
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe(III) phytate metallogel as a prototype anhydrous, intermediate temperature proton conductor†
Harshitha Barike
Aiyappa‡
ab,
Subhadeep
Saha‡
ab,
Pritish
Wadge
a,
Rahul
Banerjee
*ab and
Sreekumar
Kurungot
*ab
aPhysical/Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India. E-mail: k.sreekumar@ncl.res.in; r.banerjee@ncl.res.in; Fax: +91-20-25902636; Tel: +91-20-25902535
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India
First published on 2nd October 2014
Abstract
A proton conducting metallogel [FNPA; ferric nitrate (FN)–phytic acid (PA)] is synthesized by immobilizing a protogenic ligand (phytic acid) using iron(III) nitrate in DMF. The xerogel shows high proton conductivity of 2.4 × 10−2 S cm−1 at 120 °C, the best value known among all metal organic materials (MOMs). Marking the first such attempt in MOMs, an electrode made using the xerogel showed a power density of 0.94 mW cm−2 at 0.6 V under dry fuel cell conditions.
Metallogels are an important class of supramolecular metal organic materials (MOMs), whose intrinsic properties stem from the non-covalent interaction between the metallic entity (metal or metal ion), and organic linker (polymer or small organic molecules) resulting in a stable extended network with voluminous immobilization of solvent molecules within it.1 Most metallogelators result from non-covalent interactions, mainly hydrogen bonding as well as metal–ligand coordination.2 An important desirable property of such metallogels would be their ability to conduct protons exploiting the inherent H-bonding. Recent fuel cell research focuses on the development of proton conducting materials with the ability to conduct protons at high temperature (>100 °C) along with high mechanical stability.3 This would essentially improve the electrode kinetics as well as resolving the flooding issues normally encountered in Nafion® based proton exchange membrane fuel cells (PEMFCs).4 An important alternative for HT-PEMFCs is a phosphoric acid doped-polybenzimidazole (PBI) membrane having high proton conductivity at temperatures up to 200 °C.5 However, one of the problems affecting the PBI membrane performance is the leaching of phosphoric acid at higher levels of doping, which limits any further improvement in its conductivity.6 Taking advantage of their high thermal stability, a large number of MOMs, especially metal–organic frameworks (MOFs), have been tried as solid electrolytes for HT-PEMFCs.7 But, most of these materials show carrier-assisted proton transport, viz., water at low temperature or guests like H2SO4, H3PO4, and heterocycles loaded into their pores for high temperature conductivity.7b,c Thus there exists a finite extent of doping/loading of such proton carriers inside the pores of MOFs which thereby limits their performance. Moreover, carriers like imidazole are observed to suffer from severe limitations, e.g. poor chemical stability and adsorption onto the catalyst surfaces which eventually results in poor performance. To be used as a solid electrolyte in a proton exchange membrane fuel cell, the prospective material needs to satisfy two criteria: (1) separate the anode and cathode components (mainly the reactant gases), and (2) conduct protons across the electrodes, thus completing the external electrical circuit and making the fuel cell operational for production of electricity. Thus, there exists a need for an intrinsically proton-conducting electrolyte which could not only fasten and selectively transport protons at high temperatures (100–200 °C) and under anhydrous conditions, but also effectively separate the electrode materials and reactant gases for an optimal overall performance.
In this perspective, thermally as well as chemically stable metallogels offer a perfect platform for immobilizing the proton conducting units via their coordination to the metal centres. However, to date, one finds limited examples of such metallogels employed as proton conductors, one being a CuA–Ox xerogel which exhibits a proton conductivity of 1.4 × 10−5 S cm−1 at 65 °C under anhydrous conditions.8 It is well known that the proton conductivity depends on the number and mobility of protons.9 Among the known protogenic molecules, phosphoric acid based phytic acid (inositol hexakisphosphate) contains 12 replaceable protons and is thus capable of easily coordinating to multivalent ions. Moreover, each phytic acid (PA) molecule contains six phosphate ester (–H2PO4) groups, well known for their amphoteric nature that allows proton conduction without any assistance from external proton carriers.10 Herein, we report the immobilization of phytic acid using Fe3+ in N,N′-dimethylformamide (DMF) which results in a stable metallogel (FNPA, FN = ferric nitrate nonahydrate; PA = phytic acid). The xerogel derived from FNPA (obtained upon slow drying of FNPA metallogel) exhibits a high proton conductivity of 2.4 × 10−2 S cm−1 at 120 °C without assistance of any external agent (moisture, acid or any heterocyclic dopants). Although there exist phosphate based inorganic crystals like CsH2PO4 which show high intrinsic conductivities at intermediate operating temperatures, they have disadvantages like high water solubility which decreases their utility in practical applications.11 The high moisture (Fig. S5†) as well as acid stability of FNPA metallogel establishes it as a unique supramolecular gel material capable of conducting protons (Fig. 3c and d). For the first time, linear fuel cell polarization studies were carried out to collect electrical data using such a metallogel material. These studies ascertain the completion of the electrical circuit of the fuel cell and thereby prove the proton conduction across the pelletized xerogel.
