
Hydrothermal synthesis of hierarchical core–shell manganese oxide nanocomposites as efficient dye adsorbents for wastewater treatment†
Wei Wangab,
Tifeng Jiao*abc,
Qingrui Zhang*b,
Xiaona Luob,
Jie Hub,
Yan Chenb,
Qiuming Penga,
Xuehai Yanc and
Bingbing Li*d
aState Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, P. R. China. E-mail: tfjiao@ysu.edu.cn
bHebei Key Laboratory of Applied Chemistry, School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, P. R. China. E-mail: zhangqr@ysu.edu.cn
cNational Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China
dDepartment of Chemistry and Biochemistry, Science of Advanced Materials Doctoral Program, Central Michigan University, Mount Pleasant, MI 48859, USA. E-mail: li3b@cmich.edu
First published on 22nd June 2015
Abstract
In this work, hierarchical core–shell manganese oxide nanocomposites (Fe3O4@MnO2 and Fe2O3@MnO2) were synthesized via a facile hydrothermal process without using any templates or organic surfactants. The effect of reaction time on the microstructure and morphology of composite samples was systemically investigated. The formation mechanism of the core–shell structure was carefully analyzed by using field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and Brunauer–Emmett–Teller (BET) surface area measurement. Both nitrogen adsorption–desorption experiments and morphological study showed that the composite obtained through 6 hour hydrothermal reaction has fairly large specific surface area, uniform pore-size distribution, and good adsorption capability. The dye adsorption experiments further demonstrated the potential applications of the manganese oxide nanocomposites as efficient dye adsorbents for wastewater treatment.
1. Introduction
In recent years, water pollution has exerted negative effects not only on species living in the water but also on the broader biological community. For instance, organic dyes are often discharged with wastewater into the local environment without adequate treatment. Rapid and convenient removal of organic dyes from wastewater has been a challenging issue faced by scientists.1–4 In particular, large-scale application requires the potential dye adsorbents to exhibit high dye removal rate within a relatively short period of time and to be environmentally friendly. In comparison to other adsorbents under investigation, nanostructured adsorbents exhibit remarkably enhanced sorption capacity owing to their high surface-to-volume ratio. During the last decades, various nanosized adsorbents have been prepared and adopted for water decontamination.5–8 However, such nanomaterials still suffer from issues involving separation inconvenience from the wastewater. Thus, the development of new nanostructured adsorbents with a facile separation property is of great interest.9–11 In order to achieve this purpose, iron oxide nanoparticles, in particular magnetite (Fe3O4) and maghemite (γ-Fe2O3), have been extensively applied in designing nanocomposite adsorbents because they have a large surface area and high magnetic susceptibility.12 These nanoparticles are attracted to an external magnetic field but lose magnetism as the applied field is removed, which allows their fast and convenient isolation in the processing and recovery stages. In the past years, research has been conducted to functionalize magnetic nanoparticles by coating them with inorganic materials, polymers, biomolecules, etc.13–19 Surface functionalization has been found to enhance the stability and sorption capacity of magnetic nanocomposites. Thus, it is highly important to develop new functional composite materials that can further enhance the adsorption property of pristine magnetic nanoparticles while exhibit a low-cost and environmentally benign nature. Nanostructured manganese oxides (MnO2) are among the most reactive minerals that have a high surface area, strong adsorption ability, and good stability.20 Therefore, the modification of the Fe3O4 surface with MnO2 to form core–shell nanostructure is anticipated to increase the removal efficiency of the magnetic nanoparticles for environmental applications.21–23 For example, Zhu et al. reported the preparation of new composite adsorbent by combining honeycomb briquette cinders (HBC) with Fe3O4 and MnO2 through a co-precipitation process and the applications of the prepared nanocomposites for As(III) and As(V) removal from aqueous solutions under different experimental conditions.22 By taking advantage of the Fe3O4 core and MnO2 shell, along with the mesoporous structure of the composites, MnO2-coated Fe3O4 nanoparticles could be an ideal material with a great potential for environmental remediation. Although previous research has highlighted the advantages of MnO2 coating for metal-ion adsorption,24,25 they looked into the sorption percentage without an in-depth investigation into the mechanism and experimental parameters. In addition, no previous study has focused on the comparison of MnO2-coated magnetic or non-magnetic particles for wastewater treatment.In this study, hierarchically structured core–shell manganese oxide nanocomposites (magnetic Fe3O4@MnO2 and non-magnetic Fe2O3@MnO2) were successfully synthesized. The morphologies and structures of the composite materials were investigated by various characterization methods, including SEM, TEM, and XRD. The obtained composites were used to adsorb different dyes, for examples, methylene blue (MB) and rhodamine B (RhB) from aqueous systems. The overall performance of nanocomposites was assessed in terms of sorption kinetics.
