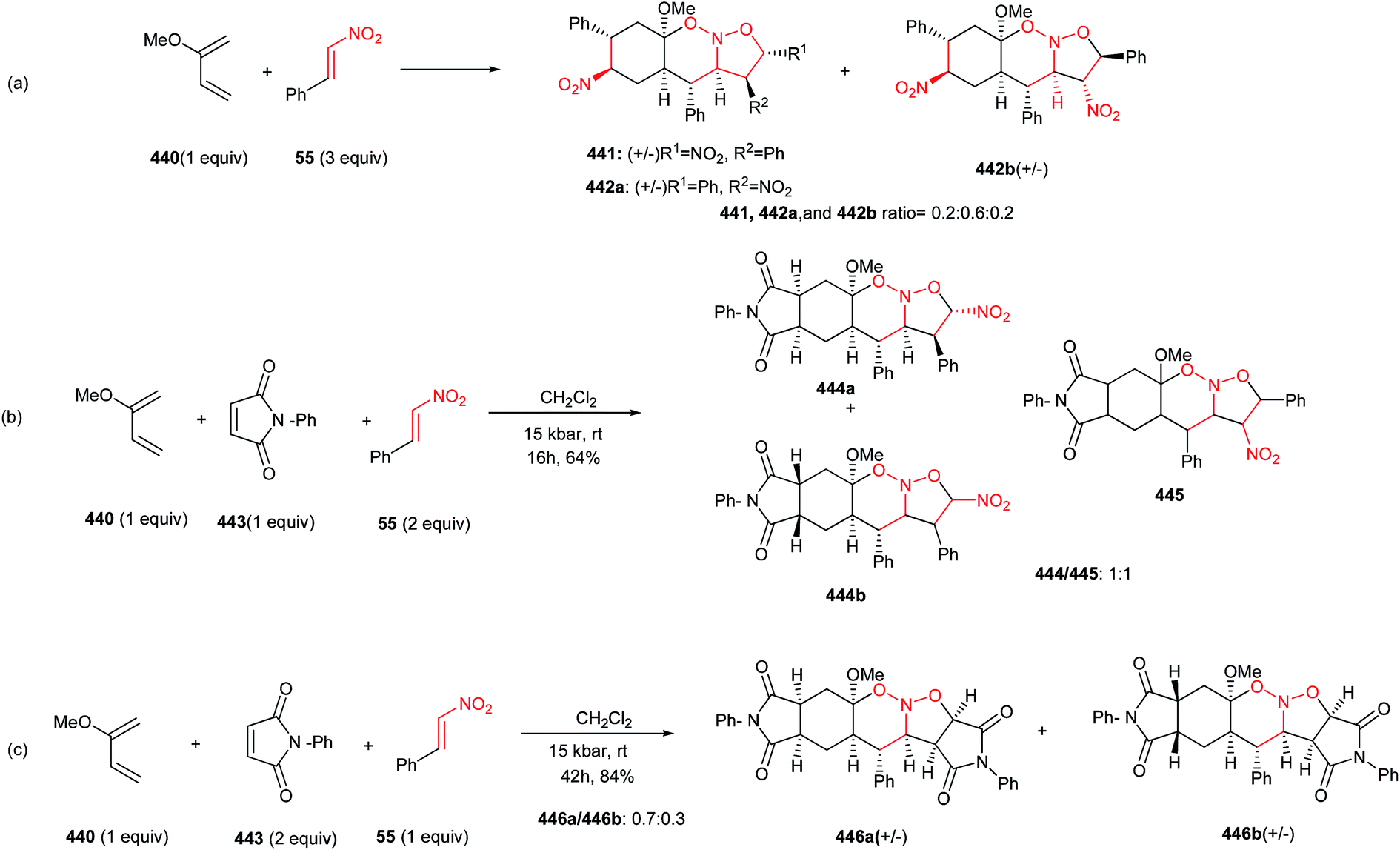
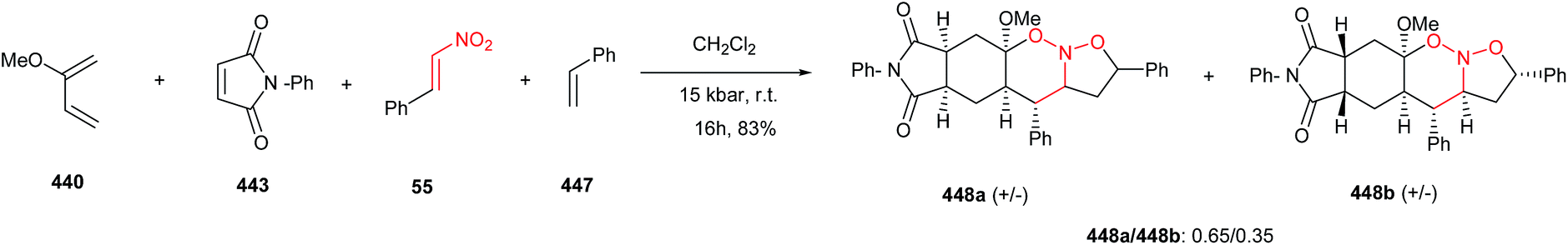
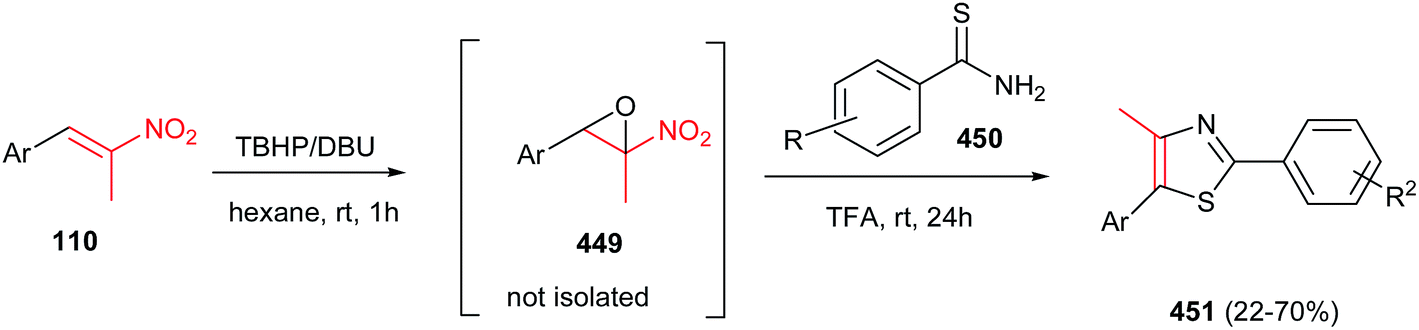
DOI:
10.1039/C4RA08828J
(Review Article)
RSC Adv., 2014,
4, 48022-48084
Part I: Nitroalkenes in the synthesis of heterocyclic compounds†
Received
18th August 2014
, Accepted 16th September 2014
First published on 17th September 2014
Abstract
The applications of nitroalkenes in the synthesis of three- to five-membered O, N and S-heterocycles, including natural products are investigated in this review. These heterocyclic compounds were synthesized from nitroalkenes with a variety of substituents at the α and β-positions and those that were part of common and medium rings via a wide variety of reactions such as Michael addition reactions, epoxidation, [3 + 2] cycloaddition and many cascade/domino/tandem reactions. In addition, the potential of nitroalkenes to take part in multi-component and cascade reactions, particularly, in diastereo- and enantioselective versions is reviewed. The high reactivity of nitroalkenes and their potential to coordinate with the metal catalysts as well as organocatalysts signify them as efficient precursors in synthetic organic chemistry. Also, the flexibility of the nitro group in functional group manipulations has expanded the scope of the nitro group, in general, and nitroalkenes, in particular, in organic synthesis.
 Azim Ziyaei Halimehjani | Azim Ziyaei Halimehjani was born in 1979 at Halimehjan, a small village in the Roudbar of Guilan, north of Iran. He obtained his B.Sc. in pure chemistry in 2001 from Shiraz University. After completing his M.Sc. in Organic Chemistry from Sharif University of Technology in 2003 under the supervision of Prof. M. R. Saidi and M. Tafazzoli, immediately he started his Ph.D. under the supervision of Prof. M. R. Saidi and advisor of Prof. J. Ipaktschi in the same University. During his Ph.D., he had two research visits from the research group of Prof. Ipaktschi (2005) and Prof. Peter R. Schreiner (2006) at Justus-Liebig-Universität Gieβen, Germany. After completing the Ph.D. in 2007, he began his academic career as Assistant Prof. of Organic Chemistry at Kharazmi University, Tehran, Iran. He has published over 35 publications. He works in the area of synthetic organic chemistry and coordination chemistry with emphasis on the chemistry of dithiocarbamates, nitroalkenes and development of new synthetic methodologies as well as green chemistry. |
 Irishi N. N. Namboothiri | Irishi N. N. Namboothiri received his MSc from Mangalore University (1988) and PhD from Indian Institute of Science (IISc), Bangalore (1994). He carried out postdoctoral research at Bar-Ilan University, Israel (1995–96), University of North Texas (1997–98) and Columbia University (1999). After a brief stint as Senior Research Scientist at Sabinsa Corporation, New Jersey (2000), he joined the faculty of Chemistry, Indian Institute of Technology Bombay, Mumbai (2001) where he is currently a professor. His research interests include organic synthesis, development of new synthetic methodologies, asymmetric catalysis, mechanistic studies and materials chemistry. He is a member of the editorial board of Journal of Chemical Sciences (2012–), an elected fellow of the National Academy of Sciences, India (2013–), and is a recipient of the Chemical Research Society of India Bronze medal (2014). He co-authored over 85 publications including two chapters and a book and is also a co-inventor of three patents. |
 Seyyed Emad Hooshmand | Seyyed Emad Hooshmand was born in Bandar Lengeh/Hormozgan, Iran, in 1988. He received his B.Sc. in chemistry from Bu-Ali Sina University, Hamedan, Iran in 2011, and his M.Sc. in organic chemistry from Kharazmi University, Tehran, Iran, under the supervision of Dr Azim Ziyaei Halimehjani, in 2013. His research interests include synthesis of novel biologically active compounds based on dithiocarbamates and synthesis of novel acid organic salts and their applications as catalyst in organic transformations. Now, he is a Ph.D. student at Shahid Beheshti University in Tehran, Iran. |
1. Introduction
Conjugated nitroalkenes constitute a unique class of electron deficient alkenes owing to their ability to take part in a wide range of organic reactions.1 These include aldol reactions,2 Michael addition reactions,3 Mannich reactions,4 (m + n) cycloadditions,5 Morita–Baylis–Hillman reactions6 and even metal mediated coupling reactions,7 to name a few. The reactivity umpolung of the nitro group as an acyl anion equivalent and nitroalkene as an acyl methyl cation equivalent is well documented in the literature.1 The exceptional ability of the nitro group to activate electrophiles and stabilize nucleophiles via co-ordination with Lewis and Bronsted acids as well as the ability of nitroalkenes to undergo Lewis base activation have elevated nitroalkenes as the substrates of choice in asymmetric reactions.3,8 The flexibility of nitro group in functional group manipulations has expanded the scope of nitro group, in general, and nitroalkenes, in particular, in synthetic organic chemistry.9 Among these, transformation to carbonyl compounds via Nef reaction, oximes, hydroxylamines and amines via reduction, nitriles via dehydration and reactive 1,3-dipoles such as nitrones, nitrile oxides and silyl nitronates with the intervention of various reagents are the prominent ones.
The pivotal role of nitroalkenes as substrates in new methodology development1–8,10 and in targeted synthesis,11 is also attributable to the ability of nitro group to survive hostile reagents and reaction conditions. The directing influence of nitro group is amply evident even in reactions where nitro group ultimately undergoes substitution or elimination (vide infra). The easy accessibility of nitroalkenes via nitro-aldol condensation and other methods such as direct nitration, nitrodecarboxylation etc. also contributed to the phenomenal growth of nitroalkene chemistry in recent decades.12
The versatility of nitroalkenes in organic synthesis has featured in numerous reviews.1–12 However, since the literature on nitroalkene chemistry currently grows by leaps and bounds, a comprehensive coverage of all the recent developments in one review article is a challenging task. Therefore, we recently reviewed the participation of nitroalkenes in the synthesis of carbocycles.13 This report surveys the central role of nitroalkenes as substrates in the synthesis of 3–5 membered O, N and S-heterocycles, including natural products. The succeeding part (part II) features the 6-membered heterocycles derived from conjugated nitroalkenes. To our knowledge, a focused review on nitroalkenes in the synthesis of heterocycles appeared 28 years ago.14
2. Synthesis of three-membered heterocycles
2.1. Epoxide derivatives
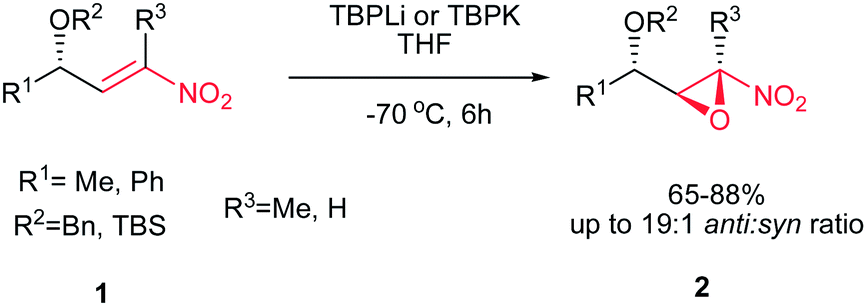
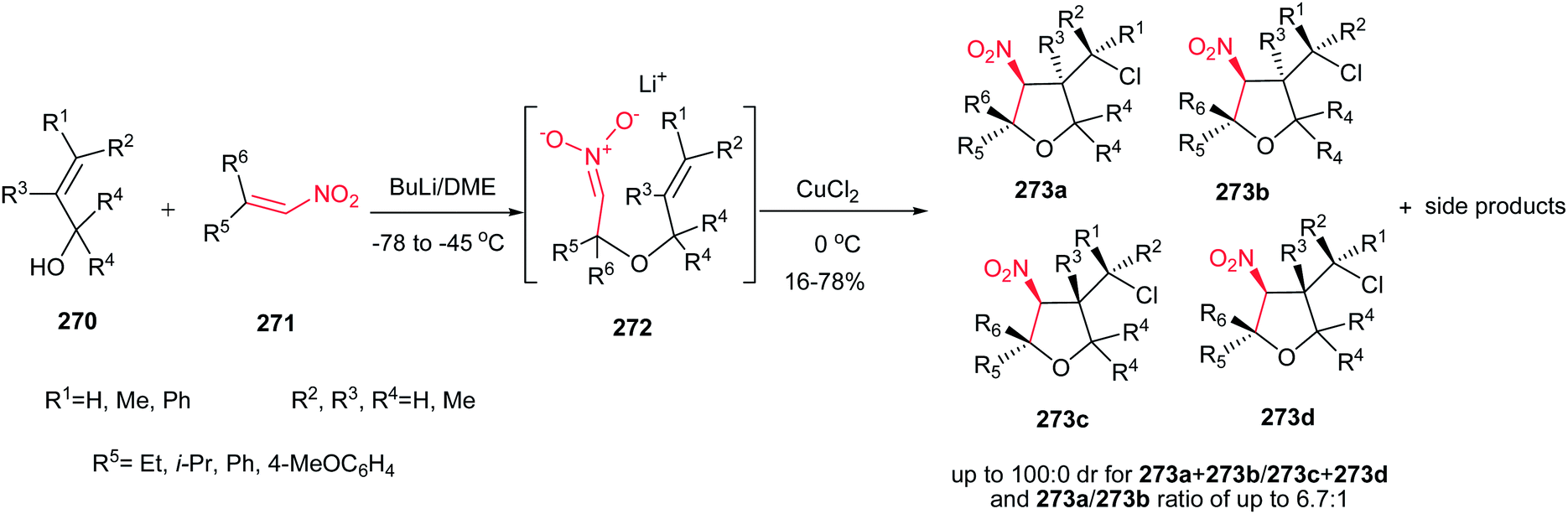
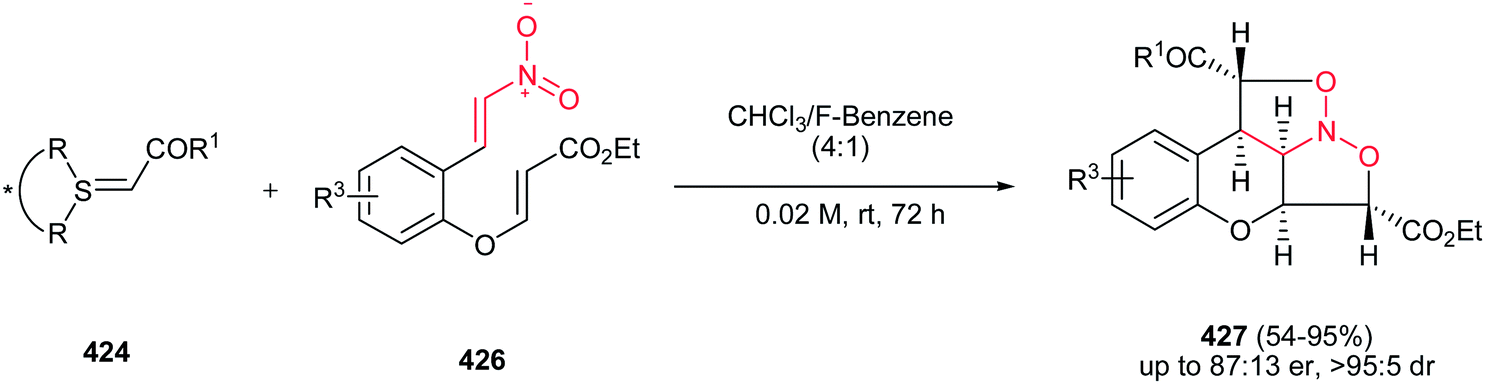
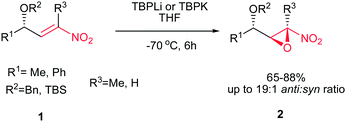
Rodríguez et al. found a versatile procedure for stereoselective epoxidation of chiral nitroalkenes 1 to provide the chiral nitroepoxides 2 (dr up to 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which are useful intermediates for synthesis of a variety of biologically active compounds. The reactions were carried out using lithium tert-butylperoxide or potassium tert-butylperoxide as the oxidizing reagent in THF as solvent at −70 °C. Nitroalkene 1 with a methyl group on the double bond also gave the corresponding product, but in lower diastereoselectivity (dr of 3.5:1). In addition, better selectivity can be enriched using potassium tert-butylperoxide as oxidant (Scheme 1).15
:1), which are useful intermediates for synthesis of a variety of biologically active compounds. The reactions were carried out using lithium tert-butylperoxide or potassium tert-butylperoxide as the oxidizing reagent in THF as solvent at −70 °C. Nitroalkene 1 with a methyl group on the double bond also gave the corresponding product, but in lower diastereoselectivity (dr of 3.5:1). In addition, better selectivity can be enriched using potassium tert-butylperoxide as oxidant (Scheme 1).15
 |
| | Scheme 1 Stereoselective epoxidation of chiral nitroalkenes. | |
2.2. Aziridine derivatives
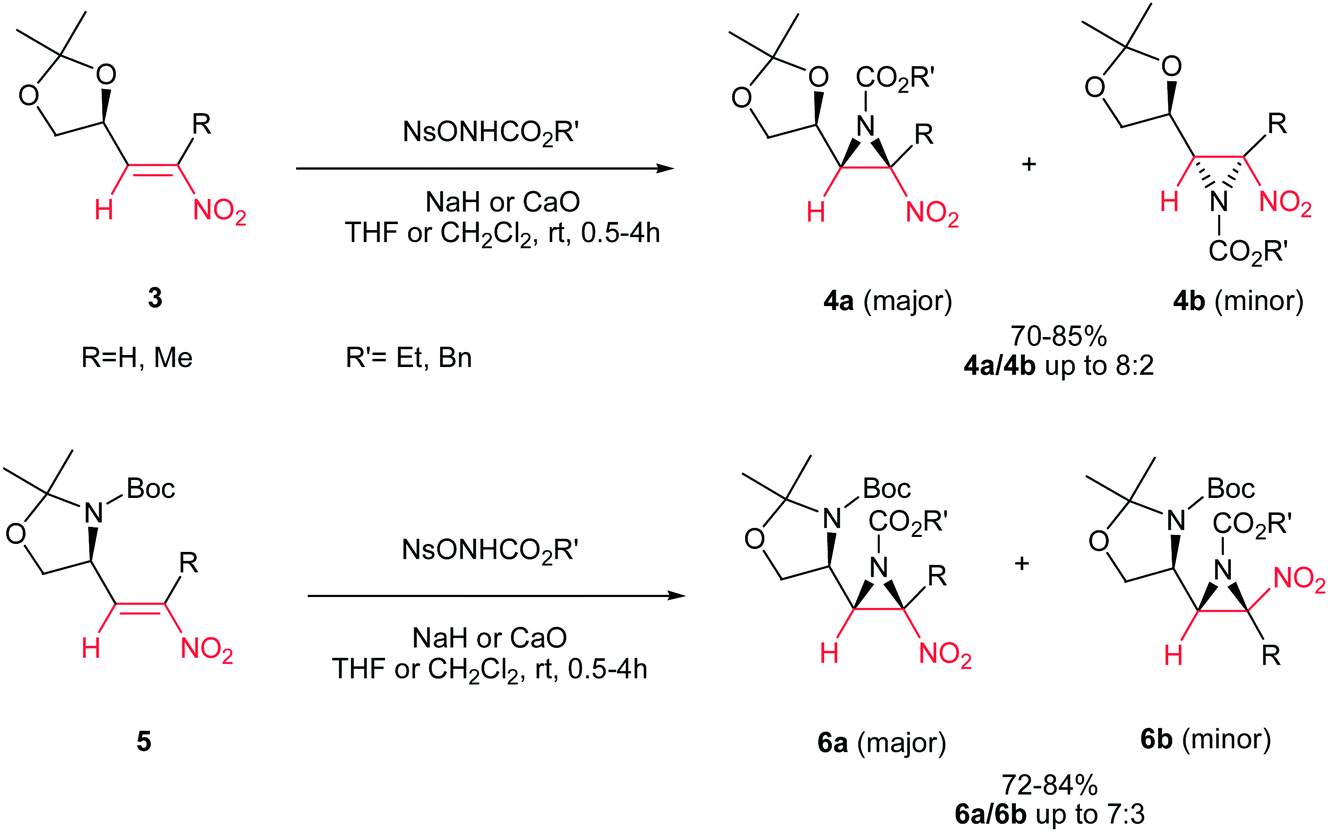
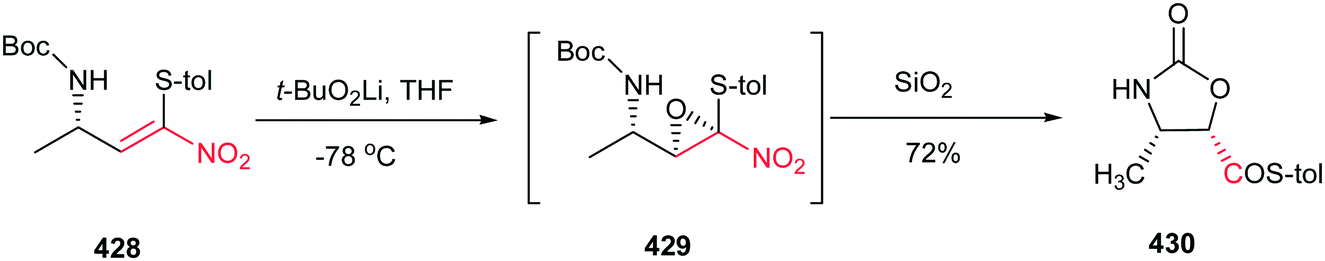
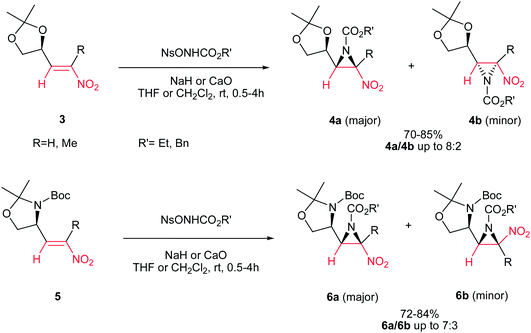
Fioravanti et al. reported an efficient protocol for asymmetric synthesis of chiral nitroaziridines 4 and 6 from optically active (E)-nitroalkenes 3 and 5 bearing a 1,3-dioxolane or 1,3-oxazolidine residue and carbamates (NsONHCO2Et or NsONHCO2Bn) via a stereoselective aza-Michael initiated ring closure reactions catalyzed by an inorganic base such as CaO or NaH. The reaction was carried out both in solution (in CH2Cl2 or THF) and solvent-free conditions with similar results. Interestingly, while nitroalkenes generated from (R)-2,2-dimethyl-1,3-dioxolane-4-carboxaldehyde 3 provided the aziridines 4a/b as products in up to 8:2 ratio, nitroalkene 5 gave stereoisomers 6a/b in up to 7:3 ratio under the same reaction conditions. Complete retention of configuration of substrates was observed during the reaction condition. Also, nitroalkenes 3 shows higher dr than 5 (Scheme 2).16
 |
| | Scheme 2 Asymmetric synthesis of chiral nitroaziridines from optically active (E)-nitroalkenes. | |
3. Synthesis of four-membered heterocycles
3.1. Substituted thietanes and azetidines
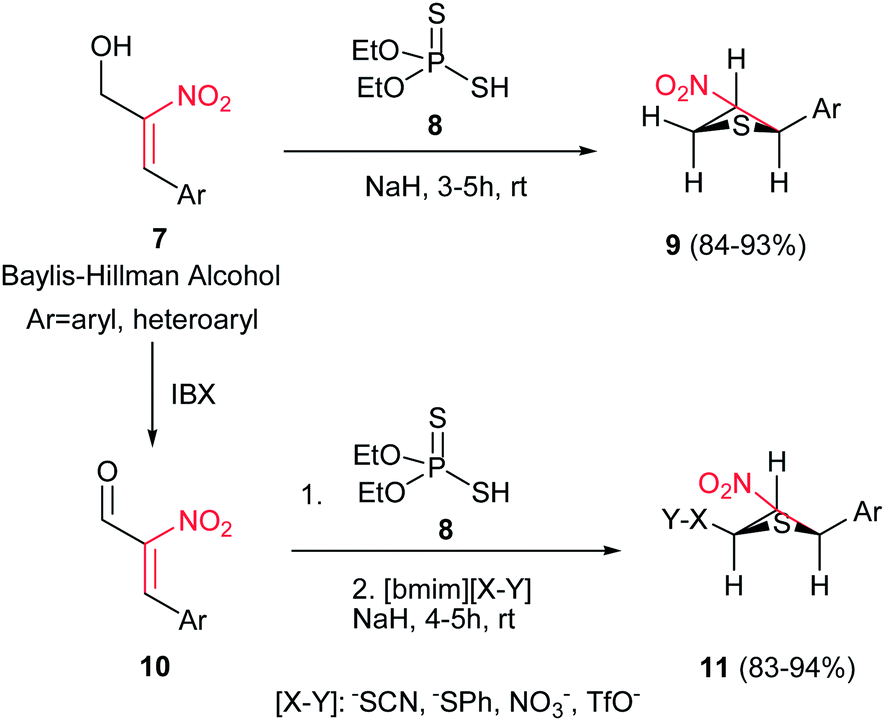
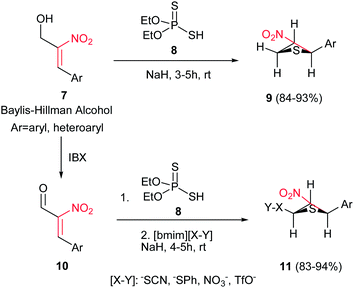
Reaction of Baylis–Hillman alcohols 7 and their aldehydes 10 with either O,O-diethyl hydrogen phosphorodithioate 8 or O,O-diethyl hydrogen phosphorodithioate in combination with a task-specific ionic liquid [bmim]X–Y afford the corresponding 2,3-di- or 2,3,4-trisubstituted thietanes 9 or 11, respectively, with complete diastereoselectivity in favor of the trans isomers as confirmed by NOE (Scheme 3).17 Diverse nucleophiles such as SCN, PhS, NO3 and TfO anions can be applied in this protocol with excellent yields. The authors have also shown that the nucleophilicty of SCN or PhS anion is considerably higher in [bmim]SCN or [bmim]SPh compared to that from KSCN or PhSNa. Another point in this report is using IBX as a mild oxidant and compatible with a variety of functional groups.
 |
| | Scheme 3 Nitroalkenes in the synthesis of substituted thietanes. | |
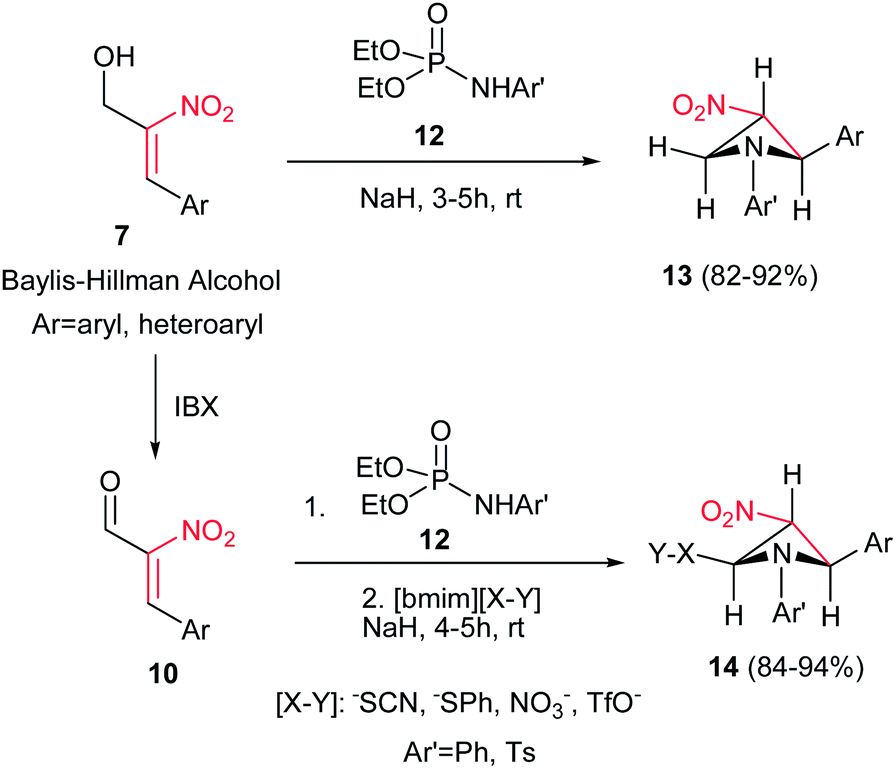
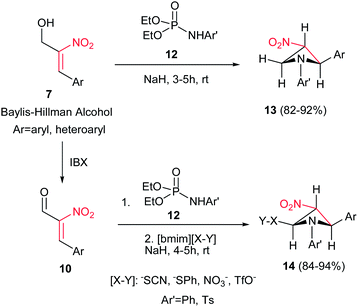
Also, the same group described that by using N-aryl/tosylphosphoramidates 12 instead of 8 under similar reaction conditions, the corresponding substituted 3-nitroazetidines 13 or 14 can be synthesized in high to excellent yields. The reaction proceeded via domino Michael/cyclization reaction (Scheme 4).18
 |
| | Scheme 4 Nitroalkenes in the synthesis of substituted 3-nitroazetidines. | |
3.2. β-Thiolactam derivatives
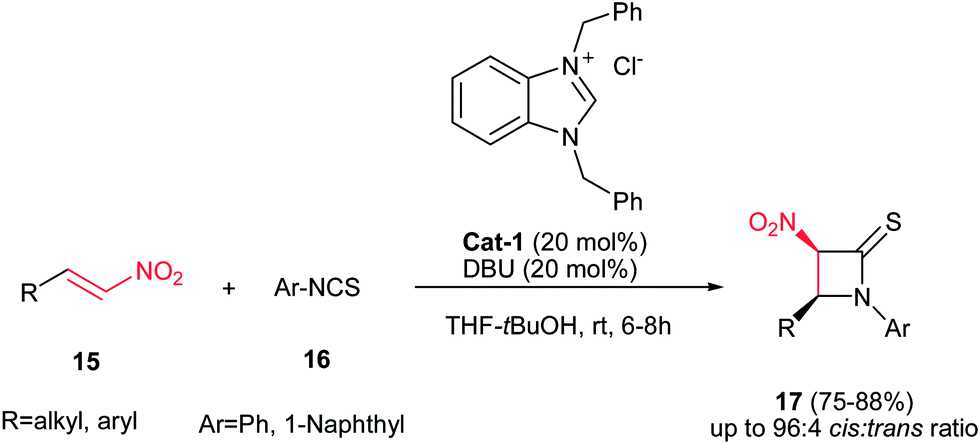
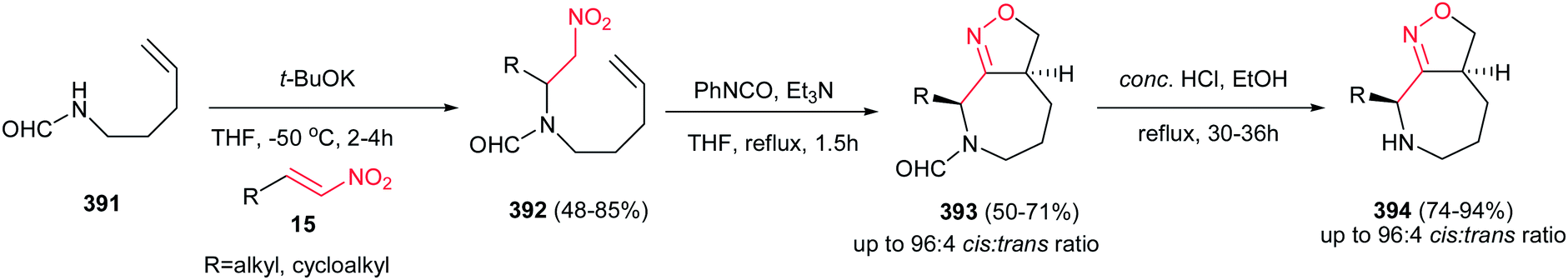
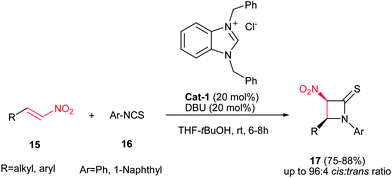
Yadav and Awasthi reported an efficient procedure for synthesis of β-thiolactams 17 from nitroalkenes 15 and aryl isothiocyanates 16 catalyzed by N-heterocyclic carbene Cat-1 (Scheme 5).19 The optimal conditions for this reaction involved stirring 15 and 16 in the presence of 20 mol% precatalyst Cat-1 and DBU, in THF-tBuOH at room temperature for 6–8 hours under a nitrogen atmosphere. Under these conditions, the β-thiolactams 17 were obtained in excellent yields with high diastereoselectivity (up to 96:4) in favor of the cis isomer. Aromatic and aliphatic nitroalkenes were compatible in this process.
 |
| | Scheme 5 N-Heterocyclic carbene-catalyzed synthesis of thiolactams from aryl isothiocyanates and nitroalkenes. | |
4. Synthesis of five-membered heterocycles
4.1. Synthesis of N-heterocyclic compounds
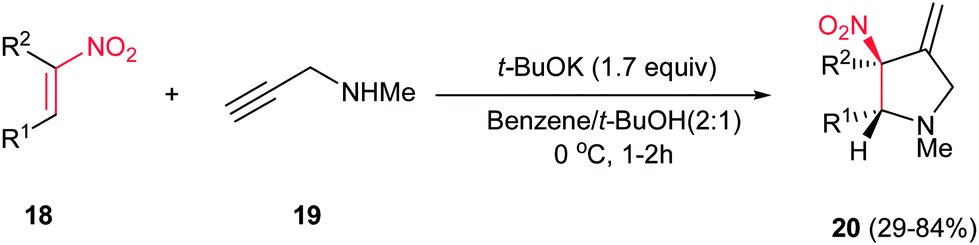
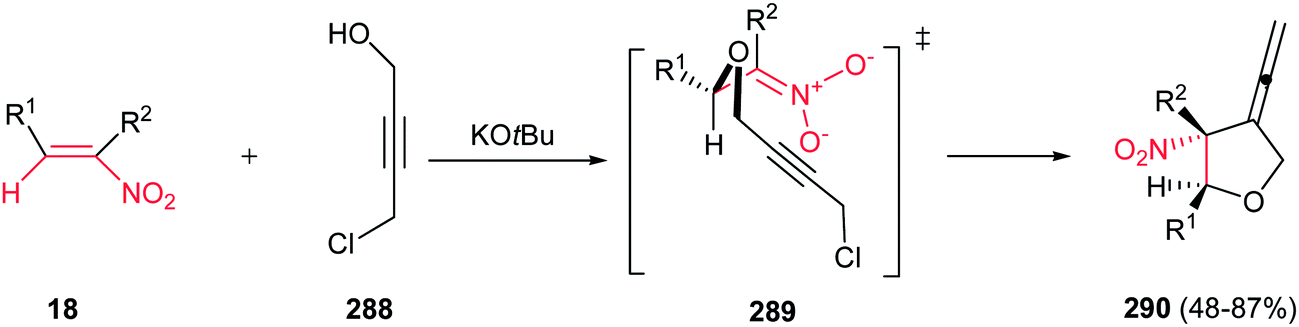
4.1.1. Pyrrolidine derivatives. Pyrrolidine rings are present in the structure of numerous natural products, pharmaceuticals, and bioactive molecules with different biological activities.20 Also, they have a wide range of applications as organocatalysts, building blocks in organic synthesis, chiral auxiliaries, and ligands for asymmetric synthesis.21 Many syntheses of pyrrolidines have been reported.Nitroalkenes were applied as efficient starting materials for synthesis of pyrrolidine derivatives. In this context, reaction of nitroalkenes 18 and N-methylprop-2-ynyl amine 19 was investigated by Rodriguez et al. for the regio- and distereoselective synthesis of the 3-methylenepyrrolidines 20 via Michael addition/5-exo intramolecular nucleophilic carbocyclization cascade (Scheme 6).22
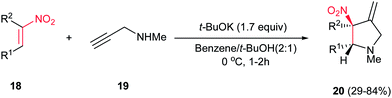 |
| | Scheme 6 3-Methylenepyrrolidines via Michael addition/5-exo intramolecular nucleophilic carbocyclization cascade | |
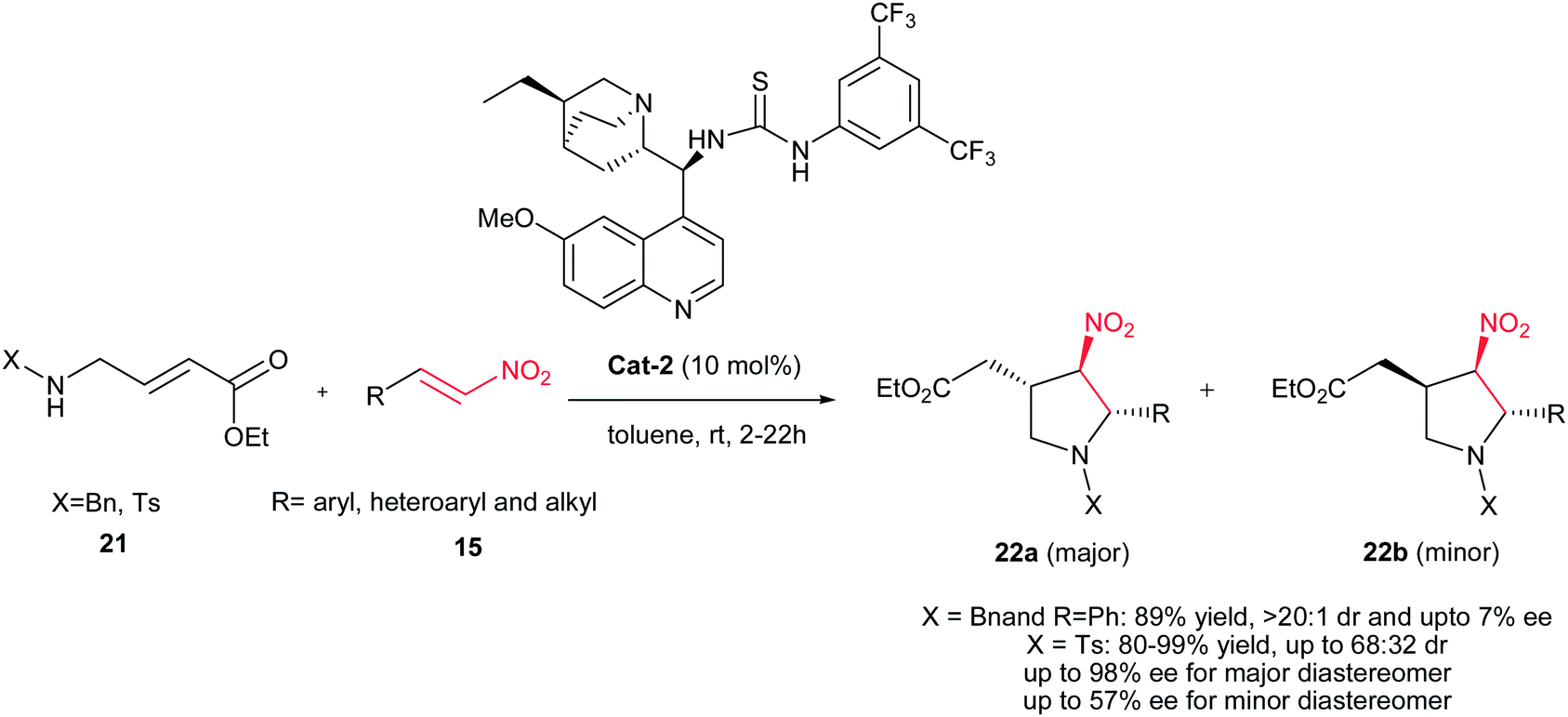
Kanger et al. demonstrated a new asymmetric synthesis of chiral pyrrolidines 22 with three stereocenters from 4-aminocrotonate 21 and nitroolefins 15 in the presence of 10 mol% bifunctional thiourea catalyst Cat-2.23 The selectivity of the reaction is highly dependent on the substituent on the nitrogen atom of the aminocrotonate. While, the reaction with N-benzyl substituted reagent proceeds in high diastereoselectivity (>20:1) toward 22 with low enantioselectivity (ee up to 7%), the N-tosyl crotonate affords products with moderate diastereoselectivity (22a:22b up to 68:32) but with high enantioselectivity in the case of major trans–trans-isomers 22a (ee from 92% to 98%) and ee up to 57% for the minor trans–cis-isomers 22b (Scheme 7). Aromatic, heteroaromatic and aliphatic nitroalkenes work equally well in this procedure. The absolute configuration of both isomers was examined via derivatization with Mosher's and mandelic acids, with the relative stereochemistry being determined via NMR analysis.
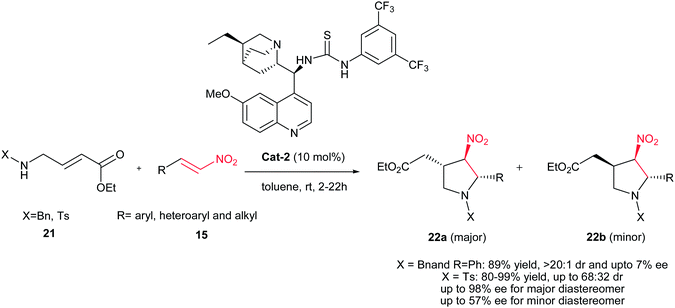 |
| | Scheme 7 Synthesis of trisubstituted chiral pyrrolidines. | |
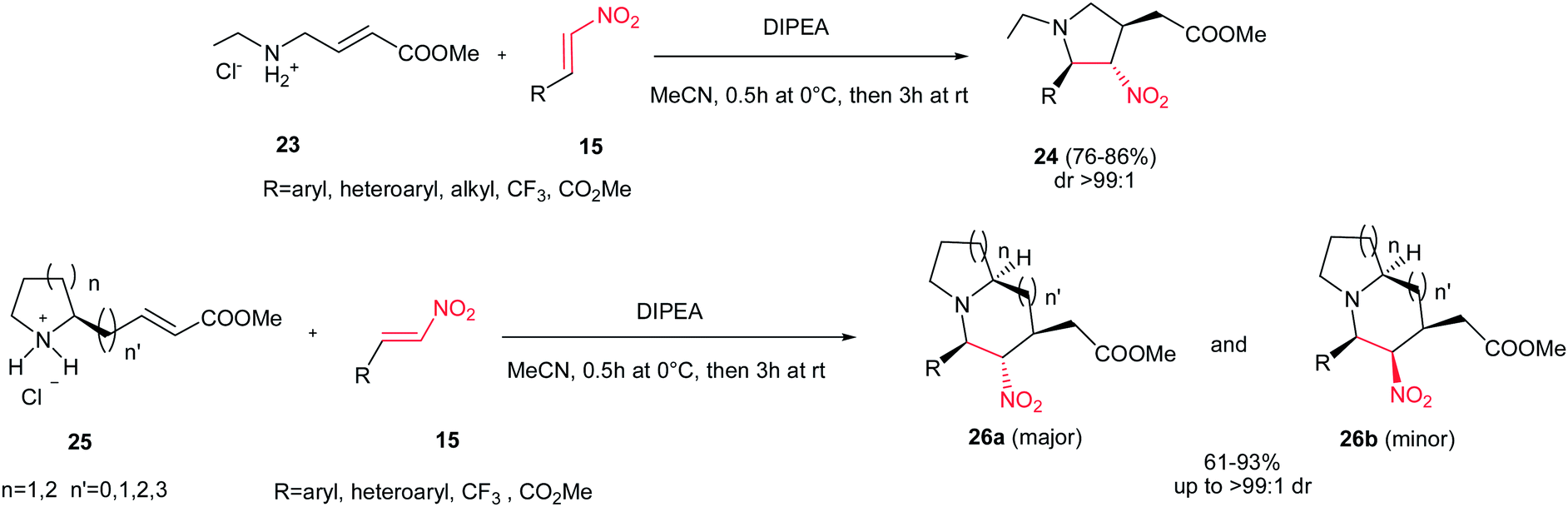
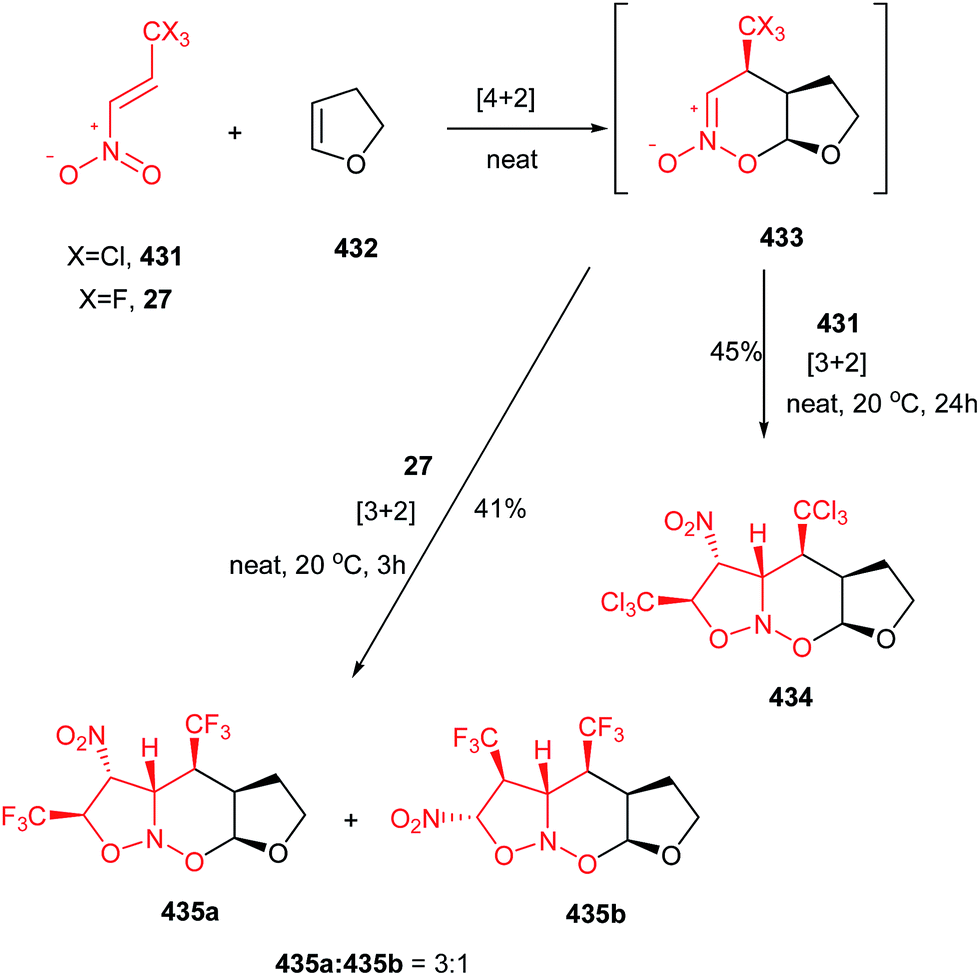
The same strategy was applied by Shi et al. for preparation of substituted pyrrolidines 24 through the reaction of amine 23 and nitroalkenes 15 with high yields and excellent diastereoselectivities (up to >99:1 dr) using Hünig's base (DIPEA) (Scheme 8).24 The reaction proceeded via aza-Michael/Michael reaction cascade. By using a cyclic amine 25, the 5-5, 5-6, 6-5 or 6-6 bicyclic structures 26 can readily be prepared in good to excellent yields. Although the products were obtained with four stereogenic centers, the reaction exhibited excellent stereoselectivity with only two stereoisomers isolated (26a and 26b) in a diastereomeric ratio of up to >99:1. The relative stereochemistry was verified by NMR and confirmed by X-ray crystallography. β-Triflouromethylnitroethene 27 (CF3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHNO2) also gave the similar cyclization products in excellent yields, which provided an interesting strategy to incorporate the CF3 group into the N-heteroycles. The formation of 7- and 8-exo-trig cyclization products is also possible with poor isolated yields (<25%).
CHNO2) also gave the similar cyclization products in excellent yields, which provided an interesting strategy to incorporate the CF3 group into the N-heteroycles. The formation of 7- and 8-exo-trig cyclization products is also possible with poor isolated yields (<25%).
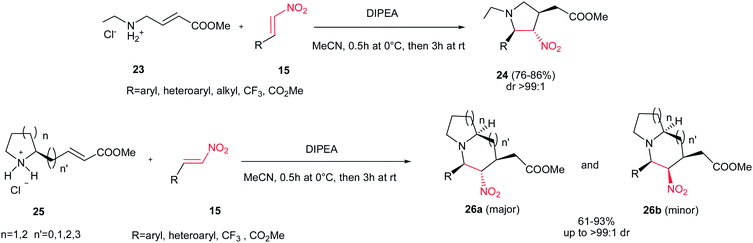 |
| | Scheme 8 Synthesis of pyrrolidine rings via cascade aza-Michael/Michael reaction. | |
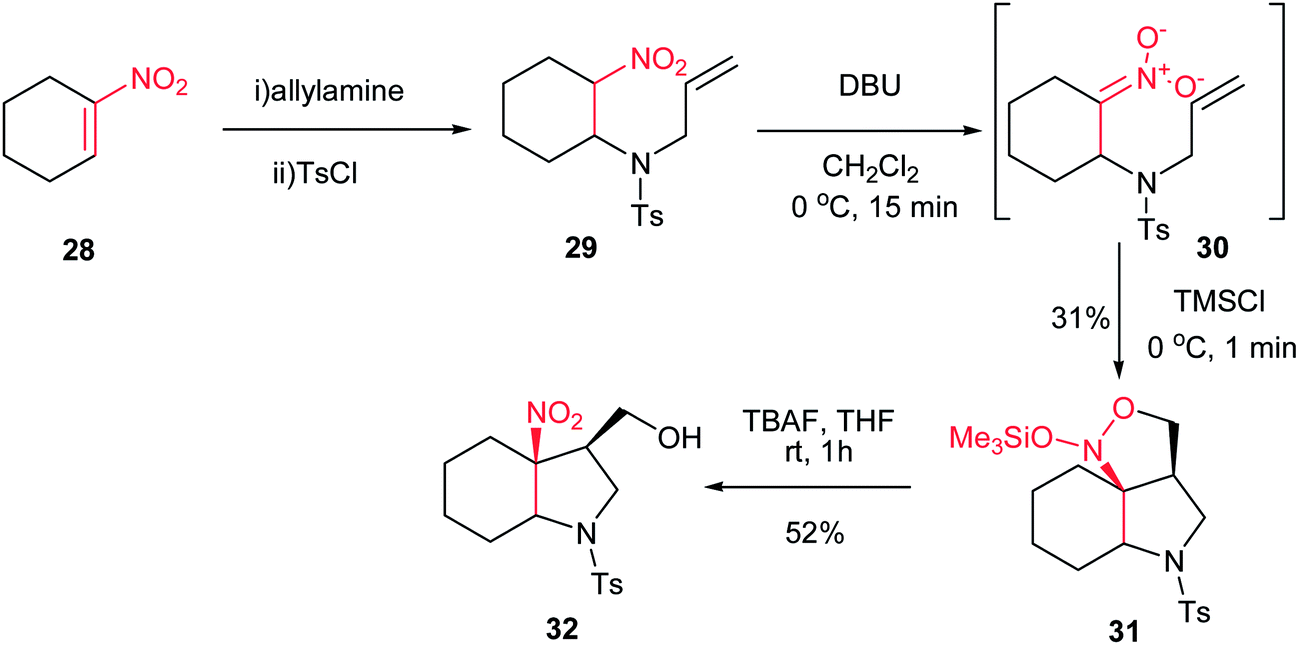
In 2005, Dulcere et al. reported a diastereoselective procedure for synthesis of fused 3-nitro-4-hydroxymethylpyrrolidine 32. They described that silylation of nitronate 30, prepared by an aza Michael addition of tosylallylamine to nitroalkene 28, provided N-(silyloxy)-isoxazolidine 31 in 31% yield, which was then diastereoselectively converted to 3-nitro-4-hydroxymethylpyrrolidine 32 in 52% yield after desilylation (Scheme 9).25
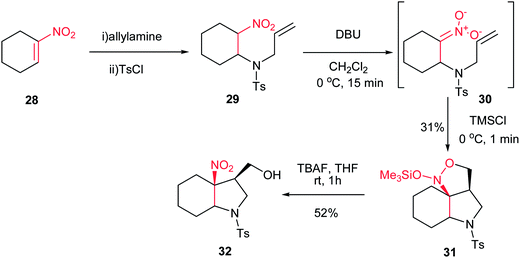 |
| | Scheme 9 Diastereoselective synthesis of fused 3-nitro-4-hydroxymethylpyrrolidine. | |
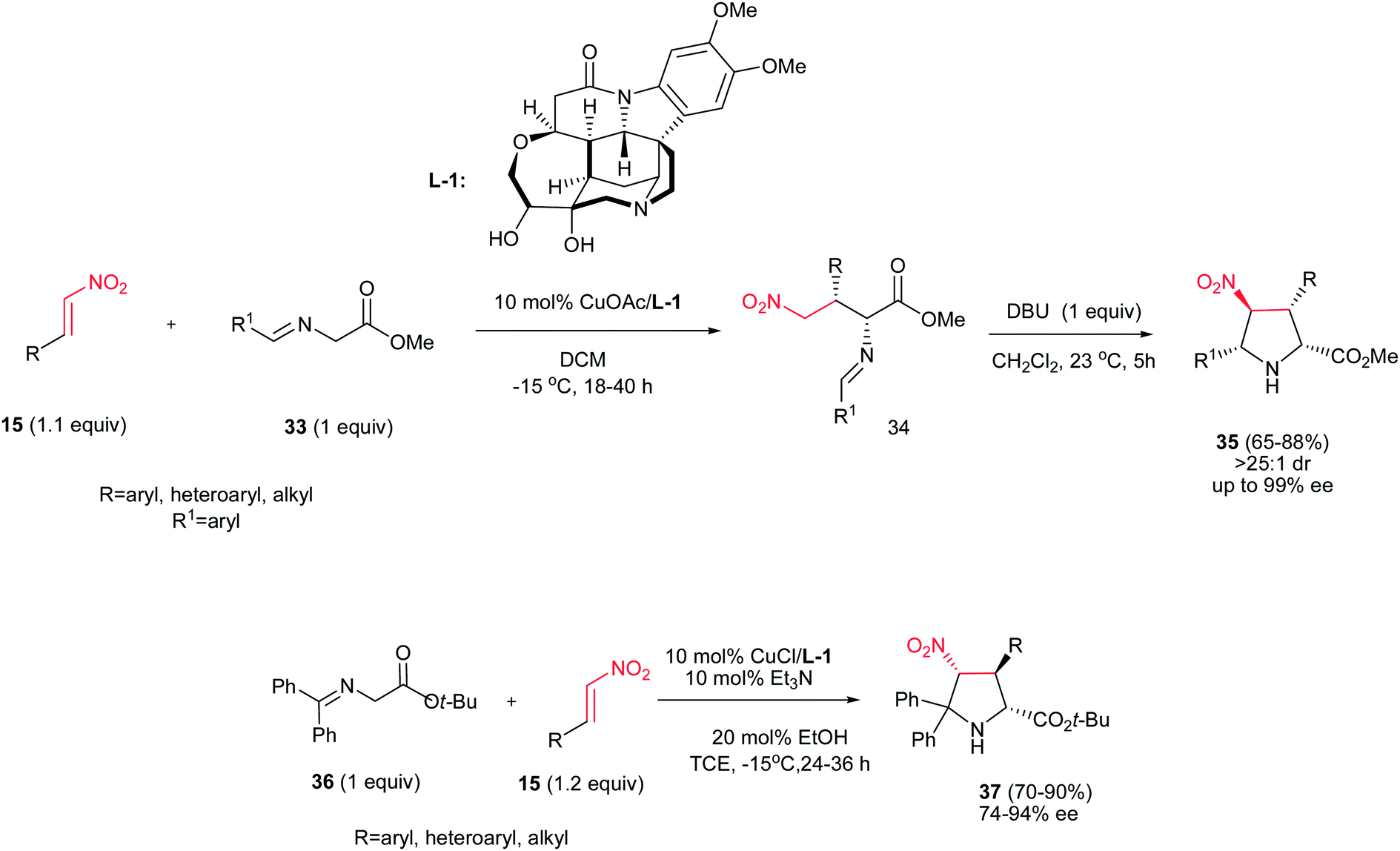
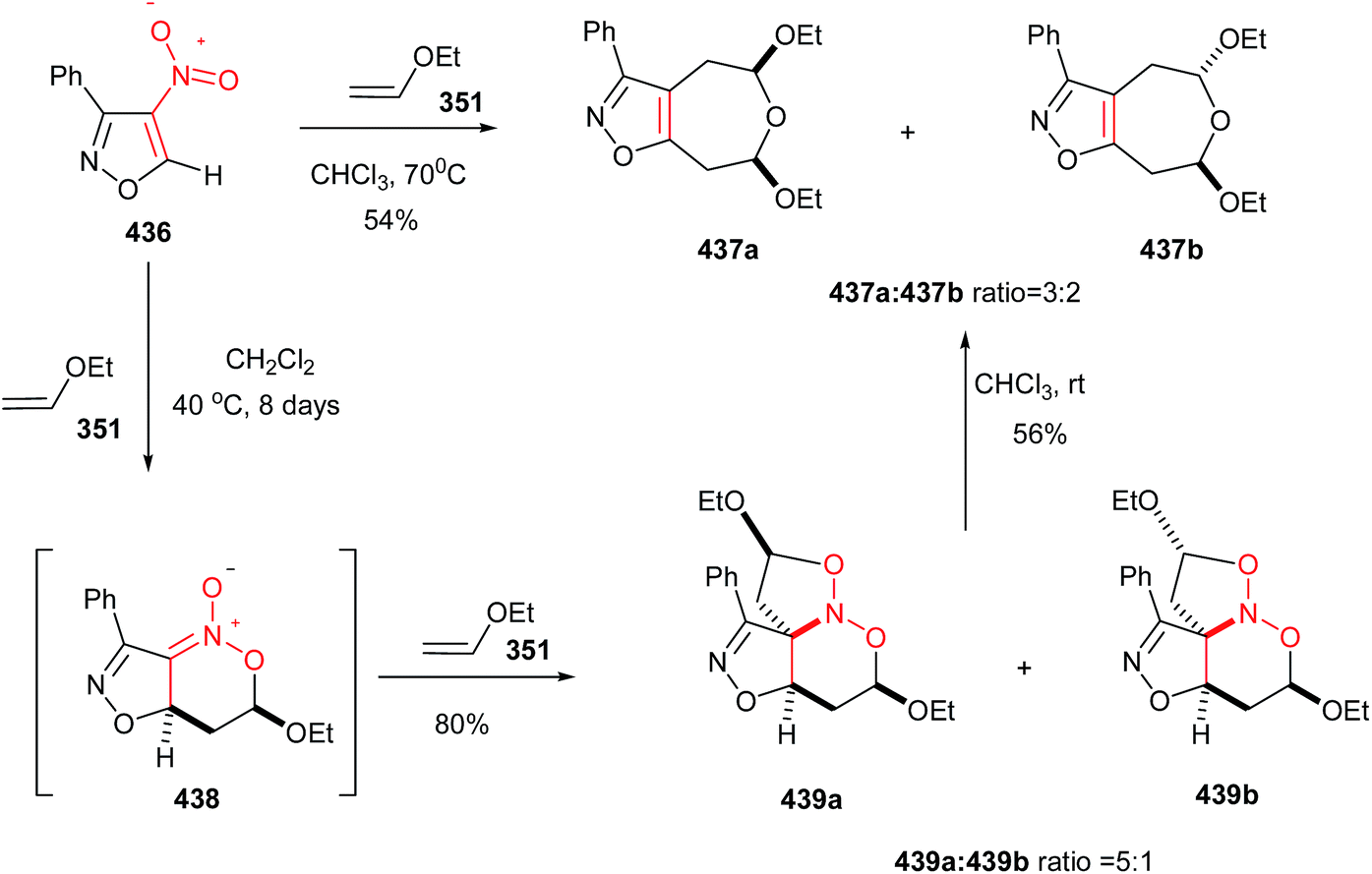
Nitroalkenes were extensively applied in [3 + 2] cycloaddition reactions for synthesis of substituted chiral pyrrolidines with different stereogenic centers. In this context, a stepwise one-pot [3 + 2] cycloaddition reaction of glycine(ket)imines 33 or 36 with nitroalkenes 15 is investigated by Oh et al. as outlined in Scheme 10.26 They described that Michael addition of glycine imines 33 to nitroalkenes 15 afforded exclusively syn-adducts 34 with excellent stereoselectivity (>25:1 dr, 97−99% ee) at −15 °C in CH2Cl2 when 10 mol% of CuOAc was used in combination with multidentate amino alcohol L-1 as ligand. Intramolecular Mannich reaction of the crude reaction mixture of syn-adducts 34 in the presence of 1 equiv. of DBU at 23 °C afforded the exo-selective chiral pyrrolidines 35 in high yields and excellent diastereo- and enantioselectivities (>25:1 dr, 93−99% ee). Surprisingly, performing the reaction with the glycine ketimines 36 instead of glycine imines in the presence of 10 mol% CuCl/L-1 and Et3N, affords the endo-selective pyrrolidines 37 in high yield (65–88%) and enantioselectivity (74–93% ee). The reaction is applicable to a wide range of aromatic, heteroaromatic, and aliphatic nitroalkenes.