The FNPA metallogel was synthesized using a simple one-pot procedure at 90 °C, wherein 0.1 mmol of Fe(NO3)3·9H2O (FN) and 0.1 mmol of phytic acid (PA) solution (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) in DMF were mixed together to form a pale yellow solution (Fig. 1). Although the gelation occurs within 30 min of heating at 90 °C (as confirmed by the tube inversion test), the rheological studies performed on the metallogel indicate it to be weak in nature. Thus, aging of the metallogel was continued for another 12 h in order to further increase the cross-linking of gelator fibres leading to a metallogel with better mechanical strength (Fig. S1†). A metallogel exhibiting similar properties could also be obtained at room temperature when the pale-yellow solution was left undisturbed for ∼6 days (Fig. 1, S2 and S4†). The off-white metallogel obtained was slowly evaporated at 70–80 °C to obtain FNPA xerogel which was powdered and pelletized for proton conductivity studies.
:1 v/v) in DMF were mixed together to form a pale yellow solution (Fig. 1). Although the gelation occurs within 30 min of heating at 90 °C (as confirmed by the tube inversion test), the rheological studies performed on the metallogel indicate it to be weak in nature. Thus, aging of the metallogel was continued for another 12 h in order to further increase the cross-linking of gelator fibres leading to a metallogel with better mechanical strength (Fig. S1†). A metallogel exhibiting similar properties could also be obtained at room temperature when the pale-yellow solution was left undisturbed for ∼6 days (Fig. 1, S2 and S4†). The off-white metallogel obtained was slowly evaporated at 70–80 °C to obtain FNPA xerogel which was powdered and pelletized for proton conductivity studies.
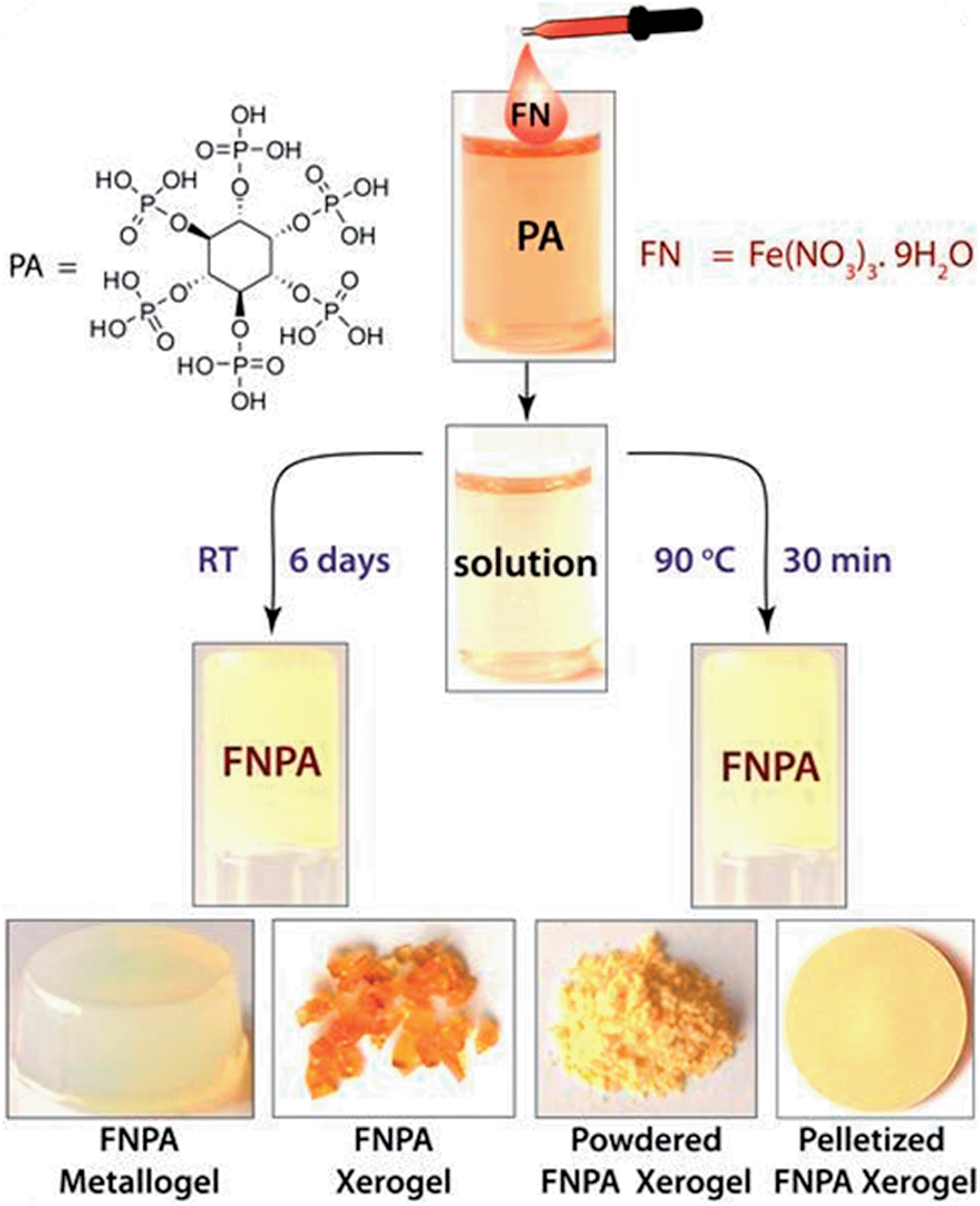 | ||
| Fig. 1 Schematic representation of the synthesis of FNPA metallogel and optical micrographs of FNPA metallogel, FNPA xerogel, powdered xerogel and pelletized FNPA xerogel used for the proton conduction studies (bottom row). | ||
