984.
- G. M. Woltermann, J. S. Magee and S. D. Griffith, in Stud. Surf. Sci. Catal., ed. S. M. John and M. M. Maurice, Elsevier, 1993, vol. 76, ch. 4, pp. 105–144 Search PubMed.
- J. Scherzer, in Stud. Surf. Sci. Catal., ed. S. M. John and M. M. Maurice, Elsevier, 1993, vol. 76, ch. 5, pp. 145–182 Search PubMed.
- A. A. Avidan, in Stud. Surf. Sci. Catal., ed. S. M. John and M. M. Maurice, Elsevier, 1993, vol. 76, ch. 1, pp. 1–39 Search PubMed.
- D. E. W. Vaughan, in Stud. Surf. Sci. Catal., ed. S. M. John and M. M. Maurice, Elsevier, 1993, vol. 76, ch. 3, pp. 83–104 Search PubMed.
- A. Corma and B. W. Wojciechowski, Catal. Rev., 1985, 27, 29–150 CAS.
- S. Mitchell, N.-L. Michels and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC.
| This journal is © The Royal Society of Chemistry 2016 |