2. Materials and methods
2.1 Materials
All the used chemical reagents, Potassium Permanganate (KMnO4, AR, Aladdin reagent Co., Ltd), ferric chloride (FeCl3·6H2O, AR, Aladdin reagent Co., Ltd), ammonium hydroxide (NH3·H2O, AR, Tianjin Fangzheng reagent factory), acetic acid (CH3COOH, AR, Tianjin kemiou chemical reagent Co., Ltd) and ethylene glycol, were of analytical grade and used without further purification. Rhodamine B and methylene blue were obtained from Beijing Chemicals (analytical reagent grade, Beijing, China). All other solvents and reagents used in this study were purchased from Sinopharm Chemical Reagent Co. Ltd (analytical reagent grade, Shanghai, China). The deionized (DI) water was obtained using a Millpore Milli-Q water purification system with a resistivity of 18.2 MΩ cm−1. All chemicals were used as received without further purification.2.2 Fabrication of core–shell nanocomposites
2.3 Dye adsorption and composite regeneration experiments
The dye adsorption capacity of hybrid materials was probed using an absorption spectroscopy. Typically, about 100 mg of nanocomposites was added to either 100 mL of MB (10 mg L−1) or 50 mL of RhB (4 mg L−1) solutions. Above dye solutions containing nanostructured adsorbents were stirred at room temperature. The gel samples were then separated by centrifugation at different time intervals and the supernatant liquid was collected for subsequent analysis using an UV-vis spectrometer (752, Sunny Hengping, Shanghai, China). The absorbance at 664 nm for MB or 554 nm for RhB was used to determine the concentration of residual dyes in the supernatant liquid. Once saturated adsorption for a given dye in an aqueous suspension was reached after 12 h in a shaking table at 25 °C, the Fe3O4@MnO2 nanocomposite was extracted from the solution through external magnetic attraction, and washed alternatively using ethylene glycol and ethanol for several times. Finally, the regenerated core–shell composites were then washed by DI water and dried in a lyophilizer for subsequent adsorption experiments.2.4 Characterization
Core–shell nanocomposites were obtained by lyophilizing at −50 °C via a lyophilizer (FD-1C-50, Beijing Boyikang Experimental Instrument Co., Ltd., China) to completely remove water over 2 to 3 days. The size and morphology of the products were characterized by using both a field-emission scanning electron microscopy (S-4800II, Hitachi, Japan; SUPRA 55 SAPPHIRE, CARL ZEISS (Shanghai), Co., Ltd) with a accelerating voltage of 5–15 kV and a transmission electron microscopy (TEM, HT7700, Hitachi High-Technologies Corporation) with commercial 300 mesh copper grids. The chemical composition of the samples was characterized by energy-dispersive X-ray spectroscopy (EDXS). EDXS analysis was typically performed at an accelerating voltage of 200 kV, using an Oxford Link-ISIS X-ray EDXS microanalysis system attached to TEM. X-ray diffraction study was carried out by using an X-ray diffractometer (SMART LAB, Rigaku) equipped with a conventional Cu Kα X-ray radiation (λ = 1.54 Å) source and a Bragg diffraction setup. The Brunauer–Emmett–Teller (BET) surface area and pore size distributions were measured by N2 adsorption and desorption using an ASAP 2010 system at 77 K. The magnetic properties of the samples were tested by a vibrating sample magnetometer (VSM) of the Physical Property Measurement System (PPMS, Quantum design Model 6000). The powders were aligned under a 10 kOe field and solidified with epoxy resin for the VSM measurements.3. Results and discussion
Fig. 1 displays typical SEM images of the as-prepared nanostructures. The SEM image shown in Fig. 1a gives a panoramic morphology of the MnO2 sample, revealing that the product is composed of many uniform siberian cocklebur-like architectures with 3–4 μm diameter. Further observation indicated that the formed structure was covered by many needle-like aggregates. It should be noted that the size of nanoparticles was evaluated by averaging 50 particles observed in different images. In addition, two types of nanoparticles (Fe3O4 and Fe2O3) have been prepared as the cores of the designed nanocomposites. As shown in Fig. 1b and c, the ranges of particle diameters were about 80–100 nm and 150–200 nm, respectively. Subsequently, two types of nanocomposites, i.e., Fe3O4@MnO2 and Fe2O3@MnO2 were prepared and investigated. In order to understand the effect of reaction time on the microstructure and morphology of