The PXRD patterns of the synthesized FNPA metallogel and FNPA xerogel indicated the amorphous nature of the material (Fig. S6†). Thermogravimetric analysis (TGA) revealed that the FNPA xerogel was stable up to ∼150 °C (Fig. S6†). This phenomenon indicates the probable participation of DMF (boiling point 153 °C) in the formation of the FNPA metallogel fibres (Fig. S7†). The HRTEM images of the FNPA metallogel reveal the formation of 20–40 nm sized nanospheres which gradually get fused together into 1D nanofibres which interweave into a 3D metallogel network (Fig. 2). Similar metamorphosis of gelator nanospheres into a gel network has been observed previously.12 The N2 sorption studies performed on the FNPA xerogel revealed a BET (Brunauer, Emmett and Teller) surface area of 124 m2 g−1 (Fig. 3a and S3†).
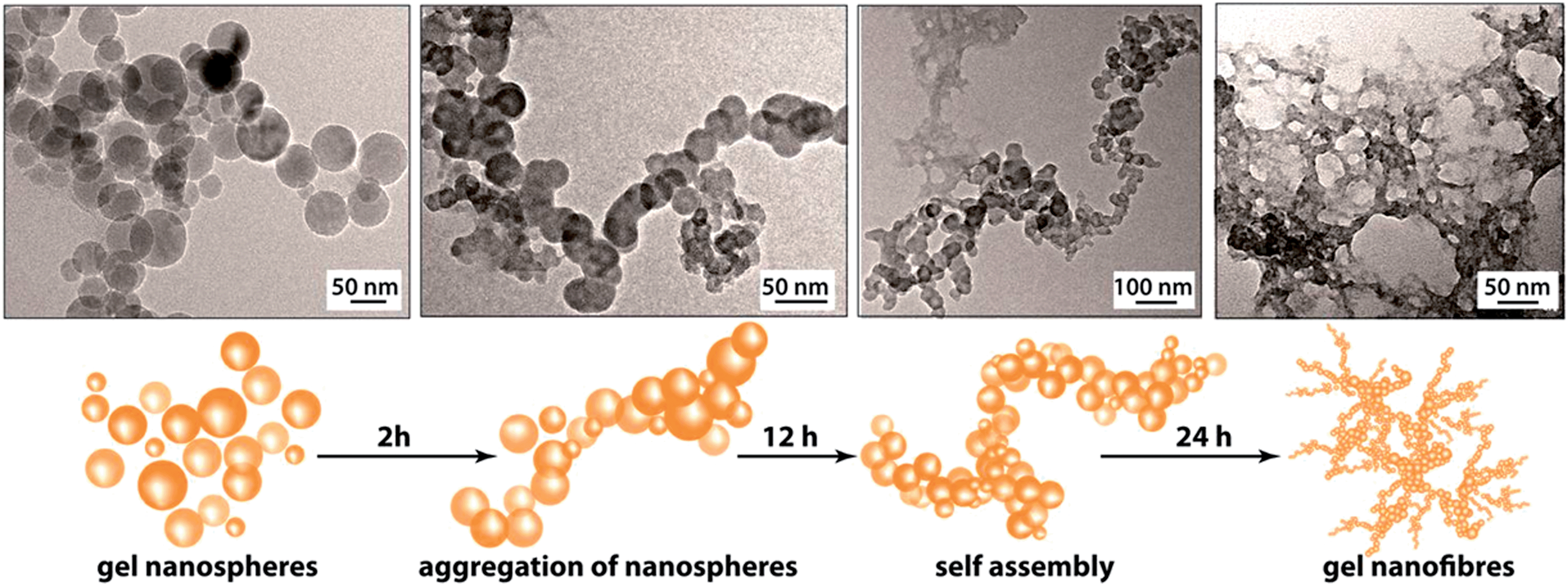 | ||
| Fig. 2 HRTEM images of gel collected at different time intervals evidencing the formation of the FNPA metallogel nanospheres and their eventual assembly into a 3D metallogel network. | ||
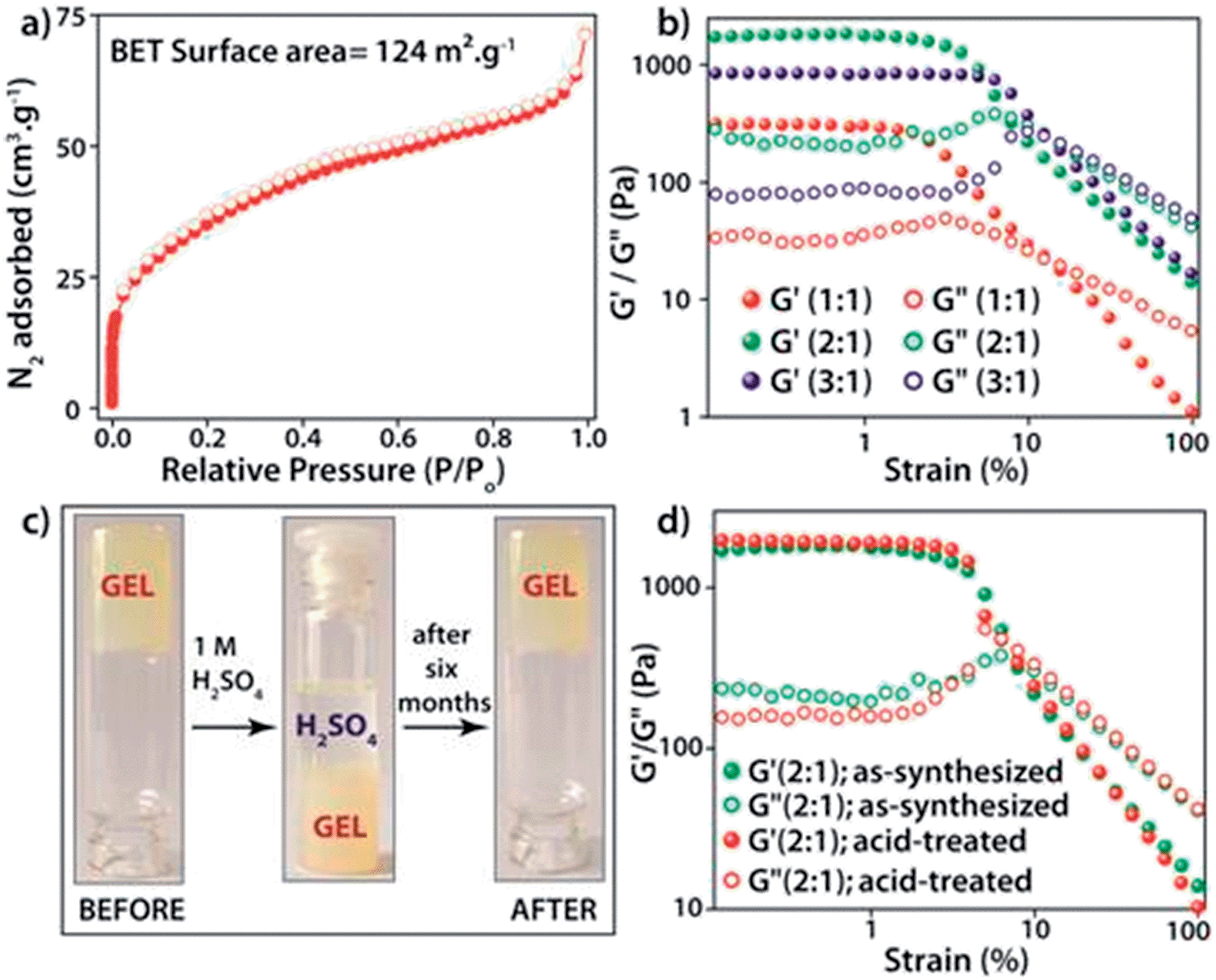 | ||
| Fig. 3 (a) N2 adsorption isotherm of FNPA (2:1) xerogel; (b) combined strain dependent studies of the FNPA metallogel (FN:PA (v/v) in DMF); (c) stability in 1 M H2SO4 for six months; (d) comparative strain sweep plot of as-synthesised and acid-treated FNPA metallogels (after six months). | ||
The metal–ligand coordination was found to be crucial for gel formation as the gel strength varied depending on the metal-to-ligand ratio. It is worth mentioning that the precursor solution having a metal-to-ligand ratio = 2:1 forms a gel at a much faster rate at room temperature and showed superior mechanical properties (Fig. 3b) compared to the gels with other ratios. The gel nature of FNPA was verified by a simple inversion-tube test (Fig. 1). FNPA metallogel was found to be robust without any considerable change over a period of 6 months in the presence of water and 0.1 M H2SO4 (Fig. 3c and d). The viscoelastic nature of this opaque metallogel was further quantified by oscillatory rheological studies. Dynamic strain sweep (DSS) test (at a constant frequency of 1 rad s−1) shows that