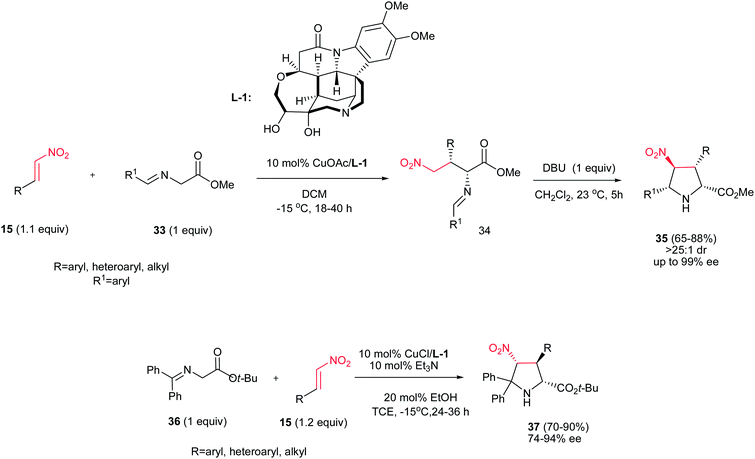 |
| | Scheme 10 Cu(I)-catalyzed asymmetric synthesis of pyrrolidine derivatives. | |
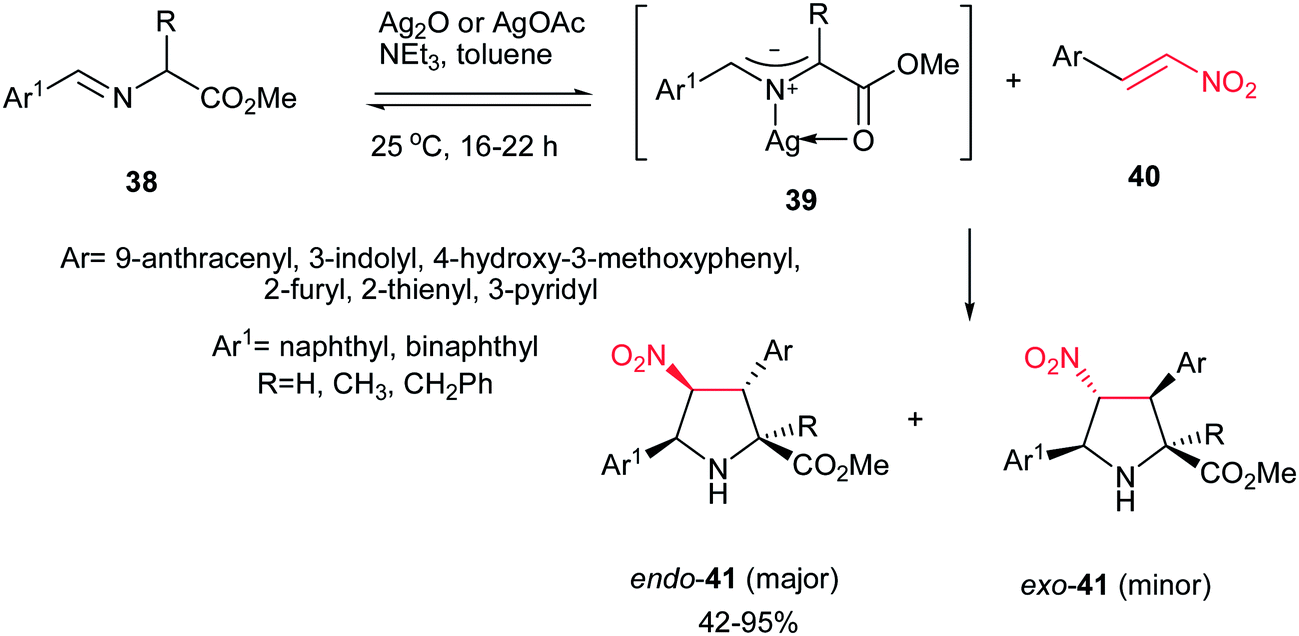
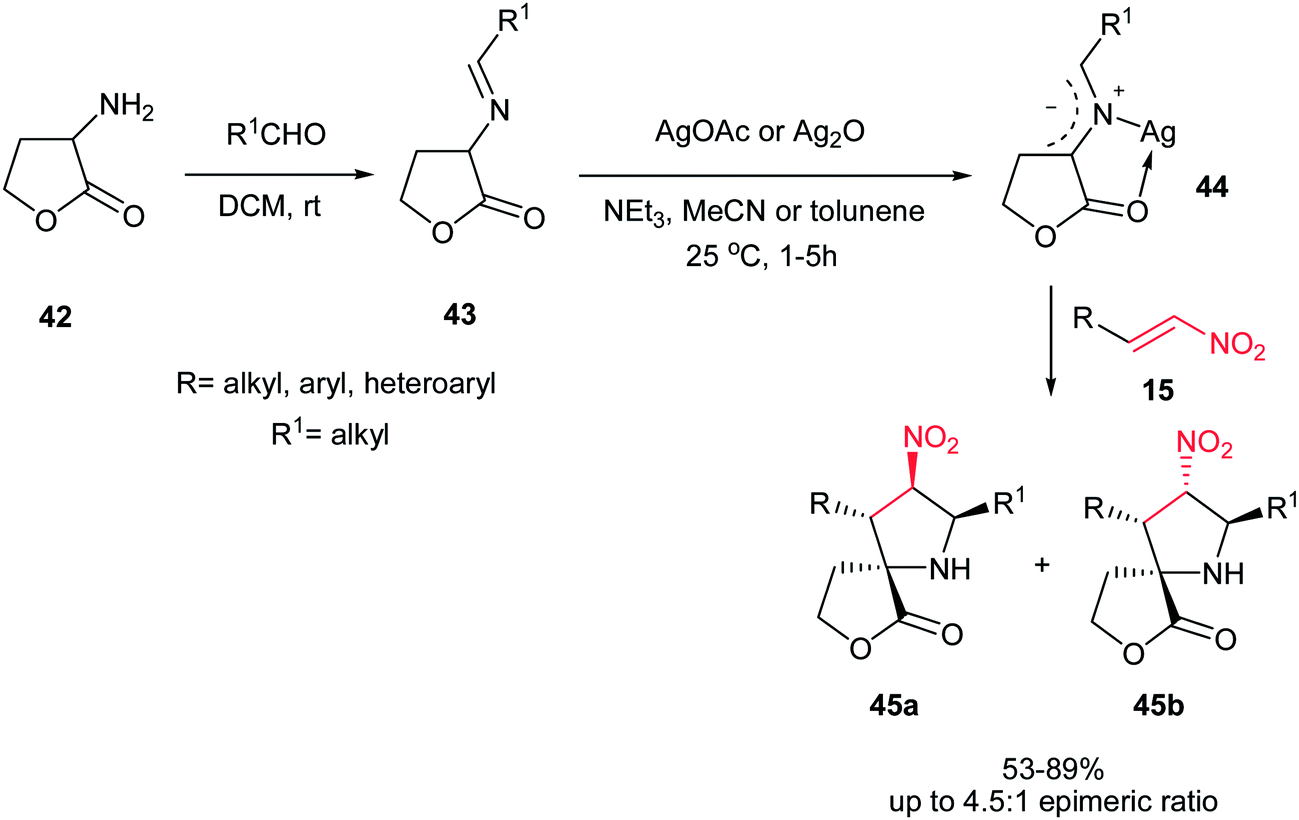
In 2008, Grigg et al. described that 1,3-dipolar cycloaddition reaction of imines 38, prepared from natural aminoacids and aldehydes, with nitroalkenes 40 in the presence of 10 mol% of AgOAc or Ag2O and NEt3 afforded the endo-selective pyrrolidine adducts 41 in high to excellent yields (Scheme 11).27 The reactions occurred via concerted cycloaddition of the in situ generated argento azomethine ylides 39 to E-nitroalkenes 40 via endo-transition states. Imines generated from glycine, alanine, and phenylalanine were used successfully in this process. Nitroalkenes with aromatic and heteroaromatic groups are well tolerated. With using the cyclic imine 43, prepared from homoserine lactone 42, the corresponding spiro nitropyrrolidines 45 were obtained in good to high yields as mixture of diastereomers 45a and 45b (Scheme 12).
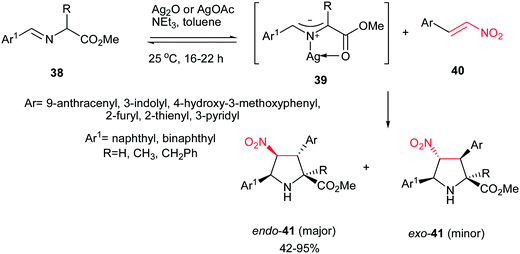 |
| | Scheme 11 Ag(I)-catalysed azomethine ylide cycloadditions with nitroalkenes. | |
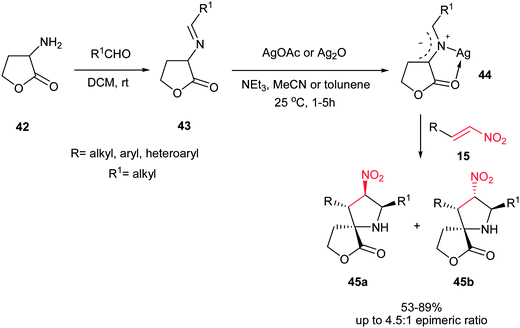 |
| | Scheme 12 Ag(I)-catalysed asymmetric synthesis of spiro nitropyrrolidines. | |
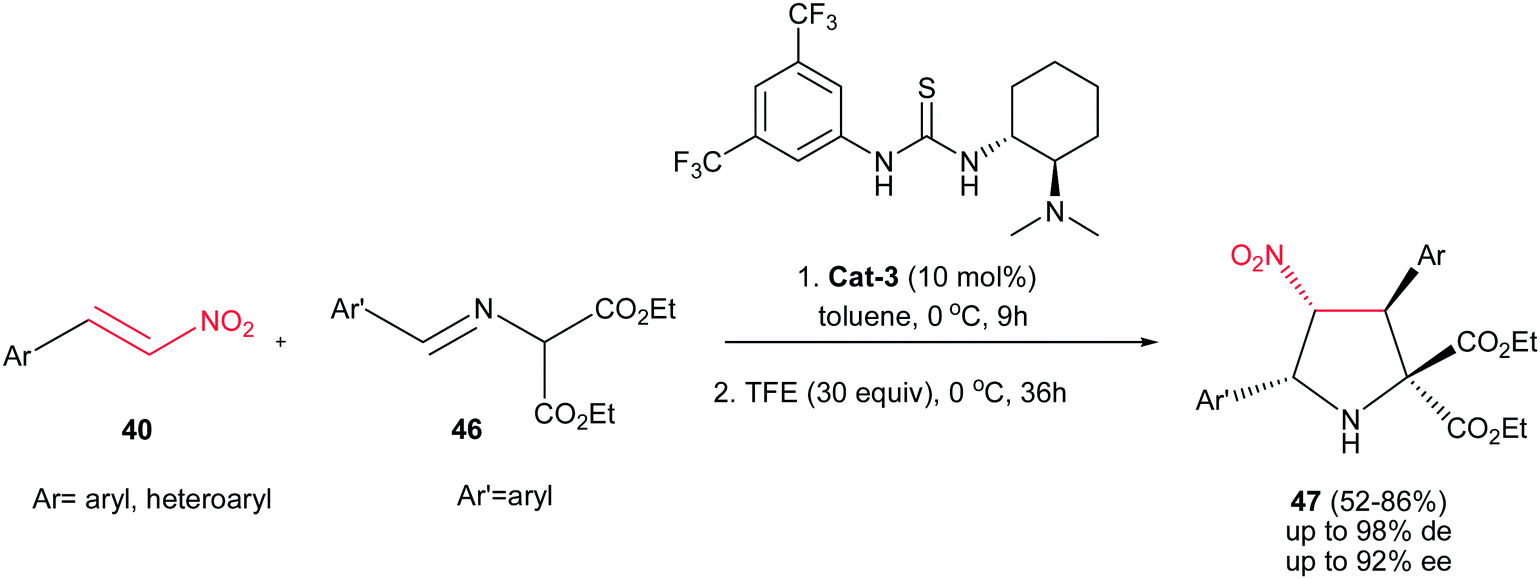
Better yields and stereoselectivities (up to 98:1:1 dr and 92% ee) were obtained in the reaction of azomethine ylides 46 with nitroalkenes 40 using Takemoto catalyst Cat-3 (Scheme 13).28 The reaction proceeds in a stepwise manner consisting of Michael addition and subsequent intramolecular aza-Henry reaction. While the first step accelerated by thiourea catalyst (10 mol%) in ethanol at 0 °C, the later step was promoted by addition of 2,2,2-trifluoroethanol (30 equiv.) to the reaction mixture and completed after stirring for additional 36 h at the same temperature. Without TFE, the reaction stopped in the first step. Although the electronic nature of substituents on the phenyl ring of nitroalkenes does not have significant effect on the outcome of the reaction, lower ee's were observed with increasing the electron-donating properties of the substituents on the imines.
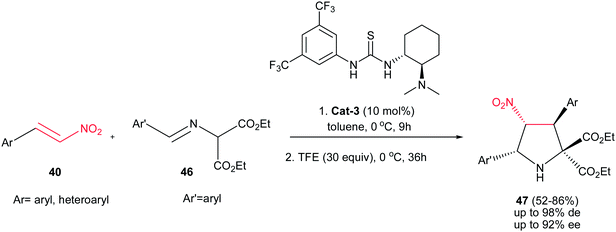 |
| | Scheme 13 Asymmetric synthesis of functionalized pyrrolidines with Takemoto's bifunctional chiral thiourea. | |
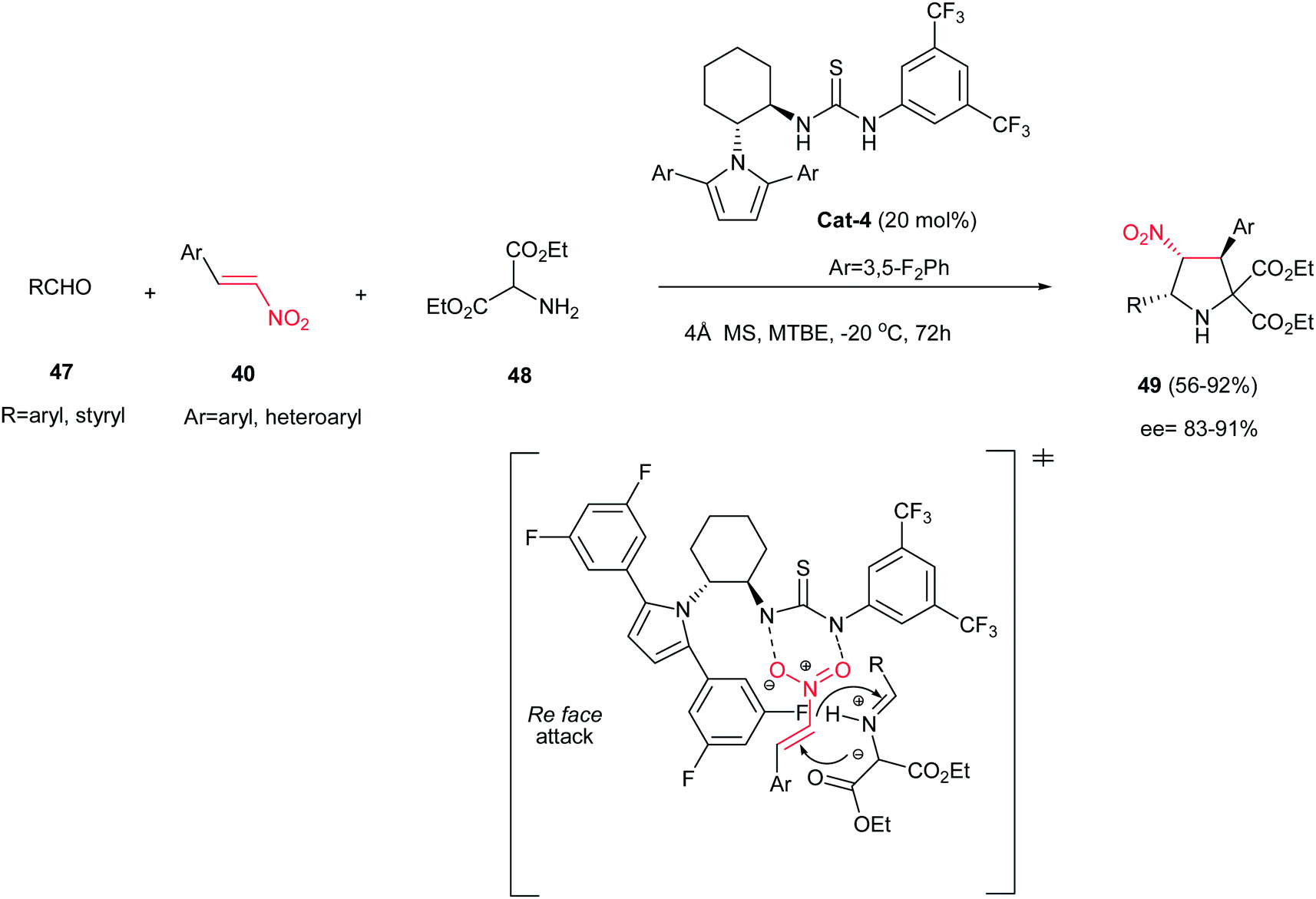
The one-pot three-component asymmetric [3 + 2] cycloaddition of aldehydes 47, diethyl α-aminomalonate 48, and nitroalkenes 40 is also investigated by Chen et al. affording highly substituted pyrrolidines 49 in high to excellent yields and stereoselectivities (Scheme 14) using 20 mol% of chiral thiourea catalyst Cat-4.29 The reaction proceeded via in situ formation of azomethine ylides from the aminomalonate and the aldehyde, followed by 1,3-dipolar cycloaddition with the nitroalkene with complete endo selectivity (>99:1). It is notable that aliphatic aldehydes do not participate in this reaction.
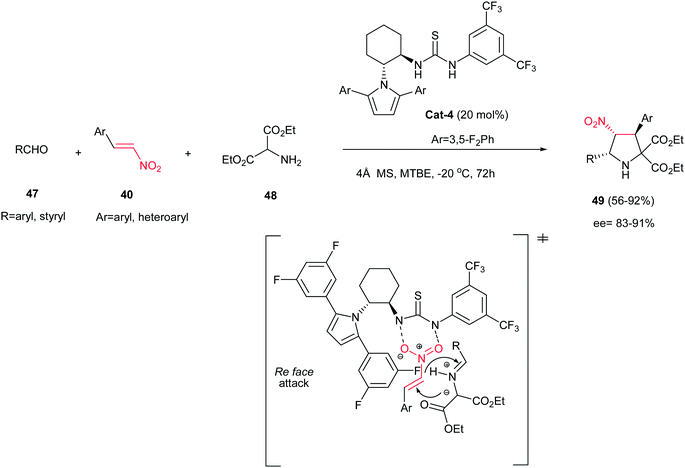 |
| | Scheme 14 Enantioselective three-component synthesis of polysubstituted pyrrolidines. | |
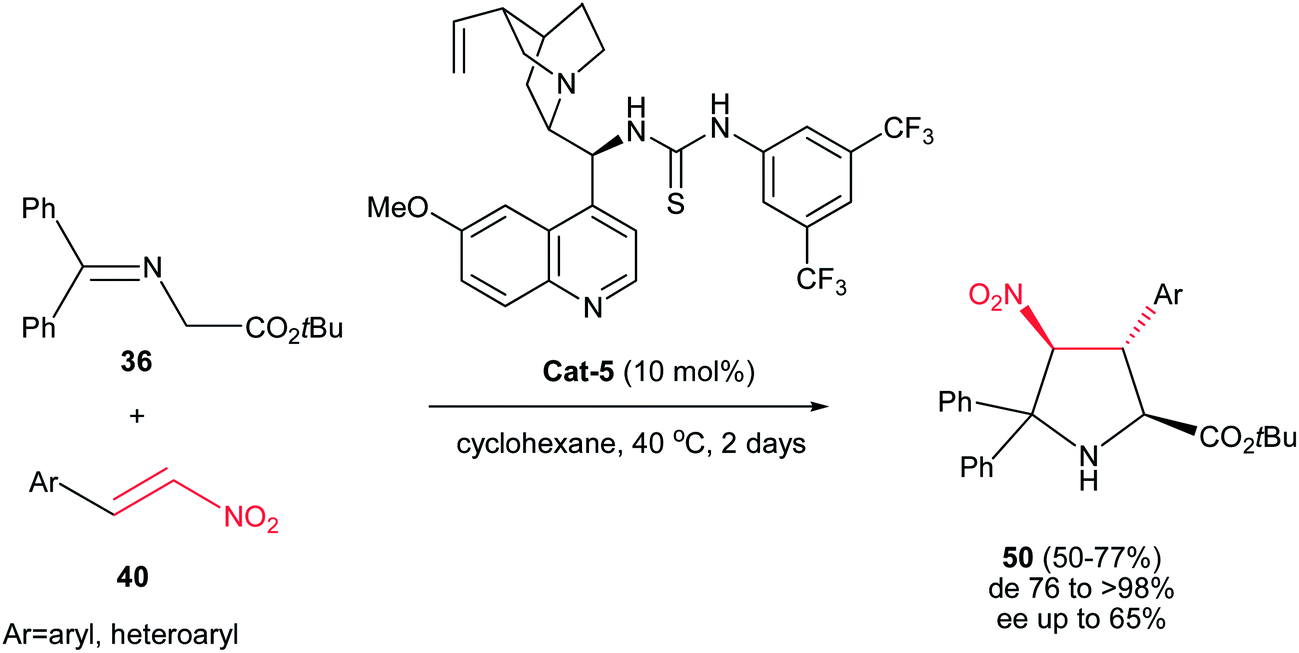
In another report, Gong et al. described the synthesis of highly substituted pyrrolidines 50 with diastereoselectivities of up to >99:1 and moderate enantioselectivities. The authors found that among the several cinchona alkaloid thiourea derivatives that were able to catalyze the reaction, catalyst Cat-5 afforded the best results in terms of catalytic activity, diastereo- and enantioselectivities. It is notable that this method is limited to the use of only the imine of benzophenone derivatives (Scheme 15).30
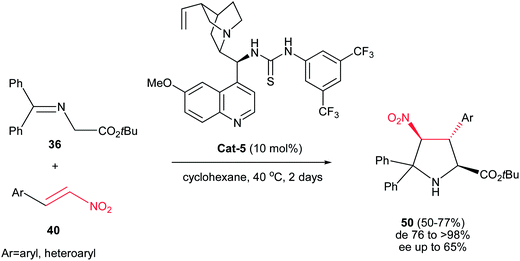 |
| | Scheme 15 The 1,3-dipolar cycloadditions of azomethine ylide generated from imine 36 with nitroalkenes 40 catalyzed by Cat-5. | |
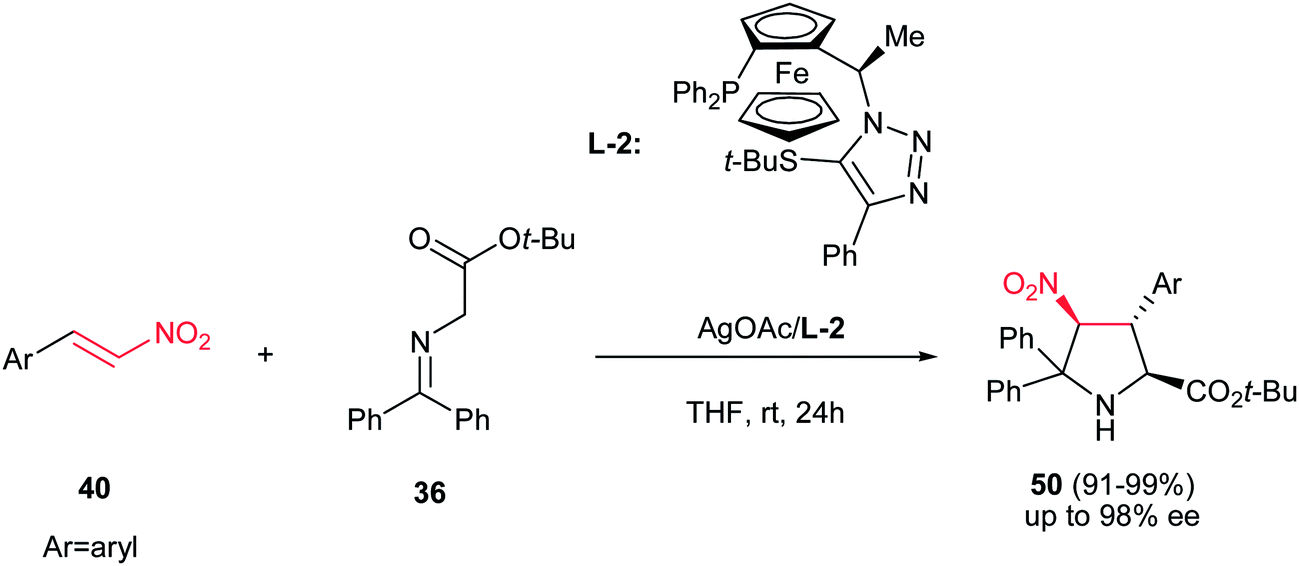
In this context, Fukuzawa et al. have shown that AgOAc engaged with the chiral ferrocenyltriazole-based P,S-ligand L-2 is another efficient catalytic system for asymmetric synthesis of pyrrolidine products 50 with the same starting materials (Scheme 16).31 The pyrrolidine products 50 (major) were obtained as sole diastereomer in good yields with high enantioselectivities (up to 96% ee) along with Michael adducts as minor products.
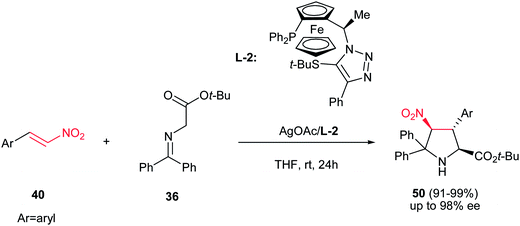 |
| | Scheme 16 AgOAc/ferrocenyl triazole-based P,S-ligand for asymmetric synthesis of functionalized pyrrolidines. | |
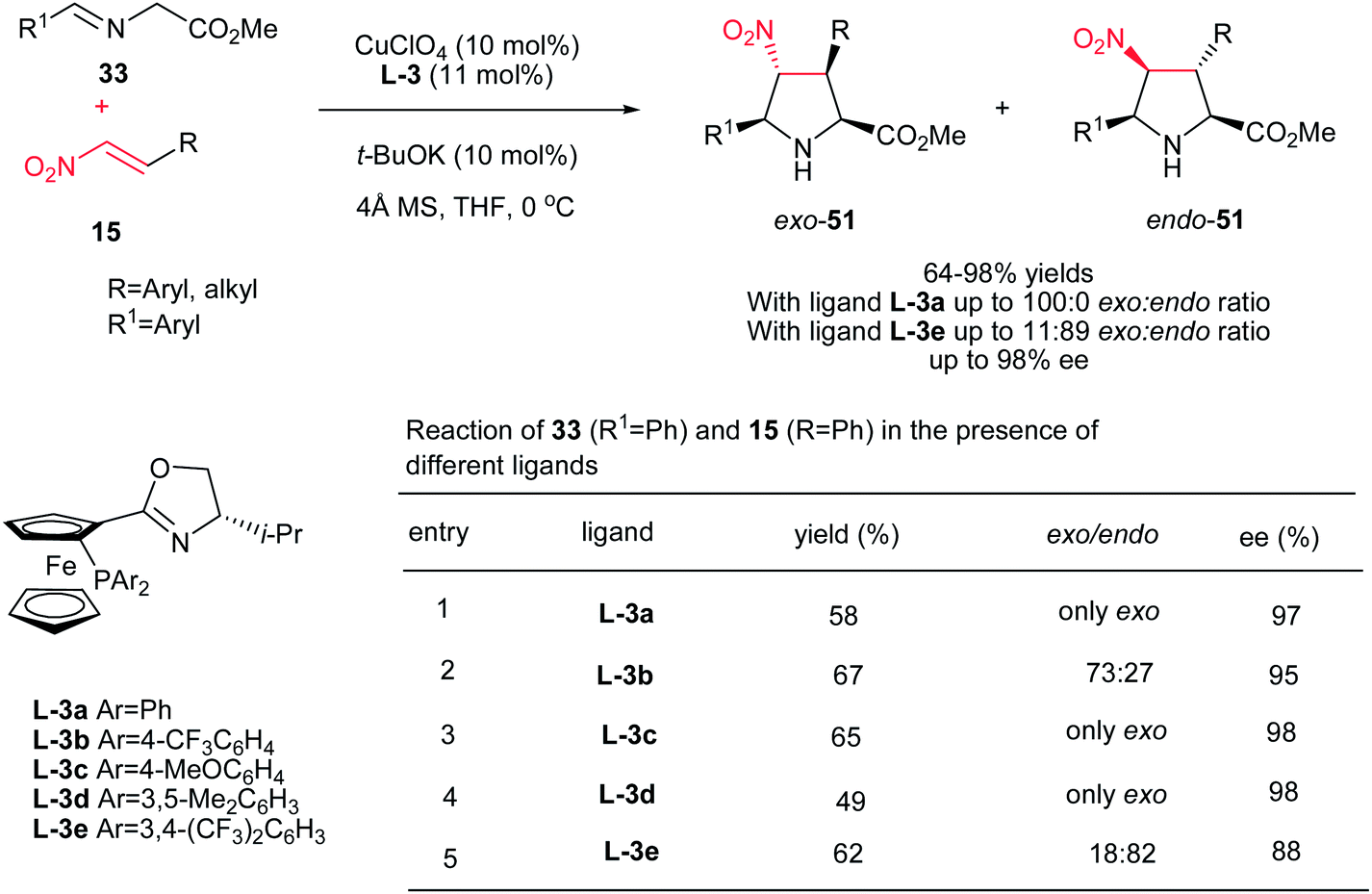
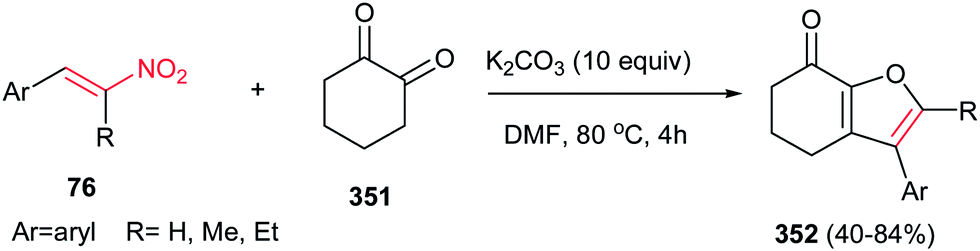
Hou et al. have shown that 10 mol% of CuClO4 in combination with a chiral P,N-ferrocene ligand L-3 is an efficient catalytic system for the asymmetric 1,3-dipolar cycloaddition of azomethine ylides 33 to nitroalkenes 15. They confirmed that by varying the groups on the phosphorus in ligands, dramatic variations in diastereoselectivity can be observed. In this context, while P,N-ferrocene ligands with electron-rich aryl group on the P atom afford exo-selective pyrrolidine adducts exo-51, the corresponding ligands with electron-deficient aryl groups provide endo-selective adducts 51. Aliphatic and aromatic nitroalkens gave similar results. The authors also examined that the Ag salts gave somewhat higher yields of the endo product compared to Cu salts, but in lower enantioselectivity (Scheme 17).32
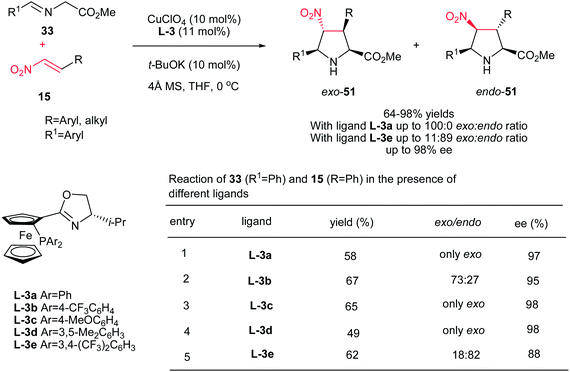 |
| | Scheme 17 Synthesis of chiral pyrrolidines promoted by CuClO4 in combination with a chiral P,N-ferrocene ligand. | |
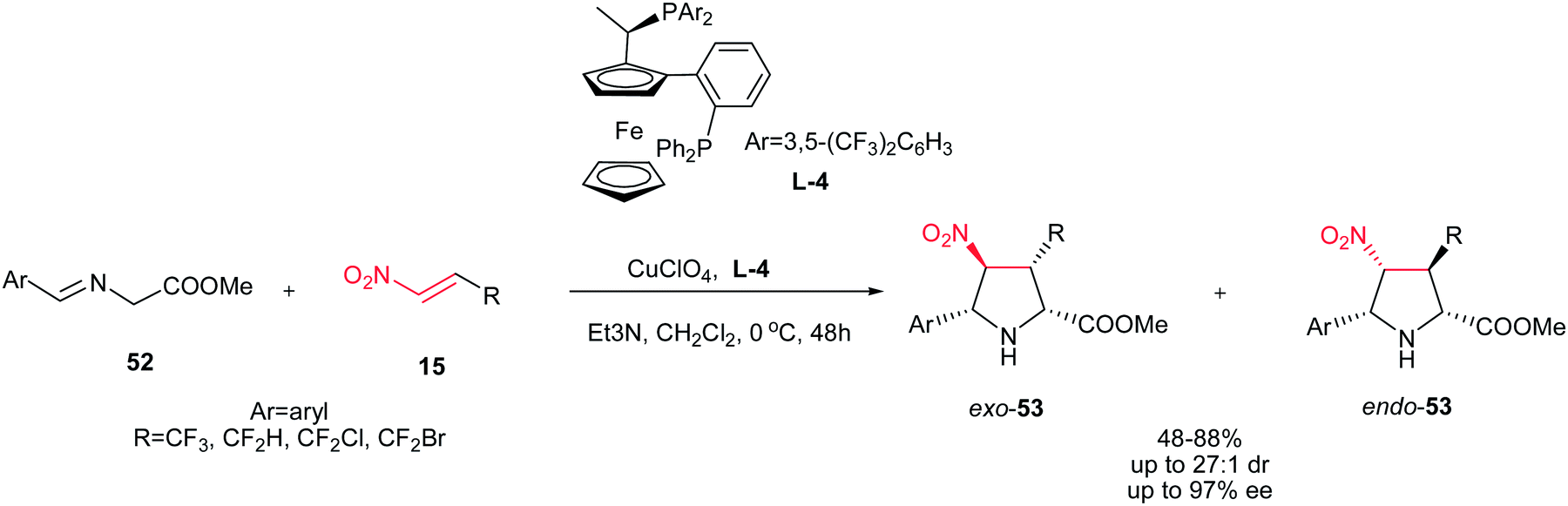
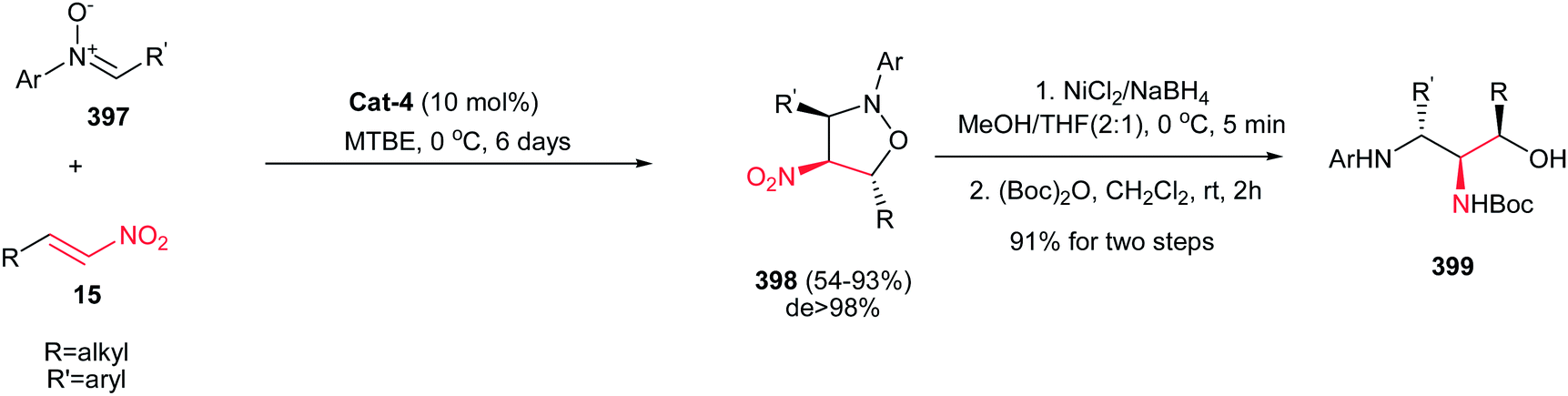
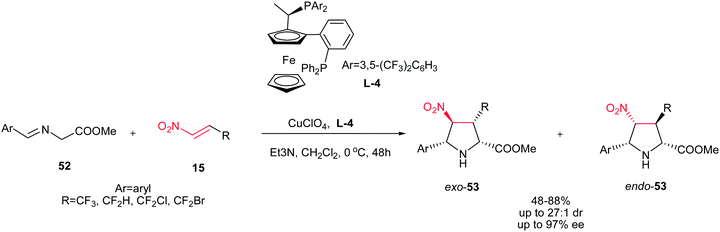
Also, the same group described that 3-(fluoromethyl)-4-nitroproline derivatives 53 can be prepared in high yields and stereoselectivities (exo:endo up to 27:1; up to 97% ee) from azomethine ylides 52 and fluoromethyl-substituted nitroalkenes 15 in the presence of copper(I) perchlorate and a commercially available chiral Walphos ligand L-4 (Scheme 18).33 They have also shown that the products can be facilely reduced with NaBH4/NiCl2 into the corresponding 4-amino-3-(fluoromethyl)proline derivatives with retention of the enantioselectivity.
 |
| | Scheme 18 Asymmetric synthesis of 3-(halogenated methyl)-4-nitroproline derivatives. | |
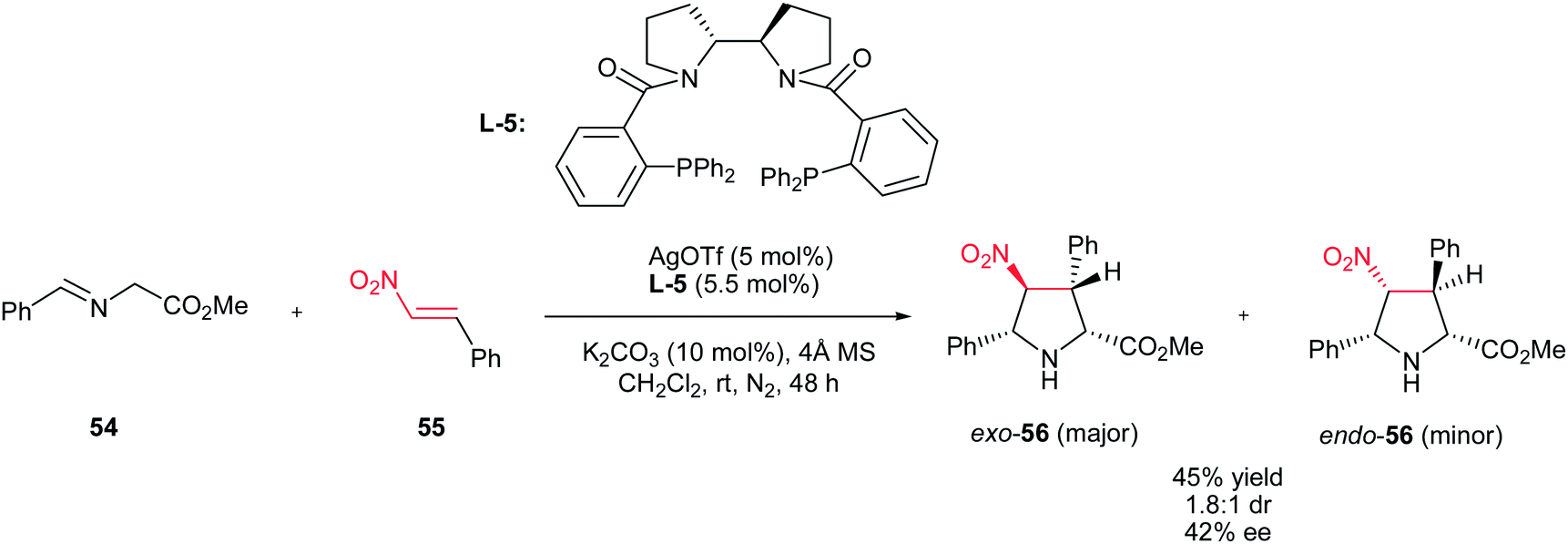
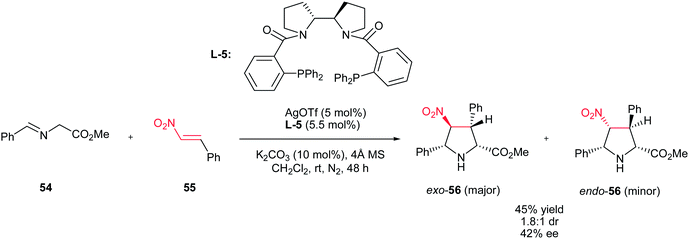
Recently, Xu et al. described that AgOTf (5 mol%) in combination with chiral bipyrrolidine-derived phosphine L-5 as ligand can also be applied for this transformation to give highly substituted pyrrolidines 56 in good yields with moderate diastereoselectivities and enantioselectivities (Scheme 19).34
 |
| | Scheme 19 Ag(I)/chiral bipyrrolidine-derived phosphine ligand for asymmetric [3 + 2] cycloaddition of azomethine ylide with nitroalkene. | |
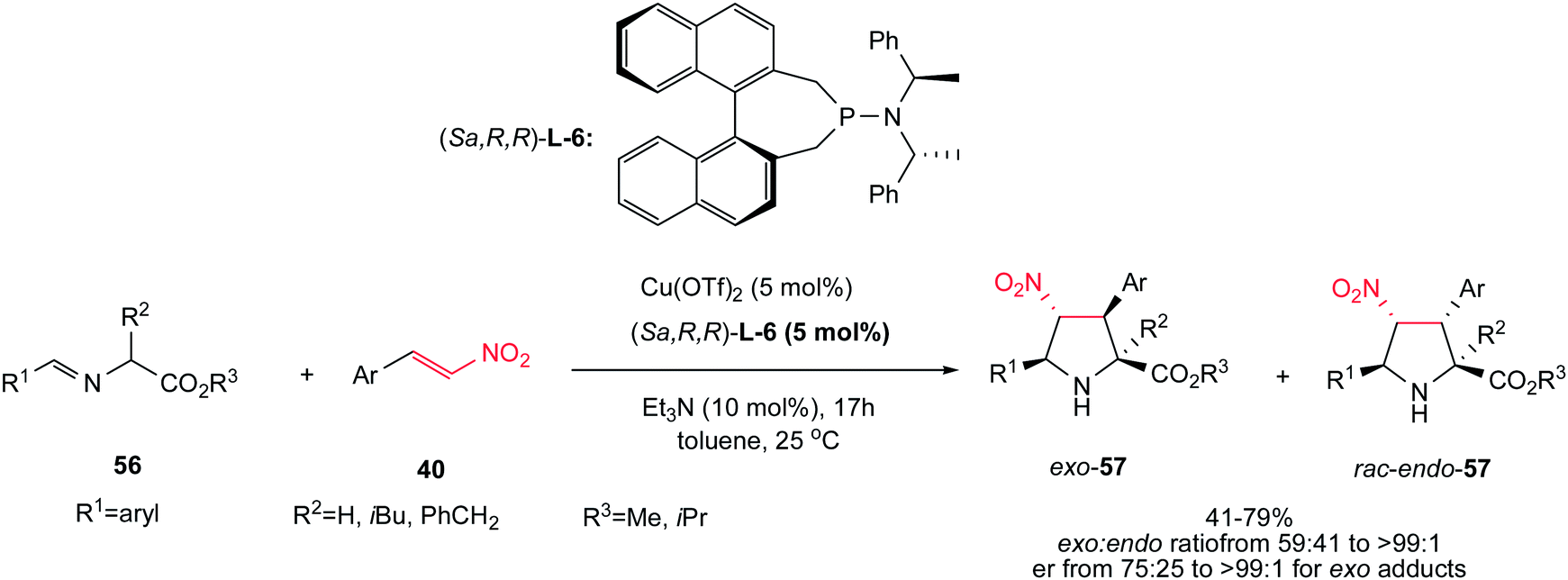
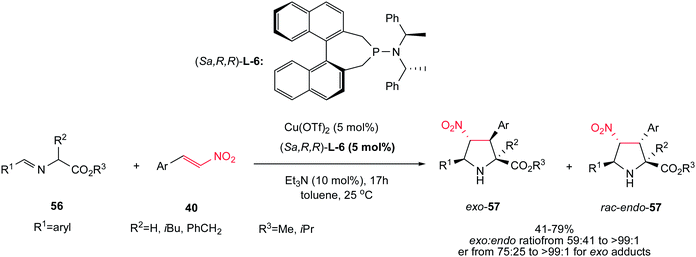
Another asymmetric protocol for synthesis of tetrasubstituted proline esters 57 is reported by Sansano and co-workers using Cu(OTf)2 as catalyst, (Sa,R,R)-phosphoramidite L-6 as chiral ligand and Et3N as external base.35 This chiral complex efficiently catalyzed the 1,3-dipolar cycloaddition reactions of azomethine ylides 56 and nitroalkenes 40 to give mainly the exo-cycloadducts exo-57 (up to >99:1) in high er (75:25 to >99:1) at room temperature (Scheme 20). According to the authors suggestion, obtaining the Michael adducts at low temperatures supported a stepwise mechanism in this protocol. Aromatic substituents in both components are compatible in this procedure. When the imino ester of leucine and phenylalanine were applied, enantiomerically enriched exo-cycloadducts (>99:1 ee) were obtained in moderate yields without further recrystallization. When the enantiomeric ligand (Ra,S,S)-L-6 was employed, the corresponding enantiomer of exo-57 was mainly isolated.
 |
| | Scheme 20 Cu(OTf)2 in combination with (Sa,R,R)-phosphoramidite for asymmetric synthesis of pyrrolidines. | |
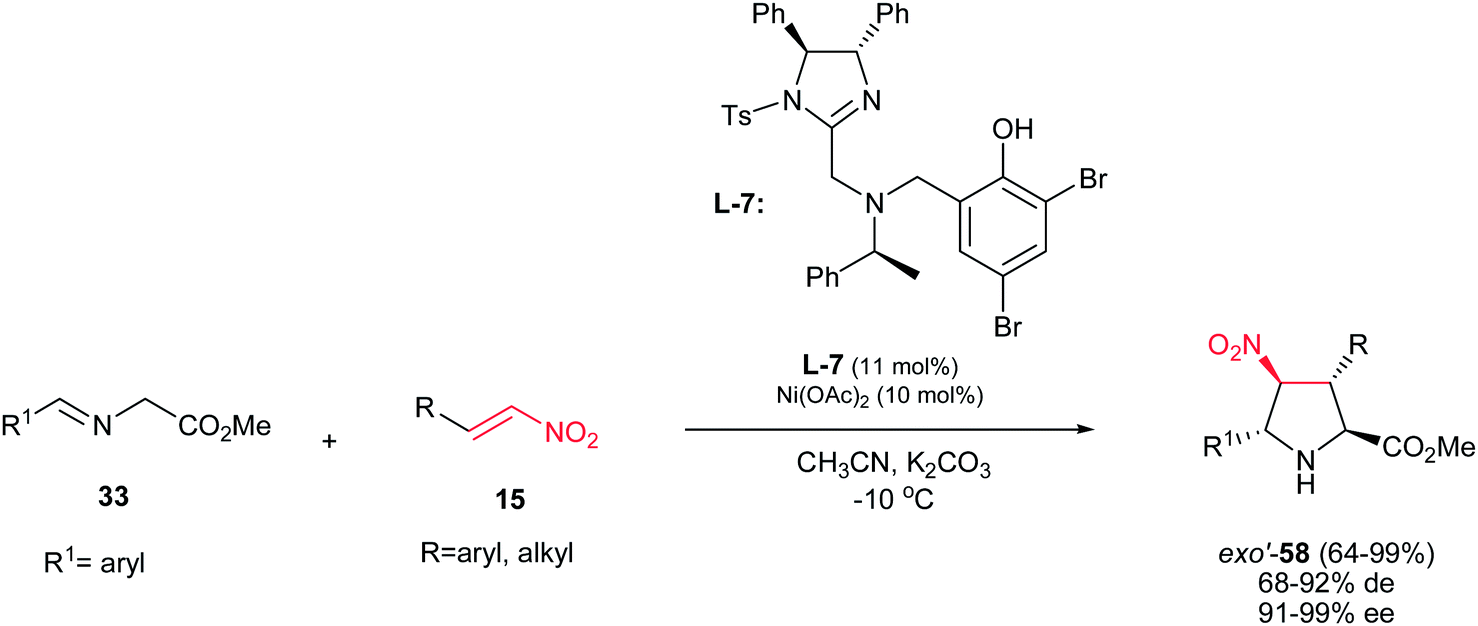
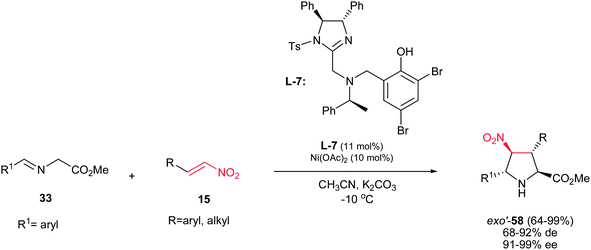
When a trans nitroalkene is used in the [3 + 2] cyclization, the stereoconjunction between the 3- and 4-positions is fixed in a trans conformation, and four diastereomers are possible, classified as endo, exo, endo′, and exo′ isomers. In contrast to previous reports, the first exo′-selective synthesis of pyrrolidine derivatives 58 is reported by Arai et al. by using the Ni(OAc)2 and ligand L-7 in the reaction of nitrostyrenes 15 and iminoesters 33. The products were obtained in high to excellent yields (64–99%), high diastereoselectivities (68–92%) and excellent ee's (91–99%) (Scheme 21).36
 |
| | Scheme 21 Ligand L-7/Ni(OAc)2-catalyzed exo′-selective [3 + 2] cycloadditions. | |
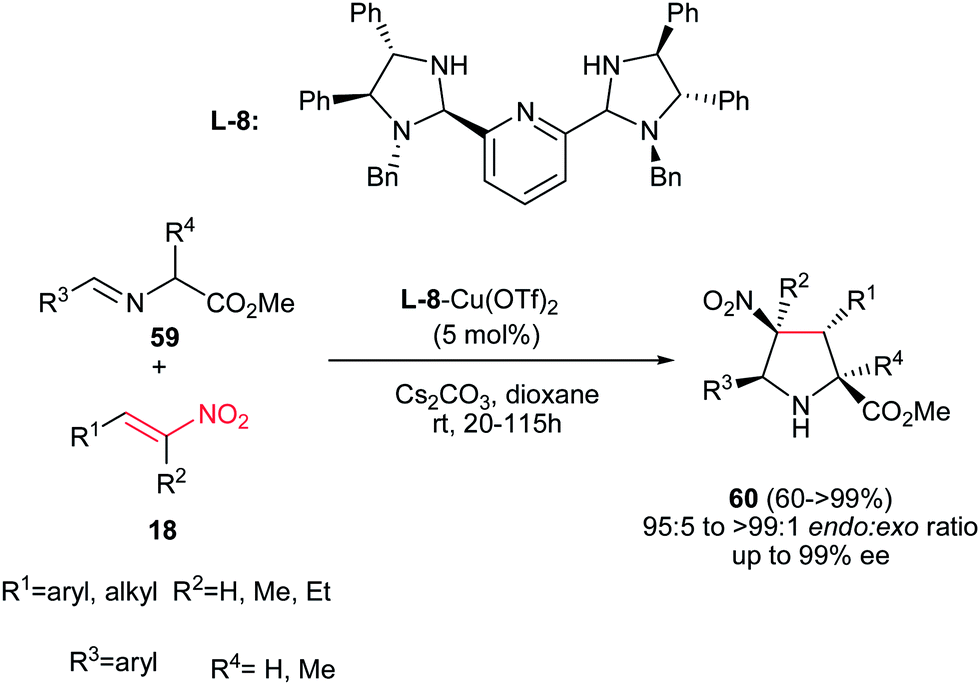
An endo-selective route for synthesis of pyrrolidine derivatives 60 was reported by Arai et al. via 1,3-dipolar cycloaddition of imino esters 59 with nitroalkenes 18 in the presence of bis(imidazolidine)pyridine (L-8)–Cu(OTf)2 complex and a basic additive. The best reaction conditions were examined using 5 mol% of L-8-Cu(OTf)2 chiral complex and Cs2CO3 in dioxane to afford the endo-selective pyrrolidines 60 in high to excellent yield.37 Electron-donating and –withdrawing substituents on the phenyl ring of both R1 and R3 were equally effective in this procedure. Aliphatic nitroalkenes also gave similar results. Furthermore, trisubstituted nitroalkenes were also well tolerated to give the corresponding products having chiral quaternary carbon centers in 99% ee. The chiral quaternary carbon center at the 2-position of the pyrrolidine ring was also generated using alanine-derived imino ester. The stereochemistry of products was assigned as (2S,3R,4S,5S) by using (S,S,S,S)-pyridine–Cu(OTf)2 catalyst (Scheme 22).
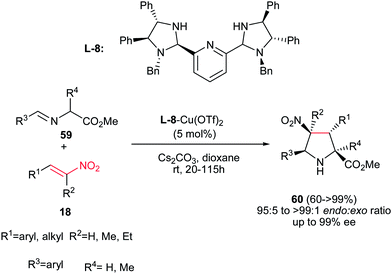 |
| | Scheme 22 Bis(imidazolidine)pyridine–Cu(OTf)2 complex promoted an endo-selective synthesis of pyrrolidines. | |
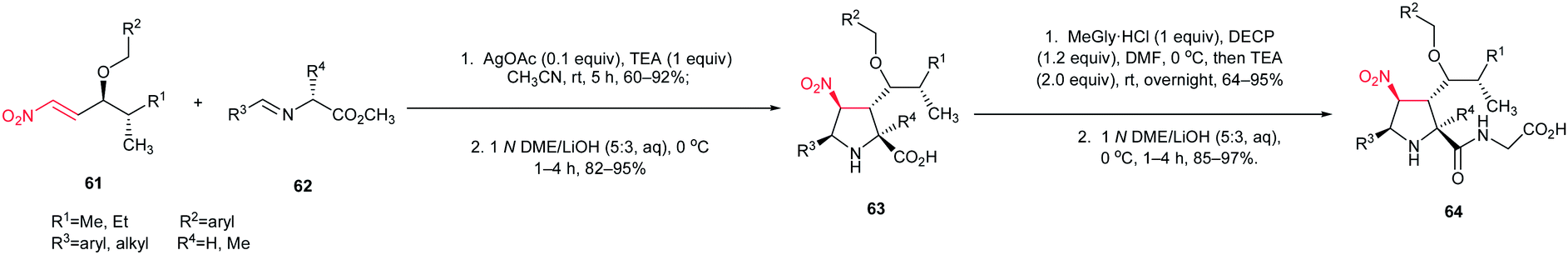
The [3 + 2] cycloaddition of azomethine ylides with nitroalkenes was successfully applied for total synthesis of inhibitors of α4β1-integrin-mediated hepatic melanoma metastasis 64 by Cossio and coworkers.38 As shown in Scheme 23, the key step in the synthetic route is the formal [3 + 2] cycloaddition between E-nitroalkenes 61 and imines 62 to yield pyrrolidines 63 which is simply converted to biologically active compound 64.
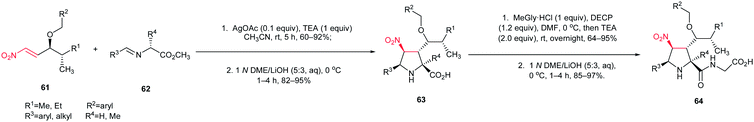 |
| | Scheme 23 Synthesis of inhibitors of α4β1-integrin-mediated hepatic melanoma metastasis 64 starting with a nitroalkene. | |
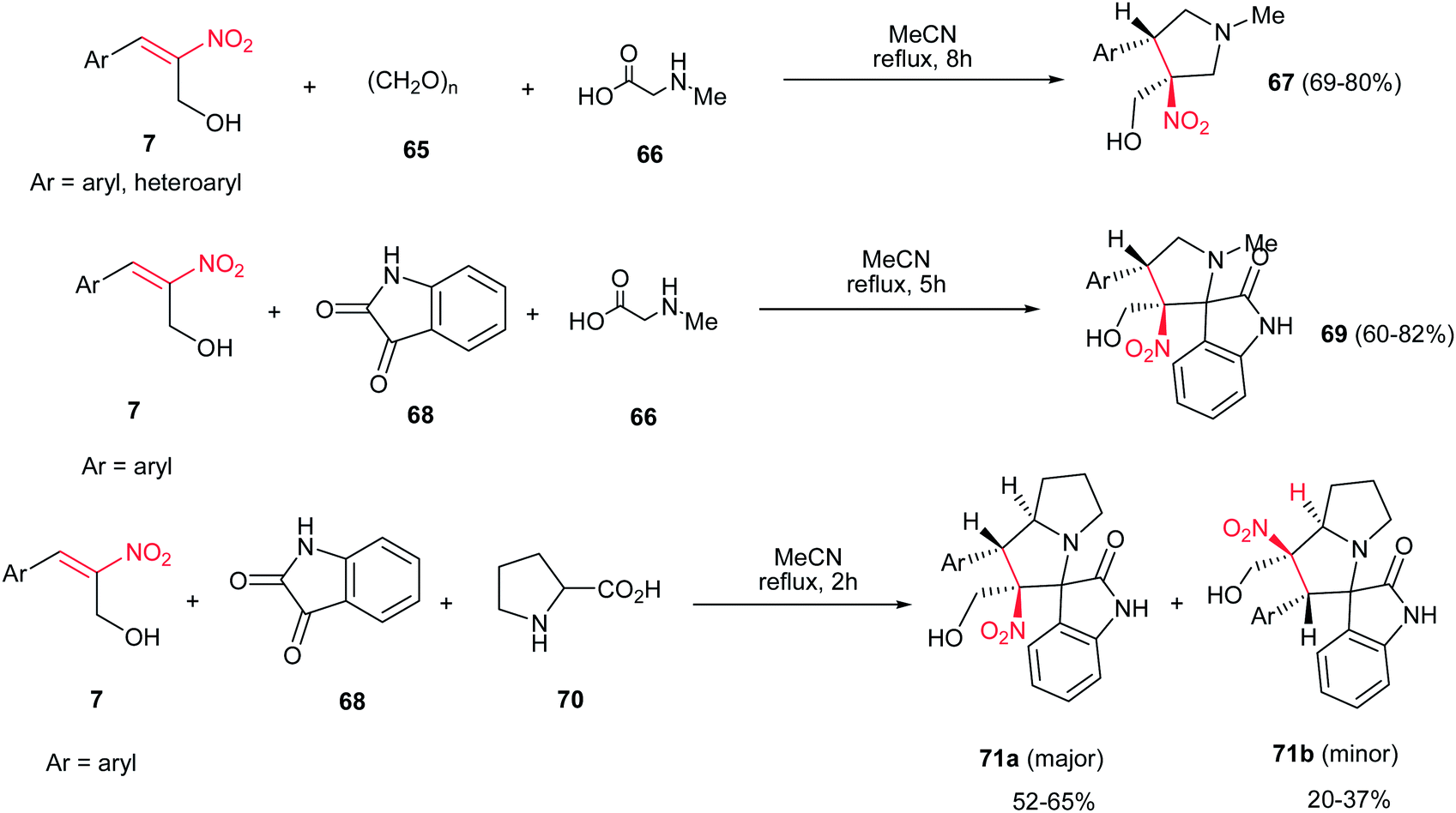
[3 + 2]-Cycloaddition reaction of (E)-2-nitro-3-phenylprop-2-en-1-ol 7, with dipoles generated from paraformaldehyde 65 and sarcosine 66 gave the desired N-methylpyrrolidines 67 in 69–80% yields under catalyst-free conditions in refluxing acetonitrile (Scheme 24).39 When isatin 68 was applied as carbonyl source, the corresponding desired 3-spiropyrrolidines 69 were obtained in very good yields (60–82%). In addition, the reaction of 7 with dipole generated from isatin 68 and proline 70 under the same reaction conditions provided the corresponding 3-spiropyrrolizidines 71a/b.
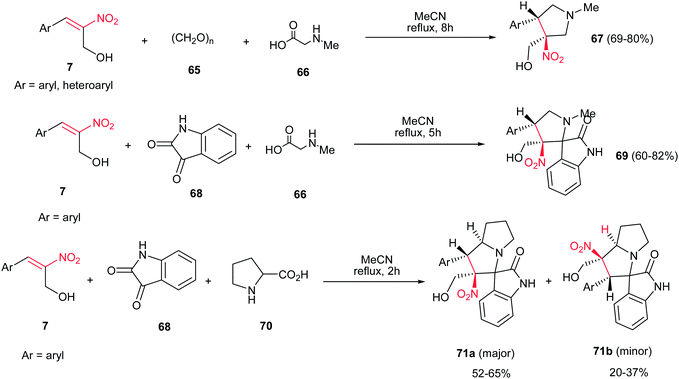 |
| | Scheme 24 (E)-2-Nitro-3-phenylprop-2-en-1-ol in [3 + 2]-cycloaddition reaction. | |
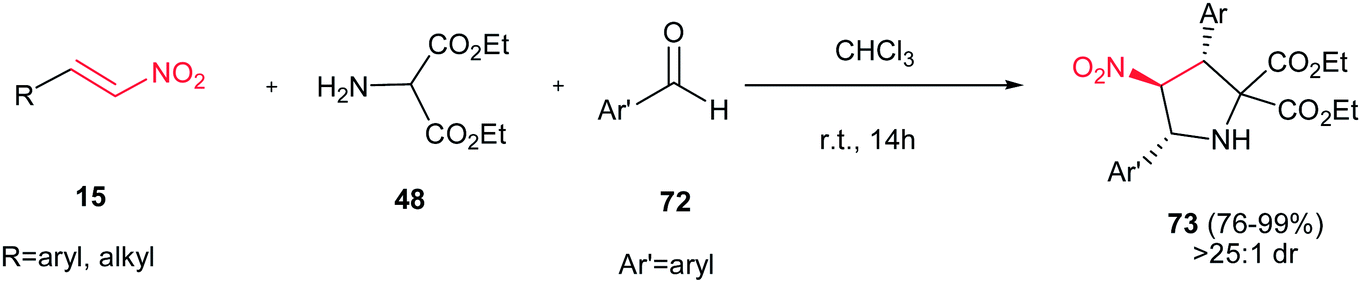
Rios and Crovetto reported another catalyst-free route toward synthesis of 2,3,4,5-tetrasubstituted pyrrolidine derivatives 73 in high yields (76–99%) and in high diastereoselectivities (>25:1) from nitroolefins 15, diethyl 2-aminomalonate 48, and different aromatic aldehydes 72 (Scheme 25).40 This is a simple and metal-free route for synthesis of pyrrolidine rings with three stereocenters. Surprisingly, the reaction in the presence of different additives such as triethylamine, thiourea, and chiral thioureas gave low diastereoselectivities.
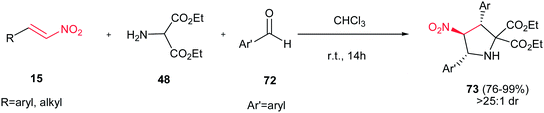 |
| | Scheme 25 Catalyst-free route toward synthesis of 2,3,4,5-tetrasubstituted pyrrolidines. | |
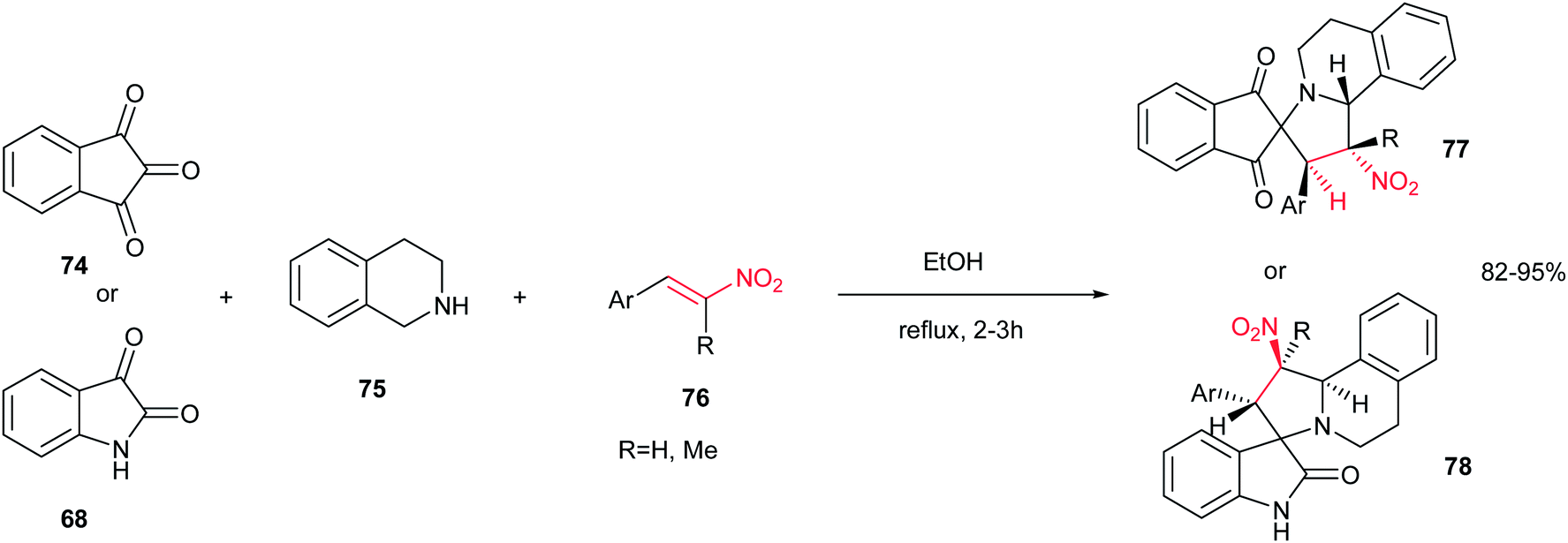
Also, Sarrafi et al. described that a one-pot endo-selective [1,3]-dipolar cycloaddition of nitrostyrenes 76 with 1,3-dipoles, generated in situ from isatin 68 or ninhydrin 74 with 1,2,3,4-tetrahydroquinoline 75, afforded a new series of spiroindolizidines 77–78 in high yield and stereoselectivity. The reaction was performed by stirring an equimolar amount of starting materials in ethanol at reflux temperature for 2–3 h (Scheme 26).41
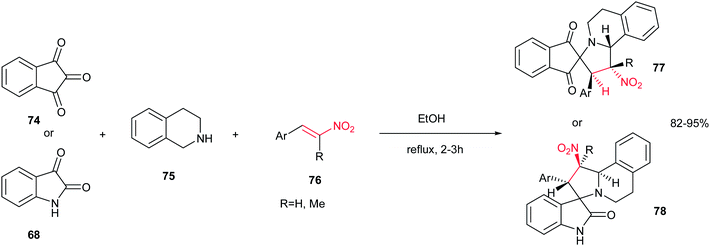 |
| | Scheme 26 Regioselective synthesis of spiroindolizidine. | |
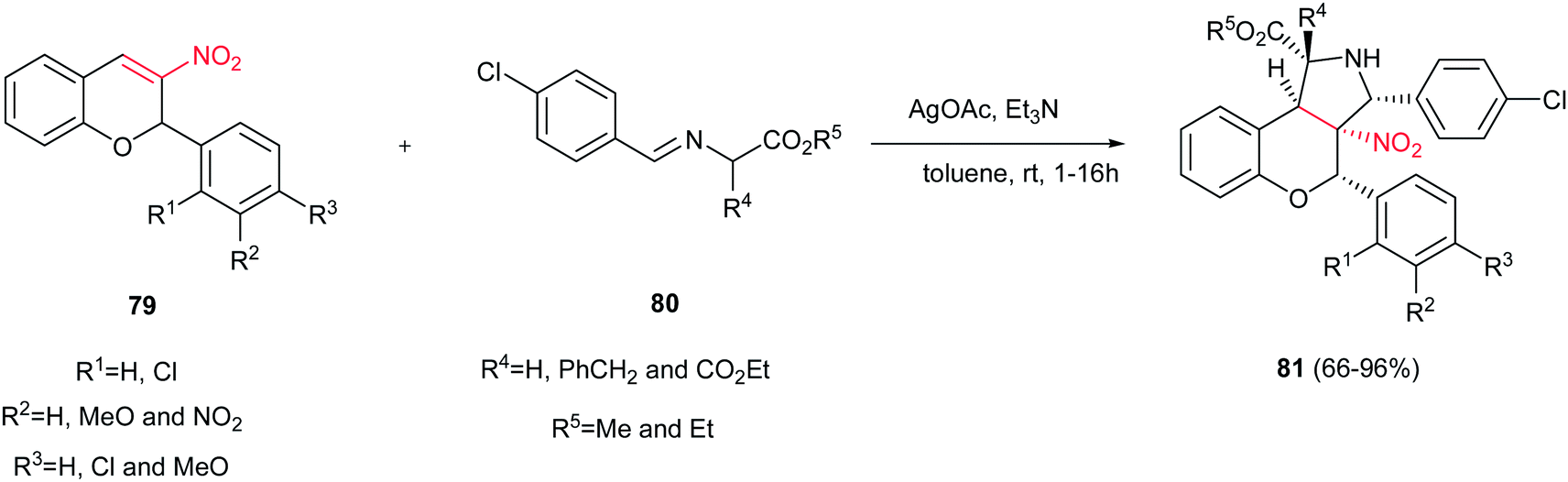
3-Nitrochromenes 79 were used as 2π components in the 1,3-dipolar cycloaddition reactions with various azomethine ylides 80 to achieve polysubstituted benzopyrano[3,4-c]-pyrrolidines 81 in the presence of AgOAc/Et3N as catalyst (Scheme 27).42 It is notable that the one-pot three-component reactions of 3-nitrochromenes, sarcosine or N-benzylglycine and aldehydes also afforded the corresponding cycloaddition products in refluxing toluene in high to excellent yields.
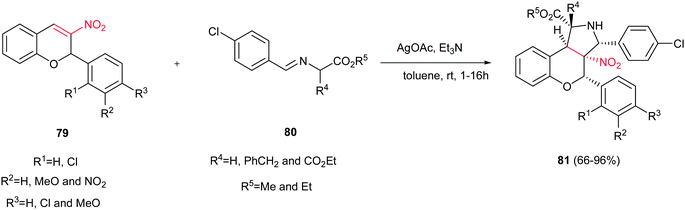 |
| | Scheme 27 3-Nitrochromenes were used as 2π components in the 1,3-dipolar cycloaddition reactions with various azomethine ylides. | |
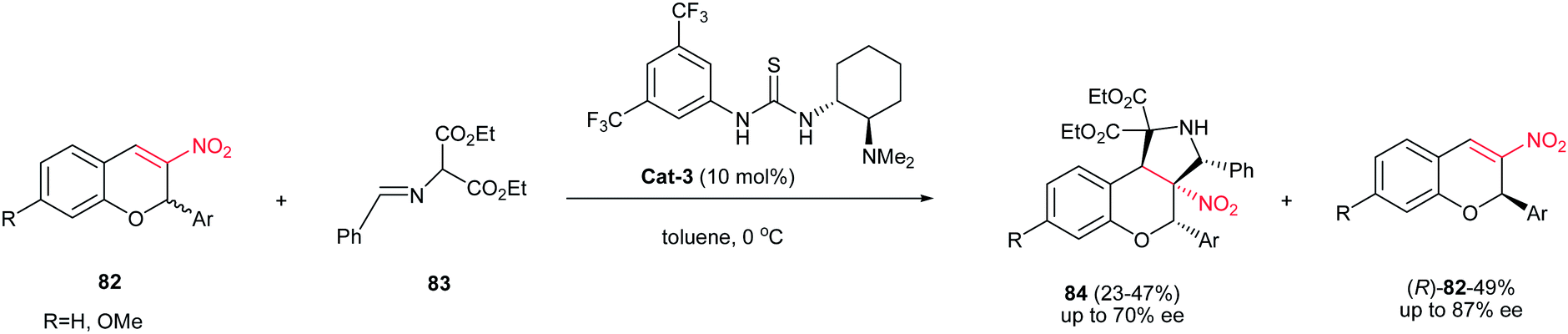
Recently, Xie et al. demonstrated that the kinetic resolution of 3-nitro-2H-chromenes 82 catalyzed by Takemoto's bifunctional chiral thiourea Cat-3 afforded the enantioenriched (R)-3-nitro-2H-chromene derivatives (R)-82 with moderate- to -good enantioselectivities (≤87% ee), along with the corresponding fused functionalized pyrrolidines 84 possessing four vicinal chiral carbon centers, with moderate enantioselectivities (≤70% ee), as shown in Scheme 28.43
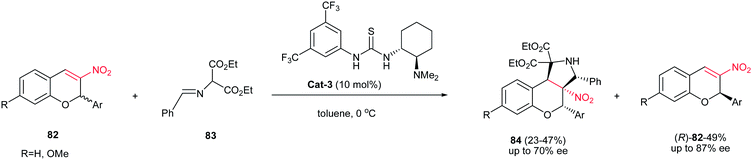 |
| | Scheme 28 Kinetic resolution of 3-nitro-2H-chromenes catalyzed by Takemoto's bifunctional chiral thiourea. | |
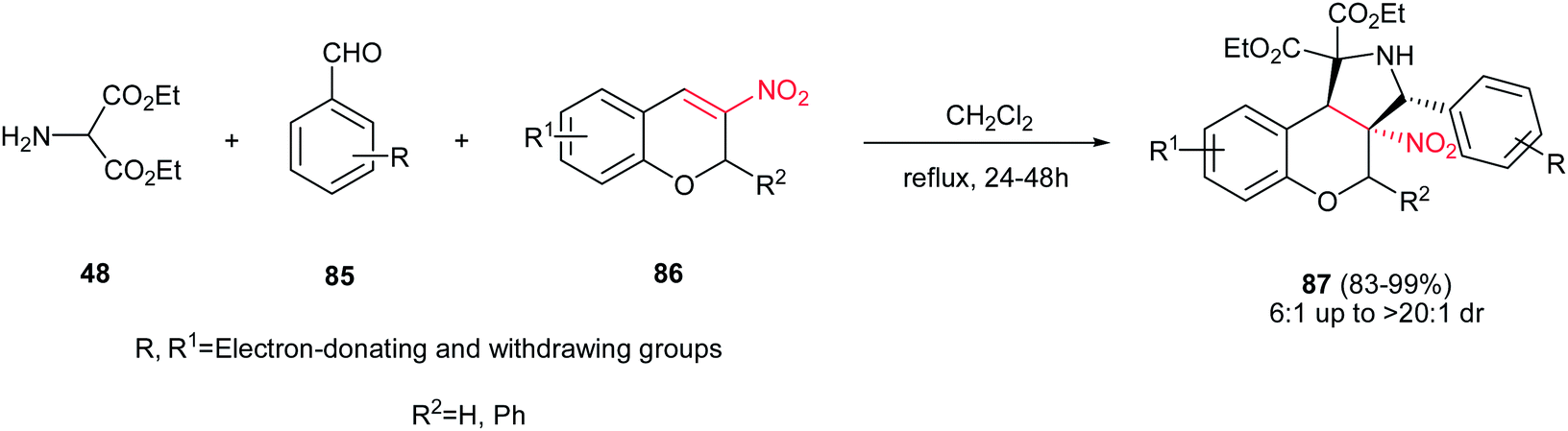
Finally, Du et al. reported a one-pot 1,3-dipolar cycloaddition of diethyl 2-aminomalonate 48, benzaldehyde derivatives 85 and 3-nitro-2H-chromenes 86 in refluxing CH2Cl2 to give polysubstituted benzopyrano [3,4-c]-pyrrolidines 87 in excellent yields (83–99%) and high diastereoselectivities (up to >20:1) under catalyst-free conditions (Scheme 29).44 It seems that the electronic and steric factors of substituent on the starting materials didn't have significant influence on the reaction yield. The configuration of the major isomer of products was determined with X-ray crystallographic analysis as cis for NO2 group and benzene ring.
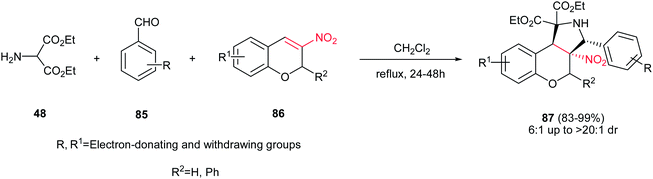 |
| | Scheme 29 One-pot 1,3-dipolar cycloaddition of diethyl 2-aminomalonate, benzaldehydes and 3-nitro-2H-chromenes. | |
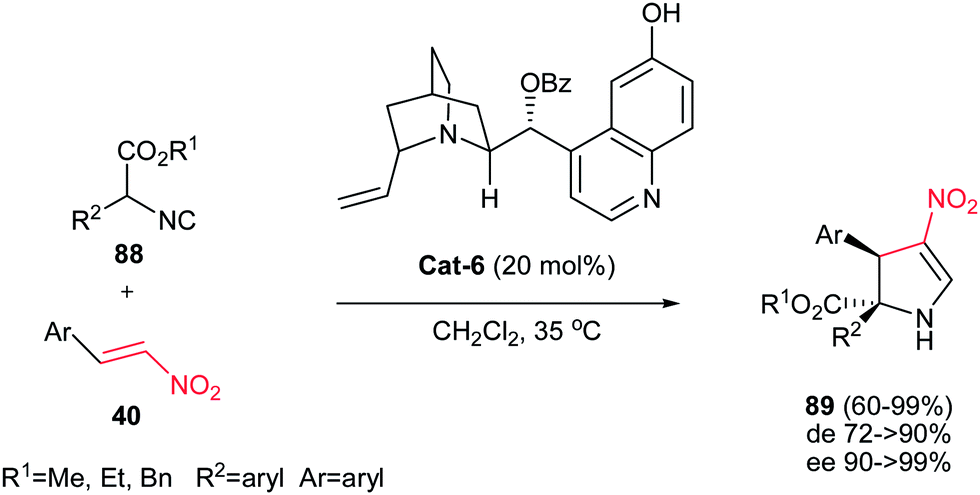
4.1.2. Dihydropyrrole derivatives. There are three kinds of pyrrolines: 1-pyrrolines, 2-pyrrolines and 3-pyrrolines. There are numerous naturally and synthetically occurring biologically active compounds containing these pyrrolines.45 Pyrrolines are versatile synthetic intermediates for synthesis of diversity of biologically active compounds especially pyrroles and pyrrolidines.46 Several methods for construction of pyrroline derivatives were reported including 1,3-dipolar cycloaddition,47 the reaction of α,β-diketones with acetamides,48 ring-closing metathesis of diallyl amines,49 nucleophilic addition of organometallic reagents,50 selective oxidation or reduction of pyrrolinones and maleimide derivatives,51 transition-metal-catalyzed coupling, oxidative or tandem process,52 domino Heck-aza-Michael53 and organocatalytic tandem Michael/cyclization sequence.54Gong et al. reported an asymmetric tandem Michael-CH insertion process (formal [3 + 2] cycloaddition) for the synthesis of dihydropyrroles from nitroolefins and α-aryl isocyanoacetates catalyzed by 20 mol% of cinchona alkaloid Cat-6.55 The corresponding chiral 2-pyrrolines were obtained in excellent enantioselectivity (90 to >99% ee) and useful diastereoselectivity. Several alkyl- and aryl-substituted nitroalkenes were examined and the best diastereoselectivity (up to 20:1) was obtained with electron-deficient aryl substituents (Scheme 30).
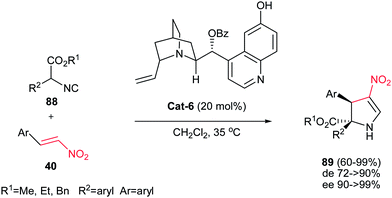 |
| | Scheme 30 [3 + 2] Cycloaddition of α-substituted isocyanoesters and nitroolefins catalysed by cinchona alkaloid. | |
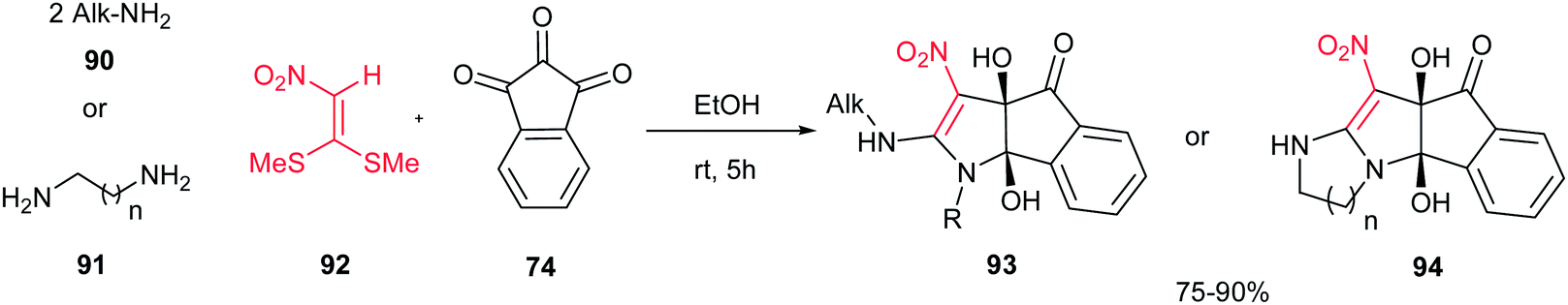
In 2011, an efficient approach for the synthesis of dihydroindeno[1,2-b]pyrroles 93 and indeno[2′,1′:4,5]pyrrolo[1,2-a]-fused 1,3-diazaheterocycles 94 was reported by Alizadeh et al. via a new and one-pot reaction between primary amines 90 or 1,n-diamines 91, 1,1-bis-(methylthio)-2-nitroethene 92 and ninhydrin 74 in aqueous ethanol under mild conditions (Scheme 31).56 The merits of this protocol are access to fairly high yields of the products without any catalyst, the ready availability of the starting materials, simple reaction conditions such as aqueous media at room temperature.
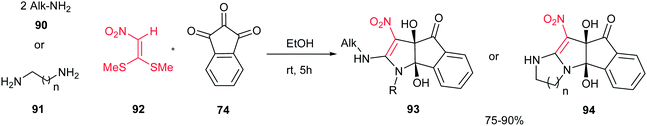 |
| | Scheme 31 Synthesis of dihyroindeno[1,2-b]pyrroles and indeno[2′,1′:4,5]pyrrolo[1,2-a]-fused 1,3-diazaheterocycles. | |
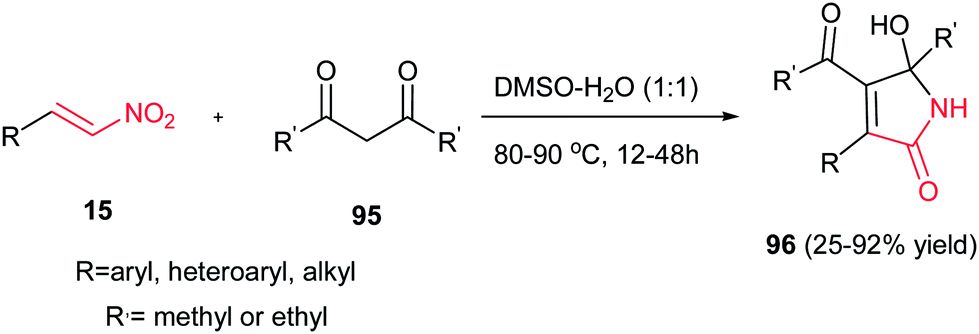
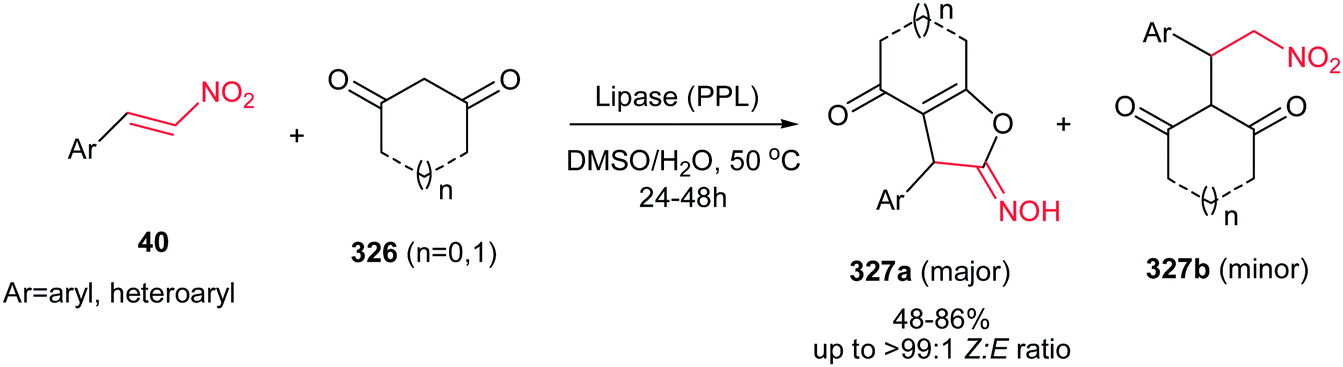
Usually, the reaction of 1,3-dicarbonyl compounds with nitroethylenes gave the Michael adducts and the hydroxyimino-substituted dihydrofuran derivatives. Surprisingly, Yu et al. demonstrated that heating the starting materials 15 and 95 in DMSO–H2O (1:1) at 80–90 °C for 12–48 h under catalyst-free conditions affords 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones 96 as major products (Scheme 32).57 They confirmed that aromatic nitroalkenes gave higher yields compared to aliphatic nitroalkenes.
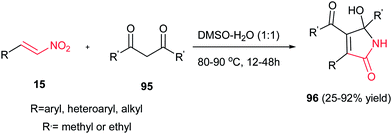 |
| | Scheme 32 A novel process for the synthesis of 5-hydroxy-1,5-dihydro-2H-pyrrol-2-ones. | |
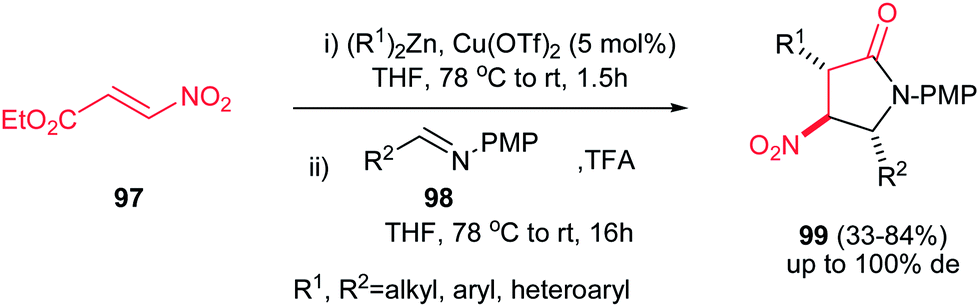
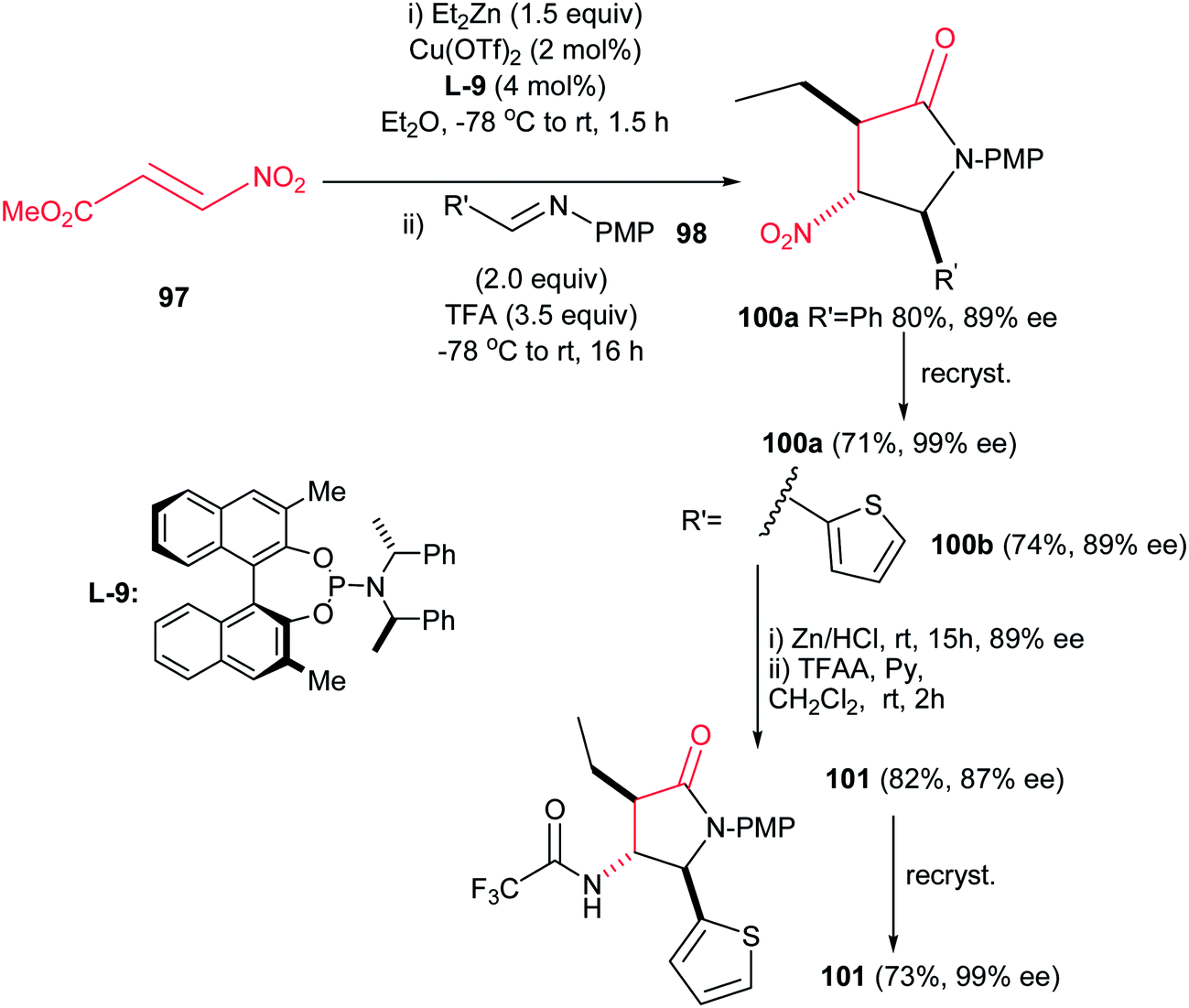
4.1.3. Pyrrolidine-2-one and pyrroloindoles. Compounds containing the 2-pyrrolidinone (γ-lactam) rings have found significant applications in the treatment of epilepsy,58 HIV,59 viral hepatitis,60 Alzheimer,61 neurodegenerative diseases and depression.62 Among the methods reported for the synthesis of 2-pyrrolidinones, intramolecular amide bond formation,63 expansion or contraction of a previously formed ring,64 radical cyclization,65 ring-closing metathesis,66 cycloaddition reactions,67 condensation of imines and cyanosuccinic anhydride68 and reaction of N-arylidene-N-alkylamines with succinic anhydride are the most applicable approaches.69Recently, Anderson et al. reported an efficient procedure for synthesis of 1,3,5-trisubstituted 4-nitropyrrolidin-2-ones 99 via Cu(OTf)2-catalyzed Michael addition of diorganozinc reagents to nitroacrylate 97 followed by a subsequent aza-Henry/lactamization reaction with N-p-(methoxy)phenyl protected aldimines 98 (Scheme 33).70 Different N-p-(methoxy)phenyl protected aldimines 98 derived from alkyl, aryl, and heteroaryl aldehydes were examined with high to excellent yields. Using the more electron-withdrawing protecting group such as N-Boc prevents the cyclization step due to the decreased availability of the nitrogen loan pair in the β-nitro-amine product. Also an enantioselective version of this reaction was performed in the presence of chiral BINOL ligand L-9 and Cu(OTf)2 to give the corresponding product 100 in high yield (74–80%) and enantioselectivity (89% ee) which could be recrystallized to afford enantiopure product in high yields (99% ee) (Scheme 34). Also, reduction of the nitro group and its protection was also performed to give the compound 101 in high yields.
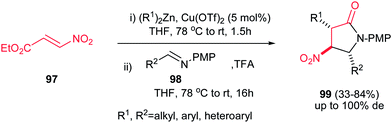 |
| | Scheme 33 Synthesis of 1,3,5-trisubstituted 4-nitropyrrolidin-2-ones. | |
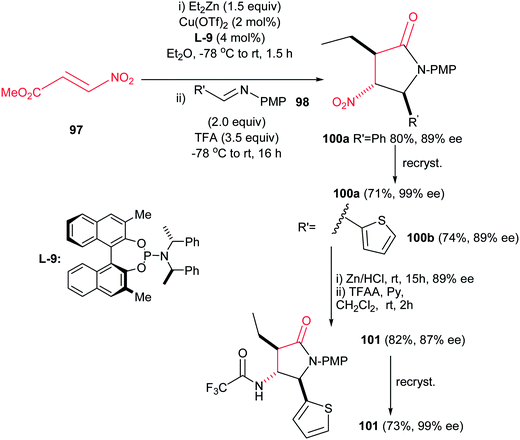 |
| | Scheme 34 Enantioselective conjugate addition/nitro-Mannich/lactamization reactions. | |
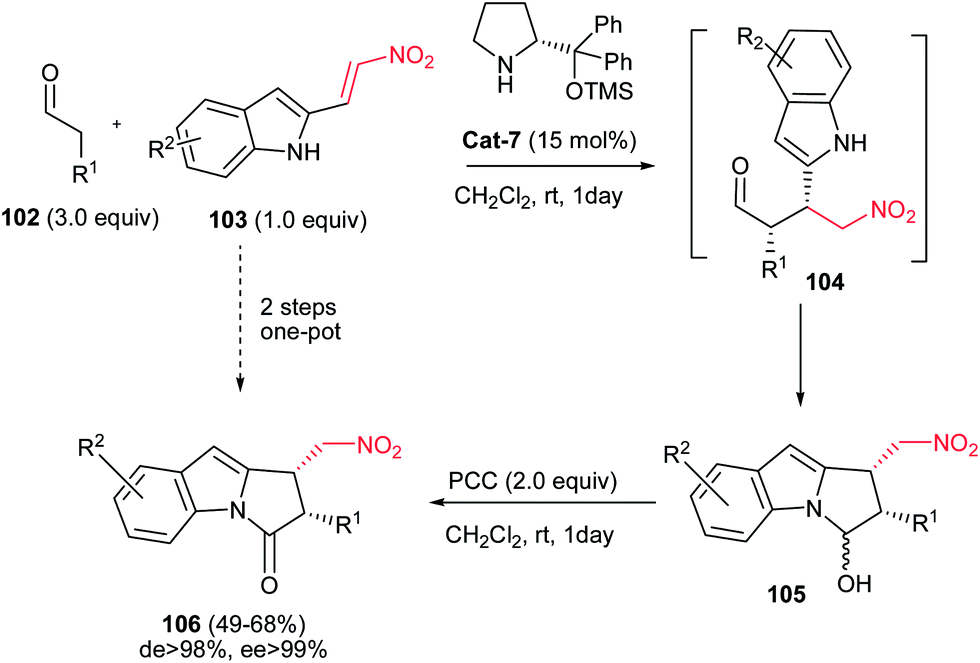
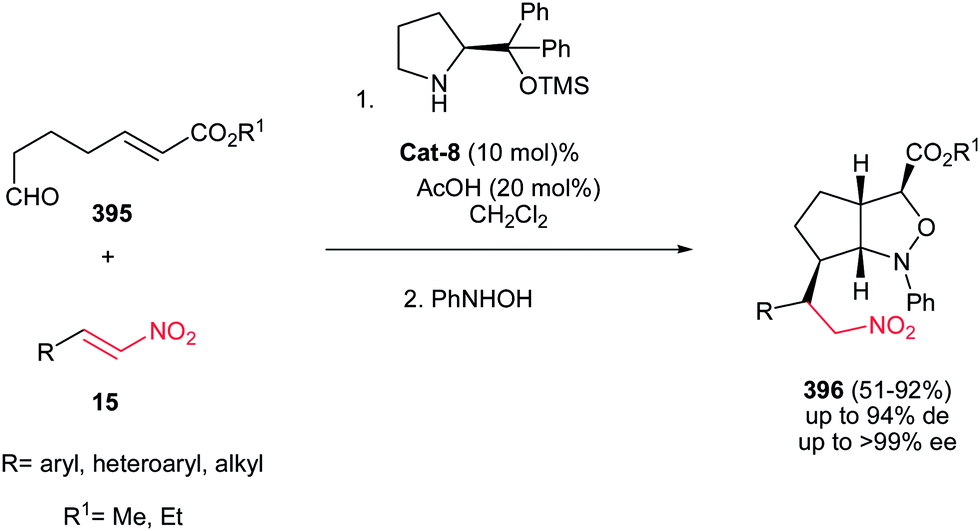
Also, 1,2-cis-disubstituted 1H-pyrrolo[1,2a]indol-3(2H)-ones 106 have been synthesized in moderate to good overall yields (49–68%) by Enders et al. via a one-pot Michael addition–hemiaminalization–oxidation reaction employing simple aldehydes 102 and 2-nitrovinyl-substituted indoles 103 as substrates (Scheme 35).71 This process is promoted by 15 mol% of (R)-diphenylprolinol TMS-ether Cat-7 and AcOH (20 mol%) as additive with very high asymmetric induction to produce the stereoisomerically pure products (>98% de, >99% ee).
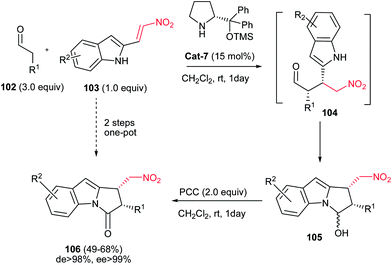 |
| | Scheme 35 Asymmetric synthesis of pyrrolo[1,2a]indolones from aldehyde and 2-nitrovinyl 1-substituted indoles. | |
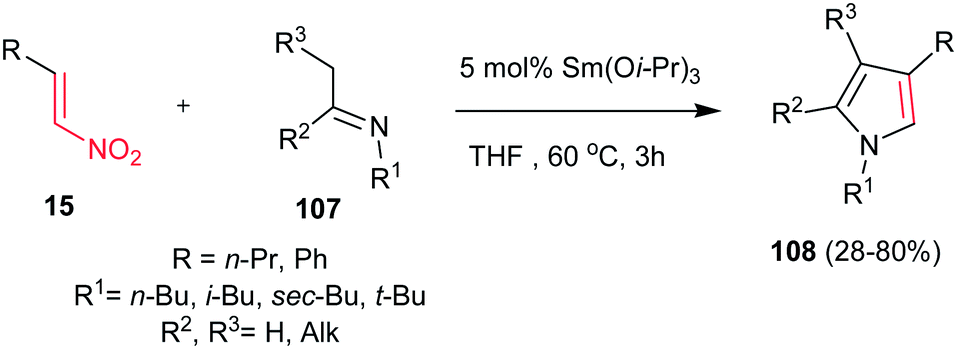
4.1.4. Pyrrole derivatives. Pyrroles are an important class of heterocycles present in the structure of many natural and synthetic bioactive molecules72 with a wide range of applications in medicinal chemistry as antibacterial,73 antiviral,74 anti-inflammatory,75 antitumour,76 antioxidant77 and antifungal78 agents. Also, recently pyrroles have found extensive applications in the field of materials chemistry and as structural elements in molecular recognition studies.79 As a result, a large number of methods have been reported for synthesis of pyrroles. Classical methods for synthesis of these nitrogen heterocycles include the Knorr,80 Hantzsch,81 and Paal–Knorr82 reactions. Several other useful routes such as 1,3-dipolar cycloadditions of acetylenes with azomethine ylides,83 metal-catalyzed,84 reductive coupling,85 aza-Wittig86 and multicomponent reactions87 were also developed for synthesis of highly substituted pyrroles.Nitroalkenes were proved to be efficient starting materials for synthesis of pyrroles with different substitution pattern. In this context, the Ishii group reported a new approach to various tri- and tetrasubstituted pyrrole derivatives 108 from nitroalkenes 15 and imines 107 with using 5 mol% of Sm(Oi-Pr)3 in refluxing THF (Scheme 36).88 The nitro-group plays a double role in this transformation as activator of the alkene moiety toward Michael addition and as a leaving group during the aromatization step.
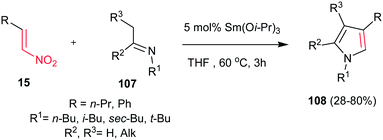 |
| | Scheme 36 Sm(Oi-Pr)3-promoted synthesis of functionalized pyrroles from imines and nitroalkenes. | |
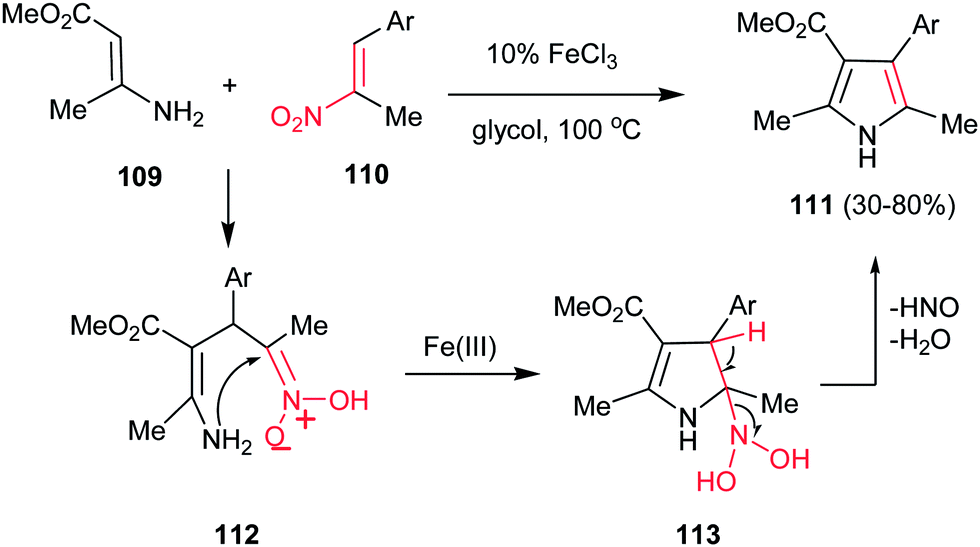
Guan et al. reported a simple and efficient procedure for synthesis of 2,3,4,5-tetra-substituted pyrroles 111 in good yields from enamine 109 and nitroalkenes 110 promoted by FeCl3 (Scheme 37).89 According to the proposed mechanism, the reaction proceeds via Michael addition of 109 to 110 to furnish the adduct 112, which undergoes cyclization into intermediate 113. Finally elimination of HNO and water provides tetrasubstituted pyrroles 111 bearing a variety of aryl substituents at the C4 atom. The one-pot four-component fashion of this work is also developed by Jana and co-workers with using FeCl3 as catalyst90 and by Pal and co-workers with using (PPh3)2PdCl2.91 They have used a nitroalkane and a carbonyl group instead of nitroalkenes.
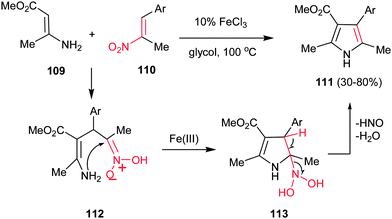 |
| | Scheme 37 FeCl3-catalyzed synthesis of functionalized pyrroles from enamines and nitroalkenes. | |
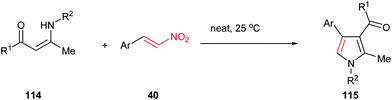
In addition, an eco-friendly procedure for synthesis of 1,2,3,4-tetrasubstituted pyrroles 115 from enaminones 114 and β-nitrostyrenes 40 under solvent- and catalyst-free conditions is developed by Yavari et al. The products were obtained in high to excellent yields (Scheme 38).92 Very recently, these products were also prepared in 68–93% yield by using Ph3PAuCl and AgOTf in methanol. This protocol tolerated enamines derived from both aliphatic and aromatic amines.93
 |
| | Scheme 38 A catalyst-free procedure for synthesis of 1,2,3,4-tetrasubstituted pyrroles. | |
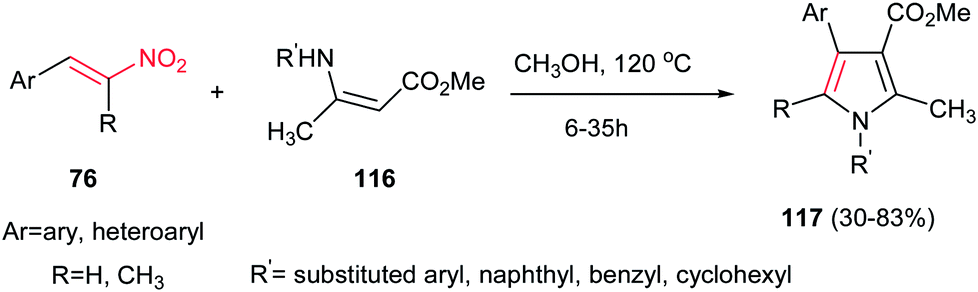
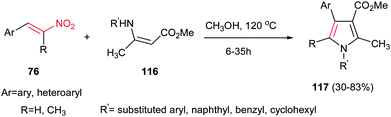
Another environmentally benign procedure for synthesis of fully substituted pyrroles 117 is reported by Guan et al. via condensation of nitroalkenes 76 and enaminoesters 116 in CH3OH at 120 °C without using any catalyst (Scheme 39).94 Diversities of nitroalkenes and enaminoesters were applied in this protocol to give the corresponding products in high to excellent yields. This protocol allows preparation of N-aryl, benzyl or cyclohexyl substituted pyrroles in high to excellent yields. The authors confirmed that electron-rich enaminoesters and nitroalkenes gave better results compare to electron-deficient ones. Also, enaminones gave lower yield than enaminoesters under similar conditions.
 |
| | Scheme 39 Catalyst-free synthesis of fully substituted pyrroles from nitroalkenes and enaminoesters. | |
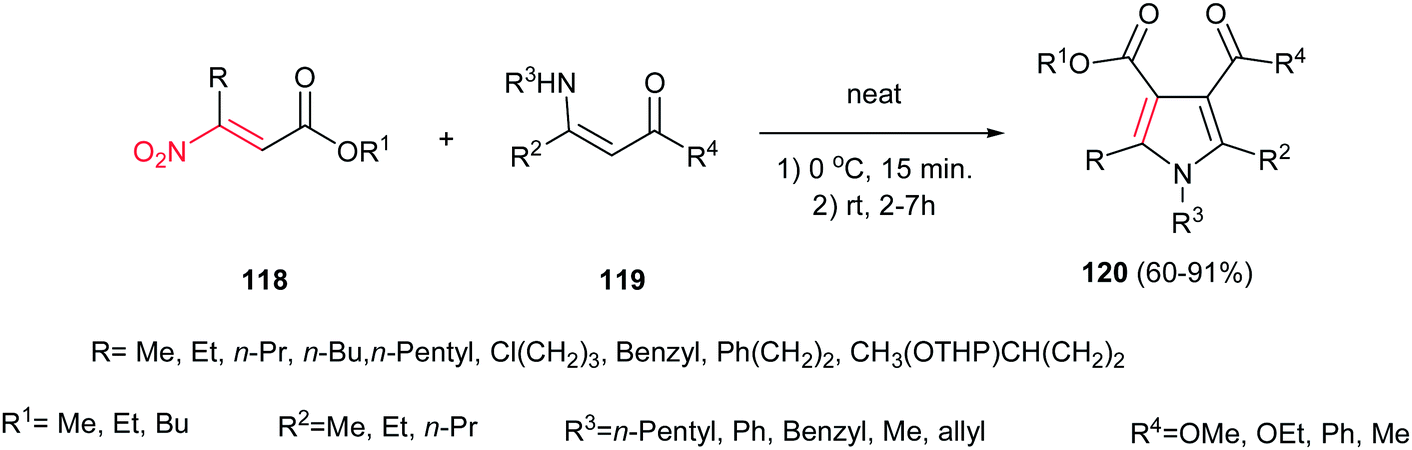
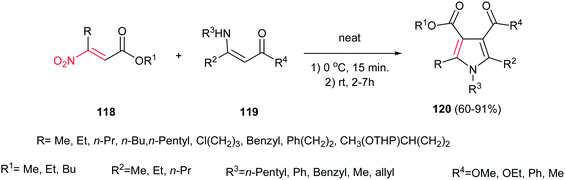
In this context, Ballini et al. described another green procedure for synthesis of pyrroles 120 in high to excellent yield with acceptable E-factor from β-nitroacrylates 118 and β-enaminones 119 under solvent- and catalyst-free conditions (Scheme 40).95 They examined that varieties of nitroacrylates with different alkyl substituents are tolerated in this protocol. Also, this procedure gave higher yield for enaminones compared to previous one.
 |
| | Scheme 40 Synthesis of pyrroles from β-nitroacrylates and β-enaminones under solvent- and catalyst-free conditions. | |
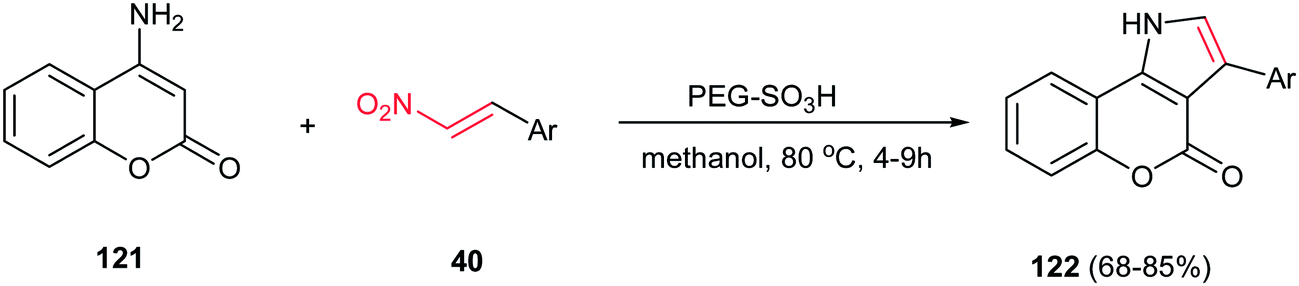
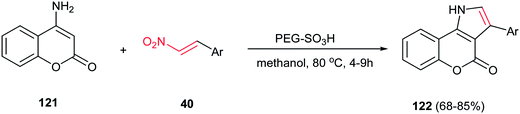
In 2012, Paul and Das described the coupling of nitroalkenes 40 with 4-aminocoumarin 121 to give the coumarin fused pyrrole derivatives 122 in high yields using PEG–SO3H, as a biodegradable, polymer supported catalyst in methanol at 80 °C (Scheme 41).96 The catalyst efficiently promoted the Michael addition and intramolecular cyclisation with the concomitant removal of the nitro group. The catalyst was recycled for five times without loss of catalytic activity. Also a wide variety of Lewis or Bronsted acids such as ZnCl2, AlCl3, I2, FeCl3, InCl3, p-toluenesulphonic acid were used, but the results were less impressive when compared to PEG–SO3H catalyzed reaction. This approach can be also applied to 6-aminouracil for synthesis of the corresponding uracil fused pyrroles. In addition, they proved that three component coupling reaction of 4-aminocoumarin, benzaldehyde and nitromethane using nitromethane as solvent is not very much efficient.
 |
| | Scheme 41 PEG–SO3H promoted synthesis of pyrrole core containing coumarins. | |
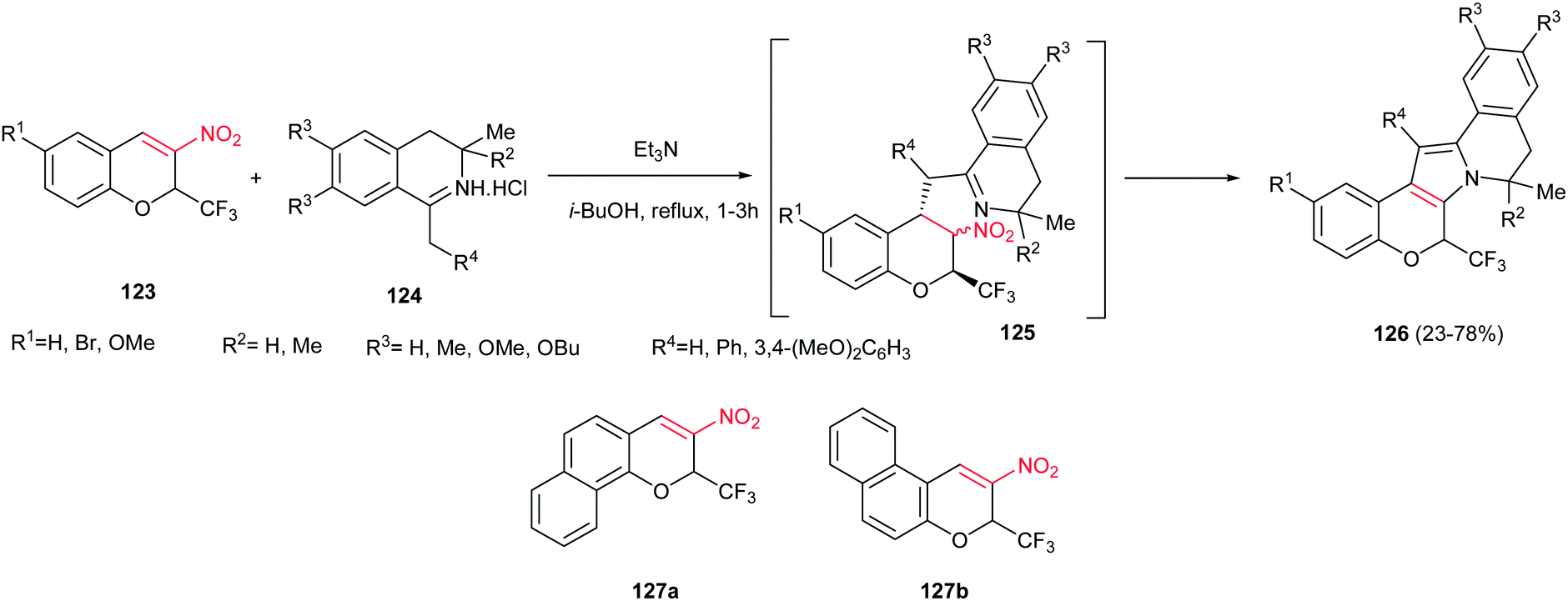
Sosnovskikh and co-workers reported an efficient approach for synthesis of 8,9-dihydro-6H-chromeno[4′,3′:4,5]pyrrolo[2,1-a] isoquinoline derivatives 126, as main skeleton of lamellarin alkaloids, from the reaction of 3-nitro-2-(trifluoromethyl)-2H-chromenes 123 and 1-methyl(benzyl)-3,4-dihydroisoquinolines 124 (Scheme 42).97 Two routes were developed for this transformation; the first path is mixing of the starting materials in toluene at room temperature to give the Michael adducts 125 and refluxing the isolated Michael adducts in isobutanol, and the second procedure is direct refluxing the starting materials in isobutanol. The first route gave higher yield compared to second one. With replacing the CF3 group with CCl3 or aryl groups, the Michael adducts were isolated as diastereomeric mixtures even in refluxing isobutanol. When benzofused chromenes 127a,b were used in the reaction with 124 (R4 = 4,5-(MeO)2C6H3), only 127a gave the desired product in 94% yield and no product was obtained with 127b, which may be the result of unfavourable steric interactions between the R4 and benzene ring in 127b.
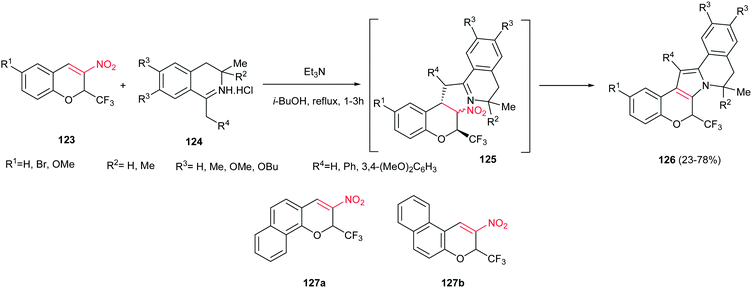 |
| | Scheme 42 Synthesis of the pentacyclic lamellarin skeleton. | |
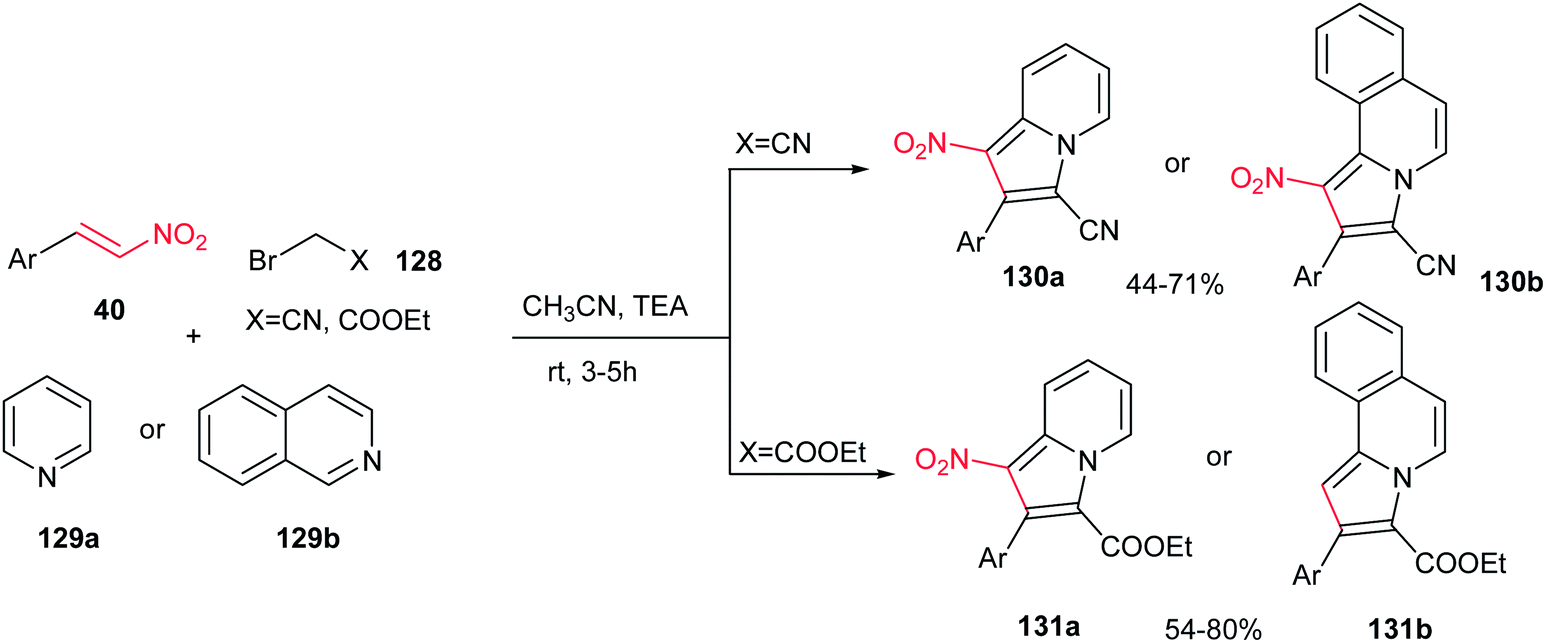
A one-pot three-component reaction of pyridinium/isoquinolinium ylide, generated in situ from pyridine/isoquinoline 129a/b with bromoacetonitrile or ethyl bromoacetate 128, with β-nitrostyrenes 40 in the presence of Et3N at ambient temperature is investigated by Perumal et al. (Scheme 43).98 They ascribed that while 128 (X = CN) afforded the 1-nitro-2-aryl-3-indolizine carbonitriles 130a and 1-nitro-2-arylpyrrolo[2,1-a]isoquinoline-3-carbonitrile 130b via reaction with pyridine and isoquinoline, respectively, the 128 (X = CO2Et) provided the ethyl 2-aryl-1-nitroindolizine-3-carboxylates 131a and ethyl 2-arylpyrrolo[2,1-a]isoquinoline-3-carboxylates 131b. Different bases such as Et3N, DBU, DMAP, piperidine and K2CO3 were examined in this procedure and the maximum yields (44–80%) were obtained employing a molar equivalent of Et3N. Nitrostyrenes with different substitutions on the phenyl ring were used successfully in this reaction. It is very interesting to note that other regioisomers were not observed. Notably, these products displayed good activity against Mycobacterium tuberculosis H37Rv.
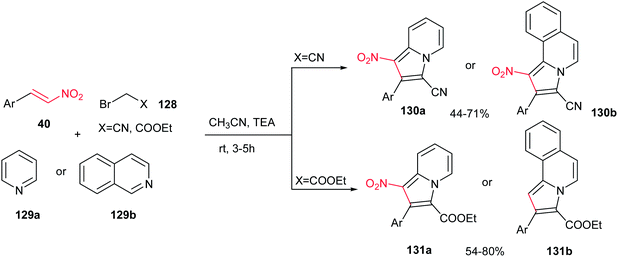 |
| | Scheme 43 Synthesis of indolizines and pyrrolo[2,1-a]isoquinolines. | |
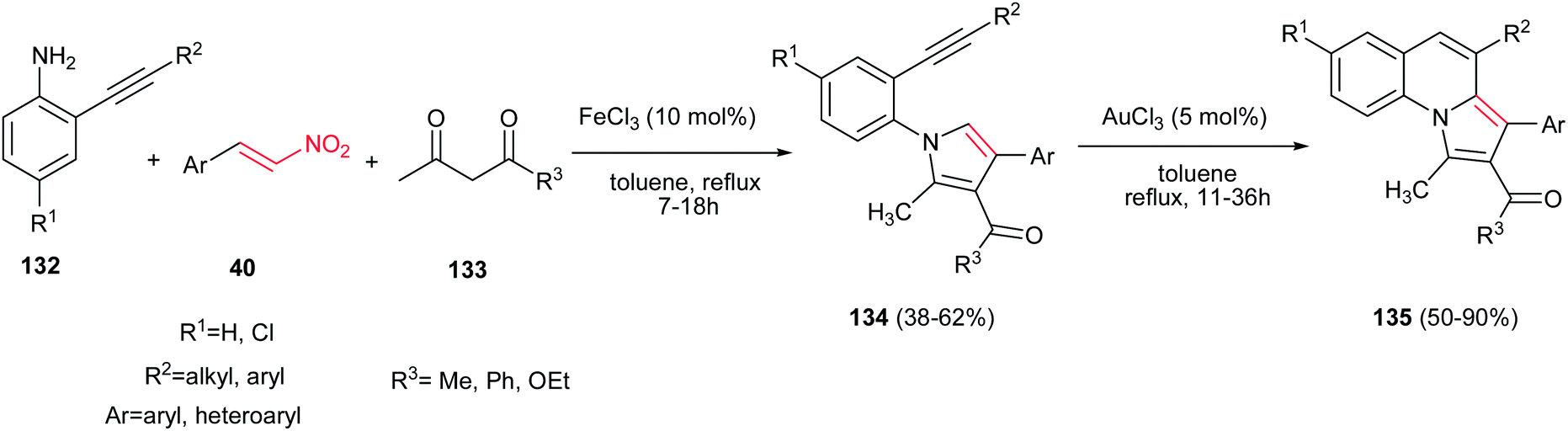
A two step procedure for synthesis of pyrrolo[1,2-a]-quinoline derivatives 135 is reported by Jana and coworkers. The first step is a one-pot three-component synthesis of N-(2-alkynylaryl)pyrroles 134 from anilines 132, 1,3-dicarbonyl compounds 133 and nitroalkenes 40 promoted by 10 mol% of FeCl3 and the second step is transformation of the pyrrol adducts 134 to pyrrolo[1,2-a]-quinoline derivatives 135 via treatment with 5 mol% of AuCl3 in toluene at reflux temperature (Scheme 44).99 The products exhibited fluorescence activity in a range from 452 to 465 nm with quantum efficiencies (Φ) ranging from 0.033 to 0.067.
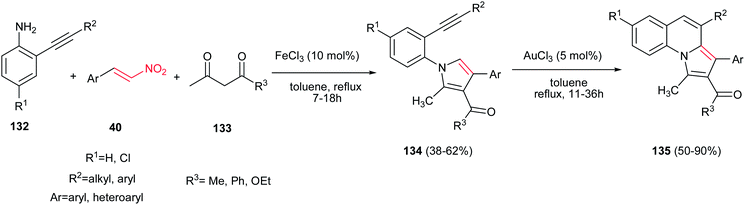 |
| | Scheme 44 Two-step synthesis of pyrrolo[1,2-a]-quinolines. | |
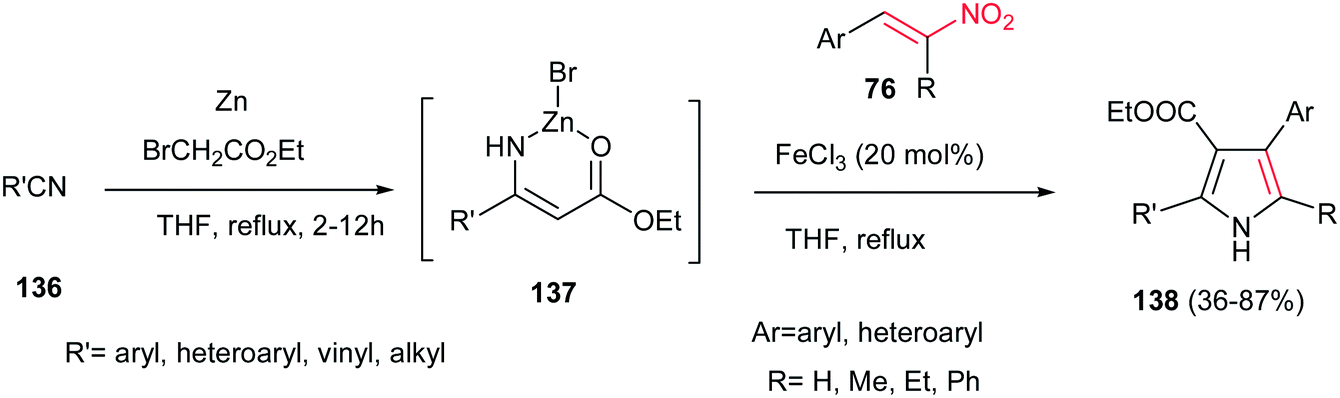
In addition, 2,3,4,5-tetrasubstituted NH-pyrroles 138 were synthesized by Guan et al. via condensation of the Blaise reaction intermediate 137 and nitroolefins 76 catalyzed by 20 mol% FeCl3 in THF at reflux temperature.100 The reaction was carried out by formation of the Blaise intermediate by nucleophilic addition of an in situ generated Reformatsky reagent to an alkyl (aryl)nitrile 136 in THF, followed by addition of nitroalkenes 76 and FeCl3 (20 mol%) and refluxing the mixture for additional times until completion (Scheme 45). Other iron salts such as FeCl2, FeCl3, FeSO4, Fe(acac)2 and Fe(acac)3 and other Lewis acids such as ZnBr2, Cu(OTf)2 and Yb(OTf)3 were also examined and afforded lower yield than FeCl3. While this protocol is insensitive to electronic effects of the aromatic nitriles and nitroalkenes, steric effects in nitriles (ortho substituted benzonitriles) had a significant influence on the reaction yield. Vinyl nitriles and aliphatic nitriles are suitable substrates for this transformation. Nitrostyrene gave lower yield (42%) than α-substituted nitroolefins.
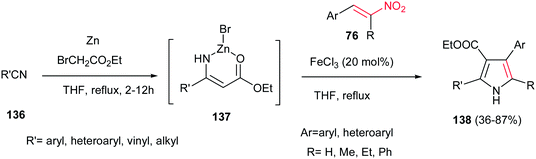 |
| | Scheme 45 2,3,4,5-Tetrasubstituted NH-pyrroles from Blaise reaction intermediate and nitroolefins. | |
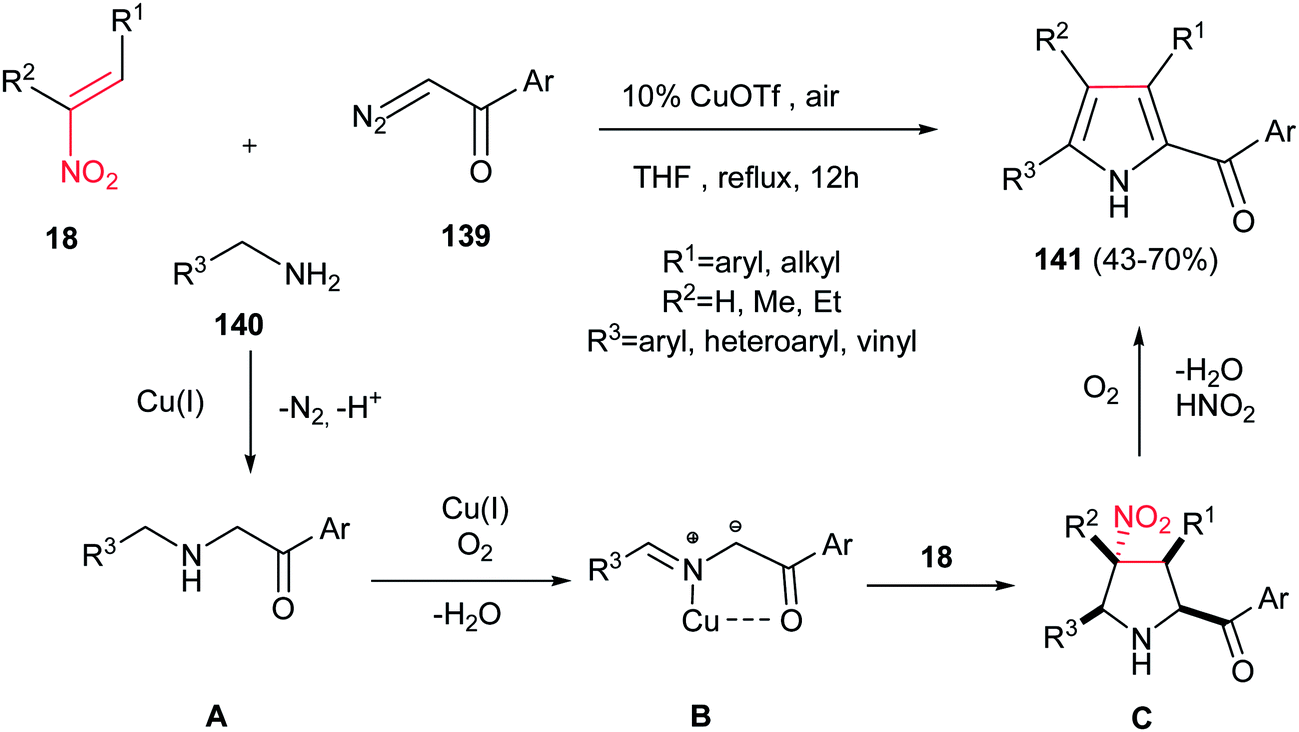
Accordingly, Lu, Wang, and co-workers reported the synthesis of highly substituted pyrroles 141 with acyl group on the C-2 position from α-diazoketones 139, nitroalkanes 18, and amines 140 in the presence of 10 mol% of CuOTf (Scheme 46).101 The authors proposed that N–H insertion of α-diazoketone 139 with amine 140 furnished intermediate A, which underwent oxidative dehydrogenation by Cu(I) under aerobic conditions to generate the azomethine ylide B. Subsequent [3 + 2] cycloaddition of the formed azomethine ylide B with nitroalkene 18 afforded the pyrrolidine ring C. Finally, thermal extrusion of HNO2 and dehydrogenative aromatization of the pyrrolidine ring gave the corresponding products 141.
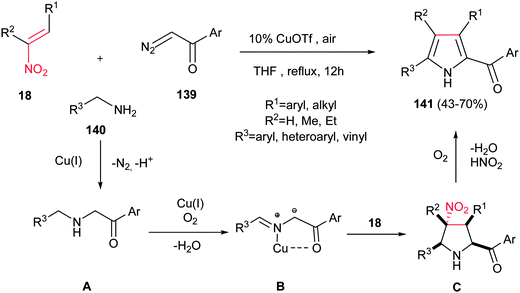 |
| | Scheme 46 CuOTf-catalyzed synthesis of substituted pyrroles from nitroalkenes, α-diazoketones and amines. | |
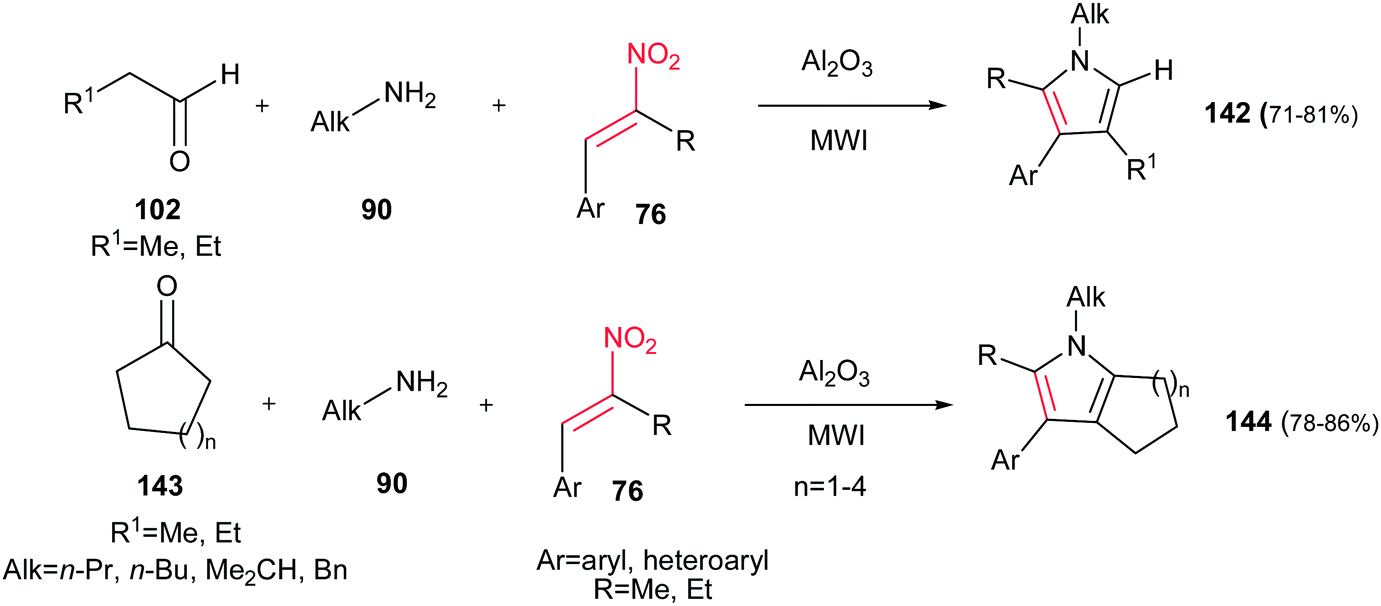
Ranu et al. developed another approach to substituted pyrroles 142 by coupling of an aldehyde/ketone 102/143, an amine 90 and an α,β-unsaturated nitroalkene 76 on the surface of alumina without any solvent under MW irradiation (Scheme 47).102 In this procedure, the presence of α-substituent on nitroalkene seems to be crucial for completion of reaction, since its absence takes the reaction along different pathways. It is also examined that open chain ketones led to different reaction products. Cyclic ketones 143 are suitable in this procedure to generate the fused pyrroles 144 in high yields. Also, the same group developed another approach for synthesis of these compounds in molten tetrabutylammonium bromide at 105 °C.103
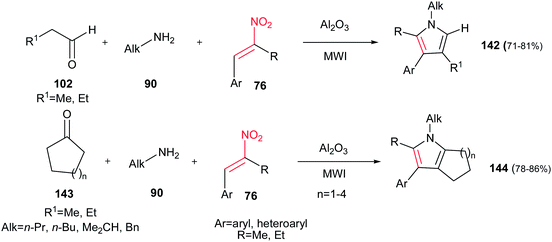 |
| | Scheme 47 Substituted pyrroles from an aldehyde/ketone, an amine and a nitroalkene. | |
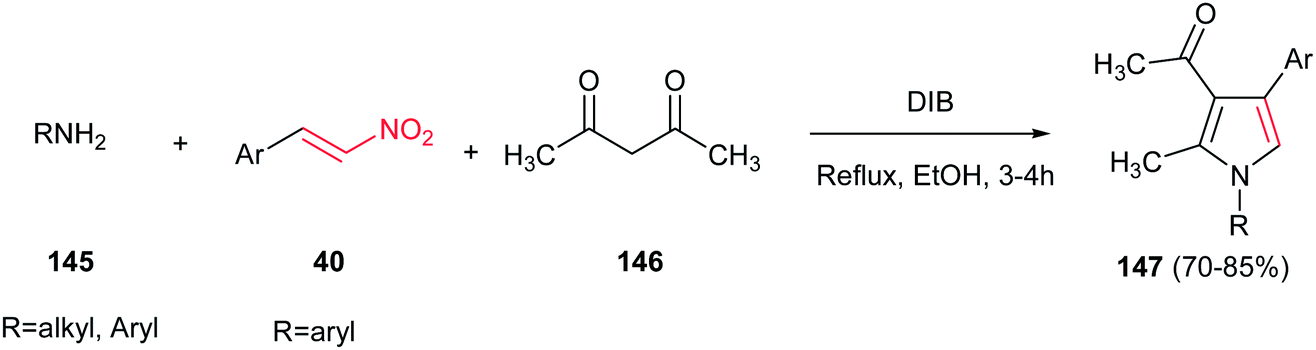
Very recently, Telvekar et al. introduced (diacetoxyiodo)benzene (DIB) as efficient catalyst for synthesis of 1,2,3,4-tetrasubstituted pyrroles 147 from amines 145, nitrostyrenes 40 and acetylacetone 146. The reaction was performed by stirring the mixture of an aniline (2 equiv.), a nitroalkene (1 equiv.), and acetylacetone (1.2 equiv.) in ethanol for 10 min, followed by addition of DIB (1 equiv.) and refluxing for 3–4 h (Scheme 48).104 Anilines, benzyl amines and cyclohexylamine were employed to afford the corresponding products in high to excellent yields. Electronic nature of the substituents on the nitrostyrenes had no effect on yields.
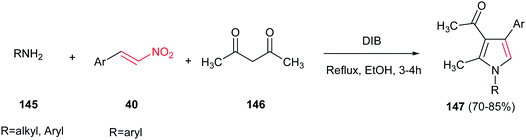 |
| | Scheme 48 Hypervalent iodine-promoted three-component direct synthesis of substituted pyrroles. | |
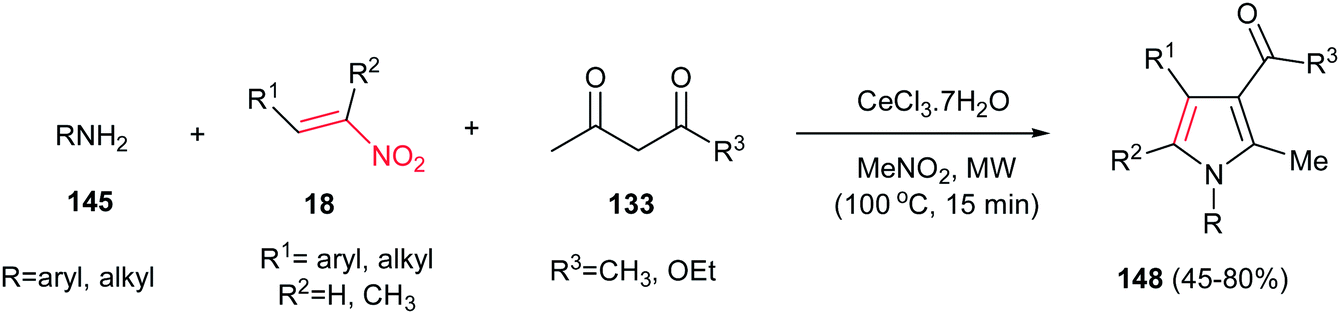
Microwave-assisted synthesis of highly substituted pyrroles 148 from nitroalkenes 18, 1,3-dicarbonyl compounds 133 and amines 145 using inexpensive CeCl3·7H2O as catalyst is reported by Silveria, Kaufman and co-workers (Scheme 49).105 Other CeIII salts such as Ce(NO3)3, Ce(OTf)3 and (NH4)2Ce(NO3)6 gave lower yield in this protocol. Nitroalkenes with electron-donating group on benzene ring afforded higher yield than those with electron-withdrawing groups. Cyclic 1,3-dicarbonyls such as dimedone, Meldrum's acid and 1,3-cyclohexanedione did not afford the expected products. With aliphatic amines, lower yields were achieved.
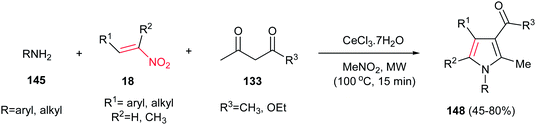 |
| | Scheme 49 CeCl3·7H2O promoted synthesis of highly substituted pyrroles. | |
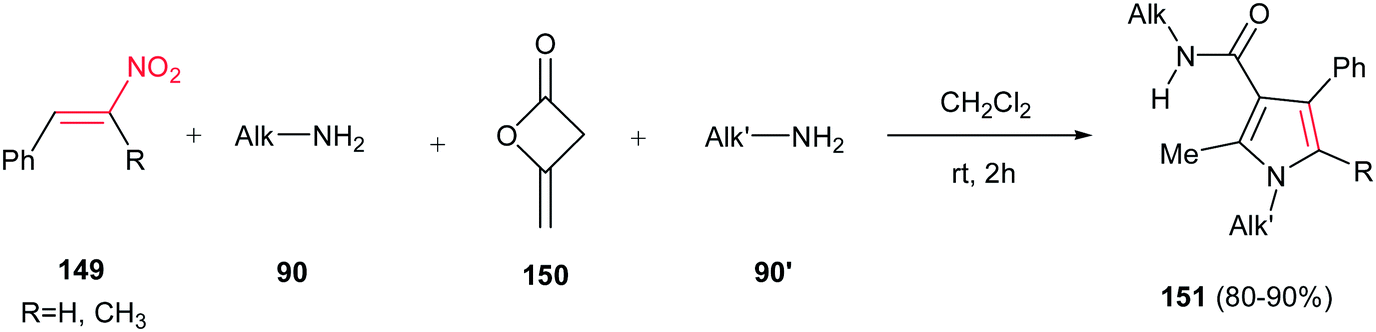
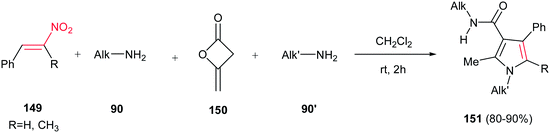
Fully substituted pyrrole derivatives 151 with a carboxamide group on the C-3 position were prepared by Alizadeh et al. via a one-pot four-component operation from primary amines 90/90′, diketene 150 and nitrostyrenes 149 (Scheme 50).106 The reaction proceeds via formation of an enaminone from two primary amines and diketene, followed by condensation of this enaminone with nitrostyrene. Not only linear amines, but also iso-butyl amine and tert-butyl amine were examined with excellent yields. The merits of this method are performing the reaction under neutral and catalyst-free conditions, simple workup, ambient reaction temperature, and excellent yields.
 |
| | Scheme 50 Fully substituted pyrroles via a one-pot four-component condensation of primary amines, diketene and nitrostyrenes. | |
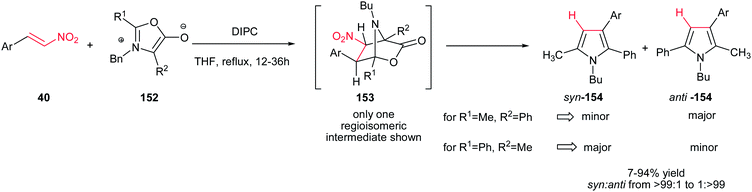
1,3-Dipolar cycloaddition of munchnones 152 and β-nitrostyrenes 40 was investigated by Gribble et al. for synthesis of substituted pyrroles 154. The reaction proceeds via 1,3-dipolar cycloaddition, followed by aromatization via elimination of CO2 and HNO2 to give the corresponding products in excellent yields and high regioselectivities using N,N′-diisopropylcarbodiimide (DIPC)in THF (Scheme 51). The reaction is insensitive to electronic nature of substituents on the phenyl ring of nitroalkenes.107
 |
| | Scheme 51 Synthesis of pyrroles via 1,3-dipolar cycloaddition of munchnones and β-nitrostyrenes. | |
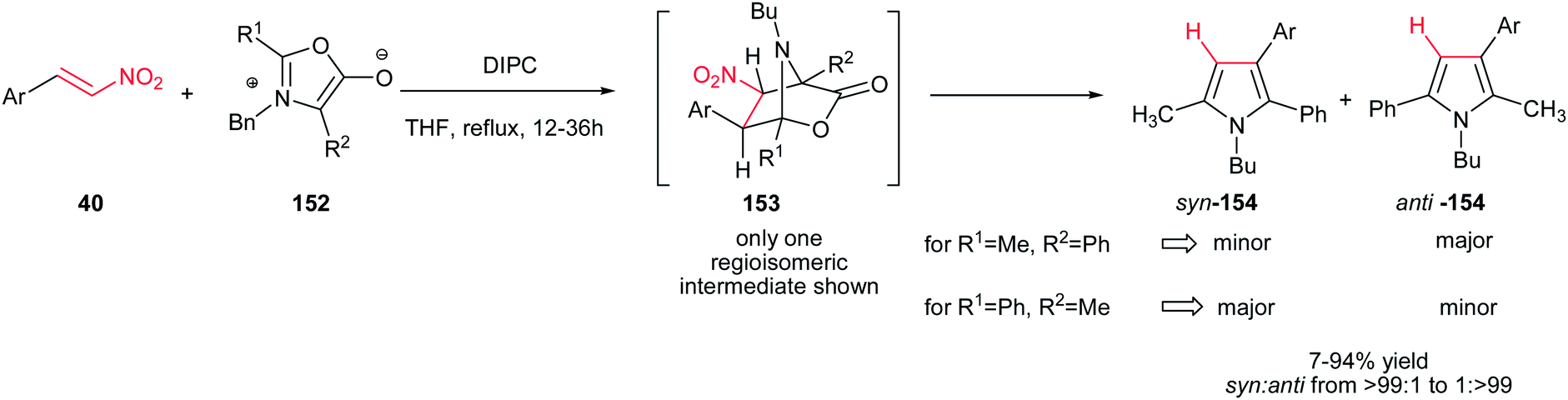
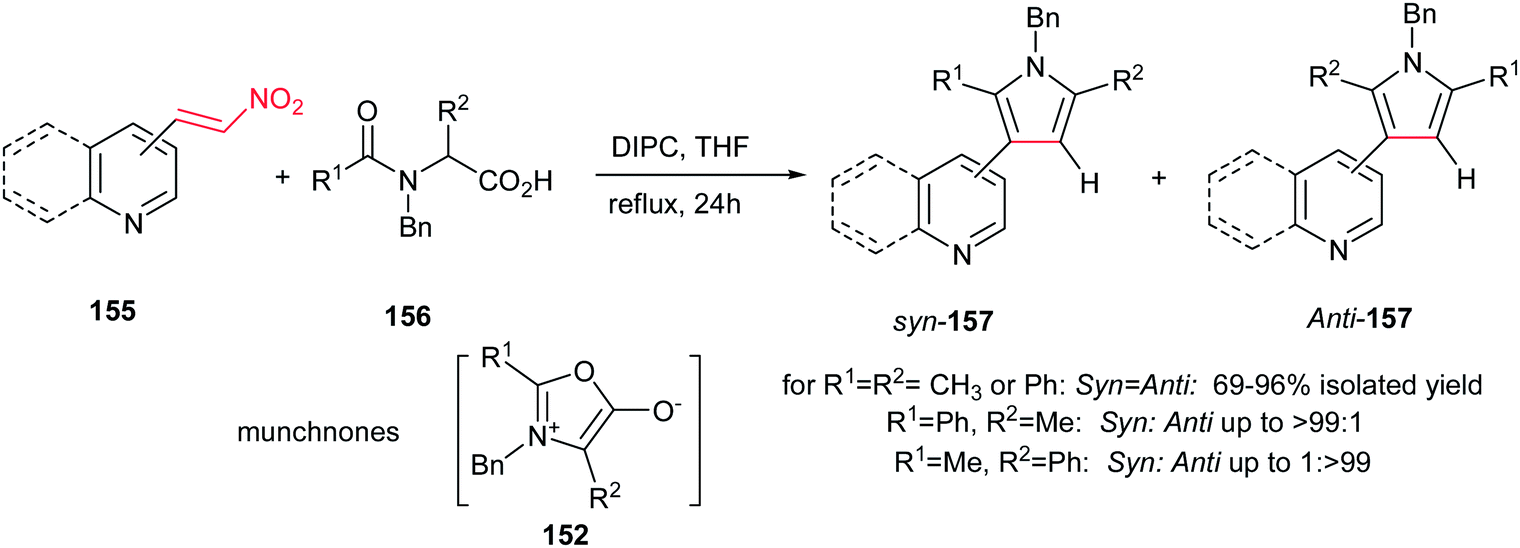
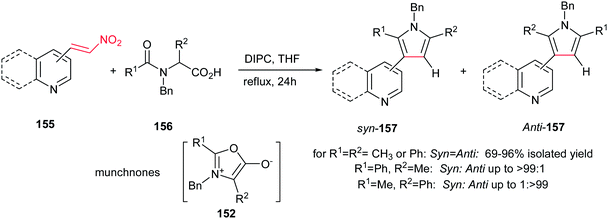
In this context, 2-, 3-, 4-pyridyl(quinolyl)pyrroles 157 were prepared by Gribble and Lopchuk from in situ prepared symmetrical and unsymmetrical münchnones 152 and nitroalkenes 155 via 1,3-dipolar cycloaddition. The Munchnones 152 were prepared in situ from their precursor 156 via treatment with DIPC. The reactions showed good regioselectivity of up to >99:1 toward syn or anti regioisomer when unsymmetrical munchnones were employed (Scheme 52).108
 |
| | Scheme 52 2-,3-,4-Pyridyl(quinolyl)pyrroles from symmetrical and unsymmetrical münchnones and nitroalkenes. | |
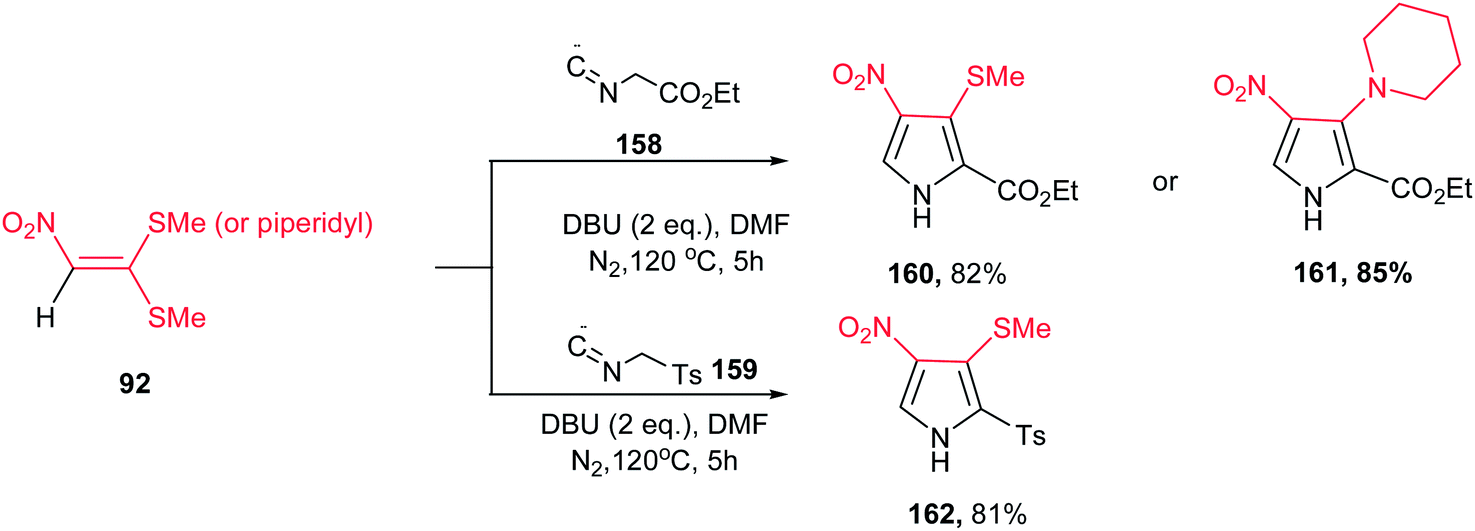
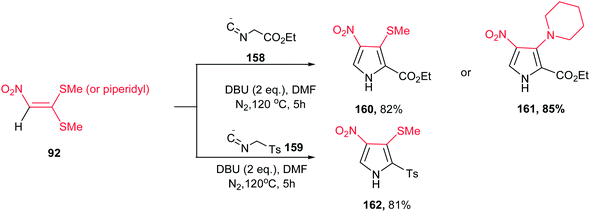
A concise route for synthesis of 2,3,4-substituted pyrroles 160–162 via a base-induced [3 + 2] cycloaddition of readily available polarized ketene S,S- and N,S-acetals 92 and 158 or 159 with activated methylene isocyanides was reported by Ila and coworkers (Scheme 53).109 A number of functional groups such as tosyl, carbalkoxy, aryl, cyano, nitro, acetyl, benzoyl, cyclic amines, etc. can be introduced at the three positions of the pyrrole ring with proper selection of starting materials. The optimized conditions for this reaction are when 92 was reacted with 158 or 159 in the presence of DBU in DMF at 120 °C. Compared to previous reports on the reaction of nitroalkenes with ethyl isocyanoacetate, the nitro group is retained in the 4-position of the pyrrole adducts 160–162 with elimination of the methylthio group which allow further constructions.
 |
| | Scheme 53 Synthesis of 2,3,4-substituted pyrroles via condensation of ketene S,S- and N,S-acetals with activated methylene isocyanides | |
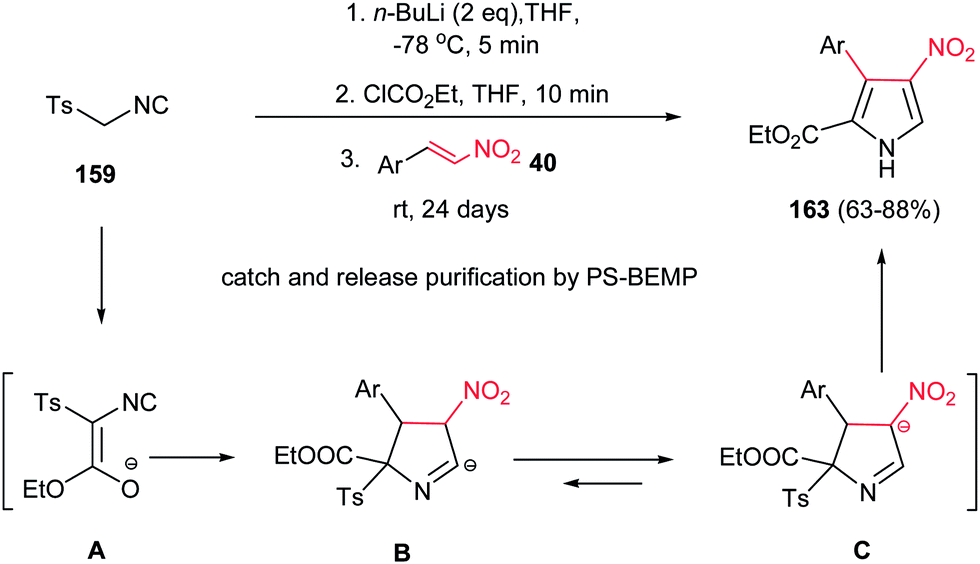
Also, Ley et al. reported a facile procedure for synthesis of nitro-substituted pyrroles 163 via a one-pot three-component reaction of tosyl isocyanide 159, ethyl chloroformate and nitrostyrenes 40 in good yield (Scheme 54).110 In this protocol, treatment of 159 with ethyl chloroformate in the presence of butyl lithium generates the intermediate isocyanoacetate A, which easily attacks nitroalkene 40 to form cyclic compound B or its isomer C. Subsequent elimination of p-toluenesulfinate from B or C and [1,5]-proton shift provides the corresponding nitropyrroles 163. The NO2 group is retained as masked amine in the target molecule. The products were isolated in good to excellent yields (63–88%) using a polymer-assisted catch-and-release workup and purification protocol using PS-BEMP (2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine on polystyrene 2%). In general, under usual workup and column chromatography conditions, the yields do not exceed 40%.
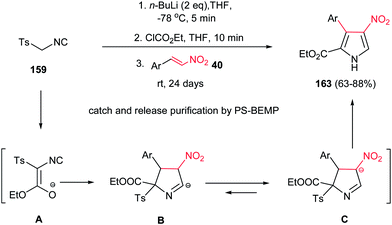 |
| | Scheme 54 Synthesis of nitro-substituted pyrroles from tosyl isocyanide, ethyl chloroformate and nitrostyrenes. | |
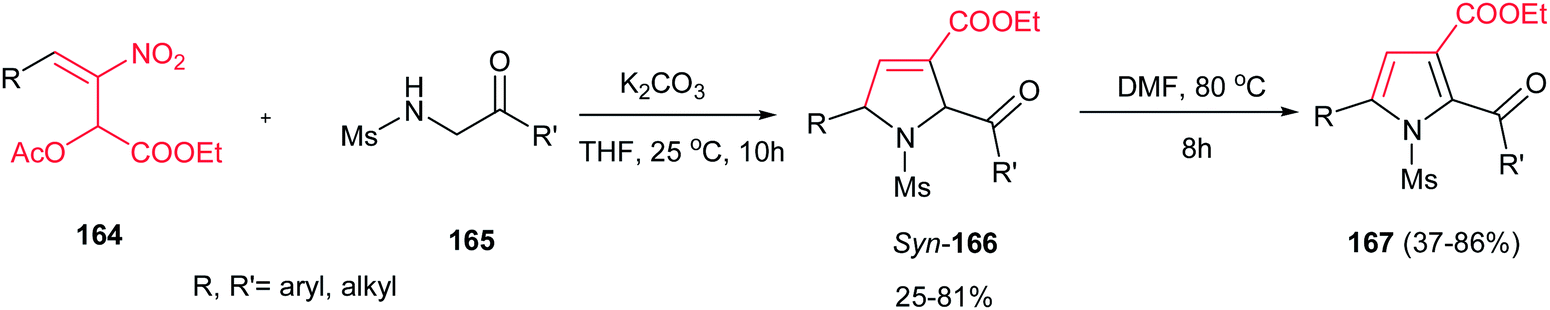
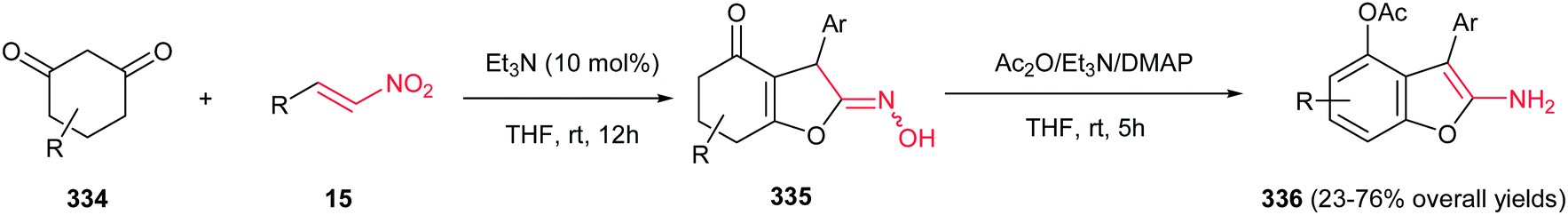
Very recently, Zou and co-workers reported a high yielding protocol for synthesis of substituted 3-pyrrolines 166 from the cascade reactions of nitroallylic acetates 164 with methanesulfonyl 2-aminoethanones 165 in the presence of a base (Scheme 55).111 Several bases and solvents were screened from which K2CO3 in THF at room temperature was selected as optimal conditions. No reaction was observed without a base. Under optimal conditions, the 3-pyrrolines were isolated with cis configuration as proved by NOESY. Electron-deficient nitroallylic acetates showed higher yields than electron-rich ones. Also, aliphatic nitroallylic acetates and methanesulfonyl 2-aminoethanones gave lower yields than aromatic substrates. In addition, it is notable that the products could be simply transformed in situ to pyrroles 167 by adding DMF to the reaction mixture and heating at 85 °C for 8 h.
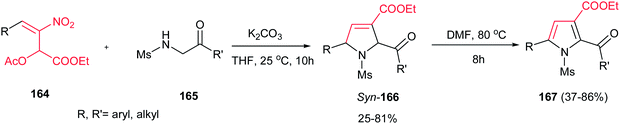 |
| | Scheme 55 Synthesis of 3-pyrrolines and pyrroles. | |
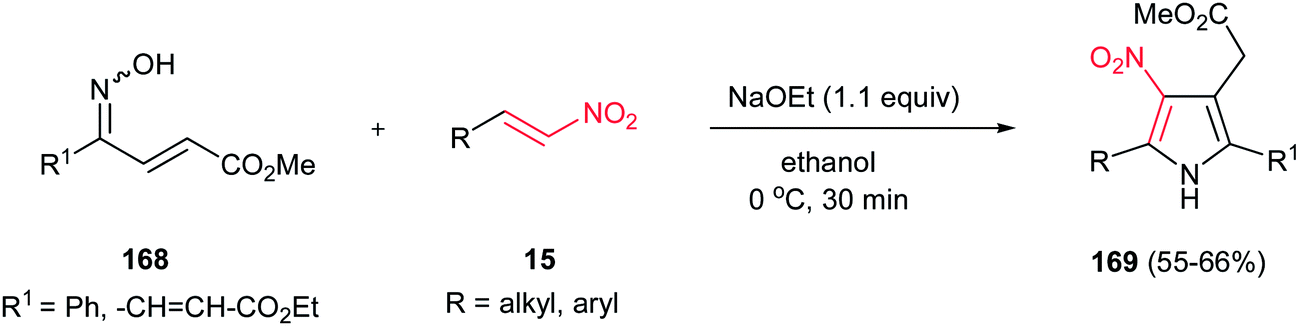
Tetrasubstituted pyrroles 169 were obtained in good to high yields from oxime-enoates 168 and nitroolefins 15 by Takasu and coworkers (Scheme 56).112 The reaction proceeds via a double Michael addition promoted by a strong base such as NaOEt, followed by dehydrative aromatization. Only ethanol was proven as suitable solvent for this reaction. In the presence of the weaker bases, such as Na2CO3 or DBU, the reaction did not proceed, even in longer reaction time. The reaction is not feasible with oxime ether. The geometry of the nitrogen lone pair of 168 is important for the formation of the pyrrole and only syn isomer provided the product.
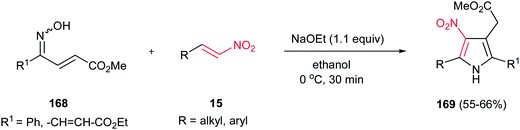 |
| | Scheme 56 Base-promoted synthesis of tetrasubstituted pyrroles from oxime-enoates and nitroolefins. | |
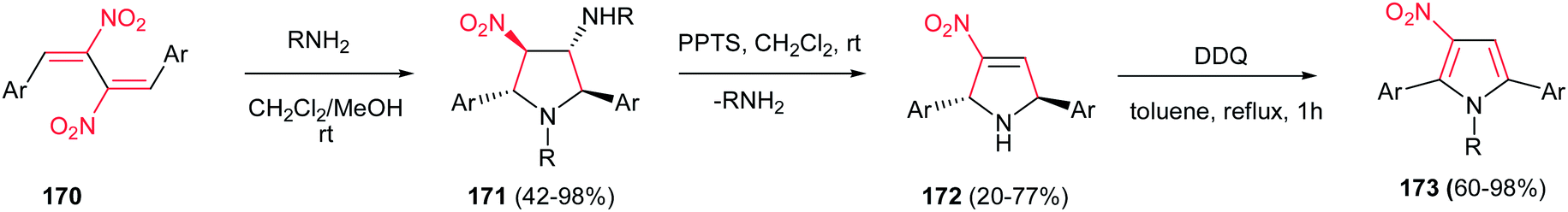
The 1,2,3,5-tetrasubstituted pyrroles 173 were synthesized by Dell'Erba in three steps from primary amines and activated dinitrobutadienes 170.113 The reaction proceeded via an unusually favored 5-endo-trig ring closure to give the corresponding pyrrolidine 171 as pure all-trans diastereomer. Then, 171 was treated with pyridinium p-toluenesulfonate (PPTS) in dichloromethane at room temperature to generate the nitropyrroline 172 via elimination of alkylamine group. Subsequent oxidation of 172 using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) affords corresponding nitropyrrole 173 (Scheme 57).
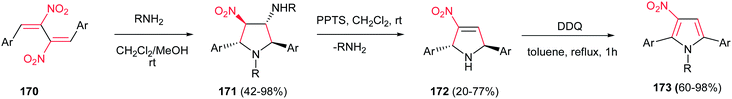 |
| | Scheme 57 1,2,3,5-Tetrasubstituted pyrroles from primary amines and dinitrobutadienes. | |
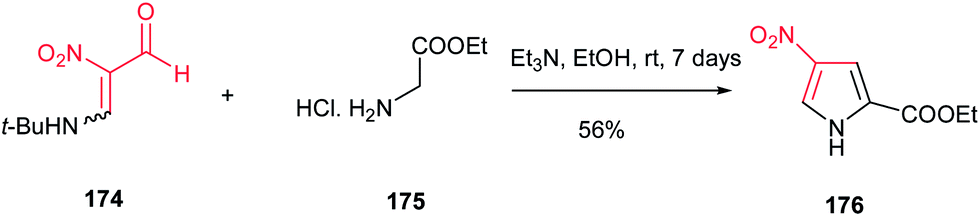
Nishiwaki et al. described that reaction of the enamine of nitromalondialdehyde 174 with glycine ethyl ester hydrochloride 175 afforded ethyl 4-nitropyrrole-2-carboxylate 176 in 56% yield (Scheme 58).114
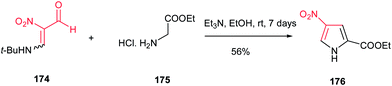 |
| | Scheme 58 Synthesis of pyrroles from enamine of nitromalondialdehyde and glycine ethyl ester. hydrochloride. | |
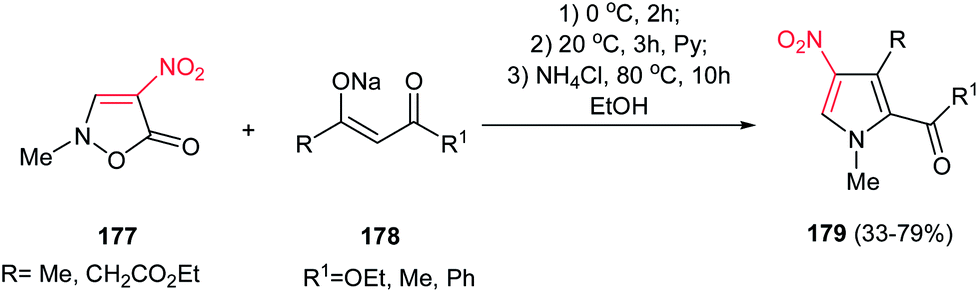
Also, the same group reported another approach for synthesis of 1,2,3,4-tetrasubstituted pyrroles 179 from nitroisoxazolone 177 and various β-ketoesters 178. The nitroisoxazolone 177 was considered by the authors as the hidden form of the substituted nitroenamine (Scheme 59).115
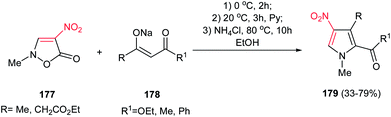 |
| | Scheme 59 Synthesis of 1,2,3,4-tetrasubstituted pyrroles from nitroisoxazolone and the enolate of various β-ketoesters. | |
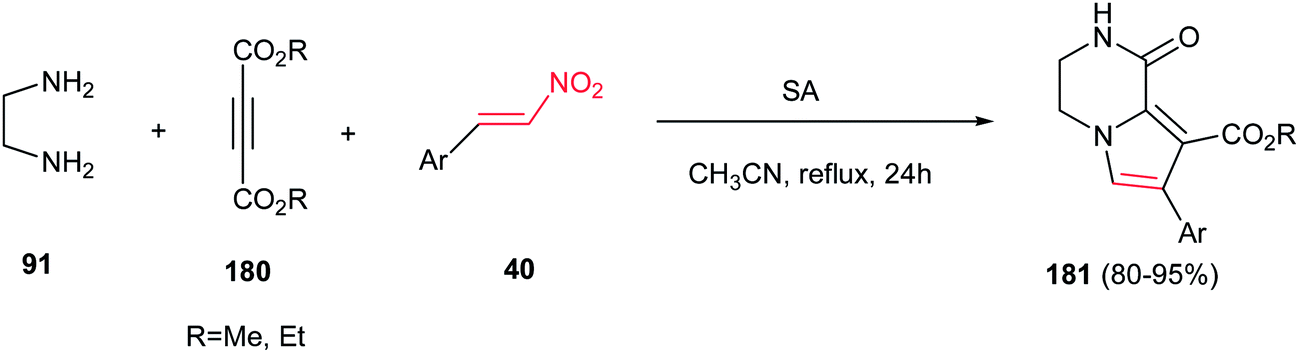
Finally, Moradi and co-workers reported a one-pot three-component synthesis of pyrrolo[1,2-a]pyrazines 181 from ethylenediamine 91, acetylenic esters 180 and nitrostyrene derivatives 40 (Scheme 60).116 The reaction was performed via initial stirring of the ethylenediamine 91 (1.2 equiv.) and an acetylenic ester 180 (1 mmol) in CH3CN at room temperature, followed by addition of a β-nitrostyrene 40 (1 mmol) and sulfamic acid (SA, 20 mol%), then refluxing the mixture for 24 h. Diversity of nitroalkenes with electron-donating and -withdrawing groups on the phenyl ring gave similar yields in this protocol.
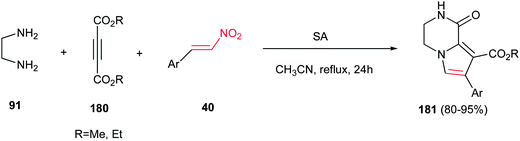 |
| | Scheme 60 One-pot three-component synthesis of pyrrolo[1,2-a]pyrazines. | |
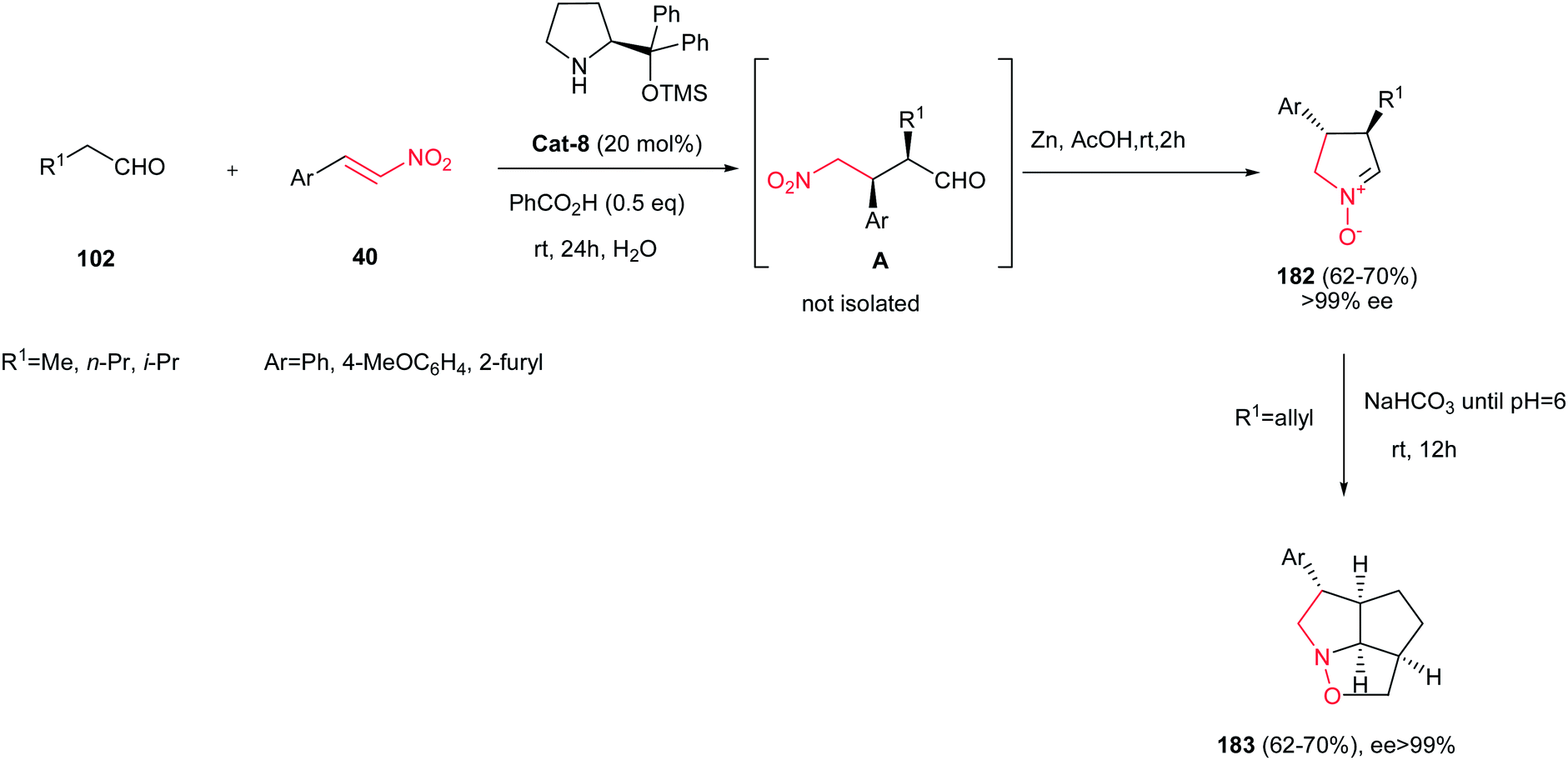
4.1.5. Five-membered cyclic nitrones. Cyclic nitrones have been used as advanced intermediates in organic synthesis for the preparation of various natural and biologically active compounds.117 Nitrones possess one of the largest dipole moments known for any functional group type (3.37–3.47 D) making them potentially useful for non-linear optic applications and control of molecular orientation. Many routes are available for synthesis of these nitrones including the oxidation of cyclic amines or hydroxylamines,118 intramolecular condensation of ω-hydroxylamino-carbonyl derivatives,119 intramolecular cyclization of a suitable leaving group (halide, epoxide, or mesylate) by the oxime nitrogen atom,120 and cyclizations of ω-unsaturated oximes.121Reductive cyclizations of γ-nitrocarbonyl derivatives have found wide applications in synthesis of cyclic nitrones. In this context, Merino et al. reported a one-pot synthesis of enantiomerically pure five-membered cyclic nitrones 182 through the organocatalytic Michael addition of aliphatic aldehydes 102 to trans-nitroalkenes 40 and in situ Zn-promoted reductive cyclization by using water as a solvent (Scheme 61).122 This methodology were also applied successfully for preparation of alkenyl cyclic nitrones (182, R1 = allyl) that undergo spontaneous intramolecular 1,3-dipolar cycloaddition to provide tricyclic derivatives 183 in water with excellent ee values (ee > 99%).
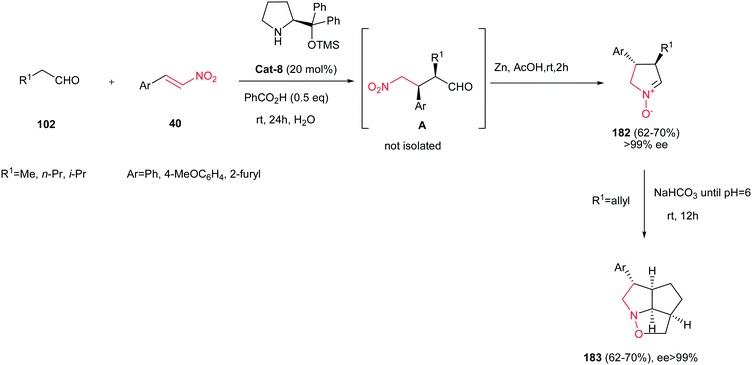 |
| | Scheme 61 Synthesis of cyclic nitrones from aldehydes and nitroolefins and their application in the synthesis of tricyclic compounds. | |
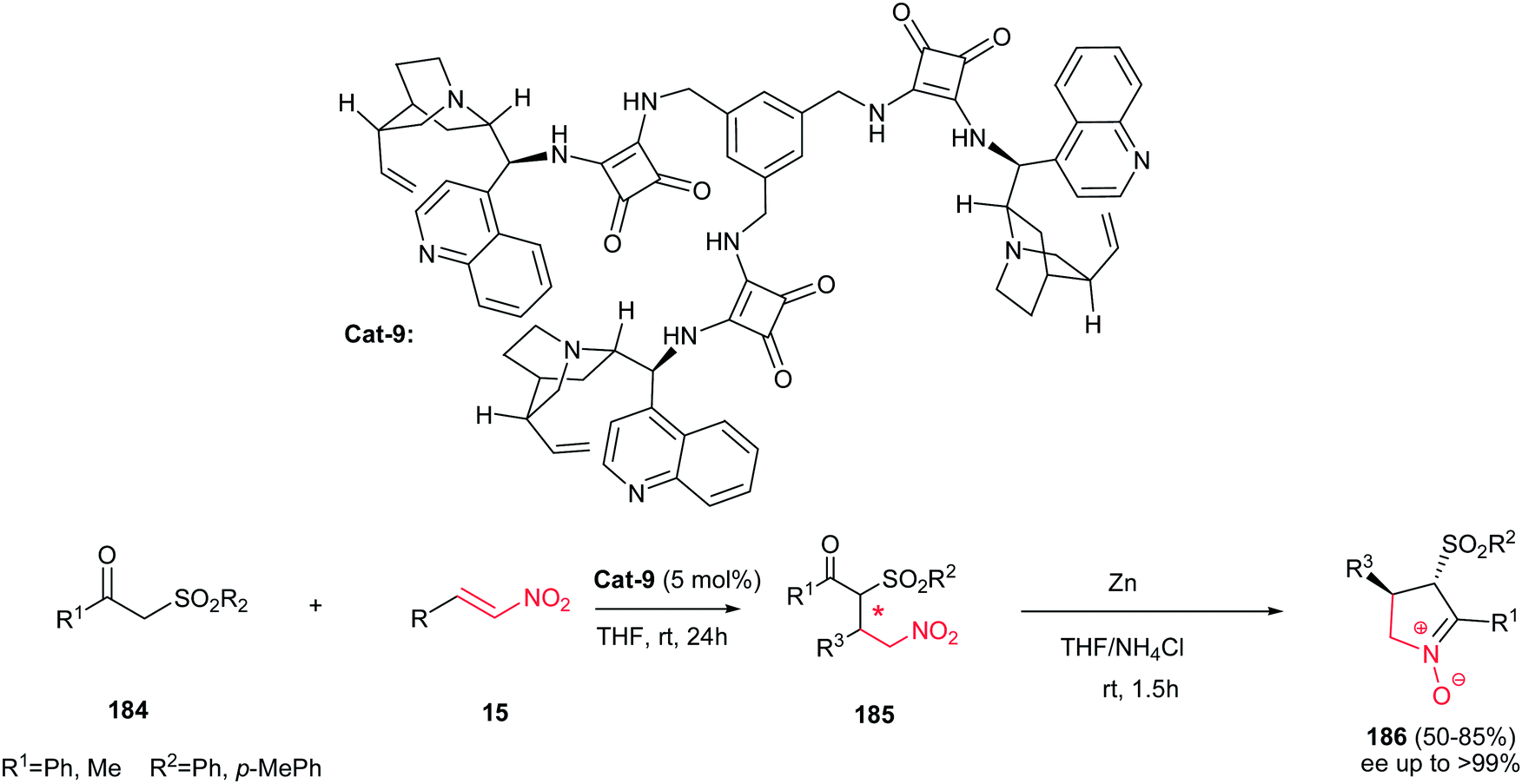
In 2012, Dong et al. demonstrated that cinchonine-squaramide Cat-9 is an efficient catalyst for asymmetric Michael addition of ketosulfones 184 to nitroalkenes 15. The Michael adducts 185 were used for subsequent transformation to chiral cyclic trans nitrones 186 via reduction with Zn/NH4Cl system with excellent results (up to 85% yield and >99% ee) (Scheme 62).123 Diverse of nitroalkenes including aliphatic, aromatic and heteroaromatic ones afforded the products in high yields. Also, it is notable that the catalyst can be recovered and reused for six cycles without losing activity and selectivity.
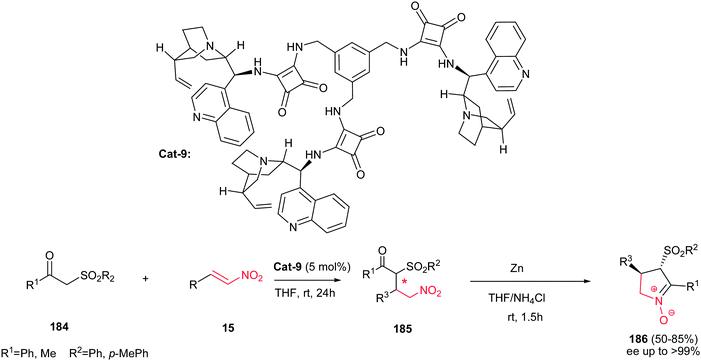 |
| | Scheme 62 A two-step synthesis of substituted chiral cyclic trans nitrones. | |
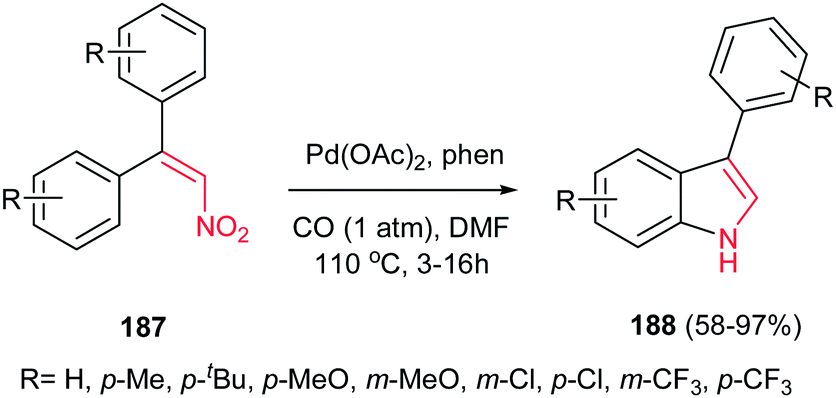
4.1.6. Indole derivatives. Indole derivatives are prevalent in numerous natural products and are extremely important in medicinal chemistry.124 According to the wide applications of indole derivatives, many reviews are published about indole synthesis125 as there are several name reactions associated with indole synthesis including Fischer, Bischler, Hinsberg, Reissert, Nenitzescu, Madelung, Bartoli, Hemetsberger, Julia, Larock, Leimgrubere-Batcho, and Sundberg approaches.126Pd-catalyzed reductive cyclization of diaryl nitroalkenes 187 with carbon monoxide as an inexpensive reductant is reported by Hsieh and Dong to give 3-arylindoles 188 in high to excellent yields (Scheme 63).127 The reactions were performed using 2 mol% Pd(OAc)2, 4 mol% 1,10-phenanthroline (phen), 1 atm of CO in DMF at 110 °C. Other transition-metal salts such as Fe3(CO)12, Rh6(CO)16, and PtCl2(PPh3)2 were also proved to be efficient for this transformation, albeit with lower yield. The reaction is sensitive to the electronic nature of substitution on the phenyl ring of nitroalkenes. Electron-rich nitroalkenes gave higher yields in shorter reaction times compared to nitroalkenes containing electron-withdrawing groups such as Cl and CF3. Nitroalkenes containing meta-substituted phenyl groups afforded both the 5- and 7-substituted indoles in excellent yields.
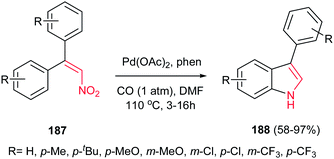 |
| | Scheme 63 Pd-catalyzed synthesis of indoles from nitroalkenes. | |
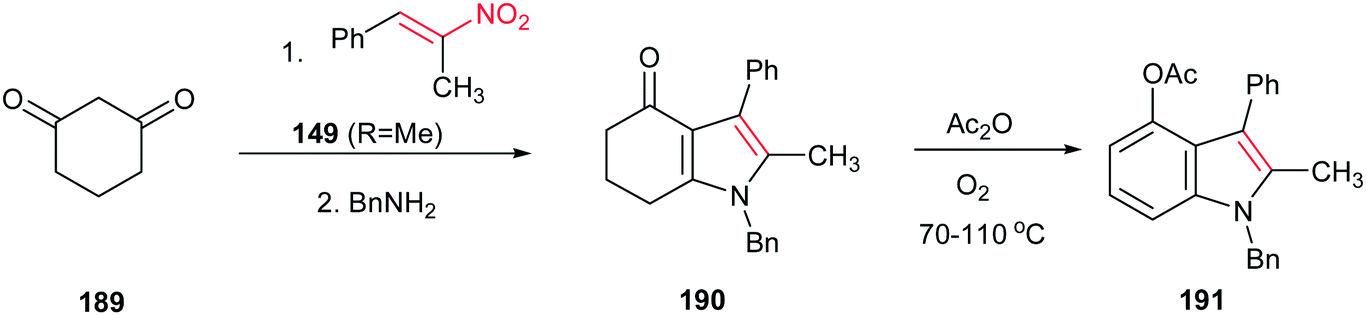
Ishikawa group also described that condensation of cyclohexane-1,3-dione 189 with the nitroalkene 149 (R = Me) provided the fused pyrrole 190 after exchange with benzylamine. Aromatization of 190 with acetic anhydride in the presence of oxygen gave the 4-oxygenated indole 191 (Scheme 64).128 In this context, very recently, Qi et al. reported another green approach for synthesis of tetrahydro-4H-indol-4-one derivatives from the same starting materials using 10 mol% of proline in water.129
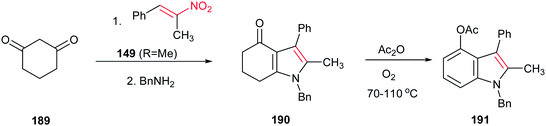 |
| | Scheme 64 Synthesis of indole from cyclohexane-1,3-dione and nitroalkene. | |
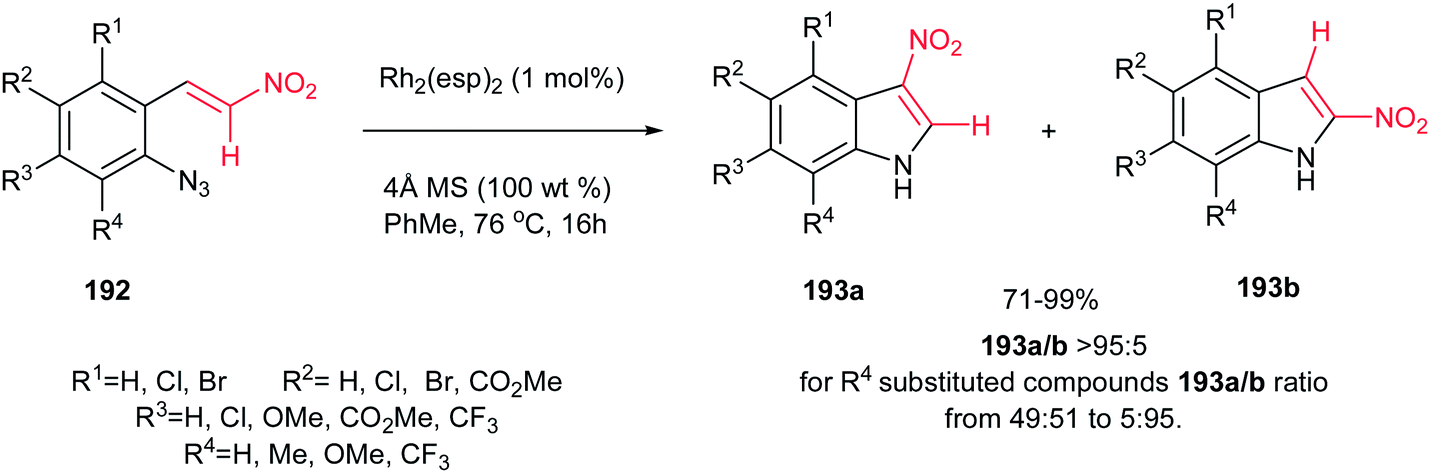
Although Pelkey and Gribble described that thermolysis of β-nitro-styrylazides 192 in xylene at 140 °C produced the 2-nitroindole 193b as the major product,130 recently, Driver et al. described that a fundamental change in reactivity of 192 can be achieved using catalytic amount of Rh2(esp)2 (1 mol%) [esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropionate] in toluene at 76 °C to give the 3-nitroindoles 193a as major products via migration of the nitro group (Scheme 65).131 The reaction gave excellent yield toward preparation of 193b. Only with an R4 substituent on azides, the 2-nitroindole 193b formation became a competitive process. This strategy is also applicable for synthesis of different 3-substituted indoles by replacing the nitro group with other electron-withdrawing groups such as ketones, esters, amides, sulfones.
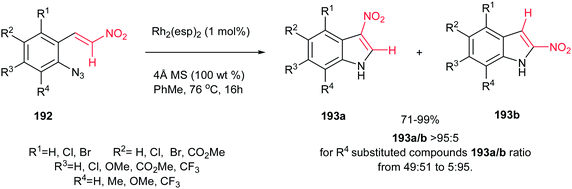 |
| | Scheme 65 Synthesis of indoles from β-nitro-styrylazides. | |
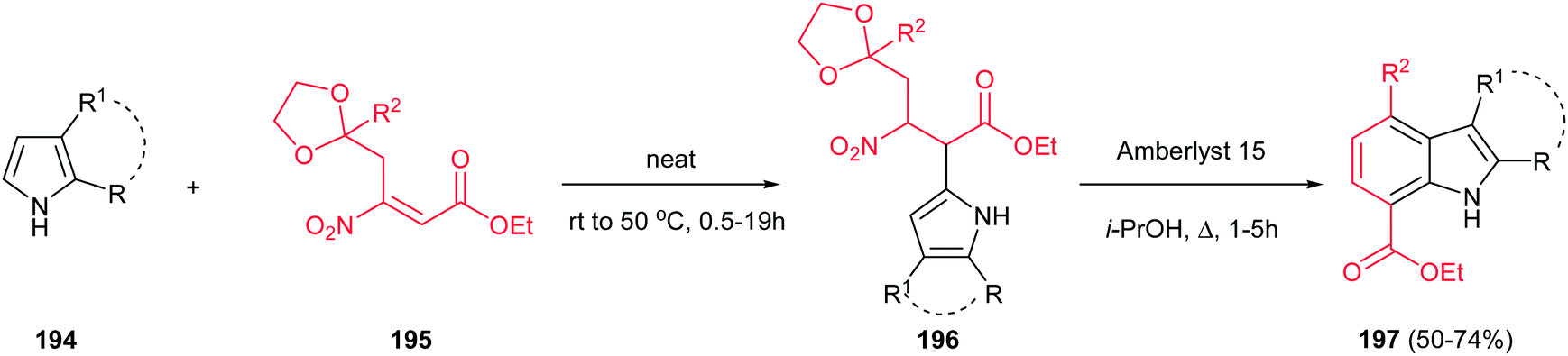
Finally, a one-pot two-step synthesis of polysubstituted indoles 197 is reported by Ballini et al. via the reaction of pyrroles 194 with β-nitroacrylates 195 under solvent- and catalyst-free conditions, followed by in situ treatment with Amberlyst 15 in refluxing isopropyl alcohol (Scheme 66).132 The reaction initiated with intermolecular Friedel–Crafts alkylation, followed by intramolecular Friedel–Crafts alkylation of the amberlyst activated oxonium cation, and subsequent losing of a molecule of both ethylene glycol and nitrous acid. It is notable that this strategy allowed synthesis of indoles 197 with different functionalities in both rings.
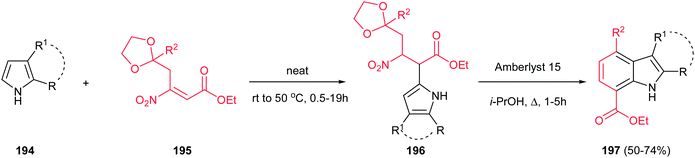 |
| | Scheme 66 A one-pot two-step synthesis of poly substituted indoles. | |
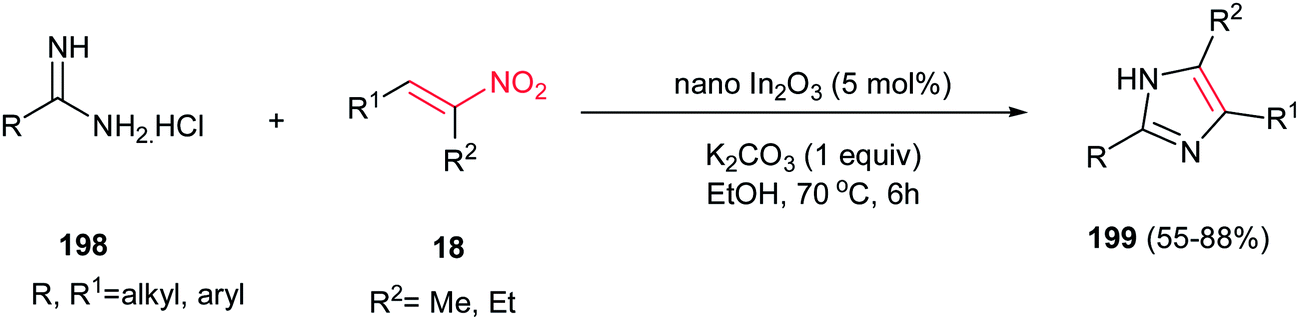
4.1.7. Imidazole derivatives. An efficient, environmentally benign and high yielding procedure for synthesis of 4,5-unsymmetrically substituted 1-H imidazoles 199 was developed by Majee, Hajra and coworkers via nano In2O3-catalyzed reaction of an amidine hydrochloride 198 and a substituted nitroalkene 18 (Scheme 67).133 The reaction proceeds via heating an equimolar amount of the starting materials in the presence of 5 mol% of In2O3 and an equivalent of K2CO3 in ethanol at 70 °C. The use of a base is necessary for progress of reaction. The catalyst can be recovered and reused for four subsequent runs without any significant loss of the activity. Other catalysts such as In(OTf)3, InCl3, nano ZnO and CuO afforded the products in lower yields under similar conditions. Also, fully substituted imidazoles were prepared by Chen et al. from the reaction of nitroalkenes and N-substituted amidines catalyzed by 20 mol% FeCl3 in DMF at 90 °C.134
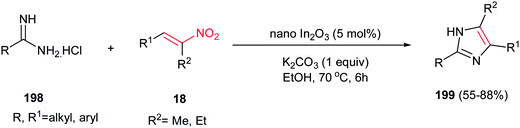 |
| | Scheme 67 Synthesis of imidazoles via tandem cyclization between amidine and nitroolefins. | |
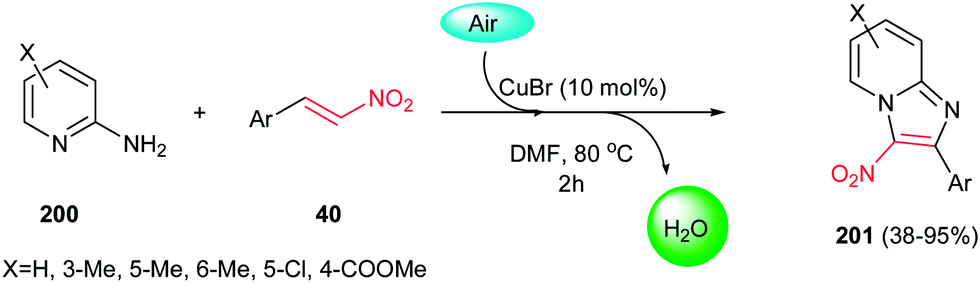
In 2012, a green procedure for synthesis of imidazo[1,2-a]pyridines 201 from 2-aminopyridines 200 and nitroolefins 40 was developed by Yan, Huang and co-workers using air as oxidant and 10 mol% CuBr as catalyst (Scheme 68).135 Among the several copper salts such as CuBr, CuCl, CuOTf, Cu(OAc)2,and Cu(OTf)2 examined for this process, CuBr afforded the highest yield. It is notable that electron-rich nitroolefins or aminopyridines showed better reactivity and gave higher yields than electron-deficient ones. More recently, Hajra et al. developed another catalytic route for this reaction by performing the reaction of nitroalkenes with 2-aminopyridines in the presence of 20 mol% FeCl3 in DMF at 80 °C.136
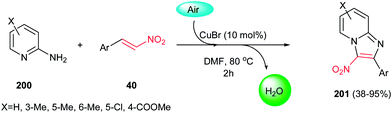 |
| | Scheme 68 Cu-catalyzed synthesis of imidazo[1,2-a]pyridines. | |
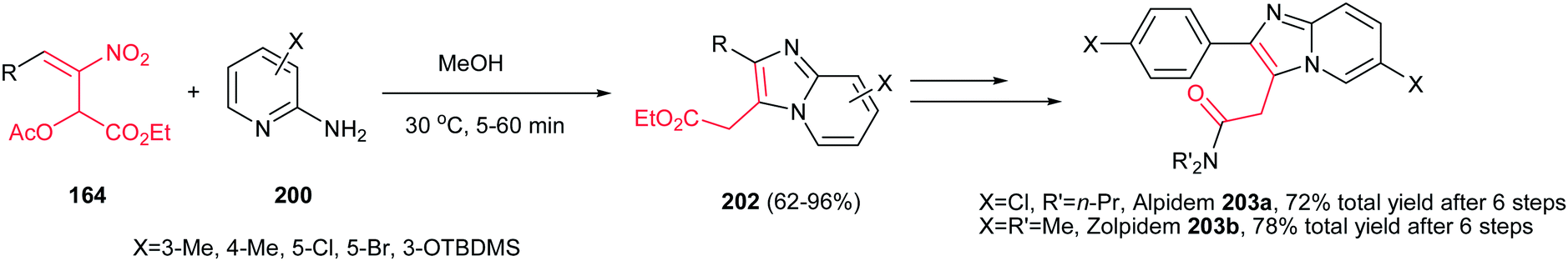
The method for synthesis of functionalized imidazo[1,2-a]pyridines 202 from reaction of MBH acetates of nitroalkenes 164 and 2-aminopyridines 200 was reported by Namboothiri et al. (Scheme 69).137 The reaction was conducted in methanol at room temperature under catalyst-free conditions to give the corresponding products in high to excellent yields (63–96%) via cascade inter-intramolecular double aza-Michael addition. While MBH acetates with unsubstituted and weakly deactivating aromatic rings gave excellent yields of products, lower yields were encountered in the case of MBH acetates with strongly electron withdrawing aromatic substituent and nitrodiene derived MBH acetates. Also aminopyridines with a substitution on the 3-position gave lower yield than those with substitution on the 4- or 5-position. In addition, aminoheterocycles such as aminopyrimidine, aminopyrazine and aminothiazole examined in this protocol, were unreactive under the above reaction conditions. This strategy was successfully applied for total synthesis of anxiolytic drug Alpidem 203a and hypnotic drug Zolpidem 203b in six steps with 72 and 78% overall yields, respectively.
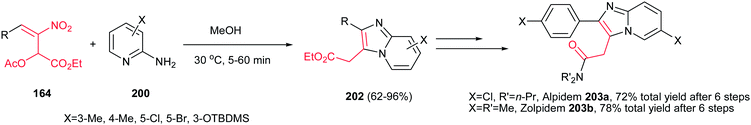 |
| | Scheme 69 Synthesis of imidazo[1,2-a]pyridines from 2-aminopyridine and MBH acetates of nitroalkenes. | |
4.1.8. Pyrazoles, pyrazolidines and pyrazolines. Pyrazoles are an important class of heterocyclic compounds with diverse biological activities as antimicrobial, antiparasitic, anti-inflammatory, antidepressant, antiviral, A2A receptor antagonist, CB1 receptor antagonist, DNA intercalating and antitumor.138 Pyrazole-containing compounds such as the blockbuster drugs Viagra, Celebrex, and Acomplia have been successfully commercialized. Also, pyrazoles have been employed as ligands for the transition-metal-catalyzed cross-coupling reactions.139 The most general and convenience methods for synthesis of pyrazole derivatives are the condensation of substituted hydrazines with 1,3-dicarbonyl compounds or their derivatives (Knorr reaction)140 and the 1,3-dipolar cycloaddition of diazoalkanes or nitrile imines with olefins or alkynes.141Pyrazoline and pyrazolidine derivatives also exhibit wide range of biological activities such as anti bacterial,13 anti depressant,14 anti tubercular15 anti amoebic16 anti inflammatory,17 herbicidal, insecticidal18 and cardiovascular19 activities.142 The most applied procedure for synthesis of pyrazolines is based on the reaction of α,β-unsaturated aldehydes and ketones with hydrazines.143 Pyrazolidines usually are accessible via reduction of pyrazolines144 or pyrazolium salts145 and by the reactions of hydrazine with 1,3-dibromides146 or phenylhydrazones with electron-deficient alkenes.147
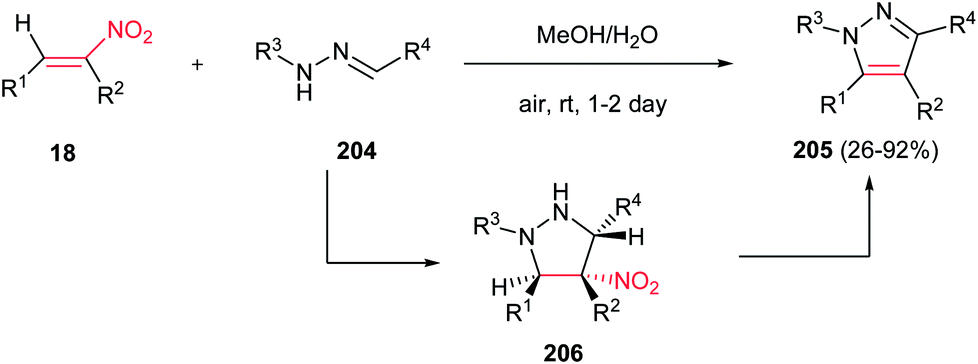
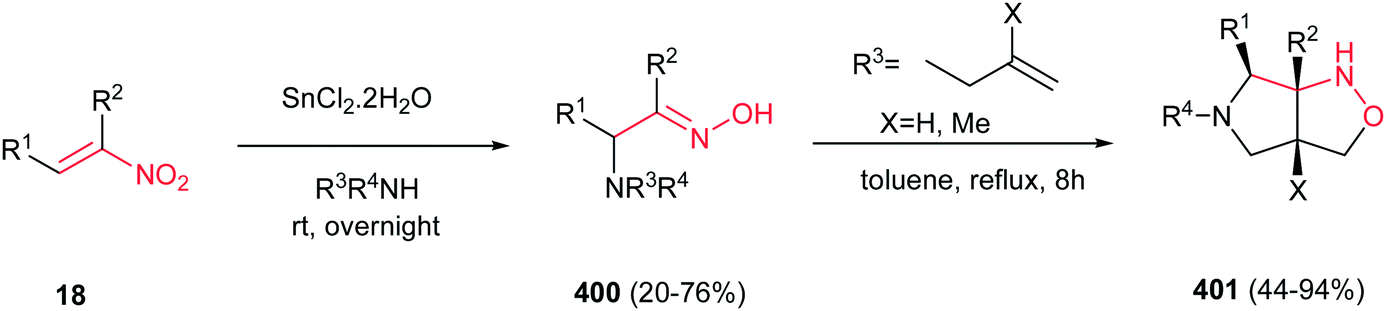
Deng and Mani developed an efficient, simple and regioselective procedure for synthesis of highly substituted pyrazoles 205 from N-substituted hydrazones 204 and nitroalkenes 18 (Scheme 70).148 This reaction is quite broad in scope, generating a diverse set of pyrazole products in moderate to excellent yields. Solvent screening showed that polar protic solvents are suitable for this transformation, while polar aprotic solvents (except dichloromethane), furnished the Michael adducts. The reaction yield is tolerated with electronic and steric effects of substituents on the hydrazone component. Hydrazones with electron deficient group on parent aldehyde favored the formation of a Michael addition product. Moreover, hydrazones with bulky group on nitrogen gave low yield. Either aryl or alkyl groups at the R3 position afford corresponding pyrazoles in excellent yields. Substitution at the R4 position of nitroolefins is also well tolerated. Mechanism study by authors revealed that the reaction proceeded via intermediate 206 which was successfully confirmed by NMR, IR and mass spectroscopy. Also, the same group developed two more practical procedures for synthesis of 1,3,5-tri- and 1,3,4,5-tetrasubstituted pyrazoles from the same starting materials. The first protocol is heating the starting materials in ethylene glycol at 120–150 °C for 16 h, while the second procedure is stirring the starting materials in triflouroethanol in the presence of 10 equiv. of TFA for two days.149
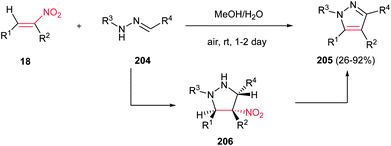 |
| | Scheme 70 Synthesis of pyrazole derivatives in aqueous methanol. | |
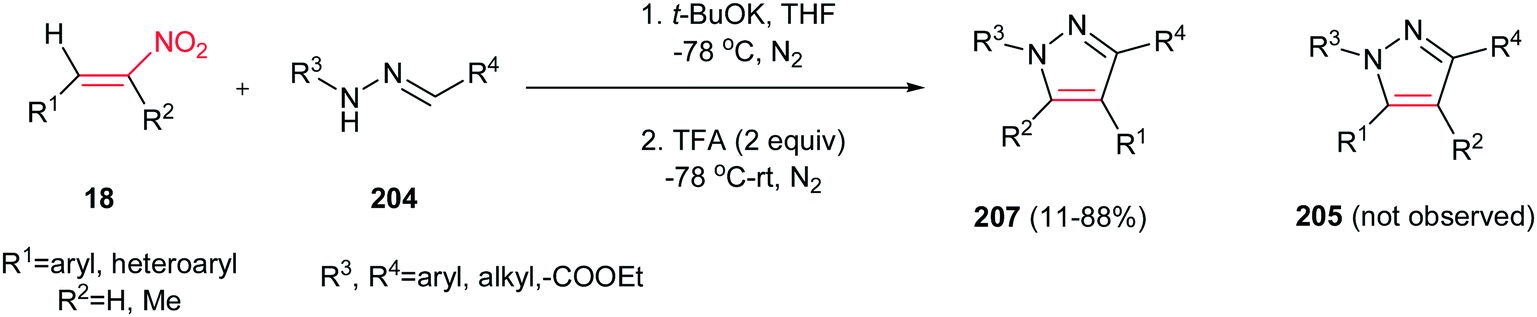
By performing the same reaction in the presence of t-BuOK at −78 °C and under atmosphere of N2, reverse regioselectivity in the pyrazoles (1,3,4-regioselectivity 207 instead of 1,3,5-regioselectivity 205) can be obtained compared to earlier protocols (Scheme 71).150 Fortunately, hydrazones with electron deficient groups on the parent aldehydes also provided high yield. In addition, 1-nitrocyclohexene gave fused pyrazole ring in 63% isolated yield.
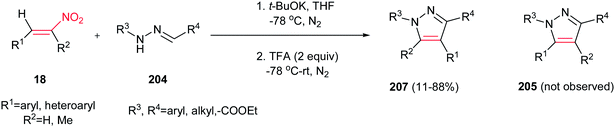 |
| | Scheme 71 Reverse regioselectivity in the synthesis of pyrazoles by using t-BuOK as catalyst. | |
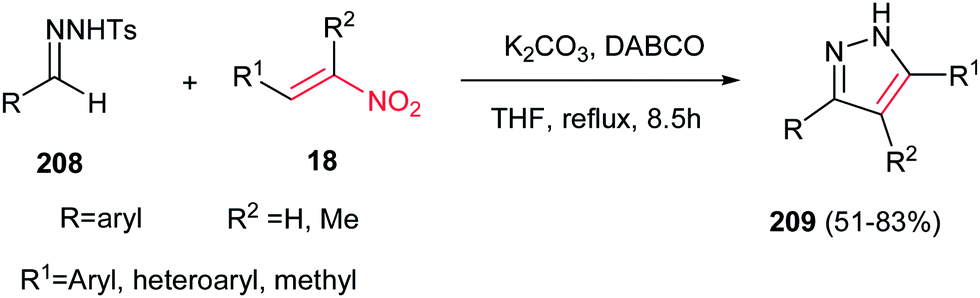
Although the previous methods provided the N-protected pyrazoles, Tang et al. described that by using tosylhydrazones 208 in the reactions with nitroalkenes 18, the corresponding 3,4,5-substituted pyrazoles 209 can be obtained in good to high yields (Scheme 72).151 Different reaction conditions were examined and the use of tosylhydrazones 208 (0.4 mmol), nitroalkenes 18 (0.8 mmol), K2CO3 (0.4 mmol) and DABCO (0.24 mmol) in THF (4 ml) under reflux temperature was selected as optimum conditions. It is noteworthy that aromatic, heteroaromatic and aliphatic nitroalkenes reacted equally in this protocol. Two mechanisms were proposed for this reaction; the first one is initiated by aza-Michael addition of deprotonated tosylhydrazone to nitroalkene, followed by sequential intramolecular cyclization, nitro-elimination and 1,3-H shift, while the second one proceeded via sequential Baylis–Hillman reaction/intramolecularcyclization/nitro-elimination and 1,3-H Shift.
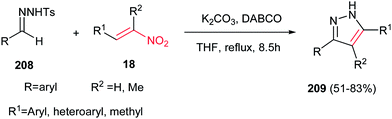 |
| | Scheme 72 Synthesis of 3,4,5-substituted pyrazoles from tosylhydrazones and nitroalkenes. | |
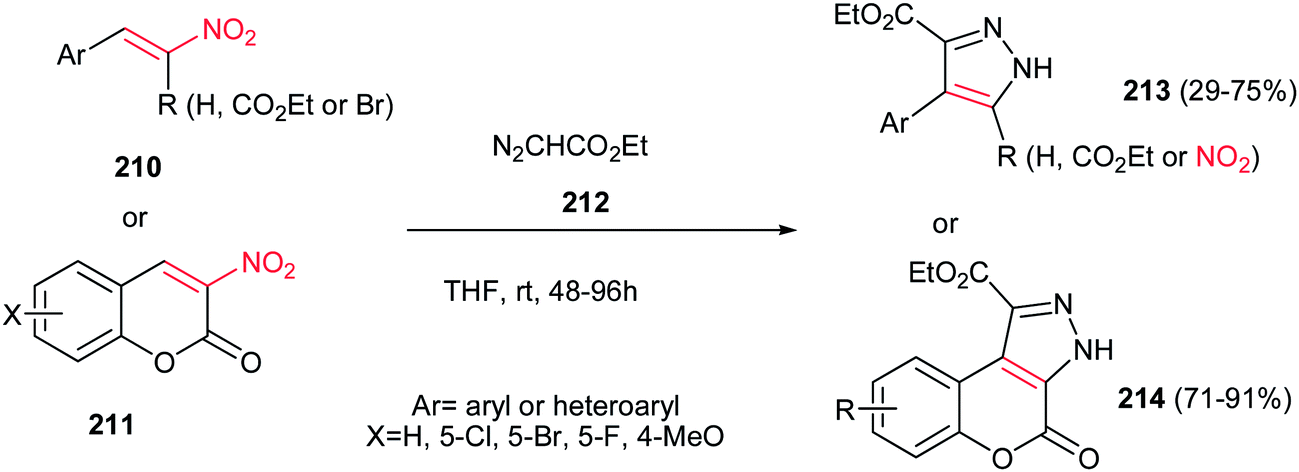
An environmentally benign procedure for synthesis of highly substituted pyrazoles 213 was reported by Xie et al. from different nitroalkenes 210 and ethyl diazoacetate 212 (Scheme 73).152 The reaction was carried out by stirring of a mixture of nitroalkenes 210 (1 equiv.) with an excess amount of ethyl diazoacetate 212 (5 equiv.) in THF for 48–96 h. α-Carbethoxy-1-nitrostyrenes gave higher yields in shorter reaction time compare to simple nitrostyrenes. Reaction of 3-nitrocumarines 211 with 212 also afforded the fused pyrazole rings 214 in high yields. By using α-bromonitroalkenes in this protocol, the nitro group could be retained in the structure of products via elimination of bromide and the corresponding nitropyrazoles could be obtained in good yields.
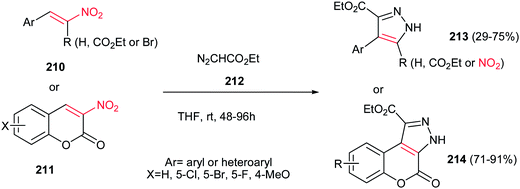 |
| | Scheme 73 Synthesis of highly substituted pyrazoles from nitroalkenes and ethyl diazoacetate. | |
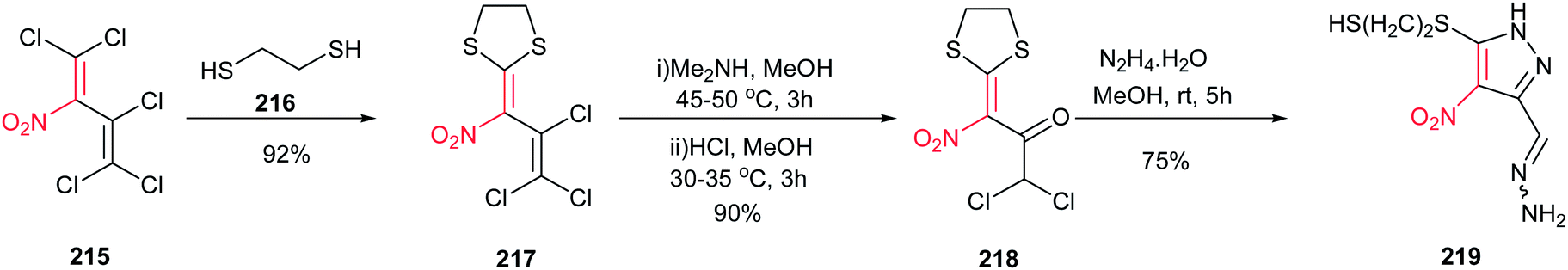
Kaufmann et al. approved that the complex ketones 218, which was prepared from polyhalogenated nitrobutadiene 215 in two steps, is a suitable building block for synthesis of pyrazole ring 219 (Scheme 74). An unexpected reaction between 218 and hydrazine in MeOH at room temperature provide the structurally interesting pyrazole 219 in 75% isolated yield.153
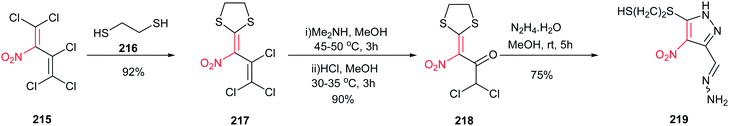 |
| | Scheme 74 Synthesis of substituted pyrazoles starting with polyhalogenated nitrobutadiene. | |
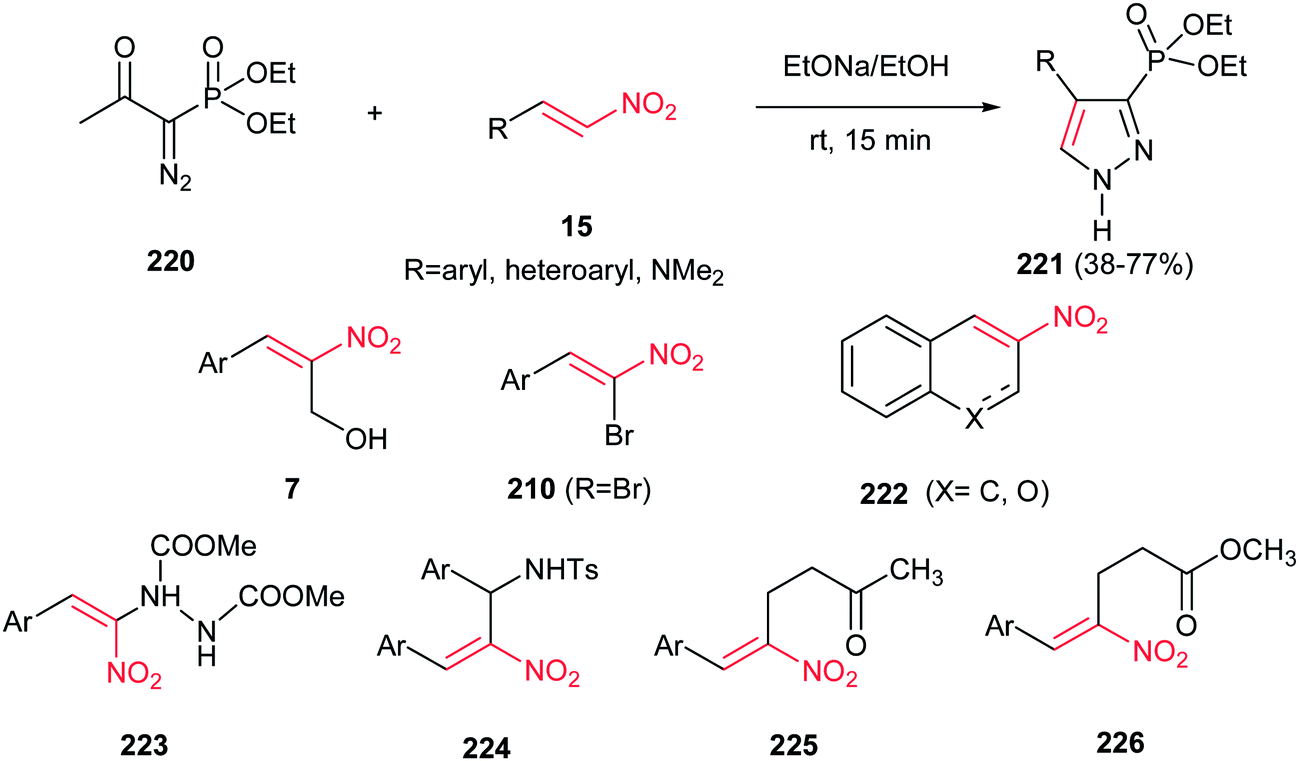
In 2007, Namboothiri and co-workers suggested an interesting one-pot route to phosphonylpyrazoles 221, based on the 1,3-dipolar cycloaddition of the anion of diethyl 1-diazomethylphosphonate, generated in situ from diethyl 1-diazo-2-oxopropylphosphonate (Bestmann–Ohira reagent) 220, with conjugated nitroalkenes 15 (Scheme 75).154 Reactions were promoted by NaOEt/EtOH at room temperature for 15 min to afford the regioisomerically pure 4-substituted 3-phosphonylpyrazoles 221 in moderate to good yields. Heteroaromatic nitroalkenes gave lower yield when compared to aromatic nitroalkenes. Also, phosphonylpyrazole with dimethyl amino group could be synthesized in 64% yield using N,N-dimethyl-2-nitroethenamine. When the 3-nitro-2H-chromene or 2-nitronaphthalene 222 was used in this protocol, the corresponding fused phosphonylpyrazole was obtained with same yields, albeit in longer reaction time. Morita–Baylis–Hillman adducts of conjugated nitroalkenes with various electrophiles 223–226 also included as partner with Bestmann–Ohira reagent. It is notable that the nitro group could be retained in the structure of phosphonylpyrazoles by using α-bromonitroalkenes 210 in the same protocol.
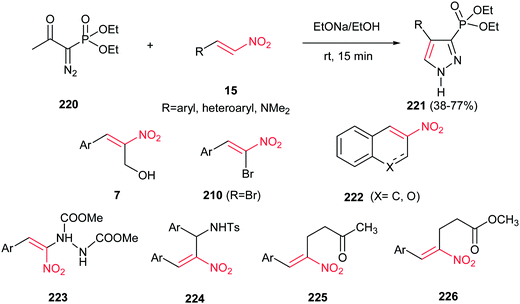 |
| | Scheme 75 Synthesis of phosphonylpyrazole from diethyl 1-diazomethylphosphonate and nitroalkenes. | |
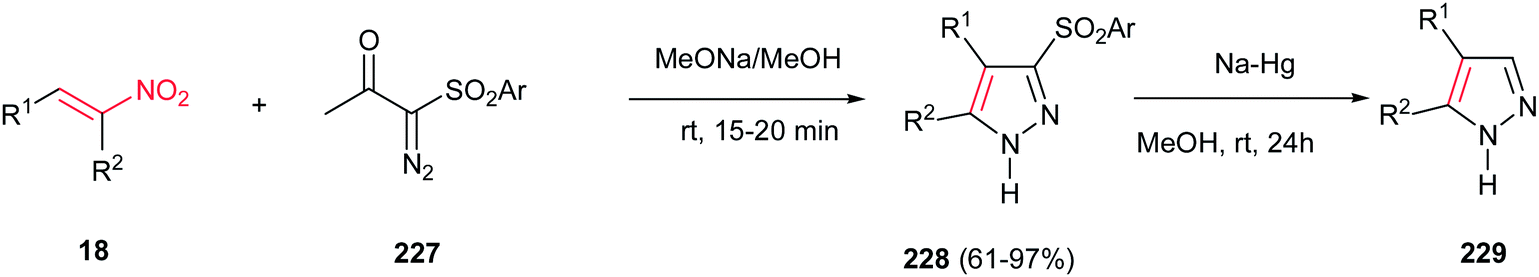
Furthermore, the same group also demonstrated that α-diazo-β-ketosulfones 227 are suitable substrates for synthesis of substituted sulfonylpyrazoles 228 in the reaction with nitroalkenes 18 (Scheme 76).155 The sulfonyl group could be simply removed via reduction with Na/Hg in methanol to give the corresponding substituted pyrazoles 229. It is notable that different groups such as aryl, heteroaryl, styrenyl, alkyl, hydroxymethyl, and hydrazinyl groups could be introduced on the pyrazole ring by the appropriate choice of nitroalkenes.
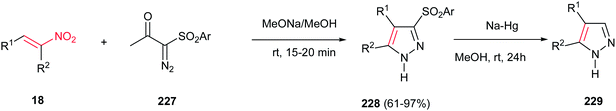 |
| | Scheme 76 Synthesis of substituted sulfonylpyrazoles from α-diazo-β-ketosulfones and nitroalkenes. | |
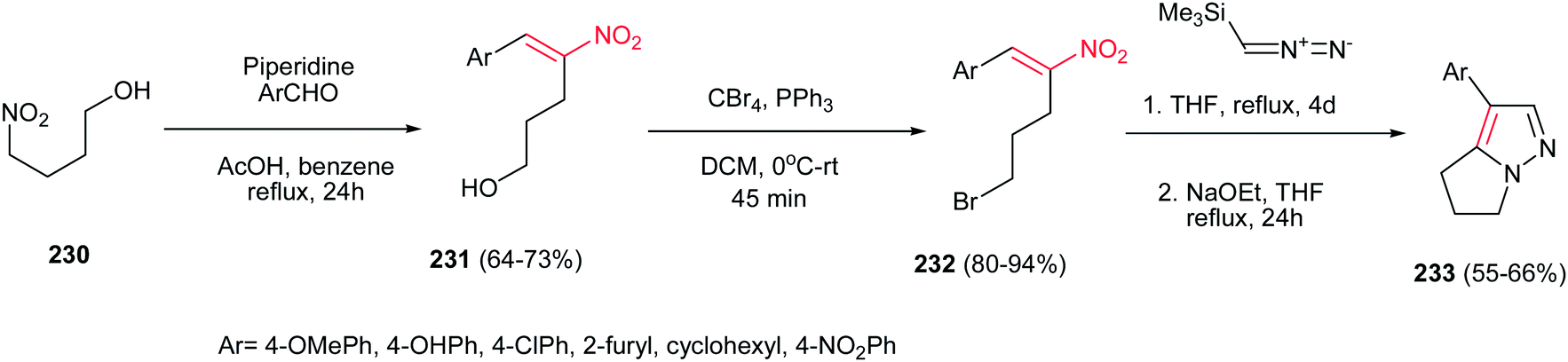
Also, reactions of bromonitroalkenes 232 with commercially available TMSCHN2 were carried out by Namboothiri et al. for synthesis of pyrazole-based withasomnine alkaloids 233 and their non-natural analogues. This one-pot protocol proceeds via 1,3-dipolar cycloaddition-elimination cascade, followed by intramolecular alkylation in the presence of NaOEt in THF at reflux temperature (Scheme 77). Bromo compound 232 was prepared in two steps from 4-nitro-1-butanol 230 via Henry condensation, followed by conversion of the OH group to Br using CBr4 and Ph3P. An overall yield of 39% was obtained for Ar = Ph from the corresponding nitroalcohol 230.156
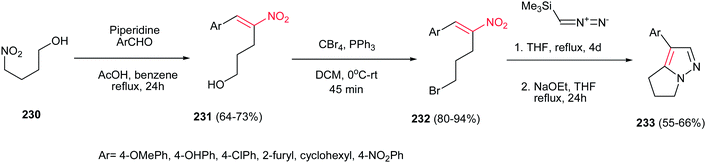 |
| | Scheme 77 Synthesis of pyrazole-based withasomnine alkaloids from bromonitroalkenes and TMSCHN2. | |
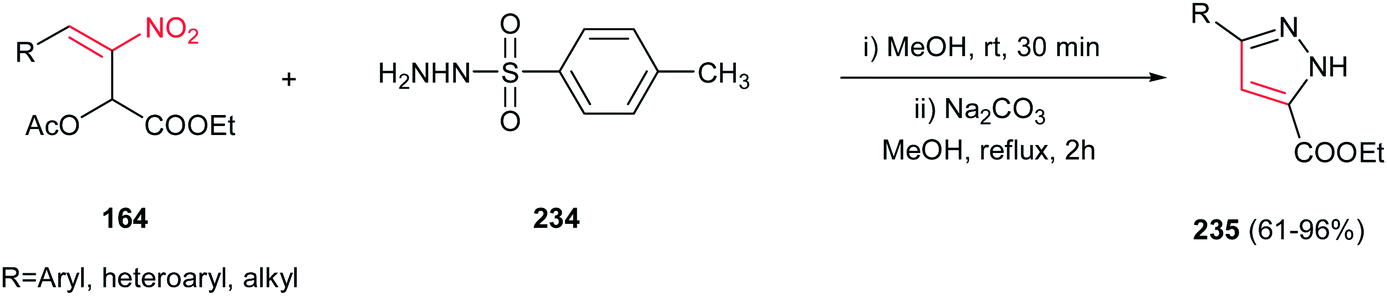
Very recently, an efficient route for regioselective synthesis of pyrazoles 235 was developed by Zou group via reaction of a nitroallylic acetate 164 and N-tosyl hydrazine 234 in methanol for 30 min, followed by addition of Na2CO3 (10 mol%) and refluxing the mixture for 2 h at 65 °C (Scheme 78).157 Aromatic, heteroaromatic and aliphatic nitroallylic acetates are compatible with this protocol to provide high to excellent yields of the products. Performing the reaction under base-free condition afforded the SN2 adducts of nitrogen attack to electrophilic γ-site. With these results in hands, the authors proposed a cascade SN2-Michael addition–aromatization mechanism for this transformation.
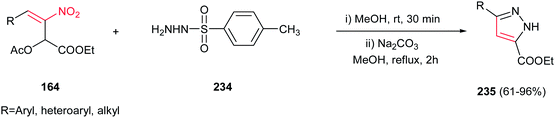 |
| | Scheme 78 Regioselective synthesis of pyrazoles from nitroallylic acetates and N-tosyl hydrazine. | |
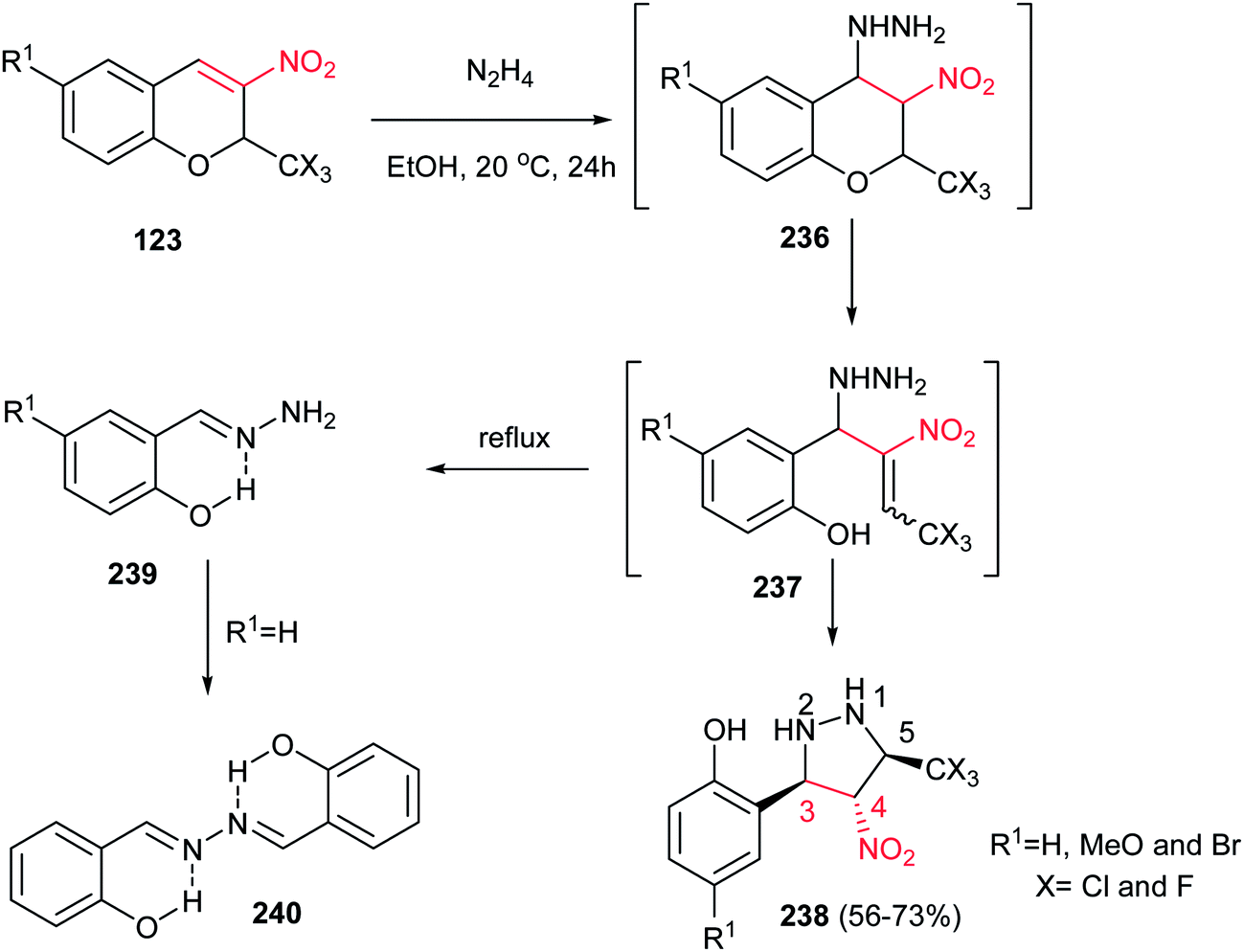
In addition, Sosnovskikh et al. demonstrated a high yielding procedure for synthesis of pyrazolidines 238 from the reaction of 3-nitro-2-trichloromethyl-2H-chromenes 123 with an equimolar amount of 60% hydrazine hydrate in ethanol at room temperature (Scheme 79).158 The corresponding products were obtained as a single, most thermodynamically stable 3,4-trans, 4,5-trans-3-(2-hydroxyaryl)-4-nitro-5-trichloromethylpyrazolidines 238 in 56–73% yields. Phenylhydrazine does not react with chromenes 123 under these conditions. If the amount of hydrazine is increased, or if the reaction is carried out in boiling ethanol, intermediate 237 decomposes to give hydrazone 240 as the predominant product.
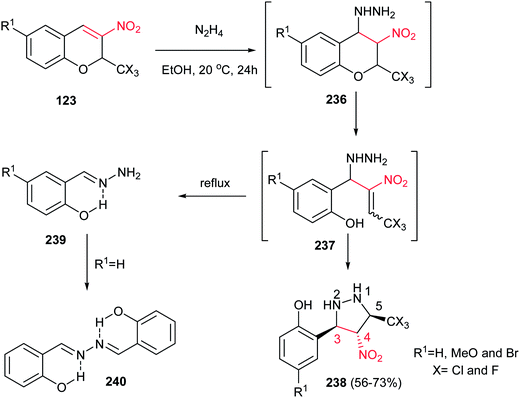 |
| | Scheme 79 Synthesis of pyrazolidines from 3-nitro-2-trichloromethyl-2H-chromenes and hydrazine hydrate. | |
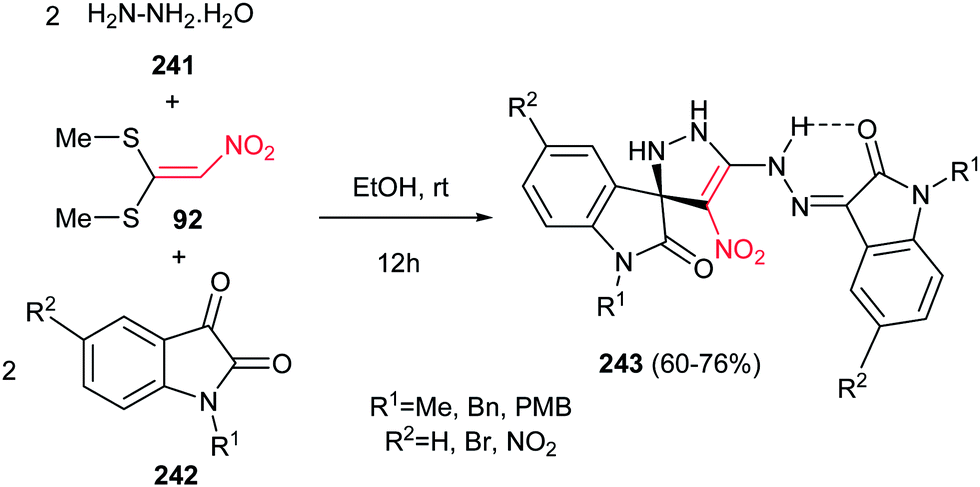
Finally, Alizadeh and Zohreh developed a novel method for preparation of spirooxindole-pyrazoline derivatives 243 in high yields via a one-pot pseudo-five-component reaction of hydrazine hydrate 241, 1,1-bis(methylthio)-2-nitroethylene 92, and isatin derivatives 242 (Scheme 80).159 The products are polynitrogen compounds with potential synthetic and pharmacological interest that will be suitable for further elaboration. The products are strongly colored with high heat resistance. The proposed mechanism shows that the reaction is initiated by formation of 1,1-bis(hydrazine)-2-nitroethylene intermediate from the addition of aqueous hydrazine to 1,1-bis(methylthio)-2-nitroethylene 92, followed by trapping this intermediate by two equivalents of isatin derivatives to give the title compounds.
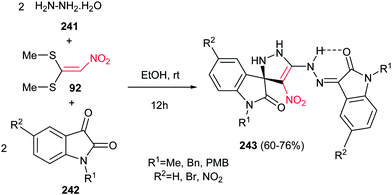 |
| | Scheme 80 A one-pot approach for synthesis of spirooxindole-pyrazolines. | |
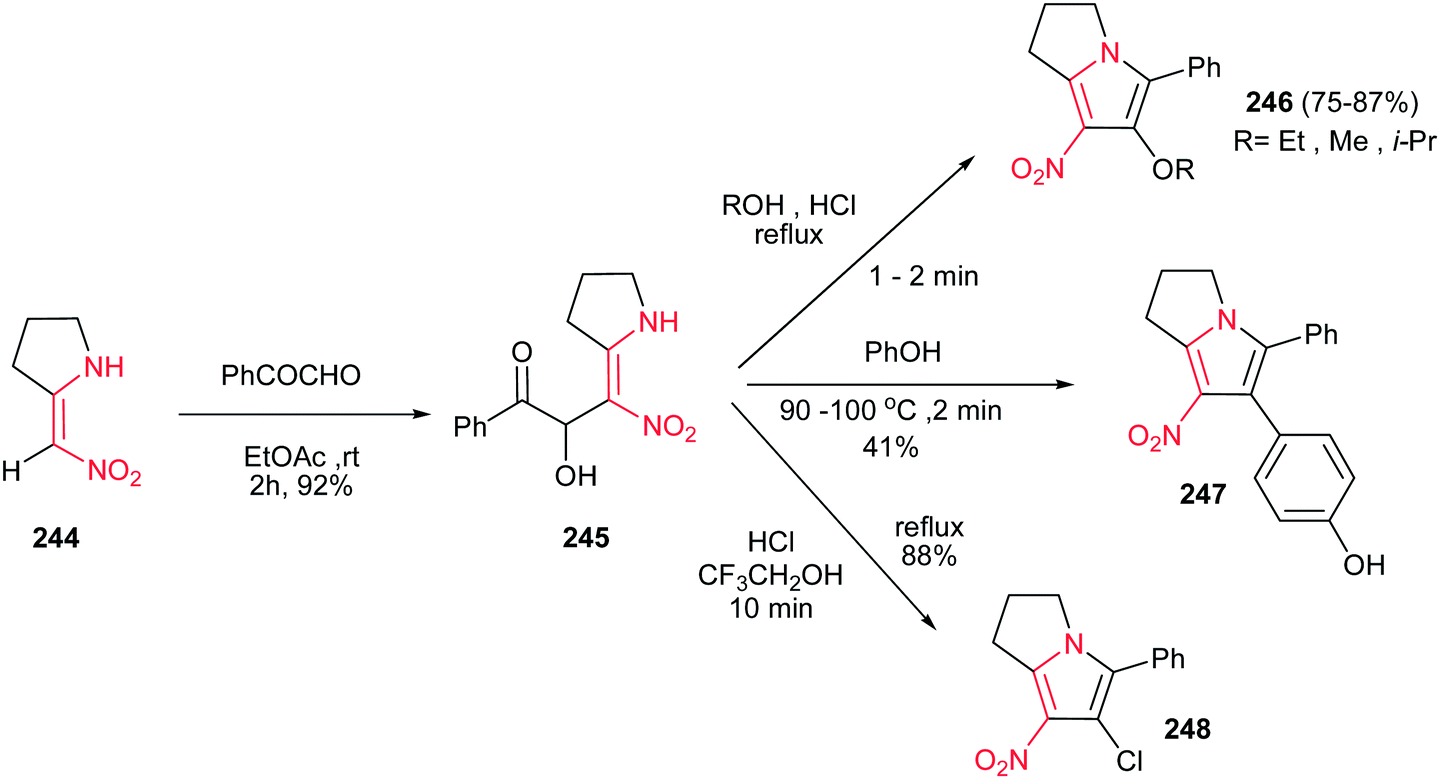
4.1.9. Pyrrolizines, pyrrolizidines and pyrrolizidinones. Pilipecz et al. described that reaction of 2-nitromethylenpyrrolidine 244 with phenylglyoxal in EtOAc at room temperature for 2 h afforded product 245 in 92% yield. These adducts were then transformed to substituted dihydro-1H-pyrrolizines 246 in 75−87% yields by heating in various alcohols in the presence of conc. HCl. Interestingly, by heating of 245 in molten phenol in the presence of HCl, 4-hydroxyphenyl substituted pyrrolizines 247 was obtained in 41% yield via Friedel–Crafts type electrophilic aromatic substitution occurred at C-4 of phenol. Performing this reaction in 2,2,2-trifluoroethanol/HCl solution afforded the chloro derivative 248 in 88% yield (Scheme 81).160
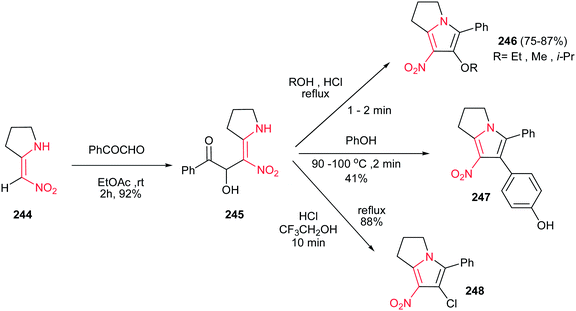 |
| | Scheme 81 Synthesis of dihydropyrrolizines from nitroalkenes. | |
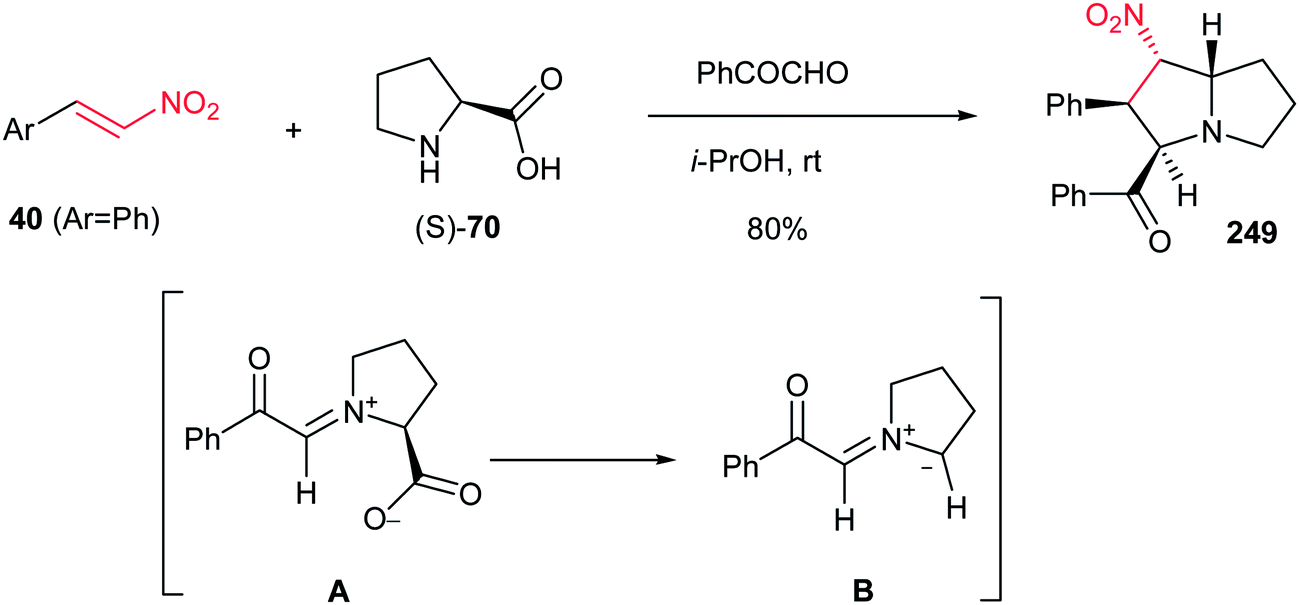
Reaction of phenylglyoxal with β-nitrostyrene 40(Ar = Ph) and an equimolar amount of L-proline (S)-70 in i-PrOH at room temperature affords substituted pyrrolizidine 249 in 80% yield as a single regioisomer. The reaction proceeds via in situ generation of 1,3-azomethine ylide A from phenylglyoxal and (S)-70 by decarboxylation, then 1,3-dipolar cycloaddition reaction with 40 (Scheme 82).161
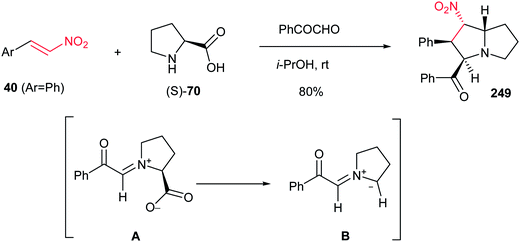 |
| | Scheme 82 Substituted pyrrolizidine from nitroalkene. | |
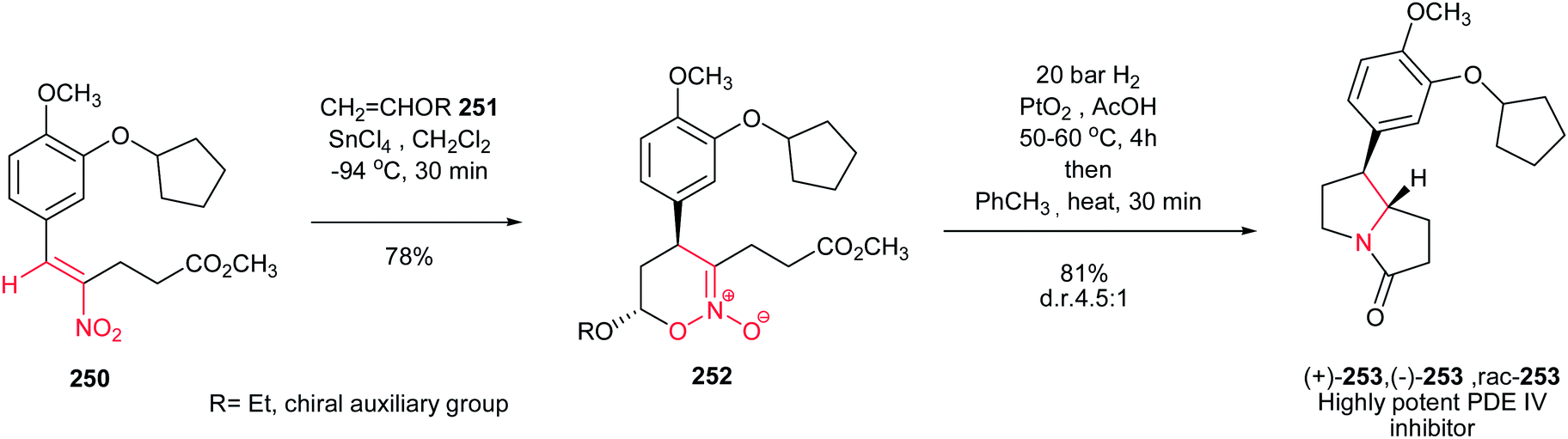
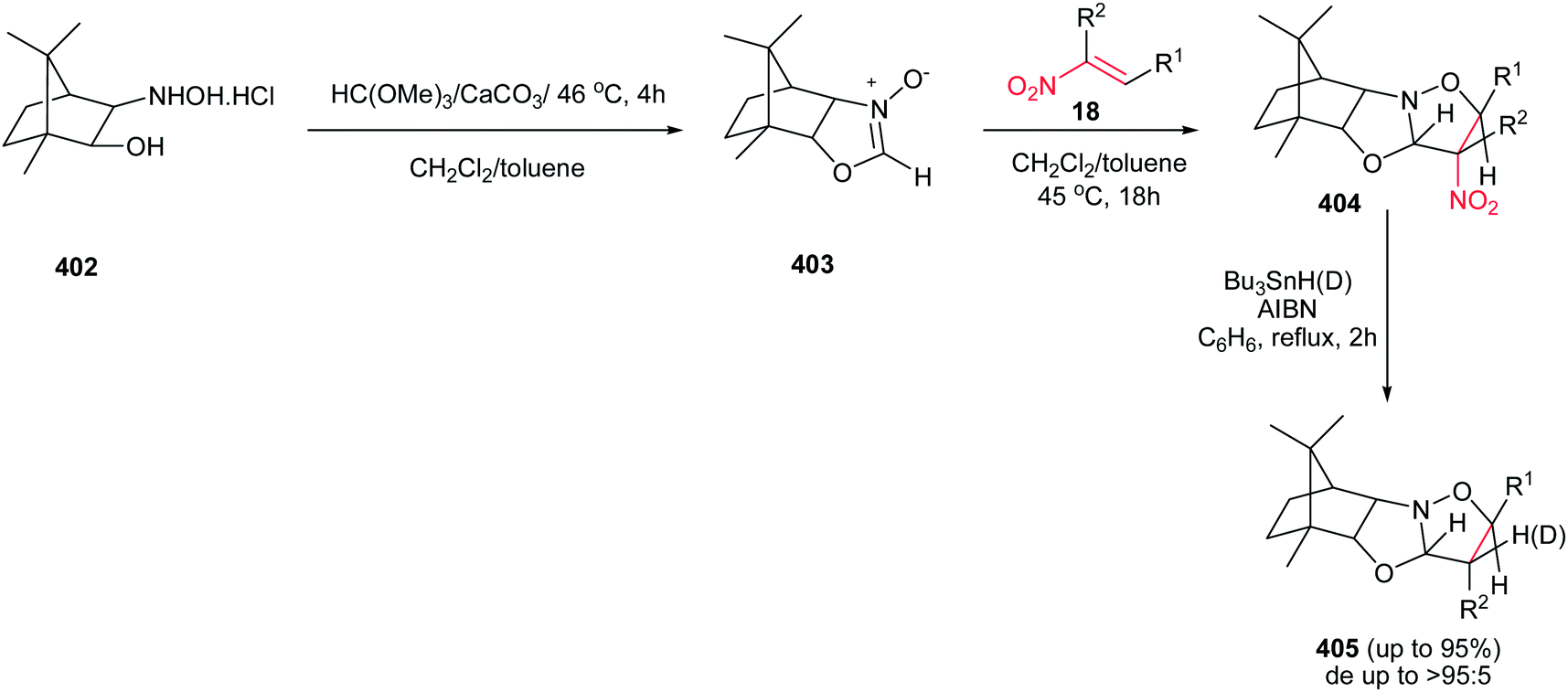
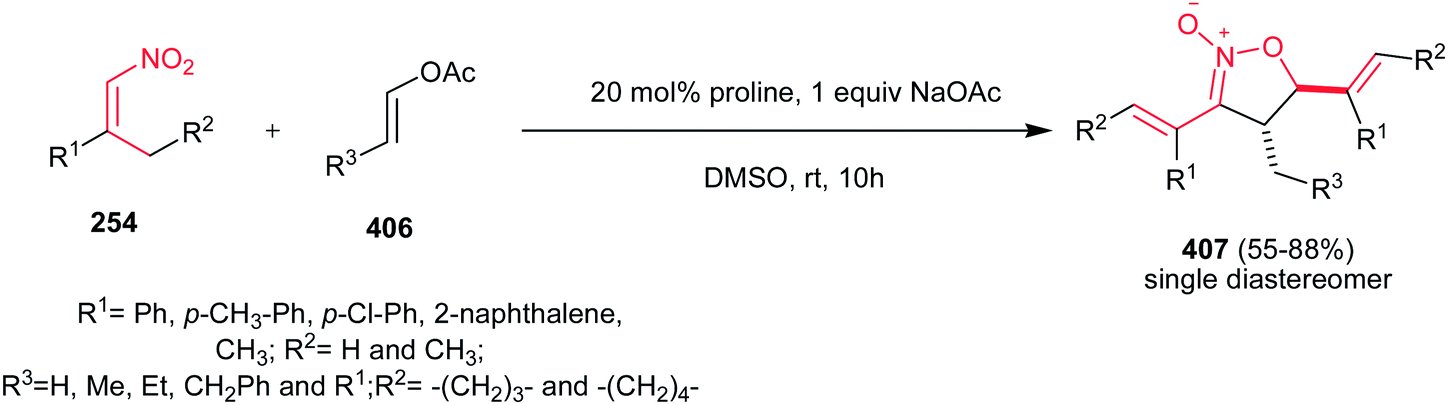
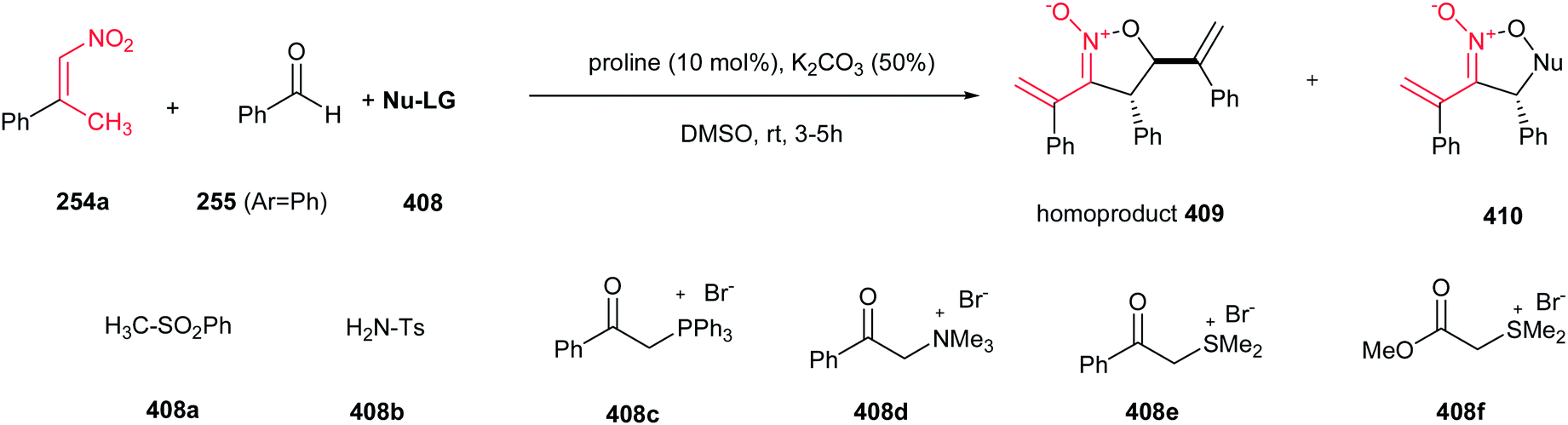
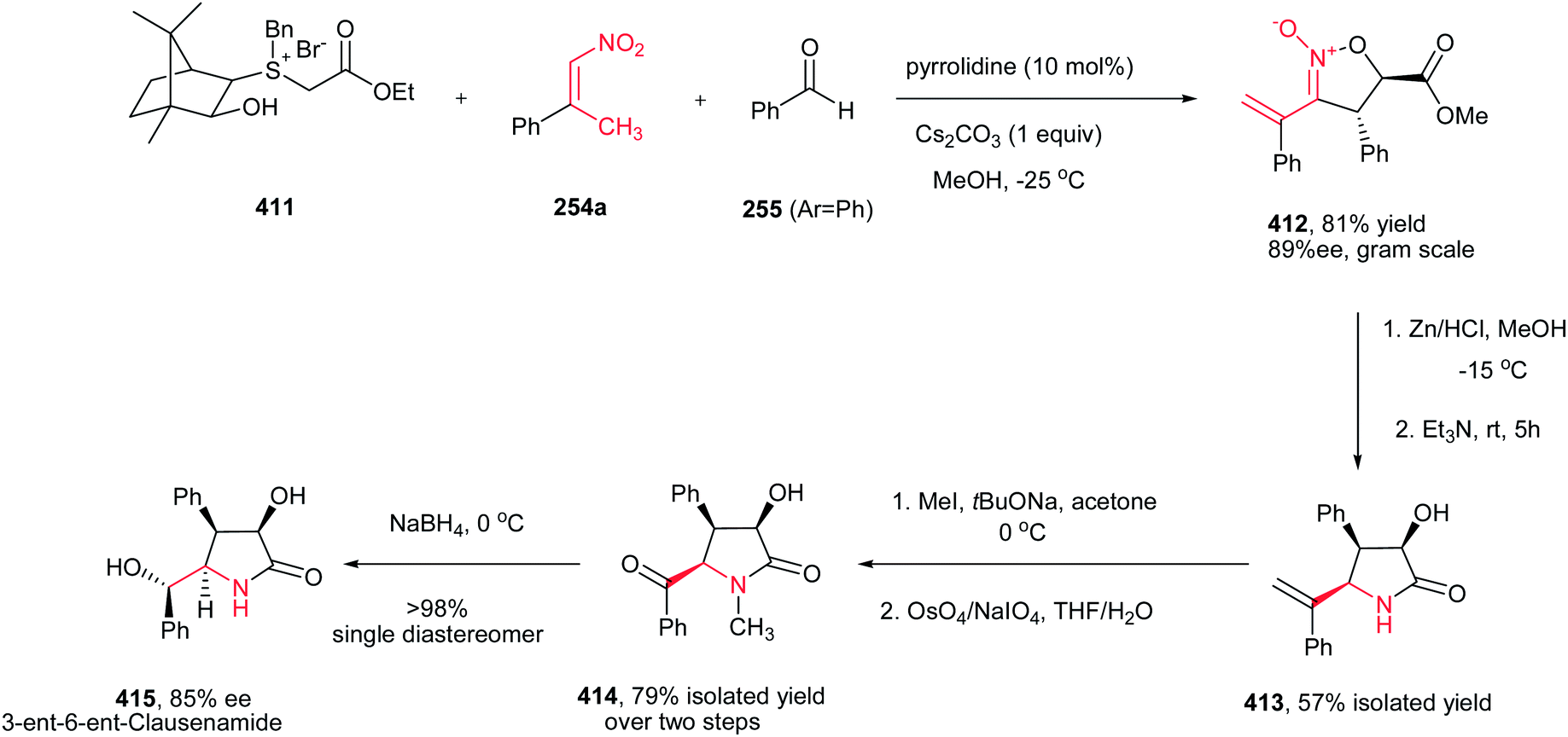
[4 + 2] Cycloaddition reaction of nitroalkene 250 to ethyl vinyl ether 251 was applied as key step for racemic synthesis of GlaxoSmithKline's highly potent PDE IVb inhibitor 253 (Scheme 83).162 Subsequent hydrogenation of nitronate 252 with Adams catalyst (PtO2/AcOH) under 20 bar H2 at 50–60 °C for 4 h afforded the racemic product 253 in high yield and diastereoselectivity. Reduction with other catalytic systems such as Ra–Ni, 5% Pd–C, 0.5% Pd/Al2O3, 5% Pd/CaCO3, 5%Rh/Al2O3 and Rh(PPh3)3Cl gave only low diastereoselectivity of 253. Also, the asymmetric version of this protocol was carried out with using a vinyl ether containing a chiral auxiliary group to give the cyclic nitronate 252 in 8.3:1 diatereomeric ratio. Subsequent reduction of the major diastereomer with Adams catalyst provided the corresponding 253 in 7:1 diastereomeric ratio.
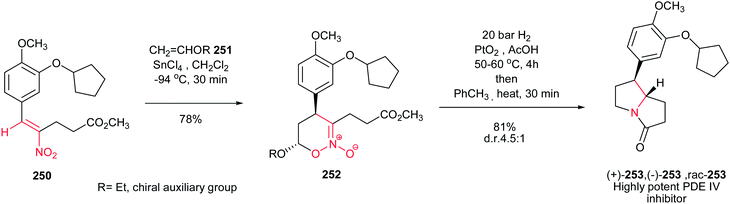 |
| | Scheme 83 [4 + 2] cycloaddition reaction of nitroalkene as key step for synthesis of GlaxoSmithKline's highly potent PDE IVb inhibitor 253. | |
4.1.10. 1,2,3-Triazole derivatives. 1,2,3-Triazoles are an important class of heterocycles with considerable interest due to their wide applications in many pharmaceuticals and their usefulness in synthetic organic chemistry. A large number of 1,2,3-triazoles exhibit various biological activities as antiviral,163 antimicrobial,164 antifungal,165 anticancer,166 anti-HIV,167 β3-selective adrenergic receptor agonists168 and kinase inhibitors.169 However, the scope of triazole chemistry is not confined to drug discovery. They have found wide applications in numerous other areas of modern chemical sciences, such as bioconjugation,170 supramolecular chemistry,171 and polymer sciences.172 In addition, they are commercially employed as anticorrosive agents, agrochemicals, photostabilizer photographic materials, and dyes.173 Substituted 1,2,3-triazoles are commonly prepared by Huisgen's 1,3-dipolar cycloaddition between organic azides and substituted alkynes.174Shi et al. developed a one-pot three-component condensation of a β-alkyl nitroalkene 254, an aldehyde 255 and sodium azide catalyzed by L-proline for synthesis of NH-1,2,3-triazole derivatives 256 (Scheme 84).175 A large number of aryl aldehydes and β-alkyl nitroalkenes 254a–e were used for this transformation and more than 25 new (NH)-triazoles 256 were prepared in good to excellent yields under mild conditions. Substituted azides (n-hexanyl azide and phenyl azide) are not suitable for this cascade process. The presence of vinyl group on the C-4 position allows further elaboration to diversity of other functionalized triazole derivatives. Proposed mechanism by the authors revealed that Henry reaction between nitroalkene 254 and aryl aldehyde 255 furnished the 2-nitro-1,3-diene, which underwent 1,3-dipolar cycloaddition with N3−. Finally aromatization occurred by elimination of NO2−.
 |
| | Scheme 84 Synthesis of NH-1,2,3-triazoles from β-alkyl nitroalkenes, aldehydes and sodium azide. | |
In 2009, reaction of nitroalkene-containing C-glycosides 257 with sodium azide was investigated by Zou et al.176 They described that while at room temperature only the 1,3-dipolar cycloaddition products 258 were obtained, at elevated reaction temperature, the 1,5-disubstituted triazole-fused sugars 259 was obtained as major product in good yield via a tandem β-elimination/cycloaddition/Michael addition (Scheme 85).
 |
| | Scheme 85 Reaction of nitroalkene-containing C-glycosides with sodium azide. | |
4-Aryl-5-cyano- or 4-aryl-5-carbethoxy-1H-1,2,3-triazoles 260 were synthesized by Fringuelli and coworkers via a [3 + 2] cycloaddition reaction of 2-aryl-1-cyano- or 2-aryl-1-carbethoxy-1-nitroethenes 210 with TMSN3 catalyzed by TBAF under solvent-free conditions (Scheme 86).177 This protocol affords good to excellent yields (70–90%) of products. While (E)-2-aryl-1-cyano-1-nitroethene gave high to excellent yield (75–90%) in the presence of 0.1 equiv. of TBAF at 30 °C with TMSN3 (2.0 equiv.) under SFC conditions in less than 3 h, the 2-aryl-1-carbethoxy-1-nitroethene needs 4 equiv. of TMSN3 and higher temperatures (50–80 °C) and longer reaction times (4–12 h) to afford the product in comparable yield. This procedure does not require anhydrous or inert atmosphere.
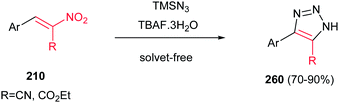 |
| | Scheme 86 Synthesis of 4-aryl-5-cyano- or 4-aryl-5-carbethoxy-1H-1,2,3-triazoles. | |
In 2005, Zard et al. have shown that reaction of nitroalkenes 40 and 262 or vicinal acetoxy nitro derivatives (as nitroalkene precursor) with sodium azide in hot dimethyl sulfoxide (80–90 °C) gave the corresponding 1,2,3-triazoles 261 in excellent yields (Scheme 87).178 Aliphatic and aromatic niroalkenes were compatible with this protocol. They reported that the amount of sodium azide is crucial for this reaction and in the presence of at least 4 eq. of sodium azide, the triazole was the only product.
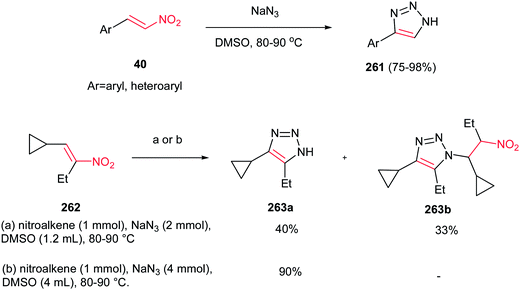 |
| | Scheme 87 Catalyst-free synthesis of triazoles from nitroalkenes and NaN3. | |
Very recently, condensation of 2-alkynyl nitroolefin 264 (obtained from 2-alkynylbenzaldehyde) and sodium azide in DMSO at 100 °C was introduced by Kundu et al. as an efficient route for synthesis of tricyclic triazoloisoquinolines 265 in excellent yields (Scheme 88).179 The reaction proceeded via a domino [3 + 2] cycloaddition/extrusion of the nitro group/regioselective 6-endo cyclization sequence. Using other solvents such as DMF, toluene, toluene:H2O and CH3CN:H2O (9:1) gave trace to low yield of products. No variation in yield was observed by varying the R1 and R2 groups, except for R2 = CN, in which case no product was observed.
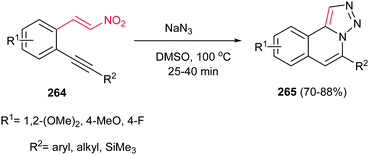 |
| | Scheme 88 Synthesis of tricyclic triazoloisoquinolines from 2-alkynyl nitroolefin. | |
4.1.11. Tetrazole derivatives. Kaufmann et al. demonstrated that the reaction of 1,1,2-trichloro-2-nitroethene 266 with excess benzotriazole (BztH) affords 1,1-bis(benzotriazol-1-yl)-2-chloro-2-nitroethene 267 in excellent yields. Then, transamination of 267 with different aniline derivatives provides the corresponding 1-(arylimino)-1-(benzotriazolyl)ethanes 268, which produces the product 269 via cycloaddition with sodium azide. By using the arenediamines instead of aniline derivatives, the corresponding bis tetrazoles were also obtained in excellent yields. The tetrazoles 269 are valuable precursors for further constructions on the side chain (Scheme 89).180
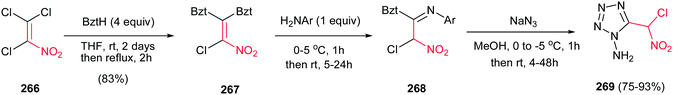 |
| | Scheme 89 Synthesis of tetrazoles starting with 1,1,2-trichloro-2-nitroethene. | |
4.2. Synthesis of O-heterocyclic compounds
4.2.1. Tetrahydrofurans derivatives. Nitroalkenes were used as efficient starting materials for synthesis of several tetrahydrofuran lignans which have found diverse biological activity in recent years. In this context, highly functionalized 4-(1-haloalkyl)-3-nitrotetrahydrofurans 273a–d were prepared by an oxidative tandem process consisting of conjugate addition reaction of lithium allyloxides of 270 to nitroalkenes 271 followed by single electron transfer oxidation of the resulting nitronates 272 in moderate to good yield and moderate to very good diastereoselectivity (Scheme 90).181 The authors found that the optimal conditions for the tandem reactions consist of using butyl lithium as the base in dimethoxyethane and using cupric halides as single electron transfer oxidants. The nitro- and chloride functionalities in the structure of products can be easily modified to other interesting structures. Also this methodology should be easily applicable to other Michael acceptors such as α,β-unsaturated esters, amides or nitriles.
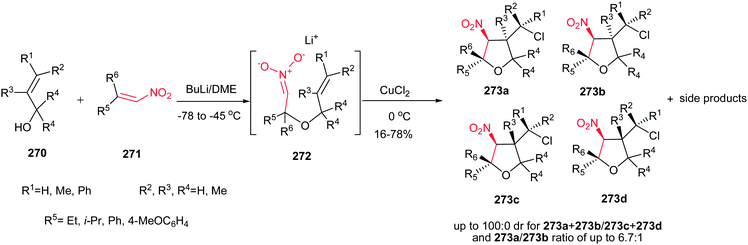 |
| | Scheme 90 Tandem alkoxide conjugate addition/radical cyclization. | |
The same strategy was applied by the same authors for synthesis of Galgravin 277a and Veraguensin 277b in only three or four steps from nitroalkenes 274 and allylic alcohols 275.182 While the corresponding bromo derivatives 276 can be directly reduced and denitrated by excess Bu3SnH/AIBN at reflux in toluene in good yield, the chloro derivatives need two steps for halide and nitro removal via treatment with Bu3SnH/AIBN followed by reduction with LiAlH4. Also β-nitro ethers 278, simply prepared from the starting materials using BuLi, undergo reductive radical cyclizations with Bu3SnH/AIBN to afford Galbelgin 279a and Ganschisandrin 279b as major products (Scheme 91).
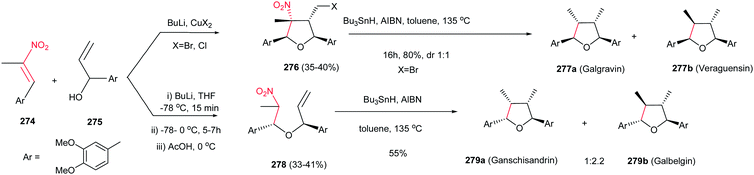 |
| | Scheme 91 Tetrahydrofuran lignans via tandem oxidative anionic−radical processesor reductive radical cyclizations. | |
Namboothiri et al. demonstrated that the Michael addition reaction of a O-, S-, N-, and C-centered nucleophiles possessing unsaturated tether 281 to β-furyl nitroethylene 280 furnish a suitable building block 282 for further construction via intramolecular Diels–Alder reaction of furan diene to give five- and six-membered carbocycles and heterocycles 283 fused to an easily cleavable oxabicycloheptene moiety. Cleavage of the oxa-bridge in the cycloadducts with BF3·OEt2 (path A) and Ac2O/H2SO4 (path B) afford novel multifunctional fused tetrahydofurans 284 and 285 in 51% and 73%, respectively (Scheme 92).183
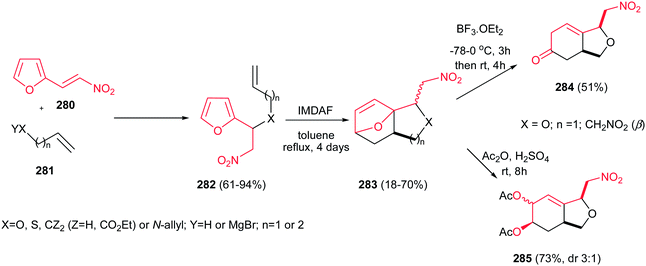 |
| | Scheme 92 Fused tetrahydrofurans via Michael-initiated intramolecular Diels–Alder furan reaction. | |
Rodrigues et al. described that addition of nitroalkenes 18 to a solution of prop-2-ynyl alcohols 286 in a mixture of benzene and t-BuOH containing t-BuOK, led to formation of 3-methylenetetrahydrofurans 287a in moderate to good yields and high diastereoselectivities, which in some cases were accompanied by the formation of corresponding dihydropyrans 287b (Scheme 93).22 The reaction proceeded with total diastereoselectivity due to allylic 1,3-strain. Among other bases examined in this transformation (BuLi, NaH, KH, K2CO3 and Cs2CO3), only Cs2CO3 gave similar results but in lower yield.
 |
| | Scheme 93 Synthesis of 3-methylenetetrahydrofurans via Michael addition/cyclization sequence. | |
In 2001, Dulcere et al. developed a two-step protocol for the construction of a series of vinylidene tetrahydrofurans 290 as a single diastereomer in high yield via KOt-Bu mediated oxy-Michael addition of propargyl alcohols 288 to nitroalkenes 18 followed by SNi′ ring-closure (Scheme 94).184 Different leaving groups such as chloride, bromide, and alkysulfonate examined, provided comparable results. The trans relationship between the nitro and the alkyl substituent R2 is due to conformer 289 which is favored to avoid allylic strain.
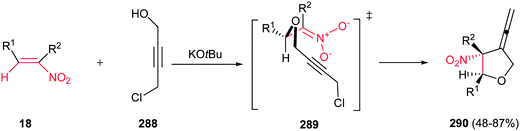 |
| | Scheme 94 Sequential Michael addition-SNi′ displacement. | |
Alexakis et al. reported an enantioselective procedure for synthesis of chiral nitrosubstituted tetrahydrofuranyl ethers 294 via a tandem Michael/acetalisation/cyclisation reaction using combination of Cat-8 and a gold complex as catalytic system (Scheme 95).185 The reaction occurred between alkyne-tethered nitroalkenes 291 and aldehyde 292 to generate the corresponding products 294 in good yields and high diastereo-and enantioselectivities of up to 94% de and >99% ee, respectively. In this work, the reaction initiated with amine-catalyzed Michael addition followed by sequential addition of Au(I) complex to achieve acetalisation/cyclisation sequence. It is notable that p-TsOH was used to prevent deactivation of the Au(I) catalyst by the secondary amine catalyst.
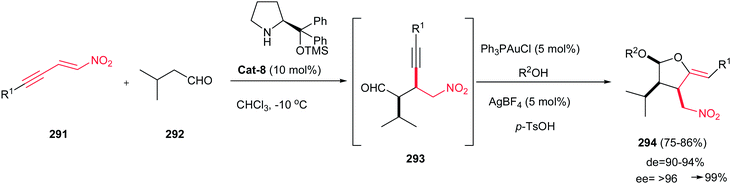 |
| | Scheme 95 Chiral nitrosubstituted tetrahydrofuranyl ethers via tandem Michael/acetalisation/cyclisation reaction. | |
In addition, Hou and coworkers reported another asymmetric procedure for synthesis of substituted tetrahydrofurans 296 in high yields and stereoselectivities via [3 + 2]-cycloaddition reaction of vinyl epoxide 295 and nitroalkenes 15 using Pd/1,1′-ferrocene-P,N-ligand (L-10) (Scheme 96). Aliphatic nitroalkenes afford the products in similar yields and enantioselectivities compared to aromatic niroalkenes, but in lower diastereoselectivities.186
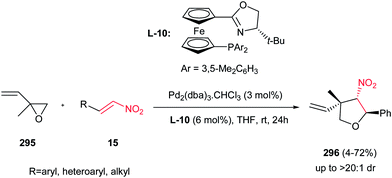 |
| | Scheme 96 Pd/1,1′-ferrocene-P,N-ligand catalyzed [3 + 2]-cycloaddition reaction of vinyl epoxide and nitroalkenes. | |
Another approach for synthesis of tetrahydrofurans is [3 + 2] cycloaddition reaction of carbonyl ylides with alkenes.187 In this context, the Rh2(OAc)4-catalyzed reaction of a nitrostyrene 40 with dimethyl diazomalonate 297 and an aldehyde 85 was investigated by Nair et al. to afford tetrahydrofurans 298 in 76% yield as a single diastereomer (Scheme 97).188 These transformations proceed via Rh-catalyzed generation of a carbonyl ylide 299 from the diazo compound and the aldehyde, which then undergoes a [3 + 2] dipolar cycloaddition with the nitroalkene. It is notable that these transformations are only effective with electron-poor alkenes.
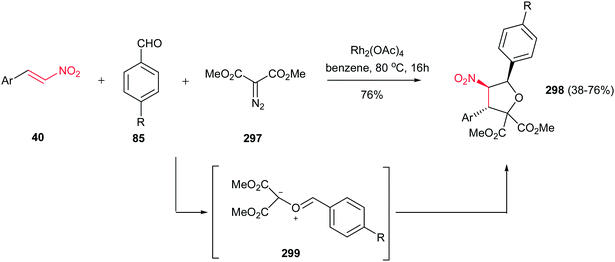 |
| | Scheme 97 Rh-promoted synthesis of tetrahydrofurans from nitrostyrenes, dimethyl diazomalonate and aldehydes. | |
4.2.2. Furans and dihydrofurans. Furans and their derivatives are an important class of compounds due to their widespread occurrence in nature and versatile applications in medicinal chemistry and pharmaceutical industry.189 The biological activities such as anti-microbial,190 anticancer,191 and tubulin binding properties192 of furan containing compounds are well-documented in the literature. The classical methods for synthesis of furan derivatives are cyclocondensation of 1,4-dicarbonyl compounds (Paal–Knorr synthesis)193 and that of α-haloketones or analogous compounds with β-dicarbonyl compounds (Feist–Benary synthesis).194 Recently, transition metal-mediated cycloisomerization of alkynyl and allenyl substrates195 and feasible cascade processes196 have emerged as an efficient strategy for the synthesis of highly substituted furanes.Dihydrofuran derivatives have also shown pharmacological properties, such as antibacterial,197 antifungal198 and anticancer activities.199 Also they are valuable potential intermediates in the synthesis of many biologically active compounds.200
Very recently, Rodriguez et al. reported a simple and one-pot procedure for synthesis of substituted furans 301 starting with a nitroalkene 210. The furan ring is formed via a formal [3 + 2] cycloaddition of a 1,2-dicarbonyl or 2-ketophosphonate ester 300 and an α-halo-α-nitroalkene 210 in one pot manner using DBU as the base in excellent yields (51–92%) in a relatively short time (10–30 min) (Scheme 98). The cycloaddition with ketophosphonate is particularly attractive because ketophosphonates can serve as synthetic handles in cross-coupling reactions.201
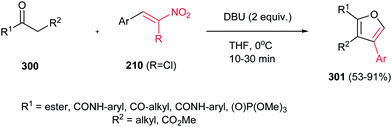 |
| | Scheme 98 Synthesis of substituted furans via [3 + 2] cycloaddition of a 1,2-dicarbonyl or 2-ketophosphonate ester and an α-halo-α-nitroalkene | |
Namboothiri et al. described a highly regioselective cascade reactions of β-dicarbonyl compounds 303 with Morita–Baylis–Hillman acetates of nitroalkenes 164 and 302 for preparation of functionalized and fused furans 304–305 in high yields (Scheme 99).202 The reaction promoted by DABCO and proceeded in a cascade Michael-5-exo-trig-oxa-Michael fashion in the case of open chain 303a and six-membered 303b cyclic β-dicarbonyl compounds afforded fused furans 304–305. For five-membered cyclic β-dicarbonyl compounds such as cyclopentane-1,3-dione 303c, a cascade Michael-5-endo-trig-oxa-Michael reaction takes place to afford fused 4H-pyrans 306. A proposed mechanism for preparation of these products is given in Scheme 100. However, Meldrum's acid and barbitoric acid did not give the corresponding products.
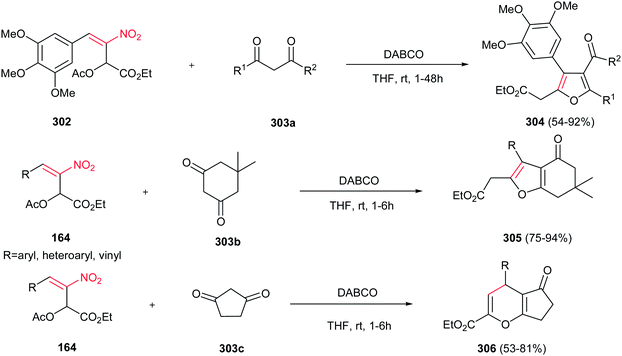 |
| | Scheme 99 Synthesis of furans and pyrans from β-dicarbonyl compounds and Morita–Baylis–Hillman acetates of nitroalkenes. | |
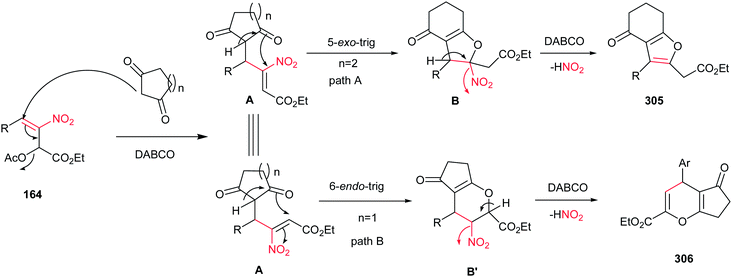 |
| | Scheme 100 Proposed mechanism for preparation of furans and pyrans. | |
A one-pot synthesis of tetrasubstituted furan derivatives 308 has been developed by Palmieri et al. starting with α-functionalized carbonyl derivatives 307 and β-nitroacrylates 118, catalyzed by acidic alumina under catalyst-free conditions (Scheme 101).203 This process gives at least the presence of two powerful functionalities in the 3- and 4-positions of furan rings.
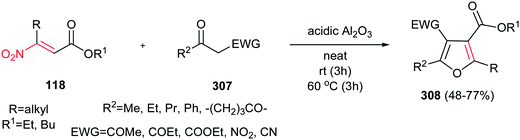 |
| | Scheme 101 Acidic alumina-catalyzed synthesis of tetrasubstituted furans from α-functionalized carbonyl derivatives and β-nitroacrylates. | |
Sosnovskikh et al. demonstrated that reaction of 1,3-dicarbonyl compounds 309 with (E)-1,1,1-trifluoro-3-nitrobut-2-ene 310 in the presence of NaOAc in ethanol at rt, afforded β-(trifluoromethyl)furans 311 in good yields (Scheme 102). Meldrum's acid and its derivatives did not react with 310 under the same conditions which may be because of the lack of enolic form in solution.204
 |
| | Scheme 102 Synthesis of fused β-(trifluoromethyl)furans from (E)-1,1,1-trifluoro-3-nitrobut-2-ene and 1,3-dicarbonyl compounds. | |
Shi et al. developed a highly efficient cascade synthesis of dihydrofurans via the same strategy as outlined in Scheme 84, but with using 1,3-diketone/β-keto-esters instead of sodium azide (Scheme 103).205 Proline (5 mol%) was examined as efficient catalyst for this approach to provide dihydrofurans in excellent yields (up to 95%) and diastereoselectivity (only trans isomers). Since one equivalent of HNO2 would be generated in this process, application of 1.0 equiv. of base (0.5 equiv. of K2CO3) significantly improves the reaction yield. With proper selection of starting materials, substituted groups on all the four positions of furan could be controlled.
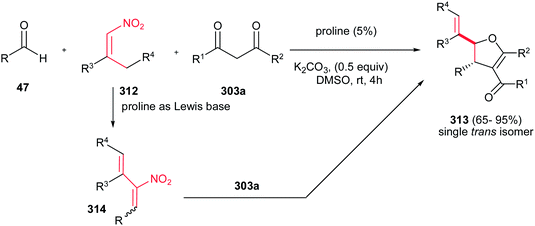 |
| | Scheme 103 Proline-catalyzed synthesis of dihydrofurans from 1,3-diketone/β-keto-esters, aldehydes and nitroalkenes. | |
Reactions of curcumins 315 with α-bromonitroalkenes 210 afford dihydrofurans 316 with two contiguous chiral centers through an intermolecular Michael addition-intramolecular nucleophilic substitution (O-alkylation), which is analogous to an ‘interrupted’ Feist–Benary reaction. No double Michael adducts were isolated in these reactions. The products were obtained as single diastereomers in excellent yields (Scheme 104).206
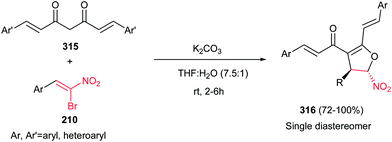 |
| | Scheme 104 Dihydrofurans from curcumins and α-bromonitroalkenes. | |
A one-pot asymmetric synthesis of tetronic acid derivatives 318 in good yields and with excellent enantioselectivities was reported by Yan et al. in 2012.207 The reaction proceeds via asymmetric conjugate addition of ethyl 4-chloro-3-oxobutanoate 317 to nitroalkenes 15 catalysed by 6′-dimethyl quinine Cat-10 and subsequent intramolecular cyclization promoted by AcOLi. Various β-aryl, heteroaryl, and alkyl nitroalkenes are generally applicable in the reaction (Scheme 105). The absolute configuration of the products was assigned as R by X-ray diffraction analysis. Furthermore, the products are synthetically useful for the preparation of chiral aza-mimics of prostaglandins and γ-lactams.
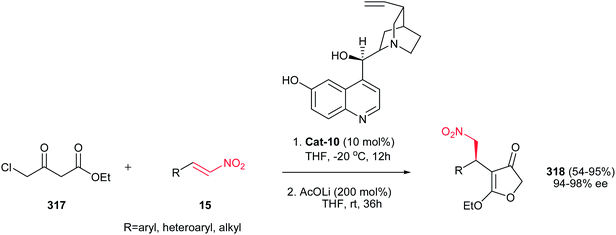 |
| | Scheme 105 Synthesis of tetronic acid derivatives. | |
Very recently, reactions of 4-hydroxycoumarin 319 and nitroolefins 40 were investigated by Wang et al. to afford the 2,3-dihydrofuro[3,2-c]-coumarin type adducts 320 (fused dihydrofurans) in moderate yields (53–75%) and satisfactory enantioselectivities (64–90% ee) (Scheme 106).208 The reaction proceeded via domino Michael addition–intramolecular cyclization, followed by dehydration and tautomerization in the presence of chiral bifunctional thiourea Cat-11 (20 mol%) and 2,6-difluorobenzoic acid (20 mol%) as additive in 1,4-dioxane for 5 days. The electronic feature and position of the substituents on the aromatic ring of nitroalkenes have only slight effects on the yields, while striking effects on enantioseletivities. In this context, Yao and coworkers described that by using 4-hydroxy-N-methylquinolinone 321 instead of 319, the corresponding fused 5-hydroxyimino-4,5-dihydrofurans 322 can be obtained as inseparable mixture of E:Z isomers (E:Z ratio of up to 40:1) (Scheme 107).209
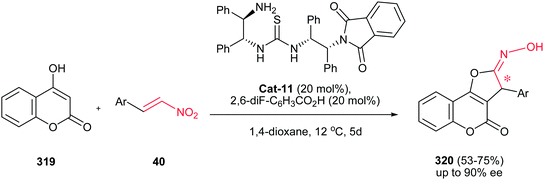 |
| | Scheme 106 Asymetric addition of 4-hydroxycoumarin to nitroalkenes. | |
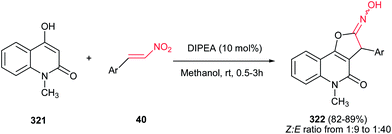 |
| | Scheme 107 Synthesis of fused 5-hydroxyimino-4,5-dihydrofurans from 4-hydroxy-N-methylquinolinone and nitroalkenes. | |
Wang and co-workers described a four-component protocol for the direct synthesis of 2-arylideneamino-3-aryl-4H-furo[3,2-c]chromen-4-ones 323 from substituted nitrostyrenes 40, aromatic aldehydes 72, coumarins 319, and ammonium acetate (Scheme 108).210 Different reaction conditions were tested and the best results were obtained when a mixture of a nitroalkene 40 (1 equiv.), 4-hydroxy coumarin (1 equiv.), and piperidine (50 mol%) were stirred in EtOH at room temperature for 12 h, followed by addition of an aldehyde 72 (1 equiv.) and ammonium acetate (1 equiv.) and stirring for 12 h at the same temperature, then refluxing the final mixture for 3 h. Nitrostyrenes and aromatic aldehydes with both electron-donating and -withdrawing groups were amenable for this reaction providing the products in moderate to good yields.
 |
| | Scheme 108 A four-component protocol for the direct synthesis of 2-arylideneamino-3-aryl-4H-furo[3,2-c]chromen-4-ones. | |
Furthermore, Parra et al. demonstrated a facile and efficient protocol for the synthesis of trans-dihydroarylfuran derivatives 325 from (Z)-bromonitroalkenes 210 and naphthol or phenol derivatives 324 in good yields and excellent enantioselectivities by using squaramide catalysis Cat-12 (Scheme 109).211 The reaction proceeds via a Michael–Friedel–Crafts reaction followed by a nucleophilic substitution on the bromide carbon. The use of a base for neutralization of generated HBr is a key point in this reaction. Nitroalkenes with bulkier alkyl groups on the β position were not compatible, due to the steric hindrance of the first step. The absolute configuration of products was determined as (1S,2S).
 |
| | Scheme 109 Synthesis of trans-dihydroarylfurans from (Z)-bromonitroalkenes and naphthols or phenols. | |
In 2011, Yu et al. reported a biocatalytic route for synthesis of 5-hydroxyimino-4,5-dihydrofurans and their fused derivatives 327a via a coupling reaction between β-nitrostyrenes 40 and 1,3-dicarbonyl compounds 326 catalyzed by the lipase derived from porcine pancreas (PPL) (Scheme 110).212 Aromatic and heteroaromatic nitroalkenes with different substitutions were successfully applied in this protocol to give the corresponding products in high yields and stereoselectivity (Z/E up to 99:1), along with the Michael adducts 327b as minor products. 1,3-Cyclohexanedione 326 (n = 1) showed better reactivity and lower stereoselectivity compared to linear 2,4-pentanedione 326 (n = 0) under similar reaction conditions. However, 1,3-cyclopentanedione, ethyl acetoacetate, and diethylmalonate could not give the corresponding cyclic products and the Michael adducts were only achieved.
 |
| | Scheme 110 A biocatalytic route for synthesis of 5-hydroxyimino-4,5-dihydrofurans. | |
Although reaction of 2-naphthols 324 with simple nitroalkenes 15 gave the Michael adducts 328a in the presence of thiourea–tertiary amine organocatalysts at −50 °C for 96 h, surprisingly, Chen et al. reported that by expanding the reaction time to 144 h, optically pure dimeric 1,2-dihydronaphtho[2,1-b]furanyl-2-hydroxylamine derivatives 328b can be obtained as major product (Scheme 111).213
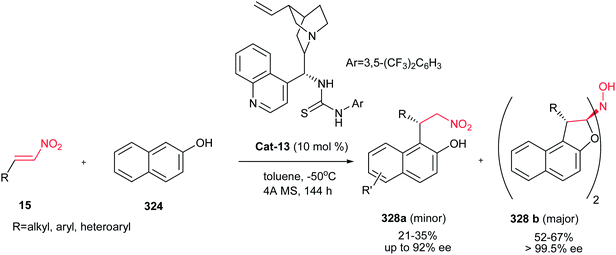 |
| | Scheme 111 Asymmetric Friedel–Crafts/cascade reaction of 2-naphthols and nitroalkenes. | |
Finally, reaction of ethyl 4,4,4-trifluoro-3-oxobutanoate 329 and nitroalkenes 40 were investigated by Song et al. to prepare trifluoromethyl-substituted furan derivatives 330a (major product) and 330b (minor product) (Scheme 112).214 The reaction proceeded via Michael addition followed by intramolecular cyclization reaction. While treatment of 330a with 1.2 equivalents of TsOH in refluxing t-BuOH afforded ethyl 2-hydroxy-5-imino-4-aryl-2-(trifluoromethyl)-2,5-dihydrofuran-3-carboxylates 330d in good yields, with less sterically hindered alcohols such as EtOH and MeOH, ethyl 2-alkoxy-5-oxo-4-aryl-2-(trifluoromethyl)-2,5-dihydrofuran-3-carboxylates 330c was formed as major products. In i-PrOH, 330c and 330d could be obtained in approximately equal amount. Furthermore, they proved that 330d could be directly synthesized by one-pot, tandem reaction of 329 with 40 in t-BuOH.
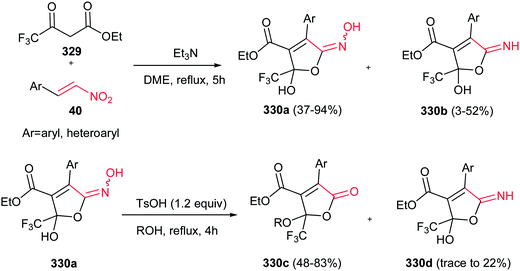 |
| | Scheme 112 Reaction of ethyl 4,4,4-trifluoro-3-oxobutanoate and nitroalkenes. | |
4.2.3. Benzofurans, naphthofurans and their reduced derivatives. The presence of nitro group in the structure of benzofurans allows further constructions leading to formation of many biologically active compounds. In this context, Liu et al. reported a two-step synthesis of various 3-alkyl-2-nitrobenzo[b]furans 333 starting with 2-((E)-2-nitrovinyl)phenols 331 via a hypervalent iodine-induced oxidative cyclization, with good to excellent yields (Scheme 113).215 The reaction start with Michael addition of a Grignard reagent to 2-((E)-2-nitrovinyl)phenols 331 to generate the alkylated 2-(2-nitroethyl)phenols 332. Oxidative cyclization of 332 were achieved using PhI(OAc)2/TBAI. The best results were obtained when 2.5 equiv. of TBAI were used with 3.0 equiv. of PhI(OAC)2 in acetonitrile at 35 °C in the presence of 2 equiv. of triethylamine. Also, indole was used instead of Grignard reagent as nucleophile in the first step to give the 3-(2-nitrobenzofuran-3-yl)-1H-indole in 52% yield.
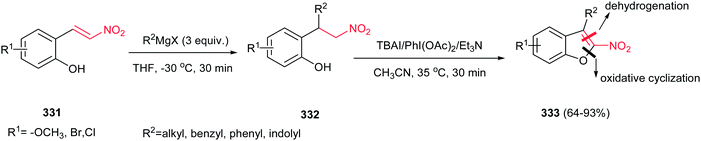 |
| | Scheme 113 Synthesis of 3-alkyl-2-nitrobenzo[b]furans starting with 2-((E)-2-nitrovinyl)phenols. | |
In 2005, Ishikawa et al. developed the synthesis of 4-acetoxy-2-amino-3-arylbenzofurans 336 from nitroalkenes 15 and cyclohexane-1,3-diones 334.216 This one-pot two-step process provided first the cyclic oxime intermediates 335 in THF in the presence of catalytic amount of Et3N (10 mol%), which was then converted to the corresponding products 336 via treatment with acetic anhydride (Ac2O), triethylamine(Et3N), and 4-(N,N-dimethylamino)pyridine (DMAP) at room temperature (Scheme 114). Is it notable that aliphatic nitroalkenes gave lower yield compared to aromatic nitroalkenes. Unsymmetrical cyclohexane-1,3-diones afforded the 5-alkyl4-acetoxy-2-amino-3-arylbenzofurans in combination with their regioisomer, 7-alkyl 4-acetoxy-2-amino-3-arylbenzofurans. In this context, Yao et al. demonstrated that similar yields and selectivity in the synthesis of 335 can be achieved using silica gel as mild acidic catalyst in combination with microwave irradiation at 60 °C in methanol.217
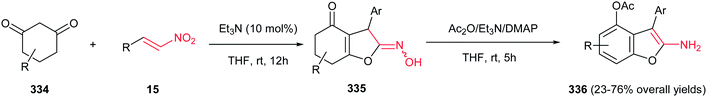 |
| | Scheme 114 Formation of 4-acetoxy-2-amino-3-arylbenzofurans from 1-aryl-2-nitroethylenes and cyclohexane-1,3-diones. | |
Very recently, Namboothiri et al. investigated the reaction of Morita–Baylis–Hillman acetates of nitroalkenes 164 with different arenols such as β-naphthol 324, α-naphthols 337, and substituted phenols 338 under basic conditions to give the corresponding arenofurans 339–341 as single regioisomers in good to excellent yield (Scheme 115).218 Several organic and inorganic bases were screened in this protocol and, finally, K2CO3 in toluene at room temperature was selected as optimal conditions with regard to yields and reaction times. While the reaction of 164 with β-naphthol 324 is not sensitive to the electronic nature of substituents on the aryl group in the acetates, for 1-naphthols, acetates164 with strongly electron donating aryl groups afforded greater yield than those with weak electron donating ability. In addition, phenols with strongly electron donating alkoxy groups reacted well with 164 and afforded the corresponding furans 341 in good to excellent yields (22–82%). Simple phenol gave the Michael adduct as the only product. Proposed mechanism by the authors involved an SN2′ reaction/intramolecular oxa-Michael addition reaction cascade. Furthermore, this strategy was successfully employed for the total synthesis of an anti fungal agent isoparvifuran 342 in six steps from nitrostyrene with 47% overall yield.
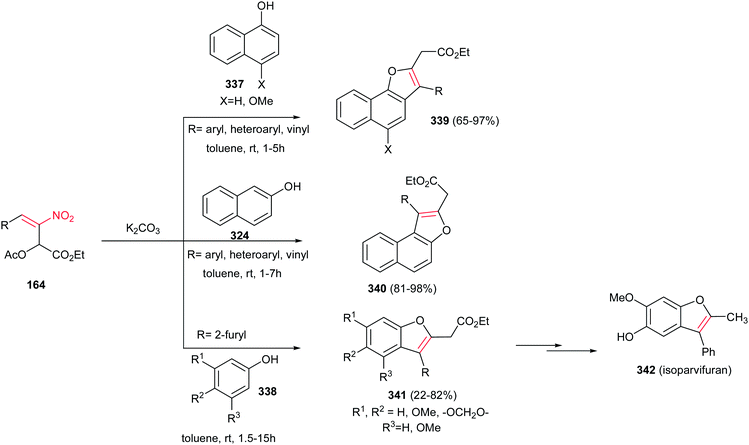 |
| | Scheme 115 Synthesis of naphthofurans and benzofurans. | |
Hajra and co-workers described the reaction of naphthols/phenol 337/324 with nitroalkenes 18 in the presence of indium(III) triflate to give the benzofuran and naphthofuran derivatives 343a/b in high yields (Scheme 116).219 The reaction was performed by stirring an equimolar amount of starting materials and 5 mol% of In (OTf)3 in DCE at reflux temperature with a CaCl2 moisture guard tube. Aliphatic nitroalkenes were used successfully in this transformation, albeit with lower yields compared to aromatic nitroalkenes. Under the reaction conditions, no Michael adduct was observed.
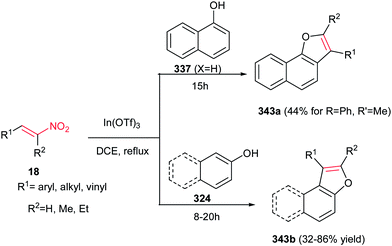 |
| | Scheme 116 Reaction of naphthols/phenols with nitroalkenes in the presence of indium(III) triflate. | |
As shown in Scheme 117, a one-pot four-component reaction between β-nitrostyrenes 40, aromatic aldehydes 85, ammonium acetate, and cyclohexane-1,3-diones 344 for synthesis of polysubstituted 3-aryl-2-arylmethylene amino-4-hydroxybenzofurans 345 was described by Wang et al.220 The reaction was carried out by slow addition of a cyclohexane-1,3-dione 334 to a mixture of a β-nitrostyrene 40, an aromatic aldehyde 85, ammonium acetate, and piperidine in DMF at room temperature, and refluxing this mixture for 7 h to afford the titled compounds 345 in 46–73% yields. Variety of aromatic aldehydes and nitroalkenes were successfully used in this protocol.
 |
| | Scheme 117 Synthesis of polysubstituted 3-aryl-2-arylmethylene amino-4-hydroxybenzofurans. | |
A series of functionalized 2,3-dihydrobenzofurans 347 was prepared by Xie and coworkers starting with 2-hydroxyarylnitroalkenes 331 and diethyl α-bromomalonate 346 via a domino oxa-Michael/aldol alkylation reaction catalyzed by K2CO3 under mild conditions (Scheme 118).221 Other bases such as KOH, KOAc and DABCO gave lower yields compared to K2CO3. The best yield was obtained when 120 mol% of K2CO3 was used in acetone at room temperature for 3 h. Also, using ethyl 2-chloroacetoacetate instead of 346 afforded the corresponding product in high yield and excellent diastereselectivity (>99:1).
 |
| | Scheme 118 Synthesis of 2,3-dihydrobenzofurans starting with 2-hydroxyarylnitroalkenes and diethyl α-bromomalonate. | |
Very recently, Yao et al. described the synthesis of substituted 2-amino-3-phenylnaphtho[2,3-b]furan-4,9-dione derivatives 349 in moderate to good yields via the NH4OAc-catalyzed reaction of 2-hydroxy-1,4-naphthoquinone 348 (1 equiv.) and nitroalkenes 40 (2 equiv.) in water at 100 °C (Scheme 119).222 Nitroalkenes with electron-donating groups afforded higher yields than those with electron-withdrawing groups. Other bases such as TEA, basic Al2O3, (NH4)2CO3, NaOAc, NH4HCO3, KOAc, K2CO3 and t-BuOK resulted in the same products in lower yields. Performing the reaction at temperatures up to 80 °C produced the Michael adducts. Mechanistic studies revealed that the ammonium acetate is not the source of the amino group in the products and the reaction proceeded via in situ reduction of the nitro group to the amino functionality without using any reducing agents during the process. Also, a one-pot three-component version of this reaction using an aldehyde 255, 2-hydroxy-1,4-naphthoquinone 348 and nitromethane 350 was developed by the same authors. Under similar conditions, the yields of the products obtained from multicomponent reactions are less than those obtained from the direct condensation of nitroalkenes and 2-hydroxy-1,4-naphthoquinone.
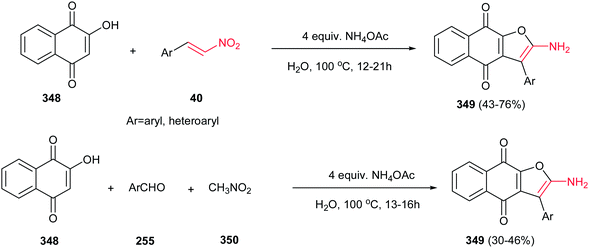 |
| | Scheme 119 NH4OAc-catalyzed preparation of 2-amino-3-phenylnaphtho[2,3-b]furan-4,9-dione derivatives in water. | |
Finally, 3-aryl-5,6-dihydrobenzofuran-7(4H)-ones 352 are prepared by Hunt and Simpkins through the reaction of β-nitrostyrenes 76 and 1,2-cyclohexanedione 351 promoted by 10 equiv. of K2CO3 as a base in DMF at 80 °C. The products were obtained in good yields (40–84%). Also, this process is highly dependent on the base stoichiometry (Scheme 120).223
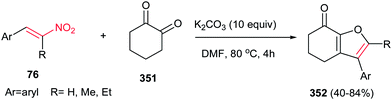 |
| | Scheme 120 K2CO3-promoted synthesis of 3-aryl-5,6-dihydrobenzofuran-7(4H)-ones. | |
4.3. Synthesis of S-heterocyclic compounds
4.3.1. Thiophenes, dihydrothiophenes and tetrahydrothiophenes. Thiophene derivatives form an integral part of many natural products and pharmaceuticals.224 They have wide applications in advanced materials such as conjugated polymers,225 organic conductors,226 semiconductors,227 and light emitting devices.228 Generally, formation of thiophenes can be divided in four main categories; (1) reaction of the 1,4-difunctional compounds with sulfides (Paal−Knorr strategy); (2) reaction of unsaturated compounds with sulfides (Gewald reaction); (3) reaction of 1,2-difunctional compounds with thiodiacetic acid and esters; and (4) reaction of aryl methyl ketones with sulfides.229 In addition, dihydro- and tetrahydrothiophenes have found extensive applications in the structure of many biologically active compounds230 and are important intermediates that form thiophene derivatives through dehydration and aromatization.Southern and coworkers developed an efficient approach to 3-nitro-2-substituted thiophenes 357 in two steps starting with commercially available 1,4-dithane-2,5-diol 353 and nitroalkenes 15.231 The reaction proceeds via formation of the thiolate anion 354 from dithiane 353 with a catalytic amount of triethylamine, and subsequent Michael addition-intramolecular Henry reaction to generate the tetrahydrothiophenes (THTs) 356 in excellent yields (Scheme 121). Subsequent dehydration and oxidation of THTs using 20 equiv. (w/w) of acidic alumina with 1.5 equiv. of chloranil under microwave irradiation (with the maximum temperature set to 125 °C and maximum power set to 200 W) form 3-nitro-2-substituted thiophenes 357 bearing a wide range of substituents (aromatic, heterocyclic, aliphatic, H, CH2OTBS) at the 2-position. Alternative dehydration and oxidation systems such as SiO2/DDQ and TFA/DDQ were also used, but gave lower yield. Both aromatic and aliphatic nitroalkenes are compatible with this protocol. Also, reactions of 1,4-dithiane-2,5-diol 353 with 2-nitroethylacetates 358, used as stable precursors for the corresponding nitroalkenes, were investigated by Risi et al. for synthesis of 4-nitrotetrahydrothiophen-3-ol scaffolds 359 (Scheme 122).232
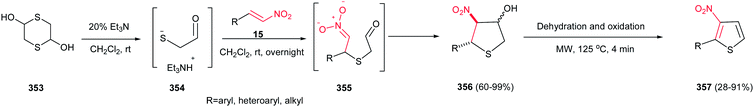 |
| | Scheme 121 Tandem Michael addition-intramolecular Henry reaction for generation of tetrahydrothiophenes and their oxidation to thiophenes. | |
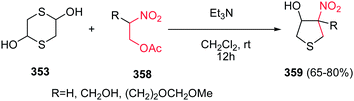 |
| | Scheme 122 Synthesis of 4-nitrotetrahydrothiophen-3-ols from 1,4-dithiane-2,5-diol and 2-nitroethylacetates. | |
Very recently, Tao et al. reported that the same reaction can be efficiently catalyzed by a tertiary amine immobilized fiber Cat-14 to give the THTs 360a/b in high to excellent yields (75–93%) (Scheme 123).233 The catalyst Cat-14 was synthesized via reaction of PANF and ethylenediamine followed by alkylation with 3-dimethylaminopropylchloride hydrochloride in the presence of K2CO3. The fiber catalyst exhibits excellent recyclability and reusability (up to 10 times) without any additional treatment.
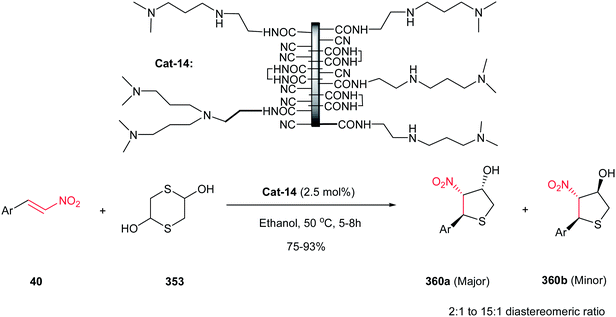 |
| | Scheme 123 Synthesis of substituted THTs from nitroalkenes and 1,4-dithiane-2,5-diol. | |
A novel asymmetric domino thia-Michael/Michael addition reaction between nitroalkenes 15 and trans-ethyl 4-mercapto-2-butenoate 361 was employed by Wang et al. for synthesis of trisubstituted THTs 362 (Scheme 124).234 Different organocatalysts, solvents and reaction temperatures were screened and using 20 mol% of Takemoto amine thiourea catalyst Cat-15 for the reaction of 1.2 mmol of 361 with 1 mmol of 15 in chloroform at −40 °C for 72 h was selected as optimum condition. Under this condition, one C–S and one C–C bond and three stereogenic centers were generated in a “one pot” fashion with high enantioselectivity (92–97% ee) and good diasteriomeric ratios (6:1 to >30:1). Notably, aromatic, heteroaromatic and aliphatic nitroalkenes afforded similar results in this transformation. Mechanistic studies by the authors revealed that the stereochemical outcomes are the results of an unprecedented activation mode of cooperative direct stereocontrol and dynamic kinetic resolution by catalyst.
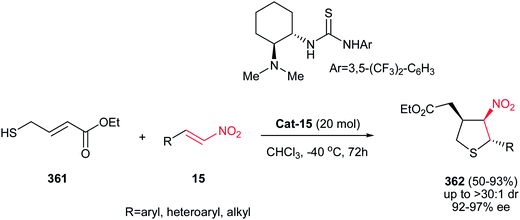 |
| | Scheme 124 Asymmetric domino thia-Michael/Michael addition approach for synthesis of THTs. | |
In 2007, Chunikhin et al. established that the reaction of nitrostyrenes 40 with cyanothioacetamide 363 in the presence of catalytic amount of a base such as tetramethylethylenediamine (TMEDA) or morpholine in ethanol at room temperature afford compounds 364 in high yields (Scheme 125).235
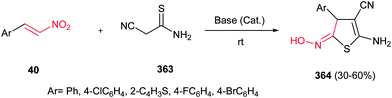 |
| | Scheme 125 Reaction of nitrostyrenes with cyanothioacetamide. | |
Reaction of trans-β-nitrostyrene 55 with mesoionics 365, prepared by the reaction of thioureas and α-chlorophenylacetic acid chloride in the presence of triethylamine, afforded a diastereomeric mixture of racemic dihydrothiophenes 367a/b in CH2Cl2 at room temperature for 48 h, which can be separated by flash chromatography (Scheme 126).236 The reaction proceeds via the bicyclic intermediate 366a and 366b, which were evidenced by NMR experiments in CDCl3 at 0 °C.
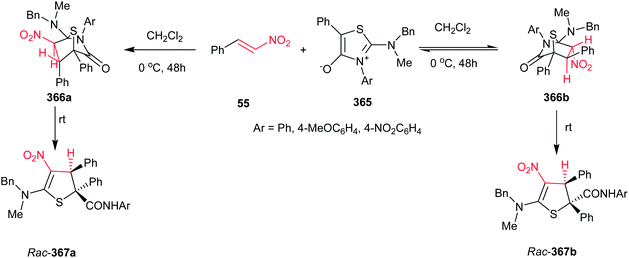 |
| | Scheme 126 Formation of dihydrothiophenes from thioisomunchnones and trans-β-nitrostyrene. | |
Finally, Bogdanowicz-Szwed and Gil in 2004 carried out the reaction of cyclic 3-oxoacid thioanilides with β-nitrostyrenes to achieve the functionalized spiro[cycloalkano-2,3-thiophenes] in boiling anhydrous ethanol in the presence of catalytic amounts of piperidine in moderate to good yields (41–86%).237 Reaction of the obtained products with acetic anhydride yielded the corresponding oxime acetates. Any attempt for transformation of the products into nitrogen heterocycles via Dimroth or Beckmann rearrangements under acidic conditions was unsuccessful (Scheme 127).
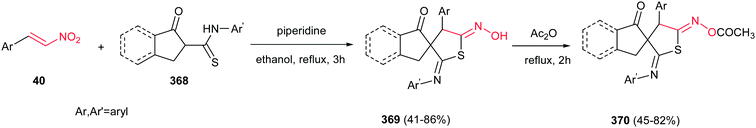 |
| | Scheme 127 Functionalized spiro[cycloalkanono-2,3-thiophenes] from cyclic 3-oxoacid thioanilides and β-nitrostyrenes. | |
4.4. N,O-Heterocyclic compounds
4.4.1. Isoxazole, Isoxazoline and isoxazolidine. Isoxazole and its reduced forms have served as useful building blocks in organic synthesis in the total synthesis of several natural and unnatural biologically active compounds such as β-lactam antibiotics, quinolizidine and indolizine tricycles, testosterone, sarkomycin, and biotin.238 In addition, these compounds can be simply transformed into a variety of 1,3-bifunctional organic compounds such as β-hydroxy ketones, α,β-unsaturated ketones and γ-amino alcohols.239 1,3-dipolar cycloaddition reactions240 and condensation of hydroxylamine with 1,3-dicarbonyl compounds and α,β-unsaturated carbonyl compounds241 are the most applied procedures for synthesis of isoxazole and its reduced derivatives.Although, the base-catalyzed reaction of nitroalkenes with isocyanoacetate is known as the Barton–Zard pyrrole synthesis, recently, Adib and co-workers demonstrated that by heating two equiv. of an isocyanide 371 with nitrostyrenes 40 in water at 80 °C, 5-(alkylamino)-4-aryl-3-isoxazolecarboxamides 372 could be obtained in 83–93% isolated yields (Scheme 128).242 This protocol is well tolerated for aromatic and heteroaromatic nitroalkenes.
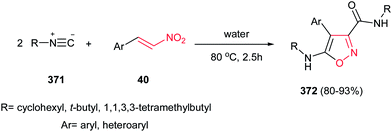 |
| | Scheme 128 Synthesis of 5-(alkylamino)-4-aryl-3-isoxazolecarboxamides from isocyanides and nitrostyrenes in water. | |
In 2010, Perumal and co-workers reported the synthesis of isoxazolobenzoxepanes 376 starting with nitroalkenes 373 derived from O-propargyl salicylaldehyde (Scheme 129).243 They have shown that Michael addition of an indole 374 to the nitroalkene moiety of 373 in the presence of KHSO4 in water afforded the corresponding nitroalkane 375, which can be simply transformed to the isoxazolobenzoxepane 376 via intramolecular nitrile oxide cycloaddition upon treatment with (Boc)2O/DMAP in methanol at 90 °C. Notably, replacing the terminal alkynes with internal alkynes gave similar yields.
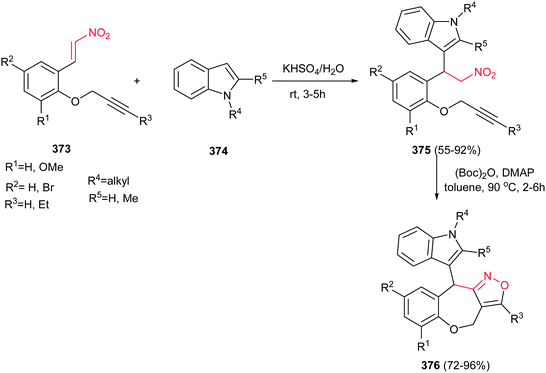 |
| | Scheme 129 Synthesis of isoxazolobenzoxepanes starting with nitroalkenes derived from O-propargyl salicylaldehyde. | |
Gao et al. reported that the bicyclic isoxazoles 378 can be prepared from nitroalkenes 15 and prop-2-ynylmalonate 377a or prop-2-ynylmalononitrile 377b via a one-pot tandem Michael addition–dehydration-[3 + 2] cycloaddition reactions in the presence of t-BuOK and TCT or TCT/ZnCl2 (Scheme 130).244 The rate of cycloaddition of nitroalkenes 15 with prop-2-ynylmalonate 377a is faster than with prop-2-ynylmalononitrile 377b, because the presence of the bulky dimethoxy carbonyl groups enables the dipolarphile to be closer to the nitrile oxide to more easily undergo the intramolecular cycloaddition.
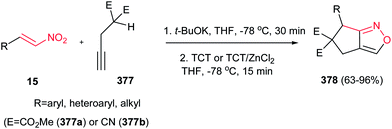 |
| | Scheme 130 Isoxazoles prepared from nitroalkenes and prop-2-ynylmalonate or prop-2-ynylmalononitrile. | |
Also, the same group described that reaction of allyl malonate 379 with nitroalkenes 15 in the presence of t-BuOK at −78 °C gave the nitronates 380 which can be simply transformed to corresponding nitrile oxides 381 via treatment with 3 eq. of TCT. Then, intramolecular [3 + 2] cyclization of the nitrile oxide with the alkene moiety produces the bicyclic isoxazolines 382 in 84–97% yield (Scheme 131).244
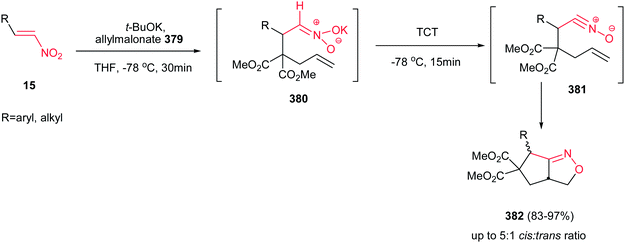 |
| | Scheme 131 One-pot and two-step synthesis of isoxazolines. | |
Furthermore, highly substituted isoxazolines 386 were synthesized by Whittle and coworkers via a one-pot four-component condensation of TMSCN 383, epoxides 384, nitroalkenes 15 and methylacrylate 385. They proposed that reaction of trimethylsilyl cyanide 383 with epoxide 384 in the presence of Pd(CN)2 generates isonitrile A which undergoes [1 + 4] cycloaddition reaction with nitroalkenes in lithium perchlorate medium to form N-(isoxazolylidene)alkylamines B. After fragmentation to nitrile oxides C, it can undergo intermolecular 1,3-dipolar cycloadditions with methyl acrylate to produce substituted isoxazolines 386 in one synthetic operation with moderate yields (Scheme 132).245
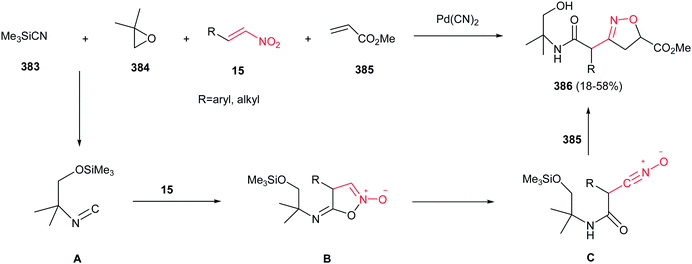 |
| | Scheme 132 One-pot four-component synthesis of substituted isoxazolines. | |
Very recently, Sabbasani and Lee reported an efficient method for synthesis isooxazolidinone derivatives 390 in good yields from α-nitro-α,β-unsaturated silyloximes 389 via treatment with TBAF. The oximes 389 were prepared by reaction of silylallenes 387 and nitrogen dioxide radical, generated from NaNO2 and AcOH (Scheme 133).246
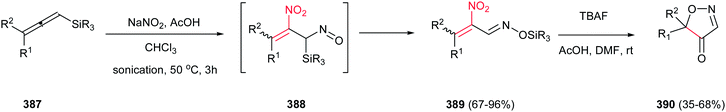 |
| | Scheme 133 Synthesis of isooxazolidinone derivatives from α-nitro-α,β-unsaturated silyloximes. | |
A two-step protocol for synthesis of fused isoxazolidines 394 starting with nitrolakenes is developed by Kamimura et al. in 2008. The first step is Michael addition of the pretreated N-(4-pentenyl)formamide 391 with t-BuOK to nitroalkenes 15 in THF at −50 °C to give the corresponding adducts in 48–85% isolated yields. The second step is treatment of these Michael adducts 392 with phenyl isocyanate in the presence of Et3N in THF at reflux temperature to afford the bicycloazepines 393 in 50–71% as diastereomeric mixtures in favor of cis isomer (cis:trans ratio of up to 96:4). The N-formyl group simply removed from the structure of products 393 via treatment with ethanolic diluted HCl solution without significant epimerization. In addition, they have shown that performing these two reactions in a one-pot manner gave the corresponding products in moderate yields (Scheme 134).247
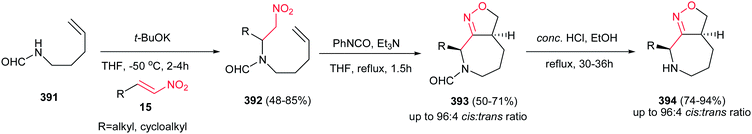 |
| | Scheme 134 Synthesis of isoxazoline fused to azepines. | |
Reaction of 7-oxohept-2-enoate derivatives 395 with nitroolefins 15 were investigated by Zhong et al. to give highly stereoselective bicyclic isoxazolidines 396 bearing five stereogenic centers (Scheme 135).248 The reaction proceeded via domino Michael addition/nitrone formation/intramolecular [3 + 2] nitrone-olefin cycloaddition catalysed by 10 mol% of chiral α,α-diphenyl prolinol trimethylsilyl ether Cat-8 and AcOH (20 mol%) as an additive. The products were obtained in good-to-excellent yields with excellent diastereo- and enantioselectivities of up to 94% de and >99% ee, respectively. While the yields and diastereoselectivities affected by substituents on the nitroolefins, for almost all nitroalkenes tested, the enantioselectivities were higher than 98% ee. Moreover, the products are valuable intermediates for preparation of α-hydroxy-γ-aminoacid derivatives, which could have potential applications in both synthetic chemistry and the pharmaceutical industry.
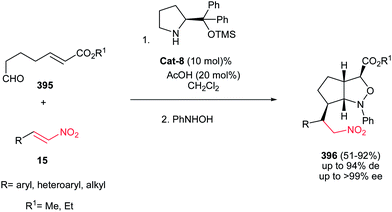 |
| | Scheme 135 Stereoselective synthesis of bicyclic isoxazolidines bearing five stereogenic centers. | |
Asymmetric 1,3-dipolar cycloaddition of nitrones 397 with β-alkyl nitroolefins 15 was reported by Chen et al. in 2008. Among the several thiourea organocatalysts examined for this reaction, Cat-4 derived from (R,R)-1,2-diaminocyclohexane exhibited excellent diastereoselectivities (generally >99:1 dr) and moderate to high enantioselectivities (up to 88% ee). The cycloadducts 398 can be simply converted to protected 2,3-diaminopropanol derivatives 399 with three contiguous chiral centers via reduction with NiCl2/NaBH4 and subsequent protection with (Boc)2O in excellent yield and enantioselectivity (Scheme 136).249
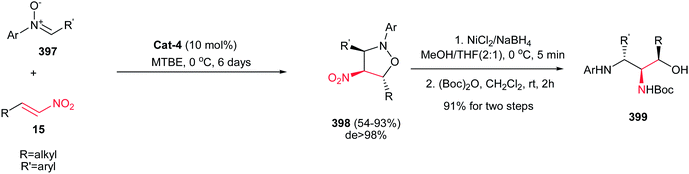 |
| | Scheme 136 [3 + 2] Cycloaddition of nitrones and nitroalkenes. | |
Gottlieb et al. described a one-pot procedure for synthesis of α-dialkylaminoaldoximes 400 from nitroalkenes 18 and secondary amines (as solvent and reagent) in the presence of tin(II) chloride. Alkyl and aryl substituted nitroalkenes undergo this transformation, however better yields were obtained with β-nitrostyrenes containing electron donating substituents. When a secondary allylamine was used, the corresponding α-allylamino aldoxime underwent an efficient intramolecular oxime–olefin cycloaddition in toluene under nitrogen at 110 °C to give bicyclic isoxazolidines 401 in good to high yields (Scheme 137).250
 |
| | Scheme 137 SnCl2·2H2O catalyzed synthesis of bicyclic isoxazolidines from nitroalkenes and secondary amines. | |
Finally, [2 + 3] cycloadditions between camphor-derived oxazoline N-oxide 403 and α,β-substituted nitroalkenes 18 afforded stereoselectively adducts 404 with isoxazolidine cycle in the structure. Denitration of products with Bu3SnH (D) in the presence of AIBN gave compounds 405 in good yield and stereoselectivity (Scheme 138).251
 |
| | Scheme 138 [2 + 3] Cycloaddition reactions between camphor-derived oxazoline N-oxide and α,β-substituted nitroalkenes. | |
4.4.2. Isoxazoline N-oxide derivatives. Recently nitroalkenes have found wide applications in the synthesis of isoxazoline N-oxides. In this context, Shi et al. reported a catalytic cascade one-step synthesis of isoxazoline-N-oxide 407 from nitroalkenes 254 and vinyl esters 406 (as aliphatic aldehyde analogue) (Scheme 139).252 This approach provides only trans isomers with good to excellent yields. The best results were obtained when 20 mol% of proline and 1 equiv. of NaOAc were used in DMSO at room temperature. Lewis bases such as DMAP and PPh3 were also used and resulted in lower yield with significant amounts of polymerization. While various 2,2-disubstituted nitroalkenes were suitable for this transformation, monoalkyl substituted nitroalkenes gave only trace amount of the desired products with a significant amount of polymerization adducts.
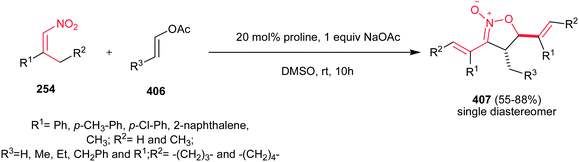 |
| | Scheme 139 A catalytic cascade one-step synthesis of isoxazoline-N-oxide from nitroalkenes and vinyl esters. | |
In another report, the same group also described that a one-pot condensation of nitroalkene (254a)-aldehyde (255)- sulfur ylide 408e/f in the presence of proline and K2CO3 afford the fully substituted isoxazoline-N-oxides 410 in high to excellent yields. The phosphine ylide 408c and amine ylide 408d also generate the desired product 410 but with significant competition reaction for homo-condensation and production of 409 (Table 1).253 This strategy was successfully used for stereoselective gram-scale total synthesis of clausenamide 415 in five steps from nitroalkene as shown in Scheme 140.
Table 1 Substrate screening for three-component condensation in the synthesis of isoxazoline-N-oxidea
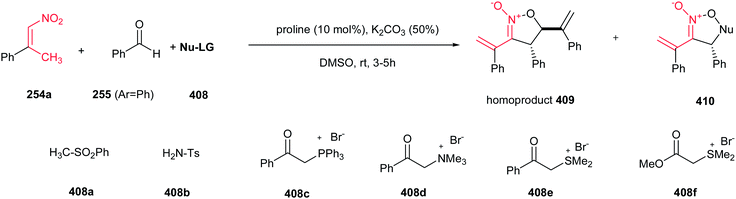
|
| Entry |
Nu-LG |
Product |
| 409 (%) |
410 (%) |
| Reaction condition: 254a (1.0 equiv.), 255 (1.0 equiv., 0.75 M), and catalyst were mixed in solvents. NMR yield with 1,3,5-trimethoxybenzene as internal standard. Isolated yield. |
| 1 |
408a |
76b |
<5 |
| 2 |
408b |
85b |
<5 |
| 3 |
408c |
58b |
35b |
| 4 |
408d |
74b |
18b |
| 5 |
408e |
<5b |
91c |
| 6 |
408f |
<5b |
92c |
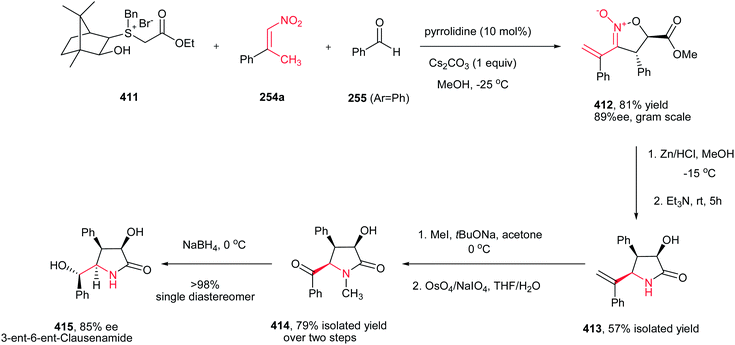 |
| | Scheme 140 Total synthesis of clausenamide. | |
Also, Tang et al. developed an efficient protocol for synthesis of isoxazoline N-oxides 418a/b in excellent yield (79–99%) and diastereomeric ratios of higher than 99/1 in favor of trans isomer from sulfonium salt 416 and substituted nitroalkenes 417 (Scheme 141).254 The reaction proceeds well in CH3CN in the presence of both inorganic bases (Cs2CO3, K2CO3, KOH, and KOtBu) and organic base (iPr2NH). No cyclopropanes formation was observed. Comparison to Shi's work, this protocol is applicable for ammonium ylides. The asymmetric version of this reaction was also investigated by the same group using cinchona alkaloid-derived ammonium salts 419a and 419b and higher than 96% ee values are achieved for products, albeit in opposite configurations. The enantiomeric excesses are nearly independent of the substituents of the aryl and heteroaryl groups. Aliphatic nitroalkene were used as well as aromatic nitroalkenes in asymmetric version with excellent ee, although both the yield and the diastereoselectivity decreased.
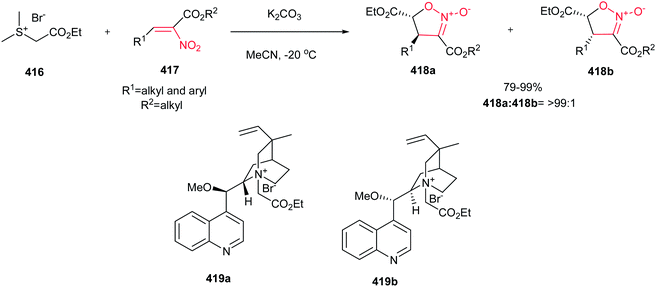 |
| | Scheme 141 Synthesis of isoxazoline N-oxides from sulfonium salt and substituted nitroalkenes. | |
Although it is well-known that the reaction of alkyl 2-bromomalonate 420 with β-nitroalkenes 18 afforded the cyclopropane rings, in 2011, Marouka et al. have shown that various chiral isoxazoline-N-oxides 421 having a tetrasubstituted carbon can be obtained in high to excellent yields and high ee's (72–87%) by asymmetric conjugate addition of bromomalonate 420 to α,β-substituted nitroolefins 18 in the presence of 70 mol% of Cs2CO3 and 1 mol% of (R,R)-Cat-16 as chiral phase-transfer catalyst (Scheme 142).255 The reaction was initiated by Michael addition, and followed by subsequent ring-closing O-alkylation.
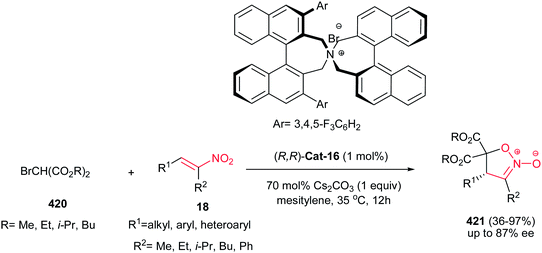 |
| | Scheme 142 Asymmetric synthesis of isoxazoline-N-oxides. | |
4.4.3. Oxazolidinone derivatives. Xiao et al. reported a new protocol for synthesis of oxazolidin-2-ones 423 from sulfur ylides 422 and nitroalkenes 15 via [4 + 1] annulations/rearrangement cascade sequentially catalyzed by thiourea and DMAP.256 The best results were obtained when 1.25 equiv. of sulfur ylide 422 reacted with 1 equiv. of nitroolefin 15 in the presence of 10 mol% of 1-(2-chlorophenyl)thiourea Cat-17 and DMAP in chloroform at room temperature for 24 h. Both aromatic and aliphatic nitroalkenes are compatible with this protocol to produce the corresponding oxazolidin-2-ones 423 in high yields (70–89%) (Scheme 143). The authors proposed that the reaction proceeded via intermediate A. Also, they have shown that by performing the same reaction in the presence of 50 mol% of the chiral bis-urea catalyst Cat-18, enantioenriched 4,5-substituted oxazolidinones were obtained in moderate to excellent isolated yields (65–96%) with excellent stereocontrol (up to more than 95:5 dr and 97:3 er).257 Accordingly, Toy et al. developed another catalytic system for this reaction using 10 mol% of bifunctional polymeric organocatalyst Cat-19 which contain both amine and thiourea catalytic groups.258
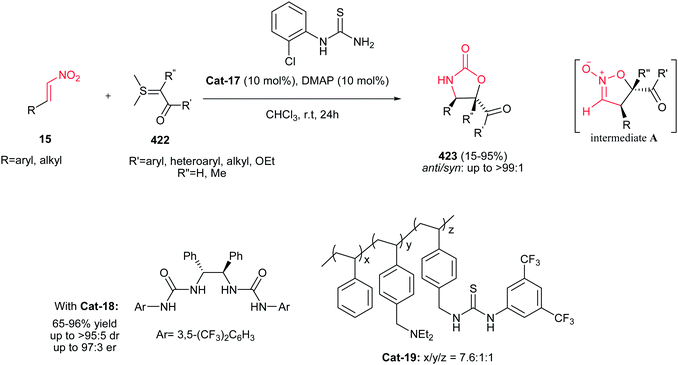 |
| | Scheme 143 Organocatalyzed cascade reaction of sulfur ylides with nitroalkenes. | |
In addition, the same authors developed another asymmetric method for this transformation by using stable chiral BINOL-derived sulfur ylides 424 and nitroalkenes 15.259 The stereochemistry of the reaction is controlled by the chiral sulfur ylide. This approach provides chiral oxazolidinones 425 in good yields and high stereoselectivities (up to 96:4 er and >95:5 dr) (Scheme 144). The absolute configuration of oxazolidinones 425 was confirmed to be (4S,5R) by X-ray crystallographic analysis. This protocol was also successfully applied to the asymmetric [4 + 1]/[3 + 2] cycloaddition cascade of sulfur ylides 424 with acrylate-tethered β-nitrostyrene 426 to afford the enantioenriched fused heterocycles 427 (up to 87:13 er and >95:5 dr) in good to excellent yields (54–95% yields) (Scheme 145).
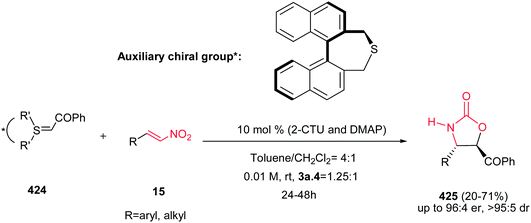 |
| | Scheme 144 Nitroolefins for the enantioselective cascade [4 + 1] annulation/rearrangement reaction. | |
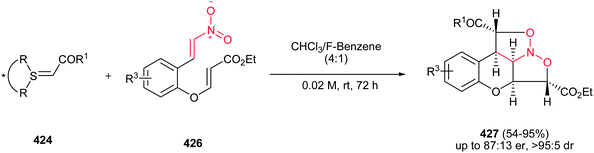 |
| | Scheme 145 Enantioselective [4 + 1]/[3 + 2] cascade reactions. | |
In 2007, Evans et al. developed an efficient procedure for the synthesis of optically active cis-oxazolidinone 430 from chiral 3-amino-substituted-1-arylthio-1-nitroalkene 428. The reaction initiated with epoxidation of 428 by t-BuO2Li to furnish the intermediate 429. Then, in the presence of SiO2, the intramolecular epoxide ring opening initiated by the carbamate group led to subsequent elimination of the nitro group, followed by migration of the thioester group to give corresponding products 430 in high yields (Scheme 146).260
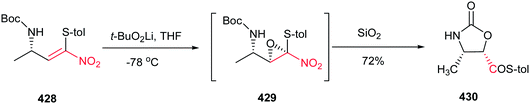 |
| | Scheme 146 Synthesis of optically active cis-oxazolidinone from chiral 3-amino-substituted-1-arylthio-1-nitroalkene. | |
4.4.4. Nitrosoacetal derivatives. The nitrosoacetals are suitable intermediates for synthesis of diversity of biologically active compounds such as pyrrolizidin-3-ones, amino acids and alkaloids.261 These compounds can be simply prepared via tandem [4 + 2]/[3 + 2] nitroalkene cycloaddition with unactivated olefins or enol ethers as dienophiles and electron-deficient alkenes as 1,3-dipolarophiles. The inter- or intamolecular fashion of this method has been investigated extensively by Denmark group and others.262 In this part, we wish to describe some of the recent key publications in the synthesis of nitrosoacetals.Domino hetero-Diels–Alder/1,3-dipolar cycloaddition reaction of 3,3,3-trichloro(trifluoro)-nitroethylenes 431 (or 27) with 2,3-dihydrofuran 432 was investigated by Sosnovskikh et al.263 They described that compound 431 (or 27) play a dual role as the heterodiene and the dipolarophile in the reaction. The trichloromethylated nitroolefin 431 gave tricyclic nitroso acetal 434 as a single regio- and stereoisomer in 45% isolated yield, while the trifluoromethylated derivative 27 afforded a 3:1 mixture of two regioisomeric cycloadducts 435a and 435b in 41% total yield (Scheme 147).
 |
| | Scheme 147 Domino hetero-Diels–Alder/1,3-dipolar cycloaddition reaction of 3,3,3-trichloro(trifluoro)-nitroethylenes with 2,3-dihydrofuran. | |
Giommi et al. demonstrated that reaction of substituted 4-nitroisoxazoline 436 with a large excess of ethyl vinyl ether 351 in anhydrous CH2Cl2 at 40 °C for 4 days afforded 5:1 mixture of the diasteremeric spiro nitrosoacetal 439a/b in excellent yield.264 By performing the reaction in refluxing chloroform, surprisingly 3:2 mixture of the diasteromeric isoxazolo-oxepines 437a/b was obtained in 54% yield. Treatment of the mixture of 439a/b in CHCl3 also afforded the products 437a/b in same yield and diastereomeric ratio (Scheme 148).
 |
| | Scheme 148 Reaction of substituted 4-nitroisoxazoline with a large excess of ethyl vinyl ether. | |
A one-pot domino (4 + 2)/(4 + 2)/(3 + 2) cycloaddition reaction of 2-methoxy buta-1,3-diene 440 with a dienophile, β-nitrostyrene and a dipolarophile under high pressure conditions gives tri, tetra and pentacyclic nitroso acetals. The reaction of 440 with 3 eq. of nitroalkenes 55 gave tricyclic nitrosoacetals as two regioisomers 441 and 442 (441/442a/442b:0.2/0.6/0.2). (Scheme 149a), while reaction of 440 with maleimide 443 in the presence of 2 eq. of niroalkene 55 gave two regioisomeric tetracyclic nitrosoacetal 444a/b and 445 (1:1 ratio) in 64% yield in which both regioisomers consisted of two diastereomers in a ratio of 0.65/0.35 (Scheme149b). The configuration of one of the stereoisomers was assigned by 2D-NOESY analysis. In addition, reaction of 440 with nitroalkene 55 and 2 eq. of maleimides 443 gave pentacyclic nitrosoacetals 446a/b (Scheme 149c). In this novel domino reaction up to six bonds and up to eight stereogenic centers are created in one step in good yield and good stereoselectivity. The results showed that N-phenylmaleimide 443 was more reactive than β-nitrostyrene 55 toward 2-methoxybuta-1,3-diene 440.265
 |
| | Scheme 149 Highly functionalized polycyclic nitrosoacetals via a one-pot domino (4 + 2)/(4 + 2)/(3 + 2) cycloaddition reactions. | |
Finally, they have examined that mixing of an equimolar of 2-methoxybuta-1,3-diene 440 (as a diene), N-phenyl maleimide 443 (as a dienophile), β-nitrostyrene 55 (as a heterodiene), and styrene 447 (as a dipolarophile) at 15 kbar and room temperature for 16 h yielded in 83% a mixture of two diastereomeric nitrosoacetals 448a and 448b (Scheme 150).265
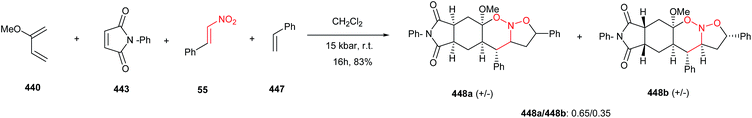 |
| | Scheme 150 A one-pot four-component route for synthesis of nitrosoacetals. | |
4.5. Synthesis of N,S-heterocyclic compounds
Tsogoeva et al. reported a one-pot two-step procedure for synthesis of 1,3-thiazoles 451 starting with nitroalkenes 110. Epoxidation of 110 with TBHP/DBU (t-BuOOH/1,8-diazabicyclo[5.4.0] undec-7-ene) gave α-nitroepoxides 449 which undergo cyclization with thioamides 450 in the presence of TFA to afford fully substituted 1,3-thiazoles 451 in good to high yields (Scheme 151).266
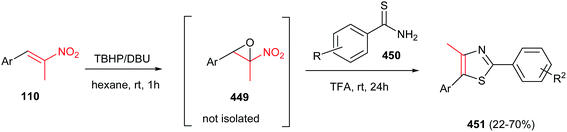 |
| | Scheme 151 One-pot two-step process for synthesis of 1,3-thiazoles from nitroalkenes. | |
5. Conclusions
Nitroalkenes are elegant substrates for the facile synthesis of 3–5 membered heterocycles. Small ring heterocycles such as epoxides, aziridines, azetidines and thietanes can be synthesized with high stereoselectivity via a Michael addition-intramolecular cyclization strategy involving nitroalkenes and an O, N or S-nucleophile bearing a suitable leaving group. Five-membered aromatic heterocycles such as pyrrole, pyrazole, imidazole, triazole, tetrazole, isoxazole, furan and thiophene are conveniently accessible using nitroalkenes as the key substrates. Synthesis of saturated heterocycles such as pyrrolidine, tetrahydrofuran and tetrahydrothiophene is also feasible, often with excellent functional group diversity and stereoselectivity.
Abbreviations
| AcOH | Acetic acid |
| Ac | Acyl |
| acac | Acetylacetonate |
| AIBN | Azobisisobutyronitrile |
| Alk | Alkyl |
| Ar | Aryl |
| BINOL1 | 1′-Bi-2-naphthol |
| bmim | 1-Butyl-3-methylimidazolium |
| Bn | Benzyl |
| Boc | t-Butyloxy carbonyl |
| Bu | Butyl |
| Bz | Benzoyl |
| BztH | Benzotriazole |
| 2-CTU | 1-(2-Chlorophenyl)thiourea |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCE | Dichloroethane |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone |
| de | Diastereomericexcess |
| DECP | Diethylcyanophosphonate |
| DIB | (Diacetoxyiodo)benzene |
| DIPC | N,N′-Diisopropylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-(N,N-Dimethylamino)pyridine |
| DME | 1,2-Dimethoxyethane |
| DMF | Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| dr | Diastereomeric ratio |
| ee | Enantiomeric excess |
| eq | Equivalent |
| er | Enantiomeric ratio |
| esp | α,α,α′,α′-Tetramethyl-1,3-benzenedipropionate |
| Et | Ethyl |
| IBX | 2-Iodoxybenzoicacid |
| MBH | Morita–Baylis–Hillman |
| MeGly | Glycine methyl ester |
| Ms | Methanesulfonyl |
| MTBE | Methyl tertiary-butyl ether |
| MW | Microwave |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| NOESY | Nuclear Overhauser effect spectroscopy |
| Ns | 4-Nitrobenzenesulfonyl |
| PANF | Polyacrylonitrile fiber |
| PCC | Pyridinium chlorochromate |
| PEG | Polyethylene glycol |
| Ph | Phenyl |
| Phen | 1,10-Phenanthroline |
| PMB | p-Methoxybenzyl |
| PMP | p-Methoxyphenyl |
| PPL | Porcine pancreas lipase |
| PPTS | Pyridinium p-toluenesulfonate |
| PS-BEMP | 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine on polystyrene |
| SA | Sulfamic acid |
| SFC | Solvent-free conditions |
| TBAI | Tetrabutylammonium iodide |
| TBAF | Tetrabutylammonium fluoride |
| TBHP | tert-Butyl hydroperoxide |
| t-BuO | tert-Butoxide |
| TBPLi | Lithium tert-butylperoxide |
| TBPK | Potassium tert-butylperoxide |
| TBS | Tributylsilyl |
| TCT | 2,4,6-Trichloro-1,3,5-triazine |
| TFA | Trifluoroacetic acid |
| TFAA | Trifluoroacetic anhydride |
| TFE | 2,2,2-Trifluoroethanol |
| Tf | Trifluoromethanesulfonyl |
| TEA | Triethylamine |
| THF | Tetrahydrofuran |
| THTs | Tetrahydrothiophenes |
| TMEDA | Tetramethylethylenediamine |
| TMS | Trimethylsilyl |
| Ts | Tosyl |
| TLC | Thin-layer chromatography |
Acknowledgements
Azim Ziyaei Halimehjani and Seyyed Emad Hooshmand thank the Faculty of Chemistry of Kharazmi University for supporting their research. Also, INNN thanks Department of Science and Technology (DST), India, for supporting research in the area of nitroalkene chemistry.
References
- General reviews/books:
(a) A. G. M. Barrett and G. G. Graboski, Chem. Rev., 1986, 86, 751 CrossRef CAS;
(b) Nitroalkanes and Nitroalkenes in Synthesis: Tetrahedron Symposia-in-Print, no. 41: [In: Tetrahedron, 1990; 46(21)], ed. A. G. M. Barrett, Pergamon, Oxford, UK, 1990, p. 285 Search PubMed;
(c) V. V. Perekalin, E. S. Lipina, V. M. Berestovitskaya and D. A. Efremov, Nitroalkenes: Conjugated Nitro Compounds, Wiley, Chichester, UK, 1994, p. 256 Search PubMed;
(d) R. Ballini, S. Gabrielli and A. Palmieri, Green Chem. Environ. Sustainability, 2011, 53 CAS;
(e) R. Ballini, S. Gabrielli and A. Palmieri, Curr. Org. Chem., 2010, 14, 65 CrossRef CAS;
(f) A. G. M. Barrett, Chem. Soc. Rev., 1991, 20, 95 RSC;
(g) K. Banert, Sci. Synth., 2006, 24, 747 Search PubMed.
- Selected recent reviews:
(a) Y. Alvarez-Casao, E. Marques-Lopez and R. P. Herrera, Symmetry, 2011, 3, 220 CrossRef CAS PubMed;
(b) S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org. Chem., 2012, 2012, 3059 CrossRef CAS;
(c) R. Ballini, M. Petrini and G. Rosini, Molecules, 2008, 13, 319 CrossRef CAS PubMed;
(d) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS;
(e) M. Shibasaki, H. Groeger and M. Kanai, Compr. Asymmetric Catal., Suppl., 2004, 1, 131 CAS.
-
(a) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 12, 1877 CrossRef;
(b) Anon, Chemtracts, 2010, 23, 58 Search PubMed;
(c) D. M. Barnes, Asymmetric Catalysis on Industrial Scale, ed. H.-U. Blaser and H.-J. Federsel, Wiley-VCH Verlag GmbH, 2nd edn, 2010, p. 439 Search PubMed;
(d) D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561 CrossRef CAS PubMed;
(e) R. Somanathan, D. Chavez, F. A. Servin, J. A. Romero, A. Navarrete, M. Parra-Hake, G. Aguirre, C. Anaya de Parrodi and J. Gonzalez, Curr. Org. Chem., 2012, 16, 2440 CrossRef CAS;
(f) A. Ziyaei- Halimehjani, M. A. Ranjbari, H. Pasha Zanoussi, M. Vahdati and M. Fattahi, J. Mol. Catal. A: Chem., 2014, 381, 21 CrossRef PubMed;
(g) A. Ziyaei-Halimehjani, N. Karimi and M. R. Saidi, Synth. Commun., 2013, 43, 744 CrossRef;
(h) A. Ziyaei-Halimehjani, F. Aryanasab and M. R. Saidi, Tetrahedron Lett., 2009, 50, 1441 CrossRef PubMed;
(i) A. Ziyaei-Halimehjani and M. R. Saidi, Tetrahedron Lett., 2008, 49, 1244 CrossRef CAS PubMed , and references are therein.
- For a comprehensive review: A. Noble and J. C. Anderson, Chem. Rev., 2013, 113, 2887 CrossRef CAS PubMed.
- Selected recent reviews:
(a) S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137 CrossRef CAS PubMed;
(b) G. K. Trivedi, J. Ind. Chem. Soc., 2004, 81, 187 CAS;
(c) N. Ono, Heterocycles, 2008, 75, 243 CrossRef CAS PubMed;
(d) I. N. N. Namboothiri and N. Rastogi, Top. Heterocycl. Chem., Synthesis of Heterocycles via Cycloadditions I, ed. A. Hassner, Springer-Verlag, Germany, 2008, vol. 12, p. 1 Search PubMed;
(e) I. N. N. Namboothiri and A. Hassner, Top. Curr. Chem., Stereoselective Heterocyclic Synthesis III, ed. P. Metz, Springer-Verlag, Germany, 2001, vol. 216, p. 1 Search PubMed.
- For a comprehensive review: K. Kaur and I. N. N. Namboothiri, Chimia, 2012, 66, 913 CrossRef CAS PubMed.
- Selected recent articles:
(a) M. Ganesh and I. N. N. Namboothiri, Tetrahedron, 2007, 63, 11973 CrossRef CAS PubMed;
(b) J. G. Greger, S. J. P. Yoon-Miller, N. R. Bechtold, S. A. Flewelling, J. P. MacDonald, C. R. Downey, E. A. Cohen and E. T. Pelkey, J. Org. Chem., 2011, 76, 8203 CrossRef CAS PubMed.
- Selected recent reviews:
(a) W. Yan, X. Shi and C. Zhong, Asian J. Org. Chem., 2013, 2, 904 CrossRef CAS;
(b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- Selected recent reviews:
(a) G. W. Kabalka and R. S. Varma, Org. Prep. Proc. Int., 1987, 19, 283 CrossRef CAS;
(b) R. Ballini, E. Marcantoni and M. Petrini, Amino Group Chemistry, ed. A. Ricci, 2008, p. 93 Search PubMed.
-
(a) A. Hassner and I. N. N. Namboothiri, Organic Syntheses Based on Named Reactions, Elsevier, Oxford, UK, 3rd edn, A Practical Guide, 2012 Search PubMed;
(b) A. X. Wang, Name Reactions for Homologations, ed. J. J. Li, John Wiley & Sons, 2009, p. 404 Search PubMed;
(c) M. Rueping, Nachr. Chem., 2007, 55, 35 CrossRef CAS.
- Selected reviews:
(a) S. V. Ley and C. E. Gutteridge, Chemtracts: Org. Chem., 1995, 8, 222 CAS;
(b) S. V. Ley and C. E. Gutteridge, Chemtracts: Inorg. Chem., 1996, 8, 166 Search PubMed.
- Recent review: G. Yan, A. J. Borah and L. Wang, Org. Biomol. Chem., 2014, 12, 6049 CAS.
- A. Ziyaei-Halimehjani, I. N. N. Namboothiri and S. E. Hooshmand, RSC Adv., 2014, 4, 31261 RSC.
- For a comprehensive review: R. S. Varma and G. W. Kabalka, Heterocycles, 1986, 24, 2645 CrossRef CAS.
- A. Jain, S. Rodriguez, I. Lopez and F. V. Gonzalez, Tetrahedron, 2009, 65, 8362 CrossRef CAS PubMed.
- S. Fioravanti, F. Marchetti, L. Pellacani, L. Ranieria and P. A. Tardella, Tetrahedron: Asymmetry, 2008, 19, 231 CrossRef CAS PubMed.
- A. Rai and L. D. S. Yadav, Tetrahedron, 2012, 68, 2459 CrossRef CAS PubMed.
- A. Rai and L. D. S. Yadav, Org. Biomol. Chem., 2011, 9, 8058 CAS.
- C. Awasthi and L. D. S. Yadav, Synlett, 2010, 1783 Search PubMed.
-
(a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 603 RSC;
(b) Y. Cheng, Z. T. Huang and M. X. Wang, Curr. Org. Chem., 2004, 8, 325 CrossRef CAS;
(c) J. M. Aurrecoechea, A. Fernández, J. M. Gorgojo and C. Saornil, Tetrahedron, 1999, 55, 7345 CrossRef CAS;
(d) J. C. Braekman and D. Daloze, in Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier, Amsterdam, 1990, vol. 6, p. 421 Search PubMed;
(e) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC;
(f) D. O'Hagan, Nat. Prod. Rep., 1997, 14, 637 RSC;
(g) N. Uchide and O. J. Kunio, Antimicrob. Chem., 2003, 52, 8 CrossRef CAS PubMed;
(h) A. Bianco, M. Maggini, G. Scorrano, C. Toniolo, G. Marconi, C. Villani and M. Prato, J. Am. Chem. Soc., 1996, 118, 4072 CrossRef CAS;
(i) B. J. L. Royles, Chem. Rev., 1995, 95, 1981 CrossRef CAS;
(j) J. M. Domagala, S. E. Hagen, T. Joannides, J. S. Kiely, E. Laborde, M. C. Schroeder, J. A. Sesnie, M. A. Shapiro, M. J. Suto and S. J. Vanderroest, Med. Chem., 1993, 36, 871 CrossRef CAS.
-
(a) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC;
(b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed;
(c) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed;
(d) F. X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 3693 CrossRef CAS;
(e) D. M. Huryn, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon: Oxford, U.K., 1991, vol. 1, p. 64 Search PubMed;
(f) D. Enders and M. Klatt, Synthesis, 1996, 1403 CrossRef CAS PubMed;
(g) E. J. Corey, P. W. Yuen, F. J. Hannon and D. A. Wierda, J. Org. Chem., 1990, 55, 784 CrossRef CAS;
(h) B. H. Kim, H. B. Lee, J. K. Hwang and Y. G. Kim, Tetrahedron: Asymmetry, 2005, 16, 1215 CrossRef CAS PubMed;
(i) F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000, 100, 2159 CrossRef CAS PubMed;
(j) J. K. Whitesell, Chem. Rev., 1989, 89, 1581 CrossRef CAS.
- E. Dumez, A.-C. Durand, M. Guillaume, P.-Y. Roger, R. Faure, J.-M. Pons, G. Herbette, J.-P. Dulcere, D. Bonne and J. Rodriguez, Chem.–Eur. J., 2009, 15, 12470 CrossRef CAS PubMed.
- A. Noole, T. Pehk, I. Jarving, M. Lopp and T. Kanger, Tetrahedron: Asymmetry, 2012, 23, 188 CrossRef CAS PubMed.
- L. N. Gautam, Q. Wang, N. G. Akhmedov, J. L. Petersen and X. Shi, Org. Biomol. Chem., 2013, 14, 1917 Search PubMed.
- P.-Y. Roger, A.-C. Durand, J. Rodriguez and J.-P. Dulcere, Org. Lett., 2004, 6, 2027 CrossRef CAS PubMed.
- H. Y. Kim, J. Y. Li, S. Kim and K. Oh, J. Am. Chem. Soc., 2011, 133, 20750 CrossRef CAS PubMed.
- R. Grigg, C. Kilner, M. A. B. Sarker, C. O. de la Cierva and H. A. Dondas, Tetrahedron, 2008, 64, 8974 CrossRef CAS PubMed.
- J. Xie, K. Yoshida, K. Takasu and Y. Takemoto, Tetrahedron Lett., 2008, 49, 6910 CrossRef CAS PubMed.
- Y.-K. Liu, H. Liu, W. Du, L. Yue and Y.-C. Chen, Chem.–Eur. J., 2008, 14, 9873 CrossRef CAS PubMed.
- M.-X. Xue, X.-M. Zhang and L.-Z. Gong, Synlett, 2008, 691 CAS.
- K. Imae, T. Konno, K. Ogata and S. Fukuzawa, Org. Lett., 2012, 14, 4410 CrossRef CAS PubMed.
- X.-X. Yan, Q. Peng, Y. Zhang, K. Zhang, W. Hong, X.-L. Hou and Y.-D. Wu, Angew. Chem., Int. Ed., 2006, 45, 1979 CrossRef CAS PubMed.
- Q. Li, C.-H. Ding, X.-H. Li, W. Weissensteiner and X.-L. Hou, Synthesis, 2012, 265 CAS.
- X. Gu, Z.-J. Xu, V. K.-Y. Lo and C.-M. Che, Synthesis, 2012, 3307 CAS.
- L. M. Castello, C. Najera, J. M. Sansano, O. Larranaga, A. de Cozar and F. P. Cossio, Org. Lett., 2013, 15, 2902 CrossRef CAS PubMed.
- T. Arai, N. Yokoyama, A. Mishiro and H. Sato, Angew. Chem., Int. Ed., 2010, 49, 7895 CrossRef CAS PubMed.
- T. Arai, A. Mishiro, N. Yokoyama, K. Suzuki and H. Sato, J. Am. Chem. Soc., 2010, 132, 5338 CrossRef CAS PubMed.
- A. Zubia, L. Mendoza, S. Vivanco, E. Aldaba, T. Carrascal, B. Lecea, A. Arrieta, T. Zimmerman, F. Vidal-Vanaclocha and F. P. Cossio, Angew. Chem., 2005, 117, 2963 CrossRef.
-
(a) M. Bakthadoss, N. Sivakumar, A. Devaraj and D. S. Sharada, Synthesis, 2011, 2136 CrossRef CAS PubMed;
(b) M. Bakthadoss and N. Sivakumar, Synlett, 2009, 1014 CrossRef CAS PubMed.
- L. Crovetto and R. Rios, Synlett, 2008, 1840 CAS.
- K. Alimohammadi, Y. Sarrafi and B. Rajabpour, C. R. Chimie, 2014, 17, 156 CrossRef CAS PubMed.
- A. Viranyi, G. Marth, A. Dancso, G. Blasko, L. Toke and M. Nyerges, Tetrahedron, 2006, 62, 8720 CrossRef CAS PubMed.
- J.-W. Xie, L.-P. Fan, H. Su, X.-S. Li and D.-C. Xu, Org. Biomol. Chem., 2010, 8, 2117 CAS.
- Y. Jia and D.-M. Du, RSC Adv., 2013, 3, 1970 RSC.
-
(a) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213 CrossRef CAS PubMed;
(b) L. Constantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65 CrossRef;
(c) M. S. J. Butler, Nat. Prod., 2004, 67, 2141 CrossRef CAS PubMed;
(d) T. A. Smith, S. J. Croker and R. S. T. Loeffler, Phytochemistry, 1986, 25, 683 CrossRef CAS;
(e) W. K. Anderson and A. S. Milowsky, J. Med. Chem., 1987, 30, 2144 CrossRef CAS;
(f) S. B. Singh, M. A. Goetz, E. T. Jones, G. F. Bills, R. A. Giacobbe, L. Herranz, S. Stevens-Miles and D. L. Williams Jr, J. Org. Chem., 1995, 60, 7040 CrossRef CAS;
(g) H. Kakeya, C. Onozawa, M. Sato, K. Arai and H. Osada, J. Med. Chem., 1997, 40, 391 CrossRef CAS PubMed.
-
(a) R. A. Batey, P. D. Simoncic, D. Lin, R. P. Smyj and A. J. Lough, Chem. Commun., 1999, 651 RSC;
(b) H. Ulbrich, B. Fiebich and G. Dannhardt, Eur. J. Med. Chem., 2002, 37, 953 CrossRef CAS.
-
(a) L. Li and J. Zhang, Org. Lett., 2011, 13, 5940 CrossRef CAS PubMed;
(b) G. Sathishkannan and K. Srinivasan, Org. Lett., 2011, 13, 6002 CrossRef CAS PubMed;
(c) E. Moreno-Clavijo, A. T. Carmona, H.-U. Reissig, A. J. Moreno-Vargas, E. Alvarez and I. Robina, Org. Lett., 2009, 11, 4778 CrossRef CAS PubMed;
(d) F. Shi, G. J. Xing, W. Tan, R. Y. Zhu and S. Tu, Org. Biomol. Chem., 2013, 11, 1482 RSC.
-
(a) E. G. Howard, R. V. Lindsey Jr and C. W. Theobald, J. Am. Chem. Soc., 1959, 81, 4355 CrossRef CAS;
(b) W. Birru, R. T. Fernley, L. D. Graham, J. Grusovin, R. J. Hill, A. Hofmann, L. Howell, P. J. James, K. E. Jarvis, W. M. Johnson, D. A. Jones, C. Leitner, A. J. Liepa, G. O. Lovrecz, L. Lu, R. H. Nearn, B. J. O. Driscoll, T. Phan, M. Pollard, K. A. Turner and D. A. Winkler, Bioorg. Med. Chem., 2010, 18, 5647 CrossRef CAS PubMed.
- Q. Yang, X. Y. Li, H. Wu and W. J. Xiao, Tetrahedron Lett., 2006, 47, 3893 CrossRef CAS PubMed.
-
(a) M. Quai, S. Frattini, U. Vendrame, M. Mondoni, S. Dossena and E. Cereda, Tetrahedron Lett., 2004, 45, 1413 CrossRef CAS PubMed;
(b) M. J. Fan, B. Qian, L. B. Zhao and Y. M. Liang, Tetrahedron, 2007, 63, 8987 CrossRef CAS PubMed;
(c) M. Adib, M. Mahdavi, M. A. Noghani and H. R. Bijanzadeh, Tetrahedron Lett., 2007, 48, 8056 CrossRef CAS PubMed.
-
(a) R. S. Coleman and P. H. Liu, Org. Lett., 2004, 6, 577 CrossRef CAS PubMed;
(b) J. Clayden, R. Turnbull and I. Pinto, Tetrahedron: Asymmetry, 2005, 16, 2235 CrossRef CAS PubMed.
-
(a) S. Ma and H. Xie, J. Org. Chem., 2002, 67, 6575 CrossRef CAS PubMed;
(b) A. J. Clark, C. P. Dell, J. M. McDonagh, J. Geden and P. Mawdsley, Org. Lett., 2003, 5, 2063 CrossRef CAS PubMed;
(c) N. Rosas, A. Cabrera, P. Sharma, J. L. Arias, J. L. Garcia and H. Arzoumanian, J. Mol. Catal. A: Chem., 2000, 156, 103 CrossRef CAS;
(d) H. Arzoumanian, M. Jean, D. Nuel, J. L. Garcia and N. Rosas, Organometallics, 1997, 16, 2726 CrossRef CAS;
(e) L. Yang, C. H. Lei, D. X. Wang, Z. T. Huang and M. X. Wang, Org. Lett., 2010, 12, 3918 CrossRef CAS PubMed.
- P. Wu, H. Liu and X. Tong, Tetrahedron Lett., 2012, 53, 4673 CrossRef CAS PubMed.
- G. Zhang, Y. Zhang, X. Jiang, W. Yan and R. Wang, Org. Lett., 2011, 13, 3806 CrossRef CAS PubMed.
- C. Guo, M.-X. Xue, M.-K. Zhu and L.-Z. Gong, Angew. Chem., Int. Ed., 2008, 47, 3414 CrossRef CAS PubMed.
- A. Alizadeh, A. Zarei and A. Rezvanian, Synthesis, 2011, 497 CrossRef CAS PubMed.
- M.-Y. Wu, K. Li, N. Wang, T. He and X.-Q. Yu, Synthesis, 2011, 1831 CAS.
-
(a) P. A. Reddy, B. C. H. Hsiang, T. N. Latifi, M. W. Hill, K. E. Woodward, S. M. Rothman, J. A. Ferrendelli and D. F. Covey, J. Med. Chem., 1996, 39, 1898 CrossRef CAS PubMed;
(b) K. Das Sarma, J. Zhang, Y. Huang and J. G. Davidson, Eur. J. Org. Chem., 2006, 3730 CrossRef CAS.
-
(a) A. Spaltenstein, M. R. Almond, W. J. Bock, D. G. Cleary, E. S. Furfine, R. J. Hazen, W. M. Kazmierski, F. G. Salituro, R. D. Tung and L. L. Wright, Bioorg. Med. Chem. Lett., 2000, 10, 1159 CrossRef CAS;
(b) W. M. Kazmierski, W. Andrews, E. Furfine, A. Spaltenstein and L. Wright, Bioorg. Med. Chem. Lett., 2004, 14, 5689 CrossRef CAS PubMed.
- J. Cook, J. Barzya, C. Brennan, D. Lowe, Y. Wang, A. Redman, W. Scott and J. Wood, Tetrahedron Lett., 2005, 46, 1525 CrossRef CAS PubMed.
- B. Winbal, CNS Drug Rev., 2005, 11, 169 Search PubMed.
-
(a) D. M. Barnes, J. Ji, M. G. Fickes, M. A. Fitzgerald, S. A. King, H. E. Morton, F. A. Plagge, M. Preskill, S. H. Wagaw, S. J. Wittenberger and J. Zhang, J. Am. Chem. Soc., 2002, 124, 13097 CrossRef CAS PubMed;
(b) K. Tang and J. T. Zhang, Neurol. Res., 2002, 24, 473 CrossRef CAS PubMed.
-
(a) H. Krawczyk, L. Albrecht, J. Wojciechowski, W. M. Wolf, U. Krajewska and M. Rozalski, Tetrahedron, 2008, 64, 6307 CrossRef CAS PubMed;
(b) R. K. Dieter and K. Lu, Tetrahedron Lett., 1999, 40, 4011 CrossRef CAS;
(c) D. Basavaiah and J. S. Rao, Tetrahedron Lett., 2004, 45, 1621 CrossRef CAS PubMed.
-
(a) W. Brabandt and N. Kimpe, J. Org. Chem., 2005, 70, 8717 CrossRef PubMed;
(b) B. Alcaide, Y. Martín-Cantalejo, J. Pérez-Castells, M. A. Sierra and A. Monge, J. Org. Chem., 1996, 61, 9156 CrossRef CAS.
-
(a) V. Rodriguez-Soria, L. Quintero and F. Sartillo-Piscil, Tetrahedron, 2008, 64, 2750 CrossRef CAS PubMed;
(b) S. Berlin, C. Ericsson and L. Engman, J. Org. Chem., 2003, 68, 8386 CrossRef CAS PubMed.
- S. Spiess, C. Berthold, R. Weihofen and G. Helmchen, Org. Biomol. Chem., 2007, 5, 2357 CAS.
- C. W. Roberson and K. A. Woerpel, J. Org. Chem., 1999, 64, 1434 CrossRef CAS.
- D. Q. Tan, A. Younai, O. Pattawong, J. C. Fettinger, P. H. Y. Cheong and J. T. Shaw, Org. Lett., 2013, 15, 5126 CrossRef CAS PubMed.
-
(a) B. Shetty, A. Mc Fadden and P. Hofer, US Patent 4476311, 1984;
(b) N. Castagnoli, J. Org. Chem., 1969, 34, 3187 CrossRef CAS;
(c) M. Cushman and N. Castagnoli, J. Org. Chem., 1972, 37, 1268 CrossRef CAS;
(d) M. Joannic, D. Humbert and M. Pesson, C. R. Acad. Sci, 1972, 275, 45 CAS.
- J. C. Anderson, L. R. Horsfall, A. S. Kalogirou, M. R. Mills, G. J. Stepney and G. J. Tizzard, J. Org. Chem., 2012, 77, 6186 CrossRef CAS PubMed.
- D. Enders, C. Wang, X. Yang and G. Raabe, Synlett, 2011, 469 CrossRef CAS PubMed.
-
(a) H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed;
(b) C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517 RSC;
(c) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582 CrossRef PubMed;
(d) D. T. Davies, Aromatic Heterocyclic Chemistry, OUP, Oxford, 1992 Search PubMed;
(e) S. Lesage, H. Xu and L. Durham, Hydrol. Sci. J., 1993, 38, 343 CrossRef CAS.
-
(a) R. W. Burli, D. McMinn, J. A. Kaizerman, W. Hu, Y. Ge, Q. Pack, V. Jiang, M. Gross, M. Garcia, R. Tanaka and H. E. Moser, Bioorg. Med. Chem. Lett., 2004, 14, 1253 CrossRef CAS PubMed;
(b) R. W. Burli, P. Jones, D. McMinn, Q. Le, J. X. Duan, J. A. Kaizerman, S. Difuntorum and H. E. Moser, Bioorg. Med. Chem. Lett., 2004, 14, 1259 CrossRef CAS PubMed.
-
(a) M. Bialer, B. Yagen, R. Mechoulam and Y. Beker, J. Med. Chem., 1980, 23, 1144 CrossRef CAS;
(b) Y. Ohba, K. Kamaike, Y. Terui, T. Oshima and E. Kawashima, Nucleic Acids Res. Suppl., 2003, 3, 29 CrossRef CAS.
-
(a) E. Toja, D. Selva and P. Schiatti, J. Med. Chem., 1984, 27, 610 CrossRef CAS;
(b) V. J. Demopoulos and E. Rekka, J. Pharm. Sci., 1995, 84, 79 CrossRef CAS.
- W. A. Denny, G. W. Rewcastle and B. C. Baguley, J. Med. Chem., 1990, 33, 814 CrossRef CAS.
- J. Lehuede, B. Fauconneau, L. Barrier, M. Ourakow, A. Piriou and J. M. Vierfond, Eur. J. Med. Chem., 1999, 34, 991 CrossRef CAS.
- M. Del Poeta, W. A. Schell, C. C. Dykstra, S. Jones, R. R. Tidwell, A. Czarny, M. Bajic, M. Bajic, A. Kumar, D. Boykin and J. R. Perfect, Antimicrob. Agents Chemother., 1998, 42, 2495 CAS.
- For reviews on pyrrole based materials, see:
(a) Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1997 Search PubMed;
(b) Y. Matano and H. Imahori, Acc. Chem. Res., 2009, 42, 1193 CrossRef CAS PubMed;
(c) S. Higgins, Chem. Soc. Rev., 1997, 26, 247 RSC.
-
(a) L. Knorr, Ber., 1884, 17, 1635 CrossRef; For recent examples:
(b) C. M. Shiner and T. D. Lash, Tetrahedron, 2005, 61, 11628 CrossRef CAS PubMed;
(c) R. K. Bellingham, J. S. Carey, N. Hussain, D. O. Morgan, P. Oxley and L. C. Powling, Org. Process Res. Dev., 2004, 8, 279 CrossRef CAS;
(d) J. M. Manley, M. J. Kalman, B. G. Conway, C. C. Ball, J. L. Havens and R. Vaidyanathan, J. Org. Chem., 2003, 68, 6447 CrossRef CAS PubMed.
-
(a) A. Hantzsch, Ber., 1890, 23, 1474 CrossRef; For recent examples:
(b) V. S. Matiychuk, R. L. Martyak, N. D. Obushak, Y. V. Ostapiuk and N. I. Pidlypnyi, Chem. Heterocycl. Comp., 2004, 40, 1218 CrossRef CAS;
(c) L. Calvo, A. González-Ortega and M. C. Sañudo, Synthesis, 2002, 2450 CAS;
(d) A. W. Trautwein, R. D. Sümuth and G. Jung, Bioorg. Med. Chem. Lett., 1998, 8, 2381 CrossRef CAS.
-
(a) C. Paal, Ber., 1885, 18, 367 CrossRef;
(b) L. Knorr, Ber., 1885, 18, 299 CrossRef; For recent examples:
(c) J. Chen, H. Wu, Z. Zheng, C. Jin, X. Zhang and W. Su, Tetrahedron Lett., 2006, 47, 5383 CrossRef CAS PubMed;
(d) G. Minetto, L. F. Raveglia, A. Sega and M. Taddei, Eur. J. Org. Chem., 2005, 5277 CrossRef CAS;
(e) B. K. Banik, I. Banik, M. Renteria and S. K. Dasgupta, Tetrahedron Lett., 2005, 46, 2643 CrossRef CAS PubMed.
-
(a) S. Lahiri, M. P. Mahajan, R. Prasad and M. V. George, Tetrahedron, 1977, 33, 3159 CrossRef CAS;
(b) W. E. McEwen, A. V. Grossi, R. J. MacDonald and A. P. Stamegna, J. Org. Chem., 1980, 45, 1301 CrossRef CAS;
(c) O. Tsuge and K. Ueno, Heterocycles, 1982, 19, 1411 CrossRef CAS;
(d) A. Padwa, G. Haffmanns and M. Thomas, J. Org. Chem., 1984, 49, 3314 CrossRef CAS;
(e) A. R. Katritzky, M. Drewniak and P. Lue, J. Org. Chem., 1988, 559, 5854 CrossRef;
(f) K. I. Washizuka, S. Minakata, I. Ryu and M. Komatsu, Tetrahedron, 1999, 55, 12969 CrossRef CAS;
(g) J. L. Bullington, R. R. Wolff and P. F. Jackson, J. Org. Chem., 2002, 67, 9439 CrossRef CAS PubMed;
(h) A. R. Katritzky, S. Zhang, M. Wang, H. C. Kolb and P. J. Steel, J. Heterocycl. Chem., 2002, 39, 759 CrossRef CAS.
-
(a) E. Benedetti, G. Lemiere, L. L. Chapellet, A. Penoni, G. Palmisano, M. Malacria, J. P. Goddard and L. Fensterband, Org. Lett., 2010, 12, 4396 CrossRef CAS PubMed;
(b) A. Saito, O. Konishi and Y. Hanzawa, Org. Lett., 2010, 12, 372 CrossRef CAS PubMed;
(c) J. T. Binder and S. F. Kirsch, Org. Lett., 2006, 8, 2151 CrossRef CAS PubMed;
(d) S. Kramer, J. L. H. Madsen, M. Rottlander and T. Skrydstrup, Org. Lett., 2010, 12, 2758 CrossRef CAS PubMed;
(e) S. Chiba, Y. F. Wang, G. Lapointe and K. Narasaka, Org. Lett., 2008, 10, 313 CrossRef CAS PubMed;
(f) V. F. Ferreira, M. C. B. V. A. De Souza, C. Cunha, L. O. R. Pereira and M. L. G. Ferreira, Org. Prep. Proced. Int., 2001, 33, 411 CrossRef CAS.
- A. Fürstner, H. Weintritt and A. Hupperts, J. Org. Chem., 1995, 60, 6637 CrossRef.
- A. Katritzky, J. Jiang and P. J. Steel, J. Org. Chem., 1994, 59, 4551 CrossRef CAS.
-
(a) V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402 RSC;
(b) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS PubMed;
(c) G. Balme, Angew. Chem., Int. Ed., 2004, 43, 6238 CrossRef CAS PubMed.
- H. Shiraishi, T. Nishitani, T. Nishihara, S. Sakaguchi and Y. Ishii, Tetrahedron, 1999, 55, 13957 CrossRef CAS.
- Z.-H. Guan, L. Li, M.-N. Zhao, Z.-H. Ren and J. Li, Synthesis, 2012, 532 CrossRef PubMed.
- S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674 CrossRef CAS PubMed.
- G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy, L. S. Reddy, P. M. Kumar, G. R. Krishna, C. M. Reddy, D. Rambabu, R. Kapavarapu, C. Lakshmi, T. Meda, K. K. Priya, K. V. L. Parsa and M. Pal, Chem. Commun., 2011, 47, 7779 RSC.
- I. Yavari, M. Ghazvini and A. Aminkhani, J. Chem. Res., 2011, 35, 558 CrossRef CAS.
- A. Abdukader, Q. Xue, A. Lin, M. Zhang, Y. Cheng and C. Zhu, Tetrahedron Lett., 2013, 54, 5898 CrossRef CAS PubMed.
- Z.-H. Guan, L. Li, Z.-H. Ren, J. Li and M.-N. Zhao, Green Chem., 2011, 13, 1664 RSC.
- A. Palmieri, S. Gabrielli, C. Cimarelli and R. Ballini, Green Chem., 2011, 13, 3333 RSC.
- S. Paul and A. R. Das, Catal. Sci. Technol., 2012, 2, 1130 CAS.
-
(a) V. Y. Korotaev, V. Y. Sosnovskikh, A. Y. Barkov, P. A. Slepukhin, M. A. Ezhikova, M. I. Kodess and Y. V. Shklyaev, Tetrahedron, 2011, 67, 8685 CrossRef CAS PubMed;
(b) V. Y. Korotaev, V. Y. Sosnovskikh, I. B. Kutyashev, A. Y. Barkov and Y. V. Shklyaev, Tetrahedron Lett., 2008, 49, 5376 CrossRef CAS PubMed.
- S. Muthusaravanan, S. Perumal, P. Yogeeswari and D. Sriram, Tetrahedron Lett., 2010, 51, 6439 CrossRef CAS PubMed.
- S. Sarkar, K. Bera, S. Jalal and U. Jana, Chem.–Eur. J., 2013, 6055 CrossRef CAS.
- M.-N. Zhao, H. Liang, Z.-H. Ren and Z. H. Guan, Adv. Synth. Catal., 2013, 355, 221 CrossRef CAS.
- D. Hong, Y. Zhu, Y. Li, X. Lin, P. Lu and Y. Wang, Org. Lett., 2011, 13, 4668 CrossRef CAS PubMed.
- B. C. Ranu and A. Hajra, Tetrahedron, 2001, 57, 4767 CrossRef CAS.
- B. C. Ranu and S. S. Dey, Tetrahedron Lett., 2003, 44, 2865 CrossRef CAS.
- N. C. Jadhav, P. B. Jagadhane, H. V. Patile and V. N. Telvekar, Tetrahedron Lett., 2013, 54, 3019 CrossRef CAS PubMed.
- C. C. Silveira, S. R. Mendes, G. M. Martins, S. C. Schlosser and T. S. Kaufman, Tetrahedron, 2013, 69, 9076 CrossRef CAS PubMed.
- A. Alizadeh, A. Rezvanian and H. R. Bijanzadeh, Synthesis, 2008, 725 CrossRef CAS PubMed.
- J. M. Lopchuk, R. P. Hughes and G. W. Gribble, Org. Lett., 2013, 15, 5218 CrossRef CAS PubMed.
- J. M. Lopchuk and G. W. Gribbe, Tetrahedron Lett., 2011, 52, 4106 CrossRef CAS PubMed.
- N. C. Misra, K. Panda, H. Ila and H. Junjappa, J. Org. Chem., 2007, 72, 1246 CrossRef CAS PubMed.
- I. R. Baxendale, C. D. Buckle, S. V. Ley and L. Tamborini, Synthesis, 2009, 1485 CAS.
- T. Chen, N. Shao, H. Zhu, B. Zhang and H. Zou, Tetrahedron, 2013, 69, 10558 CrossRef CAS PubMed.
- Y. Kuroda, K. Imaizumi, K. Yamada, Y. Yamaoka and K. Takasu, Tetrahedron Lett., 2013, 54, 4073 CrossRef CAS PubMed.
- C. Dell'Erba, A. Mugnoli, M. Novi, M. Pani, G. Petrillo and C. Tavani, Eur. J. Org. Chem., 2000, 903 CrossRef.
- N. Nishiwaki, T. Ogihara, T. Takami, M. Tamura and M. Ariga, J. Org. Chem., 2004, 69, 8382 CrossRef CAS PubMed.
- N. Nishiwaki, M. Nakanishi, T. Hida, Y. Miwa, M. Tamura, K. Hori, Y. Tohda and M. Ariga, J. Org. Chem., 2001, 66, 7535 CrossRef CAS.
- L. Moradi, M. Piltan, H. Rostami and G. Abasi, Chin. Chem. Lett., 2013, 24, 740 CrossRef CAS PubMed.
-
(a) P. Merino, in Science of Synthesis, ed. A. Padwa, Georg Thieme Verlag, Stuttgart, Germany, 2004, vol. 27, ch. 13 (Nitrones and Analogues), p. 511 Search PubMed;
(b) R. C. F. Jones, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Towards Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Wiley, New York, USA, 2002, ch. 1 (Nitrones), p. 1 Search PubMed;
(c) K. B. G. Torssell, Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, VCH, Weinheim, Germany, 1988 Search PubMed;
(d) R. Revuelta, S. Cicchi, A. Goti and A. Brandi, Synthesis, 2007, 485 Search PubMed;
(e) I. Delso, E. Marca, V. Mannucci, T. Tejero, A. Goti and P. Merino, Chem.–Eur. J., 2010, 16, 9910 CrossRef CAS PubMed;
(f) J. K. Su, Y. M. Jia, R. He, P. X. Rui, N. Han, X. He, J. Xiang, X. Chen, J. Zhu and C. Y. Yu, Synlett, 2010, 1609 CAS;
(g) O. P. Bande, V. H. Jashav, V. G. Puranik, D. D. Dhavale and M. Lombardo, Tetrahedron Lett., 2009, 50, 6906 CrossRef CAS PubMed;
(h) P. Merino, I. Delso, T. Tejero, F. Cardona, M. Marradi, E. Faggi, C. Parmeggiani and A. Goti, Eur. J. Org. Chem., 2008, 2929 CrossRef CAS;
(i) I. Delso, T. Tejero, A. Goti and P. Merino, J. Org. Chem., 2011, 76, 4139 CrossRef CAS PubMed.
- A. E. McCaig and R. H. Wightman, Tetrahedron Lett., 1993, 24, 3939 CrossRef.
-
(a) A. Peer and A. Vasella, Helv. Chim. Acta, 1999, 82, 1044 CrossRef CAS.
-
(a) J. Markandu, H. A. Dondas, M. Frederickson and R. Grigg, Tetrahedron, 1997, 38, 13165 CrossRef;
(b) H. A. Dondas, M. Frederickson, R. Grigg, J. Markandu and M. Thornton-Pett, Tetrahedron, 1997, 42, 14339 CrossRef;
(c) C. W. Holzapfel and R. Crous, Heterocycles, 1998, 48, 1337 CrossRef CAS;
(d) M. Closa and R. H. Wightman, Synth. Commun., 1998, 28, 3443 CrossRef CAS;
(e) C. Chevrier, D. LeNouen, M. Neuburger, A. Defoin and C. Tarnus, Tetrahedron Lett., 2004, 45, 5363 CrossRef CAS PubMed.
-
(a) P. Armstrong, R. Grigg, S. Surendrakumar and W. J. Warnock, J. Chem. Soc., Chem. Commun., 1987, 1327 RSC;
(b) A. Hall, K. P. Meldrum, P. R. Therond and R. H. Wightman, Synlett, 1997, 123 CrossRef CAS PubMed;
(c) R. Grigg, J. Markandu, T. Perrior, S. Surendrakumar and W. J. Warnock, Tetrahedron, 1992, 48, 6929 CrossRef CAS;
(d) T. Ishikawa, Y. Tajima, M. Fukui and S. Saito, Angew. Chem., Int. Ed., 1996, 35, 1863 CrossRef CAS;
(e) P. Herzegh, I. Korvács, L. Szilágyi, T. Varga, Z. Dinya and F. Sztaricskai, Tetrahedron Lett., 1993, 34, 1211 CrossRef.
- D. Sadaba, I. Delso, T. Tejero and P. Merino, Tetrahedron Lett., 2011, 52, 5976 CrossRef CAS PubMed.
- X. Han, X. Wu, C. Min, H. B. Zhou and C. Dong, RSC Adv., 2012, 2, 7501 RSC.
-
(a) J. E. Saxton, Nat. Prod. Rep., 1986, 3, 357 RSC;
(b) J. Ezquerra, C. Pedregal and C. Lamas, J. Org. Chem., 1996, 61, 5804 CrossRef CAS;
(c) A. M. Lobo and S. J. Prabhakar, Heterocycl. Chem., 2002, 39, 429 CrossRef CAS;
(d) W. N. Xiong, C. G. Yang and B. Jiang, Bioorg. Med. Chem., 2001, 9, 1773 CrossRef CAS;
(e) H. Tokuyama and T. Fukuyama, Kagaku Kogyo, 2001, 52, 416 CAS;
(f) M. Toyota and M. Ihara, Nat. Prod. Rep., 1998, 15, 327 RSC.
-
(a) S. A. Patil, R. Patil and D. D. Miller, Curr. Med. Chem., 2011, 18, 615 CrossRef CAS;
(b) S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC;
(c) J. J. Song, J. T. Reeves, D. R. Fandrick, Z. Tan, N. K. Yee and C. H. Senanayake, ARKIVOC, 2010, 390 CrossRef CAS;
(d) G. Palmisano, A. Penoni, M. Sisti, F. Tibiletti, S. Tollari and K. M. Nicholas, Curr. Org. Chem., 2010, 14, 2409 CrossRef CAS;
(e) S. A. Patil, R. Patil and D. D. Miller, Curr. Med. Chem., 2009, 16, 2531 CrossRef CAS;
(f) J. Barluenga, F. Rodriguez and F. J. Fananas, Chem.–Asian J., 2009, 4, 1036 CrossRef CAS PubMed;
(g) J. S. Russel and E. T. Pelkey, Prog. Heterocycl. Chem., 2009, 20, 122 CAS;
(h) K. Kruger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef;
(i) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed;
(j) G. W. Gribble, Pure Appl. Chem., 2003, 75, 1417 CrossRef CAS;
(k) G. W. J. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045 RSC.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 719 Search PubMed ; and the references are therein.
- T. H. H. Hsieh and V. M. Dong, Tetrahedron, 2009, 65, 3062 CrossRef CAS PubMed.
- M. Arai, Y. Miyauchi, T. Miyahara, T. Ishikawa and S. Saito, Synlett, 2009, 122 CAS.
- F. Zhang, C. Li and C. Qi, Synthesis, 2013, 3007 CAS.
- T. Pelkey and G. W. Gribble, Tetrahedron Lett., 1997, 38, 5603 CrossRef.
- B. J. Stokes, S. Liu and T. G. Driver, J. Am. Chem. Soc., 2011, 133, 4702 CrossRef CAS PubMed.
- A. Palmieri, S. Gabrielli, D. Lanari, L. Vaccaro and R. Ballini, Adv. Synth. Catal., 2011, 353, 1425 CrossRef CAS.
- S. Mitra, A. K. Bagdi, A. Majee and A. Hajra, Tetrahedron Lett., 2013, 54, 4982 CrossRef CAS PubMed.
- X. Liu, D. Wang and B. Chen, Tetrahedron, 2013, 69, 9417 CrossRef CAS PubMed.
- R. L. Yan, H. Yan, C. Ma, Z.-Y. Ren, X.-A. Gao, G. S. Huang and Y. M. Liang, J. Org. Chem., 2012, 77, 2024 CrossRef CAS PubMed.
- S. Santra, A. K. Bagdi, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 1065 CrossRef CAS.
- D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2012, 14, 4580 CrossRef CAS PubMed.
-
(a) S. Bondock, W. Fadaly and M. A. Metwally, J. Eur. Med. Chem., 2010, 45, 3692 CrossRef CAS PubMed;
(b) P. Rathelot, N. Azas, H. EI-Kashef, F. Delmas, C. D. Giorgio, P. Timon-David, J. Maldonado and P. Vanelle, J. Eur. Med. Chem., 2002, 37, 671 CrossRef CAS;
(c) M. A. A. El-Sayed, N. L. Abdel-Aziz, A. A. M. Abdel-Aziz, A. S. El-Azab and K. E. H. El Tahir, Bioorg. Med. Chem., 2012, 20, 3306 CrossRef CAS PubMed;
(d) M. Abdel-Aziz, A. G. Abuo-Rahma EI-Din and A. A. Hassan, J. Eur. Med. Chem., 2009, 44, 3480 CrossRef CAS PubMed;
(e) A. I. Hashem, A. S. A. Youssef, K. A. Kandeel and W. S. I. Abou-Elmagd, J. Eur. Med. Chem., 2007, 42, 934 CrossRef CAS PubMed;
(f) R. J. Gillespie, J. A. Cliffe, C. E. Dawson, C. T. Dourish, S. Gaur, A. M. Jordan, A. R. Knight, J. Lerpiniere, A. Misra, R. M. Pratt, J. Roffey, G. C. Stratton, R. Upton, S. M. Weiss and D. S. Williamson, Bioorg. Med. Chem. Lett., 2008, 18, 2924 CrossRef CAS PubMed;
(g) F. Teixeira-Clerc, B. Julien, P. Grenard, J. Tran Van Nhieu, V. Deveaux, L. Li, V. Serriere-Lanneau, C. Ledent, A. Mallat and S. Lotersztajn, Nat. Med., 2006, 12, 671 CrossRef CAS PubMed;
(h) R. Zhang, X. Wu, J. C. Yalowich and B. B. Hasinoff, Bioorg. Med. Chem., 2011, 19, 7023 CrossRef CAS PubMed;
(i) P. C. Lv, H. Q. Li, J. Sun, Y. Zhou and H. L. Zhu, Bioorg. Med. Chem., 2010, 18, 4606 CrossRef CAS PubMed.
-
(a) R. A. Singer, S. Caron, R. E. McDermott, P. Arpin and N. M. Do, Synthesis, 2003, 1727 CrossRef CAS PubMed;
(b) R. A. Singer, M. Dore, J. E. Sieser and M. A. Berliner, Tetrahedron Lett., 2006, 47, 3727 CrossRef CAS PubMed.
-
(a) L. Knorr, Ber., 1883, 16, 2587 CrossRef;
(b) M. V. Patel, R. Bell, S. Majest, R. Henry and T. Kolasa, J. Org. Chem., 2004, 69, 7058 CrossRef CAS PubMed;
(c) S. Peruncheralathan, T. A. Khan, H. Ila and H. Junjappa, J. Org. Chem., 2005, 70, 10030 CrossRef CAS PubMed.
-
(a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef;
(b) A. Padwa, 1,3-Dipolar Cycloaddition Chemistry, John Wiley & Sons, New York, 1984, vol. I Search PubMed;
(c) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, New York, 2002 Search PubMed.
-
(a) M. D. Ankhiwala and M. V. Hathi, Indian J. Heterocycl. Chem., 1996, 5, 229 CAS;
(b) G. Turan-zitouni, A. Ozdemir and K. Guven, Arch. Pharm., 2005, 338, 96 CrossRef CAS PubMed;
(c) Y. R. Prasad, A. L. Rao and P. Ravi, Bioorg. Med. Chem. Lett., 2005, 5, 5030 CrossRef PubMed;
(d) M. Abid and A. Azam, Bioorg. Med. Chem. Lett., 2006, 16, 2812 CrossRef CAS PubMed;
(e) A. C. Grosscurt, R. Vanher and K. Wellinga, J. Agric. Food Chem., 1979, 27, 406 CrossRef CAS;
(f) R. A. Nugent, M. Murphy, S. T. Schlachter, C. J. Dunn, R. J. Smith, N. D. Staite, L. A. Galinet, S. K. Shields, D. G. Aspar, K. A. Richard and N. A. J. Rohloff, Med. Chem., 1993, 36, 134 CrossRef CAS;
(g) R. Van Hes, K. Wellinga and A. C. Grosscurt, J. Agric. Food Chem., 1978, 26, 915 CrossRef CAS;
(h) J. H. H. Eussen, K. Wellinga and B. Stork, Pestic. Sci., 1990, 29, 101 CrossRef CAS;
(i) A. Burger, Burger's Medicinal Chemistry and drug discovery, John Wiley Publications Inc., 5th edn, 1995, vol. 2–6 Search PubMed.
-
(a) M. Shaharyar, A. A. Siddiqui, M. A. Ali, D. Sriram and P. Yogeeswari, Bioorg. Med. Chem. Lett., 2006, 16, 3947 CrossRef CAS PubMed;
(b) A. Levai, Heterocycl. Commun., 2003, 9, 287 CrossRef CAS;
(c) F. Chimenti, A. Bolasco, F. Manna, D. Secci, P. Chimenti, O. Befani, P. Turini, V. Giovannini, B. Mondovi, R. Cirilli and F. Torre, J. Med. Chem., 2004, 47, 2071 CrossRef CAS PubMed;
(d) F. Chimenti, B. Bizzarri, F. Manna, A. Boalsco, D. Secci, P. Chimenti, A. Granese, D. Rivanera, D. Lilli, M. M. Scaltrito and M. I. Brenciaglia, Bioorg. Med. Chem. Lett., 2005, 15, 603 CrossRef CAS PubMed;
(e) A. Levai, J. Jeko and D. I. J. Brahmbhatt, Heterocycl. Chem., 2005, 42, 1231 CrossRef CAS;
(f) A. Levai, J. Heterocyclic Chem., 2005, 39, 1 CrossRef;
(g) A. Levai and J. Jeko, J. Heterocyclic Chem., 2006, 43, 111 CrossRef CAS.
- R. J. Crawford, A. Mishra and R. J. Dummel, J. Am. Chem. Soc., 1966, 88, 3959 CrossRef CAS.
-
(a) L. A. Bañuelos, P. Cuadrado, A. M. González-Nogal, I. López-Solera, F. J. Pulido and P. R. Raithby, Tetrahedron, 1996, 52, 9193 CrossRef;
(b) P. Cuadrado, A. M. González-Nogal and S. Martínez, Tetrahedron, 1997, 53, 8585 CrossRef CAS.
- R. J. Crawford and A. Mishra, J. Am. Chem. Soc., 1965, 87, 3768 CrossRef CAS.
- B. B. Snider, R. S. E. Conn and S. Sealfon, J. Org. Chem., 1979, 44, 218 CrossRef CAS.
- X. Deng and N. S. Mani, Org. Lett., 2006, 8, 3505 CrossRef CAS PubMed.
- X. Deng and N. S. Mani, J. Org. Chem., 2008, 73, 2412 CrossRef CAS PubMed.
- X. Deng and N. S. Mani, Org. Lett., 2008, 10, 1307 CrossRef CAS PubMed.
- M. Tang, W. Zhang and Y. Kong, Org. Biomol. Chem., 2013, 11, 6250 CAS.
- J. W. Xie, Z. Wang, W.-J. Yang, L.-C. Kong and D.-C. Xu, Org. Biomol. Chem., 2009, 7, 4352 CAS.
- V. A. Zapol'skii, J. C. Namyslo, M. Gjikaj and D. E. Kaufmann, Synlett, 2007, 1507 Search PubMed.
-
(a) R. Muruganantham, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2007, 9, 1125 CrossRef CAS PubMed;
(b) R. Muruganantham and I. N. N. Namboothiri, J. Org. Chem., 2010, 75, 2197 CrossRef CAS PubMed.
- R. Kumar and I. N. N. Namboothiri, Org. Lett., 2011, 13, 4016 CrossRef CAS PubMed.
- D. Verma, R. Kumar and I. N. N. Namboothiri, J. Org. Chem., 2013, 78, 3482 CrossRef CAS PubMed.
- N. Shao, T. Chen, T. Zhang, H. Zhu, Q. Zheng and H. Zou, Tetrahedron, 2014, 70, 795 CrossRef CAS PubMed.
- V. Y. Korotaev, I. B. Kutyashev, V. Y. Sosnovskikh and M. I. Kodess, Mendeleev Commun., 2007, 17, 52 CrossRef CAS PubMed.
- A. Alizadeh and N. Zohreh, Synlett, 2012, 428 CrossRef CAS PubMed.
- M. V. Pilipecz, Z. Mucsi, P. Nemes and P. Scheiber, Heterocycles, 2007, 71, 1919 CrossRef CAS.
- F. Felluga, C. Forzato, P. Nitti, G. Pitacco, E. Valentin and E. Zangrando, J. Heterocycl. Chem., 2010, 47, 664 CAS.
- A. Y. Sukhorukov, Y. D. Boyko, Y. V. Nelyubina, S. Gerard, S. L. Ioffe and V. A. Tartakovsky, J. Org. Chem., 2012, 77, 5465 CrossRef CAS PubMed.
-
(a) S. Palhagen, R. Canger, O. Henriksen, J. A. van Parys, M. E. Riviere and M. A. Karolchyk, Epilepsy Res., 2001, 43, 115 CrossRef CAS;
(b) S. Velazquez, R. Alvarez, C. Perez, F. Gago, C. E. De, J. Balzarini and J. M. Camarasa, Antiviral Chem. Chemother., 1998, 9, 481 CAS.
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, J. Med. Chem., 2000, 43, 953 CrossRef CAS PubMed.
- N. G. Aher, V. S. Pore, N. N. Mishra, A. Kumar, P. K. Shukla, A. Sharma and M. K. Bhat, Bioorg. Med. Chem. Lett., 2009, 19, 759 CrossRef CAS PubMed.
- M. J. Soltis, H. J. Yeh, K. A. Cole, N. Whittaker, R. P. Wersto and E. C. Kohn, Drug Metab. Dispos., 1996, 24, 799 CAS.
-
(a) R. Alvarez, S. Velazquez, R. San, S. Aquaro, C. E. De, C. F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185 CrossRef CAS;
(b) M. Whiting, J. Muldoon, Y. C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, B. K. Sharpless, J. H. Elder and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 1435 CrossRef CAS PubMed;
(c) A. Brik, J. Muldoon, Y. C. Lin, J. C. Elder, D. S. Goodsell, A. J. Olson, V. V. Fokin, B. K. Sharpless and C. H. Wong, ChemBioChem., 2003, 4, 1246 CrossRef CAS PubMed.
- L. L. Brockunier, E. R. Parmee, H. O. Ok, M. R. Candelore, M. A. Cascieri, L. F. Colwell, L. Deng, W. P. Feeney, M. J. Forrest, G. J. Hom, D. E. MacIntyre, L. Tota, M. J. Wyvratt, M. H. Fisher and A. E. Weber, Bioorg. Med. Chem. Lett., 2000, 10, 2111 CrossRef CAS.
-
(a) V. Pande and M. J. Ramos, Bioorg. Med. Chem. Lett., 2005, 15, 5129 CrossRef CAS PubMed;
(b) P. H. Olesen, A. R. Sørensen, B. Ursö, P. Kurtzhals, A. N. Bowler, U. Ehrbar and B. F. Hansen, J. Med. Chem., 2003, 46, 3333 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388 RSC.
- A. C. Fahrenbach and J. F. Stoddart, Chem.–Asian J., 2011, 6, 2660 CrossRef CAS PubMed.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC.
-
(a) N. H. Morgan, (CHOMERICS, Inc.), Eur. Pat. Appl., EP 437979 A2 19910724, 1991;
(b) R. J. Willis and I. D. Marlow, Eur. Pat Appl., 400842, 1990, Chem. Abstr., 1991, 114, 164247b.
- L. Ackermann and H. K. Potukuchi, Org. Biomol. Chem., 2010, 8, 4503 CAS.
- S. Sengupta, H. Duan, W. Lu, J. L. Petersen and X. Shi, Org. Lett., 2008, 10, 1493 CrossRef CAS PubMed.
- W. Zou, M. Bhasin, K. Vembaiyan and D. T. Williams, Carbohydr. Res., 2009, 344, 1024 CrossRef CAS PubMed.
- D. Amantini, F. Fringuelli, O. Piermatti, F. Pizzo, E. Zunino and L. Vaccaro, J. Org. Chem., 2005, 70, 6526 CrossRef CAS PubMed.
- B. Quiclet-Sire and S. Z. Zard, Synthesis, 2005, 3319 CrossRef CAS PubMed.
- R. K. Arigela, S. Samala, R. Mahar, S. K. Shukla and B. Kundu, J. Org. Chem., 2013, 78, 10476 CrossRef CAS PubMed.
- V. A. Zapol'skii, X. Yang, J. C. Namyslo, M. Gjikaj and D. E. Kaufmann, Synthesis, 2012, 885 Search PubMed.
-
(a) U. Jahn and D. Rudakov, Synlett, 2004, 1207 CrossRef CAS PubMed;
(b) U. Jahn, D. Rudakov and P. G. Jones, Tetrahedron, 2012, 68, 1521 CrossRef CAS PubMed.
- U. Jahn and D. Rudakov, Org. Lett., 2006, 8, 4481 CrossRef CAS PubMed.
- I. N. N. Namboothiri, M. Ganesh, S. M. Mobin and M. Cojocaru, J. Org. Chem., 2005, 70, 2235 CrossRef CAS PubMed.
- E. Dumez, R. Faure and J.-P. Dulcere, Eur. J. Org. Chem., 2001, 2577 CrossRef CAS.
- S. Belot, A. Quintard, N. Krause and A. Alexakis, Adv. Synth. Catal., 2010, 352, 667 CrossRef CAS.
- W.-Q. Wu, C.-H. Ding and X.-L. Hou, Synlett, 2012, 1035 CAS.
-
(a) R. Huisgen and P. de March, J. Am. Chem. Soc., 1982, 104, 4953 CrossRef CAS;
(b) M. Alt and G. Maas, Tetrahedron, 1994, 50, 7435 CrossRef CAS;
(c) C.-D. Lu, Z.-Y. Chen, H. Liu, W.-H. Hu, A.-Q. Mi and M. P. Doyle, J. Org. Chem., 2004, 69, 4856 CrossRef CAS PubMed.
- V. Nair, S. Mathai and R. L. Varma, J. Org. Chem., 2004, 69, 1413 CrossRef CAS PubMed.
-
(a) B. A. Keay and P. W. Dibble, Furans and their benzo derivatives: Applications. In Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Elsevier, Oxford, 1996, p. 395 Search PubMed;
(b) B. Figadere, Acc. Chem. Res., 1995, 28, 359 CrossRef CAS;
(c) A. Roy and S. Saraf, Biol. Pharm. Bull., 2006, 29, 191 CrossRef CAS.
- N. Zanatta, S. H. Alves, H. S. Coelho, D. M. Borchhardt, P. Machado, K. M. Flores, F. M. Silva, T. B. Spader, J. M. Santurio, H. G. Bonacorso and M. A. P. Martins, Bioorg. Med. Chem., 2007, 15, 1947 CrossRef CAS PubMed.
- R. R. Rao, V. Chaturvedi, K. S. Babu, P. P. Reddy, V. R. S. Rao, P. Sreekanth, A. S. Sreedhar and J. M. Rao, Med. Chem. Res., 2012, 21, 634 CrossRef CAS.
- D. J. Kerr, E. Hamel, M. K. Jung and B. L. Flynn, Bioorg. Med. Chem., 2007, 15, 3290 CrossRef CAS PubMed.
- V. Amarnath and K. Amarnath, J. Org. Chem., 1995, 60, 301 CrossRef CAS.
-
(a) F. Feist, Chem. Ber., 1902, 35, 1537 CrossRef CAS;
(b) E. Benary, Chem. Ber., 1911, 44, 489 CrossRef CAS; For recent examples, see:
(c) M. A. Calter and C. Zhu, Org. Lett., 2002, 4, 205 CrossRef CAS PubMed;
(d) M. A. Calter, R. M. Phillips and C. Flaschenriem, J. Am. Chem. Soc., 2005, 127, 14566 CrossRef CAS PubMed;
(e) E. Holtz and P. Langer, Synlett, 2004, 1805 CAS.
-
(a) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285 CrossRef CAS;
(b) S. Ma, J. Zhang and L. Lu, Chem.–Eur. J., 2003, 9, 2447 CrossRef CAS PubMed;
(c) J. A. Marshall and G. S. Bartley, J. Org. Chem., 1994, 59, 7169 CrossRef CAS;
(d) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 5195 CrossRef CAS PubMed;
(e) A. S. K. Hashmi, Angew. Chem., Int. Ed., 1995, 34, 1581 CrossRef.
-
(a) R. Shen and X. Huang, Org. Lett., 2008, 10, 3283 CrossRef CAS PubMed;
(b) G.-W. Wang, Y.-W. Dong, P. Wu, T.-T. Yuan and Y.-B. Shen, J. Org. Chem., 2008, 73, 7088 CrossRef CAS PubMed;
(c) R. Shen, S. Zhu and X. Huang, J. Org. Chem., 2009, 74, 4118 CrossRef CAS PubMed;
(d) Q. F. Wang, H. Hou, L. Hui and C.-G. Yan, J. Org. Chem., 2009, 74, 7403 CrossRef CAS PubMed.
- A. A. Avetisyan, L. V. Karapetyan, R. V. Paronikyan and H. M. Stepanyan, Pharm. Chem. J., 2011, 45, 156 CrossRef CAS.
- E. Logoglu, M. Yilmaz, H. Katircioglu, M. Yakut and S. Mercan, Med. Chem. Res., 2010, 19, 490 CrossRef CAS.
- Y. Zhang, H. Zhong, T. Wang, D. Geng, M. Zhang and K. Li, Eur. J. Med. Chem., 2012, 48, 69 CrossRef CAS PubMed.
- T. G. Kilroy, T. P. O'Sullivan and P. J. Guiry, Eur. J. Org. Chem., 2005, 23, 4929 CrossRef.
- W. Raimondi, D. Dauzonne, T. Constantieux, D. Bonne and J. Rodriguez, Eur. J. Org. Chem., 2012, 6119 CrossRef CAS.
- D. K. Nair, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2012, 53, 3349 CrossRef CAS PubMed.
- R. Ballini, S. Gabrielli and A. Palmieri, Synlett, 2010, 2468 CAS.
- A. Y. Barkov, V. Y. Korotaev and V. Y. Sosnovskikh, Tetrahedron Lett., 2013, 54, 4181 CrossRef CAS PubMed.
- C. Zhong, T. Liao, O. Tuguldur and X. Shi, Org. Lett., 2010, 12, 2064 CrossRef CAS PubMed.
- N. Ayyagari, D. Jose, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron Lett., 2011, 52, 258 CrossRef CAS PubMed.
- Y.-Y. Yan, R.-J. Lu, J.-J. Wang, Y.-N. Xuan and M. Yan, Tetrahedron, 2012, 68, 6123 CrossRef CAS PubMed.
- R.-Q. Mei, X.-Y. Xu, L. Peng, F. Wang, F. Tiana and L.-X. Wang, Org. Biomol. Chem., 2013, 11, 1286 CAS.
- D. K. Barange, V. Kavala, C.-W. Kuo, P.-M. Lei and C.-F. Yao, Tetrahedron, 2011, 67, 2870 CrossRef CAS PubMed.
- Z. Zhou, H. Liu, Y. Li, J. Liu, Y. Li, J. Liu, J. Yao and C. Wang, ACS Comb. Sci., 2013, 7, 363 CrossRef PubMed.
- C. Jarava-Barrera, F. Esteban, C. Navarro-Ranninger, A. Parra and J. Aleman, Chem. Commun., 2013, 2001 RSC.
- M.-Y. Wu, K. Li, T. He, X.-W. Feng, N. Wang, X.-Y. Wang and X.-Q. Yu, Tetrahedron, 2011, 67, 2681 CrossRef CAS PubMed.
- T.-Y. Liu, H.-L. Cui, Q. Chai, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Chem. Commun., 2007, 2228 RSC.
- L. Song, X. Li, C. Xing, D. Li, S. Zhu, H. Deng and M. Shao, Synlett, 2010, 830 CrossRef CAS PubMed.
- S.-C. Lu, P.-R. Zheng and G. Liu, J. Org. Chem., 2012, 77, 7711 CrossRef CAS PubMed.
- T. Ishikawa, T. Miyahara, M. Asakura, S. Higuchi, Y. Miyauchi and S. Saito, Org. Lett., 2005, 7, 1211 CrossRef CAS PubMed.
- D. K. Barange, B. R. Raju, V. Kavala, C.-W. Kuo, Y.-C. Tu and C.-F. Yao, Tetrahedron, 2010, 66, 3754 CrossRef CAS PubMed.
- T. Kumar, S. M. Mobin and I. N. N. Namboothiri, Tetrahedron, 2013, 69, 4964 CrossRef CAS PubMed.
- D. Kundu, M. Samim, A. Majee and A. Hajra, Chem.–Asian J., 2011, 6, 406 CrossRef CAS PubMed.
- Y. Li, H. Liu, L. Sun, J. Liu, Z. Xue, J. YaO and C. Wang, Synlett, 2013, 1851 Search PubMed.
- Q.-B. Li, F.-T. Zhou, Z.-G. Liu, X.-F. Li, W.-D. Zhu and J.-W. Xie, J. Org. Chem., 2011, 76, 7222 CrossRef CAS PubMed.
- M. R. Zanwar, V. Kavala, S. D. Gawande, C. W. Kuo, T.-S. Kuo, M.-L. Chen, C.-H. He and C.-F. Yao, Eur. J. Org. Chem., 2013, 8288 CrossRef CAS.
- C. M. Simpkins and D. A. Hunt, Tetrahedron Lett., 2013, 54, 3371 CrossRef CAS PubMed.
- K. Koike, Z. Jia, T. Nikaido, Y. Liu, Y. Zhao and D. Guo, Org. Lett., 1999, 1, 197 CrossRef CAS , and references are therein.
- G. Schopf and G. Koßmehl, Adv. Polym. Sci., 1997, 129, 1 CrossRef CAS; D. Ekinci, F. Tumer and U. Demir, Eur. Polym. J., 2002, 38, 1837 CrossRef.
-
(a) M. S. Kiani and G. R. Mitchell, Synth. Met., 1992, 46, 293 CrossRef CAS;
(b) C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- X.-C. Li, H. Sirringhaus, F. Garnier, A. B. Holmes, S. C. Moratti, N. Feeder, W. Clegg, S. J. Teat and R. H. Friend, J. Am. Chem. Soc., 1998, 120, 2206 CrossRef CAS.
- N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628 CrossRef CAS.
-
(a) D. E. Wolf and K. Folkers, The Preparation of Thiophenes and Tetrahydrothiophenes, Organic Reactions, 2011, p. 410 Search PubMed;
(b) S. Gronowitz, Thiophene and its Derivatives, ed. S. Gronowitz, John Wiley and Sons, New York, 1985, Part 1, p. 42 Search PubMed;
(c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS;
(d) G. Revelant, S. Dunand, S. Hesse and G. Kirsch, Synthesis, 2011, 2935 CAS;
(e) T. Wang, X. G. Huang, J. Liu, B. Li, J. J. Wu, K. X. Chen, W. L. Zhu, X. Y. Xu and B. B. Zeng, Synlett, 2010, 1351 CAS;
(f) W. You, X. Yan, Q. Liao and C. Xi, Org. Lett., 2010, 12, 3930 CrossRef CAS PubMed;
(g) G. C. Nandi, S. Samai and M. S. Singh, J. Org. Chem., 2011, 76, 8009 CrossRef CAS PubMed.
-
(a) S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57 CrossRef CAS PubMed;
(b) J. C. Aloup, J. Bouchaudon, D. Farge, C. James, J. Deregnaucourt and M. Hardy-Houis, J. Med. Chem., 1987, 30, 24 CrossRef CAS;
(c) M. W. Harrold, R. A. Wallace, T. Farooqui, L. J. Wallace, N. Uretsky and D. D. Miller, J. Med. Chem., 1989, 32, 874 CrossRef CAS.
- C. J. O' Connor, M. D. Roydhouse, A. M. Przybyz, M. D. Wall and J. M. Southern, J. Org. Chem., 2010, 75, 2534 CrossRef PubMed.
- N. Baricordi, S. Benetti, V. Bertolasi, C. De Risi, G. P. Pollini, F. Zamberlan and V. Zanirato, Tetrahedron, 2012, 68, 208 CrossRef CAS PubMed.
- C. Xu, J. Du, L. Ma, G. Li, M. Tao and W. Zhang, Tetrahedron, 2013, 69, 4749 CrossRef CAS PubMed.
- C. Yu, Y. Zhang, A. Song, Y. Ji and W. Wang, Chem.–Eur. J., 2011, 17, 770 CrossRef CAS PubMed.
- K. S. Chunikhin, L. A. Rodinovskaya and A. M. Shestopalov, Chem. Heterocycl. Comp., 2007, 43, 1247 CrossRef CAS.
- M. Avalos, R. Babiano, P. Cintas, J. L. Jimenez and J. C. Palacios, Acc. Chem. Res., 2005, 38, 460 CrossRef CAS PubMed.
- K. Bogdanowicz-Szwed and R. Gil, Monatsh. Chem., 2004, 135, 1415 CrossRef CAS PubMed.
-
(a) P. Caramella and P. Grunanger, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, Wiley, New York, 1984, vol. 1, p. 291 Search PubMed;
(b) K. B. G. Torsell, Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis, VCH, New York, 1988 Search PubMed;
(c) P. Conti, C. Dallanoce, M. D. Amici, C. D. Micheli and K. N. Klotz, Bioorg. Med. Chem., 1998, 6, 401 CrossRef CAS;
(d) S. Srirastara, L. K. Bajpai, S. Batra, A. P. Bhaduri, J. P. Maikhuri, G. Gupta and J. D. Dhar, Bioorg. Med. Chem., 1999, 7, 2607 CrossRef.
-
(a) A. A. Fuller, B. Chen, A. R. Minter and A. K. Mapp, J. Am. Chem. Soc., 2005, 127, 5376 CrossRef CAS PubMed;
(b) J. W. Bode and E. M. Carreira, Org. Lett., 2001, 3, 1587 CrossRef CAS PubMed;
(c) A. P. Kozikowski and P. D. Stein, J. Am. Chem. Soc., 1982, 104, 4023 CrossRef CAS;
(d) D. P. Curran, J. Am. Chem. Soc., 1983, 105, 5826 CrossRef CAS;
(e) S. H. Andersen, K. K. Sharma and K. B. G. Torsell, Tetrahedron, 1983, 39, 2241 CrossRef CAS;
(f) P. G. Baraldi, A. Barco, S. Benetti, S. Manfredini and D. Simoni, Synthesis, 1987, 276 CrossRef CAS PubMed.
-
(a) A. Padwa and W. H. Pearson, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Inc., New Jersey, 2003, p. 437 Search PubMed;
(b) S. E. Denmark and J. M. Kallemeyn, J. Org. Chem., 2005, 70, 2839 CrossRef CAS PubMed;
(c) V. Jaeger and P. A. Colinas, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Chemistry of Heterocyclic Compounds, Wiley, Hoboken, NJ, 2002, vol. 59, p. 361 Search PubMed;
(d) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed;
(e) T. V. Hansen, P. Wu and V. V. Fokin, J. Org. Chem., 2005, 70, 7761 CrossRef CAS PubMed.
-
(a) P. Cuadrado, A. M. González-Nogal and R. Valero, Tetrahedron, 2002, 58, 4975 CrossRef CAS;
(b) T. Bandiera, P. Grunanger and F. M. Albini, J. Heterocycl. Chem., 1992, 29, 1423 CrossRef CAS.
- M. Adib, M. Mahdavi, S. Ansari, F. Malihi, L.-G. Zhu and H. R. Bijanzadeh, Tetrahedron Lett., 2009, 50, 7246 CrossRef CAS PubMed.
- K. Ramachandiran, K. Karthikeyan, D. Muralidharan and P. T. Perumal, Tetrahedron Lett., 2010, 51, 3006 CrossRef CAS PubMed.
- S. Gao, Z. Tu, C.-W. Kuo, J.-T. Liu, C.-M. Chu and C.-F. Yao, Org. Biomol. Chem., 2006, 4, 2851 CAS.
- N. M. Fedou, P. J. Parsons, E. M. E. Viseux and A. J. Whittle, Org. Lett., 2005, 7, 3179 CrossRef CAS PubMed.
- V. R. Sabbasani and D. Lee, Org. Lett., 2013, 15, 3954 CrossRef CAS PubMed.
- A. Kamimura, T. Yoshida and H. Uno, Tetrahedron, 2008, 64, 11081 CrossRef CAS PubMed.
- D. Zhu, M. Lu, L. Dai and G. Zhong, Angew. Chem., Int. Ed., 2009, 48, 6089 CrossRef CAS PubMed.
- W. Du, Y.-K. Liu, L. Yue and Y.-C. Chen, Synlett, 2008, 2997 CAS.
- L. Gottlieb, A. Hassner and H. E. Gottlieb, Synth. Commun., 2000, 30, 2445 CrossRef CAS.
- A. Voituriez, J. Moulinas, C. Kouklovsky and Y. Langlois, Synthesis, 2003, 1419 CAS.
- H. Duan, X. Sun, W. Liao, J. L. Petersen and X. Shi, Org. Lett., 2008, 10, 4113 CrossRef CAS PubMed.
- C. Zhong, L. N. S. Gautam, D. Wang, N. G. Akhmedov, J. L. Petersen and X. Shi, Tetrahedron, 2011, 67, 4402 CrossRef CAS PubMed.
-
(a) C.-Y. Zhu, X.-M. Deng, X.-L. Sun, J.-C. Zheng and Y. Tang, Chem. Commun., 2008, 738 RSC;
(b) C.-Y. Zhu, X.-L. Sun, X.-M. Deng, J.-C. Zheng and Y. Tang, Tetrahedron, 2008, 64, 5583 CrossRef CAS PubMed.
- T. Kano, A. Yamamoto, S. Song and K. Maruoka, Bull. Chem. Soc. Jpn., 2011, 84, 1057 CrossRef CAS.
- L.-Q. Lu, Y.-J. Cao, X.-P. Liu, J. An, C.-J. Yao, Z.-H. Ming and W.-J. Xiao, J. Am. Chem. Soc., 2008, 130, 6946 CrossRef CAS PubMed.
- L.-Q. Lu, F. Li, J. An, Y. Cheng, J.-R. Chen and W.-J. Xiao, Chem.–Eur. J., 2012, 18, 4073 CrossRef CAS PubMed.
- J. Lu and P. H. Toy, Synlett, 2011, 2985 CAS.
- L.-Q. Lu, Z. H. Ming, J. An, C. Li, J.-R. Chen and W.-J. Xiao, J. Org. Chem., 2012, 77, 1072 CrossRef CAS PubMed.
- L. A. Evans, H. Adams, C. G. Barber, L. Caggiano and R. F. W. Jackson, Org. Biomol. Chem., 2007, 5, 3156 CAS.
-
(a) S. E. Denmark and J. J. Cottell, Nitronates, in The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, Hoboken, NJ, 2002, p. 83 Search PubMed;
(b) S. E. Denmark, V. Guagnano and J. Vaugeois, Can. J. Chem., 2001, 79, 1606 CrossRef CAS PubMed;
(c) S. E. Denmark, R. Y. Baiazitov and S. T. Nguyen, Tetrahedron, 2009, 65, 6535 CrossRef CAS PubMed;
(d) G. S. Creech and O. Kwon, J. Am. Chem. Soc., 2010, 132, 8876 CrossRef CAS PubMed.
-
(a) S. E. Denmark, M. Seierstad and B. Herbert, J. Org. Chem., 1999, 64, 884 CrossRef CAS PubMed, and references are therein;
(b) M. Avalos, R. Babiano, J. L. Bravo, P. Cintas, J. L. Jiménez, J. C. Palacios and M. A. Silva, Chem.–Eur. J., 2000, 6, 267 CrossRef CAS;
(c) S. E. Denmark and L. Gomez, Org. Lett., 2001, 3, 2907 CrossRef CAS PubMed;
(d) L. L. de Carvalho, R. A. Burrow and V. L. P. Pereira, Beilstein J. Org. Chem., 2013, 9, 838 CrossRef CAS PubMed.
- V. Y. Korotaev, V. Y. Sosnovskikh, M. A. Barabanov, A. Y. Barkov and M. I. Kodess, Mendeleev Commun., 2010, 20, 17 CrossRef CAS PubMed.
- D. Giommi, S. Turchi, A. Danesi and C. Faggi, Tetrahedron, 2001, 57, 4237 CrossRef.
- L. W. A. van Berkom, G. J. T. Kuster, F. Kalmoua, R. Gelder and H. W. Scheeren, Tetrahedron Lett., 2003, 44, 5091 CrossRef CAS.
-
(a) S. Wei, L. M. Weiß and S. B. Tsogoeva, Synthesis, 2012, 3441 CAS;
(b) K. M. Weiß, S. Wei and S. B. Tsogoeva, Org. Biomol. Chem., 2011, 9, 3457 RSC.
Footnote |
| † Dedicated to Professor Mohammad Reza Saidi on the occasion of his 70th birthday. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.