samples, time-dependent experiments in hydrothermal reaction were carried out and the resultant products were analyzed at time of 3 h, the obtained MnO2 sample has a spherical morphology with 2.5–3 μm diameter. The SEM image (Fig. 1d) demonstrates that the composites are covered by many interleaving thin nanoplates. These nanoplates seemed to grow perpendicularly from a center. With increasing the reaction time to 6 h, the diameter of flower-like structures changed to about 4 μm with similar morphology. When reaction time reached 9 h, the spherical structures merged together, leaving little holes or defects. Finally, the aggregates continuously grew in size to form hollow urchin-like structures with visible cavities after 15 h (Fig. 1g). In addition, Fe2O3@MnO2 nanocomposite achieved after 6 h reaction was also prepared, as shown in Fig. 1h. Uniform siberian cocklebur-like structures with a 2–3 μm diameter and partially non-coated Fe2O3 nanoparticles were also clearly observed. In addition, the typical EDXS of Fe3O4@MnO2 nanocomposites prepared at 6 h showed the existence of Fe and Mn elements, also confirming the successful preparation of the composite materials. The above time-dependent morphological evolution could be correlated to an “Ostwald ripening process”.29–31 During the reaction process, a large number of nuclei are first formed in a short period of time through a well-known “Ostwald ripening process”, followed by a slow crystal growth process. The aggregates continuously grow in size and density to form spheres with solid cores. This stage could last for several hours, and then an interior cavity is gradually formed via a core evacuation process due to higher surface energies. A subsequent increase in the hydrothermal time not only leads to the complete damage of the urchin structure but also increases the size of the individual nanorods, which confirms that Ostwald ripening is the underlying mechanism in this hollowing process. Such a core evacuation process is also observed in the similar synthesis of titania and NiO hollow spheres.32,33 Present experimental results also reveal that 6 h would be enough for the nanocomposites to reach their equilibrium size of flower-like structures. In addition, the interesting morphology and the above microstructures was further studied using TEM technique, as shown in Fig. 2. Several obvious stages of morphological evolution could be clearly observed for Fe3O4@MnO2 nanocomposites prepared at different times. The TEM image in Fig. 2f further confirmed that the Fe3O4@MnO2 nanocomposites obtained after 15 h hydrothermal reaction were composed of intercrossed nano-needles. It should be noted that the TEM samples were prepared on copper grids and the obtained lyophilized core–shell nanocomposites were ultrasonated in water for several minutes and dropped on copper grid carefully. So the dispersion of nanocomposites in TEM images seems not very well and the core–shell structures could exhibit deformation in TEM images. Neverless, the above results strongly indicate that the reaction time plays an important role for the fabrication of core–shell structured MnO2 nanocomposites. | ||
| Fig. 1 SEM images of as-prepared nanostructures. (a) MnO2; (b) Fe3O4; (c) Fe2O3; (d)–(g) Fe3O4@MnO2 prepared at times of 3 h, 6 h, 9 h, and 15 h, respectively; (h) Fe2O3@MnO2 prepared at time of 6 h; (i) typical EDXS of Fe3O4@MnO2 nanocomposites prepared at 6 h. | ||
 | ||
| Fig. 2 TEM images of as-prepared nanostructures. (a), MnO2; (b), Fe3O4; (c)–(f), Fe3O4@MnO2 nanocomposites prepared at times of 3 h, 6 h, 9 h, and 15 h, respectively. | ||
XRD patterns of bare MnO2 and as-prepared nanocomposites are shown in Fig. 3. Firstly, the characteristic peaks of bare MnO2 material is in well accordance with the standard card (PDF#72-1982). In addition, the curves of the prepared Fe3O4 and Fe2O3 nanoparticles were face centered cubic phases (PDF#88-0315 and PDF#89-0599). As for the obtained two kinds of nanocomposites, the results demonstrated that increasing MnO2 coating did not result in a phase change in the structure of the Fe3O4 and Fe2O3 nanoparticles. The newly appeared peaks for the samples obtained with prolonged reaction time can be attributed to the crystalline nature of MnO2 structures in outer shell layers. The large surface area and mesoporous structures of the obtained composites make them very promising candidates for the adsorption of pollutants in water purification.