the average storage modulus (G′) was one order of magnitude higher than the loss modulus (G′′) within the linear viscoelastic regime (Fig. 3b). The storage modulus presented almost zero dependence on the frequency (at a constant strain value of 1%) which is characteristic of a viscoelastic fibrillar nature. A concentration dependent HRTEM study of the FNPA metallogel indicated that the diameter of the nanospheres (and the diameter of the gelator fibre as well) is directly proportional to the concentration of the gelator (Fig. S9†). MALDI-TOF spectra of the FNPA gel as well as the xerogel exhibit an abundance of peaks in the low m/z values (m/z < 1000) region, suggesting non-covalent self-assembly of gelator units (Fig. S10†). Moreover, a prominent peak at m/z = 781.93 (2Fe3+ + 1phytic acid + 8H+) strongly supports the presence of the 2:1 complex (Fe3+:phytic acid) as the gelator unit.
The above-mentioned intriguing observations regarding the mechanical stability and the rate of formation of FNPA gel based on the metal ion (Fe3+) to ligand (phytic acid) ratio suggest that in the smallest gelator unit only two out of six phosphate ester groups chelate to two Fe3+ cations. The presence of identical groups in the phytic acid molecules gives all phosphate ester sites an equal propensity to bind with Fe3+ thereby initiating a non-directional spherical supramolecular assembly of the components (viz. Fe–phytate complex, water, DMF etc., Fig. 2). The nanospheres found in the initial phase of gelation are evidence of the ready complexation of multidentate three-dimensional phytic acid with Fe3+ in DMF. These nanospheres later aggregate via supramolecular forces (H-bonding or coordinate bonds) giving rise to a nanofibrillar network structure which eventually ensnares the whole solvent (DMF) to form a monolithic gel structure. This mechanism further helps to establish the reason behind the prospective proton conduction. On average, four out of six phosphate ester groups of a phytic acid molecule remain free after chelating with two Fe3+ ions. These free phosphate ester (–H2PO4) moieties either take part in H-bonding (with another phosphate ester group or DMF or water) or remain free. In both of the forms, they are capable of conducting protons efficiently. Moreover, the nanoscopic fibrillar structure of the gel network further assists the streamlined conduction of protons.13
The intrinsic proton conducting ability of the FNPA metallogel was analyzed using quasi-four-probe A.C. impedance measurements in anhydrous conditions. The sample was evacuated at 120 °C to eliminate any residual water molecules present in the xerogel. In the first set of preliminary experiments, pelletized xerogel was manually pressed between two stainless steel electrodes (Fig. 4c) and the entire set-up was placed inside a N2-flushed, temperature-controlled incubator (SH-241, ESPEC Co. Ltd., Japan) connected to the electrochemical work station. The sample was kept at each temperature for ∼1 h in order to attain thermal equilibrium. The impedance measurements were performed in the frequency range of 1 MHz–100 Hz with an input voltage amplitude of 10 mV. The Nyquist plots showed a partial semicircle at high frequency with a pronounced tail in the low frequency region which could be attributed to the diffusion limitations resulting due to the blocking effects experienced by protons at the electrodes (Fig. 4b).14 Thus the conductivity obtained can be solely attributed to the proton transport as the two-probe D.C. conductivity studies prove the material to be a good electrical insulator with an electrical resistivity of 6 × 107 Ω cm−1. The presence of a single semicircle in the Nyquist plot indicates bulk polarization and excludes any grain boundary contribution owing to the highly amorphous nature of the material as well as the fibrillar nature of the xerogel. High proton conductivity up to 1.2 × 10−2 S cm−1 was obtained at 130 °C. On continued heating, the conductivity decreased to 3.6 × 10−3 S cm−1 at 150 °C due to possible degradation of the material thereafter (as indicated by isothermal TGA analysis, Fig. S7†). However, the sample retained its conductivity when cooled to RT from 130 °C and then reheated (Fig. 4b).