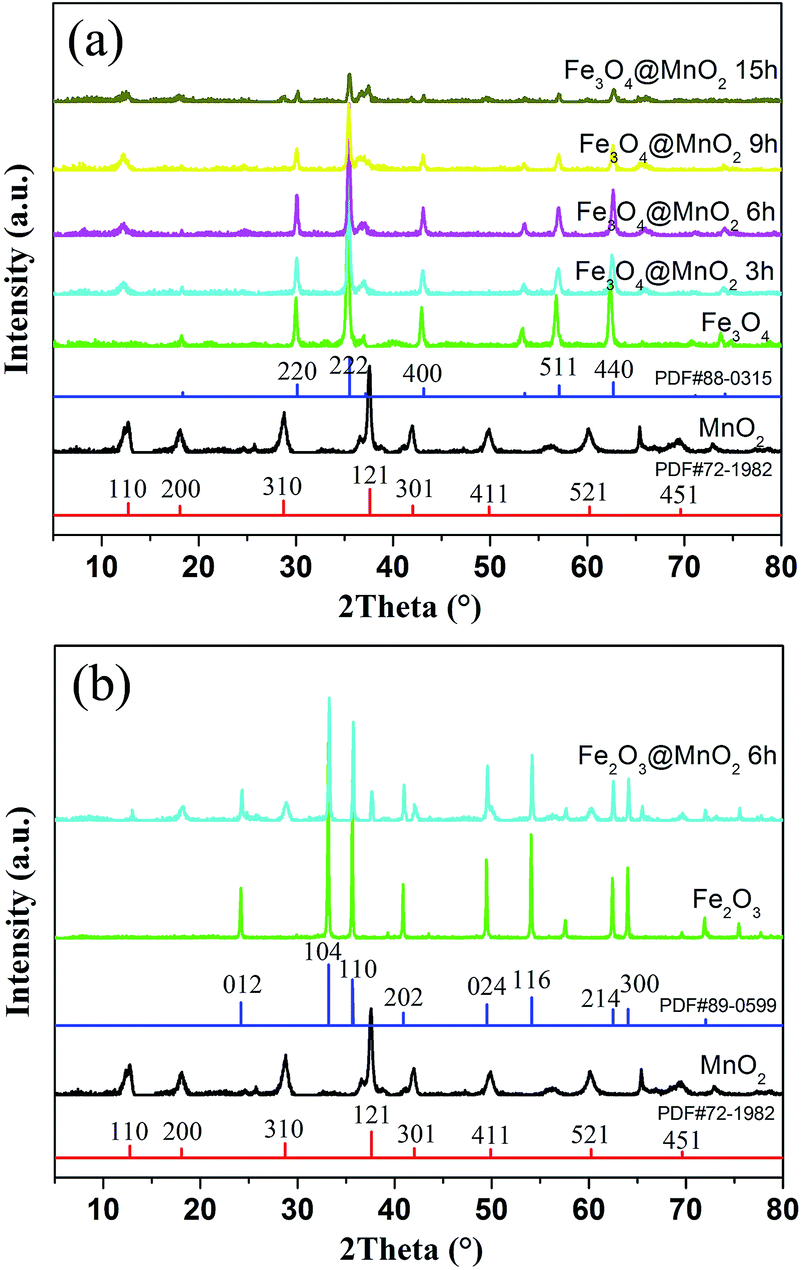 | ||
| Fig. 3 XRD patterns of as-prepared MnO2, Fe3O4@MnO2 (a) and Fe2O3@MnO2 (b) nanocomposites. | ||
Fig. 4 displays the nitrogen adsorption–desorption isotherms and pore size distribution curves calculated by the Barrett–Joyner–Halenda (BJH) method. At low relative pressure (P/P0 < 0.45), the adsorption and desorption curves coincide because of reversible monolayer adsorption. At a higher relative pressure region (P/P0 0.45–0.95), all of the isotherms have significant hysteresis, which can be ascribed to the presence of a mesoporous structure in the interleaving nanoplates or cavities of the hollow spheres, and the hysteresis loop at P/P0 = 0.9 to 1 could result from the internanorods space.34,35 The specific surface area for each sample was obtained by the BET method. The physical properties deduced from N2 adsorption at 77 K of as-prepared nanocomposites samples were listed in Table S1 in ESI.† The specific surface areas of MnO2, Fe3O4@MnO2, and Fe2O3@MnO2 nanocomposites prepared at time of 6 h were 40.5, 72.5, and 52.2 m2 g−1, respectively. The larger specific surface-area of Fe3O4@MnO2 sample than that of Fe2O3@MnO2 sample could be considered as the result of smaller particle diameter of Fe3O4 and more MnO2 aggregate in shell part of sphere composites. The high specific surface area and the mesoporous nature of the hollow spheres and urchins can effectively absorb the dye, thus increasing the contact probability of pollutant molecule and catalyst. BJH analysis for mesopore distribution is also shown. The pore sizes of the MnO2, Fe3O4@MnO2, and Fe2O3@MnO2 nanocomposites prepared upon 6 h are within the range of 7–16 nm, respectively.