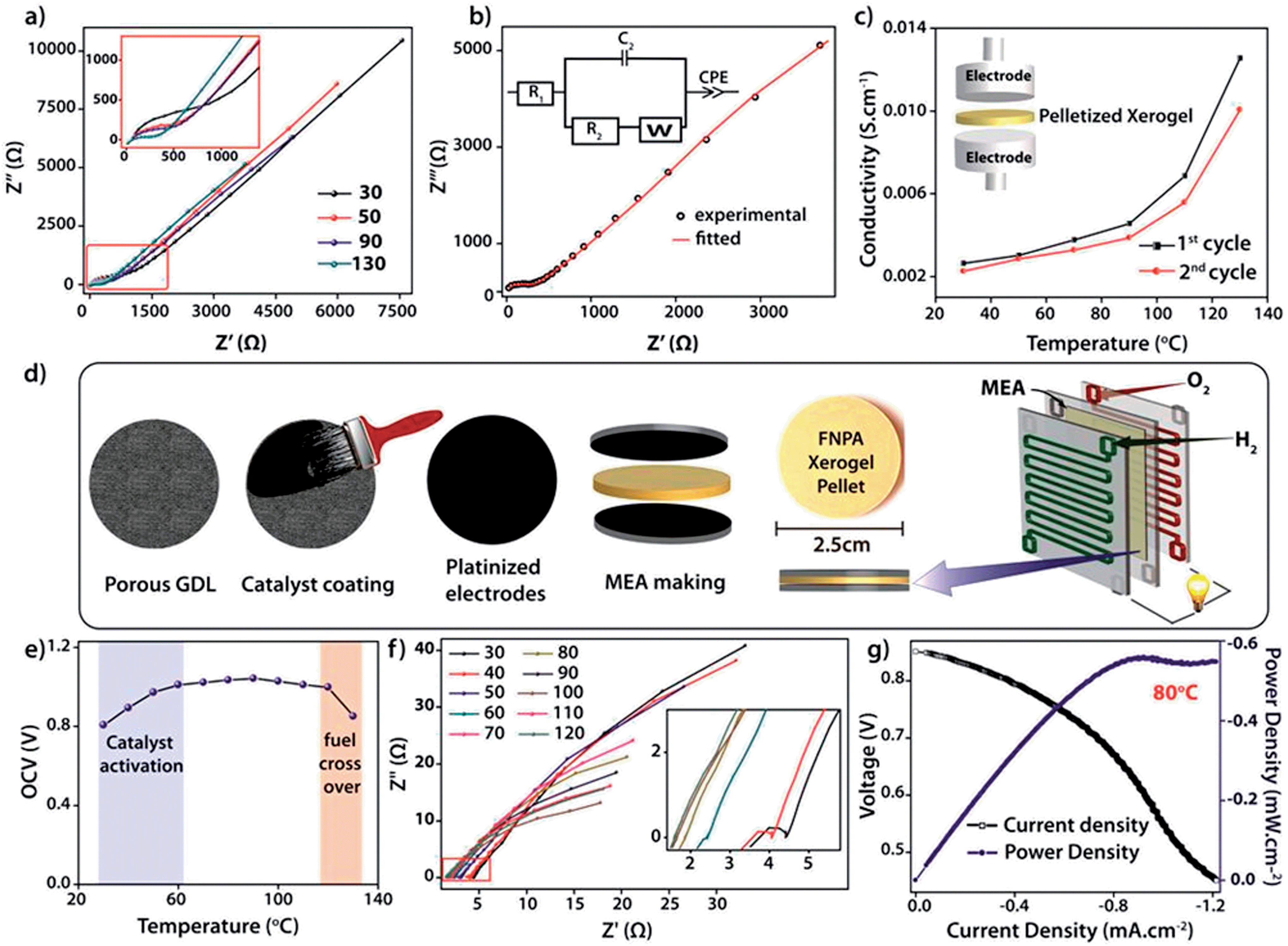 | ||
| Fig. 4 (a) Nyquist plots obtained at different temperatures (in °C); (b) equivalent circuit determined for the Nyquist plot obtained at 130˚C with circuit model used for data fitting (inset); (c) proton conductivity measured during first and second run and representation of the Stainless Steel (SS) electrode assembly (inset); (d) schematic representation of MEA made using the pelletized FNPA xerogel as a solid electrolyte; (e) OCV measurement; (f) fuel cell polarization plot obtained at 80 °C using dry H2, Pt, C/FNPA xerogel/Pt, C, dry O2 electrochemical cell; (g) linear polarization plot obtained using the electrochemical cell at 80 °C. | ||
For direct realization of the material as a solid electrolyte and separating membrane for PEMFC, as a proof of concept, a second set of experiments were carried out under dry H2/O2 fuel cell conditions. D.C. measurements were carried out by fabricating a gas-tight 2 × 2 cm2 MEA (membrane electrode assembly) using dry H2 and dry O2 gases as reactants, Pt–C gas-diffusion electrodes (1 mg of Pt cm−2) and pelletized FNPA xerogel (cold pressed at 1000 kg N for 2 min) as solid electrolyte (Fig. 3d and S12†). The electromotive force (emf) measurements carried out on the MEA showed a starting open circuit voltage (OCV) of 0.807 V at 30 °C followed by an increment to a maximum of 1.02 V ± 0.02 at 100 °C on thermal activation of the Pt catalyst. The OCV remained constant thereafter on further increasing the temperature to 120 °C (Fig. 4f and S13†). However, at 130 °C the OCV dropped to 0.85 V owing to the fuel cross-over effect. Extrapolating the studies further, an in situ impedance study was carried out on the MEA to monitor the entire fuel cell reaction at each temperature (Fig. 4f). The impedance response included a combination of the responses of the cathode (Pt/C, O2) and anode half-cells (Pt/C, H2). The presence of a single semicircle inferred that the time constants (product of interfacial resistance and capacitance) of the two half cells were comparable. At 30 °C, the low frequency region of the Nyquist plot clearly showed the presence of a distinct charge transfer resistance (R2) due to the catalyst layer, and a proton conductivity of 8.6 × 10−3 S cm−1. With increasing temperature, an improvement in the interfacial charge transfer was observed which de-escalates the time constant, as evidenced by the disappearance of the semicircle. A high proton conductivity value of 2.4 × 10−2 S cm−1 was obtained at 120 °C before the OCV started decreasing due to the fuel cross flow (Fig. S15†). The activation energy calculated using the Arrhenius equation was found to be 0.19 eV, indicative of a highly efficient Grotthuss pathway for proton conduction (Fig. S16†).15
Marking the first such attempt in MOMs, current (I)–voltage (V) polarization plots were obtained by driving the fuel cell reaction using FNPA xerogel as solid electrolyte (pellet thickness = 1615 μm) and Pt/C electrodes by passing dry H2 (35–50 sccm) and O2 gases (35–50 sccm) at the anode and cathode respectively. At 0.6 V (standard operating potential of PEMFC fuel cells), a power density of 0.55 mW cm−2 was achieved at 80 °C (Fig. 4g). A similar polarization test was carried out using a pellet with much lower thickness (735 μm). A power density of 0.94 mW cm−2 at 0.6 V verified the crucial role of pellet thickness in determining the MEA performance (Fig. S17†). It is worth noting that although the present fuel cell performance is poor compared to that of Nafion® based fuel cells, the deciding factor is largely the membrane thickness of Nafion® (usually ∼50 μm compared to the present 735 μm). It was observed that, for an optimal performance, the pellet should be dense enough to prevent fuel cross flow, and also thin enough to keep the cell resistance at a minimum (as power density = current density × voltage; V = 0.6 V). The FNPA metallogel by itself showed a maximum proton conductivity of 2.5 × 10−3 S cm−1 at 90 °C. This shows the evident effect of dilution of proton conducting units due to the reduced connectivity among the phosphate ester functions owing to the copious amount of solvent molecules trapped inside the gel network.
In conclusion, this work presents a directed design for the synthesis of a highly proton conducting metal organic material constituting a phosphate ester based ligand immobilized via gelation with Fe3+ ions in DMF. The xerogel obtained from the drying of such a metallogel belongs to a distinct class of supramolecular polymeric solids, whose properties carry the advantage of a 1D nanofibrillar structure which favours propagation of protons using minimum activation energy. The resulting xerogel was tested for its gas separating ability as well as proton conduction in dry PEMFC fuel cell conditions at 120 °C. The solid electrolyte results in an OCV of 1.02 ± 0.02 V with an inherent proton conductivity of 2.4 × 10−2 S cm−1 at 120 °C. To the best of our knowledge, this is the first attempt to studythe performance of such metallogel derived xerogels using fuel cell polarization plots. An achievement of 0.94 mW cm−2 power density at 0.6 V validates the electrical circuit completion of the fuel cell brought about by virtue of proton transport along the xerogel pellet. To the best of our knowledge, this is the first attempt to study the performance of such metal organic materials using fuel cell polarization plots. Although improvements in power density could be still expected with optimized pellet thickness, these studies undoubtedly prove the inherent ability of the gel derived materials to conduct protons at elevated temperature under dry conditions, which is an immediate requisite for intermediate temperature PEMFC.