 | ||
| Fig. 4 The nitrogen adsorption–desorption isotherms and pore size distributions of as-prepared MnO2, Fe3O4@MnO2 and Fe2O3@MnO2 nanocomposites samples at time of 6 h calculated by BJH method. | ||
Fig. 5 demonstrates the hysteresis loops of Fe3O4/MnO2 composites at room temperature. The saturation magnetization of the prepared Fe3O4 nanoparticles was 83.0 emu g−1, which was more than the value reported for another pure Fe3O4 nanocrystals.36 As for the Fe3O4@MnO2 nanocomposites prepared upon 3 h, 6 h, 9 h, and 15 h reaction, respectively, the decrement tendency with time was clearly observed. The values of saturation magnetization were 74.2, 66.4, 36.6, and 18.5 emu g−1, respectively. The decrease in the saturation magnetization with increasing hydrothermal treatment time can be attributed to the increase in the non-magnetic MnO2 content in the composites with hydrothermal treatment time. However, this value was strong enough to achieve a facile magnetic separation. The as-prepared Fe3O4/MnO2 composites were rapidly collected within seconds from an aqueous suspension by an external magnetic field. In addition, the separated particles were easily redispersed with gentle shaking to a fairly stable suspension. This is essentially important for the convenient reuse of composites.
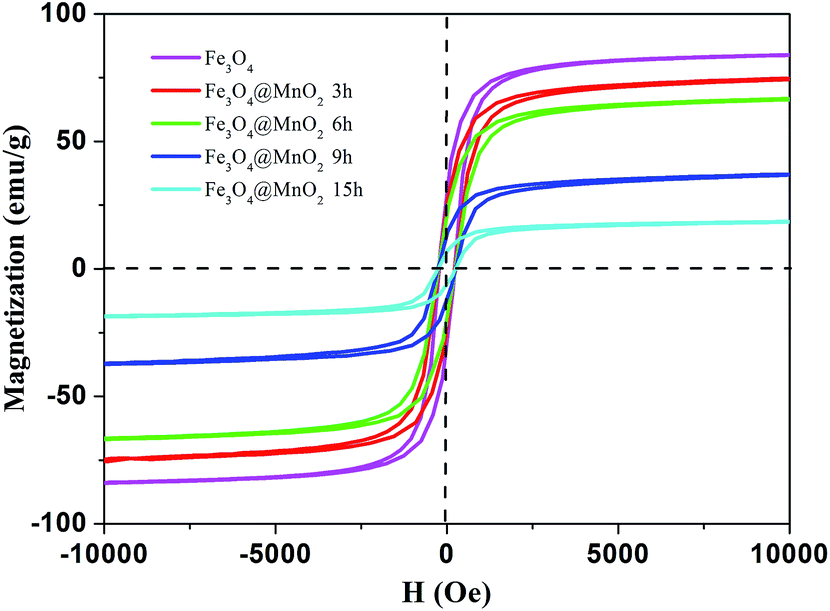 | ||
| Fig. 5 Hysteresis loops of as-prepared Fe3O4 nanoparticle and Fe3O4@MnO2 nanocomposites at different time intervals at 298 K. | ||
The dye adsorption capacity was evaluated by placing the as-prepared Fe3O4@MnO2 and Fe2O3@MnO2 nanocomposites prepared at 6 h in MB and RhB aqueous solutions, respectively. The absorbance at 662 nm (for MB) and 554 nm (for RhB) was used to determine the concentration of residual dyes for samples collected at different time intervals. The thermodynamic behaviors of the as-prepared nanocomposites for dye adsorption from aqueous solutions were reported and investigated in details,19 and the results are shown in Fig. 6. The composites exhibit a continuous adsorption process, with equilibrium time of approximately 20 and 150 min for MB and RhB, respectively. The equilibrium time thus obtained is acceptable for efficient adsorption applications. Such kinetic behaviour can be also associated with the unique structure of nanocomposites, i.e., the large three-dimensional interleaving thin nanoplates nanostructure as the adsorption sites.
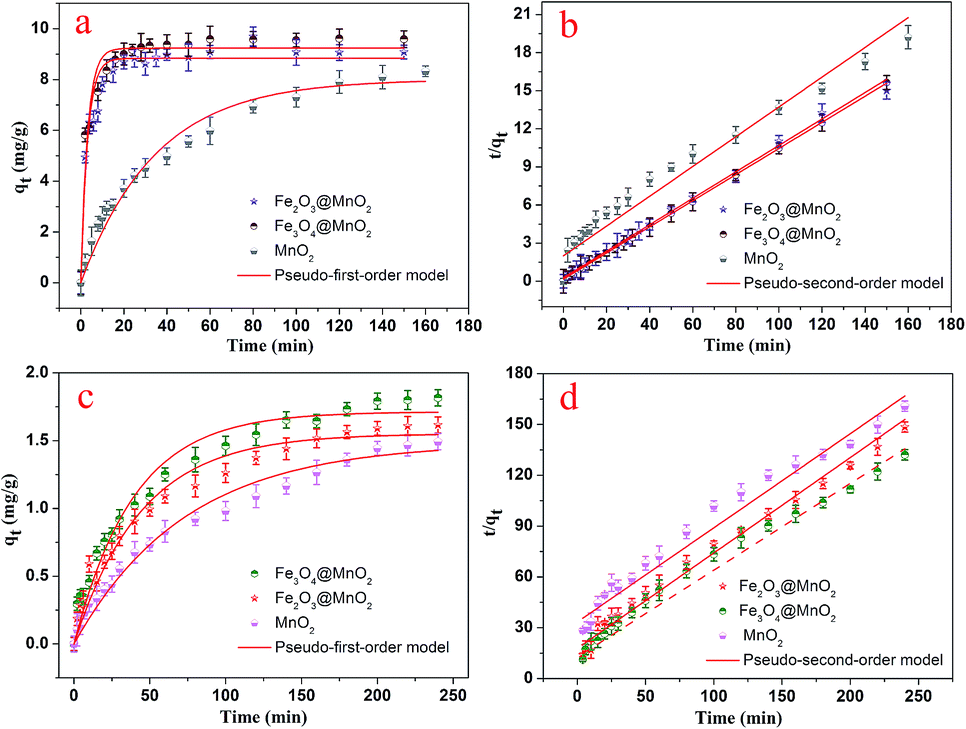 | ||
| Fig. 6 Adsorption kinetics curves of as-prepared Fe3O4@MnO2 and Fe2O3@MnO2 nanocomposites onto MB (a and b) and RhB (c and d) at 298 K. | ||
The pseudo-first-order model:
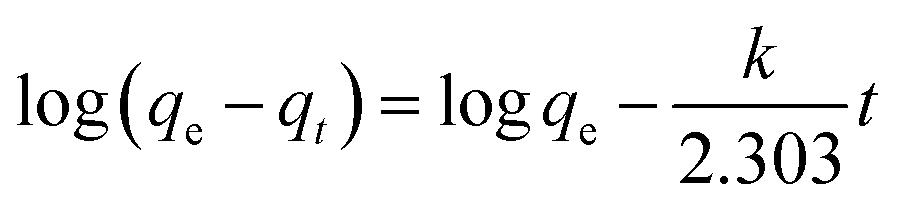 | (1) |
The pseudo-second-order model:
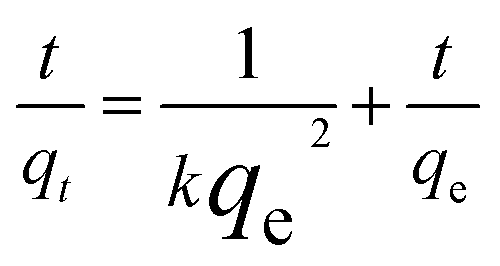 | (2) |
The adsorption–desorption cycles for MB were repeated five times using the same batch of Fe3O4@MnO2 nanocomposites prepared at 6 h, as shown in Fig. 7. As compared to the first adsorption, the removal efficiency at the fifth cycle decreased by about 9% to a value of 86%, indicating good regeneration capacity and reusability. The slight decrease in the adsorption capacity was attributed to the incomplete desorption of dyes from the surface of Fe3O4@MnO2 nanocomposites. For MB molecules that go into the vacant site of MnO2, desorption may be more difficult. After five cycles, the magnetic intensity of nanocomposites did not decrease and the adsorbent could be separated from solution rapidly. Overall, the prepared magnetic Fe3O4@MnO2 composite can be easily recycled and reused several times, which supports their long term use in water purification.
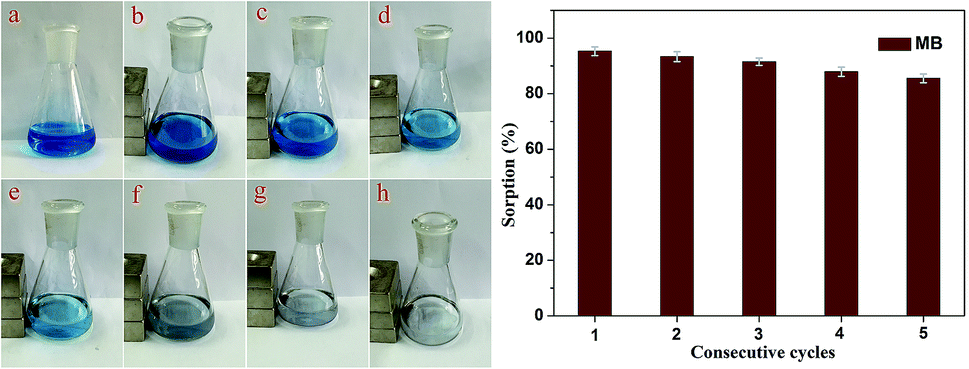 | ||
| Fig. 7 Regeneration studies of as-prepared Fe3O4@MnO2 nanocomposites in the removal of MB. The photographs showed the MB solution (a) and adsorption process under magnetic separation at different time intervals ((b)–(h) 0, 10, 20, 30, 40, 50, and 60 min). | ||
4. Conclusions
In summary, two types of hollow hierarchical core–shell MnO2 nanocomposites have been synthesized by a simple and facile hydrothermal method. The obtained novel sphere or urchin structured composites possess a high loosely mesoporous cluster structure consisting of thin plates and exhibit enhanced adsorption capacity for the dyes used in this study. The microstructures and good adsorption capability coupled with the low cost and environmentally benign nature of manganese may make these composite materials attractive for broad spectrum of applications. In addition, the magnetic Fe3O4@MnO2 nanocomposite can be easily recycled and reused several times, which supports the long term use in water purification. Furthermore, the hydrothermal method has excellent reliability, selectivity, and efficiency for synthesizing inorganic materials with uniform and distinct morphologies.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant nos 21473153 and 21207112), the Natural Science Foundation of Hebei Province (grant no. B2013203108), the Science Foundation for the Excellent Youth Scholars from Universities and Colleges of Hebei Province (grant nos Y2011113, YQ2013026), the Support Program for the Top Young Talents of Hebei Province, and the Open Foundation of National Key Laboratory of Biochemical Engineering (Institute of Process Engineering, Chinese Academy of Sciences).Notes and references
- S. Dadfarnia, A. M. Haji Shabani, S. E. Moradi and S. Emami, Appl. Surf. Sci., 2015, 330, 85–93 CrossRef CAS PubMed.
- Y. R. Zhang, S. L. Shen, S. Q. Wang, J. Huang, P. Su, Q. R. Wang and B. X. Zhao, Chem. Eng. J., 2014, 239, 250–256 CrossRef CAS PubMed.
- Y. Wang, Z. Li, Y. He, F. Li, X. Q. Liu and J. B. Yang, Mater. Lett., 2014, 134, 115–118 CrossRef CAS PubMed.
- C. Namasivayam and D. Sangeetha, J. Hazard. Mater., 2006, 135, 449–452 CrossRef CAS PubMed.
- X. Peng, Z. Luan, J. Ding, Z. Di, Y. Li and B. Tian, Mater. Lett., 2005, 59, 399–403 CrossRef CAS PubMed.
- M. Gallego, Y. P. De Pena and M. Valcarcel, Anal. Chem., 1994, 66, 4074–4078 CrossRef CAS.
- R. Rostamian, M. Najafi and A. A. Rafati, Chem. Eng. J., 2011, 171, 1004–1011 CrossRef CAS PubMed.
- S. Mahdavi, M. Jalali and A. Afkhami, Chem. Eng. Commun., 2013, 200, 448–470 CrossRef CAS PubMed.
- D. Guo, W. J. Ren, Z. Chen, M. L. Mao, Q. H. Li and T. H. Wang, RSC Adv., 2015, 5, 10681–10687 RSC.
- V. Yathindranath, M. Worden, Z. Z. Sun, D. W. Miller and T. Hegmann, RSC Adv., 2013, 3, 23722–23729 RSC.
- M. N. Nadagouda, C. Bennett-Stamper, C. White and D. Lytle, RSC Adv., 2012, 2, 4198–4204 RSC.
- Y. Liu, G. Su, B. Zhang, G. Jiang and B. Yan, Analyst, 2011, 136, 872–877 RSC.
- K. Mandel, F. Hutter, C. Gellermann and G. Sextl, ACS Appl. Mater. Interfaces, 2012, 4, 5633–5642 CAS.
- A. Farrukh, A. Akram, A. Ghaffar, S. Hanif, A. Hamid, H. Duran and B. Yameen, ACS Appl. Mater. Interfaces, 2013, 5, 3784–3793 CAS.
- S. Shin and J. Jang, Chem. Commun., 2007, 4230–4232 RSC.
- S. Shi, Y. Fan and Y. Huang, Ind. Eng. Chem. Res., 2013, 52, 2604–2612 CrossRef CAS.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2011, 46, 977–985 CrossRef PubMed.
- J. F. Liu, Z. S. Zhao and G. B. Jiang, Environ. Sci. Technol., 2008, 42, 6949–6954 CrossRef CAS.
- X. Liu, Q. Hu, Z. Fang, X. Zhang and B. Zhang, Langmuir, 2008, 25, 3–8 CrossRef PubMed.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS.
- T. Pal, R. Sahoo, M. Pradhan, A. Roy, S. Dutta, C. Ray, Y. Negishi and A. Pal, Chem.–Asian J., 2015 DOI:10.1002/asia.201500200 , in press.
- J. Zhu, S. A. Baig, T. Sheng, Z. Lou, Z. Wang and X. Xu, J. Hazard. Mater., 2015, 286, 220–228 CrossRef CAS PubMed.
- L. Tan, J. Wang, Q. Liu, Y. Sun, X. Jing, L. Liu, J. Liu and D. Song, New J. Chem., 2015, 39, 868–876 RSC.
- Z. Zhao, J. Liu, F. Cui, H. Feng and L. Zhang, J. Mater. Chem., 2012, 22, 9052–9057 RSC.
- E. J. Kim, C. S. Lee, Y. Y. Chang and Y. S. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 9628–9634 CAS.
- Q. Han, Z. Wang, J. Xia, S. Chen, X. Zhang and M. Ding, Talanta, 2012, 101, 388–395 CrossRef CAS PubMed.
- H. Jiao and J. Wang, J. Alloys Compd., 2013, 577, 402–408 CrossRef CAS PubMed.
- M. Xu, L. Kong, W. Zhou and H. Li, J. Phys. Chem. C, 2007, 111, 19141–19147 CAS.
- W. Ostwald, Z. Phys. Chem., 1900, 34, 495–503 Search PubMed.
- S. J. Bao, Q. L. Bao, C. M. Li, T. P. Chen, C. Q. Sun, Z. L. Dong, Y. Gan and J. Zhang, Small, 2007, 3, 1174–1177 CrossRef CAS PubMed.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404–6410 CrossRef CAS PubMed.
- H. G. Yang and H. C. Zeng, J. Phys. Chem. B, 2004, 108, 3492–3495 CrossRef CAS.
- Y. Wang, Q. Zhu and H. Zhang, Chem. Commun., 2005, 5231–5233 RSC.
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896–910 CrossRef CAS PubMed.
- Y. Meng, D. Chen and X. Jiao, J. Phys. Chem. B, 2006, 110, 15212–15217 CrossRef CAS PubMed.
- D. Caruntu, G. Carnutu and C. J. O'Connor, J. Phys. D: Appl. Phys., 2007, 40, 5801–5809 CrossRef CAS.
- Q. Zhang, Z. Zhang, J. Teng, H. Huang, Q. Peng, T. Jiao, L. Hou and B. Li, Ind. Eng. Chem. Res., 2015, 54, 2940–2949 CrossRef CAS.
- Q. Zhang, Q. Du, T. Jiao, B. Pan, Z. Zhang, Q. Sun, S. Wang, T. Wang and F. Gao, Chem. Eng. J., 2013, 221, 315–321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Physical properties deduced from N2 adsorption was presented in Table S1. The kinetic data for fitting was presented in Table S2. See DOI: 10.1039/c5ra08678g |
| This journal is © The Royal Society of Chemistry 2015 |