Notes and references
- (a) D. D. Diaz, D. Kuhbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427 RSC; (b) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed; (c) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC; (d) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697 RSC.
- (a) M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed; (b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- (a) H. Steininger, M. Schuster, K. D. Kreuer, A. Kaltbeitzel, B. Bingol, W. H. Meyer, S. Schauff, G. Brunklaus, J. Maier and H. W. Spiess, Phys. Chem. Chem. Phys., 2007, 9, 1764 RSC; (b) M. F. H. Schuster, W. H. Meyer, M. Schuster and K. D. Kreuer, Chem. Mater., 2004, 16, 329 CrossRef CAS.
- (a) M. Schuster, C. C. de Araujo, V. Atanasov, H. T. Andersen, K. D. Kreuer and J. Maier, Macromolecules, 2009, 42, 3129 CrossRef CAS; (b) C. L. Robert, K. Valle, F. Pereirab and C. Sancheza, Chem. Soc. Rev., 2011, 40, 961 RSC.
- (a) L. Xiao, H. Zhang, E. Scanlon, L. S. Ramanathan, E.-W. Choe, D. Rogers, T. Apple and B. C. Benicewicz, Chem. Mater., 2005, 17, 5328 CrossRef CAS; (b) S.-I. Lee, K.-H. Yoon, M. Song, H. Peng, K. A. Page, C. L. Soles and D. Y. Yoon, Chem. Mater., 2012, 24, 115 CrossRef CAS.
- (a) Y. Oono, T. Fukuda, A. Sounai and M. Hori, J. Power Sources, 2010, 195, 1007 CrossRef CAS PubMed; (b) Y. Oono, T. Fukuda and M. Hori, J. Power Sources, 2009, 189, 943 CrossRef CAS PubMed.
- (a) P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913 RSC; (b) V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640 CrossRef CAS PubMed; (c) S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831 CrossRef CAS PubMed.
- (a) S. Saha, E.-M. Schon, C. Cativiela, D. D. Diaz and R. Banerjee, Chem. - Eur. J., 2013, 19, 9562 CrossRef CAS PubMed.
- (a) S. Beyazyildirim, K. D. Kreuer, M. Schuster, A. J. Bhattacharyya and J. Maier, Adv. Mater., 2008, 20, 1274 CrossRef CAS; (b) K. Miyatake, E. Shouji, K. Yamamoto and E. Tsuchida, Macromolecules, 1997, 30, 2941 CrossRef CAS.
- (a) L. Xie, H. Liu, S. Han, B. Yue and L. Yan, J. Phys. Chem. B, 2013, 117, 16345 CrossRef CAS PubMed; (b) J. M. Taylor, K. W. Dawson and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 1193 CrossRef CAS PubMed.
- (a) D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 12780 CrossRef CAS PubMed; (b) L. J. -Garcia, A. Kaltbeitzel, W. Pisula, J. S. Gutmann, M. Klapper and K. Mullen, Angew. Chem., Int. Ed., 2009, 48, 9951 CrossRef PubMed.
- (a) T. D. Hamilton, D.-K. Bučar, J. Baltrusaitis, D. R. Flanagan, Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365 CrossRef CAS PubMed; (b) S. Varghese, N. S. S. Kumar, A. Krishna, D. S. S. Rao, S. K. Prasad and S. Das, Adv. Funct. Mater., 2009, 19, 2064 CrossRef.
- (a) Y. Chen, M. Thorn, S. Christensen, C. Versek, A. Poe, R. C. Hayward, M. T. Tuominen and S. Thayumanavan, Nat. Chem., 2010, 2, 508 CrossRef PubMed; (b) J. Weber, K.-D. Kreuer, J. Maier and A. Thomas, Adv. Mater., 2008, 20, 2595 CrossRef CAS; (c) M.-A. Kakimoto, S. J. Grunzinger and T. Hayakawa, Polym. J., 2010, 42, 697 CrossRef CAS.
- (a) J. M. Taylor, K. W. Dawson and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 1193 CrossRef CAS PubMed; (b) S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376 CrossRef CAS PubMed; (c) J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705 CrossRef CAS PubMed.
- (a) N. Agmon, Chem. Phys. Lett., 1995, 244, 456 CrossRef CAS; (b) T. Yamada, M. Sadakiyo and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 3144 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of experimental details of impedance measurements, including MEA making, supplementary figures, including TGA, rheology plots, SEM images, TEM images and gas adsorption study. See DOI: 10.1039/c4sc02294g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |