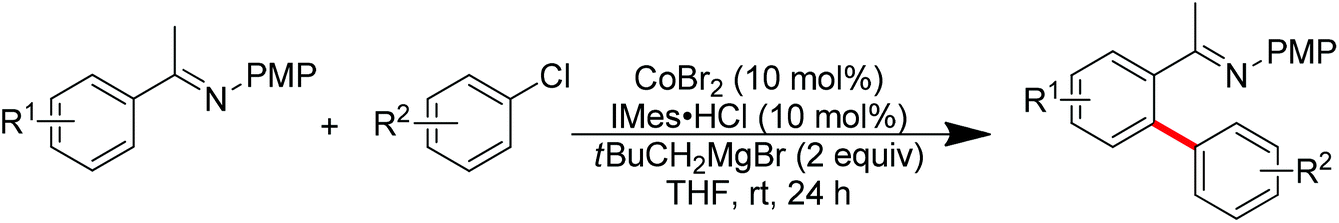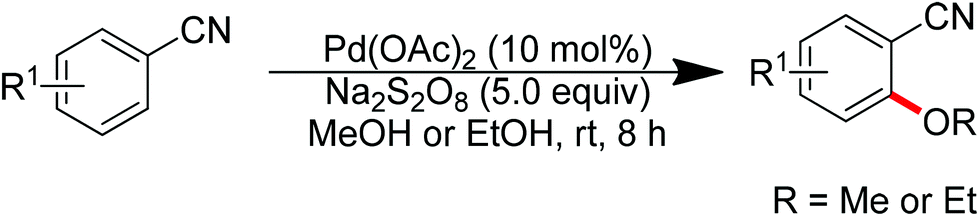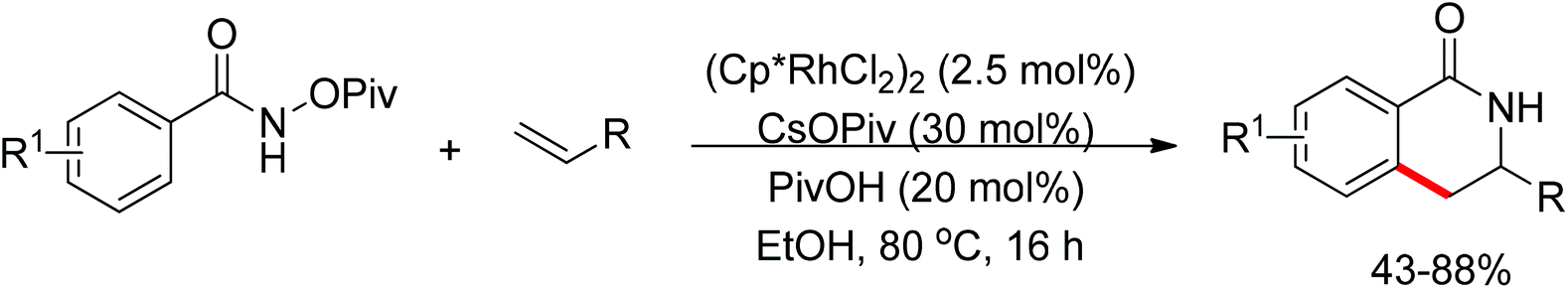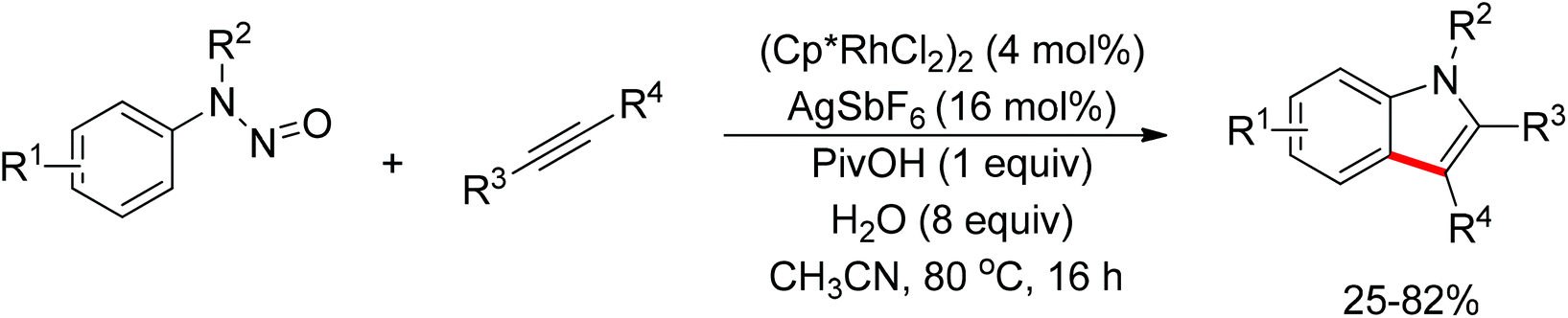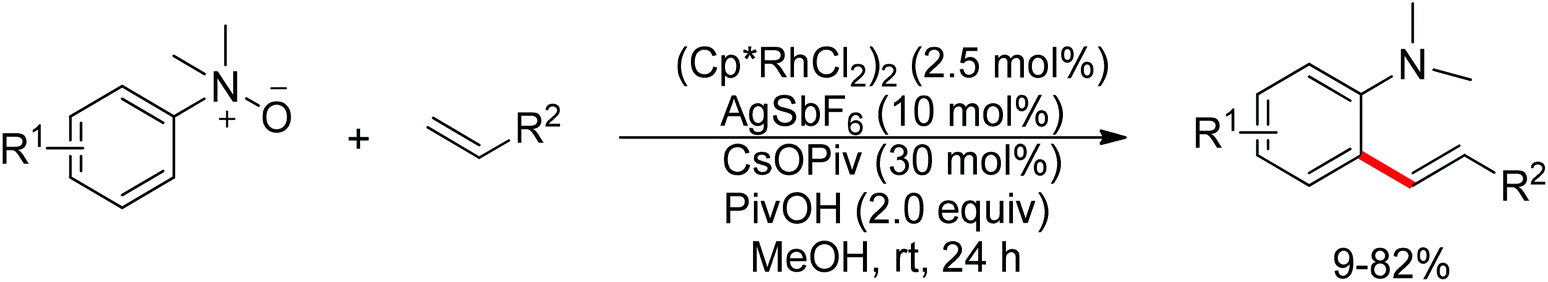DOI:
10.1039/C4QO00068D
(Review Article)
Org. Chem. Front., 2014,
1, 843-895
Recent advances in directed C–H functionalizations using monodentate nitrogen-based directing groups
Received
11th March 2014
, Accepted 14th May 2014
First published on 14th May 2014
Abstract
The use of directing groups has proven to be a successful strategy to enhance reactivity and control selectivity in C–H functionalization reactions. In the past decade, a multitude of new transformations and new directing groups have been explored, and several recent reviews have discussed directing group approaches for C–H functionalization. This review focuses specifically on the use of monodentate nitrogen-based directing groups published during the past two years, with the aim of covering a body of literature that is complementary to existing reviews.
 Min Zhang | Min Zhang was born in 1984 in Henan, China. She received her B.Sc. from Xinyang Normal University in 2007 and obtained her Ph.D. at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences with Prof. Weiping Su in 2012. After her doctoral studies, she joined Prof. Weiping Su's group as a research assistant. Her current research focuses on the development of transition metal-catalyzed C–H bond activation reactions. |
 Yuanfei Zhang | Yuanfei Zhang was born in 1988 in Chongqing, China. He obtained his B.Sc. in pharmacy from Chongqing University in 2010. After a year studying chemistry at the University of Chinese Academy of Sciences, he then joined Su's group as a Ph.D. candidate at the Fujian Institute of Research on the Structure of Matter in 2011. His current research interests focus on transition metal-catalyzed C–H bond functionalization. |
 Xiaoming Jie | Xiaoming Jie was born in 1985 in Hebei Province, China. He received his B.Sc. in chemistry from Nanjing Agricultural University in 2008. He obtained his Ph.D. at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences under the supervision of Prof. Weiping Su in 2013. He then joined Prof. Weiping Su's group as a research assistant. His current work is focused on the development of novel C–H bond functionalization reactions. |
 Huaiqing Zhao | Huaiqing Zhao obtained his B.Sc. in chemistry from Liaocheng University in 2005. He received his Ph.D. at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences in 2010 supervised by Prof. Weiping Su. Subsequently, he completed a postdoctoral stint in the lab of Prof. Huaqiang Zeng at National University of Singapore (2010–2012). In 2013, he joined, as an associate professor, Weiping Su's group at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. His current research interests focus on the development of new synthetic methodologies based on C–H bond activation. |
 Gang Li | Gang Li was born in 1983 in Fujian, China. He received his B.Sc. from the University of Science and Technology of China (USTC) in 2005. After one year graduate study at the University of Minnesota–Twin Cities, he transferred to the University of Wisconsin–Madison, where he obtained his Ph.D. with Professor Richard P. Hsung in 2009. Following one year of postdoctoral work at Emory University with Professor Albert Padwa, he moved to The Scripps Research Institute (TSRI) as a research associate with Professor Jin-Quan Yu in 2010. In the winter of 2013 he left TSRI and started his independent research at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, where he is now a professor of chemistry. Research in his group includes the development of new transition metal-catalyzed reactions and efficient strategies for the synthesis of biologically active natural products. |
 Weiping Su | Weiping Su graduated from the Anhui Education Institute in 1987 and earned his Ph.D. at the Fujian Institute of Research on the Structure of Matter (FJIRSM) in 1999 under the supervision of Prof. Maochun Hong. After one year working as an assistant professor at FJIRSM, he moved to the United States to carry out postdoctoral studies with Prof. Richard H. Holm at Harvard University (2000–2001), Prof. Jin Li at Rutgers University (2001–2002) and Prof. John G. Verkade at Iowa State University (2002–2005). He then joined the faculty at FJIRSM in 2006. His research interests include synthetic methodology, discoveries of metal complex-based homogeneous catalysts and nanoparticle-based recyclable catalysts and the structure–property relationships of catalysts. |
1. Introduction
A present focus of organic chemistry is the development of ideal transformations that are efficient, economical, and environmentally friendly to synthesize complex molecules.1 Transition metal-catalyzed carbon–hydrogen (C–H) bond functionalization reactions that convert ubiquitous but generally inert carbon–hydrogen bonds directly into carbon–carbon (C–C) bonds and carbon–heteroatom (e.g., C–O, C–N or C–X) bonds are very promising as one class of such ideal transformations. In contrast to traditional methods for these types of bond formation, C–H functionalization obviates the need to pre-functionalize (e.g., halogenate or borylate) the starting material, which lowers step count and reduces the amount of undesired waste of synthetic sequences. In this way, synthetic “shortcuts” with improved step and atom economy can be achieved.2 For this reason, transition metal-catalyzed C–H bond functionalization is often considered a “Holy Grail” in organometallic chemistry and has been extensively studied for the past few decades, leading to significant advances in the field.3
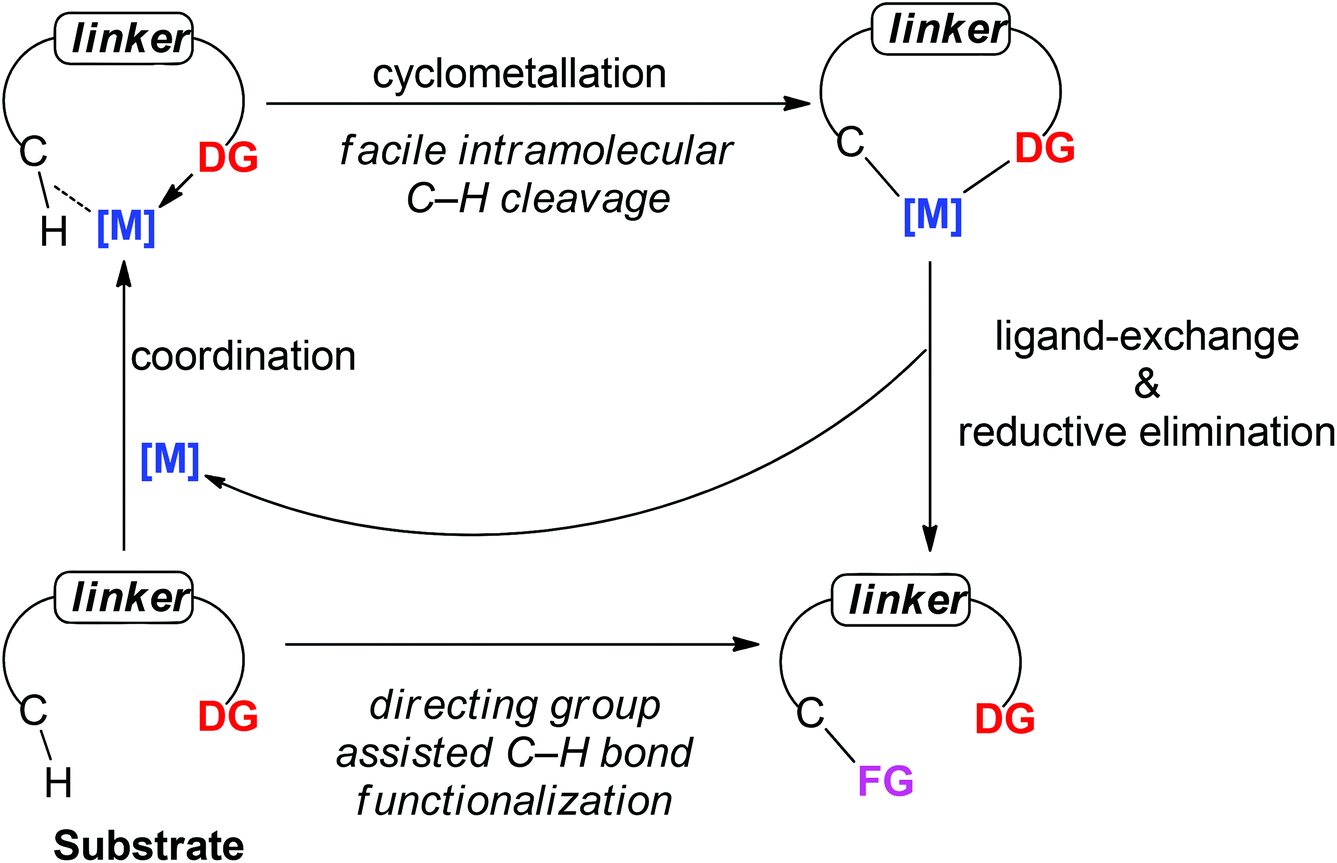
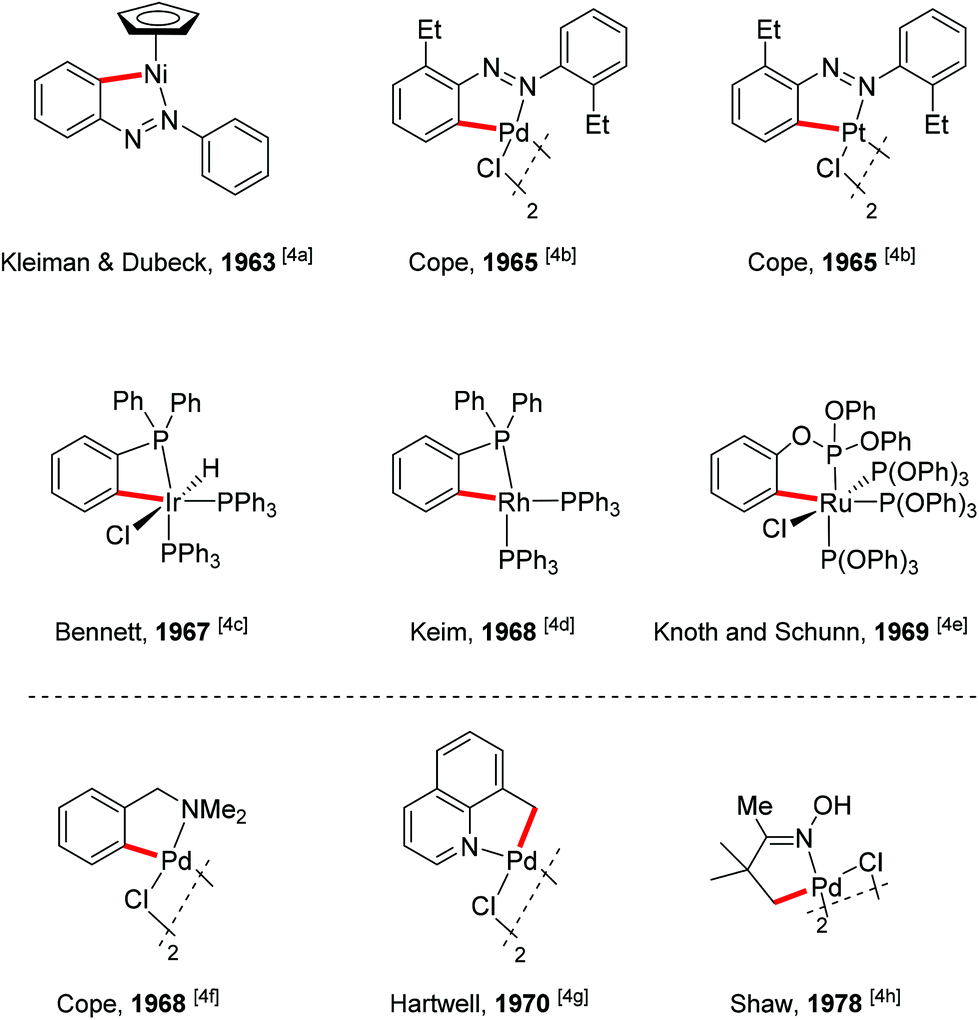
To realize the promise of transition metal-catalyzed C–H functionalization as a practical synthetic tool, however, two principal challenges must be overcome. The first challenge is the low reactivity of C–H bonds, which are generally considered to be chemically inert. The second challenge is controlling the site-selectivity, such that the reaction functionalizes only a single C–H bond in the presence of many proximal C–H bonds with similar reactivity. Among various strategies employed to address these challenges, the use of directing groups has proven to be the most successful approach (Scheme 1).3 Transition metal-catalyzed C–H functionalization reactions typically proceed by a mechanism involving initial formation of a [C–H⋯M] σ-complex.3c,d The presence of a directing group, a Lewis basic functional group containing non-bonding lone pair electrons, facilitates C–H bond cleavage by inducing pre-association between the metal and substrate. Binding of the directing group promotes formation of the agostic interaction through an increase in effective concentration between the metal and the C–H bond. Moreover, the directing group helps to arrange the geometry of the pre-transition state coordination structure in a favorable way for C–H cleavage.3w The directing group also serves to control the site selectivity of the reaction since it positions the catalyst in close proximity to a single C–H bond in the substrate, facilitating the formation of a complex intermediate via cyclometalation.3 Coordination-assisted cyclometalation was instrumental in elucidating the fundamental mechanisms of C–H bond cleavage with many different transition metals, including Ni, Pd, Pt, Ir, Rh and Ru in stoichiometric reactions. Stable and isolable cyclometalated complexes, many of which are palladacycles that were reactive towards various reagents,3a have been characterized (Scheme 2).4 In catalytic chemistry, directing groups have also played a major role in the development of the field. In 1993, Murai et al. reported an efficient C–H alkylation reaction, where a Ru(0) catalyst coordinates to a ketone directing group and activates the ortho-C–H bond of the aromatic ketone substrate.5 Since this breakthrough report, C–H functionalization research has benefited tremendously from directing groups, which have enabled the discovery of a myriad of new reactions.3g–ad Thus, to some extent, directing groups are the hands that bring the “Holy Grail” into reach for ordinary synthetic chemists.
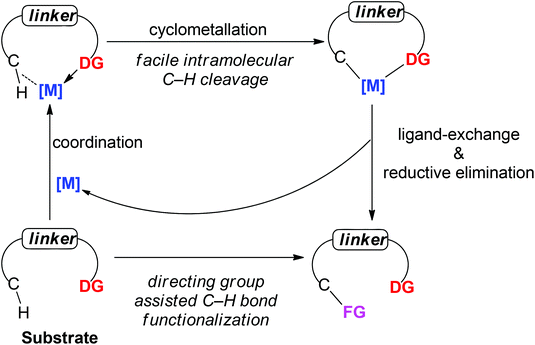 |
| | Scheme 1 General depiction of directing group (DG) assisted C–H bond functionalization. | |
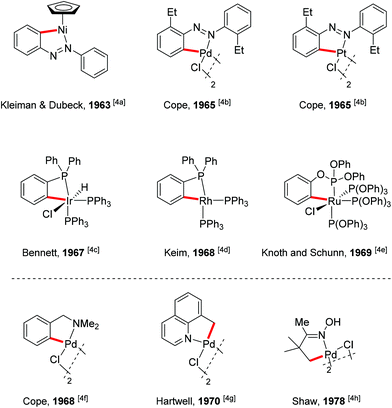 |
| | Scheme 2 Early examples of metallacycles generated from metalation of a proximal C–H bond. | |
A multitude of functionalities, including amides, amines, ketones, esters, alcohols, pyridines and oxazolines, have been reported as directing groups in C–H functionalization.3g–ad Generally speaking, a good directing group satisfies the following criteria. Firstly, it coordinates the metal in a strong yet reversible manner, such that it promotes selective C–H cleavage but allows for catalyst dissociation at the end of the reaction. Traditionally, strong directing groups (i.e., those that readily form well-defined stoichiometric cyclometalated complexes) have been used and played an important historical role in establishing the fundamentals of C–H cleavage at a transition metal center, and later they proved instrumental in the development of catalytic reactions. Recently, the use of weakly coordinating directing groups in catalytic reactions has become widespread. These directing groups may form cyclometalated intermediates that possess higher reactivity than traditional metallacycles with strong directing groups.3w Weak coordination may also facilitate transmetalation (and/or other subsequent functionalization steps) by providing a readily exchangeable coordination site at the metal center. Secondly, the directing group should ideally be an intrinsic part of the starting material and product of interest. If that is not possible, the directing group should be readily installed or removed, ideally being generated in situ and/or used catalytically.6 However, directing groups that are difficult to manipulate synthetically in further transformations can be a good platform for evaluating new concepts and discovering new transformations, despite being synthetically restrictive.
While recent reviews have been devoted to advances in employing directing groups for C–H functionalization,3k–ad the current review focuses specifically on the work using monodentate nitrogen-based directing groups and covers papers published during the past 2 years (2012–2013). Compared with other directing groups, such as oxygen-based directing groups, nitrogen-based directing groups represent the largest class of directing groups, which is largely attributed to the fact that nitrogen possesses several binding modes and can also accommodate up to three substituents that allow for fine tuning of its Lewis basicity. The stability of nitrogen-based directing groups and the ease of their installation also contribute to their widespread use. It is envisioned that this review will be complementary to previous ones. Examples using bidentate nitrogen-based directing groups are not included, since an excellent review on this topic was recently published.3aa In general, work published prior to 2012 is not covered in this review; however, exceptions to this rule are made in cases where discussion of earlier reports is necessary for providing background information. Section 2 of this review provides a survey of ortho C–H functionalization reactions assisted with directing groups. Section 3 discusses new modes of site selectivity controlled by the directing groups. The final section, section 4, deals with reactions using an oxidizing directing group, leading to redox-neutral C–H functionalizations.
2. Directing groups assisted ortho C–H functionalization
At present, most directing group-assisted C–H bond functionalization reactions involve the cleavage of C–H bonds at the ortho position to the directing group, most of which could be attributed to the stability of five or six-membered metallacycles. As an efficient way to enhance the reactivity and control the selectivity of C–H functionalization, the discovery of new nitrogen-based directing groups, as well as the development of new transformations based on existing nitrogen-based directing groups, has always been a focus of chemical attention. In the past two years, a large number of studies based on nitrogen-based directing groups appeared in the literature and various types of reaction were developed.
2.1. Carbon–carbon bond formation
2.1.1. Alkenylation and arylation of C–H bonds.
The cyclometalation of benzene derivatives bearing amino groups with stoichiometric palladium has been extensively studied. Although the N,N-dimethylaminomethyl group has been used as a directing group for the ortho-metalation by transition metal complexes to form metallacycles,4f metal-catalyzed amine-directed C–H functionalization remained a challenge until very recently. The difficulty of using amines as directing groups for catalytic C–H transformations results from the strong σ-donating ability of amino groups. In the catalytic systems, the substrates bearing amino groups, which are in excess relative to the metal catalyst, strongly bind to the metal catalyst, preventing the formation of empty coordinating sites around the catalyst centers that are crucial for C–H bond activation and cyclometalation.
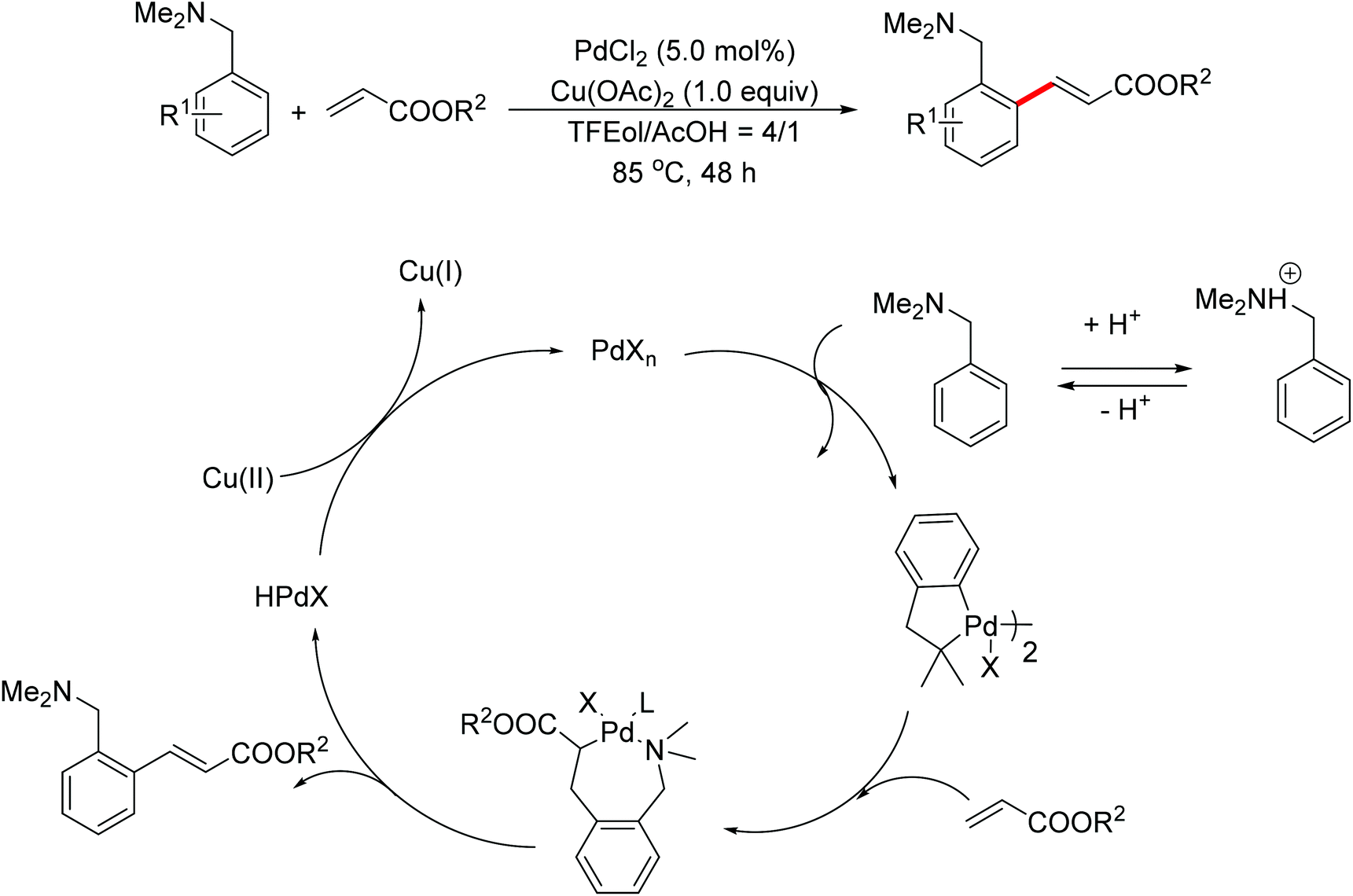
In 2007, Shi and co-workers successfully accomplished a palladium-catalyzed ortho C–H olefination directed by an N,N-dimethylaminomethyl group, presenting the first example of palladium-catalyzed amine-directed C–H functionalization reaction (Scheme 3).7 The success was attributed to using acetic acid (AcOH)–2,2,2-trifluoroethanol (TFEol) (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) as the solvent system. Acetic acid played a dual role: (1) tuning the concentration of free amines through acid–base adducts balance; (2) assisting the dissociation of the anionic ligand from the catalyst center to generate coordinately unsaturated and reactive catalyst species for C–H activation. A possible reaction mechanism involving a five-membered palladacycle was proposed by the author.
:4) as the solvent system. Acetic acid played a dual role: (1) tuning the concentration of free amines through acid–base adducts balance; (2) assisting the dissociation of the anionic ligand from the catalyst center to generate coordinately unsaturated and reactive catalyst species for C–H activation. A possible reaction mechanism involving a five-membered palladacycle was proposed by the author.
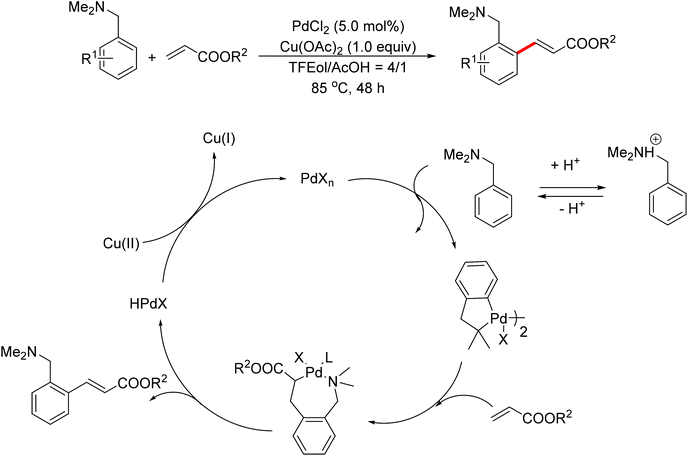 |
| | Scheme 3
ortho-Olefination of N,N-dimethylbenzylamines with alkenes. | |
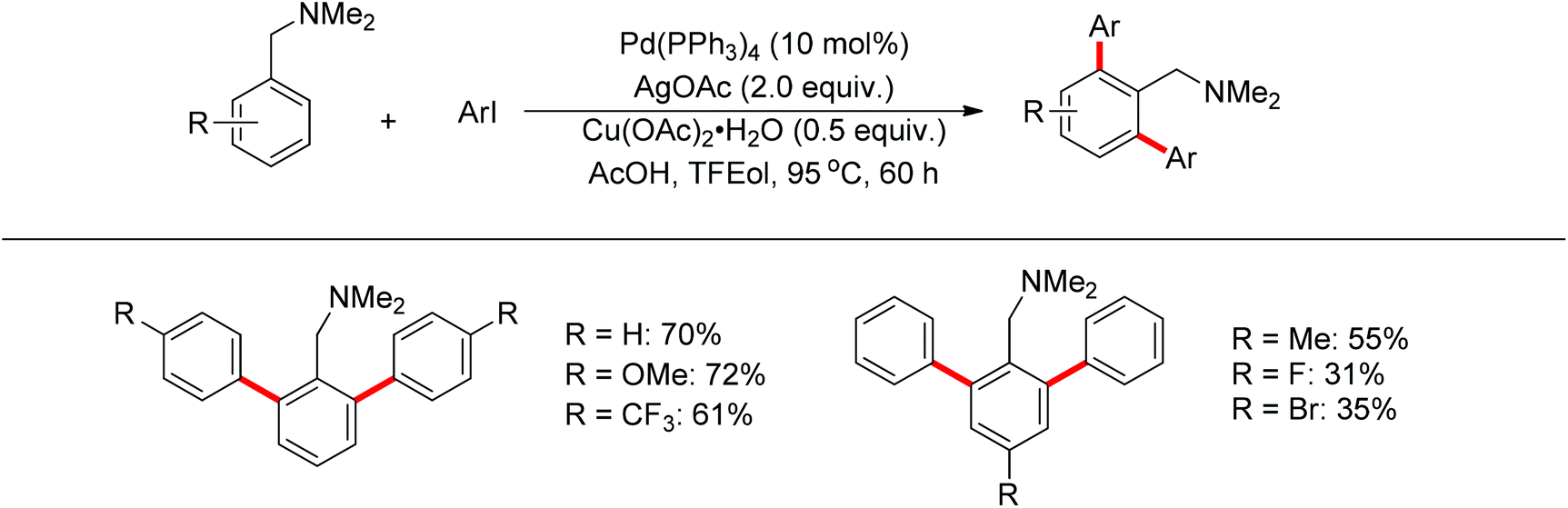
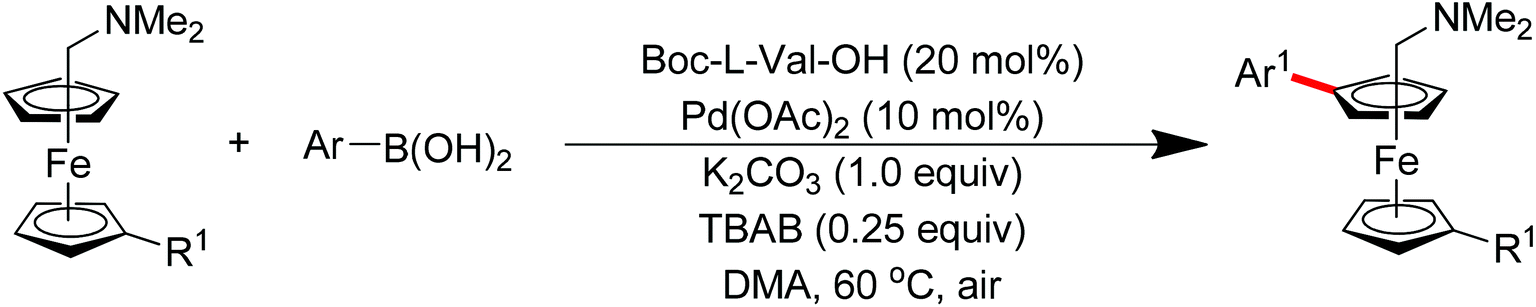
Subsequently, Zhang and co-workers achieved the palladium-catalyzed C–H ortho-arylation with N,N-dimethylbenzylamines as directing groups in 2013 (Scheme 4).8 Similarly to Shi's method, AcOH–TFEol was used as a solvent system. Various aryl iodides were used as arylating reagents in moderate to good yields for the synthesis of the desired ortho diarylated arenes in moderate to good yields. In addition, AgOAc acted as a halide scavenger to promote formation of the reactive palladium intermediate, and Cu(OAc)2·H2O was observed to be an efficient additive to improve the reaction. Meanwhile, You and co-workers realized the palladium-catalyzed direct arylation of aminomethylferrocene derivatives with arylboronic acids (Scheme 5).9 The protocol provided the planar-chiral ferrocenes with high enantioselectivity using a commercially available mono-N-protected amino acid as the ligand and air as the oxidant, which makes the process especially practical.
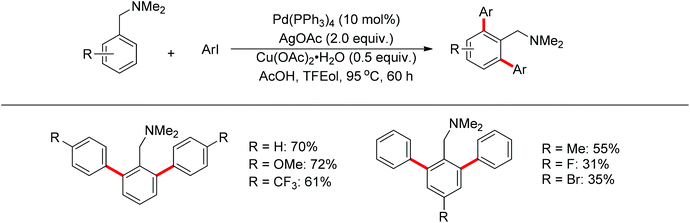 |
| | Scheme 4 Diarylation of N,N-dimethylbenzylamines with aryl iodides. | |
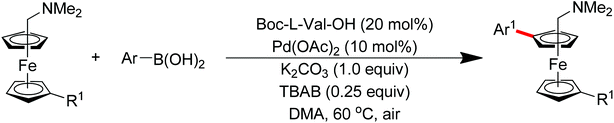 |
| | Scheme 5
ortho-Arylation of aminomethylferrocene derivatives with arylboronic acids. | |
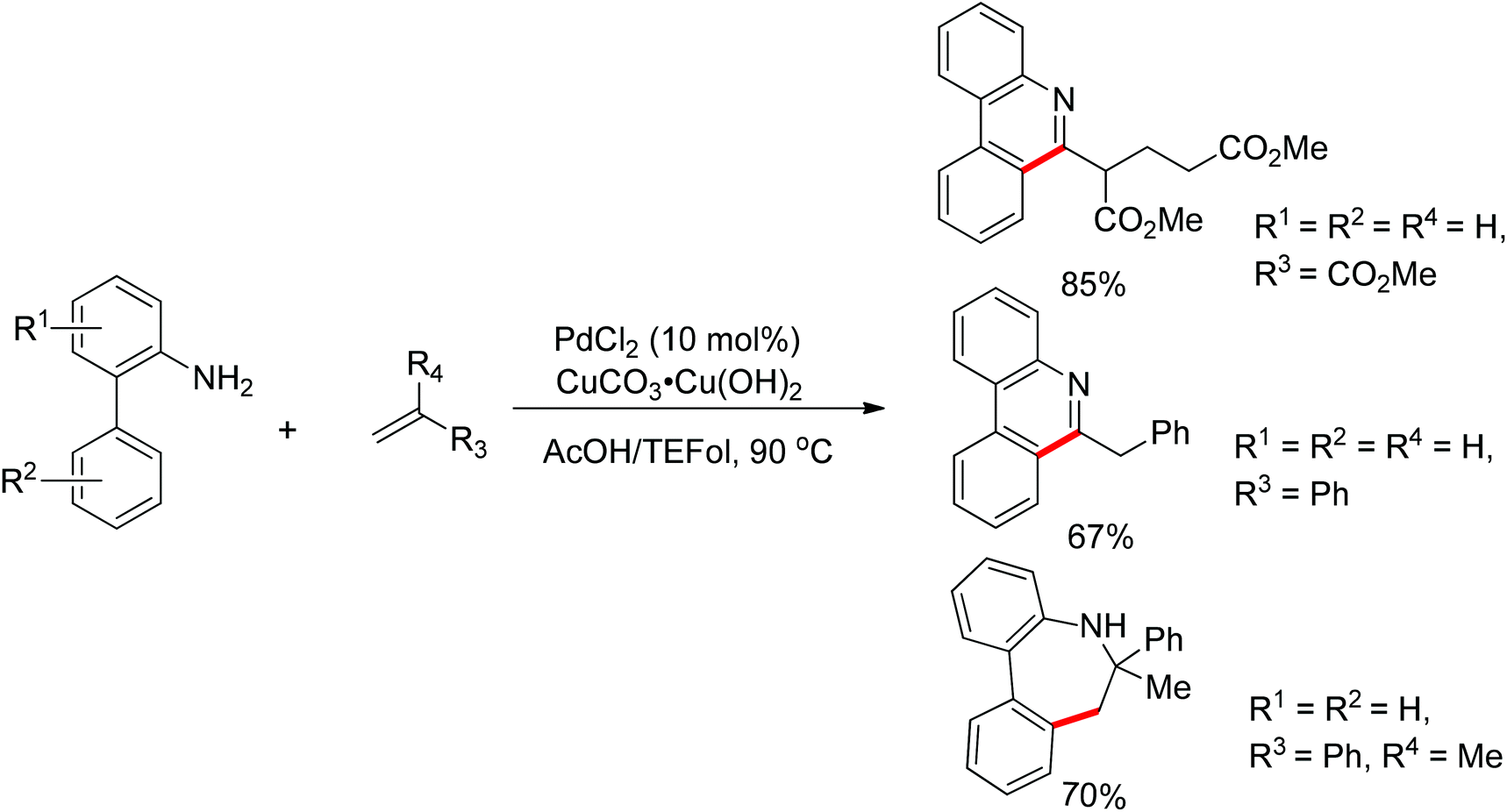
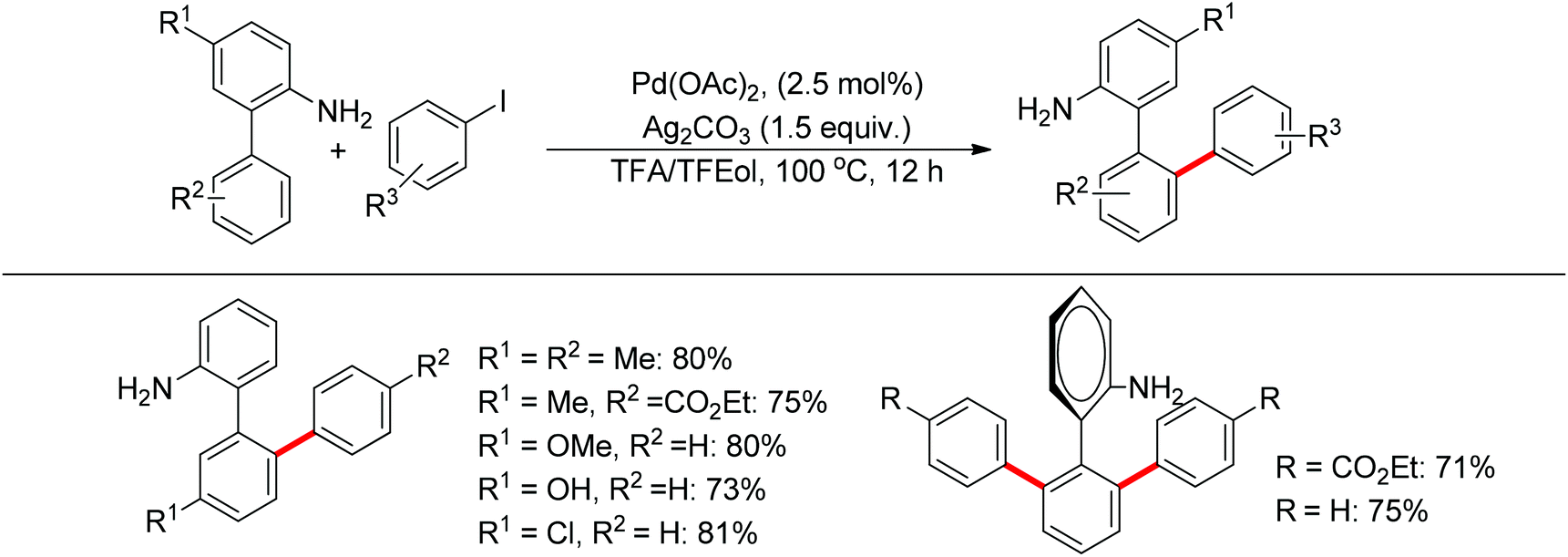
Free amines were also established as directing groups in catalytic C–H functionalizations. In 2012, Zhang et al. reported a palladium-catalyzed free amine-directed alkylation of sp2 C–H bonds and cycloamination (Scheme 6).10 In search for a balance between acid, amine and palladium for controlling the concentration of strongly coordinating free amine, the authors screened different acids and observed that acetic acid furnished the corresponding nitrogen-based heterocycles. This protocol can provide different heterocycles with various alkenes and biarylamines. Notably, this reaction can be applied to the synthesis of tricyclic compounds with a seven-membered ring. The palladium-catalyzed free amine-directed ortho C–H bond arylation of biaryl-2-amines with aryl iodides was further developed by the same group (Scheme 7).11 The choice of Brønsted acid was still important to realize the transformation and trifluoroacetic acid (TFA) was a suitable additive for this reaction. The catalytic system allowed the reaction of a series of biaryl-2-amines with a broad scope of aryl iodides to give the desired ortho monoarylated or diarylated products with exclusive regioselectivity.
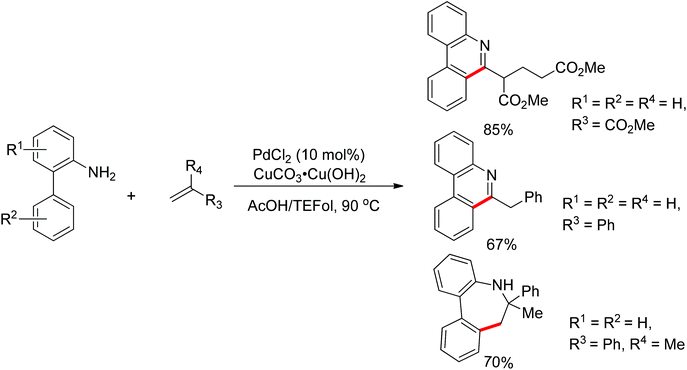 |
| | Scheme 6 Reactions of various biarylamines with alkenes. | |
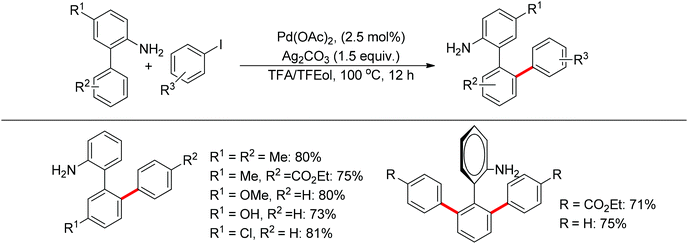 |
| | Scheme 7 Monoarylation and diarylation of biphenylamine. | |
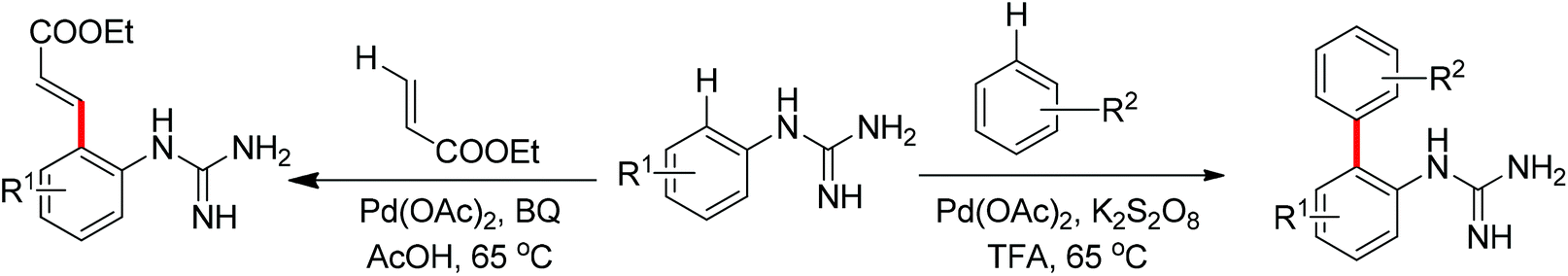
Guanidines bearing amino groups were amenable to catalytic C–H bond functionalization with free amine or imine as directing groups. Yu's group reported the palladium-catalyzed ortho C–H bond arylation and olefination of guanidine with simple unactivated arenes or ethyl acrylate (Scheme 8).12 Given the presence of a free amine or imine in guanidine, the authors considered the introduction of an acid into the catalytic system to achieve this transformation. Screening different reaction conditions, they found that the combination of TFA and K2S2O8 was an efficient system for the target arylation reactions. But for the olefination of guanidines, BQ instead of K2S2O8 offered good yields in AcOH. The difference between these two transformations was speculated to derive from distinct mechanisms. The arylation of guanidines may proceed via a Pd(IV)/Pd(II) cycle while the olefination involves Pd(II)/Pd(0) catalysis.
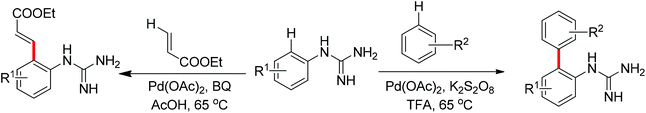 |
| | Scheme 8
ortho-Arylation and olefination of guanidines. | |
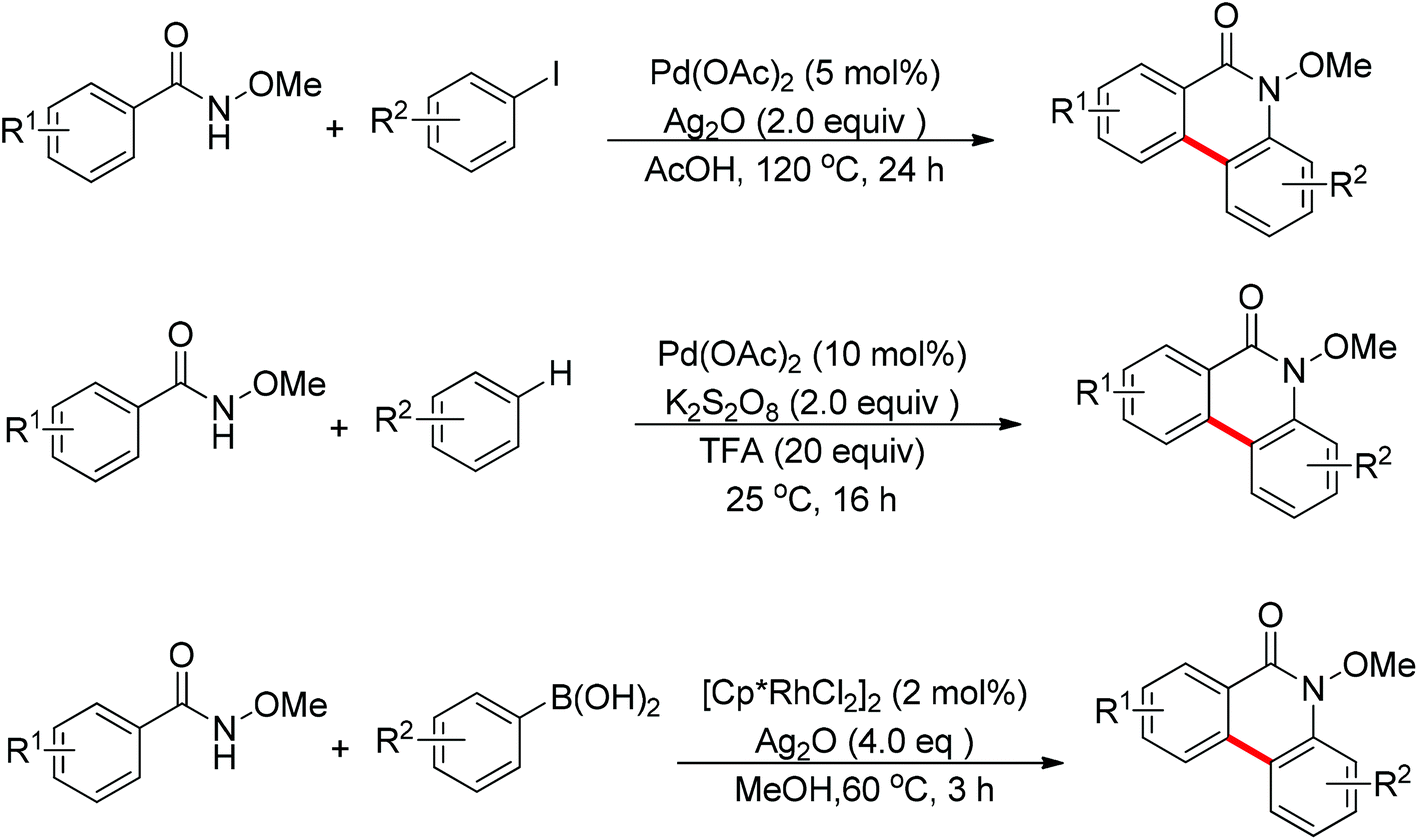
In recent years, N-substituted amides (CONHR) as directing groups have received extensive attention.13 By using CONHOMe as the directing group,14 Wang and Cheng realized the palladium-catalyzed one-pot cascade synthesis of phenanthridinone derivatives from N-methoxybenzamides with iodoarenes15 and simple arenes.16 Shortly afterwards, Cheng and co-workers further disclosed the Rh(III)-catalyzed oxidative cyclization of N-methoxybenzamides with aryl boronic acids to synthesize phenanthridinone derivatives (Scheme 9).17 The presence of a silver salt was crucial for the reaction. The reaction had excellent functional group tolerance and was highly selective with respect to both coupling partners. The C–H activation of N-methoxybenzamides mainly occurred at the less hindered position.
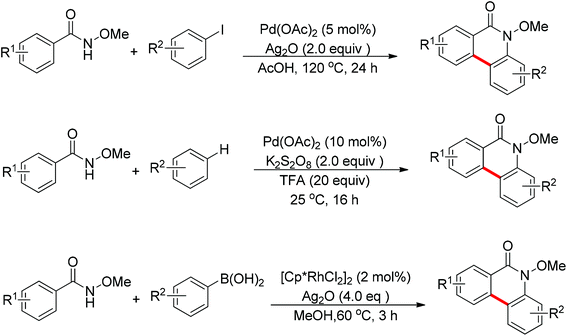 |
| | Scheme 9 Oxidative cyclization of N-methoxybenzamides with aryl boronic acids. | |
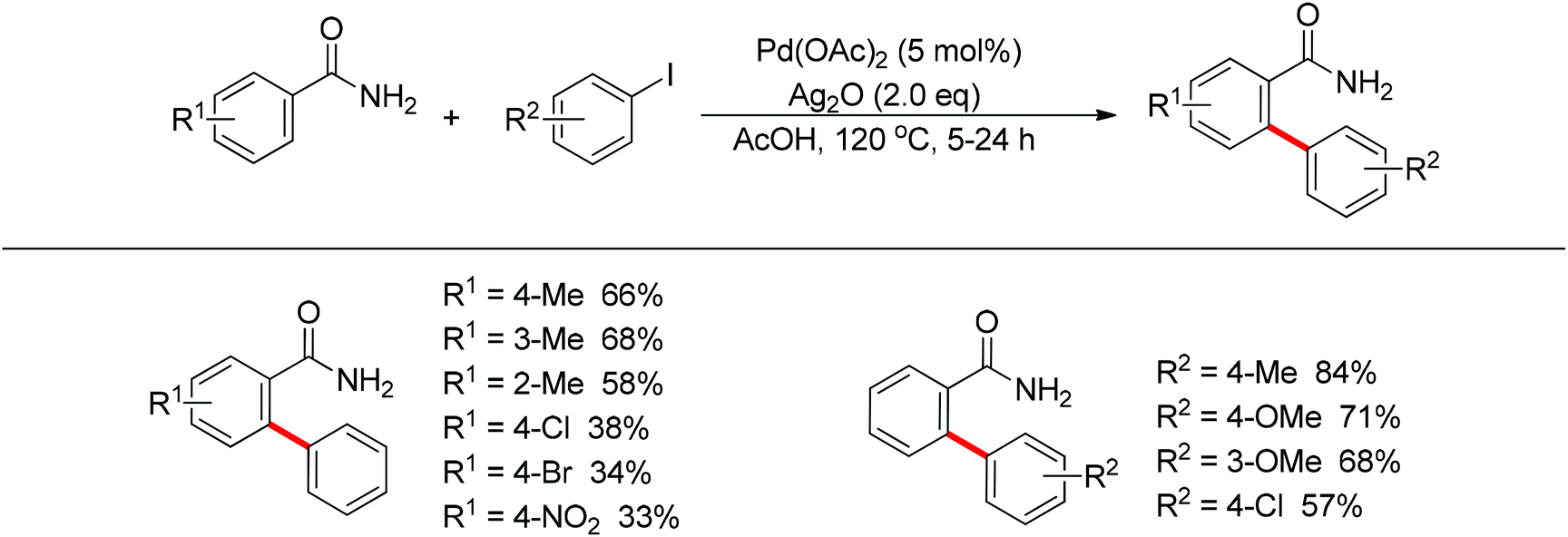
In 2012, Wang and co-workers disclosed the palladium-catalyzed ortho-arylation of benzamides with aryl iodides, where a primary amide (CONH2) was employed as the directing group for the first time to promote the C–H cleavage at the ortho position (Scheme 10).18 AcOH was used as the solvent to facilitate the electrophilic attack of the palladium(II) catalyst to benzamide. The process showed a wide substrate scope with respect to both coupling partners.
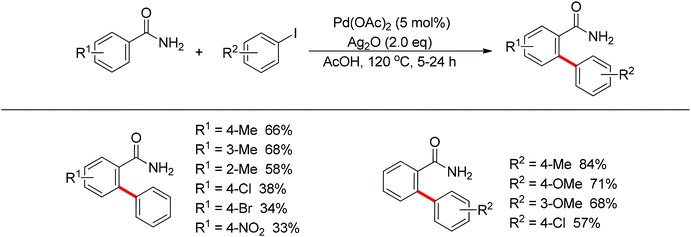 |
| | Scheme 10 Pd(II)-catalyzed ortho-arylation of benzamides with aryl iodides. | |
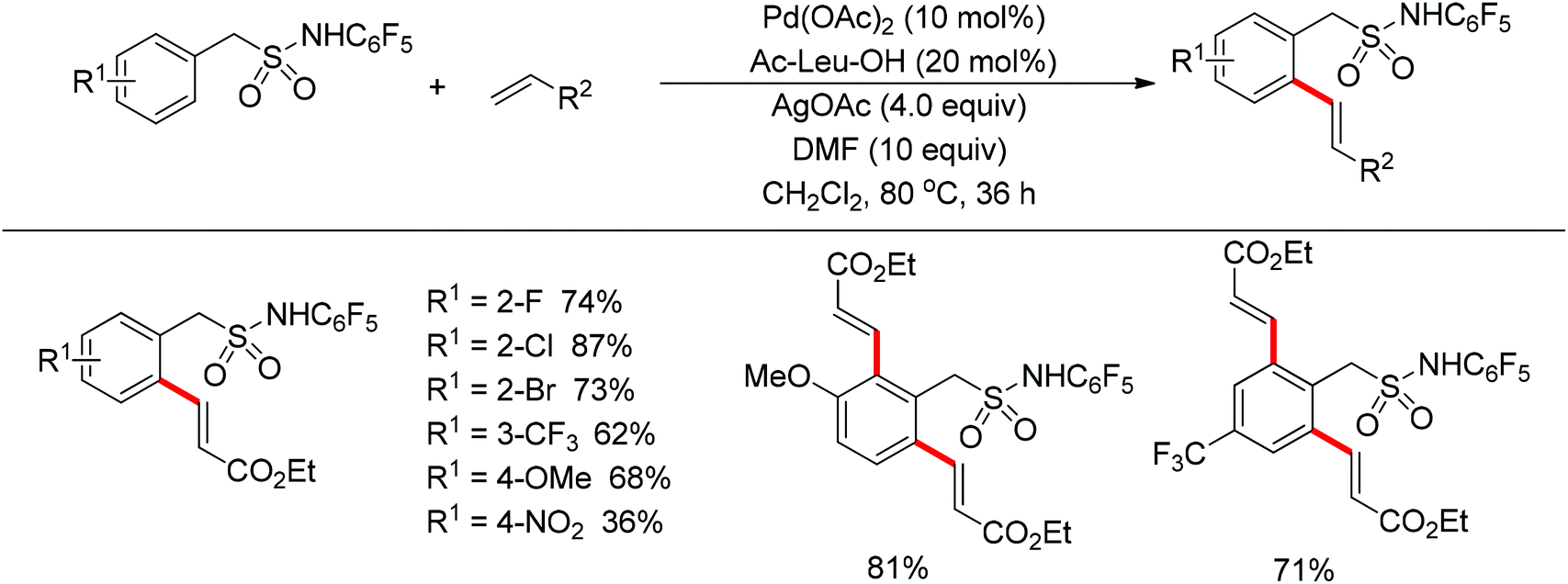
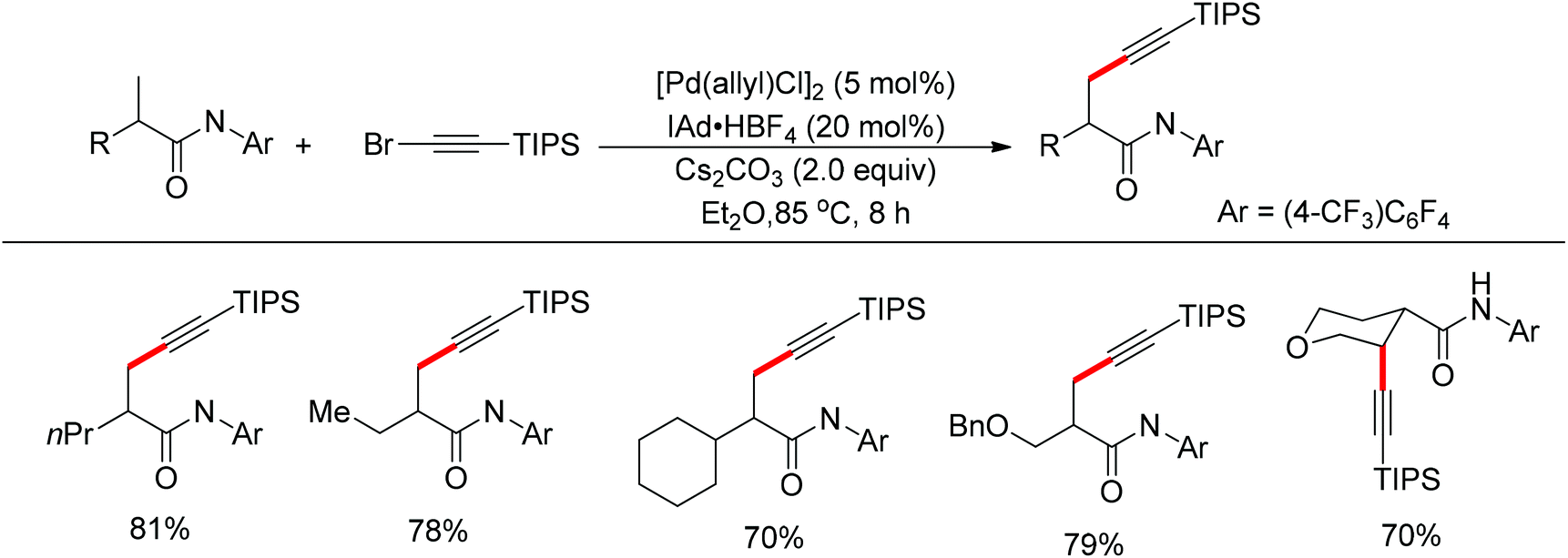
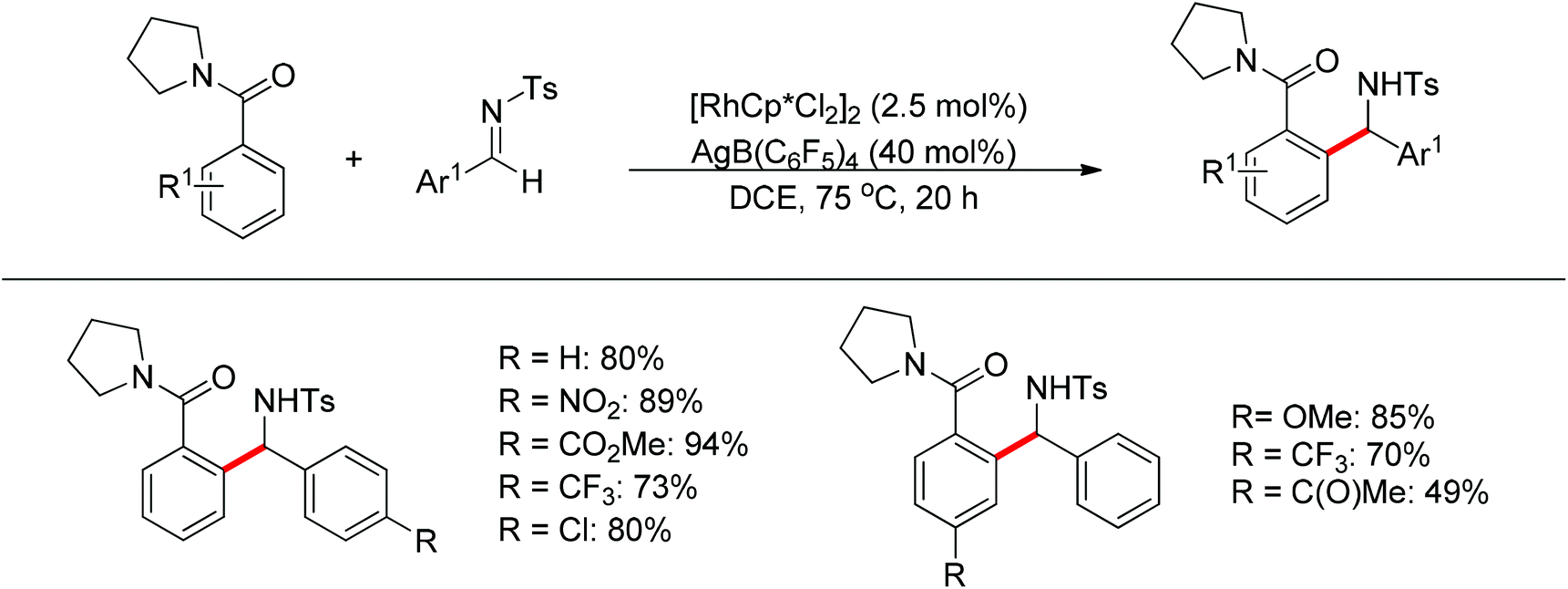
The sulfonamide moiety also proved to be an efficient directing group in the direct C–H functionalization process. A pioneering report by Yu and co-workers described the palladium(II)-catalyzed direct ortho-olefination of perfluoroaryl-substituted benzylsulfonamides (Scheme 11).19 The introduction of mono-N-protected amino acid ligand Ac-Leu-OH into the reaction system was important for achieving the transformation with high efficiency. The authors also demonstrated versatile C–H functionalizations including olefination, carboxylation, carbonylation, iodination, arylation and alkylation directed by the perfluoroaryl-substituted benzosulfonamide.
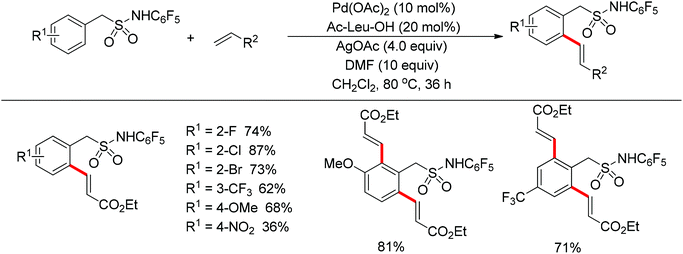 |
| | Scheme 11
ortho-Olefination of perfluoroaryl-substituted benzylsulfonamides. | |
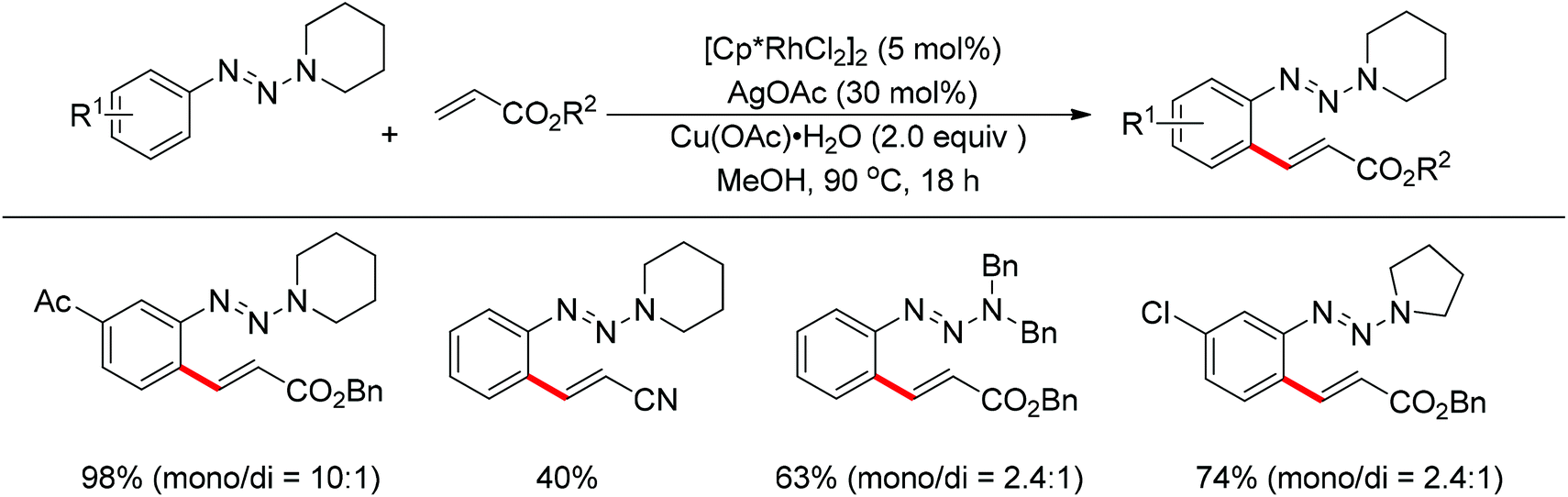
The Rh(III)-catalyzed ortho-olefination of arenes directed by a triazene group was developed by employing 2.0 equiv. Cu(OAc)2·H2O as the oxidant.20 The presence of the acetate anion, which facilitated the cyclometalation and regeneration of the rhodium catalyst, was found to be crucial for obtaining an efficient catalyst system. The protocol was applicable to an array of triazene-substituted arenes with either electron-donating or electron-withdrawing groups on the phenyl rings (Scheme 12). Moreover, the triazene moiety is versatile in undergoing further transformations and is easily removed at room temperature, which makes such a protocol particularly attractive.
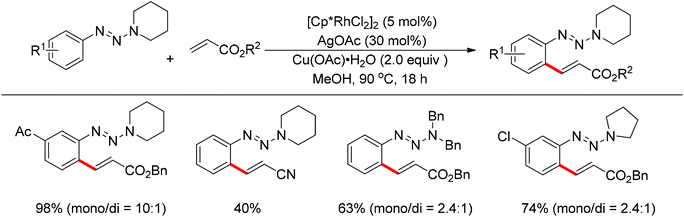 |
| | Scheme 12
ortho-Olefination of arenes directed by triazene group. | |
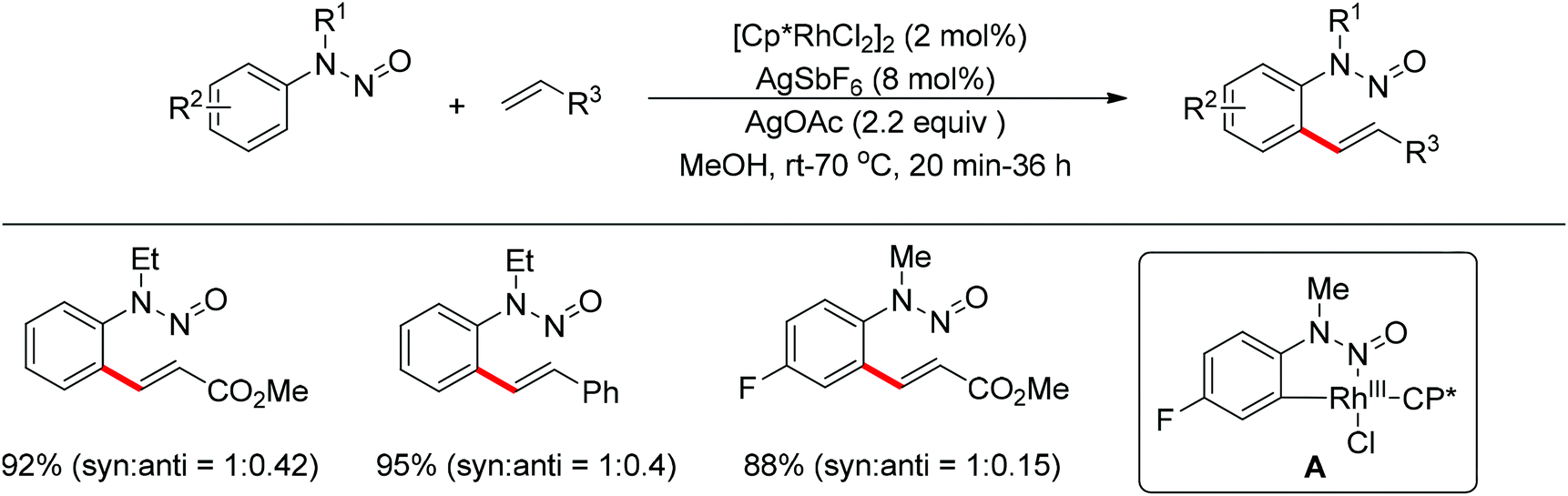
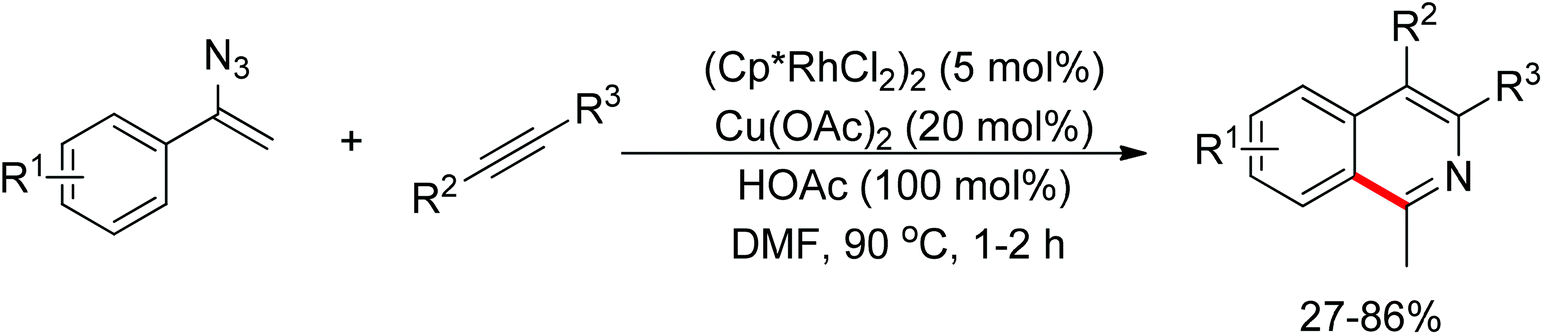
Meanwhile, Zhu and co-workers demonstrated the Rh(III)-catalyzed ortho-olefination of arenes directed by N-nitroso groups (Scheme 13).21 This method featured mild conditions, high yields and broad substrate scopes. Unactivated olefins such as 1-octene could also be employed as suitable substrates for this transformation. Comprehensive mechanistic studies indicated that the reaction pathway involves [RhCp*]2+ as the catalyst resting state and the five-membered rhodacycle A was proposed as the key intermediate in the catalytic cycle.
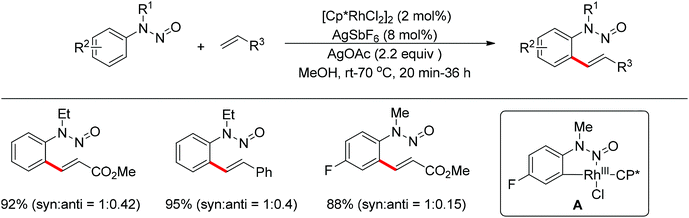 |
| | Scheme 13 Rh(III)-catalyzed ortho-olefination of arenes directed by N-nitroso group. | |
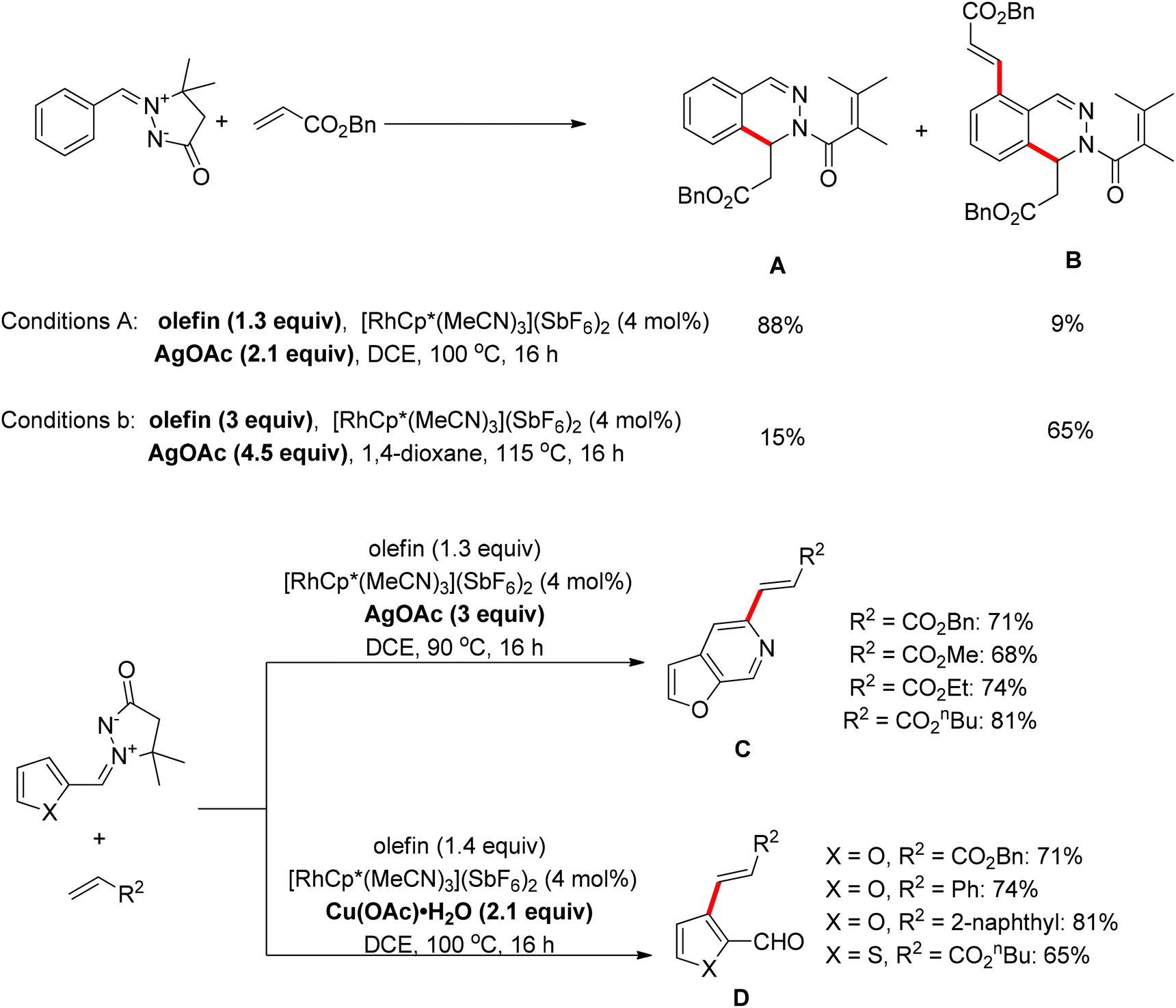
A protocol for the oxidative coupling of azomethine ylides with olefins was developed using [RhCp*(MeCN)3](SbF6)2 as the catalyst.22 The mono-olefination/cyclization product A was predominant when a slight excess of the olefin (1.3 equiv.) and AgOAc (2.1 equiv.) were used, however, if the equivalents of olefin and AgOAc were increased to a greater excess, the method mainly afforded the diolefination/monocyclization product B. For the azomethine compounds functionalized with a 2-furanyl group, a furo[2,3-c]pyridine C resulting from the combination of C–H olefination and N–N cleavage was isolated in the presence of AgOAc as the oxidant, while an olefinated aldehyde D generated from C–H olefination accompanied by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N cleavage was formed when Cu(OAc)2·H2O was used as the oxidant (Scheme 14).22
N cleavage was formed when Cu(OAc)2·H2O was used as the oxidant (Scheme 14).22
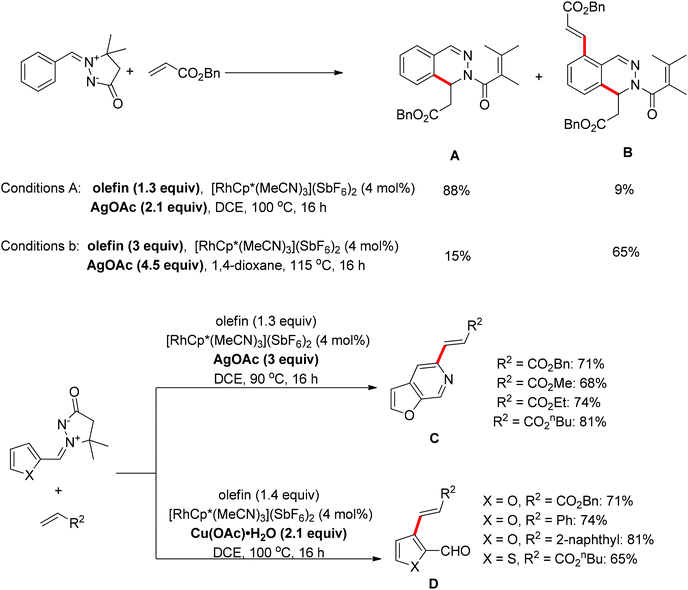 |
| | Scheme 14 Rh(III)-catalyzed oxidative coupling of azomethine ylides with olefins. | |
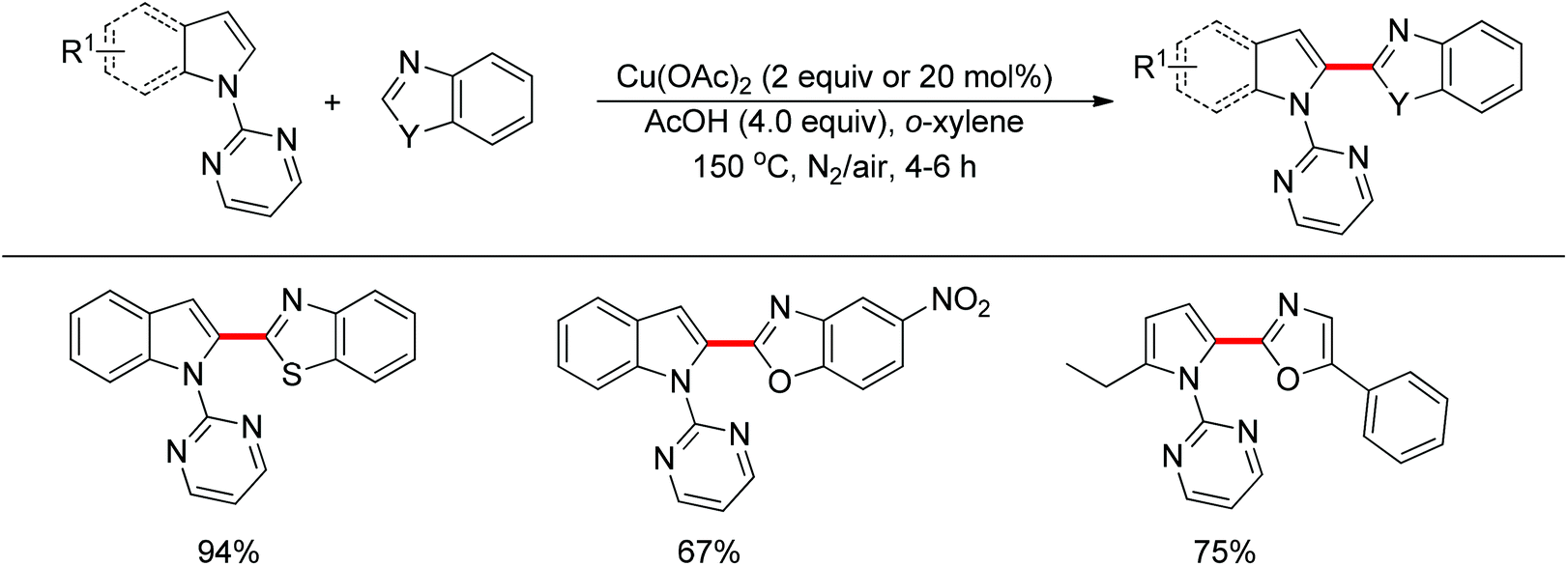
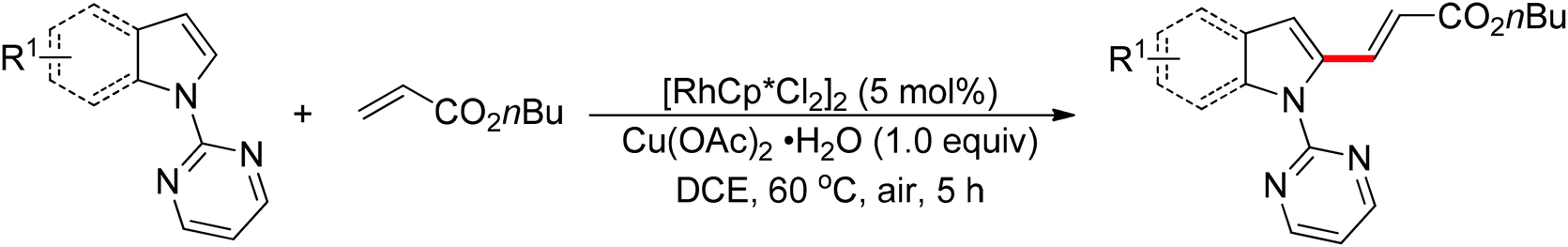
The direct cross-coupling of two non-preactivated arenes via double C–H bond cleavage represents an ideal approach to biaryl motifs since such oxidative C–H/C–H cross-coupling reaction obviates the need for preactivation of the starting materials, therefore reducing the amount of undesired by-product. In the past decade, palladium catalysts played a major role in the development of double C–H activation reactions. Recently, significant attention has been focused on the development of new reactions mediated by other transition metals such as rhodium and copper. In 2012, Miura and co-workers disclosed the oxidative C–H/C–H cross-coupling of N-(2-pyrimidyl)indoles and 1,3-azoles in the presence of a stoichiometric amount of Cu(OAc)2 or a catalytic amount of Cu(OAc)2 with air as the co-oxidant (Scheme 15).23 The 2-pyrimidyl served as the directing group to allow the reaction to occur preferentially at the C2-position of indoles, which provided a complementary method to the precedent palladium-based methodology. Substituted benzoxazoles, mono-cyclic 5-aryloxazoles, benzothiazole and N-methylbenzimidazole could all be employed as suitable substrates. The 2-pyrimidyl could be easily removed by treating the coupling products with NaOMe in DMSO. The Rh(III)-catalyzed C2-alkenylation of N-(2-pyrimidyl)indoles with acrylates was also achieved recently (Scheme 16).24
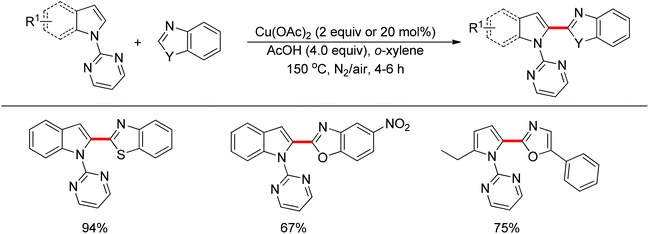 |
| | Scheme 15 Oxidative cross-coupling of N-(2-pyrimidyl)indoles with 1,3-azoles. | |
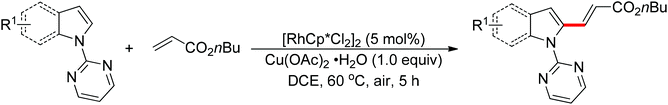 |
| | Scheme 16 Rh(III)-catalyzed C2-alkenylation of N-(2-pyrimidyl)indoles with acrylates. | |
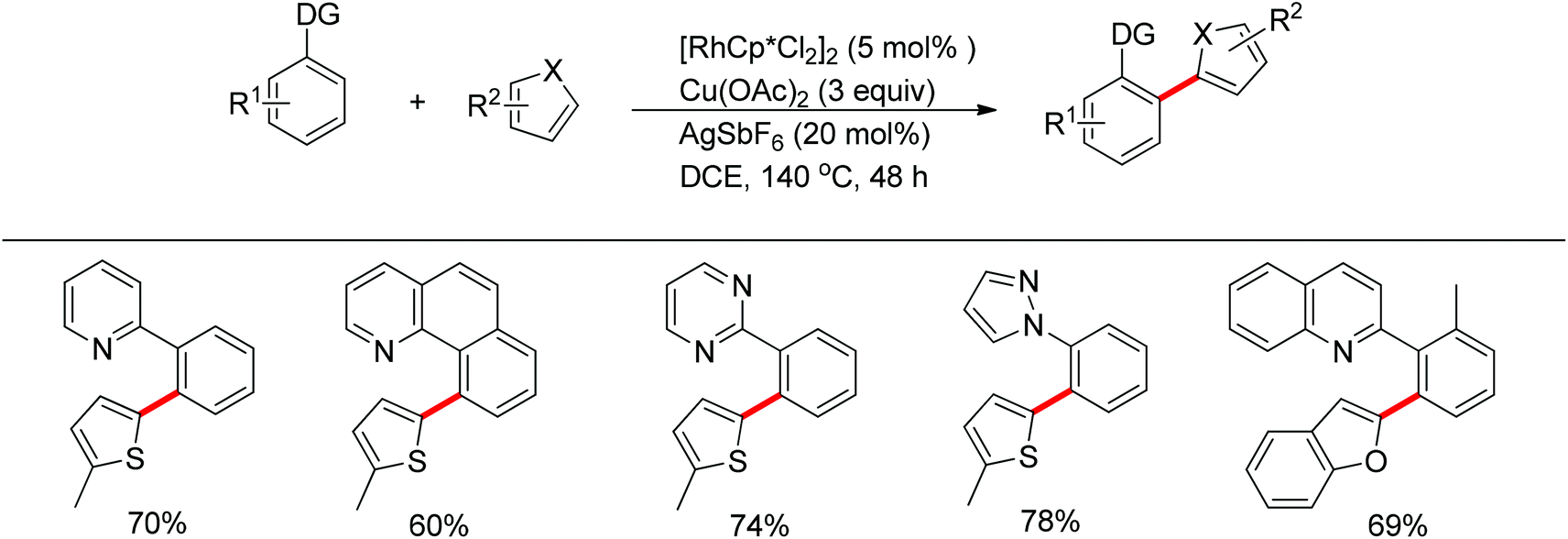
The You group developed a Rh(III)-catalyzed oxidative C–H/C–H cross-coupling of biaryl containing N-heteroarenes such as pyridine, quinolone, pyrimidine and pyrazole with five-membered heterocycles (Scheme 17).25 The C–H functionalization of five-membered heterocycles took place at the C2-position with high selectivity. A range of N-containing biaryls smoothly underwent oxidative cross-coupling with five-membered heterocycles such as thiophenes, furans, thiazoles and oxazoles, providing the desired products in good yields. The inexpensive ruthenium catalyst together with KPF6 also induced the reaction.
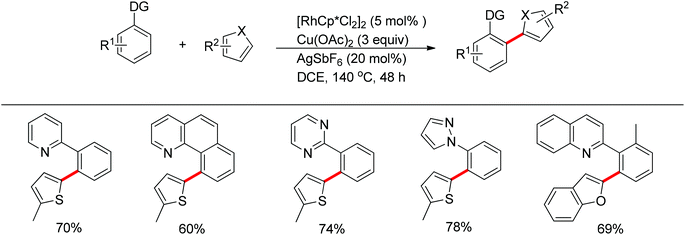 |
| | Scheme 17 Oxidative cross-coupling of N-containing heteroarenes with heterocycles. | |
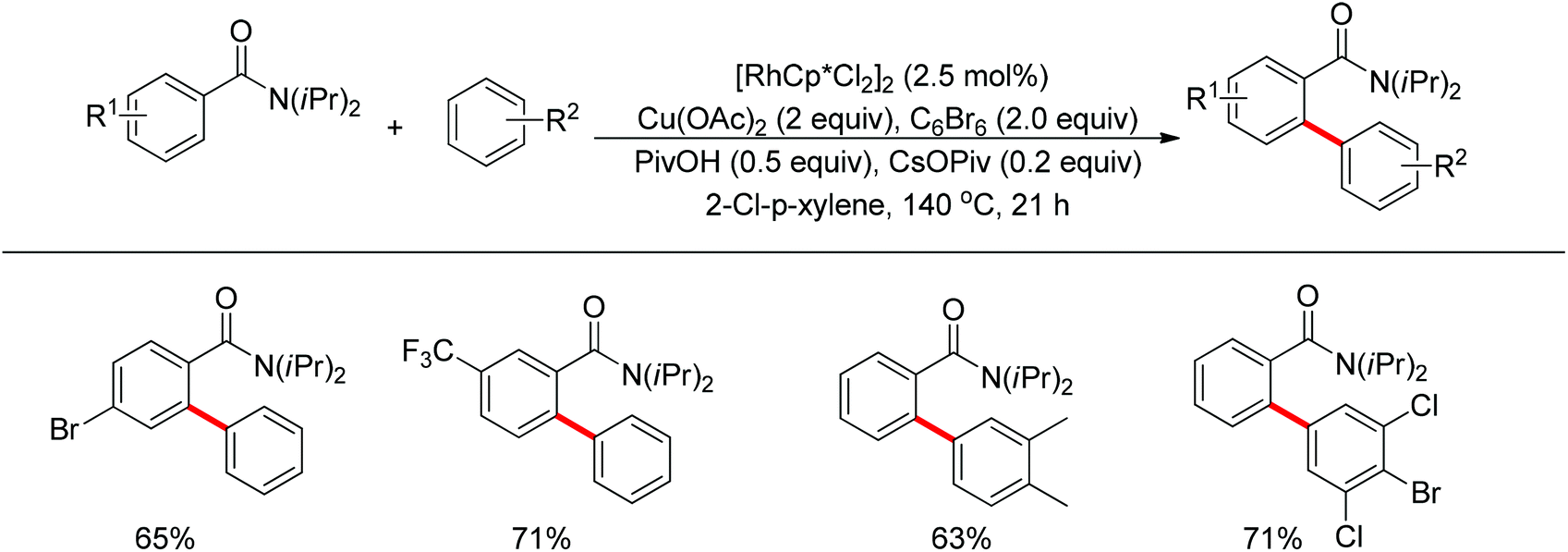
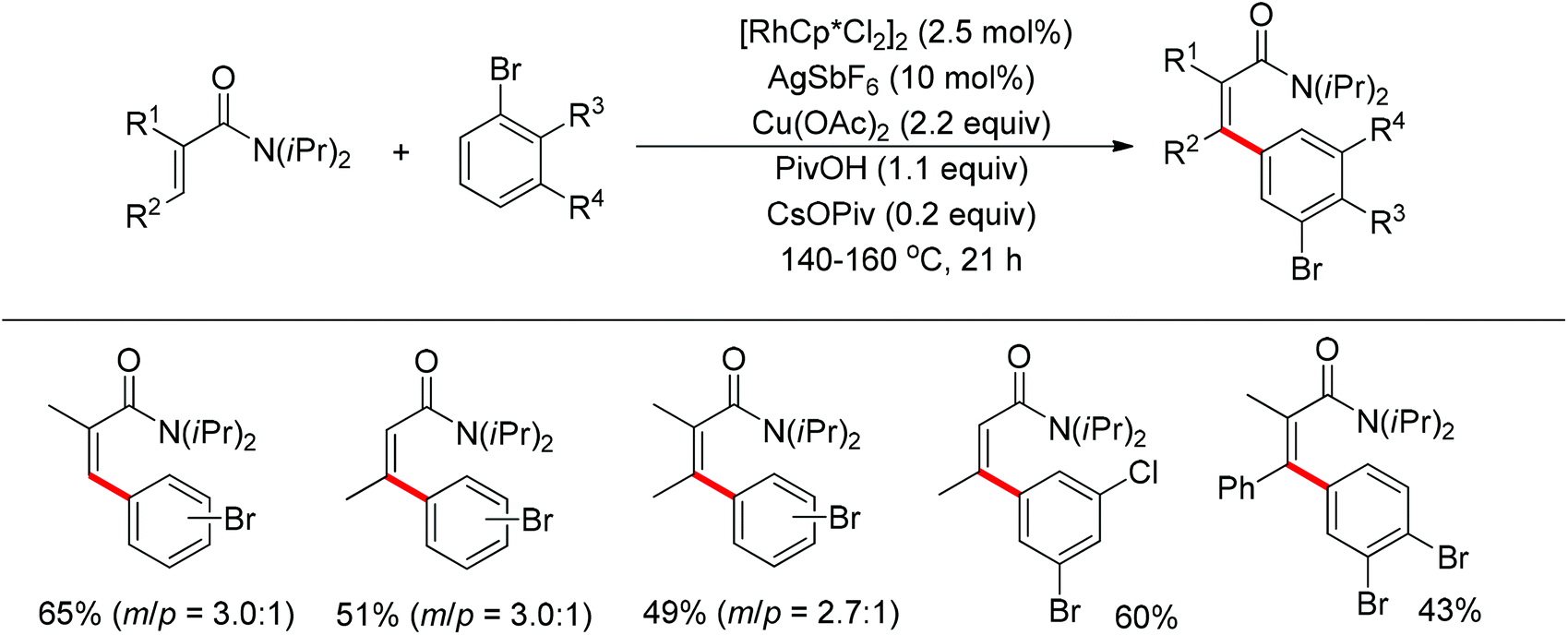
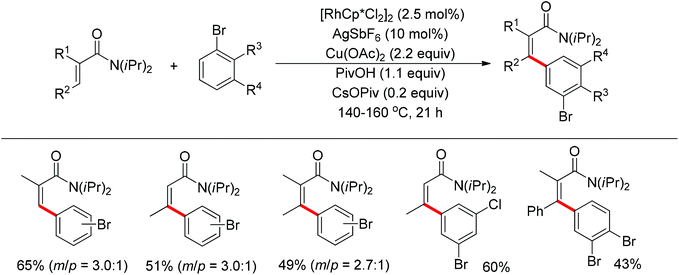
The Glorius group described the Rh(III)-catalyzed oxidative C–H/C–H cross-coupling of benzamides and simple arenes by employing Cu(OAc)2 together with C6Br6 as the oxidants (Scheme 18).26 The amide group acted as the directing group to control the regioselectivity of the reaction. Under the standard conditions, an array of benzamides with both electron-donating and -withdrawing groups at either the meta or para position of the aromatic ring underwent the reaction smoothly with various substituted arenes. The same group also developed a novel dehydrogenative Rh(III)-catalyzed cross-coupling reaction between bromoarenes and vinylic substrates bearing amides as the directing group (Scheme 19).27 This protocol occurred with complete Z selectivity because the vinyl groups were activated by the rhodium via amide-assisted C–H bond activation, not the traditional migratory insertion.
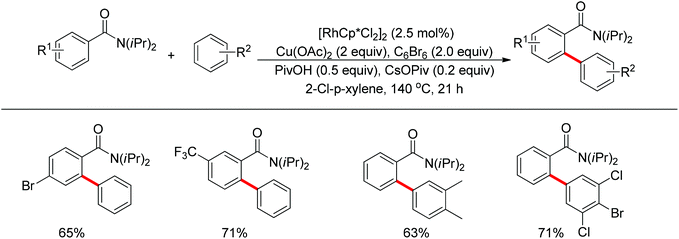 |
| | Scheme 18 Oxidative cross-coupling of benzamides with simple arenes. | |
 |
| | Scheme 19 Amide-assisted oxidative cross-coupling of bromoarenes with vinyl groups. | |
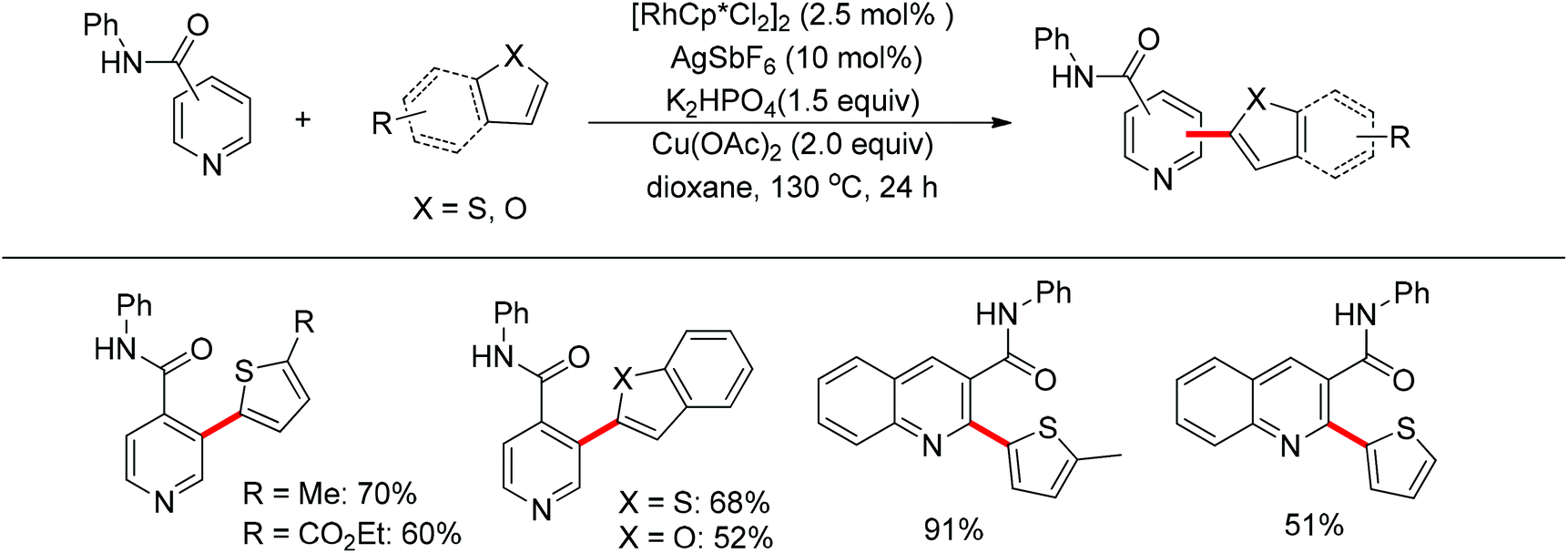
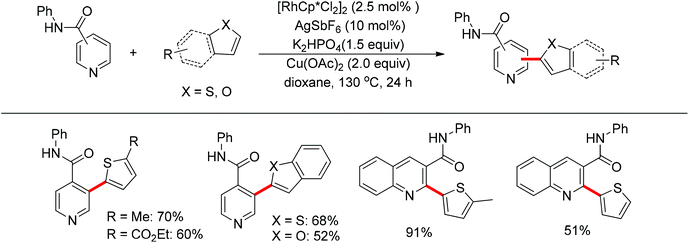
You’s research group developed a Pd(II)-catalyzed direct C2-heteroarylation of pyridines, but the required large excess of pyridine as substrate rendered this protocol less appealing.28 In view of this, a novel method for the heteroarylation of pyridines was described based on the Rh(III)-catalyzed dehydrogenative cross-coupling reaction of aromatic heterocycles with amide-substituted pyridines. This protocol significantly expanded the substrate scope together with excellent regioselectivity at the C2 or C3 position, complementing the methods for pyridine C–H heteroarylation (Scheme 20).29
 |
| | Scheme 20 Oxidative cross-coupling of aromatic heterocycles with amide-directed pyridines. | |
Nitrogen-based groups have provided a useful platform for the development of directed C–H functionalization with inexpensive first-row transition metal catalysts.3m Cobalt catalysts attracted extensive attention because of cobalt’s similar properties to its homologue, rhodium. The use of cobalt catalysts in C–H functionalization dates back to the work of Murashashi in the 1950s.30 In the past several years, C–H bond functionalization catalyzed by Co complexes has developed rapidly. Yoshikai and co-workers achieved the ortho-arylation of aromatic imines with aryl chlorides using a cobalt-N-heterocyclic carbene catalyst in combination with a neopentyl Grignard reagent (Scheme 21).31 The reactions were conducted at room temperature, and the neopentyl Grignard reagent was found to play a critical role for the reactivity of the catalyst.
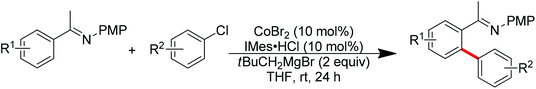 |
| | Scheme 21 Cobalt-catalyzed ortho-arylation of aromatic imines with aryl chlorides. | |
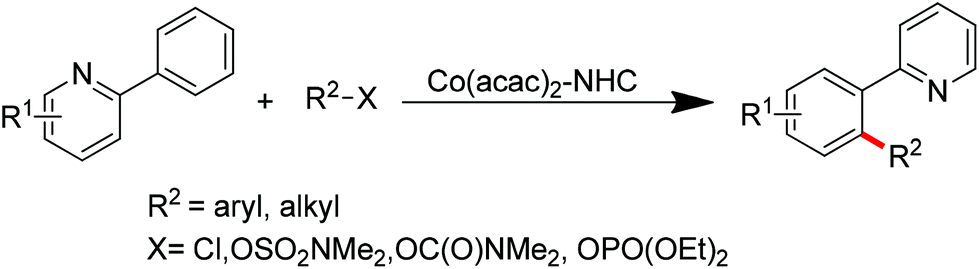
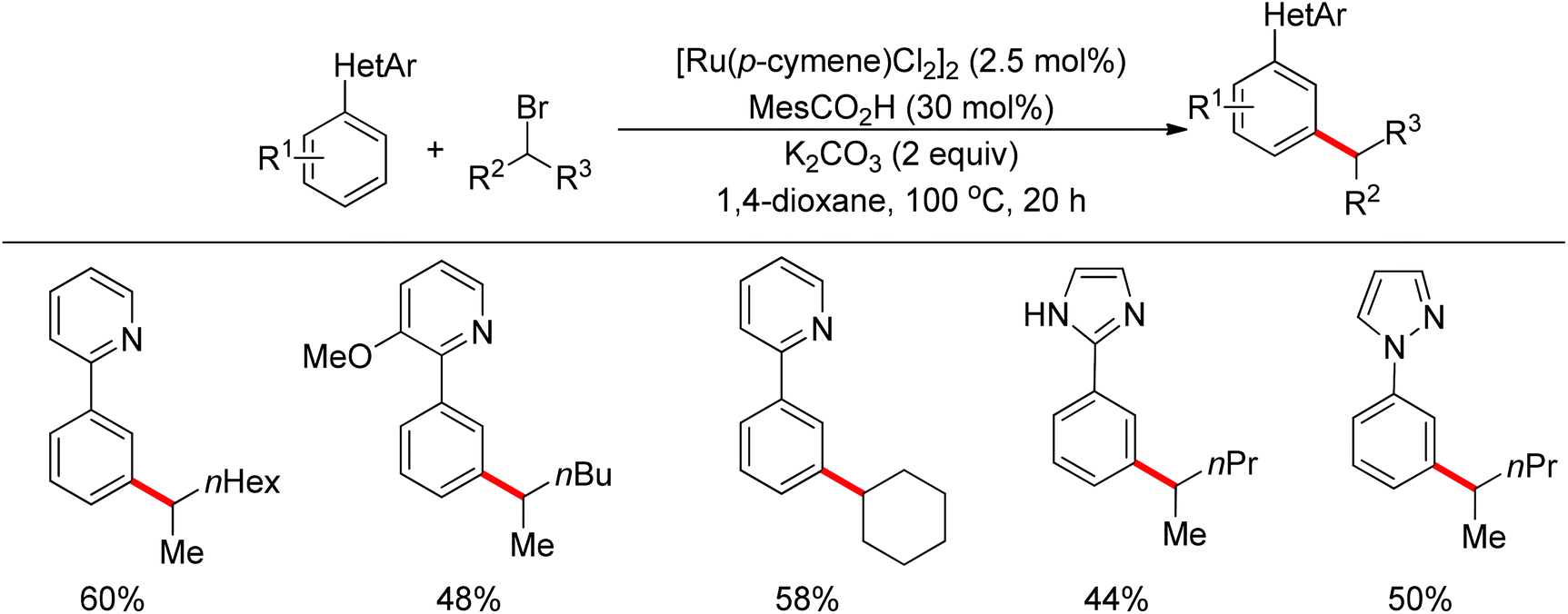
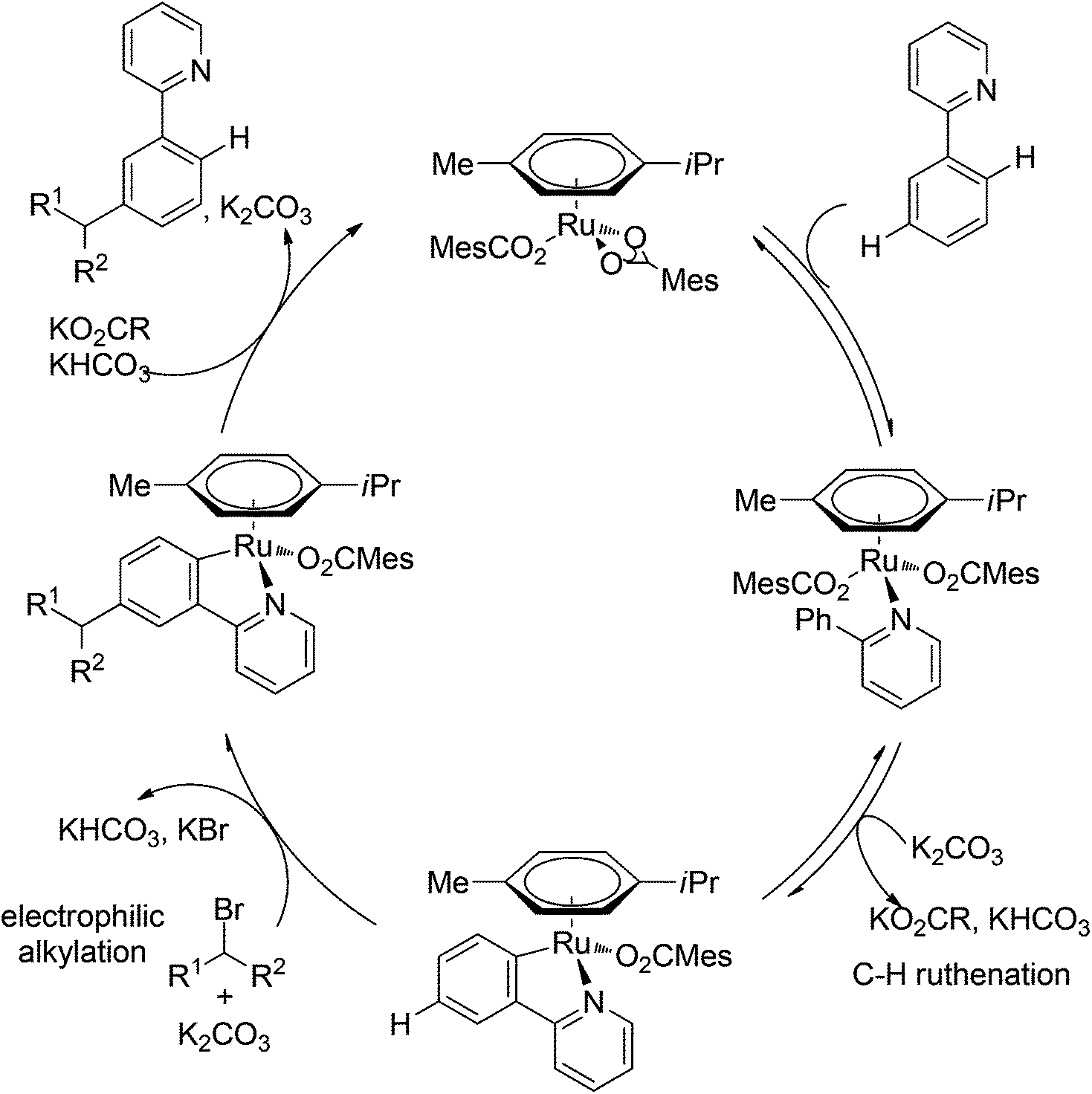
Ackermann and co-workers also disclosed the pyridine-directed aromatic C–H arylations and alkylations of a variety of arenes and heteroarenes with aryl and alkyl chlorides using a cobalt-N-heterocyclic carbene complex as the catalyst.32 Furthermore, the same group realized cobalt-catalyzed biaryl syntheses through combination of pyridine-directed C–H bond activation and C–O bond cleavage.33 Sulfamates, carbamates and phosphates were used as arylating reagents (Scheme 22).32,33
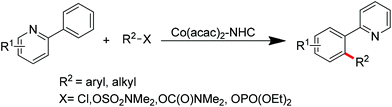 |
| | Scheme 22 Cobalt-catalyzed ortho-arylations and alkylations of 2-phenylpyridines. | |
Although transition metal catalyzed sp2 C–H bond olefination is a widely used synthetic method, the olefination of sp3 C–H bonds is rarely reported.34 The challenges in such a transformation come from the following two aspects: (1) alkenes can compete for the coordination site at the metal catalyst center, which impedes the metal-promoted sp3 C–H bond activation; (2) β-hydride elimination easily occurs after C–H activation. Sanford and co-workers realized a palladium-catalyzed pyridine directed sp3 C–H olefination utilizing air as the oxidant, and AcOH as the solvent (Scheme 23).35 In the reaction, the strongly coordinating pyridine avoided β-hydride elimination. Additionally, the products further underwent reversible intramolecular Michael addition to protect the mono-alkenylated products from over functionalization.
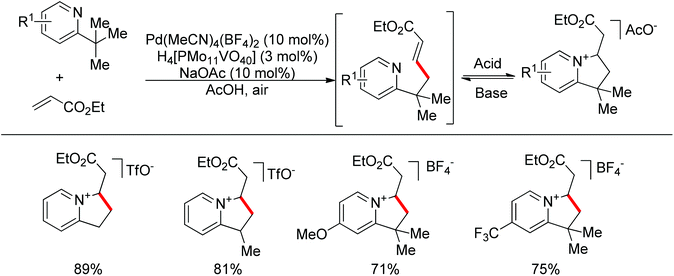 |
| | Scheme 23 Olefination and cyclization between various pyridines and ethyl acrylate. | |
Yu and co-workers realized the Pd(II)-catalyzed methylene sp3 C–H bond arylation directed by amides with aryl iodides in the presence of a mutually repulsive and electron-rich quinolone ligand (Scheme 24).36 Aryl substituted amides were found to be efficient directing groups for the C–H activation of methylene. Detailed ligand designs were carried out in order to seek the appropriate ligand. Both acyclic and cyclic amides containing β-methylene sp3 C–H bonds proved to be effective substrates for this transformation.
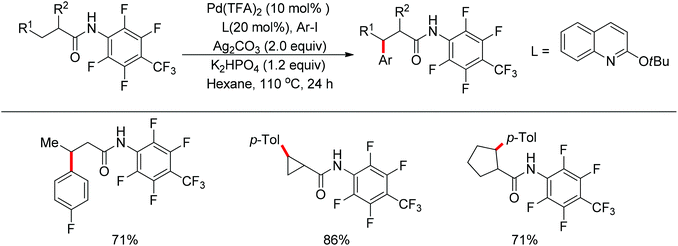 |
| | Scheme 24 Amides directed C–H activation of methylene. | |
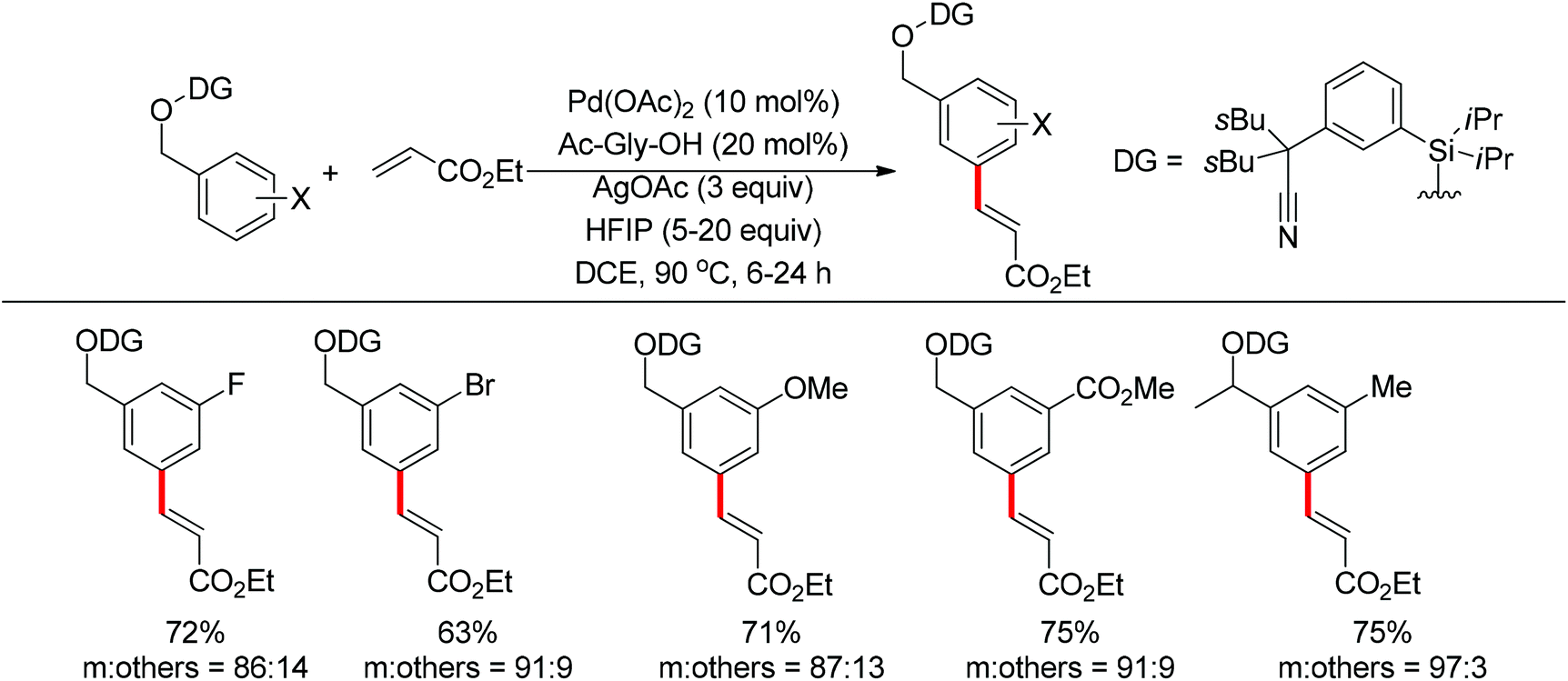
2.1.2. Alkylation of C–H bonds.
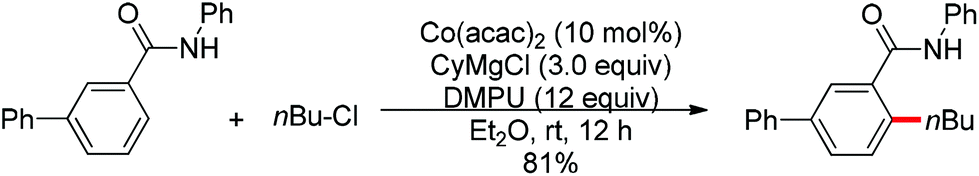
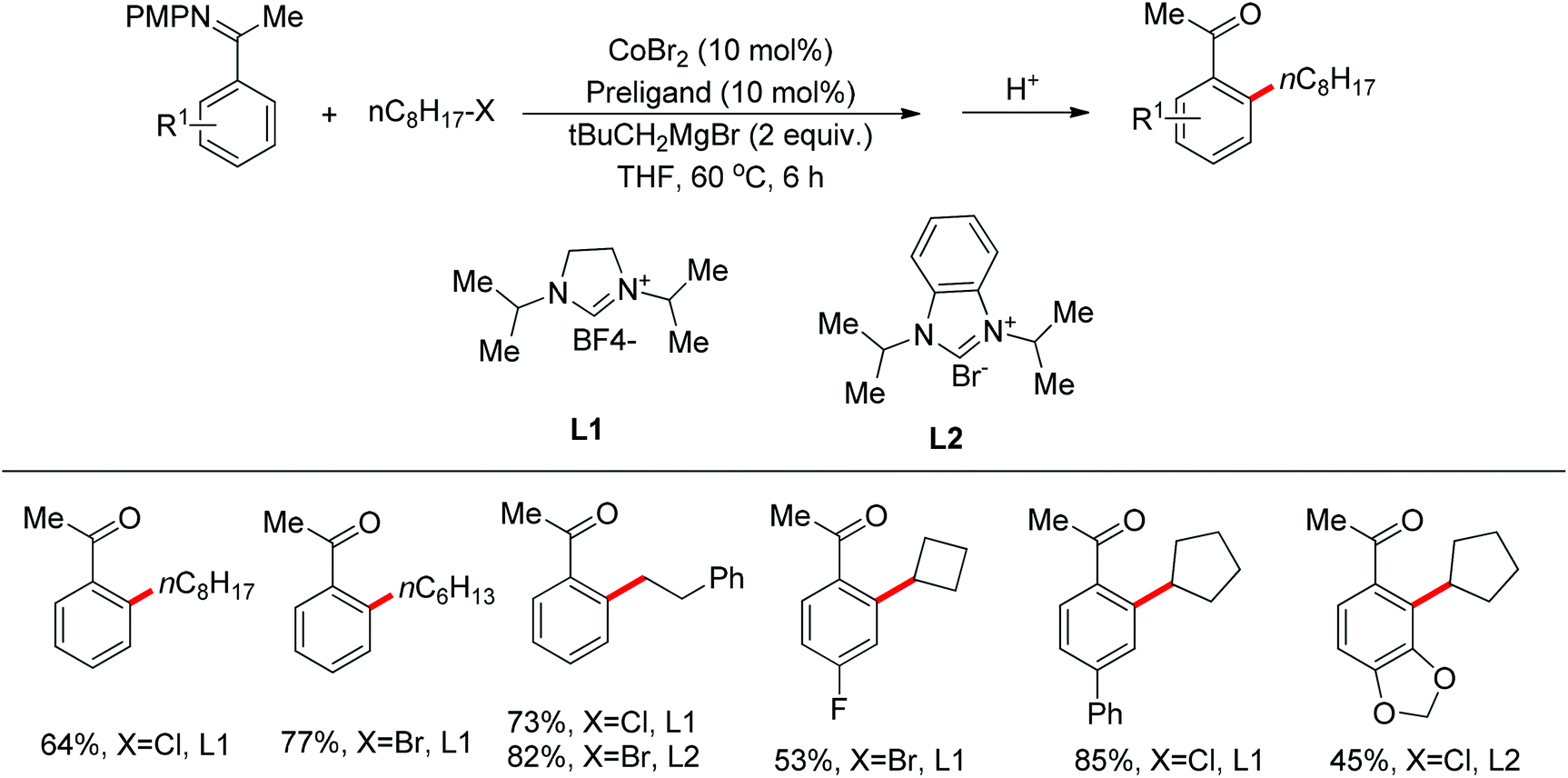
Transition metal-catalyzed cross-couplings with unactivated alkyl (pseudo)halides bearing β-hydrogens are particularly challenging, since the alkyl-metal intermediates are more prone to undergo undesired β-hydride elimination. The directing group assisted ortho-alkylation of arenes with unactivated alkyl halides has been achieved in the presence of palladium, nickel or ruthenium catalysts.37 In 2011, the application of cobalt catalysts was extended to the ortho C–H bond alkylation of secondary benzamides with alkyl chlorides at room temperature (Scheme 25).38 It was stated that the deprotonated amide acts as the directing group. This protocol was mainly applicable to primary alkyl chlorides. In 2013, Yoshikai and co-workers described the ortho C–H bond alkylation of aromatic imines with alkyl halides. After screening various ligands, the authors identified two N-heterocyclic carbene ligands to support the active Co-catalyst system for the imine-directed ortho alkylation of aromatic imines under mild conditions (Scheme 26).39 The Co-N-heterocyclic carbene catalyst system allowed the use of both primary and secondary alkyl halides as alkylating agents, and the choice of N-heterocyclic carbene ligand depended on the alkyl halide used. Based on the stereochemical outcomes for secondary alkyl halides, the authors proposed that the reaction involves single-electron transfer from a cobalt species to the alkyl halide to generate the corresponding alkyl radical, followed by recombination of the cobalt center with the resulting radical and reductive elimination to form the C–C bond.
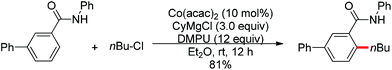 |
| | Scheme 25 Cobalt-catalyzed ortho alkylation of secondary benzamides with alkyl chlorides. | |
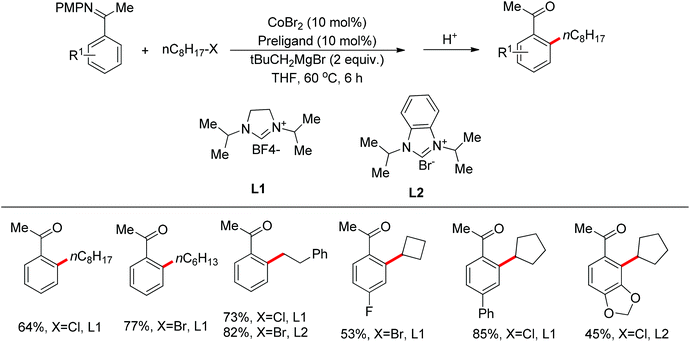 |
| | Scheme 26 Cobalt-catalyzed alkylation of aromatic imines with alkyl halides. | |
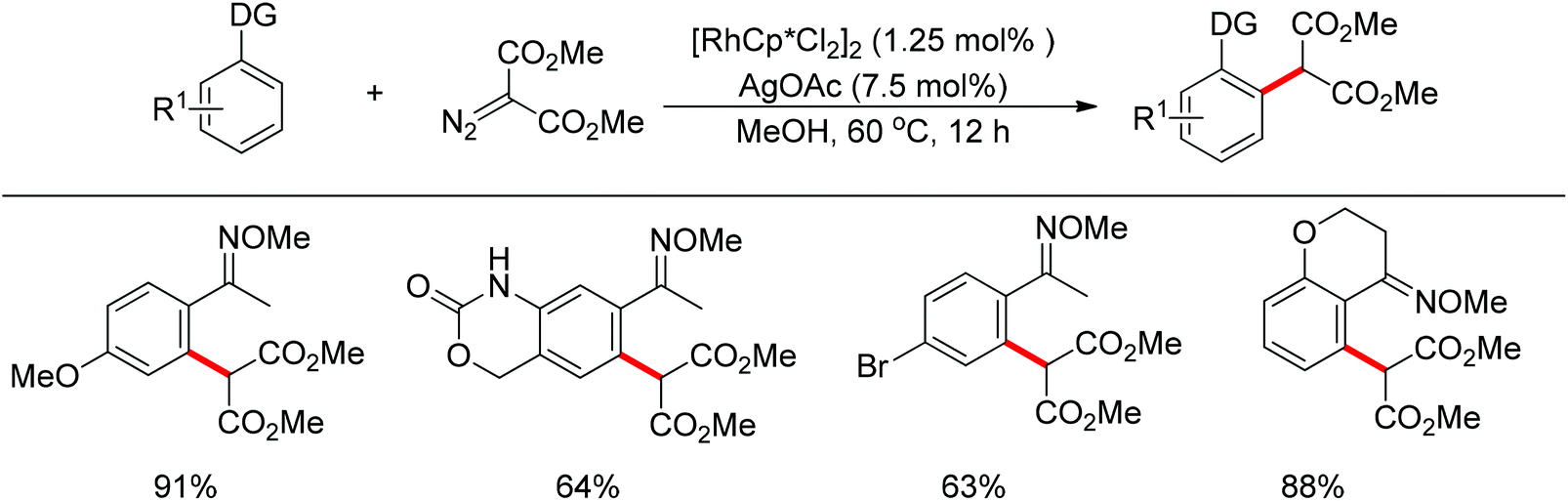
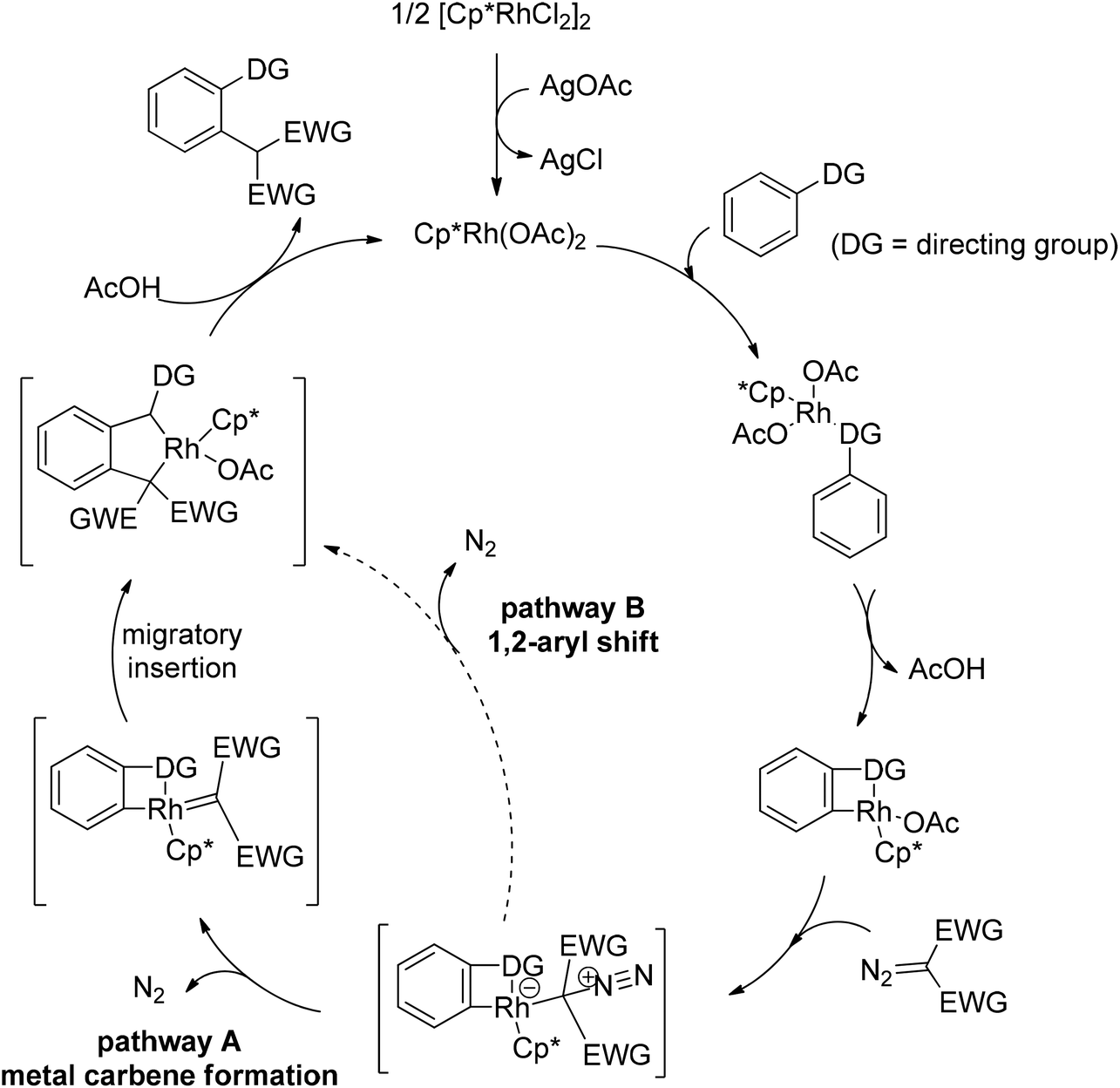
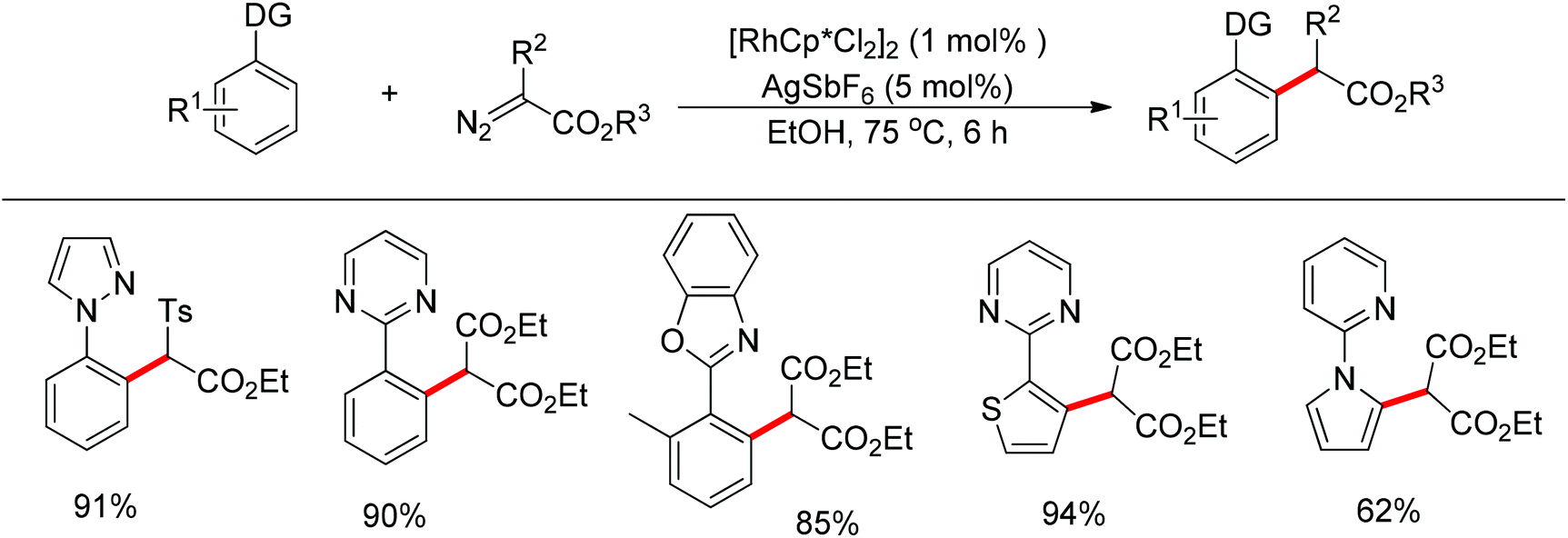
In addition to alkyl halides, the diazo group could also be used as alkylating agent via a carbene intermediate. Yu and co-workers developed the rhodium-catalyzed insertion of aromatic C–H bonds into α-diazocarbonyl compounds via migratory carbene insertion with excellent regioselectivity and functional group compatibility (Scheme 27).40 Ketone oximes, amines and carboxyl groups could all be used as effective directing groups. This protocol released benign N2 as the only by-product and thus provided an efficient approach to α-aryl carbonyl compounds. The reaction was proposed to be initiated by electrophilic C–H metalation, followed by coupling of the diazo compound with the arylrhodium complex (Scheme 28).40 At the same time, Li and co-workers realized a similar process which extended the scope to arenes bearing pyrazole, pyrimidine or oxazole as the directing group (Scheme 29).41
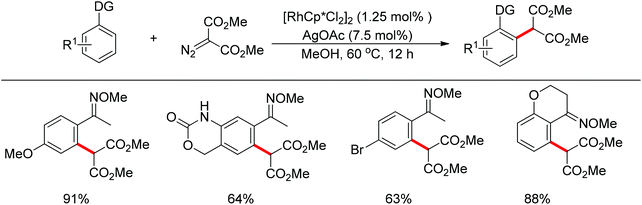 |
| | Scheme 27 Rh(III)-catalyzed alkylation of oximes with diazo compounds. | |
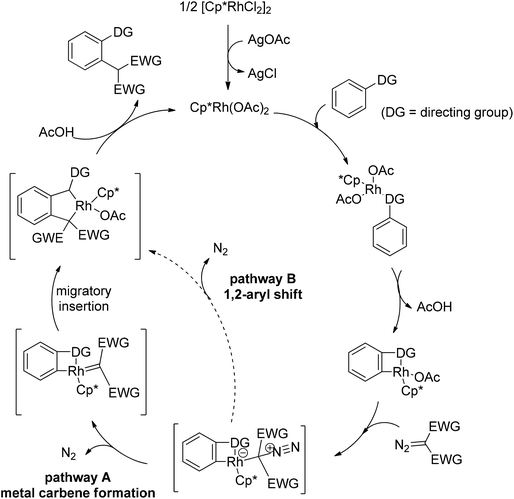 |
| | Scheme 28 Proposed mechanism for the alkylation of aromatic C–H bonds with diazo compounds. | |
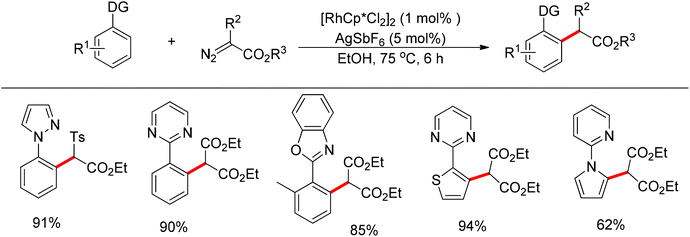 |
| | Scheme 29 Rh(III)-catalyzed alkylation of N-based arenes with diazo compounds. | |
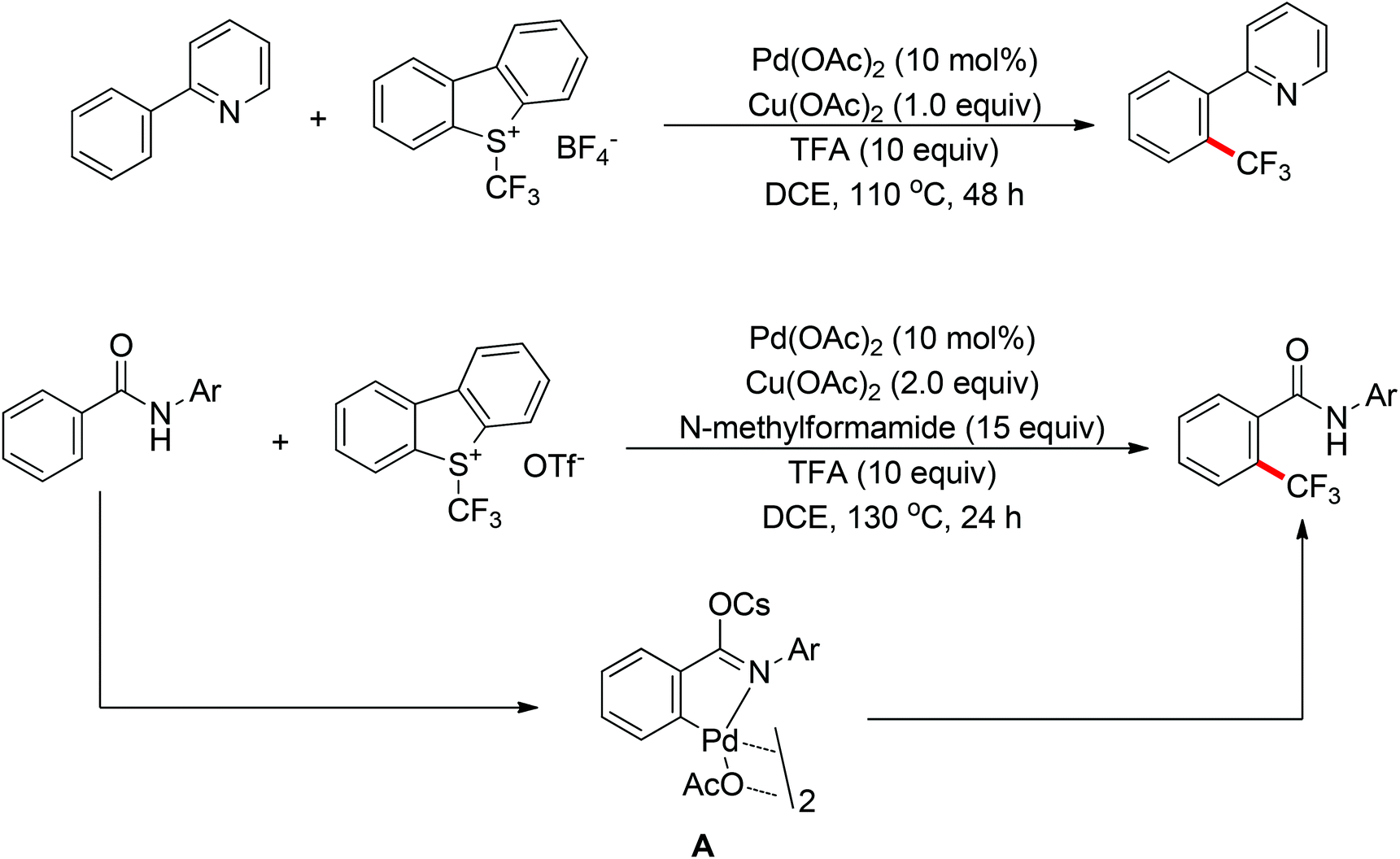
The trifluoromethyl group is widely present in pharmaceuticals, agrochemicals, dyes and polymers. As a consequence, intensive efforts have been devoted to the discovery of approaches for the trifluoromethylation of C–H bonds. Yu and co-workers described the palladium-catalyzed ortho trifluoromethylation of 2-phenylpyridines42 and N-arylbenzamides (Scheme 30).43 For the amide-directed C–H bond trifluoromethylation process, the X-ray characterization of C–H insertion intermediate A indicated that the sp2-hybridized nitrogen atom instead of the sp3-hybridized nitrogen atom is involved in this directed C–H activation. This protocol provided a novel insight into the coordination mode of acidic amides. Subsequently, the authors further described the palladium-catalyzed ortho-trifluoromethylation of benzylamines (Scheme 31).44 The addition of H2O and Ag2O was critical for this transformation. The trifluoromethylating agents also acted as the oxidants in these reactions.
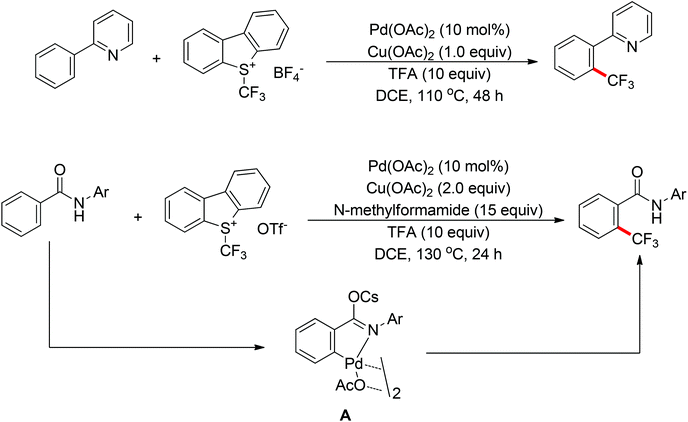 |
| | Scheme 30 Pd(II)-catalyzed ortho trifluoromethylation of N-arylbenzamides and benzylamines. | |
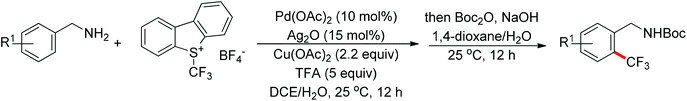 |
| | Scheme 31 Pd(II)-catalyzed ortho trifluoromethylation of 2-phenylpyridines and acetanilides. | |
2.1.4. Cyanation of C–H bonds.
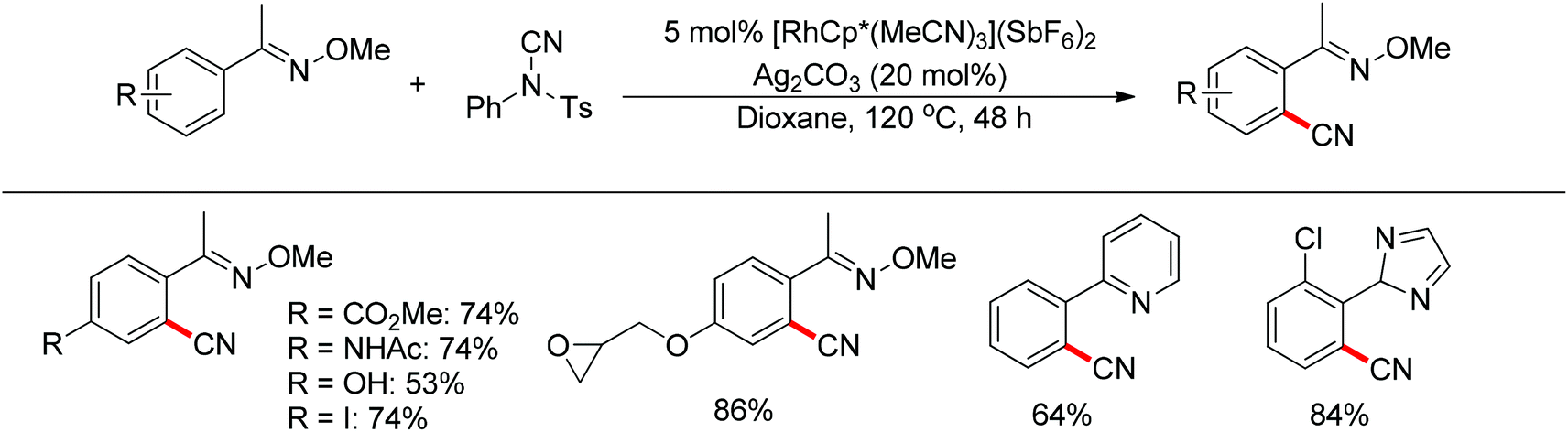
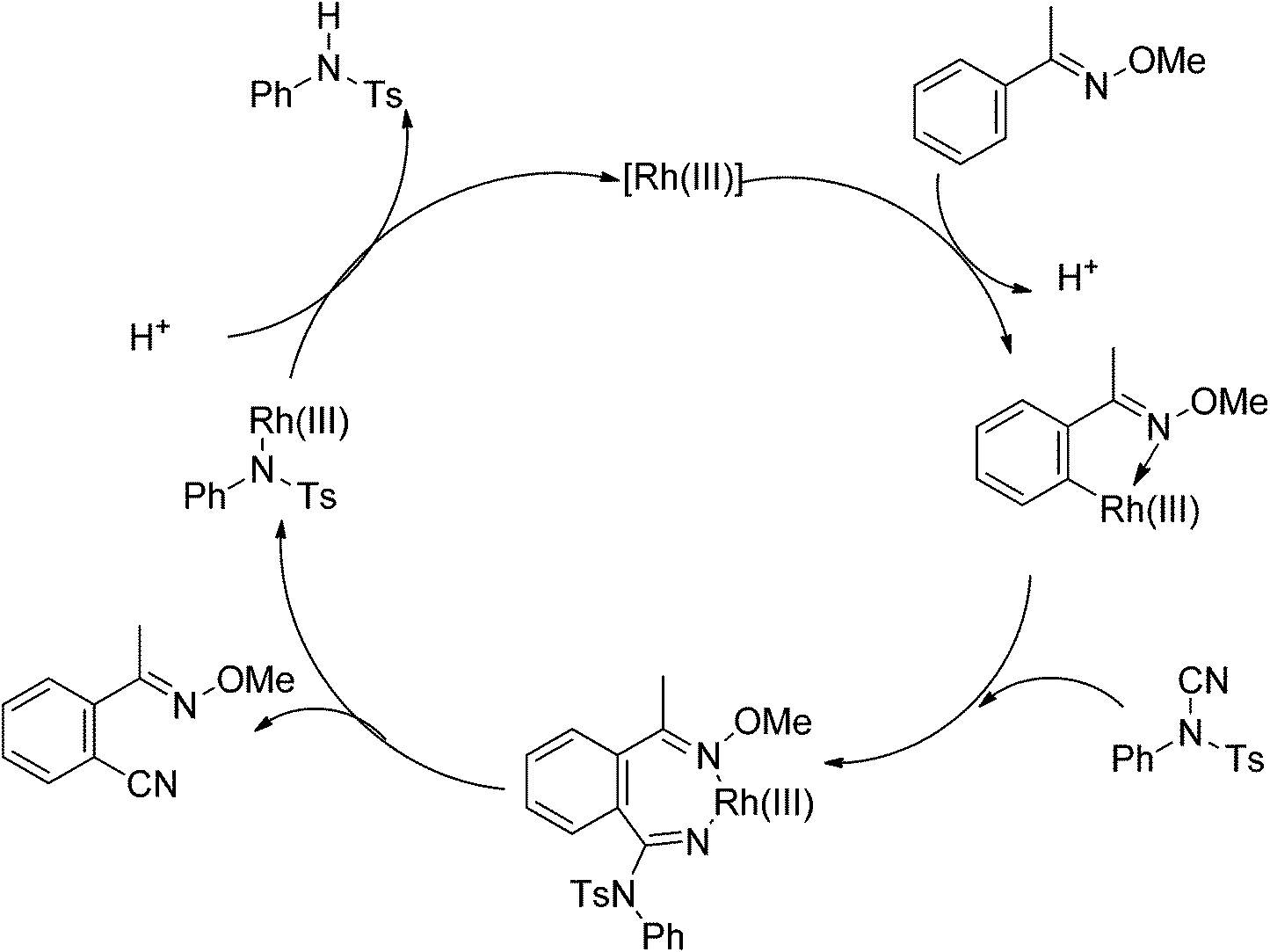
Aryl nitriles are versatile building blocks in organic synthesis since the cyano group is an important precursor for diverse transformations into other functional groups. Yu and Chang described the Cu(II)-48 and palladium(II)-catalyzed49ortho cyanation of 2-phenylpyridines using MeNO2 and DMF/NH3, respectively. In 2013, Fu and co-workers developed the rhodium-catalyzed C–H cyanation of oximes using less toxic and readily available N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) as the cyanating agent (Scheme 35).50 Many nitrogen-based directing groups such as O-methyl oximes, pyridine and pyrazole could be used as the directing group. A variety of functional groups, even sensitive esters, iodides, epoxides and unprotected phenols were tolerated. Based on the experiments and calculations, a general mechanism was postulated (Scheme 36).
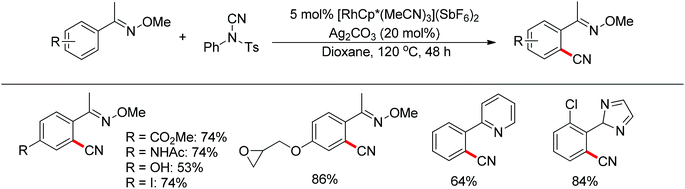 |
| | Scheme 35 Rh-catalyzed C–H cyanation of oximes. | |
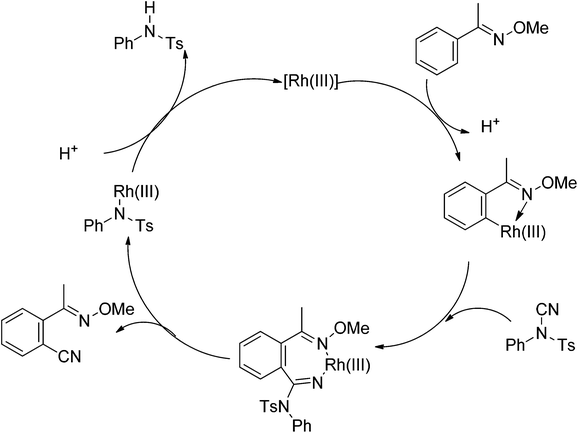 |
| | Scheme 36 Proposed mechanism for the Rh-catalyzed C–H cyanation of oximes. | |
2.1.5. Direct addition of C–H bonds.
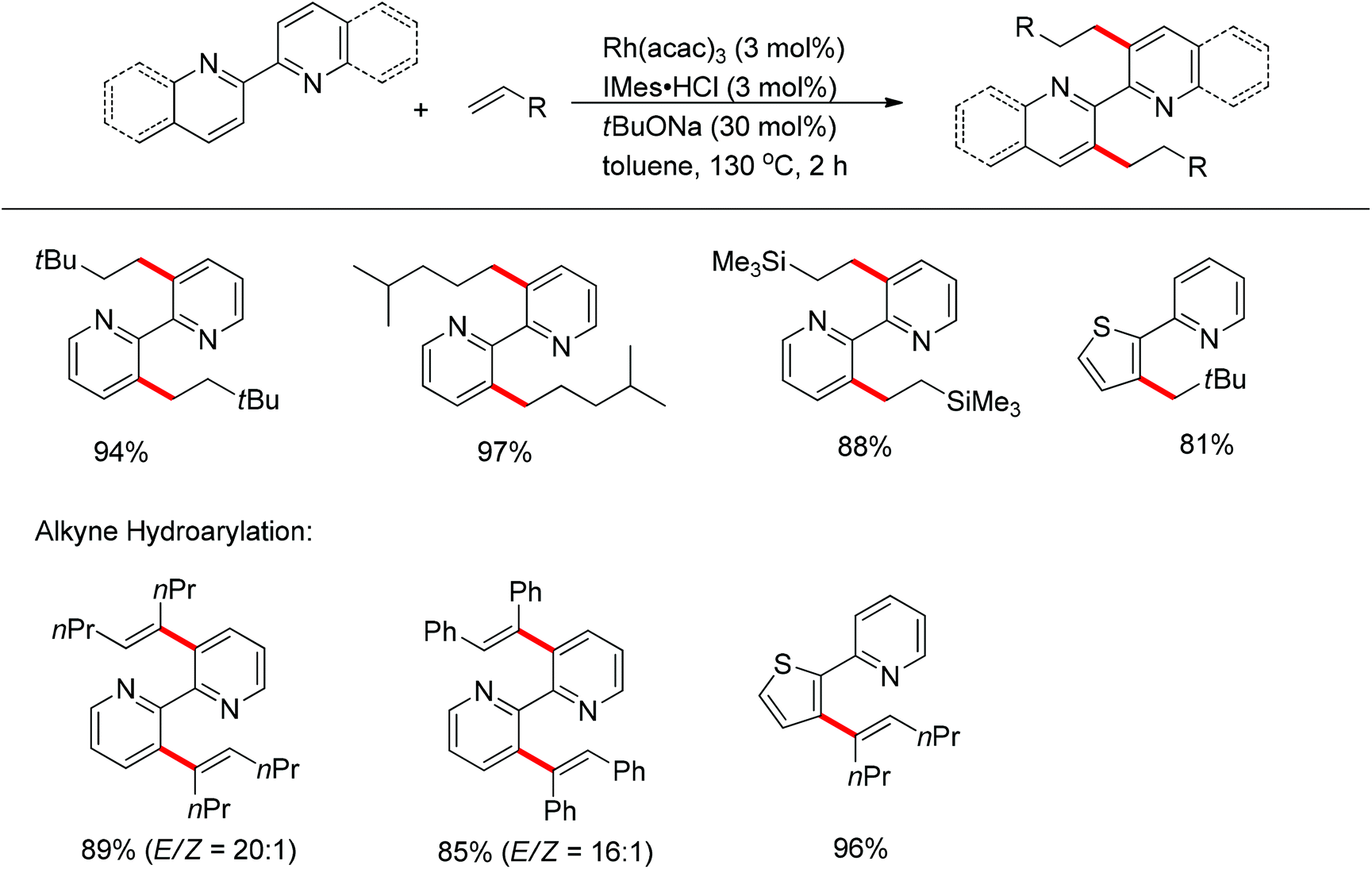
The transition-metal-catalyzed addition of C–H bonds to unsaturated bonds has been extensively investigated. In 2012, the Chang group developed the intermolecular rhodium-catalyzed hydroarylation of alkenes and alkynes with 2,2′-bipyridines (bipy) and 2,2′-biquinolines, which are representative chelating molecules (Scheme 37).51 The stable bidentate metal chelates easily formed upon the coordination of the metal center to the two nitrogen atoms in a catalytic manner, and their stability was such that it was extremely difficult for them to undergo decomplexation and inhibit the cyclometalation of 2,2′-bipyridine and analogues. The strong σ-donating N-heterocyclic carbene (NHCs) ligands displayed significant effect on the activity of the catalyst system. It was proposed that the ligand NHCs would bind to the metal center to generate the (bipy)Rh(NHC) chelates, and the strong trans-effect of NHCs would weaken the Rh–pyridyl bond trans to the ligated NHC, facilitating the C–H activation of bipyridyls. Under the established conditions, the hydroarylation took place exclusively at the 3,3′-position of 2,2′-bipyridines and 2,2′-biquinolines.
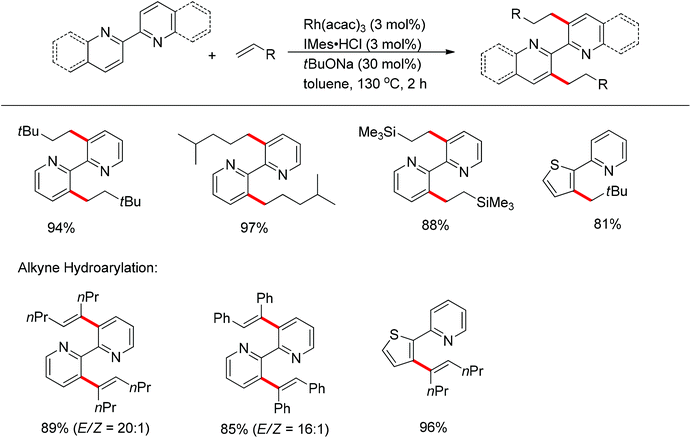 |
| | Scheme 37 Rhodium-catalyzed hydroarylation of alkenes and alkynes with 2,2′-bipyridines. | |
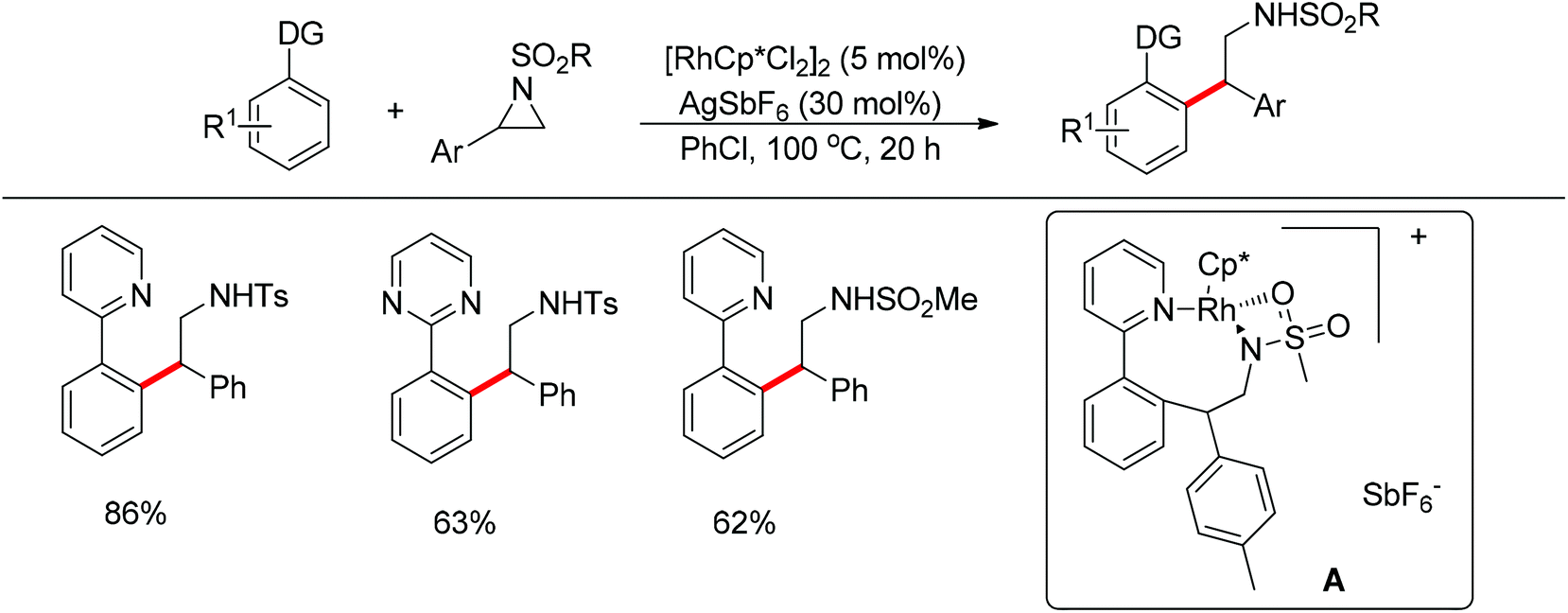
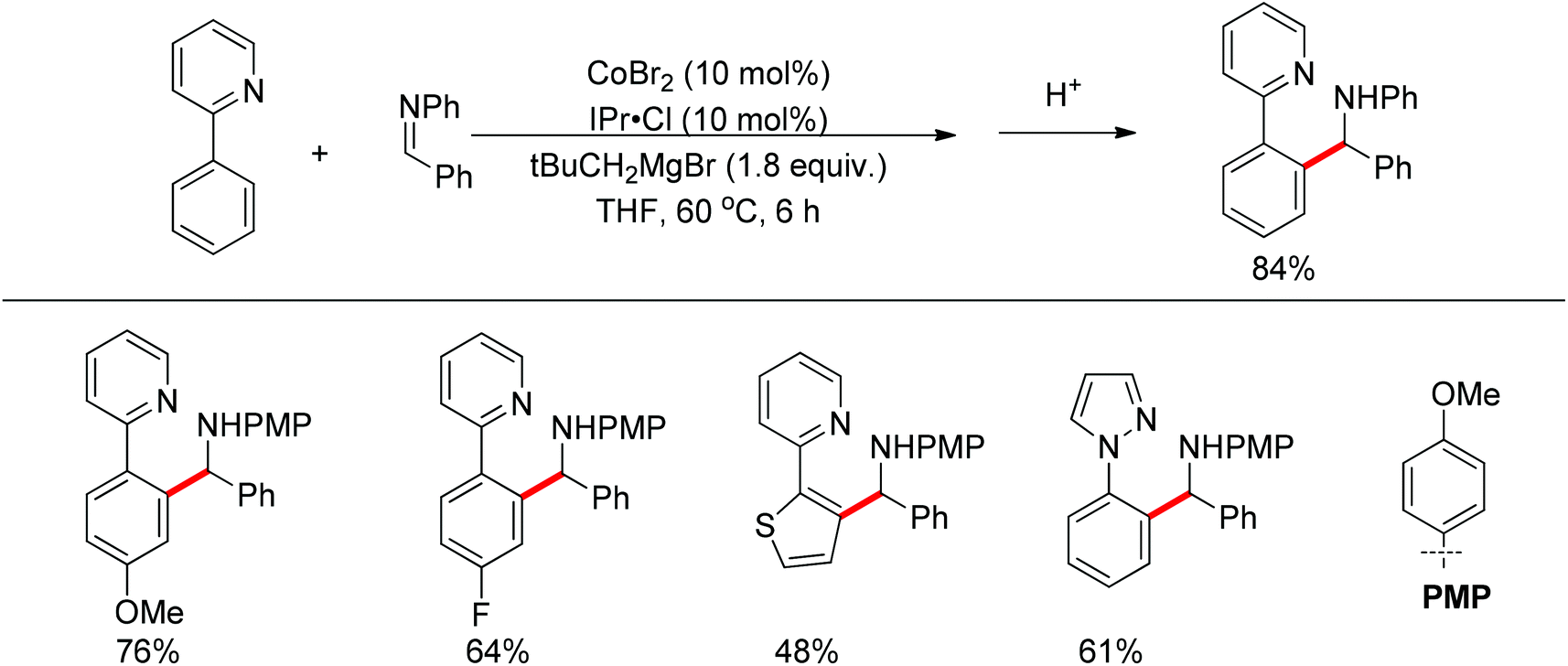
Li and co-workers developed the rhodium-catalyzed C–C coupling of arenes bearing a nitrogen-based directing group with 2-aryl substituted aziridines for the synthesis of β-branched amines (Scheme 38).52 Pyridines, N-pyrazyls and 2-pyrimidyls could be used as effective directing groups for the C–H activation reaction. The cyclometalated rhodium complex intermediate A, in which the catalytic rhodium center is stabilized by a nitrogen atom from pyridine and a nitrogen as well as an oxygen atom from aziridine, was isolated and characterised by X-ray crystallography.
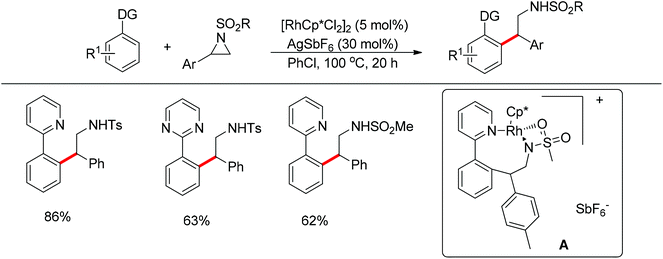 |
| | Scheme 38 Cross-coupling of N-containing heteroarenes with 2-aryl substituted aziridines. | |
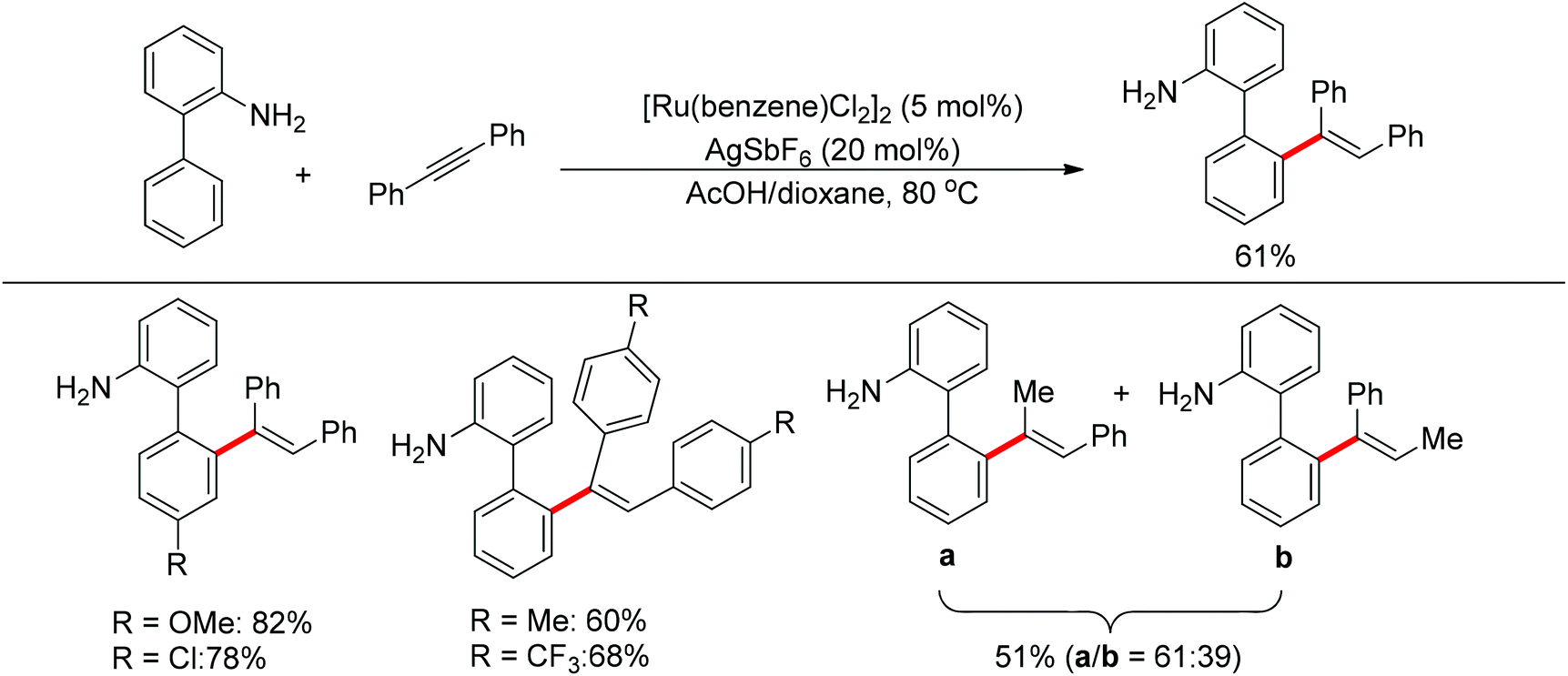
Satoh and Miura's group developed a ruthenium-catalyzed ortho C–H alkenylation directed by a free amine in 2013 (Scheme 39).53 By using acetic acid as the co-solvent, the reaction of 2-aminobiphenyl with diphenylacetylene proceeded smoothly, affording the desired product in generally good yields.
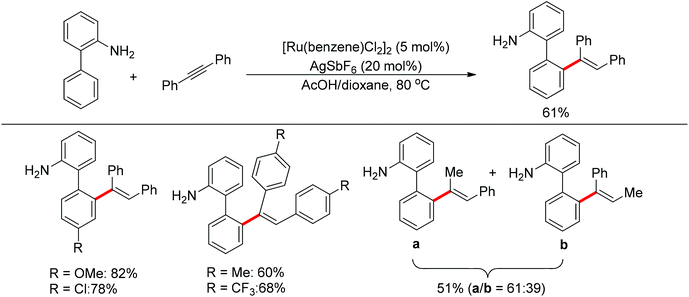 |
| | Scheme 39 Reactions of 2-aminobiphenyls and cumylamines with alkynes. | |
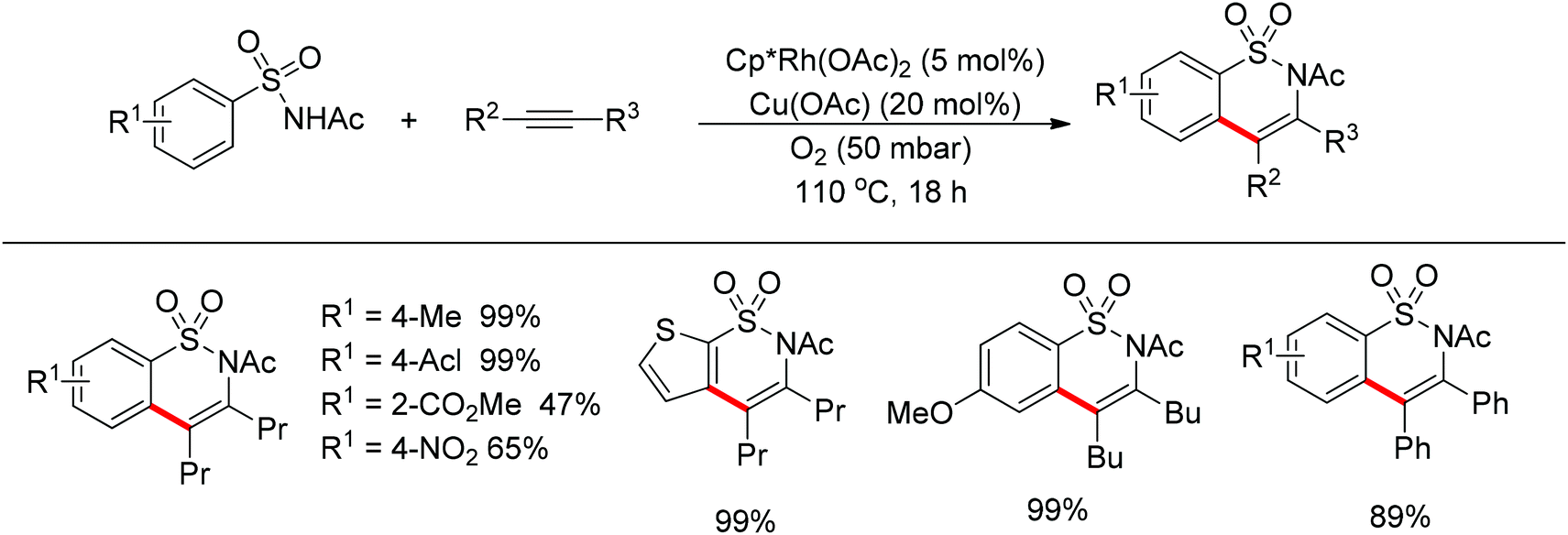
Cramer and co-workers developed the Rh(III)-catalyzed oxidative alkynylation of acylated sulfonamides followed by addition of internal alkynes to approach benzosultams (Scheme 40).54 The protocol was applicable to an array of acylated sulfonamides with either electron-donating or -withdrawing groups on the phenyl ring.
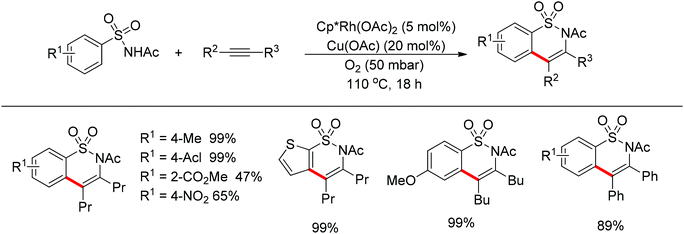 |
| | Scheme 40 Oxidative cyclization of sulfonamides with alkynes to approach benzosultams. | |
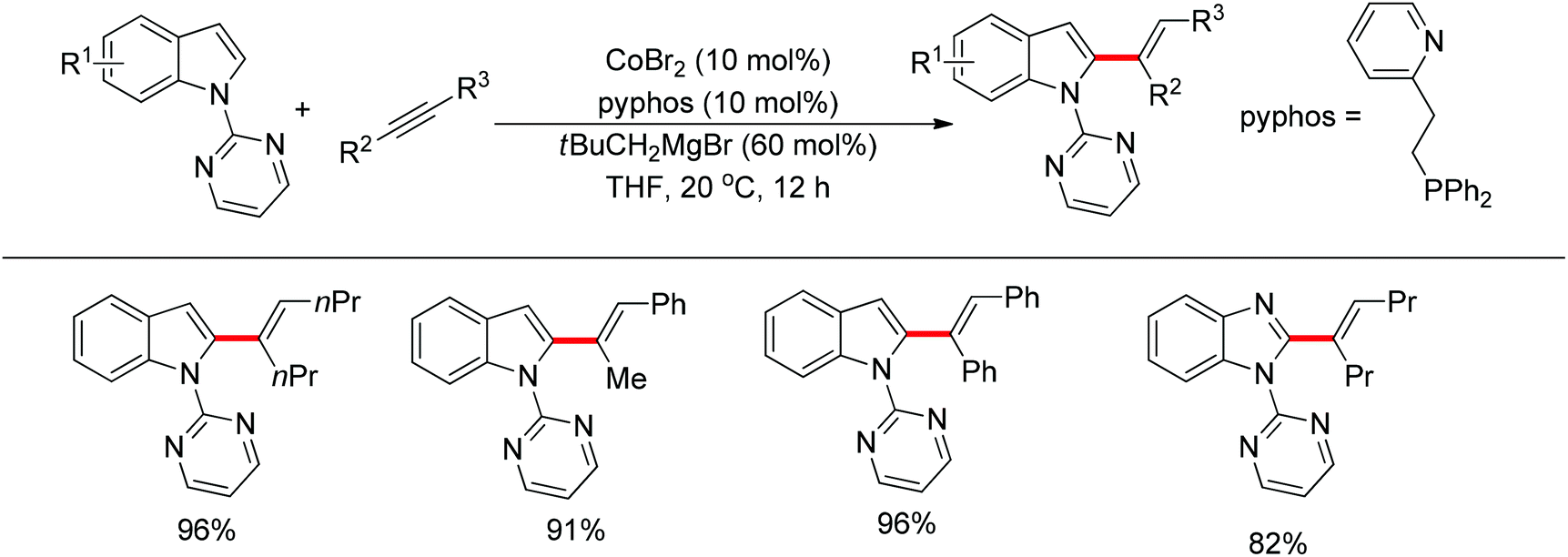
In the past several years, C–H bond functionalization catalyzed by cobalt complexes has developed rapidly. In 2013, Yoshikai's group developed an efficient C2-alkenylation of indoles containing an easily removable N-pyrimidyl group under mild reaction conditions using a cobalt catalyst (Scheme 41).55 The N-pyrimidyl directing group induced the reaction exclusively at the C2 position rather than at the traditionally preferred C3 position of the indole ring. Optimization studies indicated that a phosphine–pyridine bidentate ligand was crucial for producing an active catalyst and the choice of Grignard reagent for reducing Co(II) to Co(I) in situ also affected the catalytic activity. This reaction was applicable to a variety of internal alkynes such as diphenylalkyne and dialkyl alkynes and afforded the corresponding products in good yields with exclusive E selectivity. In the case of unsymmetrical alkynes, the reaction demonstrated a high level of regioselectivity for the C–C bond formation occurring at the less hindered acetylene carbon.
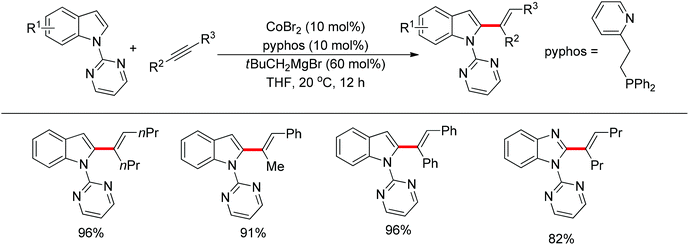 |
| | Scheme 41 Cobalt-catalyzed addition of indoles to alkynes. | |
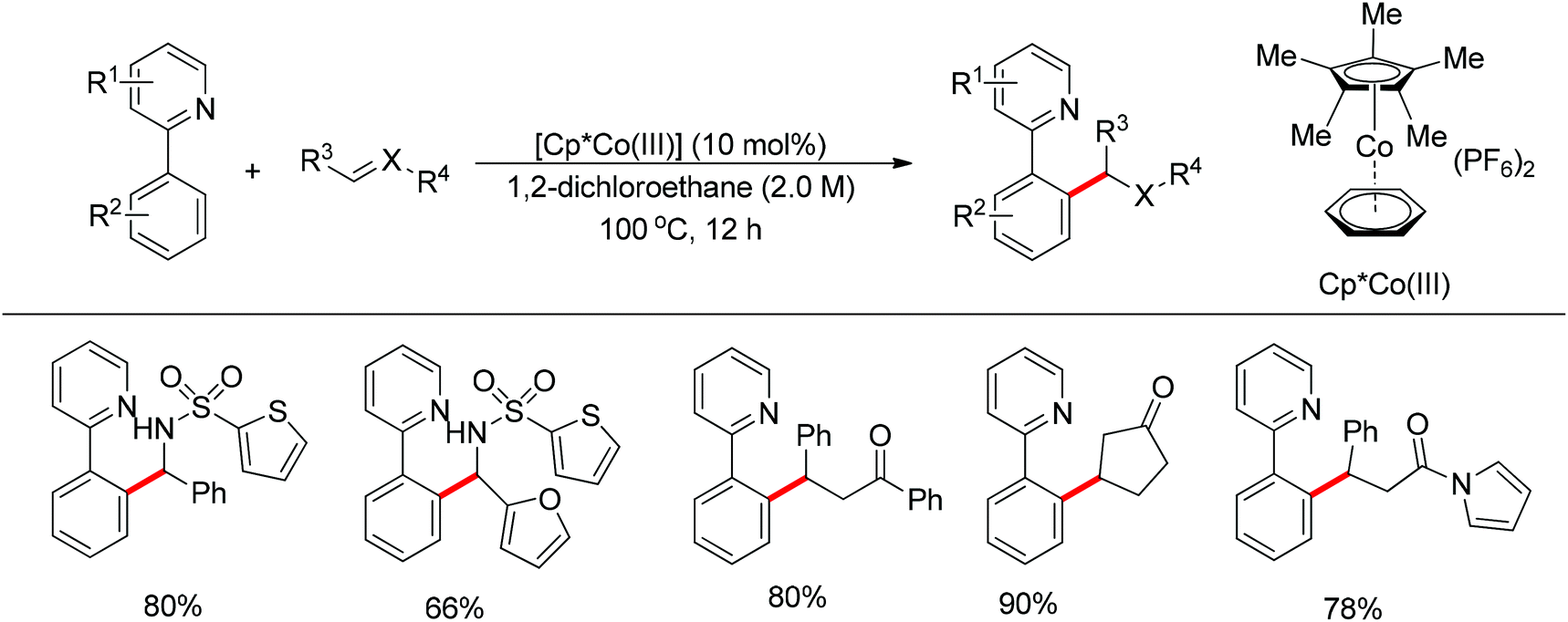
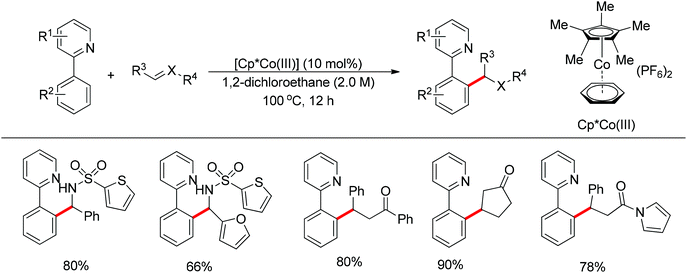
In contrast to the aforementioned Co-catalyzed methods in which a low-valent Co(I) catalyst species was formed in situ to trigger the catalytic cycle, the cationic [Cp*CoIII(arene)](PF6)2 complex was employed by Matsunaga and Kanai to effect the addition of C–H bond in 2-aryl pyridines to imines, enones, and α,β-unsaturated N-acyl pyrroles as ester and amide surrogates (Scheme 42).56 Such a cationic high-valent cobalt catalyst generated the nucleophilic species in situ without additional reagents and afforded the directed C–H bond addition products in an atom-economical manner. According to the authors, the reaction pathway involves the initial Co-mediated C–H cleavage via a concerted metalation–deprotonation (CMD) mechanism to form a Co–C bond, and the subsequent insertion of the Co–C bond into a polar double bond. Matsunaga and Kanai's work demonstrated for the first time that Co(III) catalysts behave like their homologue Rh(III) catalysts, and are able to mediate C–H activation through a CMD process. After a short time, a similar catalytic system was further expanded to the directed C2-selective addition of indoles bearing a removable 2-pyrimidyl group to imines by the same group.57 This method was applicable to a broad scope of substrates including both indoles and imines and including an isomerizable aliphatic imine, affording the desired products in moderate to excellent yields.
 |
| | Scheme 42 Cp*Co(III)-catalyzed directed C–H bond activation. | |
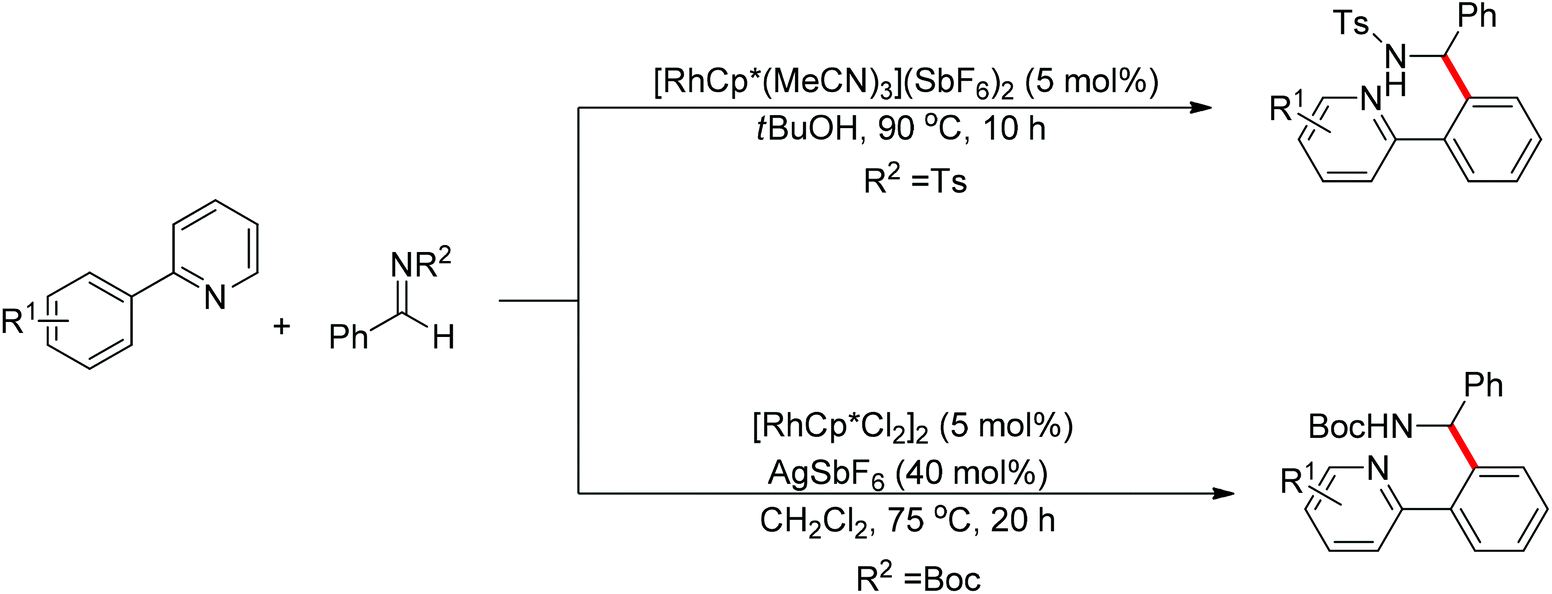
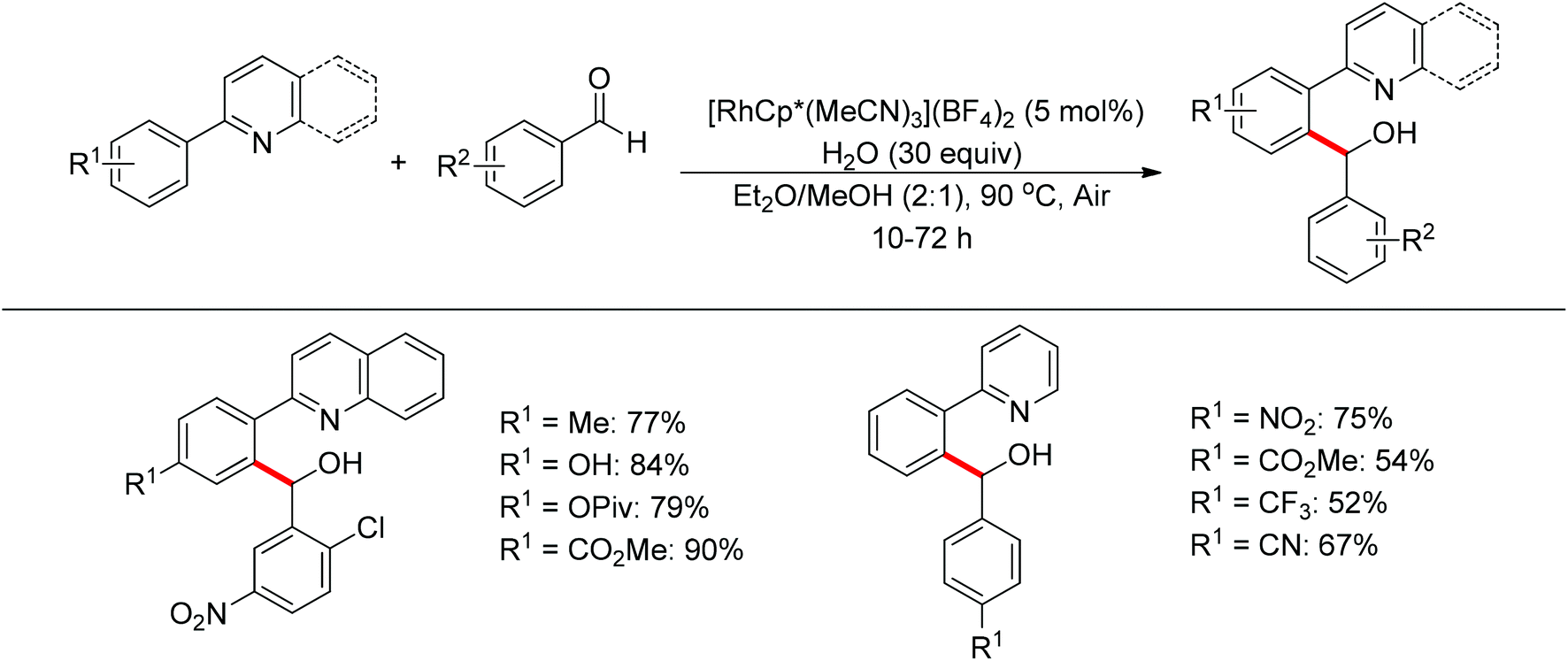
Nucleophilic addition of aryl C–H bonds to a CX group (X = O, N) is an important method for the construction of C–C bonds. Shi and Ellman independently developed the rhodium-catalyzed nucleophilic addition of aryl C–H bonds to N-sulfonyl and N-Boc aldimines using pre-prepared [Cp*Rh(MeCN)3][SbF6]258 or [Cp*RhCl2]2 in combination with AgSbF6 (Scheme 43)59 as the catalyst, respectively. An N-based directing group such as pyridinyl or quinolinyl was installed on the phenyl ring of the arene to control the selectivity of the C–H activation. The reaction was conducted under mild conditions with good functional group tolerance. Based on this observation, Shi and co-workers subsequently described the addition of aryl C–H bonds to aryl aldehydes to produce diarylmethanols (Scheme 44).60 The aldehydes bearing an electron-withdrawing group such as nitro, cyano, or chloride exhibited a higher reactivity. Moreover, the reaction was carried out under air in the presence of 30 equiv. of H2O. Shortly afterwards, they further developed the addition of olefin C–H bonds to N-sulfonylaldimines and aryl aldehydes (Scheme 45).61 Meanwhile, Ellman and co-workers successfully developed the rhodium-catalyzed addition of benzamide C–H bonds to N-sulfonyl aldimines, which employs amides as the directing group (Scheme 46). This reaction proceeded with good functional group compatibility and expanded the synthetic utility of this protocol since amides are an important motif in organic synthesis owing to various transformations of such structural units.
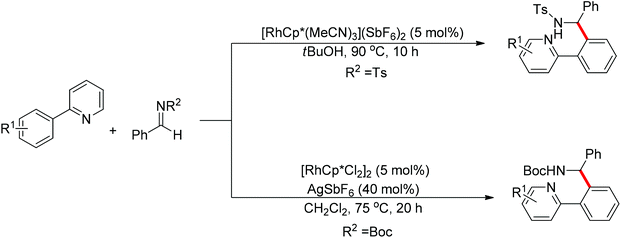 |
| | Scheme 43 Addition of 2-phenylpyridines C–H bonds to N-Boc aldimines. | |
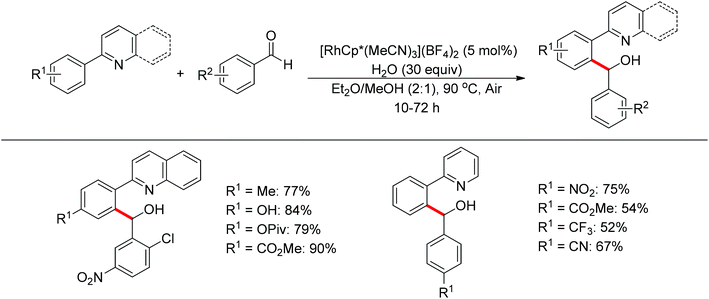 |
| | Scheme 44 Addition of aryl C–H bonds to aryl aldehydes. | |
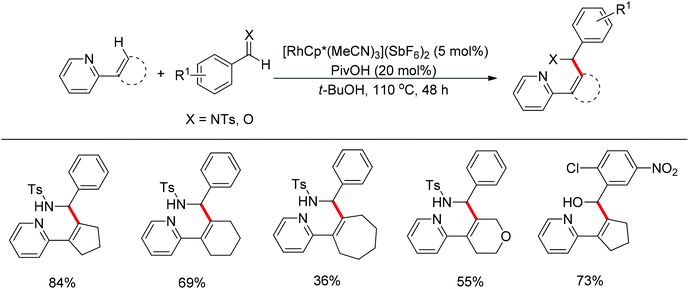 |
| | Scheme 45 Addition of olefin C–H bonds to N-sulfonylaldimines and aryl aldehydes. | |
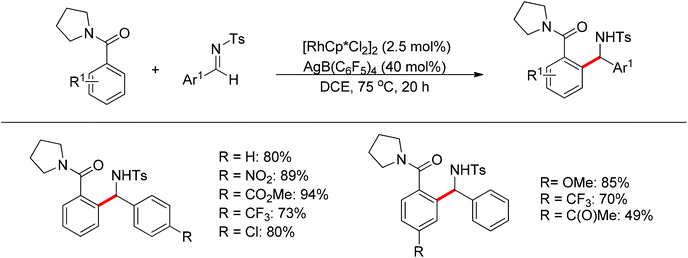 |
| | Scheme 46 Addition of benzamide C–H bonds to N-sulfonyl aldimines. | |
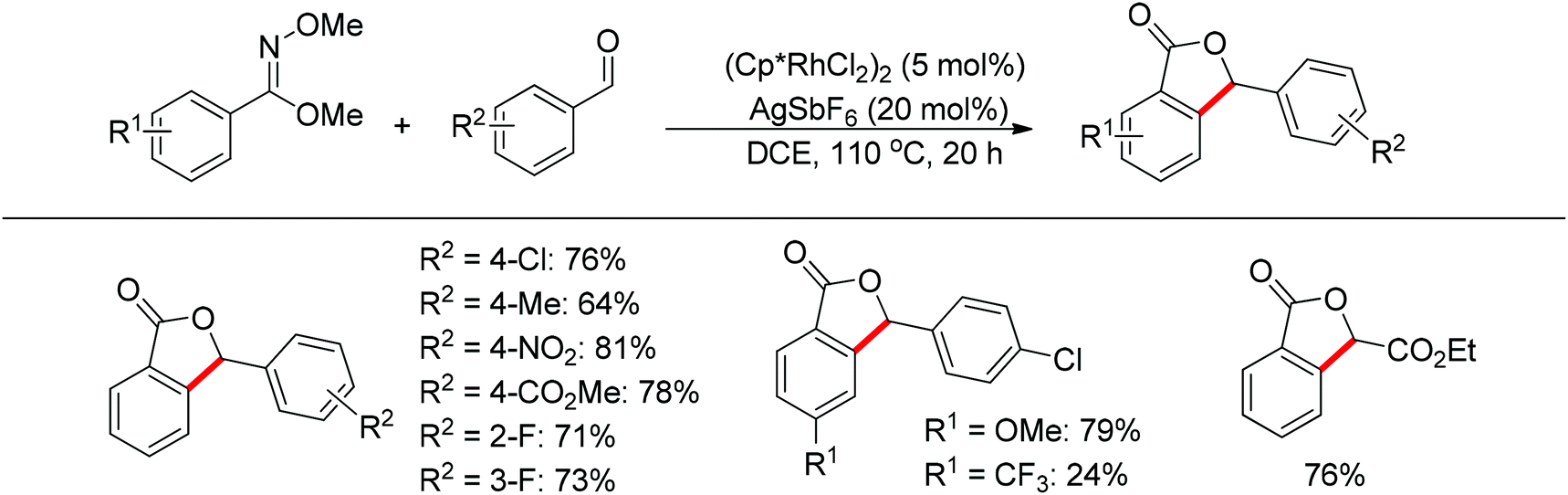
The one-pot synthesis of phthalides via the rhodium-catalyzed cascade addition and cyclization of benzimidates with aldehydes was realized (Scheme 47).62 The imidate not only acted as the directing group to promote the C–H activation of benzimidates, but also served to capture the formed alcohol intermediate to give lactones. The reaction exhibited excellent functional group compatibility. Benzaldehydes bearing chloro, fluoro, nitro, ester, and methoxy groups on the phenyl ring, together with unactivated aliphatic aldehydes, provided the desired products in high yields. Electron-deficient aldehydes often provided the products in higher yields.
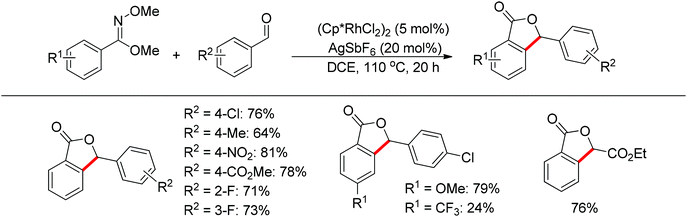 |
| | Scheme 47 Rh(III)-catalyzed cascade addition and cyclization of benzimidates with aldehydes. | |
Yoshikai and co-workers reported the cobalt-catalyzed arylation of aldimines via pyridine-directed cleavage of an aromatic C–H bond and addition of C–H across the aldimine double bond (Scheme 48).63 In this reaction, the N-heterocyclic carbene (NHC) supported Co-catalyst could insert into the ortho C–H bond of 2-arylpyridine derivatives via oxidative addition and couple with aromatic aldimines to afford diarylmethylamines. However, aromatic aldimines bearing Boc and Ts groups were not reactive.
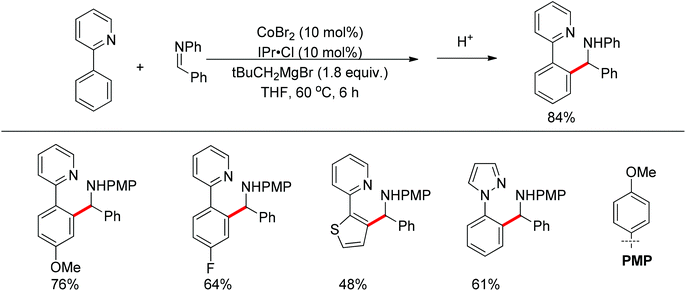 |
| | Scheme 48 Addition of 2-arylpyridines to aromatic aldimines. | |
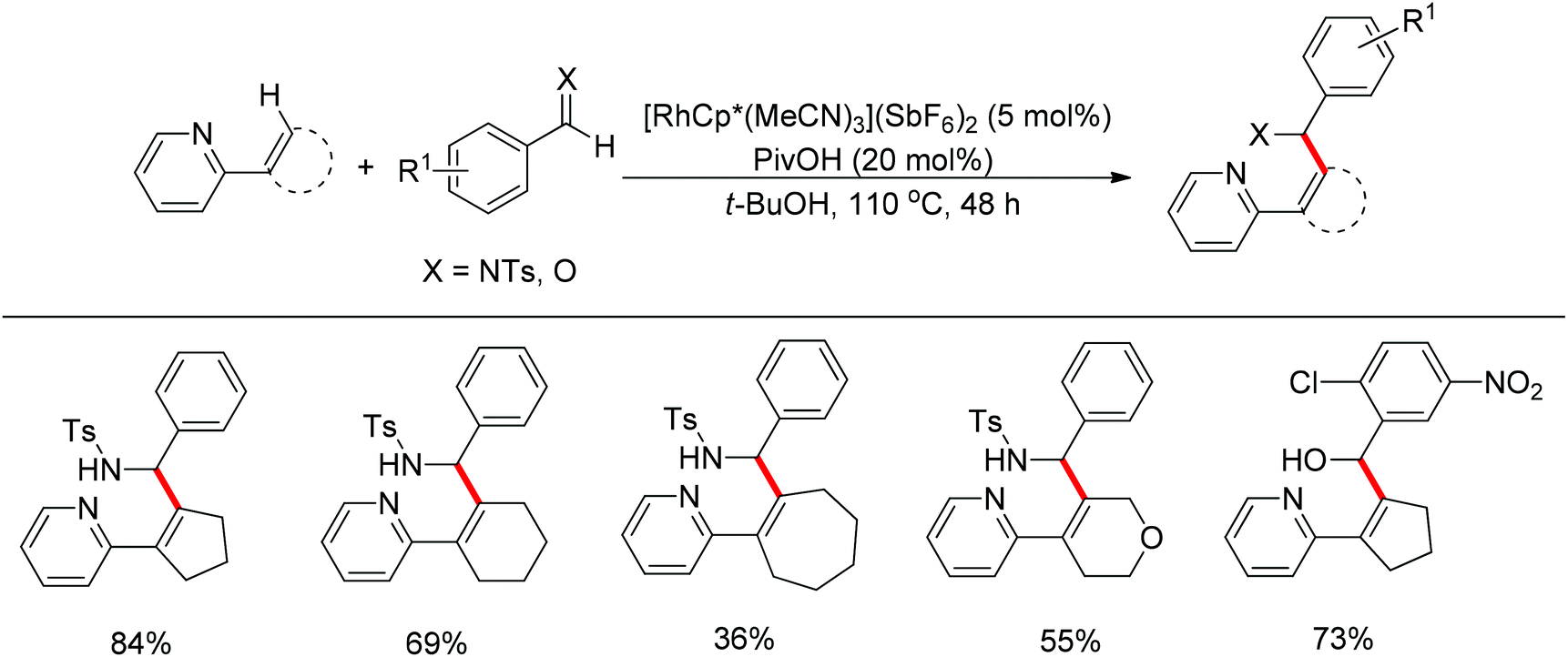
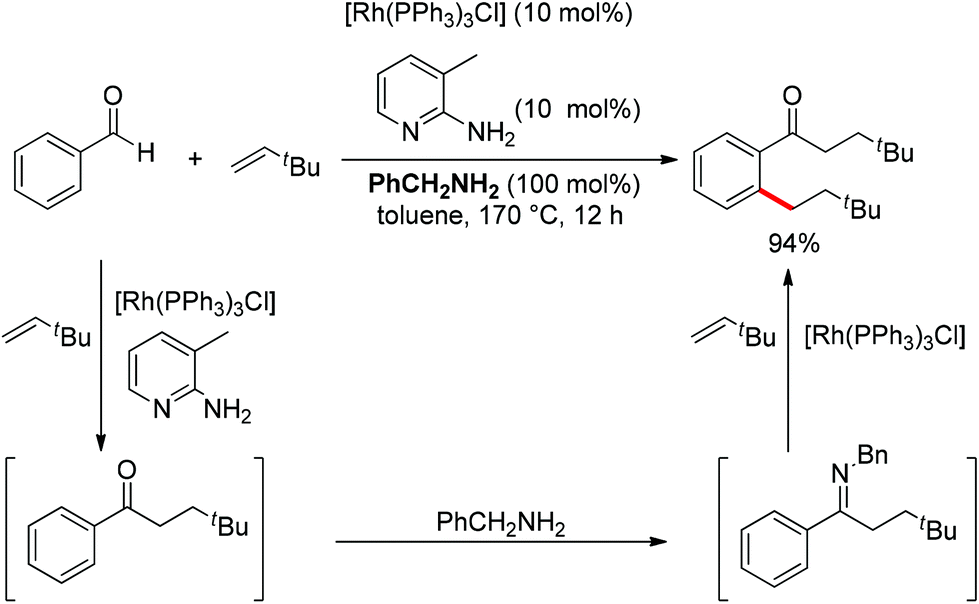
Although almost all the directing groups used at present are prepared stoichiometrically before C–H functionalization, a few research groups reported their generation in situ and even in a catalytic amount. In 2000, a pioneering report by Jun et al. disclosed that ketimine could be generated in situ and undergo ortho-alkylation with olefins (Scheme 49).64 It was proposed that metal–organic cooperative catalysis between Wilkinson's catalyst and 2-amino-3-picoline would induce hydroacylation of olefins with benzaldehyde to afford a ketone intermediate, which would react with benzylamine to generate the ketimine that directed the ortho-alkylation to produce an ortho-alkylated ketone. It should be noted that only hydroacylation would occur without benzylamine. 2-Amino-3-picoline would probably coordinate the Rh catalyst to suppress ortho-alkylation.
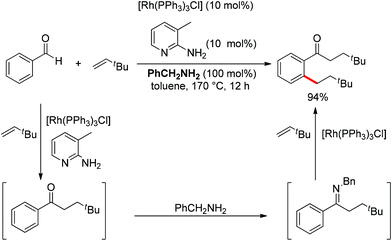 |
| | Scheme 49 Rh-catalyzed ortho-alkylation of ketones directed by in situ generated ketimine. | |
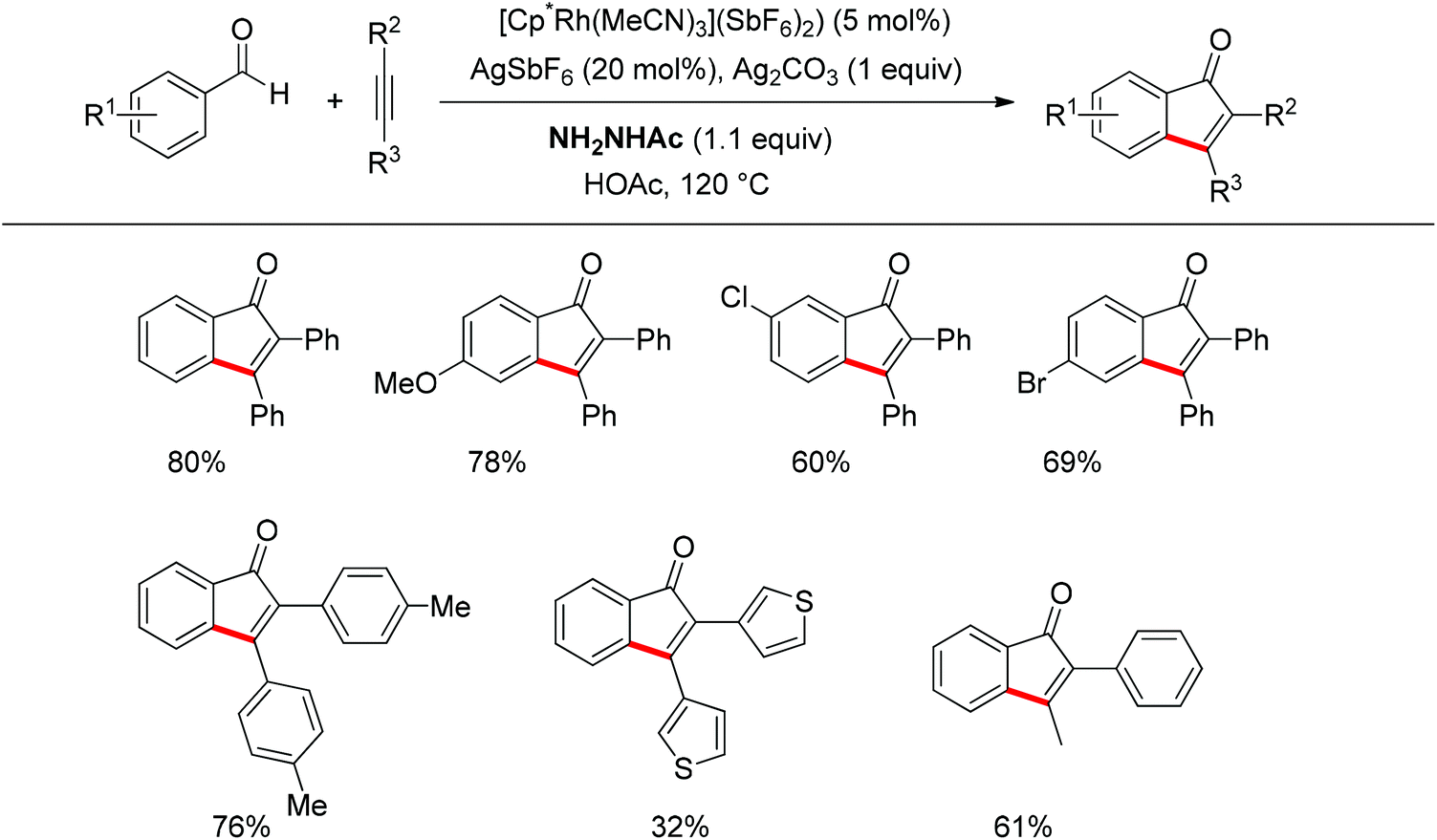
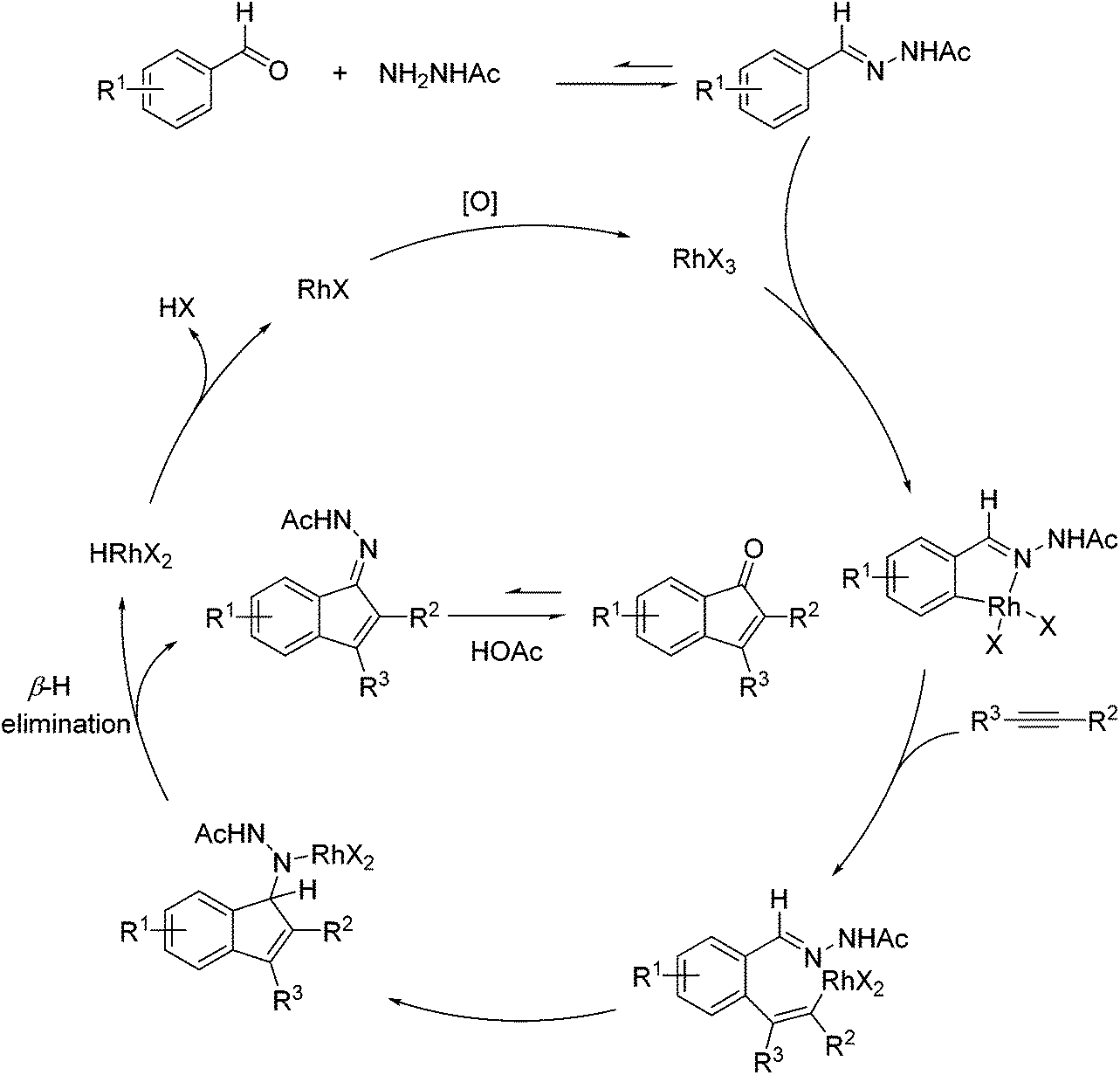
Inspired by Jun's work, Cheng and co-workers developed Rh(III) [Cp*Rh(MeCN)3](SbF6)2 (Cp* = pentamethylcyclopentadienyl) catalyzed direct annulations of aldehydes with alkynes to produce indenones in 2013 (Scheme 50).65 In this reaction a temporary directing group, namely benzaldehyde acetylhydrazone, was proposed to be formed in situ facilitating the efficient ortho-C–H bond activation (Scheme 51). As such, the directing group is traceless. In addition, functional group tolerance and substrate scope were good as depicted by the selected examples (Scheme 50).
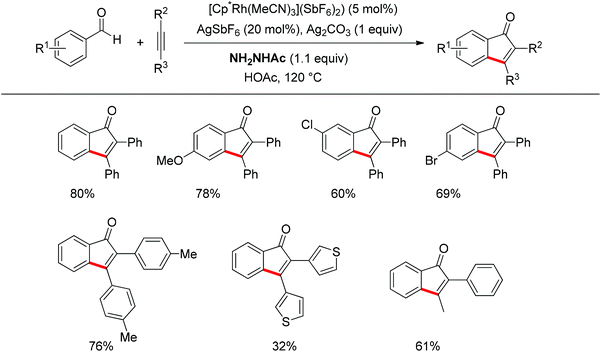 |
| | Scheme 50 Rh-catalyzed direct annulations of aldehydes with alkynes directed by a temporary acetylhydrazone directing group generated in situ. | |
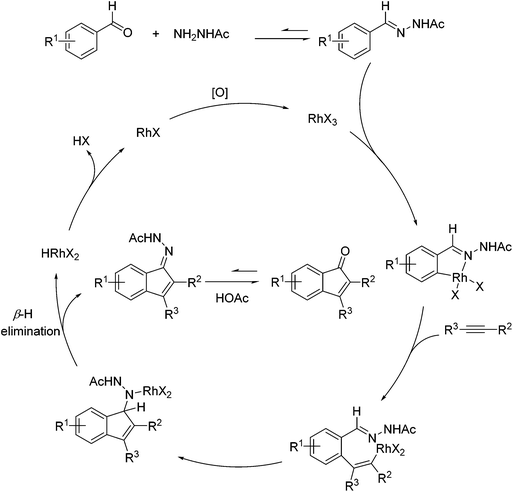 |
| | Scheme 51 A proposed catalytic cycle for Rh-catalyzed direct annulations of aldehydes with alkynes assisted by a temporary acetylhydrazone directing group generated in situ. | |
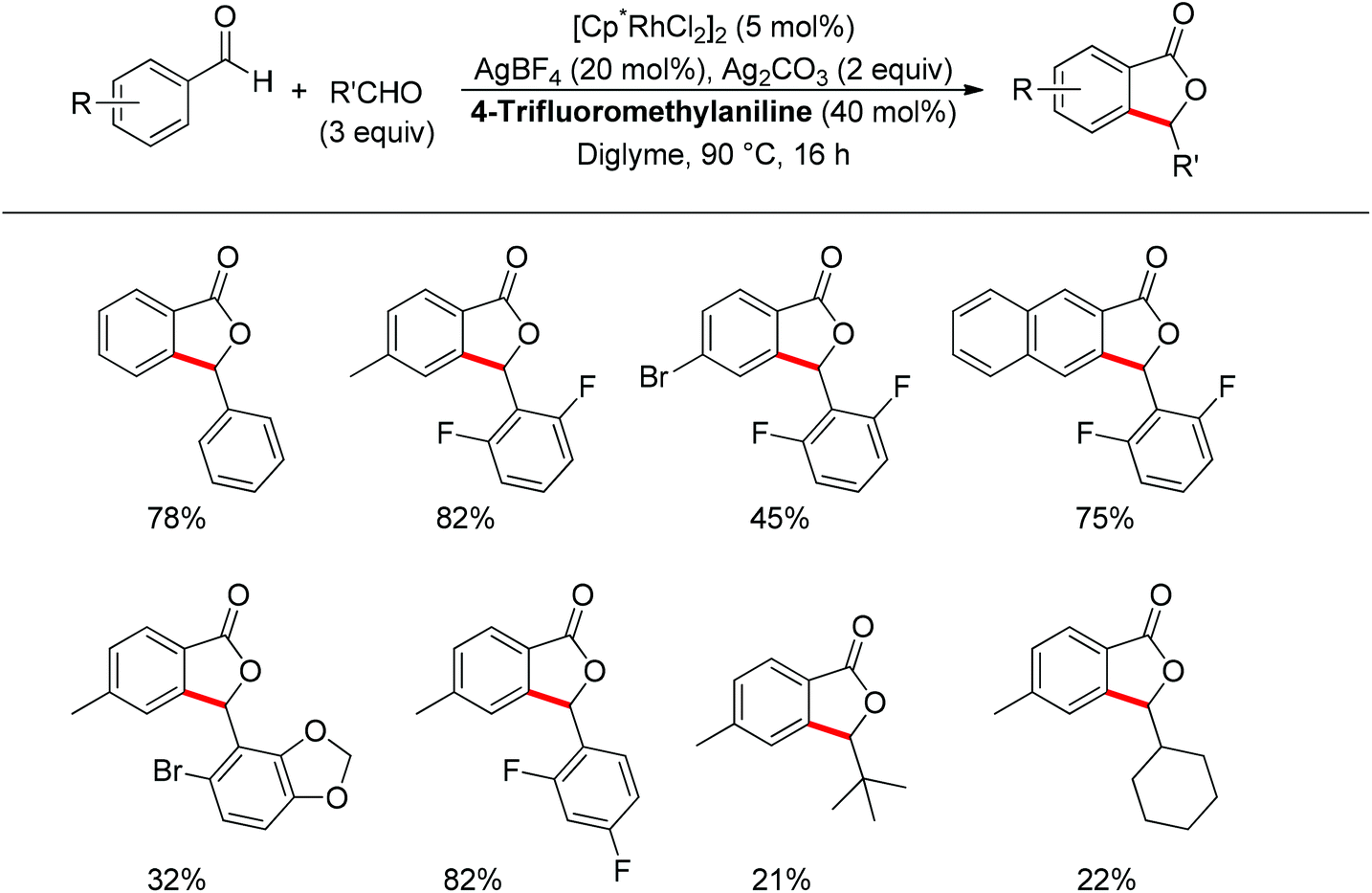
Almost at the same time, the Seayad group disclosed their work on the oxidative coupling of aldehydes for the synthesis of phthalide catalyzed cooperatively by a Rh(III) catalyst [Cp*RhCl2]2 and 4-trifluoromethylaniline (Scheme 52).66 Amazingly, only a catalytic amount of 4-trifluoromethylaniline was required to form the imine directing group in situ, leading to an effective ortho-C–H activation. The transformation was not only suitable for the homocoupling of aldehydes but also good for heterocoupling. Interestingly, aliphatic aldehydes were also applicable for such oxidative coupling. Importantly, an asymmetric version of this reaction could be foreseen as an attractive type of C–H functionalization.
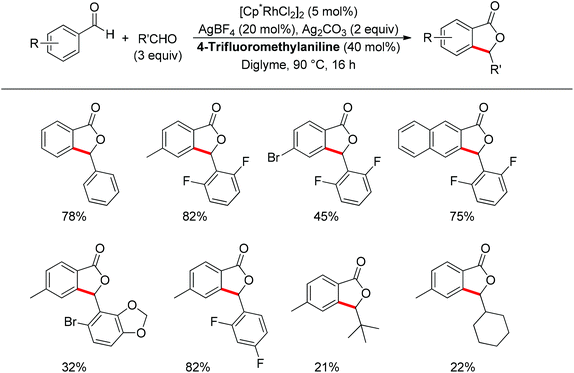 |
| | Scheme 52 Rh(III) and amine cooperatively catalyzed oxidative coupling of aldehydes to phthalides. | |
2.2. Carbon–heteroatom bond formation
2.2.1. C–N bond formation.
During the past few years, significant progress has been made in directing group assisted nitrogenation and hydroxylation of C–H bonds catalyzed by ruthenium, and this research has been reviewed by Ackermann and co-workers.67 Here and in the next section (section 2.2.2), we wish to emphasize the recent progress in Rh or Pd-catalyzed C–H bonds nitrogenation and alkoxylation as well as acyloxyation of arenes using nitrogen-based directing groups.
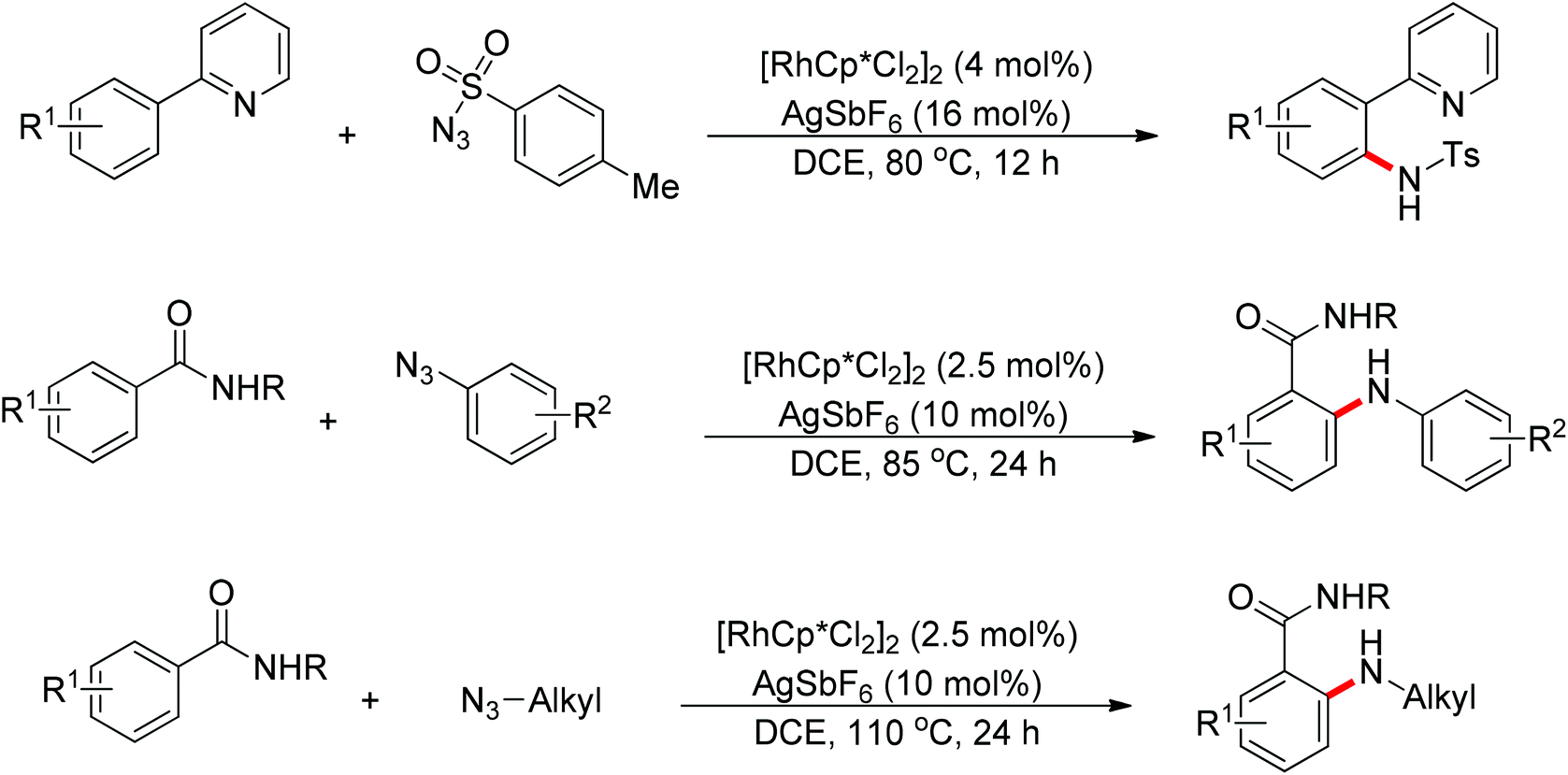
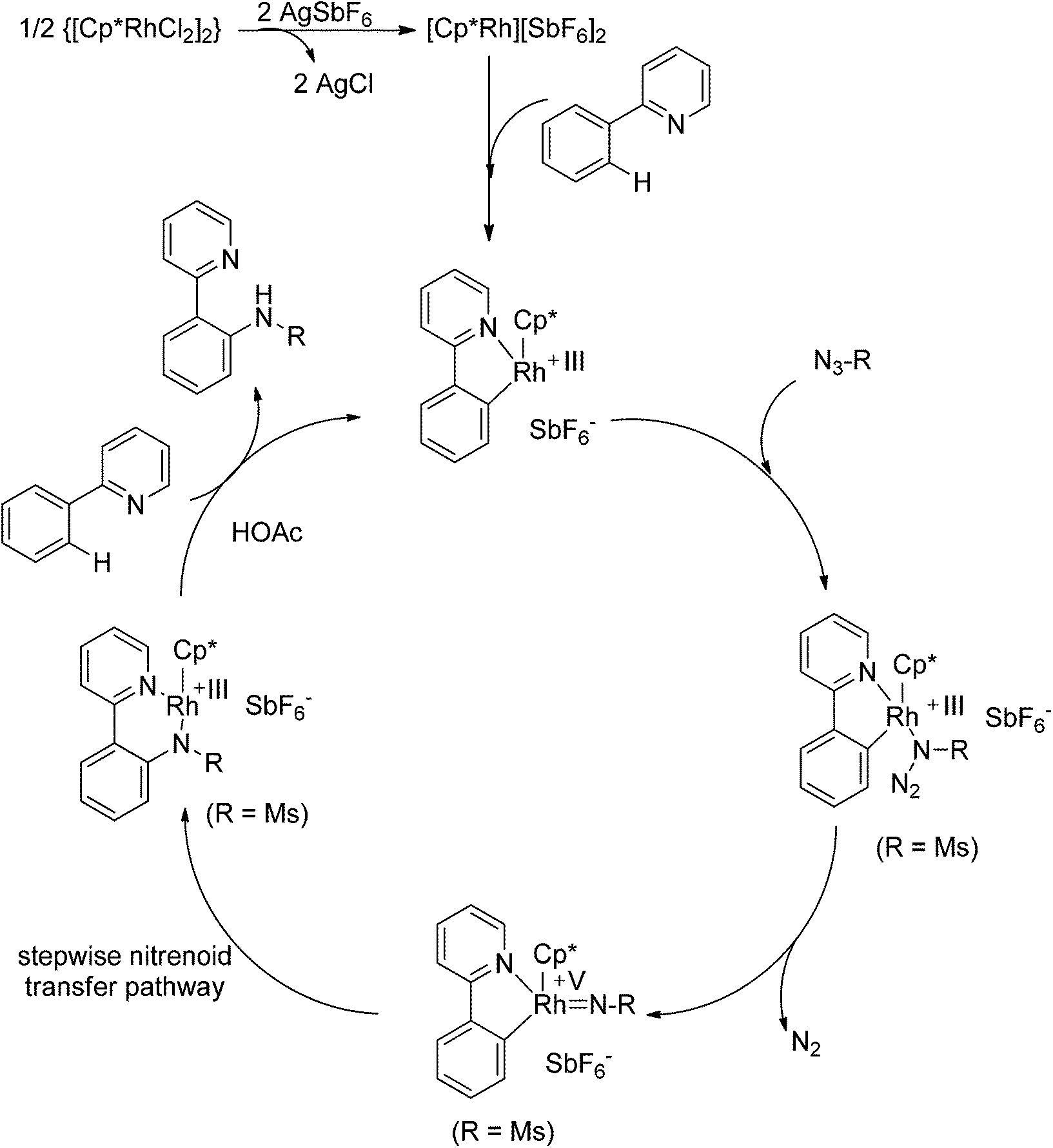
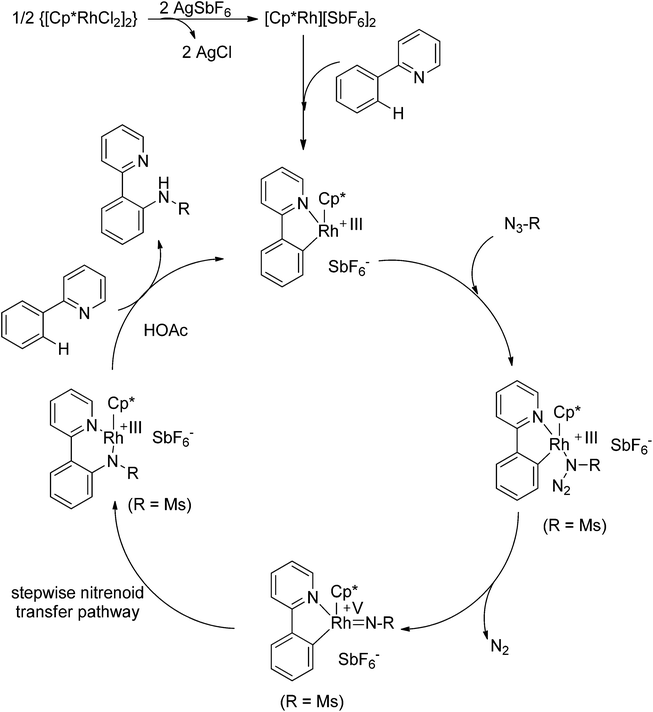
Recently, Chang and co-workers made a series of achievements in the Rh(III)-catalyzed C–H bond nitrogenation of arenes assisted by a nitrogen-based directing group. Using aryl azides as the amino source, they realized the ortho-amidation of 2-phenylpyridines with sulfonyl azides,68 the ortho-amination of benzamides and ketoximes with aryl azides,69 and the ortho-amination of benzamides with alkyl azides70 (Scheme 53). Pyridines and amides as well as the oxime group served as directing groups to control the regioselectivity of the C–H activation. These reactions exhibited excellent functional group tolerance with respect to both coupling partners. Moreover, these reactions proceeded in the absence of any external oxidants and released only N2 as the by-product, thus providing an environmentally benign process. Detailed experiments and DFT calculations were carried out to probe the mechanism of Rh(III)-catalyzed ortho-amidation of 2-phenylpyridines with sulfonyl azides, and it was proposed that the reaction proceeds via a stepwise pathway involving a key Rh(V)-nitrenoid intermediate that subsequently undergoes amido insertion (Scheme 54).71
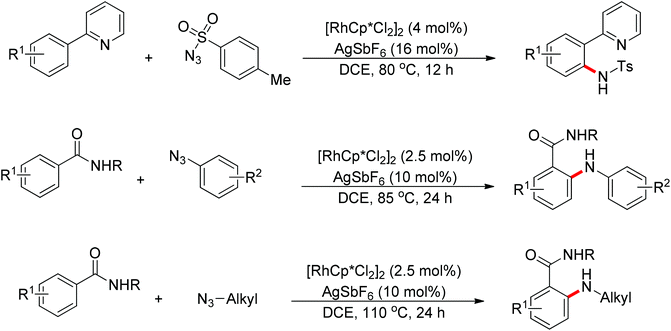 |
| | Scheme 53
ortho-Nitrogenation of pyridines and benzamides with aryl azides. | |
 |
| | Scheme 54 Proposed mechanism for the Rh-catalyzed direct C–H amination. | |
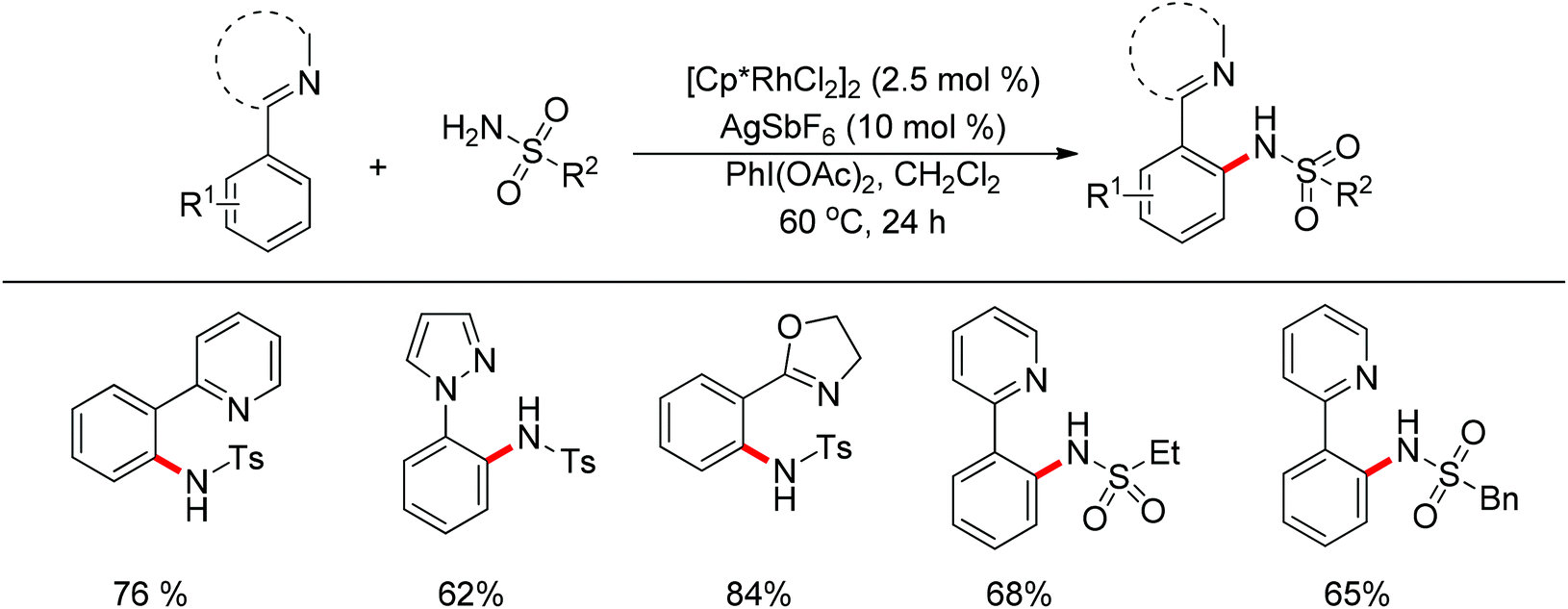
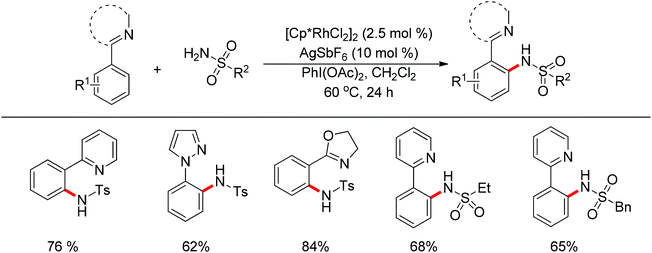
Su’s group successfully achieved a Rh(III)-catalyzed ortho sp2 C–H bond amidation directed by nitrogen-based heterocycles with sulfonamides for the first time (Scheme 55).72 This efficient protocol provided a complement to the existing amidation methods. According to the experimental results, the reaction pathway was proposed to proceed through an initial Rh-mediated C–H cleavage via a CMD mechanism to form a Rh–C bond, and the subsequent process may involve a nitrene and a Rh(V) intermediate to afford the amidated products. This method enabled a series of arenes to be amidated in good to excellent yields with both aromatic and aliphatic sulfonamides and with high functional group tolerance and selectivity.
 |
| | Scheme 55 Rh(III)-catalyzed ortho-amidation of 2-phenylpyridines with amides. | |
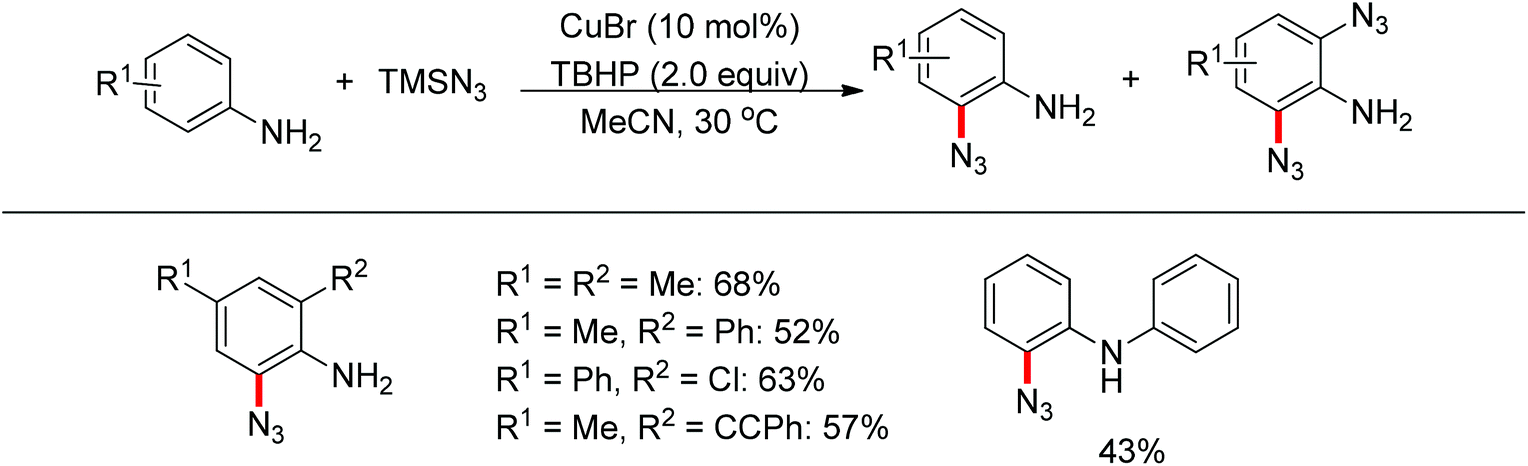
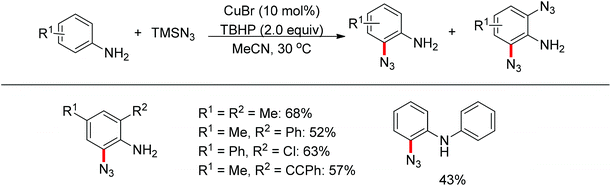
Jiao and co-workers reported a novel copper-catalyzed ortho-azidation of anilines in which the free amine was used as the directing group (Scheme 56).73 This method enabled the ortho-azidation of primary and secondary amines under mild conditions, but not tertiary amines. Preliminary mechanistic studies revealed that the reaction may involve a radical process, which is different from the regularly directing-group governed ortho-C–H functionalizations.
 |
| | Scheme 56 Copper-catalyzed ortho-azidation of anilines. | |
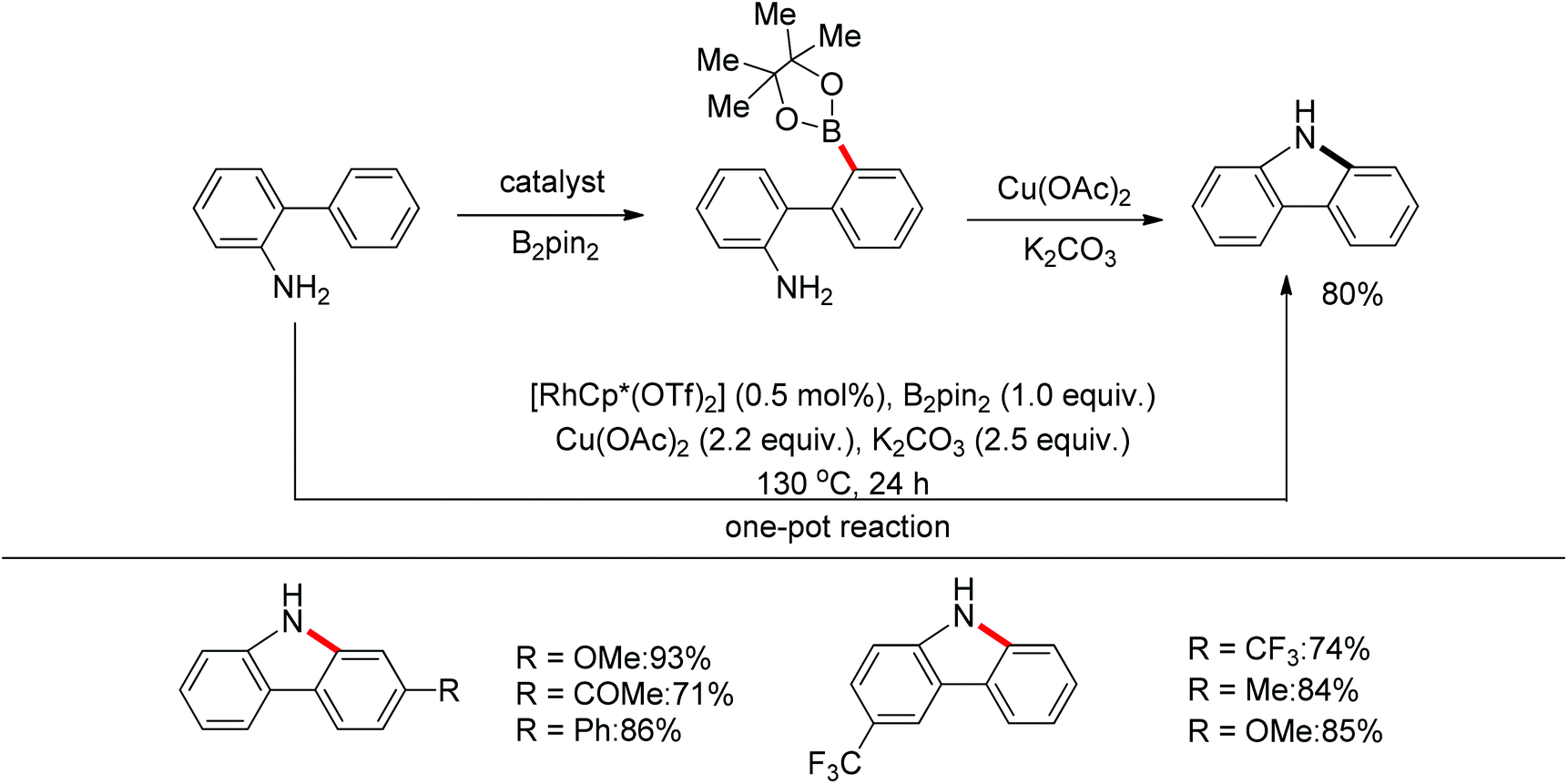
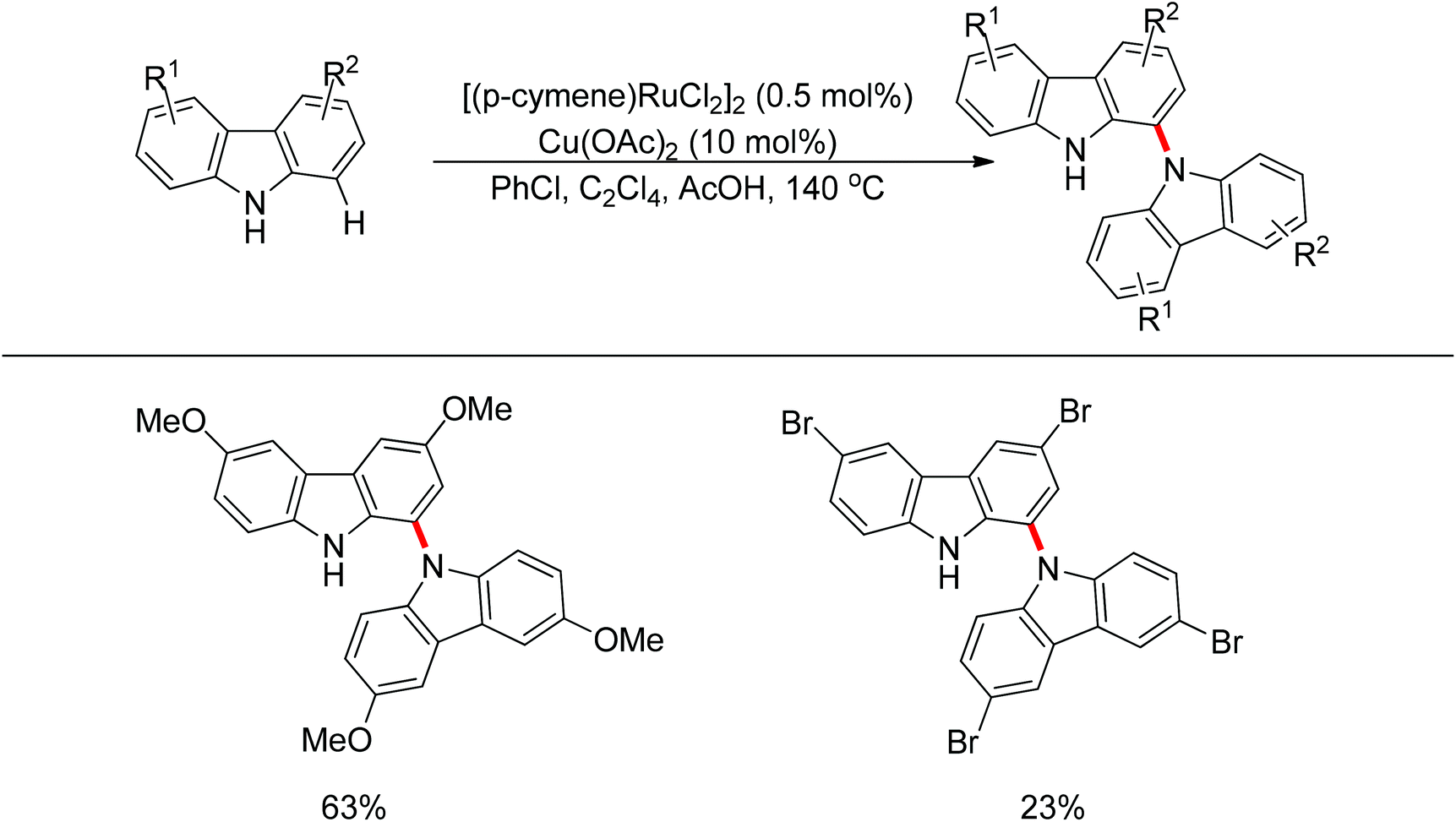
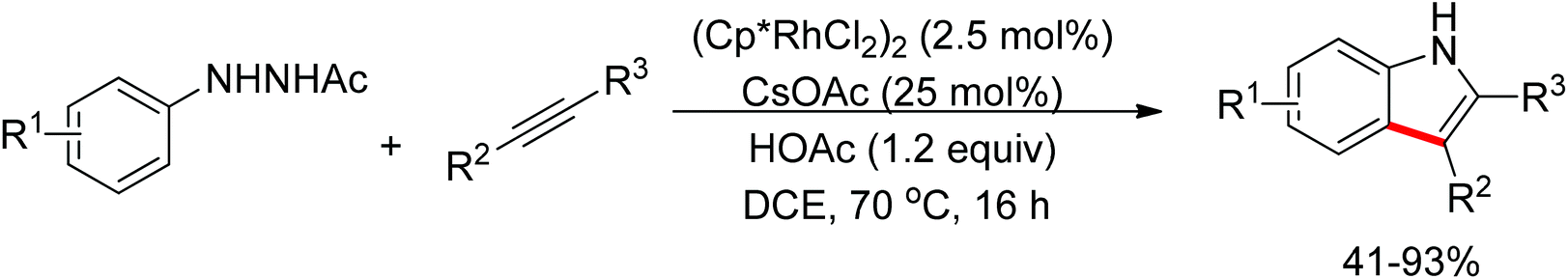
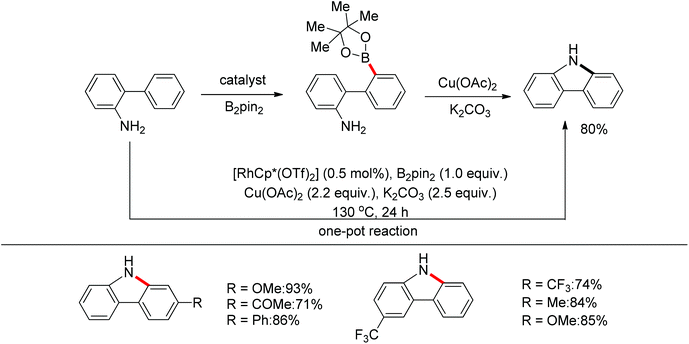
Yan and co-workers developed a Rh(III)-catalyzed method for the intramolecular amino-directed C–H activation and amination sequence to construct carbazole products (Scheme 57).74 The experiments indicated that this transformation occurred through a one-pot tandem process. Firstly, the rhodium catalyst initiated the reaction of the free 2-aminobiphenyl with bis(pinacolato)diboron (B2pin2) with a free-amino group as the directing group to form a C–B bond. Then copper mediated the formation of an intramolecular C–N bond. In this catalytic system, Cu(OAc)2 was proposed to serve as both an oxidant to oxidize Rh(I) to Rh(III), and a catalyst for C–N bond formation. However, the possibility cannot be excluded that the reaction involves amine-directed C–H activation and subsequent intramolecular amination, in which B2pin2 serves as a Lewis acid for the acid–base adduct to control the free amine concentration.
 |
| | Scheme 57 One-pot synthesis of N–H carbazoles. | |
2.2.2. C–O bond formation.
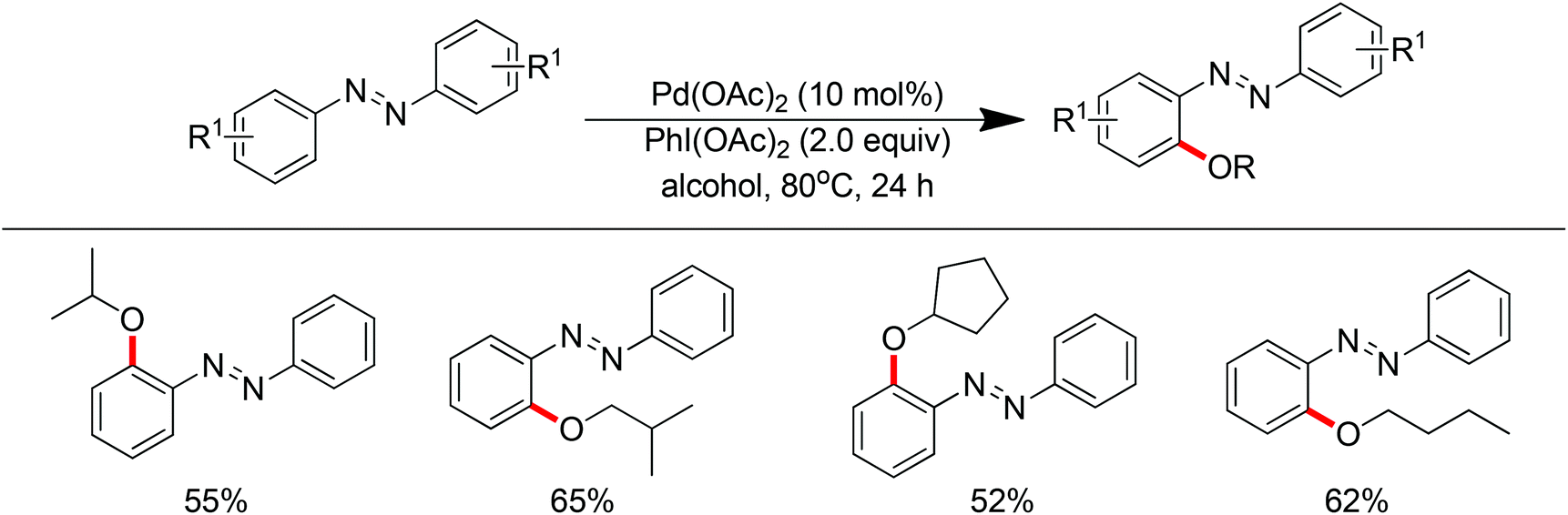
In 2012, Sun and co-workers developed the Pd(OAc)2-catalyzed ortho-alkoxylation of arylnitriles with alcohols (Scheme 58).75 Cyano was used as the directing group for the first time. It was proposed that the cyano group coordinated with palladium using its π-electrons between carbon and nitrogen, which is different from the σ-coordination fashion observed for most other directing groups. Inexpensive Na2S2O8 proved to be the most efficient oxidant for this transformation. The established conditions exhibited good functional group tolerance with respect to aromatic nitriles, but only primary methanol and ethanol could be used as the alkoxylating agents. In 2013, the same group described the Pd(II)-catalyzed ortho-alkoxylation of arenes directed by the azo group with alcohols in the presence of PhI(OAc)2 as the oxidant (Scheme 59).75 In addition to primary alcohols, this protocol was applicable to secondary alcohols such as isopropanol, isobutanol and cyclopentanol.
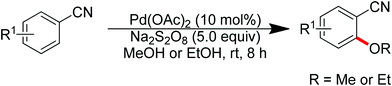 |
| | Scheme 58
ortho-Alkoxylation of arylnitriles with alcohols. | |
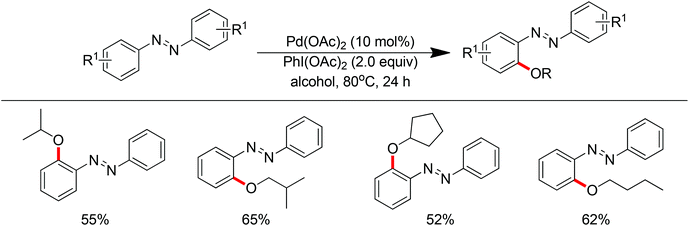 |
| | Scheme 59
ortho-Alkoxylation of aromatic azo compounds with alcohols. | |
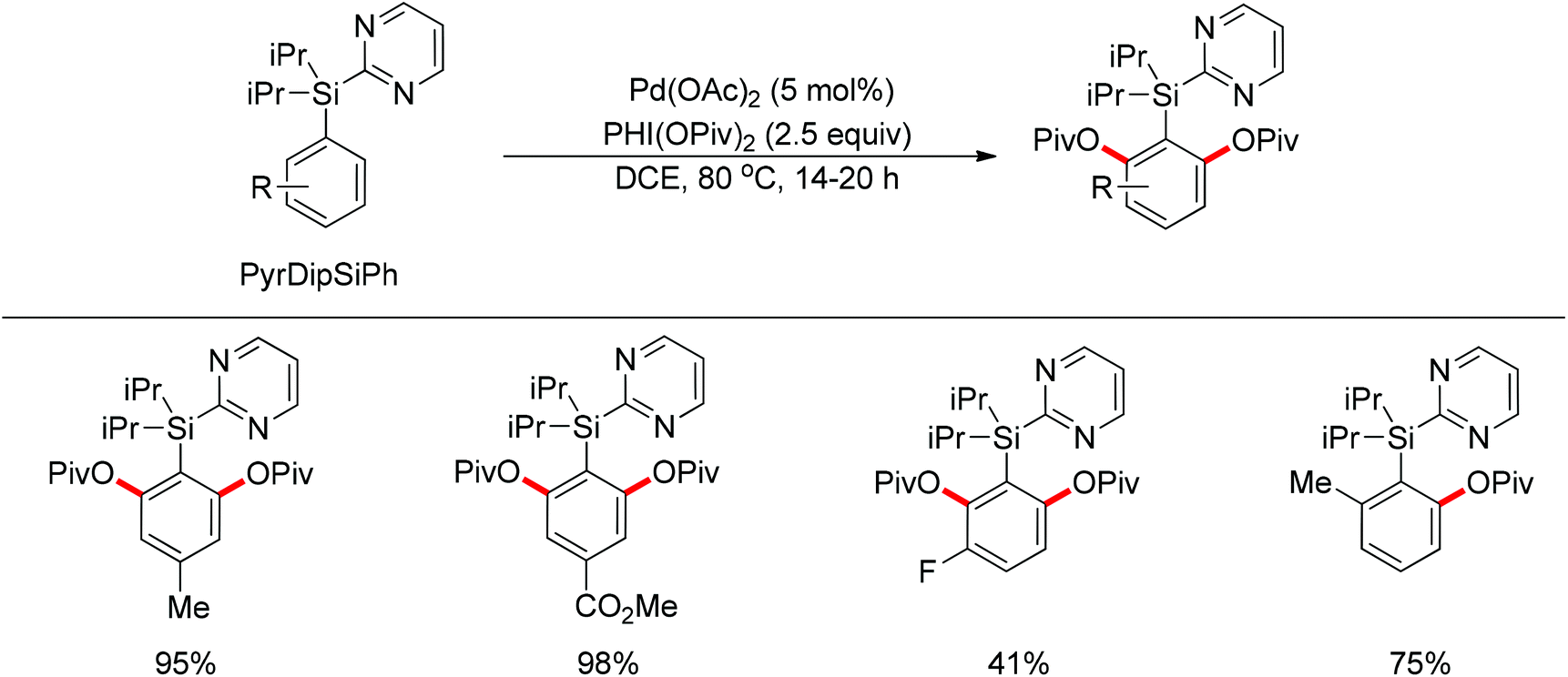
By using 2-pyridyldiisopropylsilyl (PyDipSi) as the directing group, Gevorgyan and co-workers reported the Pd(II)-catalyzed mono C–H acyloxylation and pivaloxylation of arenes.76 In 2012, they further developed the pyrimidyl-based 2-pyrimidyldiisopropylsilyl (PyrDipSi) as the directing group for the Pd(II)-catalyzed double C–H pivaloxylation of arenes (Scheme 60).77 This reaction allowed a broad range of arenes bearing both electron-donating and -withdrawing groups, producing the desired products in good yields. The established methods could also be applied to the monopivaloxylation reaction of ortho-substituted PyrDipSi arenes. Noteworthily, it was shown that the PyrDipSi group can be easily installed into arene molecules as well as efficiently removed or modified into other valuable functional groups.
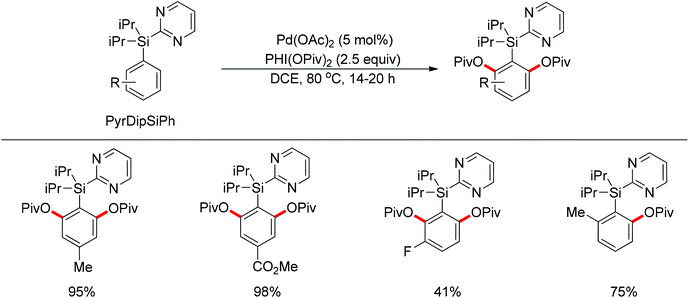 |
| | Scheme 60 Pd(II)-catalyzed double C–H pivaloxylation of arenes. | |
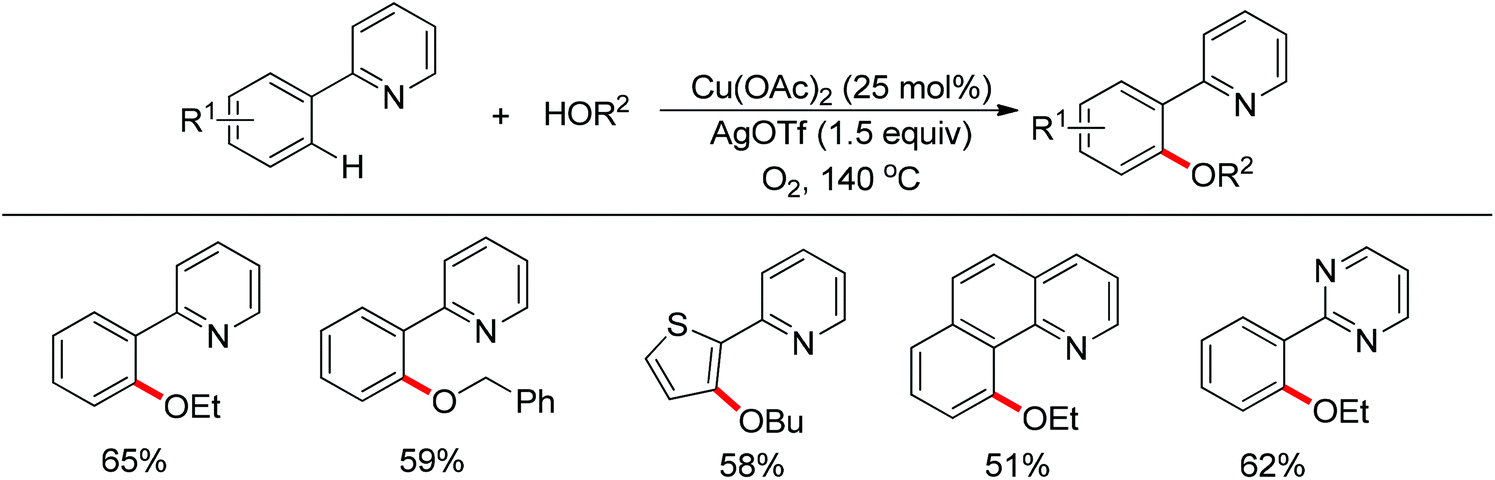
In 2013 a bimetallic Cu/Ag catalyst system was developed by Goossen and co-workers for the dehydrogenative cross-coupling of arenes bearing a pyridine group with alcohols (Scheme 61).78 This protocol provided an efficient tool for the construction of C–O bonds. According to the authors, copper was responsible for the chelation-assisted C–H activation and the silver species served as the oxidant for the oxidation of alcohols to form the alkoxy radical which transferred to the copper complex intermediate. Then reductive elimination of the resulting CuIII intermediate furnished the alkoxyarene product in moderate yields. The scope of alcohols was limited to alkyl and benzylic alcohols. In contrast, phenols were easily oxidized to form self-coupling products under the standard conditions.
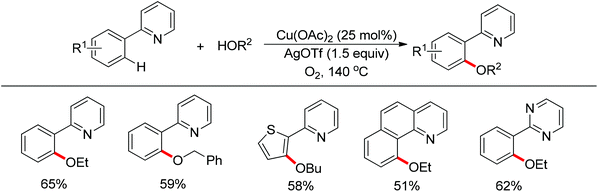 |
| | Scheme 61 Copper-catalyzed dehydrogenative coupling of arenes with alcohols. | |
2.2.4. C–P bond formation.
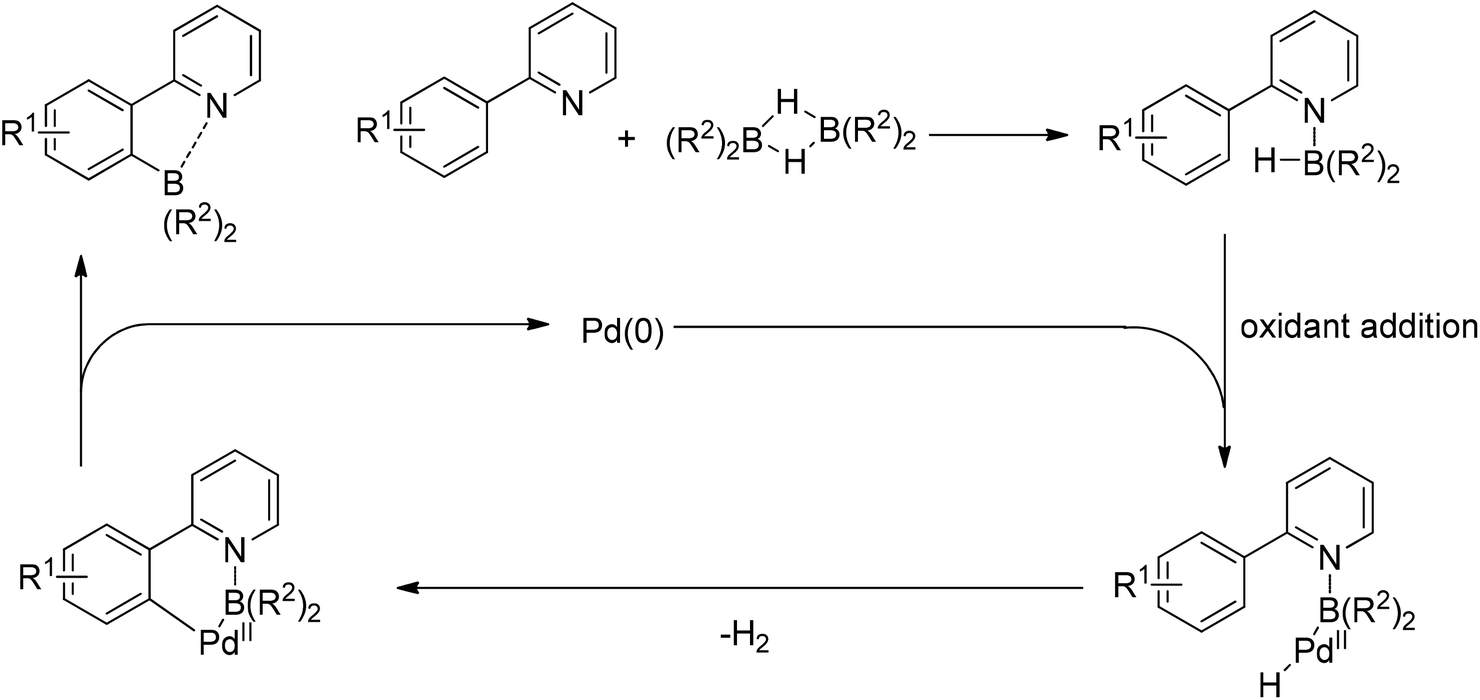
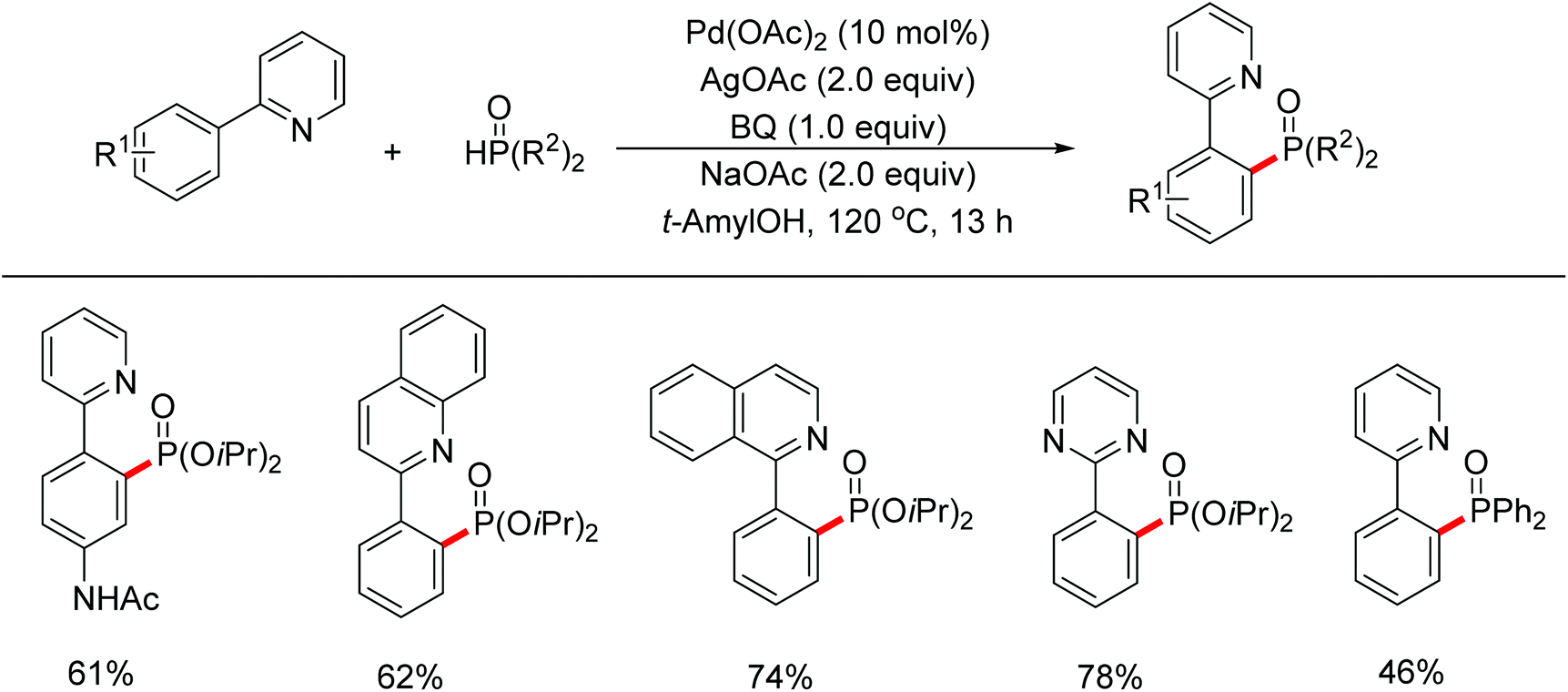
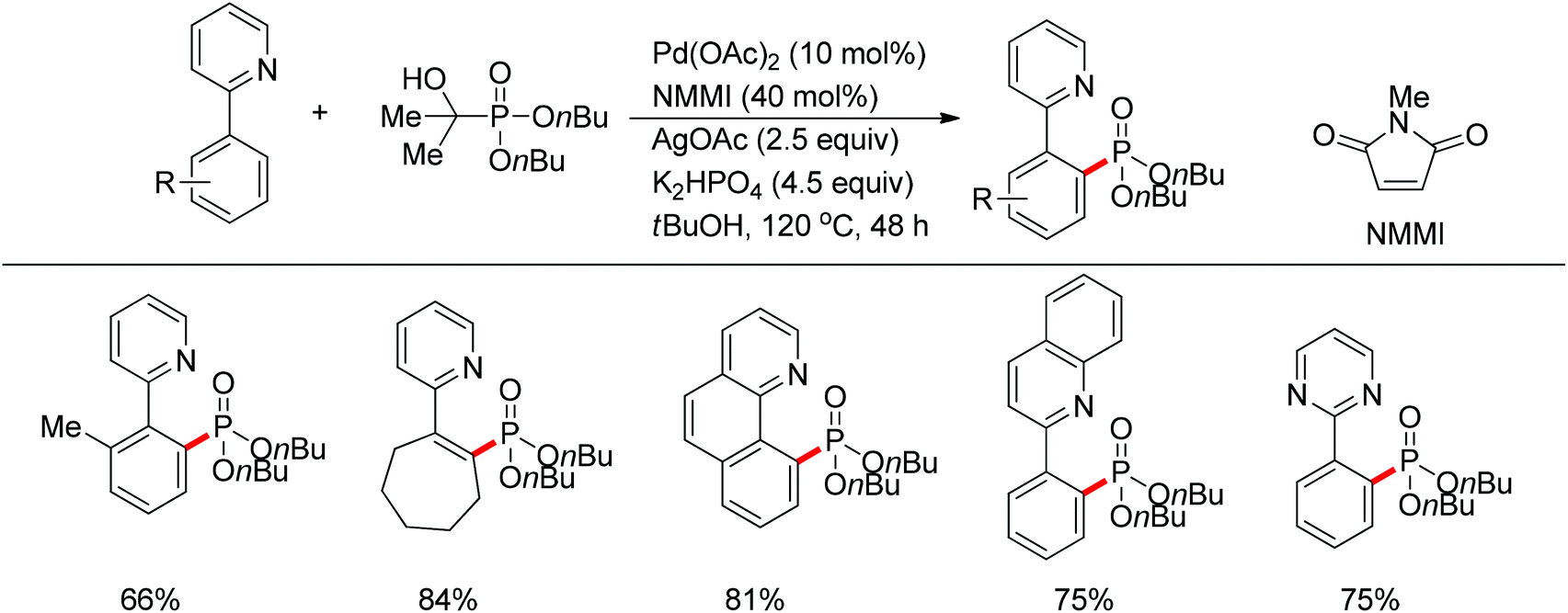
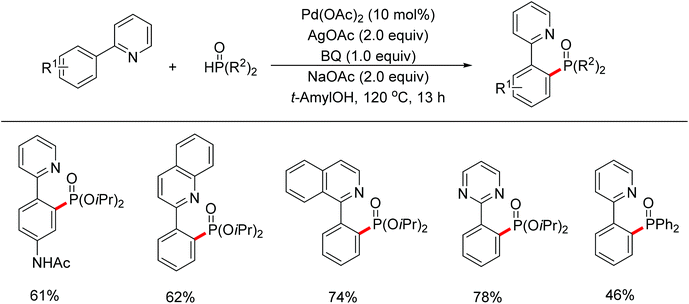
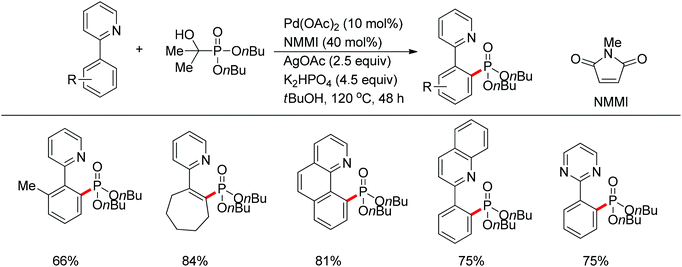
In 2013, Yu and co-workers reported the Pd(II)-catalyzed phosphonation of 2-arylpyridines with H-phosphonate or triarylphosphine oxides (Scheme 67).85 The use of BQ, which might facilitate the reduction elimination process,86 was found to be essential for the formation of the products. Both pyridine and quinoline could act as the directing group to assist the activation of ortho C–H bonds. Slowly adding H-phosphonate was required to avoid deactivation of the catalyst. Meanwhile, Murakami and co-workers reported the Pd(II)-catalyzed phosphonation of 2-arylpyridines using α-hydroxyalkylphosphonate as the phosphonating agent (Scheme 68).87 It was proposed that the α-hydroxyalkylphosphonate could gradually generate H-phosphonate which served as the masked phosphonating agent, while protecting the catalyst from deactivation at the same time. Various substituted 2-arylpyridines, as well as quinoline and pyrimidine, smoothly underwent the phosphonation reaction in good yields with high selectivity.
 |
| | Scheme 67 Pd(II)-catalyzed phosphonation of 2-arylpyridines with H-phosphonate and triarylphosphine oxides. | |
 |
| | Scheme 68 Pd(II)-catalyzed phosphonation of 2-arylpyridine with α-hydroxyalkylphosphonate. | |
2.2.5. C–X (X = I, Br, Cl and F) bond formation.
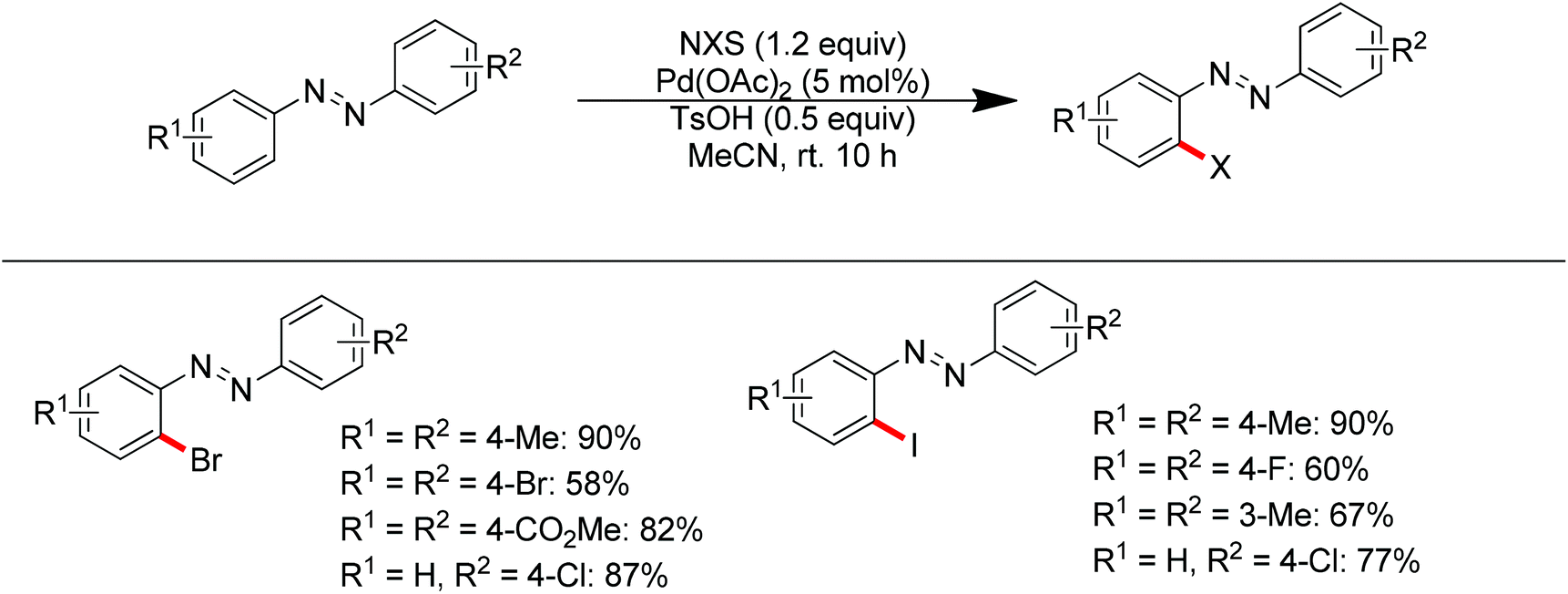
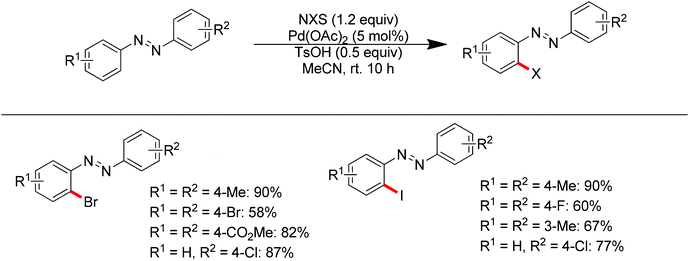
Aryl halides are the most widely used starting materials for synthetic elaboration especially in transition metal-catalyzed cross-coupling reactions. In the past decade, several palladium-catalyzed direct halogenations of C–H bonds using nitrogen-based directing groups have appeared, including iodination,88 chlorination and bromination.13c,89 In 2013, Tian and co-workers developed the palladium-catalyzed ortho-bromination and iodination of aromatic azo compounds under mild conditions (Scheme 69).90 The azo group was used as the directing group to assist the activation of the ortho C–H bond. The use of p-toluenesulfonic acid as the additive, which resulted in the formation of Pd(OTs)2 in the presence of Pd(OAc)2, was critical for this transformation. When a strong electron-donating group such as methoxy was introduced into the aromatic ring of an aromatic azo compound, the regioselectivity was switched and the directing ability of the methoxy group completely overrode the azo group (Scheme 70).
 |
| | Scheme 69 Pd(II)-catalyzed ortho-bromination and iodination of aromatic azo compounds. | |
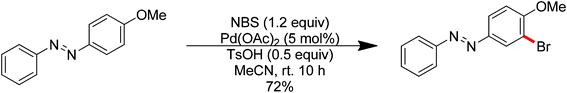 |
| | Scheme 70 Pd(II)-catalyzed ortho-bromination directed by the methoxy group. | |
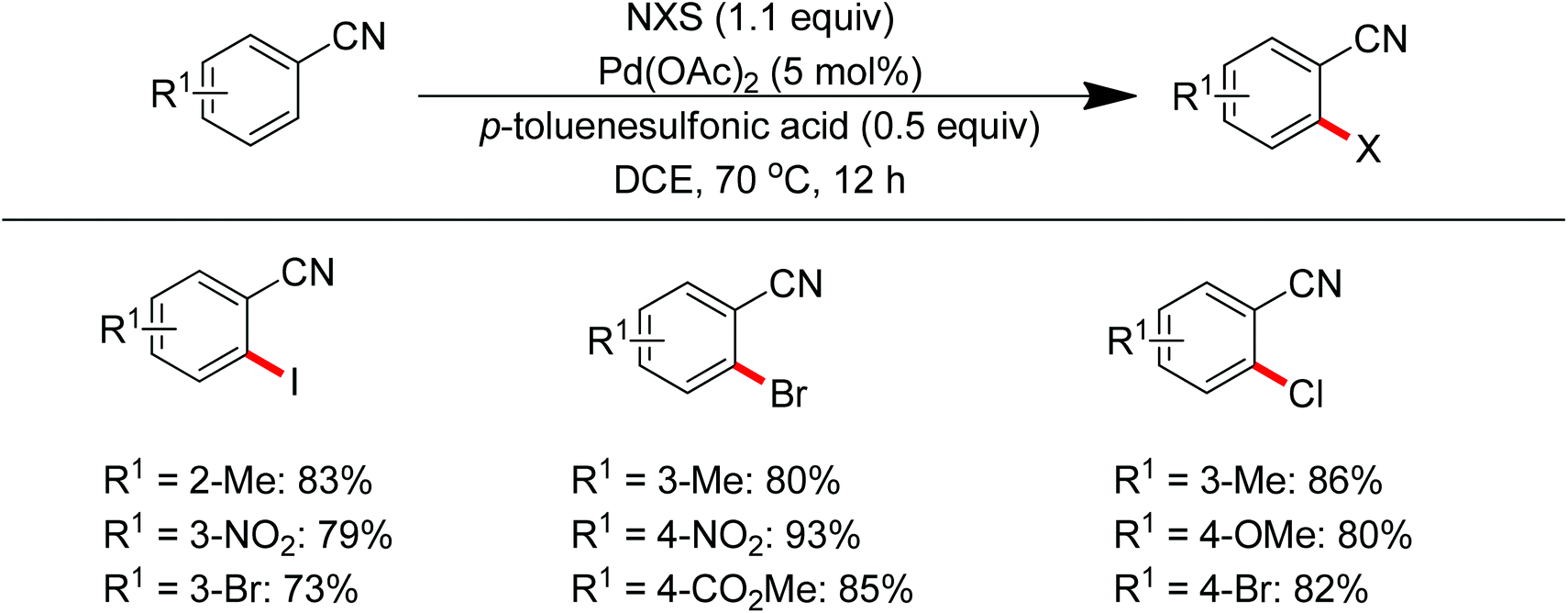
Sun and co-workers described the palladium-catalyzed ortho-halogenations of arylnitriles with NXS (X = I, Br, Cl) employing cyano as the directing group (Scheme 71).91 The established method could be applicable to iodination, bromination and chlorination of a wide range of arylnitriles with either electron-withdrawing or electron-donating groups. This protocol provided an efficient approach to ortho-halogenated arylnitriles which could not be obtained via the electrophilic aromatic substitution of arylnitriles.
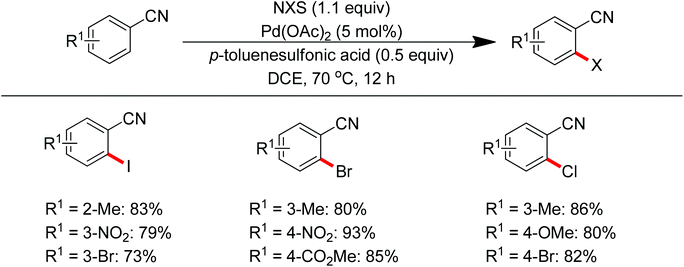 |
| | Scheme 71 Palladium-catalyzed ortho-halogenations of arylnitriles with NXS. | |
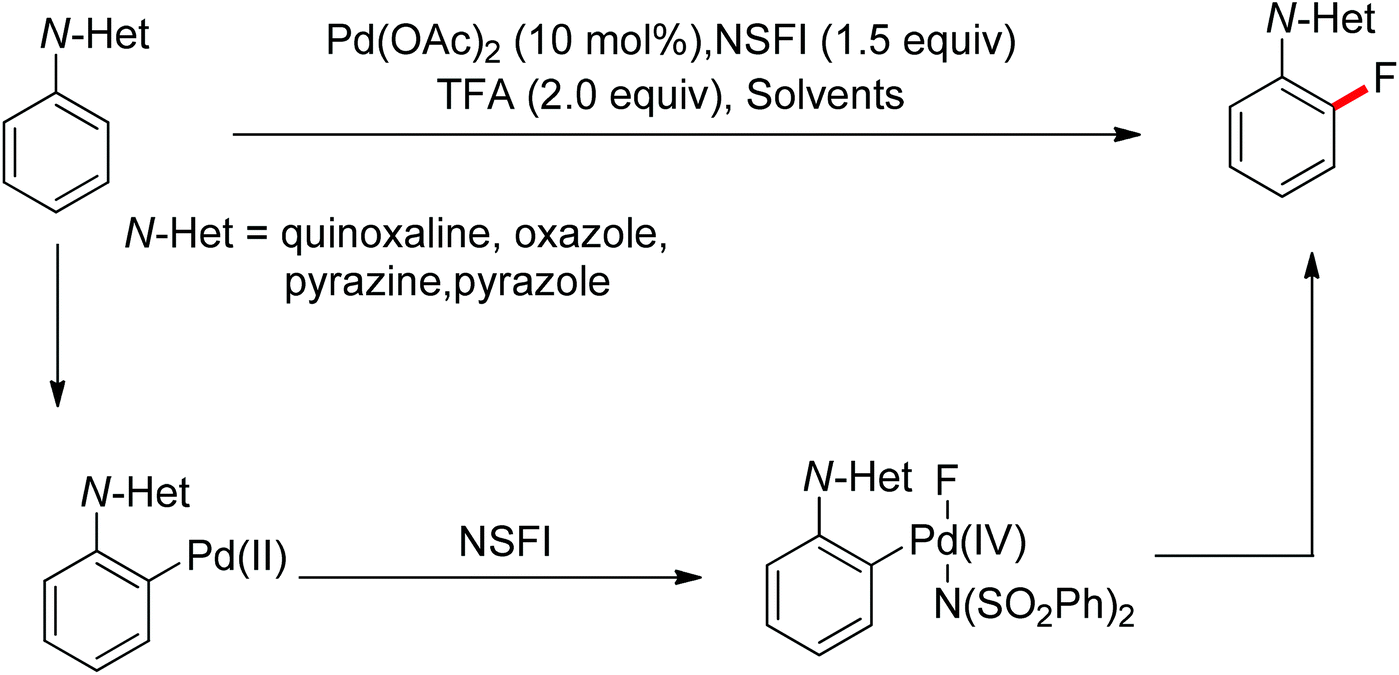
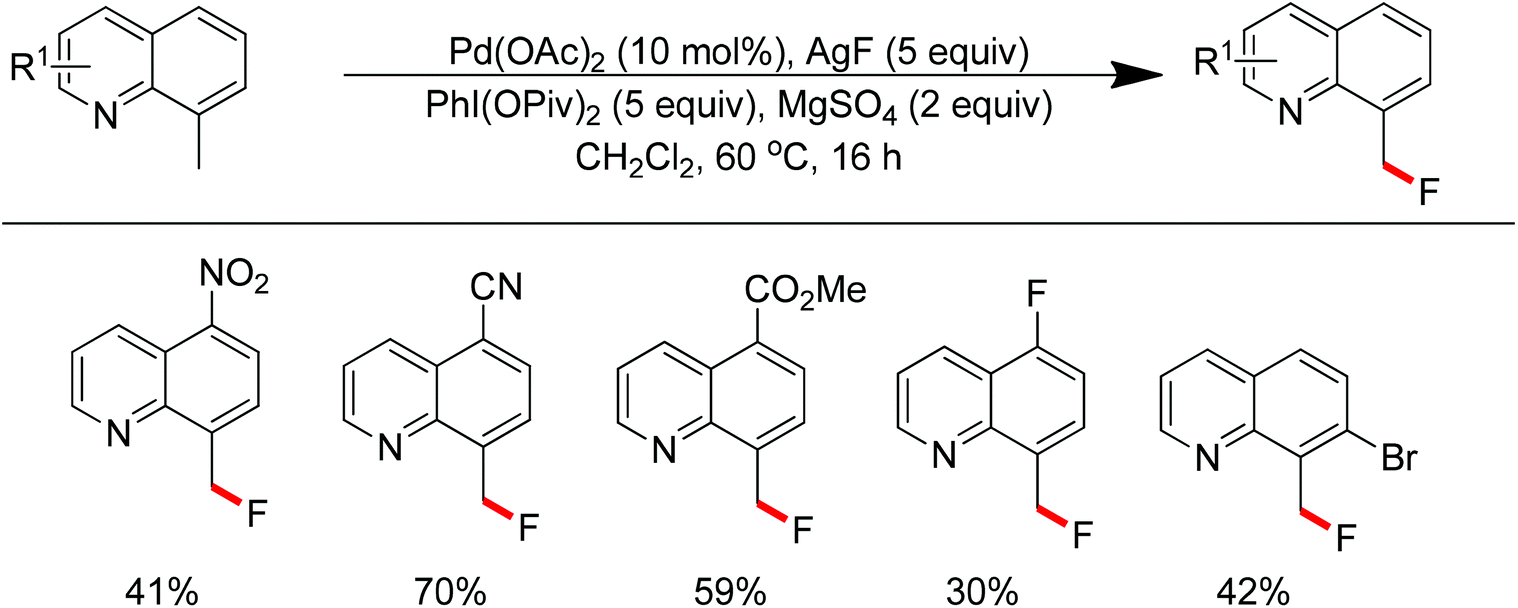
The conversion of C–H bonds to C–F bonds is extremely desirable since the introduction of fluorine has a profound effect on the biological activity, metabolism, solubility, and hydrophobicity of organic substances. The pioneering studies by Sanford and Yu realized the Pd(II)-catalyzed fluorination of C–H bonds with electrophilic fluorinating reagents using pyridine92 and benzoic amides as the directing group.93 In 2013, Xu and co-workers described the Pd(II)-catalyzed ortho-fluorination directed by various aryl-N-heterocyclic directing groups such as quinoxaline, pyrazole, benzo[d]oxazole and pyrazine derivatives (Scheme 72).94 These reactions were proposed to proceed via a Pd(II)/Pd(IV) process, in which the electrophilic fluorinating reagents served as both the oxidant and the source of fluorine. On the other hand, Sanford and co-workers developed Pd(II)-catalyzed fluorination of 8-methylquinoline derivatives with nucleophilic fluoride salts in combination with an hypervalent iodine as an external oxidant (Scheme 73).95 Both AgF and CsF could be used as the fluoride source. The presence of Ag(I) was critical for the success of the desired transformation.
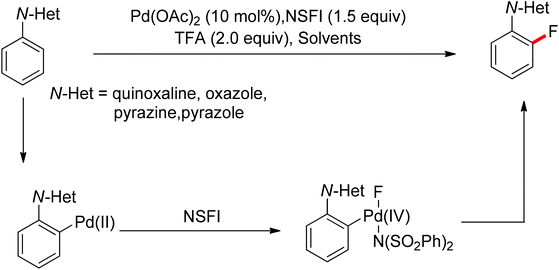 |
| | Scheme 72 Pd(II)-catalyzed ortho-fluorination of N-based arenes with electrophilic fluorinating reagents. | |
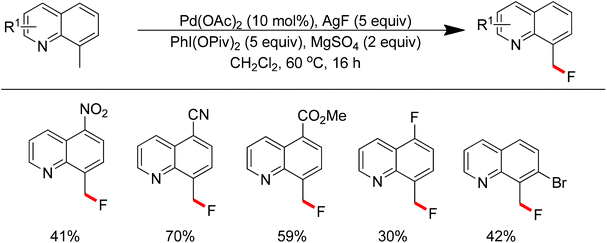 |
| | Scheme 73 Pd(II)-catalyzed fluorination of 8-methylquinoline derivatives with fluoride salts. | |
3. Directing groups govern site-selectivity beyond ortho selectivity
The application potential of C–H transformations in academia and industry largely depends on the available and predictable site-selectivities. At present, the most available proximity-driven ortho-selectivity is often achieved via five- or six-membered metallacycles. In contrast, the elusive meta- and para-selective C–H transformations may require larger metallacycles, which are often difficult to generate. Traditionally, meta-selective C–H transformations have been realized by steric control96 or ligand promotion.97 Recently, the Gaunt group also disclosed a few meta and para-selective C–H functionalizations in the presence or without copper catalyst.98 Stimulated by these studies, several new meta or para-selective transformations and new strategies appeared over the past two years.
3.1.
meta-C–H bonds functionalization controlled by electronic effects
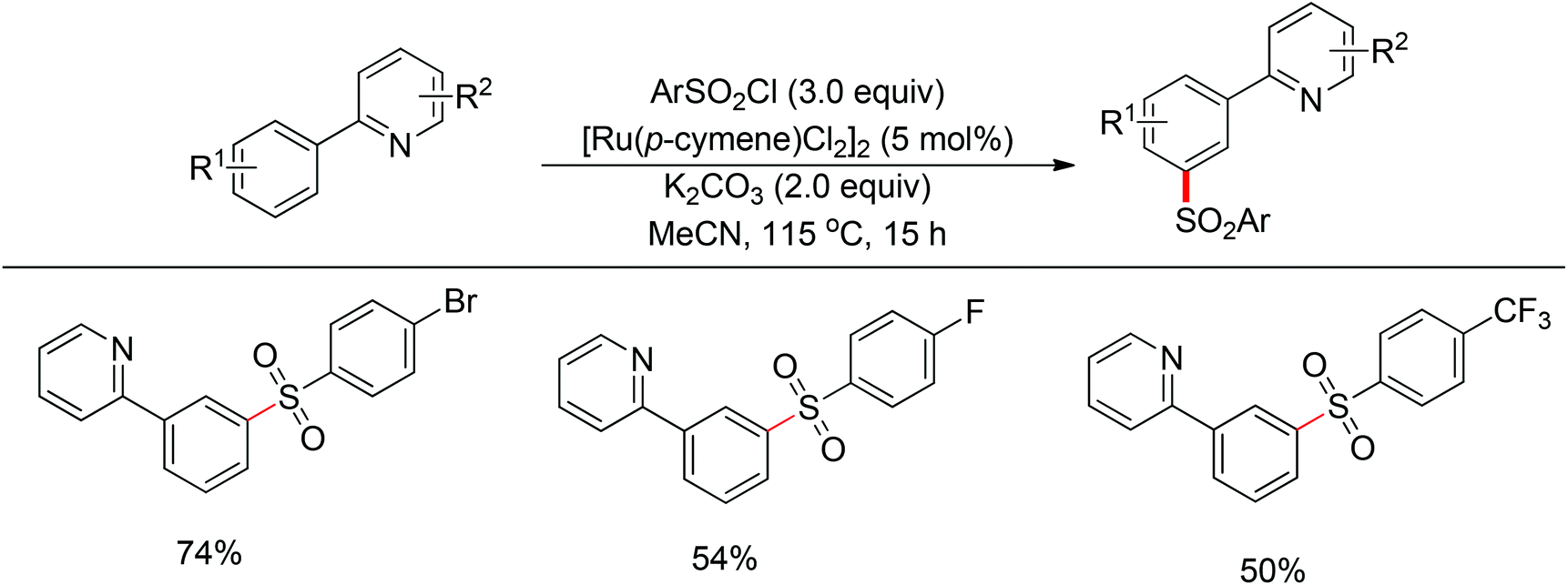
Although directing groups normally induce ortho-C–H bond functionalization, direct meta-C–H bond functionalization has also been achieved by taking advantage of directing groups. Recently, Frost and co-workers reported a strategy for a meta-selective C–H functionalization, namely the sulfonation of 2-phenylpyridines, with the assistance of a ruthenium catalyst (Scheme 74).99 The authors proposed that the chelating group initially forms a cyclometalated intermediate with the ruthenium catalyst, which then induces a strong para-directing effect. This protocol represents the first example of a catalytic σ-activation process. Preliminary kinetic studies supported this cyclometalation hypothesis and revealed that C–H bond cleavage was kinetically significant in the catalytic cycle.
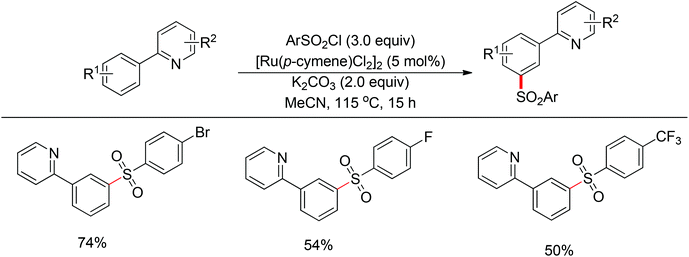 |
| | Scheme 74 Ruthenium-catalyzed meta-selective C–H sulfonation. | |
Based on Frost's work, Ackermann and co-workers reported the meta-selective C–H alkylation of N-heterocycle-containing arenes with secondary alkyl bromides (Scheme 75).100 Detailed mechanistic studies were carried out to support an initial reversible cycloruthenation which increased the reactivity of arenes to undergo electrophilic-type substitutions. The independently prepared ruthena(II)cycles also highlighted their importance as crucial intermediates. Based on these mechanistic investigations, a catalytic cycle was proposed (Scheme 76). It should be noted that only ortho-alkylation occurred with primary alkyl halides, implying that the difference of steric interaction and the electrophilicity of primary and secondary alkyl halides also played an important role in determining the selectivity of this reaction.
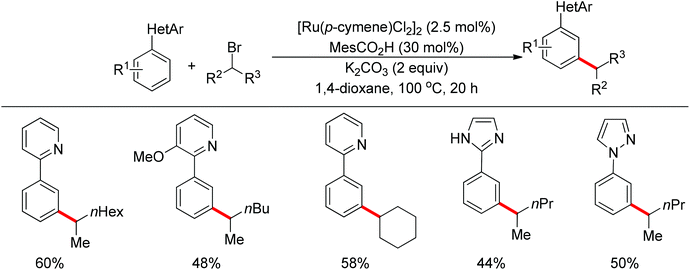 |
| | Scheme 75 Ruthenium-catalyzed meta-selective C–H alkylation. | |
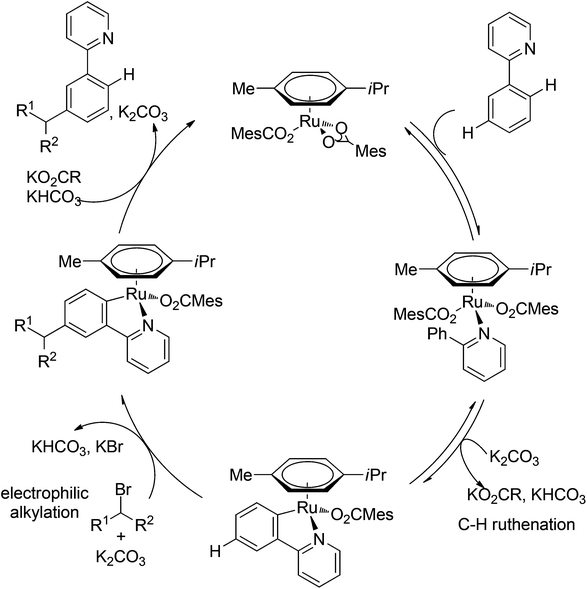 |
| | Scheme 76 Proposed catalytic cycle for the ruthenium-catalyzed meta-selective C–H alkylation. | |
3.2.
meta-selective C–H bonds functionalization with assistance of directing groups
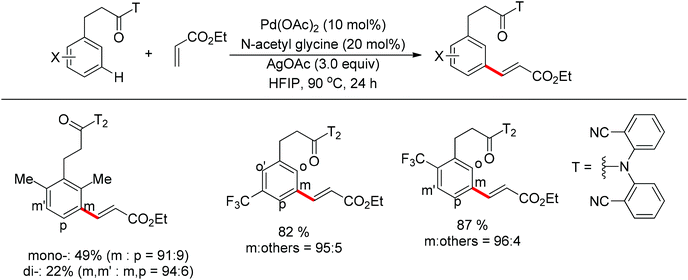
In contrast to achieving meta-selectivity by altering the electronic properties of arenes, the Yu group chose to take advantage of spatial proximity for obtaining such selectivity by employing an elaborately designed template. In 2012, Yu and co-workers reported the palladium-catalyzed direct meta-C–H olefination of toluene derivatives using a removable nitrile-based directing group (Scheme 77).101 In this seminal work, the long-range directing group was thought to coordinate the palladium catalyst via a cyclophane-type transition state (Scheme 78), which could facilitate the approach of the catalyst to induce the meta-C–H functionalization. Besides various electron-deficient α,β-unsaturated esters, ketones, phosphonates, and a number of di- and cyclic tri-substituted olefins all proved to be suitable substrates. To further show the generality of this novel meta-selective strategy, the alkenylation of hydrocinnamic acid derivatives was also studied in the same paper (Scheme 79). Similar to the benzyl ethers described above, this meta-olefination proceeded smoothly overriding any ortho-directing effects as well as the electronic and steric biases of the arene substrates.
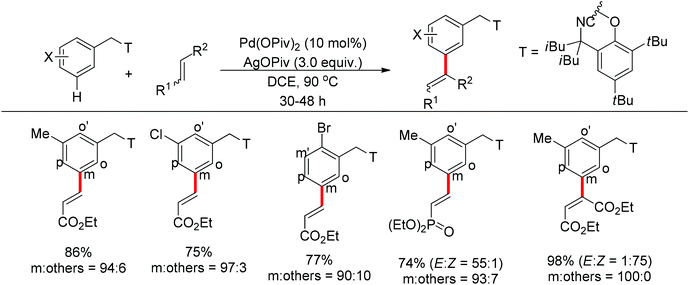 |
| | Scheme 77 Pd-catalyzed meta-selective olefination of toluene derivatives. | |
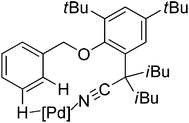 |
| | Scheme 78 Pd-catalyzed remote meta-C–H activation using a nitrile template. | |
 |
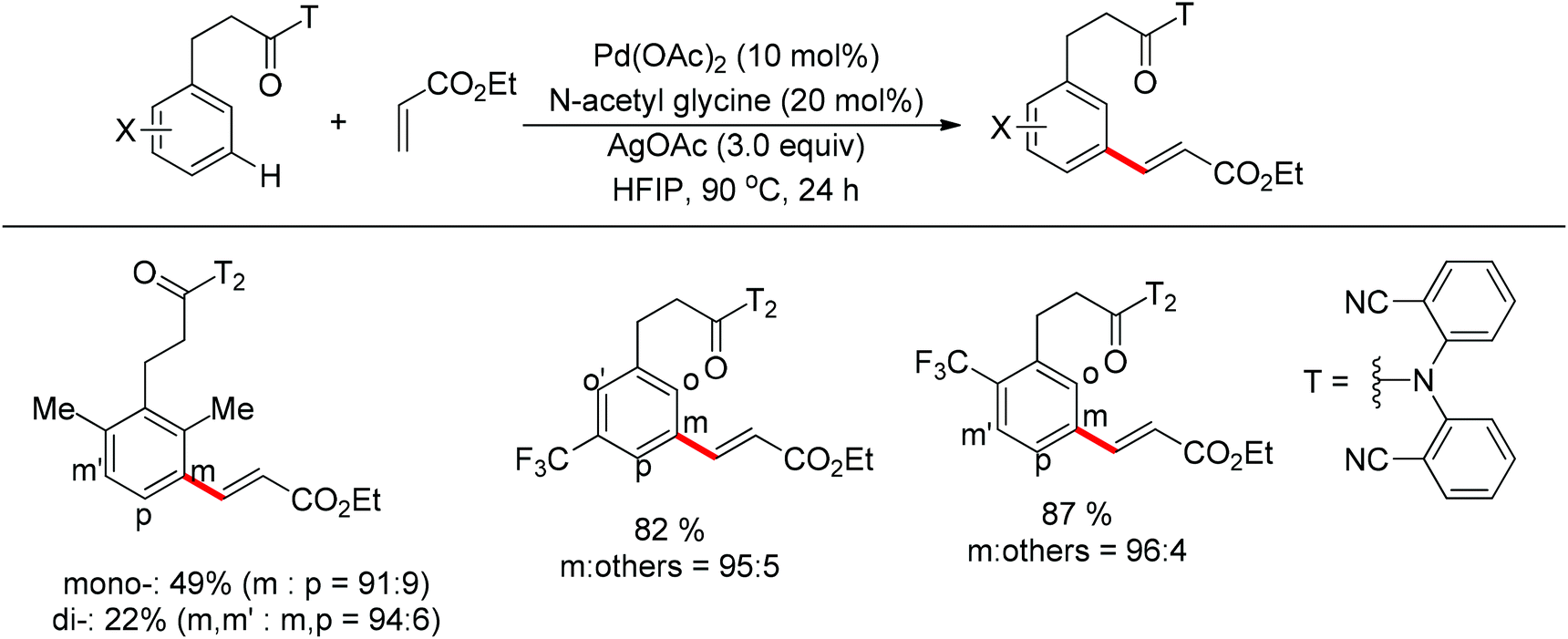
| | Scheme 79 Pd-catalyzed meta-selective olefination of hydrocinnamic acid derivatives. | |
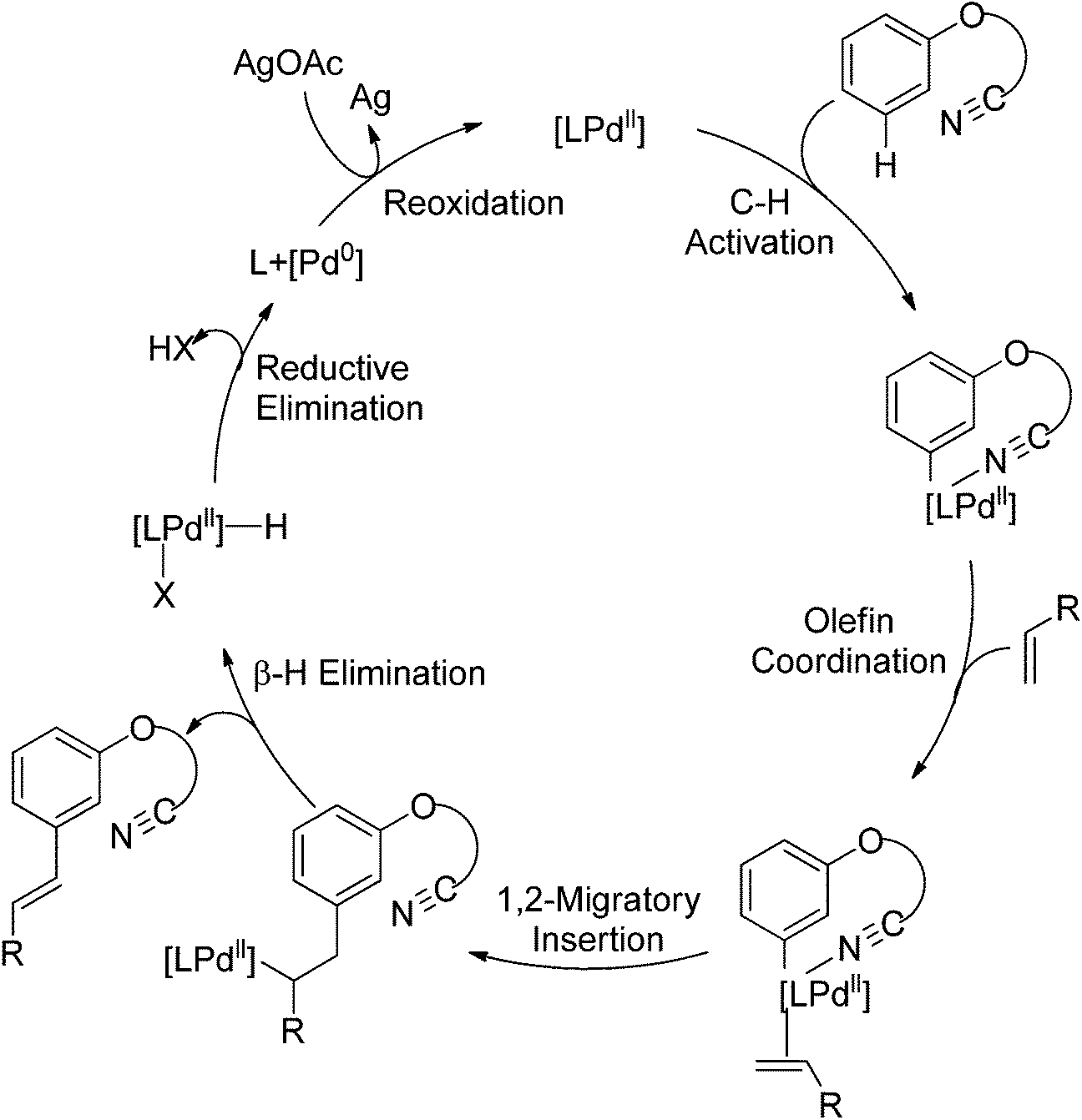
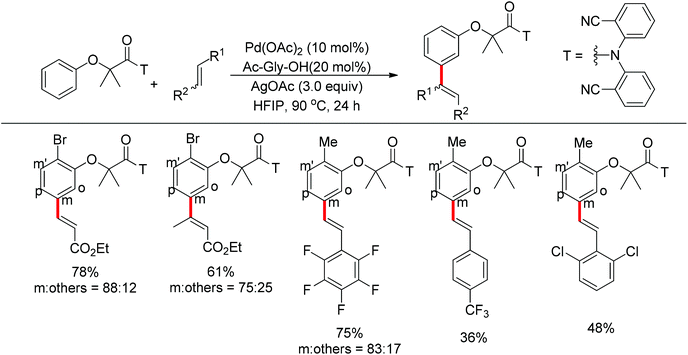
In their following work, Yu and co-workers reported the meta-C–H olefination of phenol derivatives by using nitrile template as the directing group (Scheme 80).102 Compared with previous reports, several electron-deficient styrenes could be used as substrates. Additionally, after the reaction finished, the nitrile template could be removed to afford medicinally useful α-phenoxyacetic acids. Preliminary kinetic studies revealed that the C–H cleavage was involved in the rate-determining step and a tentative catalytic cycle was proposed (Scheme 81).
 |
| | Scheme 80 Pd-catalyzed meta-selective olefination of phenol derivatives. | |
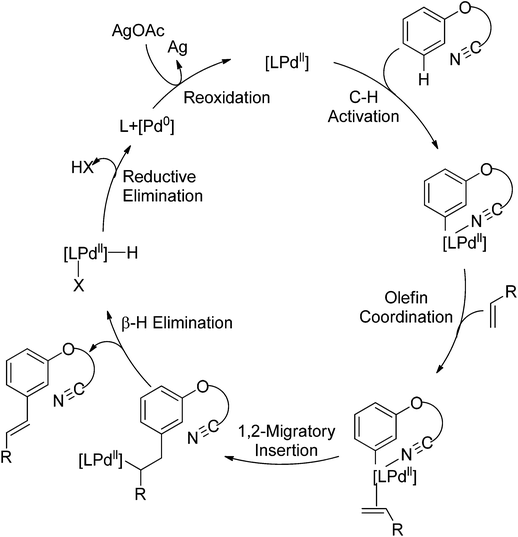 |
| | Scheme 81 Catalytic cycle of the meta-C–H olefination of phenol derivatives. | |
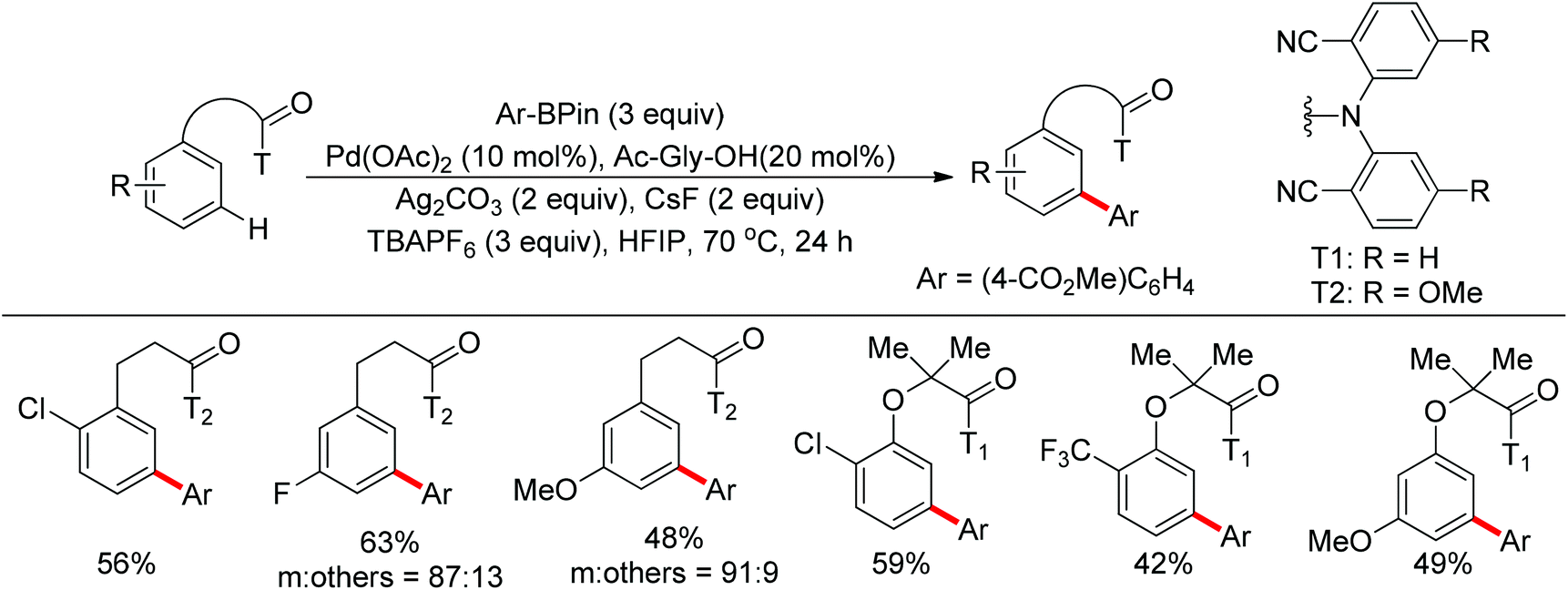
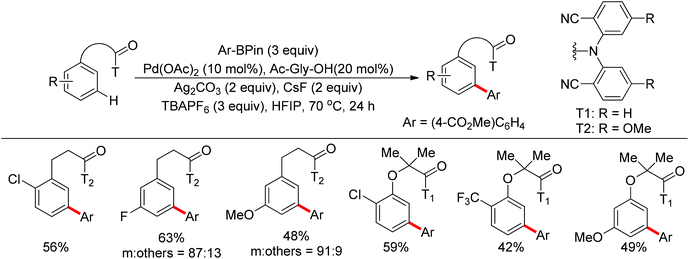
Later on, the same research group reported the meta-C–H arylation of 3-phenylpropanoic acid and phenol derivatives using arylboronic acids as coupling partners (Scheme 82).103 The addition of a monoprotected amino acid as ligand was crucial to promote the cross-coupling. A single example of meta-C–H methylation was also illustrated, which held promise for employing a broader range of alkylborons in the future.
 |
| | Scheme 82 Pd-catalyzed meta-C–H arylation of 3-phenylpropanoic acid and phenol derivatives. | |
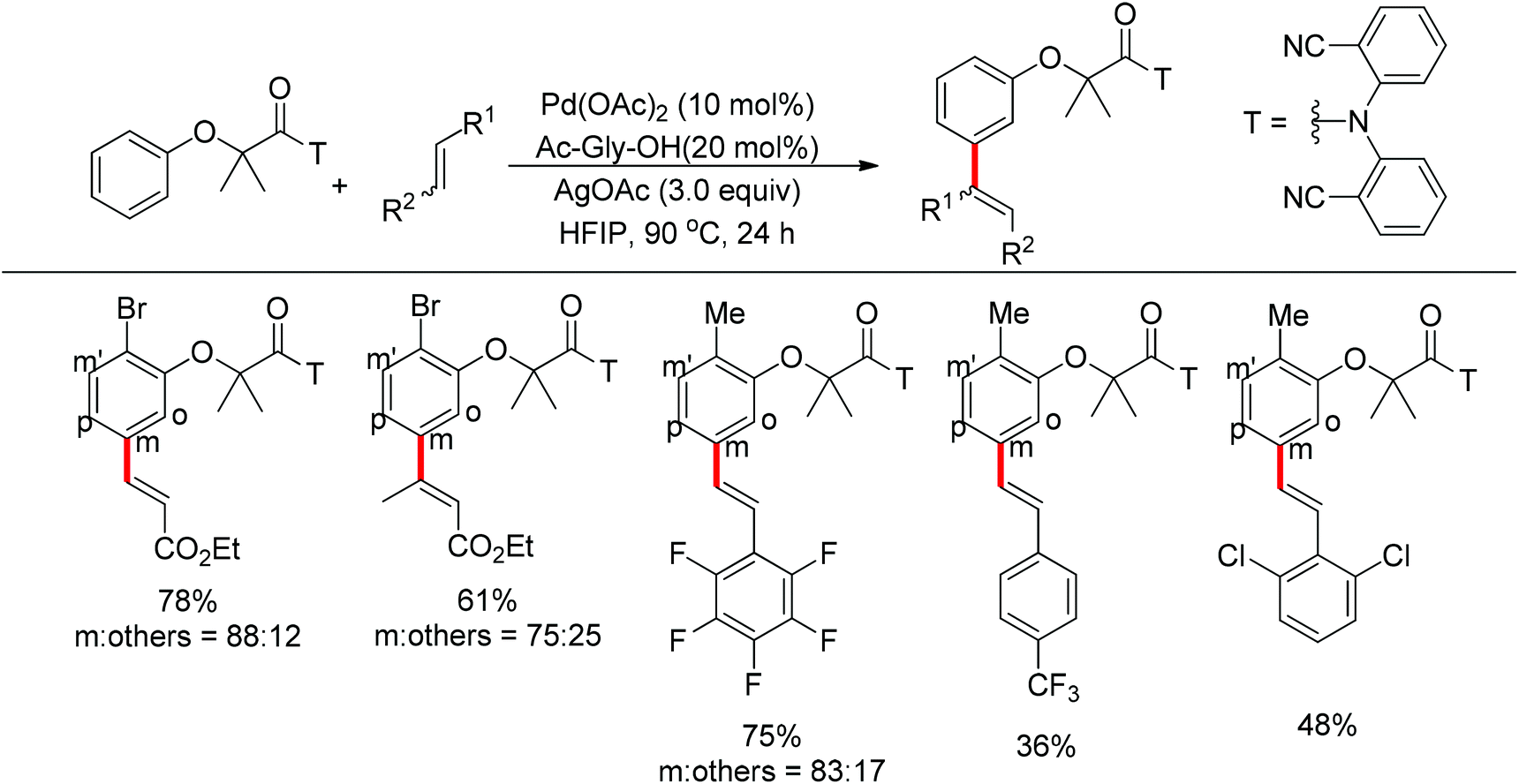
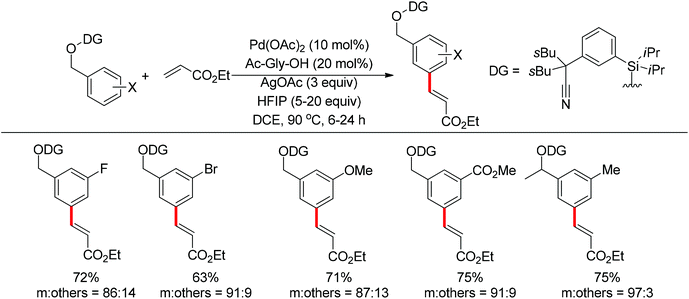
Based on Yu's nitrile-based directing groups, Tan and co-workers developed a silicon-tethered template to realize the meta-selective olefination of various benzyl alcohol derivatives (Scheme 83).104 By using this protocol, alcohol-based substrates could be used as coupling partners and the silicon-based directing group was recyclable. Similar to previous reports, mechanistic studies revealed that C–H bond activation was the rate-determining step and a bent transition state was probably involved.
 |
| | Scheme 83 Pd-catalyzed meta-C–H olefination directed by silicon-tethered directing groups. | |
3.3.
para-C–H bond functionalization controlled by electronic effects
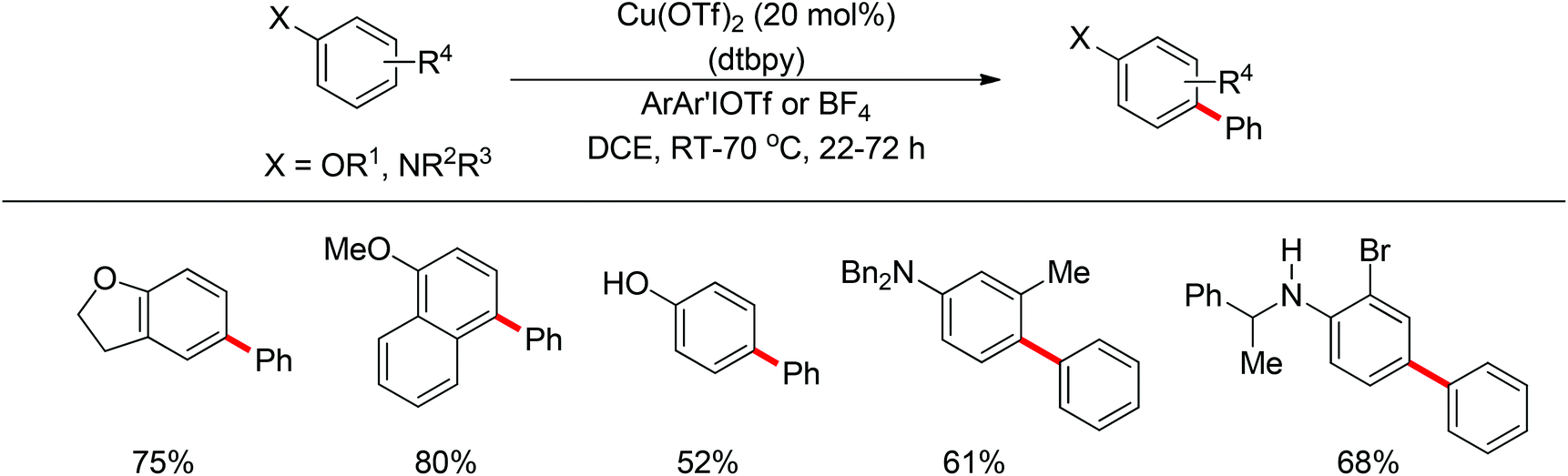
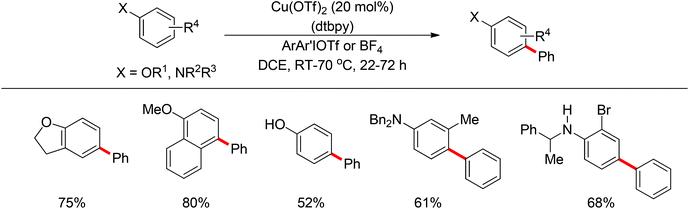
In 2011, Gaunt and co-workers developed a copper-catalyzed para-selective arylation of aniline and phenol derivatives (Scheme 84).98c This transformation occurred in the absence of copper catalyst, albeit in lower yields. The authors proposed that a Friedel–Crafts-type process was involved.
 |
| | Scheme 84 Copper-catalyzed para-selective arylation of aniline and phenol derivatives. | |
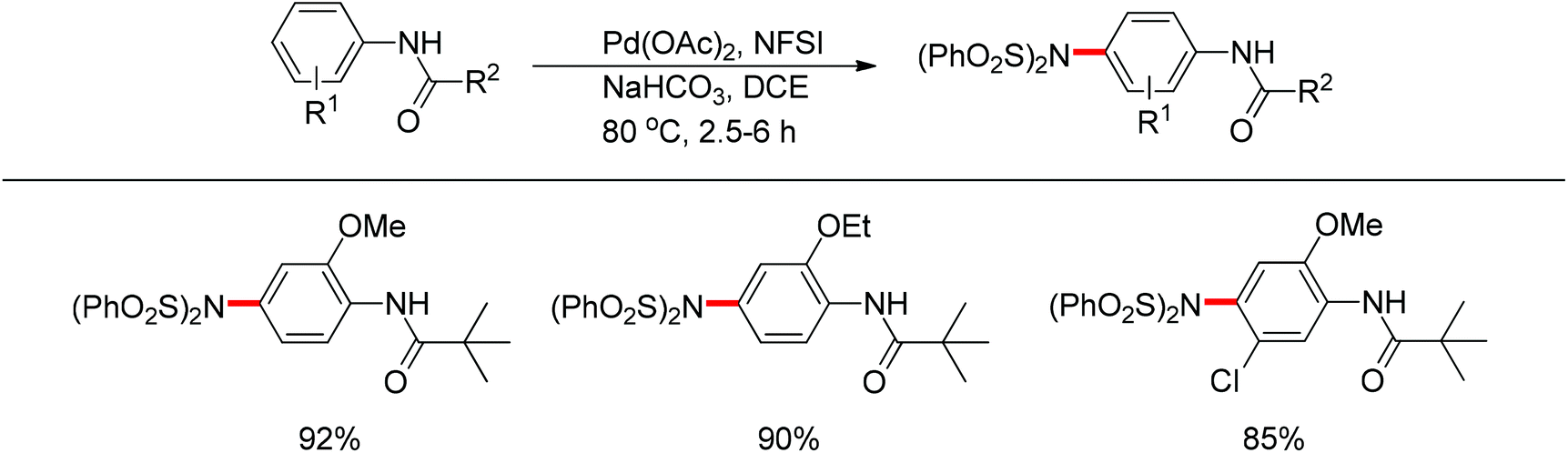
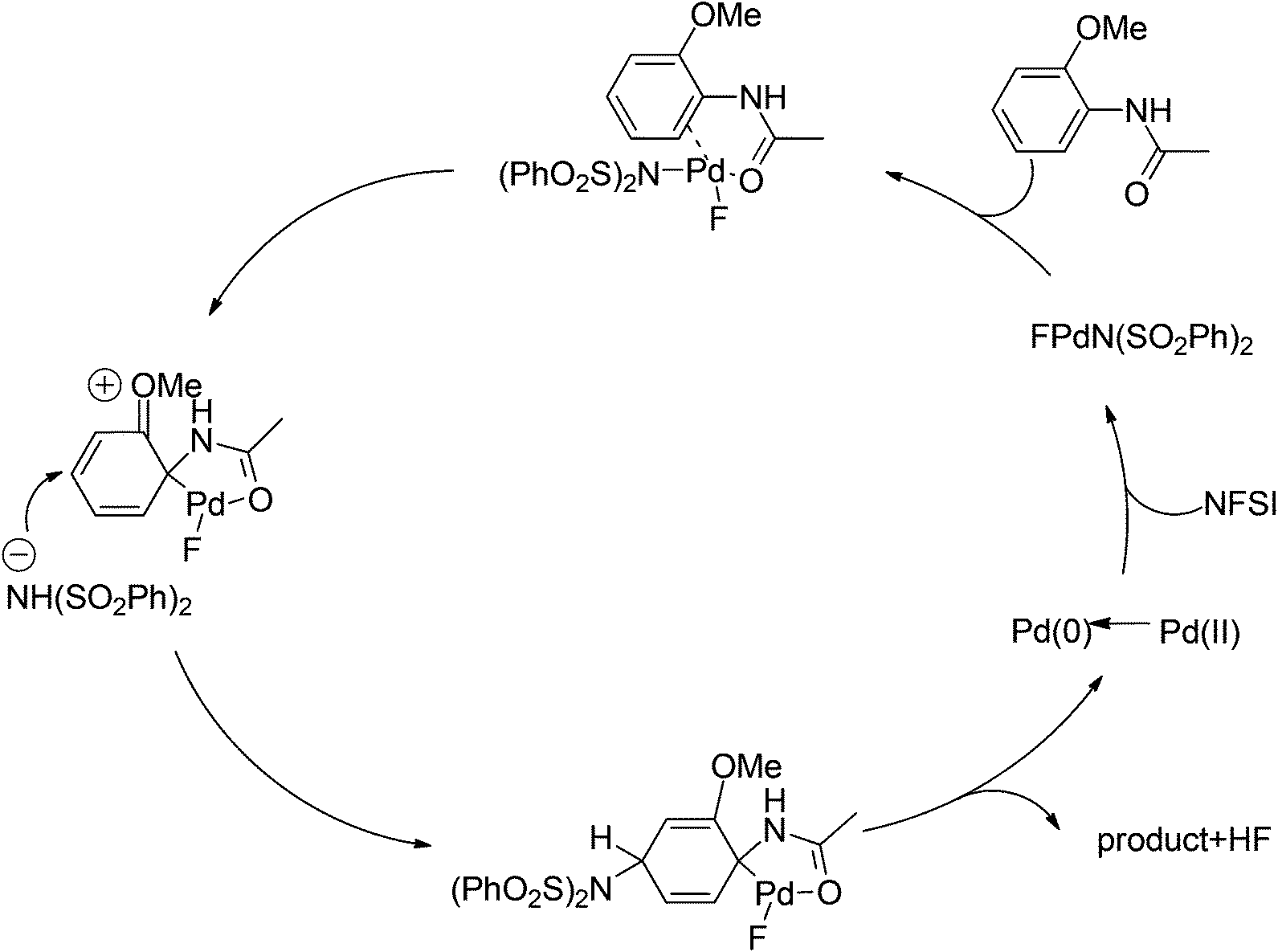
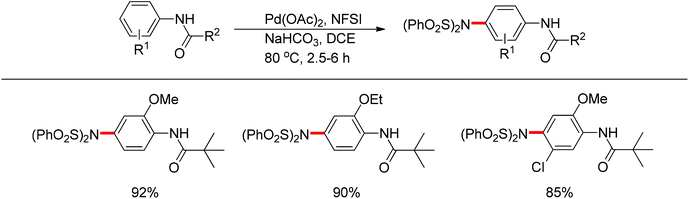
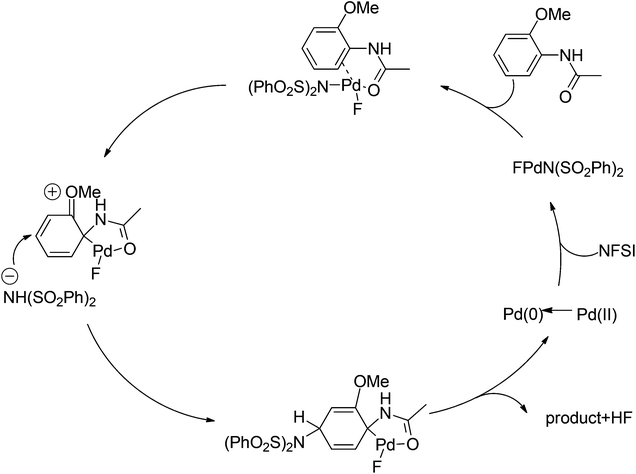
Later on, The investigation carried out by Zhang and co-workers illustrated that the ortho- and para-selective C–H amination of anilides could be realized by using N-fluorobenzenesulfonimide (NFSI), in which the 2-OR-substituted substrates afforded the para-amination products in good to excellent yields (Scheme 85).105 A possible reaction mechanism with involvement of a spiro-palladacycle intermediate was proposed by the authors (Scheme 86).
 |
| | Scheme 85 Pd-catalyzed para-selective C–H amination of anilides. | |
 |
| | Scheme 86 Proposed mechanism of the Pd-catalyzed para-selective C–H amination of anilides. | |
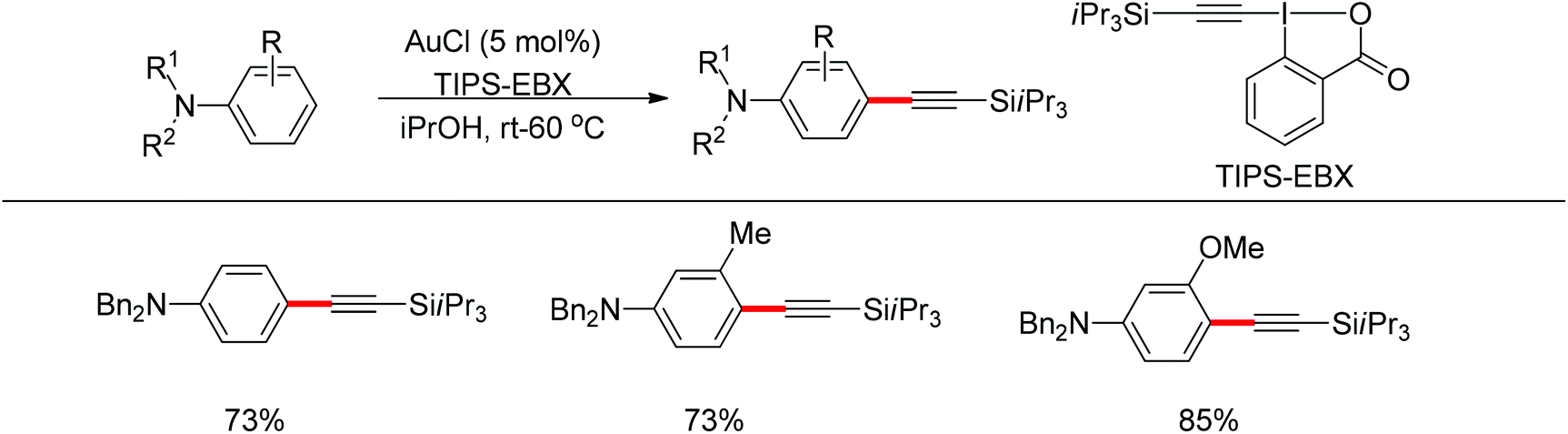
Beside palladium, gold would also enable para-selective C–H transformations. Brand and Waser reported the gold-catalyzed para-selective alkynylation of anilines in 2012 (Scheme 87).106 This protocol provided simple and straightforward access to the useful para-alkynyl anilines.
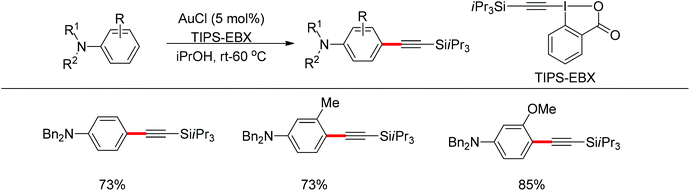 |
| | Scheme 87 Gold-catalyzed para-selective alkynylation of anilines. | |
3.4. Norbornene-mediated/catalyzed C–H bond activation
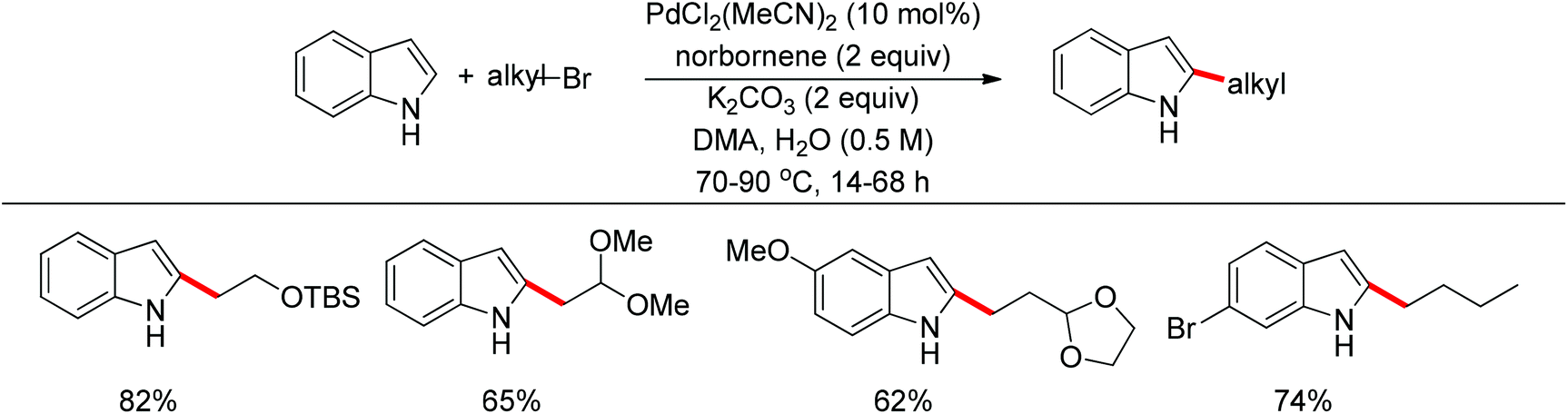
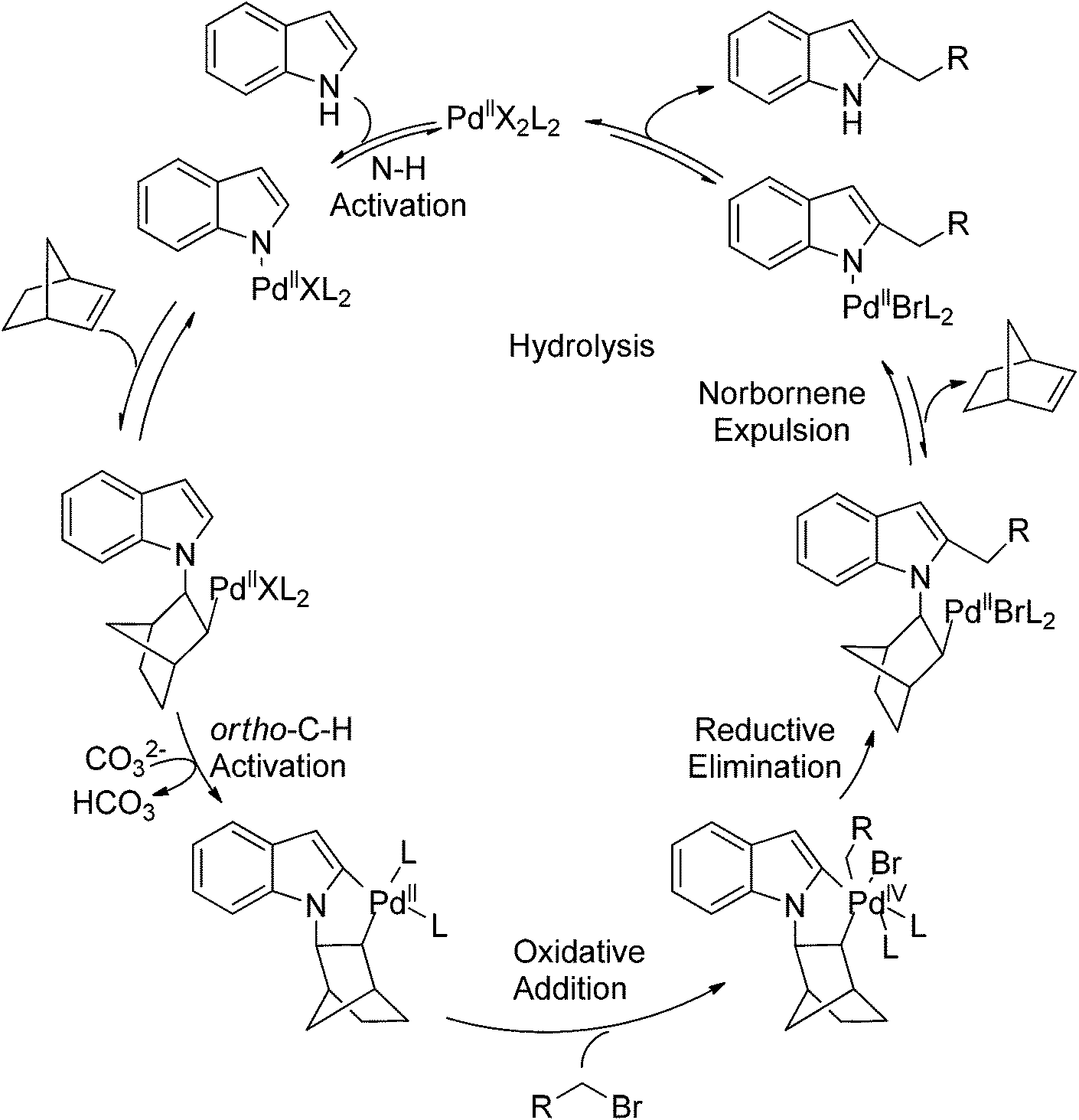
Recently, uncommon selectivity in C–H functionalization has also been observed with the cooperative assistance of a directing group and norbornene. Inspired by the Catellani reaction,107 where ortho C–H bonds of aryl halides are activated with the assistance of norbornene, Bach and co-workers reported a Palladium-catalyzed/norbornene-mediated direct 2-alkylation of indoles (Scheme 88).108 This protocol was compatible with a broad range of primary alkyl bromides and various substituted indoles, thus providing facile access to 2-alkylindole derivatives. The mechanism originally proposed by the authors was incorrect, whereas in their following research the detailed reaction mechanism was investigated with a revised catalytic cycle given (Scheme 89).109 It was revealed that the key intermediate was an N-norbornene type palladacycle and the rate-determining step was the oxidative addition of alkyl bromides to the palladium center.
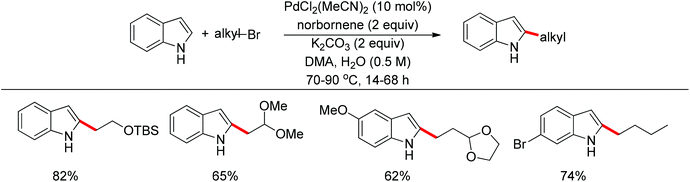 |
| | Scheme 88 Pd-catalyzed regioselective 2-alkylation of indoles. | |
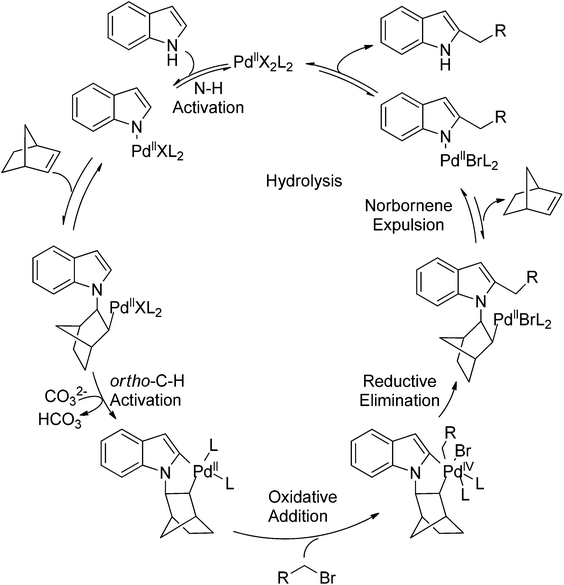 |
| | Scheme 89 The catalytic cycle of Pd-catalyzed regioselective 2-alkylation of indoles. | |
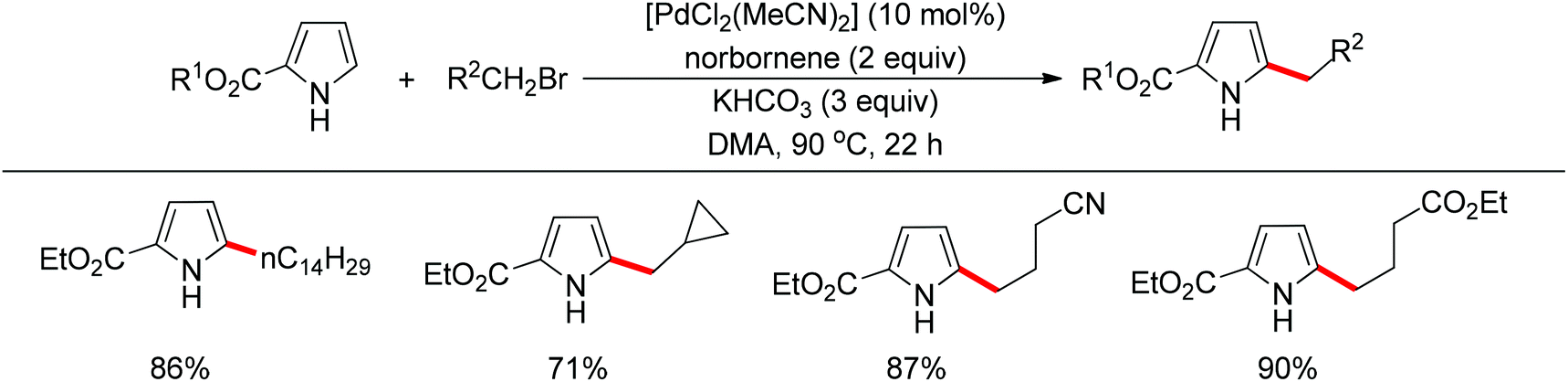
Subsequently, the same research group extended their methodology to the alkylation of pyrrole derivatives (Scheme 90).110 This protocol was similar to the one applied to indoles; however, due to electronic and acidic differences, only electron-deficient pyrroles were suitable substrates.
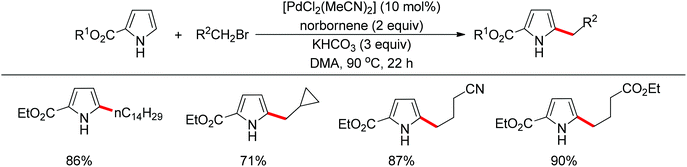 |
| | Scheme 90 Pd-catalyzed regioselective 2-alkylation of pyrroles. | |
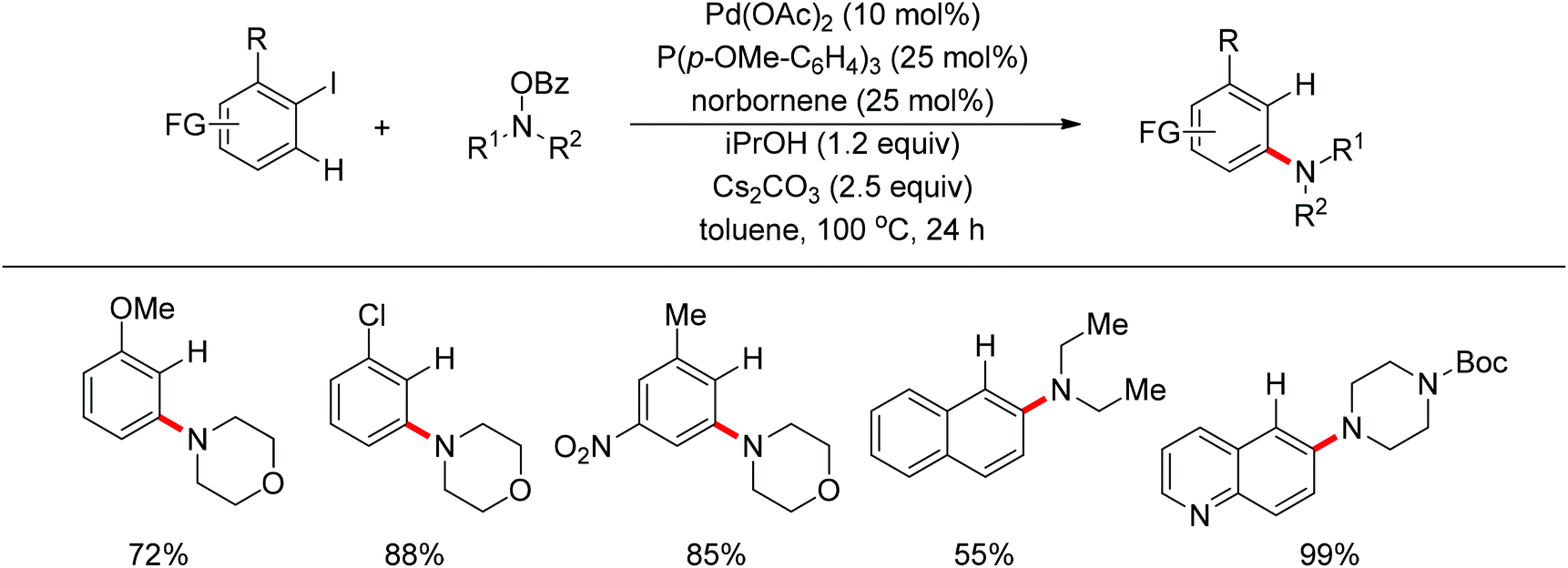
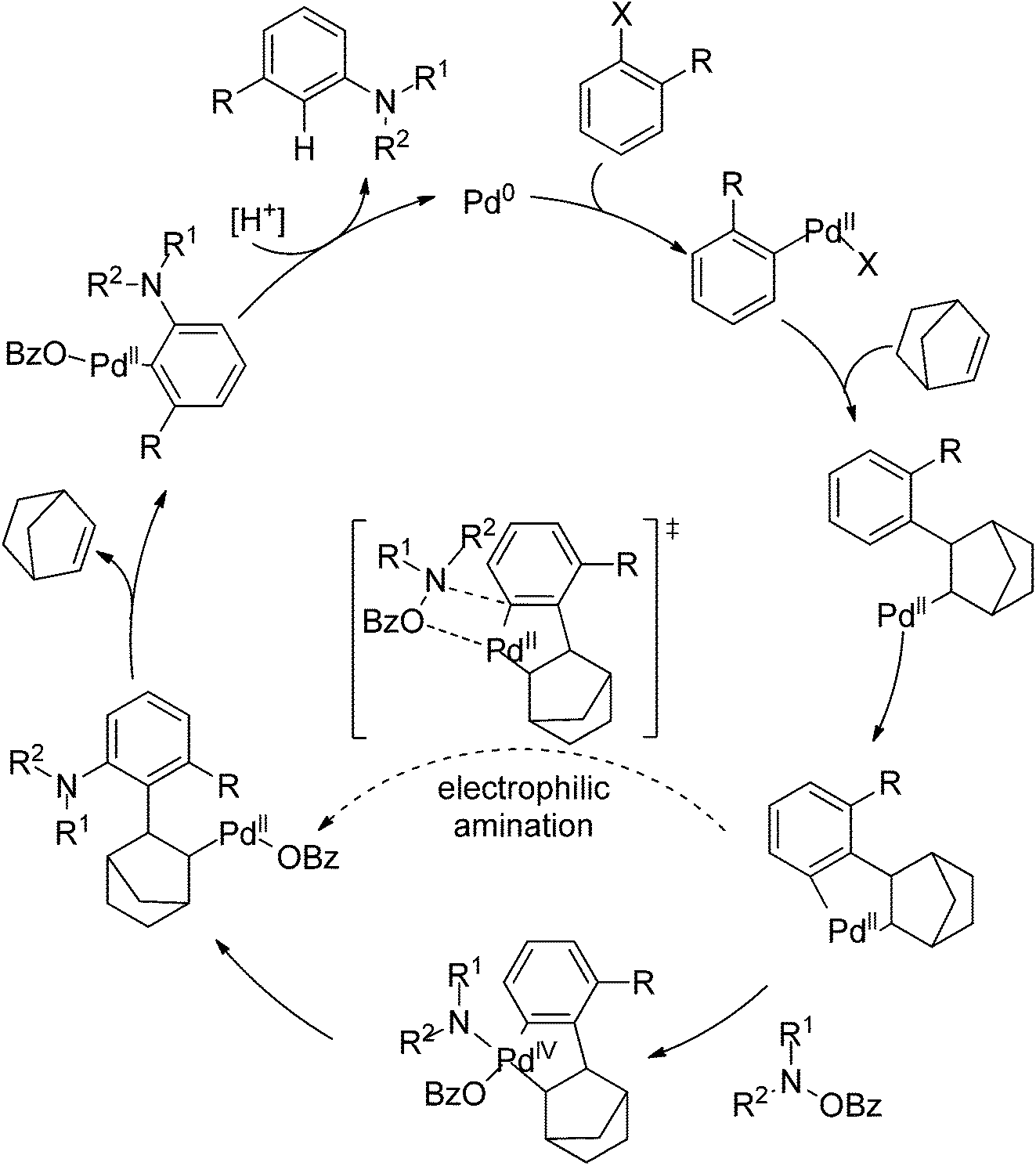
Very recently, Dong and co-workers reported a palladium and norbornene-catalyzed ortho-amination of aryl halides via Catellani-type C–H functionalization (Scheme 91).111 The use of N-benzoyloxyamines as the amine source (oxidant) and isopropanol as reductant furnished the amination products exclusively at the ortho position of halides instead of ipso. The proposed catalytic cycle was also depicted by the authors (Scheme 92).
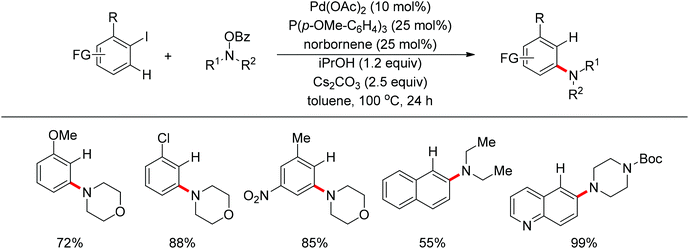 |
| | Scheme 91 Pd and norbornene-catalyzed ortho-arene amination of aryl halides. | |
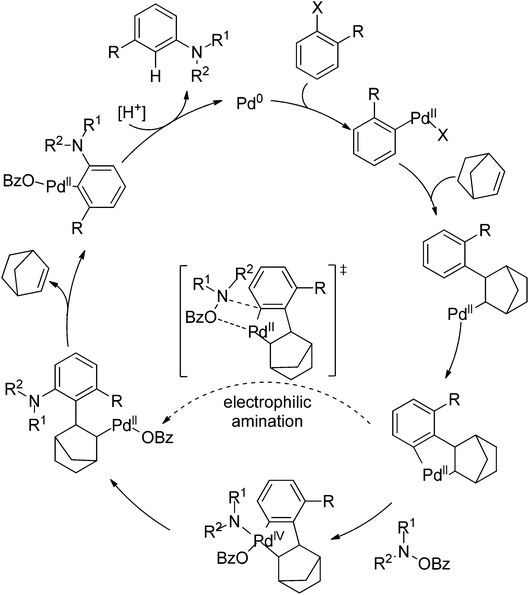 |
| | Scheme 92 The proposed catalytic cycle of palladium and norbornene-catalyzed ortho-arene amination of aryl halides. | |
3.5. C–H bond functionalization directed by polynuclear metal cluster with novel regioselectivity
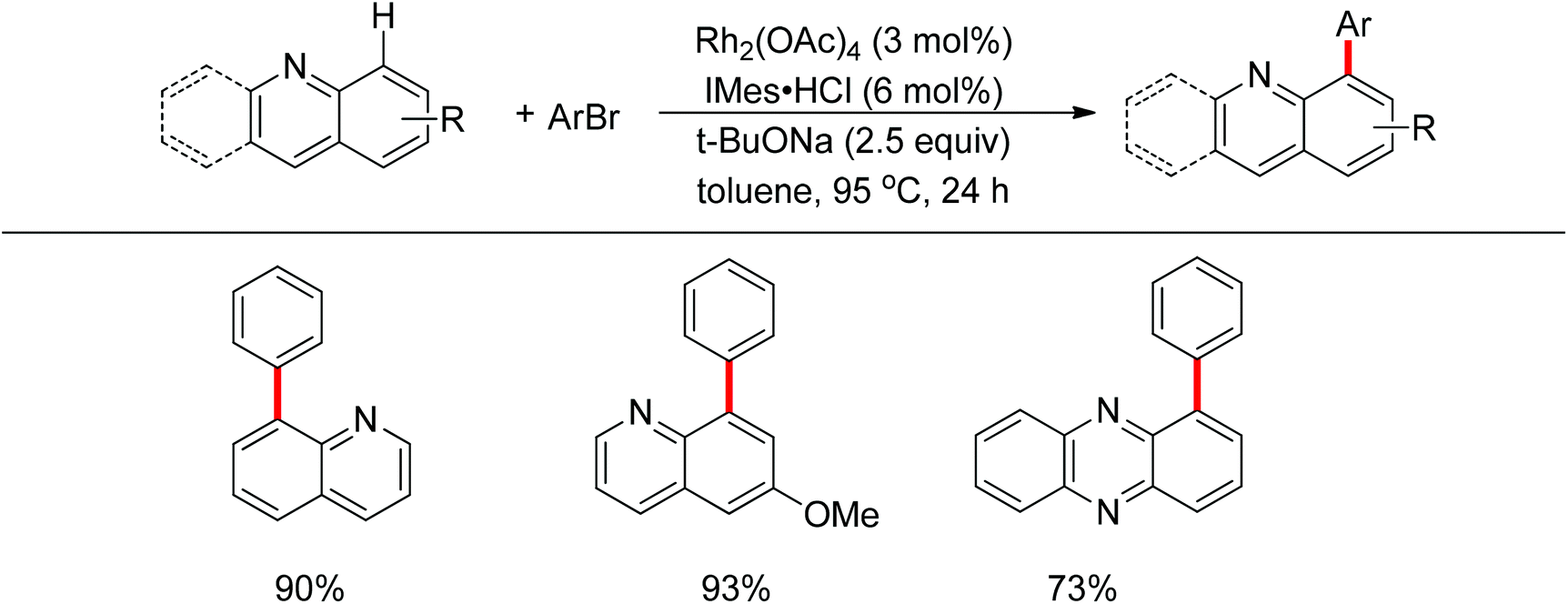
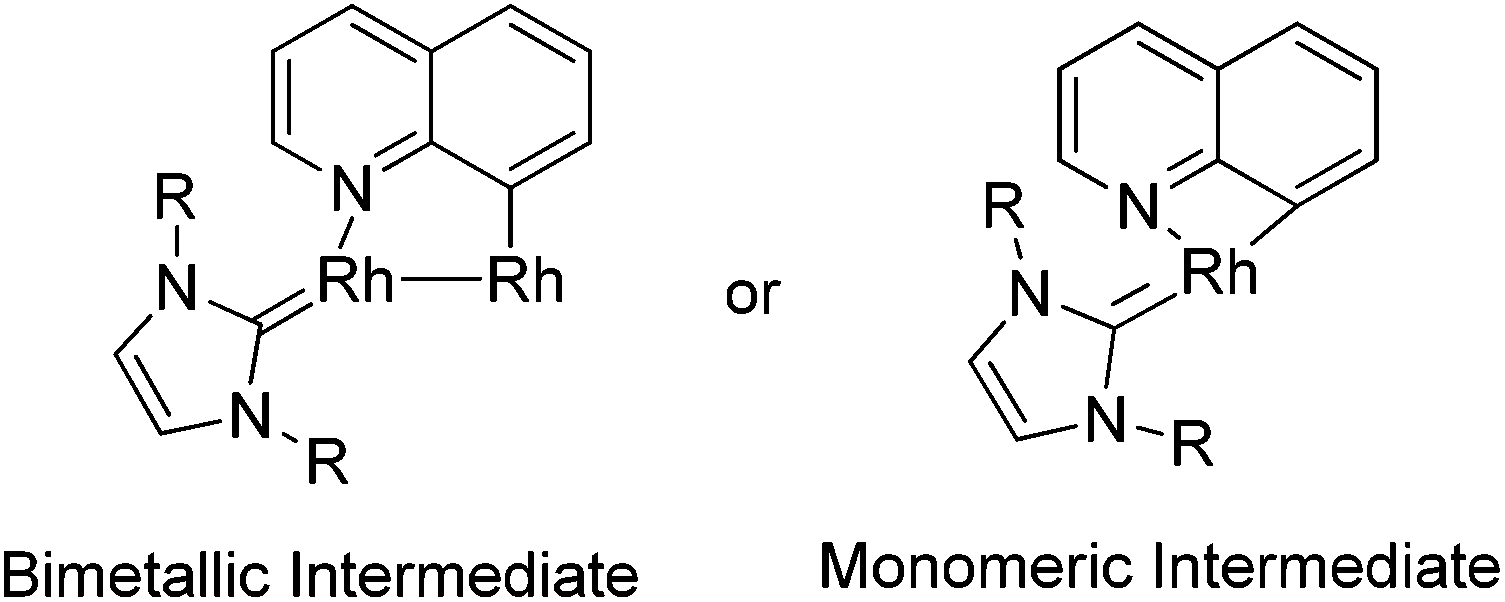
A few examples with unusual selectivities were also reported involving a polynuclear metal cluster and a directing nitrogen atom. In 2011, Chang and co-workers reported the Rh-catalyzed direct C8-selective arylation of quinolines (Scheme 93).112 The use of a suitable rhodium source and NHC ligand were crucial for achieving high reactivity and selectivity. A bimetallic Rh species bound to the NHC ligand or a monomeric Rh–NHC species were proposed as plausible intermediates (Scheme 94).
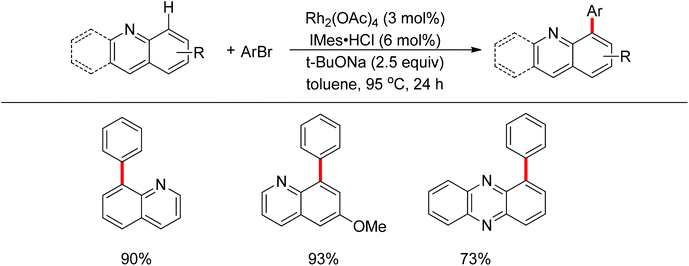 |
| | Scheme 93 Rh(NHC)-catalyzed direct C8-arylation of quinolines. | |
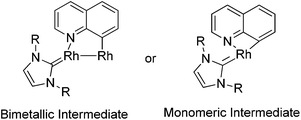 |
| | Scheme 94 Possible intermediates in the Rh(NHC)-catalyzed direct C8-arylation of quinolines. | |
One year later, Patureau and co-workers reported Ru–Cu catalyzed direct C–H amination using carbazoles (Scheme 95).113 Similar to Chang's report, in the authors’ hypothesis, the regioselectivity of this reaction was attributed to the intermediate in which a Ru(II)–Cu(II) dinuclear cluster N-coordinated with carbazole.
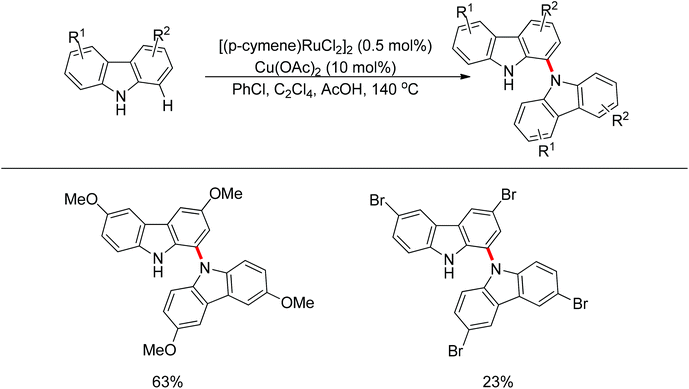 |
| | Scheme 95 Ru–Cu catalyzed direct C–H amination of carbazoles. | |
4. Oxidizing group directed redox-neutral C–H bond functionalization
Compared with the well-established oxidative C–H bond functionalization using external oxidants, mainly silver or copper salts, oxidizing group directed redox-neutral C–H bond functionalization presents unparalleled advantages: (1) no external oxidants required for the catalytic cycle, avoiding waste produced from oxidants compared with using external oxidants, (2) simple and mild reaction conditions, (3) high regioselectivity, (4) very good functional group tolerance.
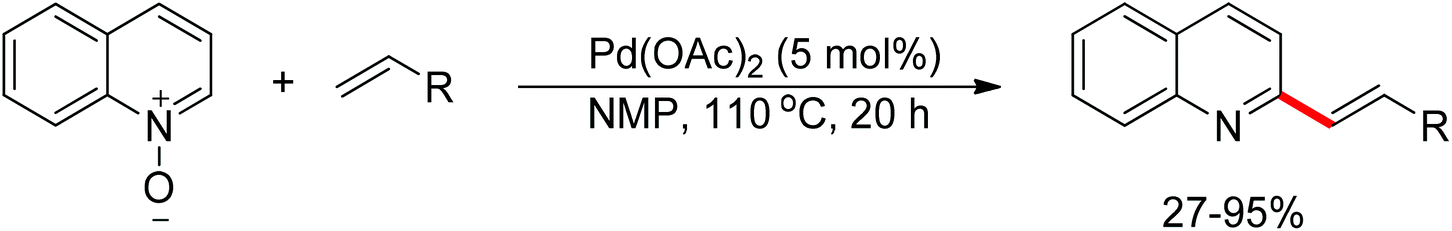
The oxidizing group directed redox-neutral strategy in C–H bond functionalization was first launched in palladium chemistry.114 In the year 2009, pioneering work was accomplished by Wu, Cui and co-workers.115 They ingeniously employed pyridine N-oxides both as substrates and oxidants in the oxidative Heck reaction (Scheme 96). Further progress was made by Tan and Hartwig.116 In their work, oxime ester acted as both directing group and internal oxidant, and indole derivatives were effectively prepared with low catalyst loading (Scheme 97).
 |
| | Scheme 96 Palladium-catalyzed olefination of pyridine N-oxides. | |
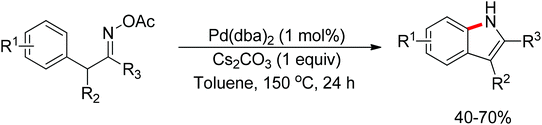 |
| | Scheme 97 Intramolecular amination of aromatic C–H bonds with oxime esters. | |
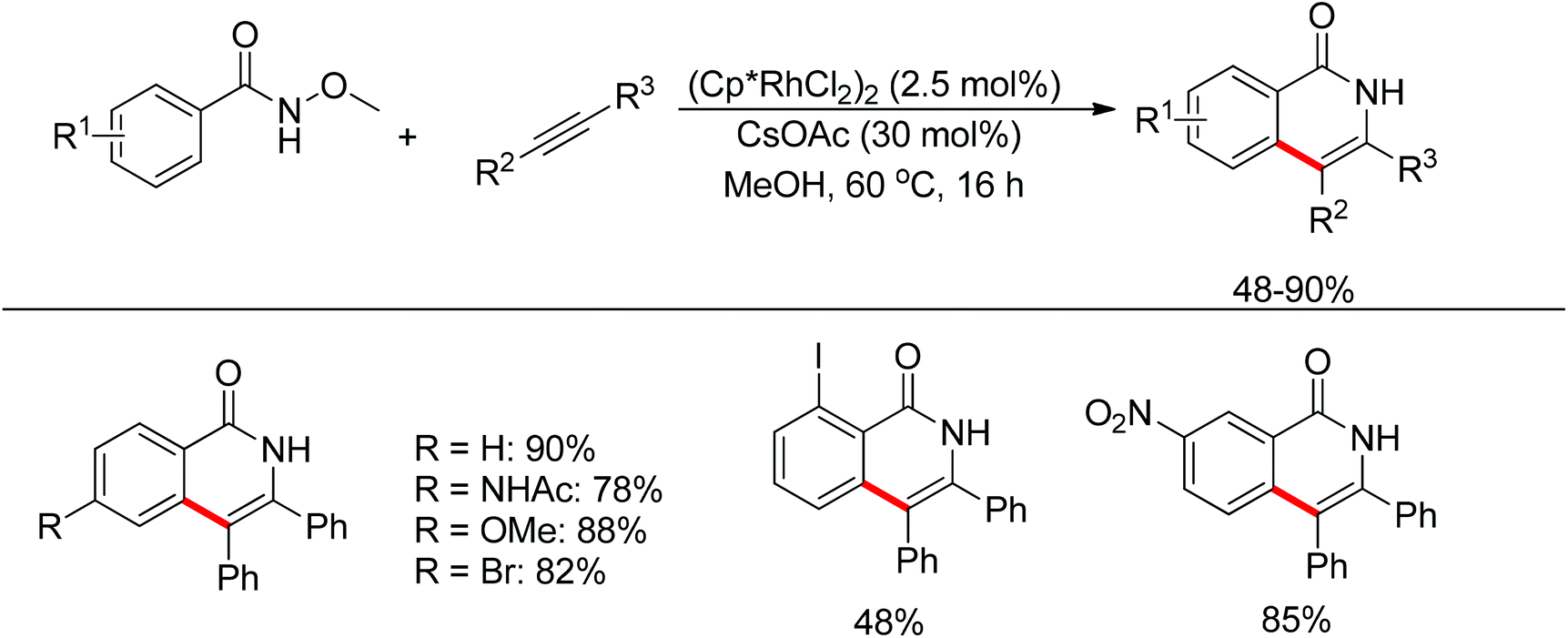
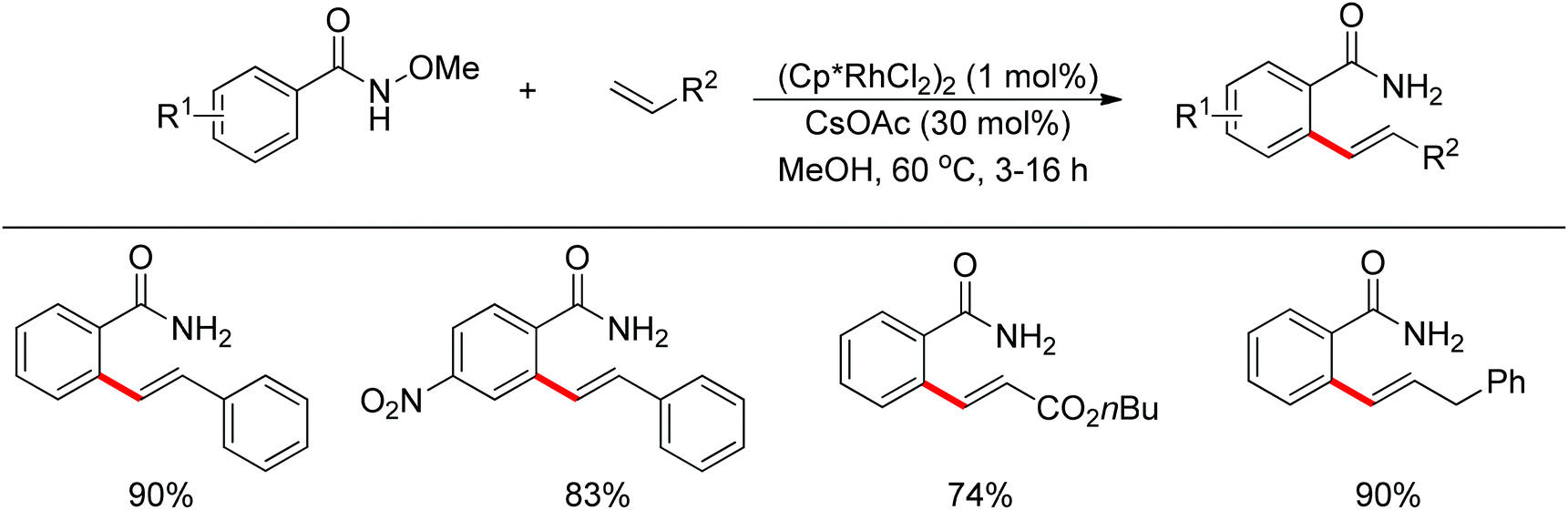
Inspired by previous work in palladium chemistry, the Fagnou group developed the novel concept of ‘redox-neutral C–H bond functionalization’ in Rh(III) chemistry.117 They elegantly reported the first example of Rh(III)-catalyzed ortho-C–H bond alkenylation of N-methoxybenzamides and C–N bond formation sequence to afford isoquinolone derivatives, in which the cleavage of the N–O bond released Rh(III) (Scheme 98).117a This isoquinolone synthesis was carried out under mild and simple reaction conditions and was high-yielding (up to 90%) with very good functional group tolerance.
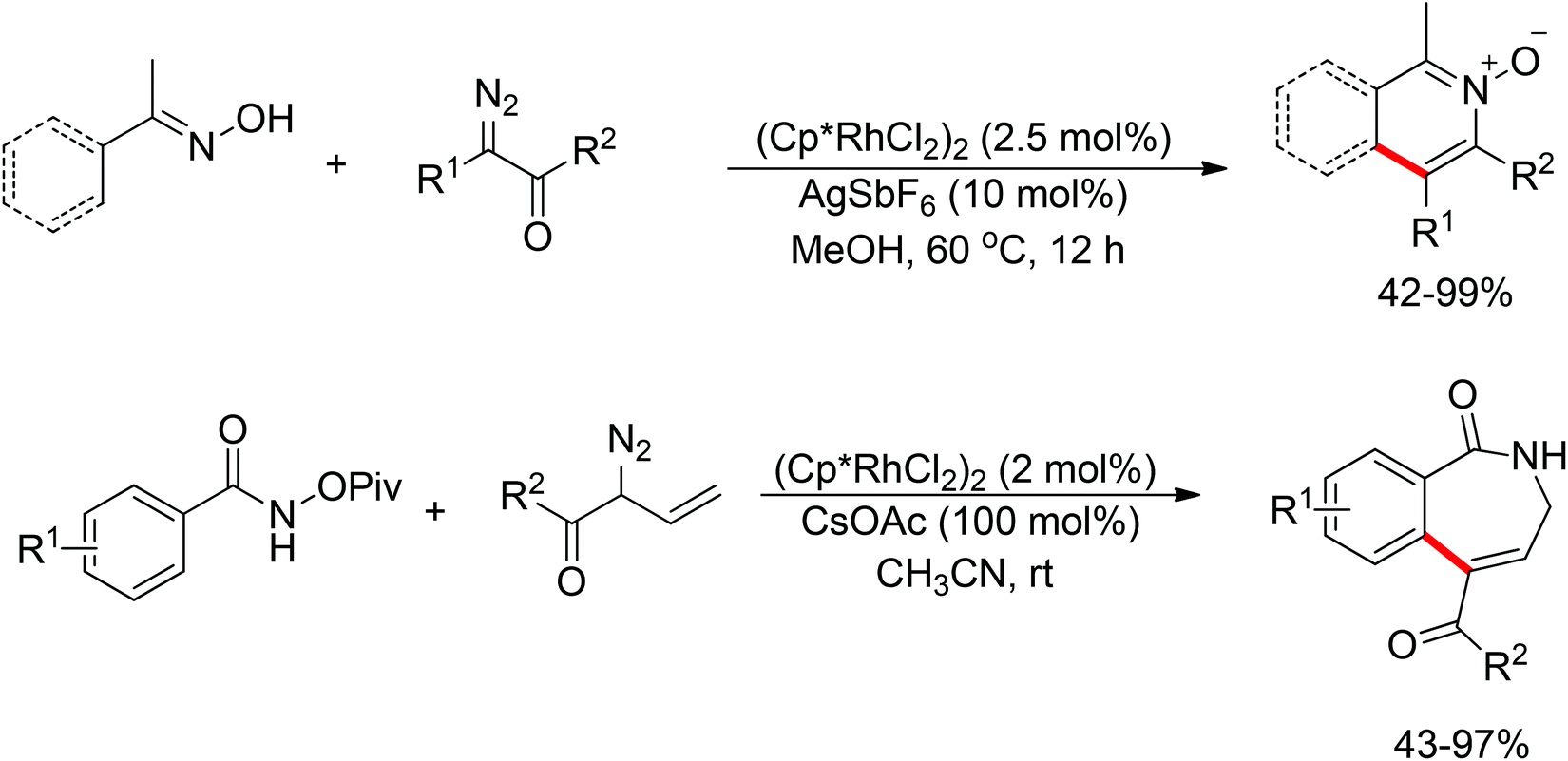
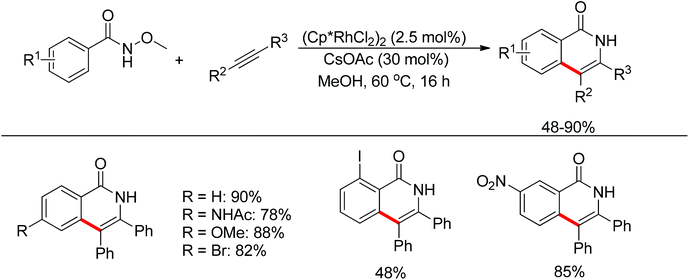 |
| | Scheme 98 Rh(III)-catalyzed redox-neutral synthesis of isoquinolone. | |
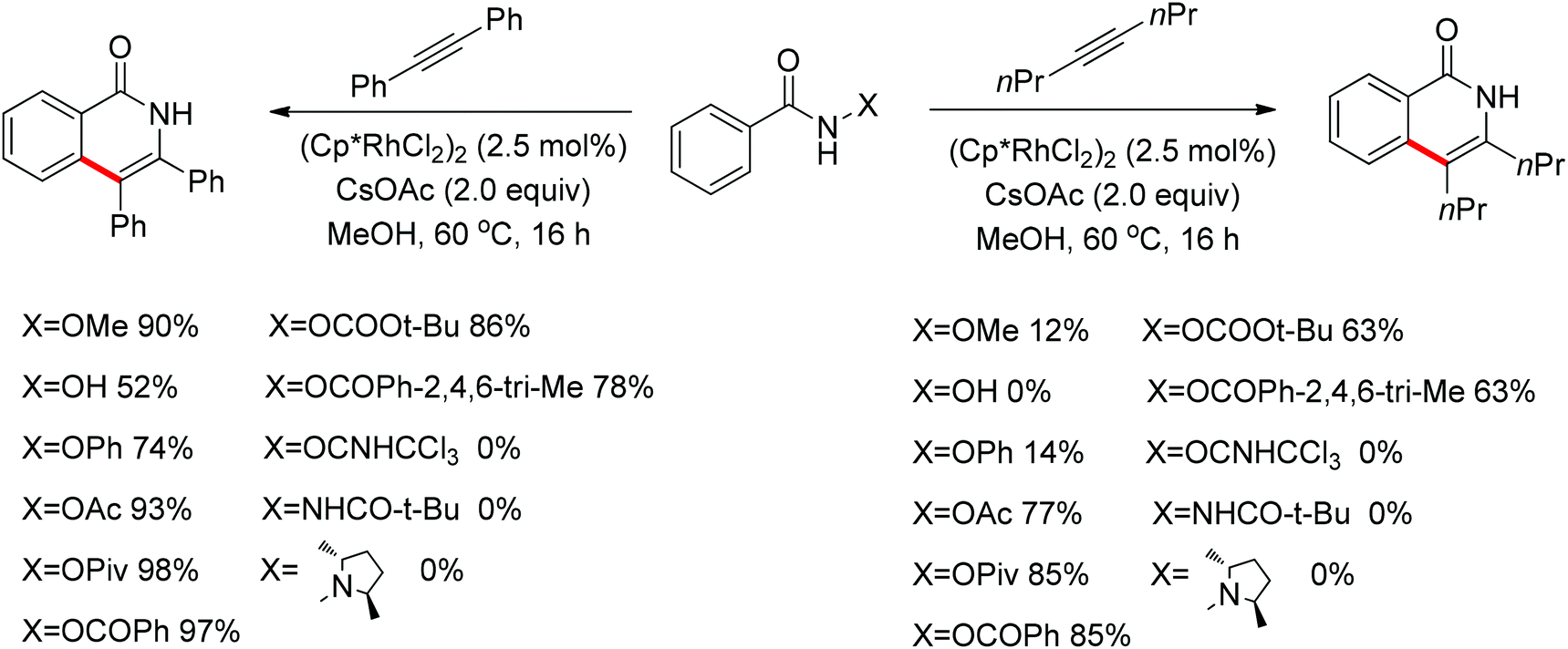
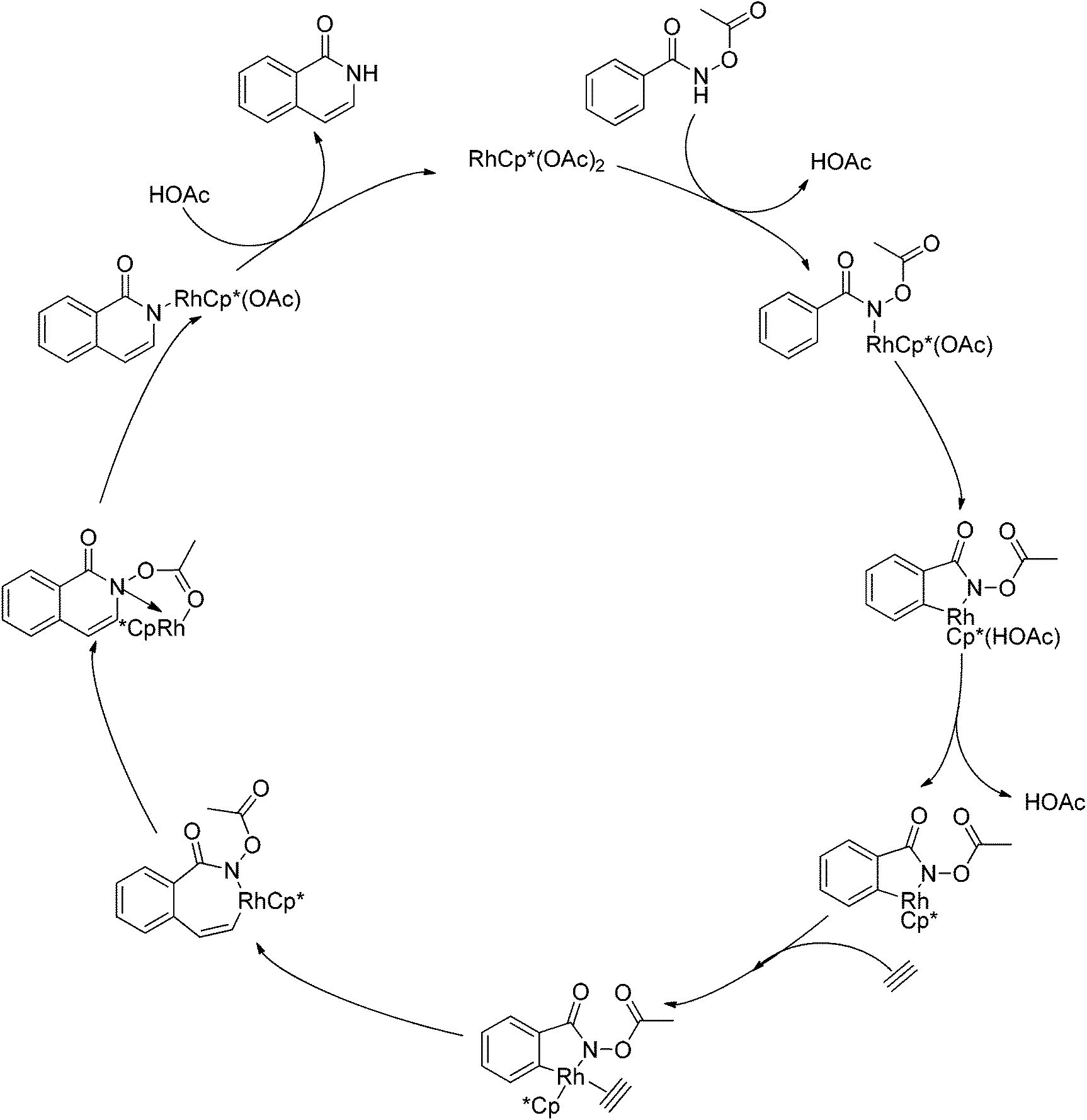
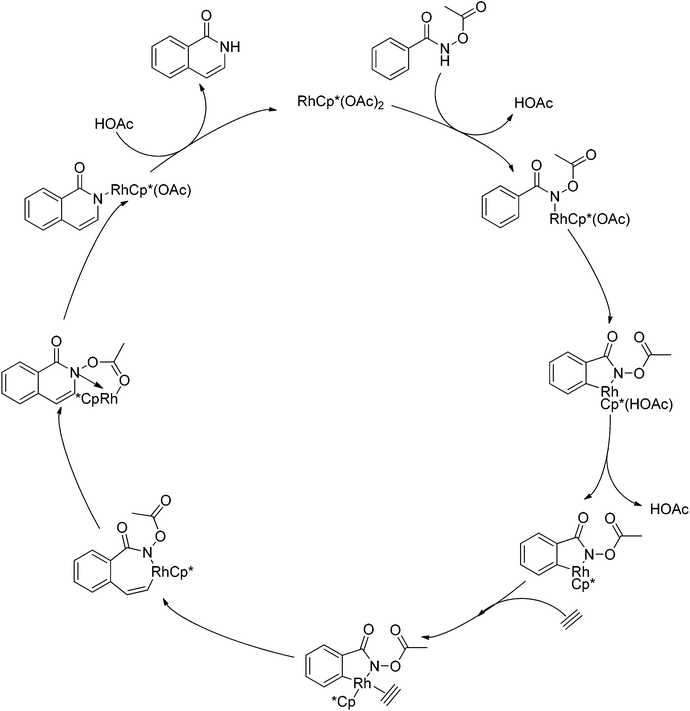
As part of systematic studies,117b the authors subsequently investigated the built-in oxidants, and found that both pivalate and benzoate gave the best results (Scheme 99). The scopes were successfully extended to terminal alkynes and alkenes with high levels of regioselectivity. Deuterium kinetic isotope effect (DKIE) studies were performed and revealed that depending on the nature of the built-in oxidants, the rate-limiting step was different. N-Methoxybenzamide showed no DKIE, while a large primary DKIE of 15 ± 1 was found with N-pivaloyloxybenzamide. DKIE studies indicated that C–H bond cleavage was the rate-limiting step when N-pivaloyloxybenzamides were used as substrates. To fully understand the reaction pathways, quantum chemical density functional theory (DFT) calculations were conducted. Based on the experiments and calculations, a general mechanism was postulated (Scheme 100). Starting from Cp*Rh(OAc)2, the catalytic cycle possesses two key steps: cyclometalation via a concerted metalation–deprotonation (CMD) transition state; and fast intramolecular oxidative addition to the Rh(I) species to generate a Rh(III) species and complete the catalytic cycle. Finally, this group also developed oxime and O-pivaloyl oxime as oxidizing directing groups in isoquinoline synthesis. Fagnou’s group's significant contribution in this type of reaction laid the benchmark for redox-neutral strategy. Since the publications of the two key papers,117 the oxidizing group directed redox-neutral C–H bond functionalization chemistry has boomed in recent years.
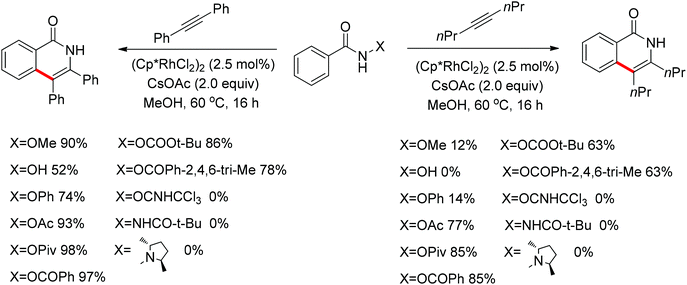 |
| | Scheme 99 Evaluation of the effect of the internal oxidant. | |
 |
| | Scheme 100 Plausible mechanism for Rh(III)-catalyzed heterocycle synthesis using an internal oxidant. | |
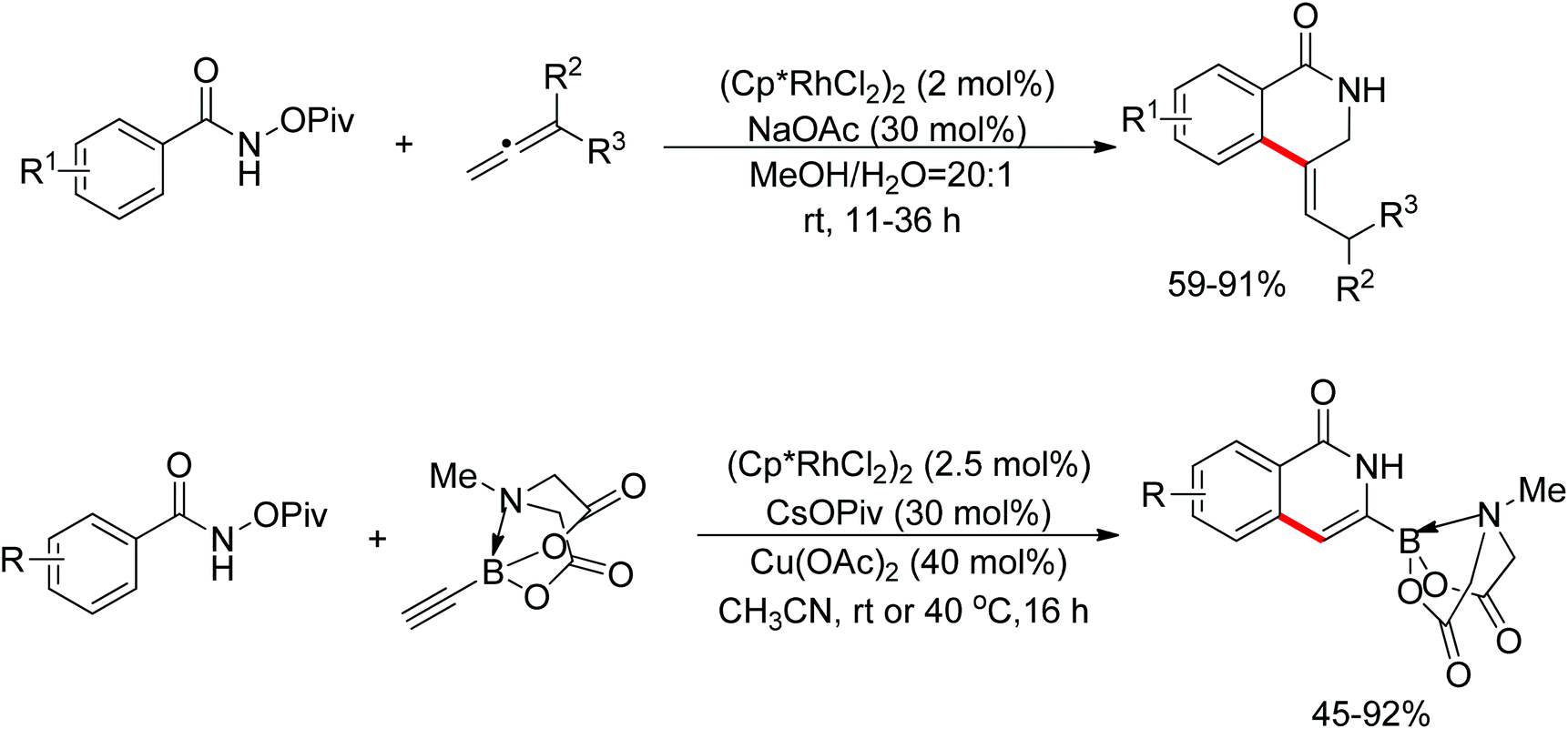
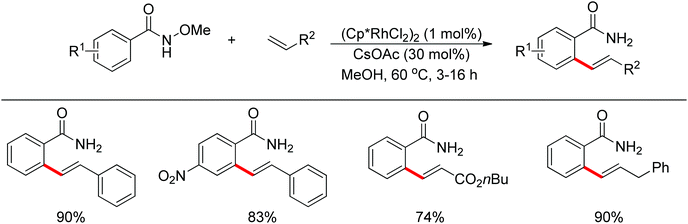
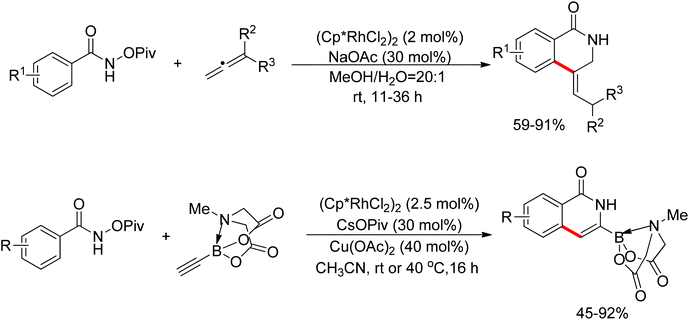
Glorius and co-workers reported a Rh(III)-catalyzed ortho-C–H olefination using an oxidizing directing group.118 Although an oxidative Heck reaction was reported by Cui and Wu,115 Glorius's work presented a powerful Cp*Rh(III) catalyst in C–H bond olefination. The reaction conditions are mild, efficient, and versatile (Scheme 101). Interestingly, annulation products bearing a chiral carbon center were formed as a racemic mixture by utilizing an N-pivaloyloxy fragment as the internal oxidant (Scheme 102). Rovis and Cramer independently applied the annulation strategy to asymmetric tetrahydroisoquinolinone synthesis by using chiral rhodium catalyst, and then to an intramolecular Heck reaction.119 Cui and Molander broadened the scope to strained alkenes and potassium vinyltrifluoroborates, respectively.120 This kind of reaction was also operable when catalyzed by less-expensive ruthenium complexes.121 Subsequently, Wang and Glorius extended the annulation strategy to allenes and alkyne MIDA boronates (Scheme 103).122
 |
| | Scheme 101 Oxidizing group directed Heck reaction. | |
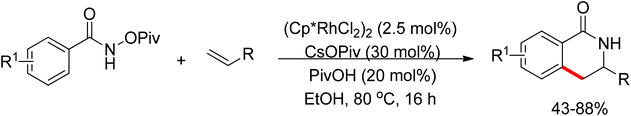 |
| | Scheme 102 Rh(III)-catalyzed annulation reaction. | |
 |
| | Scheme 103 Rh(III)-catalyzed annulation of allenes and alkyne MIDA boronates. | |
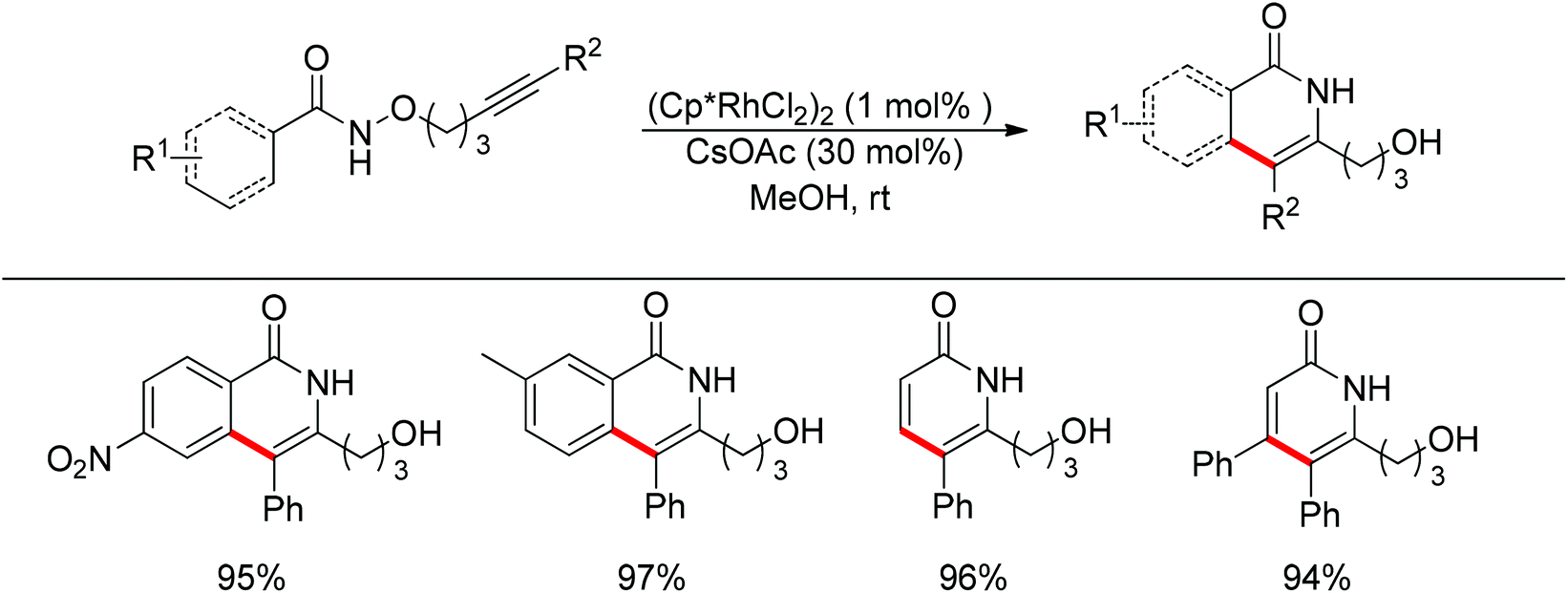
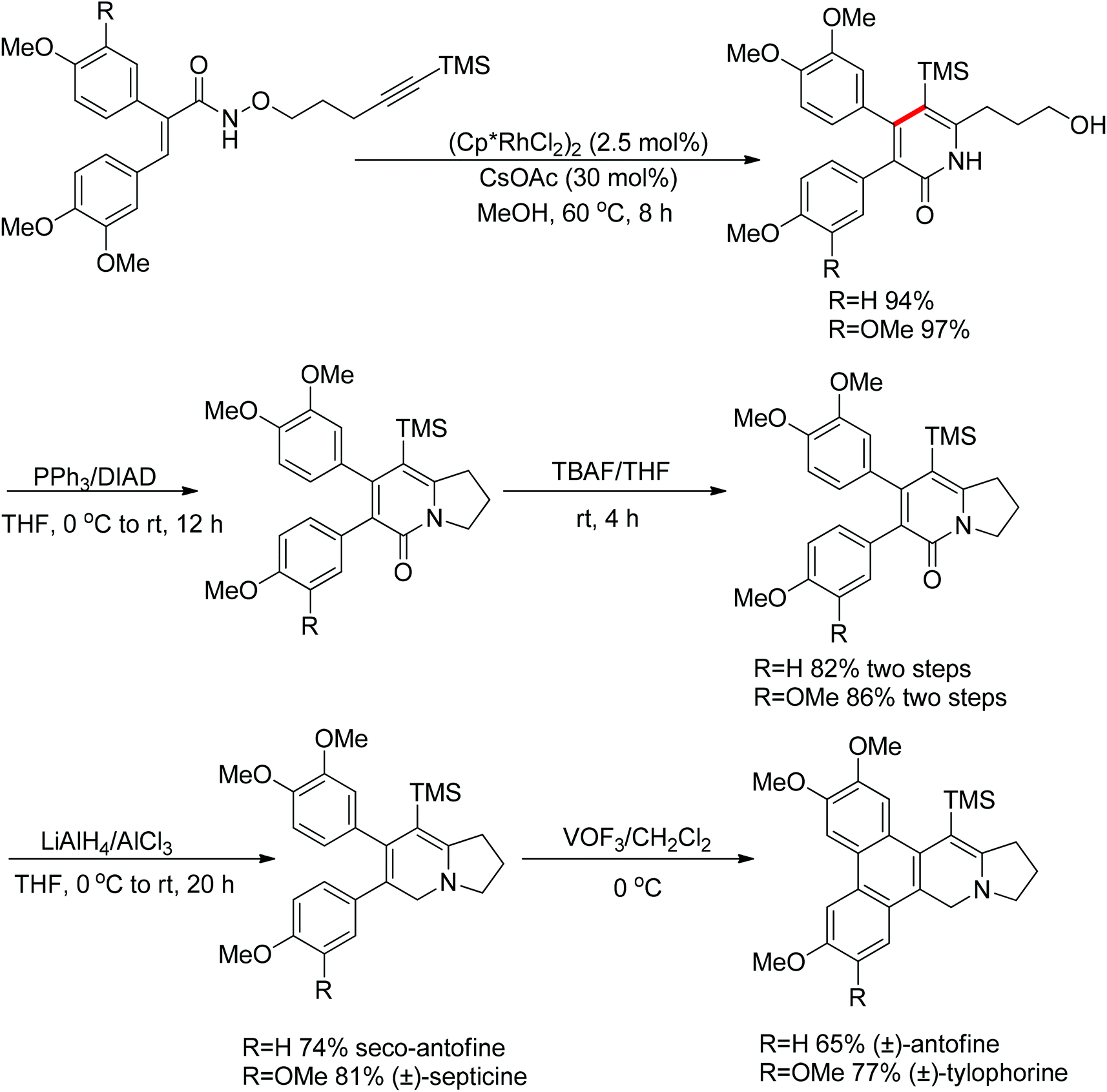
Park and co-workers skilfully applied the oxidizing directing group strategy to the intermolecular reaction that incorporated the leaving group into the product.123 In this reaction, annulation products bearing a hydroxyl group were generated in excellent yields (Scheme 104). On the basis of the newly formed hydroxyl group, this prototype reaction was used as a key step for the total synthesis of (±)-septicine, (±)-antofine, and (±)-tylophorine (Scheme 105).
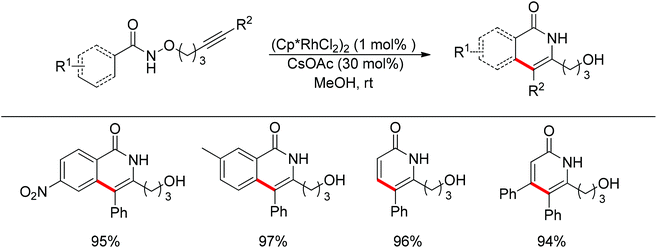 |
| | Scheme 104 Rh(III)-catalyzed intramolecular annulation. | |
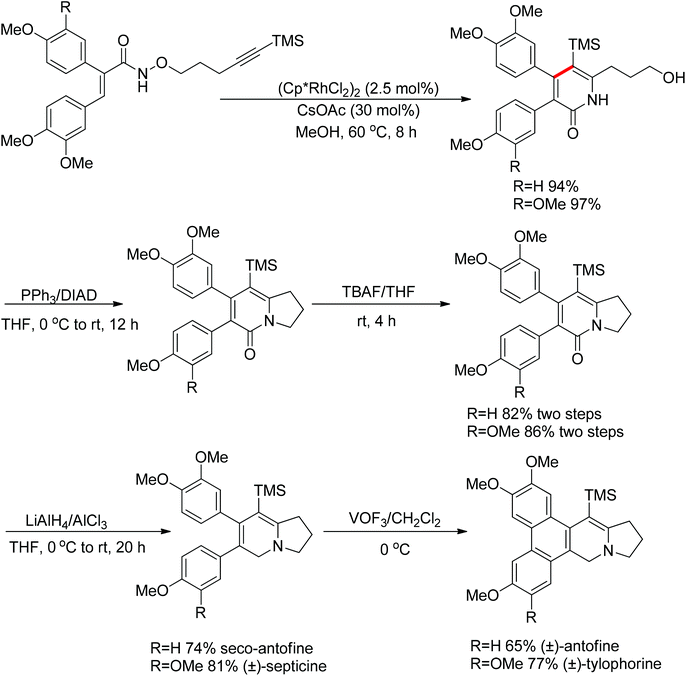 |
| | Scheme 105 Total synthesis of (±)-septicine, (±)-antofine, and (±)-tylophorine. | |
By taking advantage of the oxidizing group directed redox-neutral C–H bond functionalization strategy, Ma and co-workers elegantly disclosed a direct arylation method of allenylsilanes that delivered trisubstituted allenylsilanes in excellent yields (Scheme 106).124 Such a Heck-type allenylation offered good selectivity for sterically less-hindered C–H bonds. However, coordinating groups could shift the selectivity probably due to coordination effects.
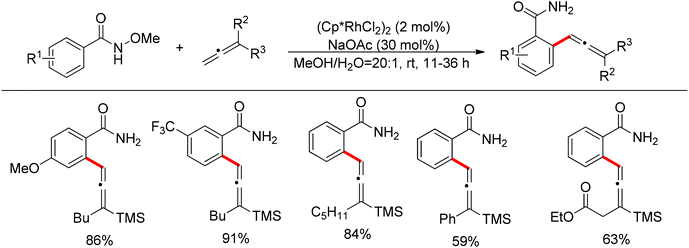 |
| | Scheme 106 Rh(III)-catalyzed Heck allenylation. | |
Although Rh(III)-catalyzed oxidizing group directed C–H bond functionalization showed good regioselectivities in most cases as aforementioned, such a strategy did not work well in the reaction of pyridine derivatives bearing oxidizing directing groups before the work of Huckins and Bercot. Taking advantage of pyridine N-oxide and amide N-oxide as double directing groups for the activation of the same C–H bond, Huckins and Bercot achieved a Rh(III)-catalyzed highly regioselective synthesis of naphthyridinone derivatives.125 Terminal alkynes and internal alkenes were effectively incorporated into the pyridine core (Scheme 107). Naphthyridinones were easily obtained by reduction of N-oxide with zinc and aqueous NH4Cl.
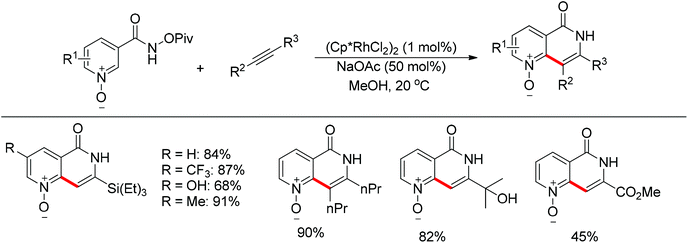 |
| | Scheme 107 Rh(III)-catalyzed regioselective synthesis of naphthyridinones. | |
Rovis and co-workers reported a new annulation reaction using diazo compounds as coupling partner and hydrogen atom acceptor to form γ-lactams (Scheme 108).126 By using the donor/acceptor diazo compounds and Cp*Rh(III) catalyst, Glorius and Cui successfully expanded the new annulation strategy to the preparation of isoquinoline N-oxides and seven-membered azepinones (Scheme 109).127
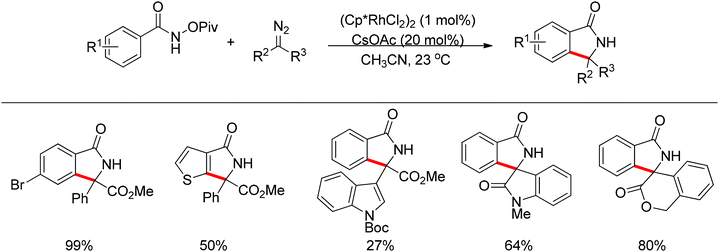 |
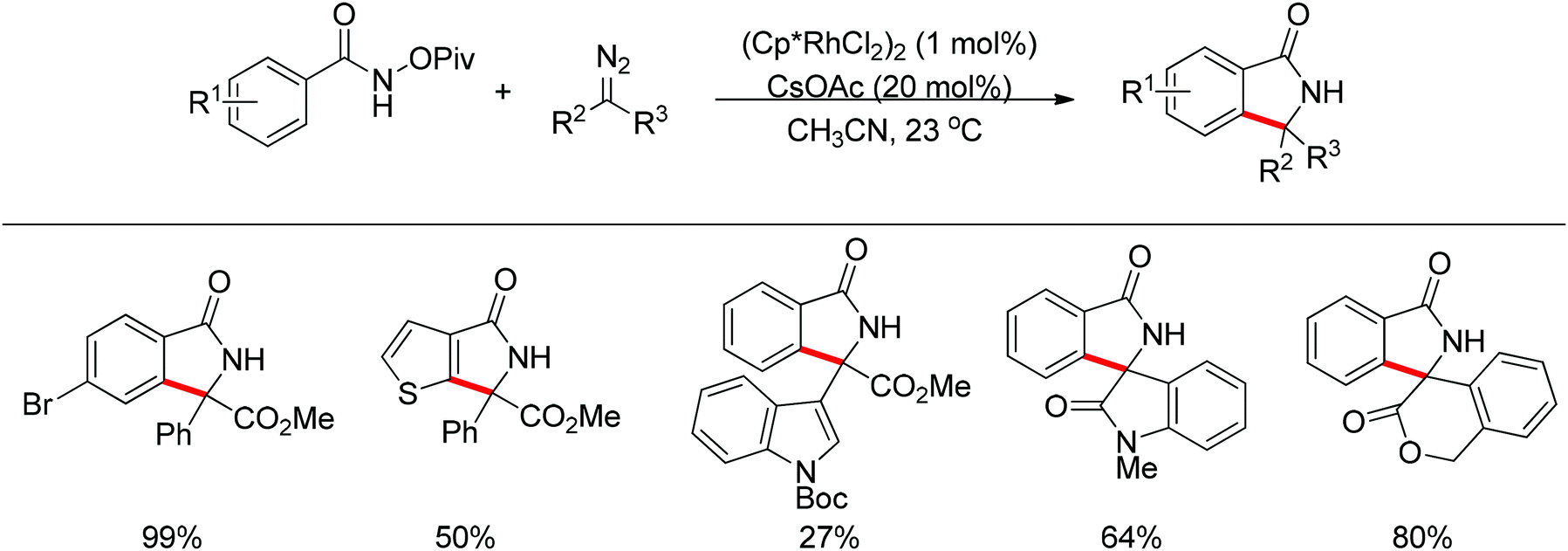
| | Scheme 108 Rh(III)-catalyzed coupling of benzamides with diazo compounds. | |
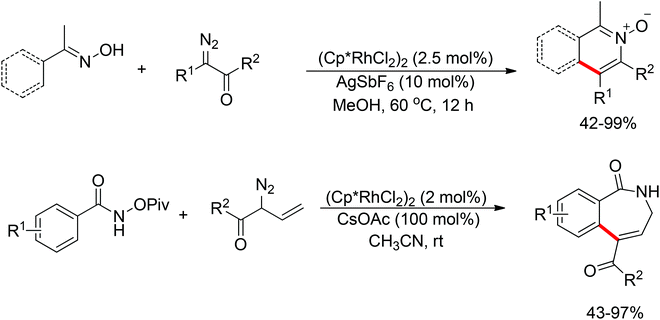 |
| | Scheme 109 Rh(III)-catalyzed annulation of oximes and benzamides with diazo compounds. | |
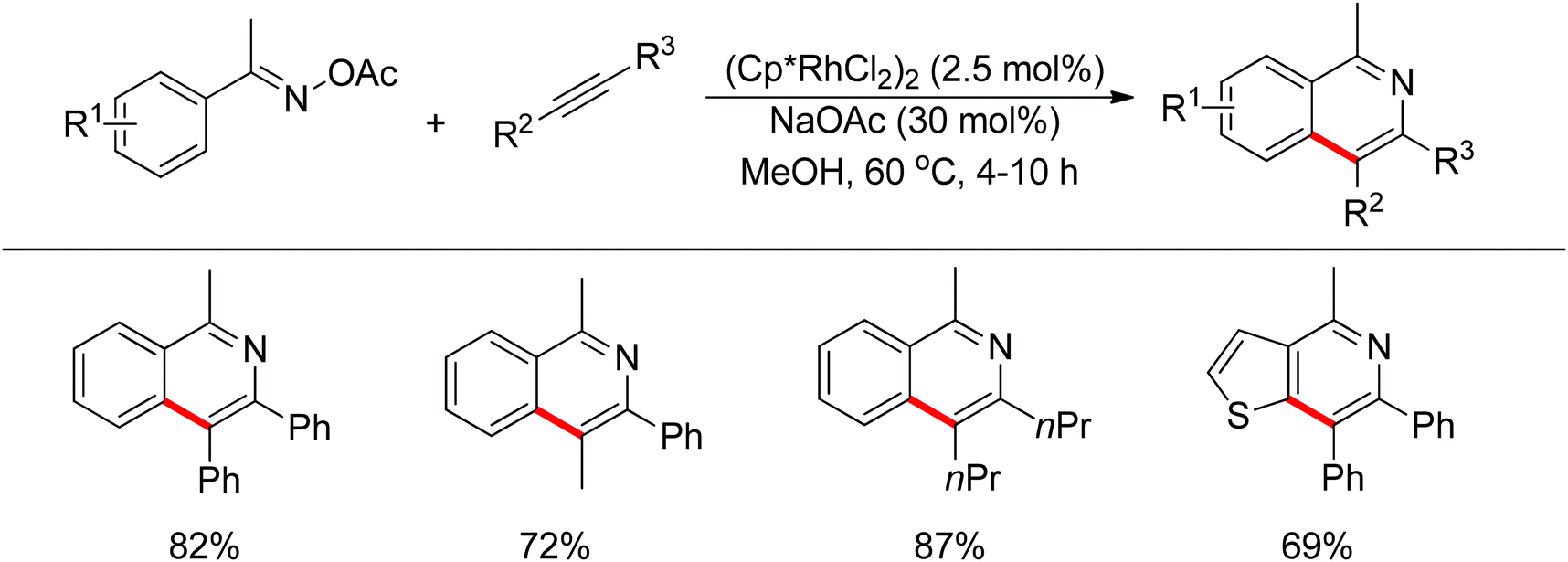
It should be mentioned that oxime ester used as directing group and internal oxidant was first introduced in Rh(III) chemistry by Chiba et al.128 Starting from aryl ketone O-acyloxime and alkynes with the aid of Cp*Rh(III) catalyst, isoquinolines were produced smoothly (Scheme 110). Later on, Chiba, Rovis and Li reported that oximines were suitable substrates in the synthesis of pyridines and isoquinolines.129 Ruthenium-catalyzed synthesis of isoquinolines using oximines as oxidizing directing groups was achieved by Jeganmohan and Ackermann as well.130 The only by-product is water, making this method a representative example of green synthesis. Oximines were previously used in the synthesis of heterocyclic compounds by Cheng, Bergman and Ellman.131
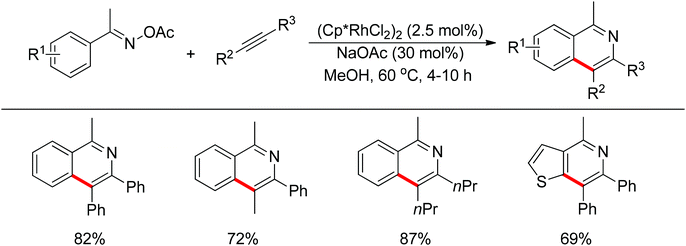 |
| | Scheme 110 Rh(III)-catalyzed synthesis of isoquinolines. | |
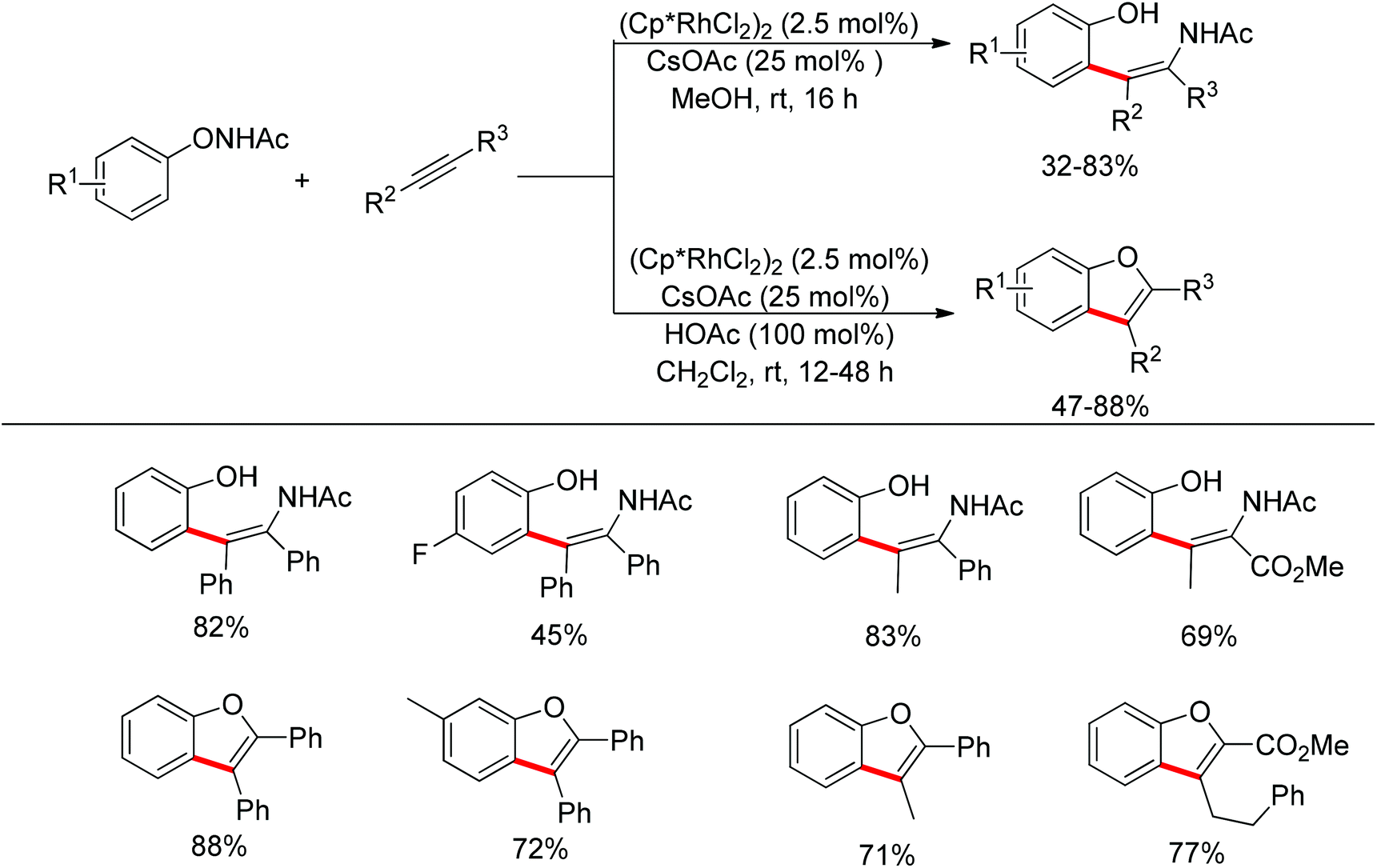
Unlike the previous reports in the transition-metal-catalyzed redox-neutral C–H functionalization, Liu and Lu developed a new oxidizing directing group that enabled tunable selectivity.132 Depending on the reaction conditions, ortho-hydroxyphenyl-substituted enamides and benzofuran derivatives could be selectively produced under mild conditions (Scheme 111). In the enamide synthesis, the amide component was incorporated into the final product while this same moiety was lost as a leaving group in the benzofuran formation. The substrate scope was expanded to alkenes.133
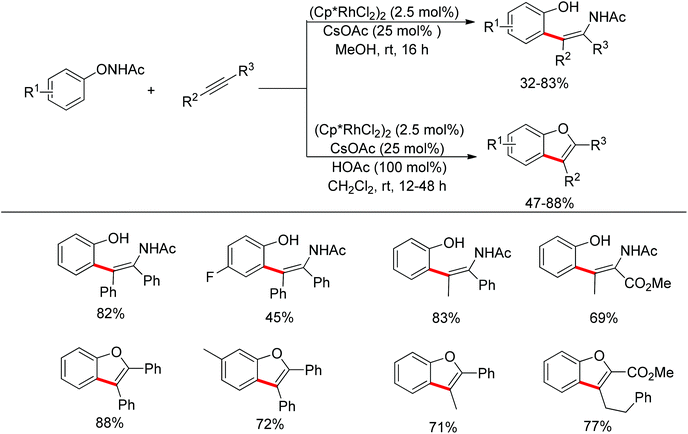 |
| | Scheme 111 Rh(III)-catalyzed selective coupling of N-phenoxyacetamides and alkynes. | |
The cleavage of N–O bonds with the departure of small molecules such as H2O, MeOH, PivOH, NH2Ac etc. features the main characters of transition-metal-catalyzed oxidizing group directed redox-neutral C–H bond functionalization.
The cleavage of N–N bonds as internal oxidant with the release of N2 in Rh(III)-catalyzed redox-neutral synthesis of isoquinolines was reported by Chiba and co-workers.134 α-Aryl vinyl azides and internal alkynes were efficiently assembled through the bimetallic cooperation of a Rh–Cu co-catalyst (Scheme 112). Chiba's work opened a new door in the rational design of oxidizing directing groups. The synthesis of isoquinolines via N–N bond cleavage was then reported by Cheng et al., who used hydrazone as both a directing group and an internal oxidant.135
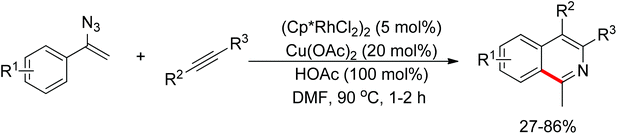 |
| | Scheme 112 Rh(III)-catalyzed synthesis of isoquinolines via N–N bond cleavage. | |
The N–N bond cleavage in redox-neutral C–H bond functionalization strategy was further developed by Zhu and Huang.136 They independently reported an N-substituted indole synthesis via Rh(III)-catalyzed N–N bond cleavage and C–C/C–N bond formation (Scheme 113). The N–N bond cleavage accompanied by the departure of NO was the key to achieve the catalyst turnover. Glorius et al. reported a similar indole synthesis based on N–N cleavage (Scheme 114).137
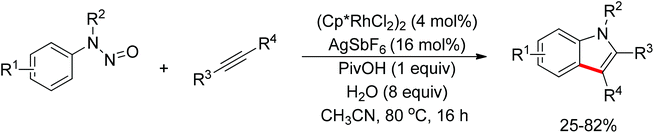 |
| | Scheme 113 Rh(III)-catalyzed indole synthesis via N–N bond cleavage. | |
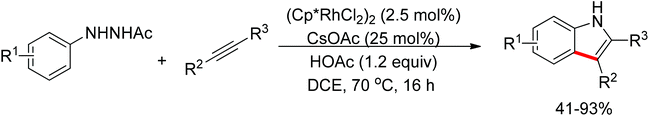 |
| | Scheme 114 Rh(III)-catalyzed hydrazine-directed synthesis of indole. | |
Very recently, You and co-workers reported a Rh(III)-catalyzed ortho-Heck reaction of aniline N-oxides.138 While N-oxides were used as internal oxidants in palladium catalysis chemistry, N-oxides as directing group and internal oxidant in rhodium chemistry had not been developed before the work of You. ortho-Alkenylated tertiary anilines were formed with H2O as the only by-product (Scheme 115).
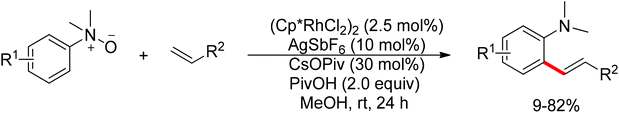 |
| | Scheme 115 Rh(III)-catalyzed olefination of aniline N-oxides. | |
The cleavage of N–O/N–N bonds as internal oxidants flourished in the redox-neutral C–H bond functionalization strategy, whereas C–N bond cleavages were scarcely investigated. To our knowledge, the only example involving C–N bond cleavage to regenerate the active catalyst was reported by Shi et al.139 Cu(OAc)2 additive was decisive to the catalyst turnover, and indenones were formed in high efficiency (Scheme 116). The mechanism was proposed by the authors as depicted in Scheme 117. Like almost all Rh(III)-catalyzed ligand directed C–H bond functionalization reactions, the catalytic cycle starts from the coordination of benzimide to the cationic rhodium species, followed by subsequent cyclometalation with the cleavage of a C–H bond. After the insertion of alkynes, this work departs from other examples of Rh(III)-catalyzed redox-neutral C–H functionalizations. An intramolecular insertion of the carbonyl group into the vinyl–Rh bond rather than intramolecular oxidative addition is carried out. According to the proposed mechanism, the authors gave a possible explanation for the beneficial effect of copper salt.
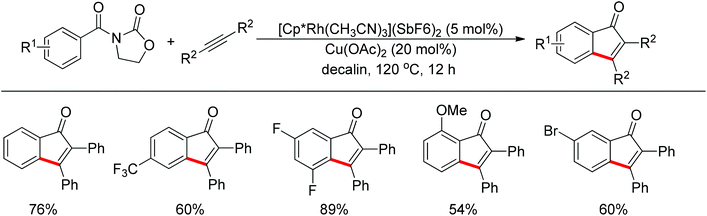 |
| | Scheme 116 Rh(III)-catalyzed synthesis of indenone via C–N bond cleavage. | |
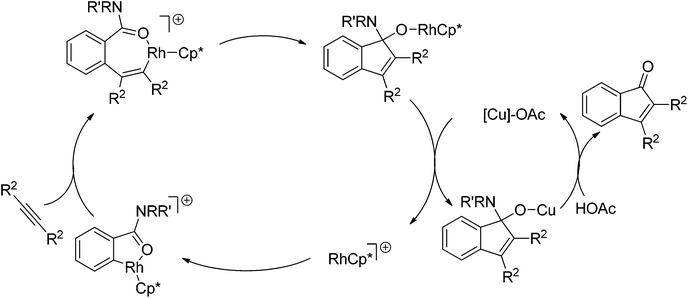 |
| | Scheme 117 Proposed reaction mechanism for Rh(III)-catalyzed synthesis of indenone via C–N bond cleavage. | |
5. Conclusion
A wide variety of monodentate nitrogen-based directing groups have been reported in the past two years. These directing groups range in structural complexity from simple to highly elaborate, and possess differing coordination strengths to metal catalysts. Collectively, this set of directing groups is compatible with many different transition metals, including Pd, Rh, Co, Cu, Ru and Ir. The field of C–H functionalization has witnessed rapid growth in the use of this class of directing groups, enabling the discovery of numerous new transformations. The key challenges of reactivity and site selectivity have been addressed to a great extent and are still driving forces for more discovery and design in this area. The increasingly widespread use of nitrogen-based directing groups can be attributed to their diverse Lewis basicity that results from various types of structures and bonding modes of nitrogen atom, stability, and ease of installation/removal. The removal of some commonly used nitrogen-based directing groups, such as acetanilides, oxazolines, and pyridines, however, is still problematic, requiring several steps or proving impossible altogether. As such, there remains a need for additional directing groups that are simple to install, remove, and/or further manipulate. An appealing work-around is generating the active directing group in situ, ideally through the use of catalytic quantities of an additive, as exemplified above. Furthermore, many nitrogen-based directing groups are presently limited by narrow scope in terms of the types of transformations that can be performed. This could be due to the strong coordination of nitrogen to various metal centers. An additional limitation at present is that many common nitrogen-based directing groups are not an intrinsic part of the substrate and/or product, which restricts practical applications. In this regard, there are tremendous opportunities to explore C–H functionalization reactions employing other common functional groups, especially alcohols, esters, ketones, carboxylic acids; these functional groups are weaker Lewis bases and are present in many organic molecules.140 Carbon-based directing groups, such as alkene,141 alkyne,142 and carbanion,143 and sulfur-based and phosphorus-based directing groups, such as thioether144 and phosphate145 are also potential new types of directing groups. Meanwhile, to reduce the cost of C–H functionalization reactions, most of which now rely on expensive precious metals, research on C–H functionalization with comparatively inexpensive first-row transition metals will become increasingly important in the coming years.3m,s,146 Achieving reactivity with these metals may require the development of novel compatible directing groups.
Selectivity, including site- and stereoselectivity, is crucial for the widespread application of C–H functionalization in academia and industry. Most of the reactions discussed above are ortho-selective or involve activation of a proximal C–H bond. Reactions that are meta- or para-selective and reactions in which a remote C–H bond is selectively functionalized hold great potential; and some excellent examples of this kind have been reviewed above. Asymmetric C–H activation, especially of methylene C–H bonds, remains in its infancy, though promising lead results have been disclosed.147 This issue remains among the most challenging in the field; to overcome it, advances in ligand design (and chiral auxiliary design) and further mechanistic studies are needed. Another potential approach is to engineer artificial metalloenzymes, which offer high control of enantioselectivity as well as site selectivity.119a Overall, numerous challenges remain in the field of C–H functionalization, and this research area promises to be fertile ground for exciting discoveries in the coming years.
Acknowledgements
Financial support from the 973 Program (2011CB932404, 2011CBA00501) and NSFC (21301173, 21332001, 21302184 and 21202164) is greatly appreciated. We gratefully thank the reviewers for their insightful comments and Dr Keary M. Engle (The California Institute of Technology) for helpful discussions.
References
-
(a) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259 CrossRef CAS;
(b) C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed;
(c) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC.
-
(a) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC;
(b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
-
(a) A. D. Ryabov, Synthesis, 1985, 233 CrossRef CAS;
(b) R. H. Crabtree, Chem. Rev., 1985, 85, 245 CrossRef CAS;
(c) A. D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS;
(d) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed;
(e) S. S. Stahl, J. A. Labinger and J. E. Bercaw, Angew. Chem., Int. Ed., 1998, 37, 2180 CrossRef;
(f) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed;
(g) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed;
(h) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS;
(i) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed;
(j) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed;
(k) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed;
(l) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed;
(m) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS;
(n) P. Thansandote and M. Lautens, Chem. – Eur. J., 2009, 15, 5874 CrossRef CAS PubMed;
(o) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed;
(p) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed;
(q) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed;
(r) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC;
(s) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed;
(t) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed;
(u) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS;
(v) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC;
(w) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed;
(x) C. Zhu, R. Wang and J. R. Falck, Chem. – Asian J., 2012, 7, 1502 CrossRef CAS PubMed;
(y) Y. Wang, G. Cheng and X. Cui, Chin. J. Org. Chem., 2012, 32, 2018 CrossRef CAS;
(z) Y. Huang and C. Wang, Synlett, 2012, 145 CrossRef PubMed;
(a
a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed;
(a
b) D. Li, C. He, H. Cai and G. Wang, Chin. J. Org. Chem., 2013, 33, 203 CrossRef CAS;
(a
c) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC;
(a
d) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC.
-
(a) J. P. Kleiman and M. Dubeck, J. Am. Chem. Soc., 1963, 85, 1544 CrossRef CAS;
(b) A. C. Cope and R. W. Siekman, J. Am. Chem. Soc., 1965, 87, 3272 CrossRef CAS;
(c) M. A. Bennett and D. L. Milner, Chem. Commun., 1967, 581 RSC;
(d) W. Keim, J. Org. Chem., 1968, 14, 179 CrossRef CAS;
(e) W. H. Knoth and R. A. Schunn, J. Am. Chem. Soc., 1969, 91, 2400 CrossRef CAS;
(f) A. C. Cope and E. C. Friedrich, J. Am. Chem. Soc., 1968, 90, 909 CrossRef CAS;
(g) G. E. Hartwell, R. V. Lawrence and M. J. Smas, J. Chem. Soc., Chem. Commun., 1970, 912 RSC;
(h) A. G. Constable, W. S. McDonald, L. C. Sawkins and B. L. Shaw, J. Chem. Soc., Chem. Commun., 1978, 1061 RSC.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed.
- G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed.
- R. Feng, J. Yao, Z. Liang, Z. Liu and Y. Zhang, J. Org. Chem., 2013, 78, 3688 CrossRef CAS PubMed.
- D.-W. Gao, Y.-C. Shi, Q. Gu, Z.-L. Zhao and S.-L. You, J. Am. Chem. Soc., 2012, 135, 86 CrossRef PubMed.
- Z. Liang, L. Ju, Y. Xie, L. Huang and Y. Zhang, Chem. – Eur. J., 2012, 18, 15816 CrossRef CAS PubMed.
- Z. Liang, R. Feng, H. Yin and Y. Zhang, Org. Lett., 2013, 15, 4544 CrossRef CAS PubMed.
- J. Shao, W. Chen, M. A. Giulianotti, R. A. Houghten and Y. Yu, Org. Lett., 2012, 14, 5452 CrossRef CAS PubMed.
-
(a) M. Miura, T. Tsuda, T. Satoh, S. Pivsa-Art and M. Nomura, J. Org. Chem., 1998, 63, 5211 CrossRef CAS;
(b) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed;
(c) X. Wan, Z. Ma, B. Li, K. Zhang, S. Cao, S. Zhang and Z. Shi, J. Am. Chem. Soc., 2006, 128, 7416 CrossRef CAS PubMed.
- D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed.
- G.-W. Wang, T.-T. Yuan and D.-D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380 CrossRef CAS PubMed.
- J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880 CrossRef CAS PubMed.
- J. Karthikeyan, R. Haridharan and C. H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 12343 CrossRef CAS PubMed.
- D. D. Li, T. T. Yuan and G. W. Wang, J. Org. Chem., 2012, 77, 3341 CrossRef CAS PubMed.
- H. X. Dai, A. F. Stepan, M. S. Plummer, Y. H. Zhang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS PubMed.
- C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242 CrossRef CAS PubMed.
- B. Liu, Y. Fan, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2013, 135, 468 CrossRef CAS PubMed.
- W. Zhen, F. Wang, M. Zhao, Z. Du and X. Li, Angew. Chem., Int. Ed., 2012, 51, 11819 CrossRef CAS PubMed.
- M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 6993 CrossRef CAS PubMed.
- B. Gong, J. Shi, X. Wang, Y. Yan, Q. Li, Y. Meng, H. E. Xu and W. Yi, Adv. Synth. Catal., 2014, 356, 137 CrossRef CAS.
- J. Dong, Z. Long, F. Song, N. Wu, Q. Guo, J. Lan and J. You, Angew. Chem., Int. Ed., 2013, 52, 580 CrossRef CAS PubMed.
- J. Wencel-Delord, C. Nimphius, H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 13001 CrossRef CAS PubMed.
- J. Wencel-Delord, C. Nimphius, F. W. Patureau and F. Glorius, Chem. – Asian J., 2012, 7, 1208 CrossRef CAS PubMed.
- B. Liu, Y. Huang, J. Lan, F. Song and J. You, Chem. Sci., 2013, 4, 2163 RSC.
- Y. Shang, X. Jie, H. Zhao, P. Hu and W. Su, Org. Lett., 2014, 16, 416 CrossRef CAS PubMed.
- S. Murahashi, J. Am. Chem. Soc., 1955, 77, 6403 CrossRef CAS.
- K. Gao, P.-S. Lee, C. Long and N. Yoshikai, Org. Lett., 2012, 14, 4234 CrossRef CAS PubMed.
- B. Punji, W. Song, G. A. Shevchenko and L. Ackermann, Chem. – Eur. J., 2013, 19, 10605 CrossRef CAS PubMed.
- W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251 CrossRef CAS PubMed.
- M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680 CrossRef CAS PubMed.
- K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541 CrossRef CAS PubMed.
- M. Wasa, K. S. Chan, X. G. Zhang, J. He, M. Miura and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570 CrossRef CAS PubMed.
- L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC.
- Q. Chen, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2010, 133, 428 CrossRef PubMed.
- K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279 CrossRef CAS PubMed.
- W. W. Chan, S. F. Lo, Z. Zhou and W. Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed.
- X. Yu, S. Yu, J. Xiao, B. Wan and X. Li, J. Org. Chem., 2013, 78, 5444 CrossRef CAS PubMed.
- X. Wang, L. Truesdale and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3648 CrossRef CAS PubMed.
- X.-G. Zhang, H.-X. Dai, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 11948 CrossRef CAS PubMed.
- M. Miura, C.-G. Feng, S. Ma and J.-Q. Yu, Org. Lett., 2013, 15, 5258 CrossRef CAS PubMed.
- N. Chatani, M. Tobisu and Y. Ano, Synlett, 2012, 2763 Search PubMed.
- S. H. Kim, S. H. Park and S. Chang, Tetrahedron, 2012, 68, 5162 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS PubMed.
- T. J. Gong, B. Xiao, W. M. Cheng, W. Su, J. Xu, Z. J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS PubMed.
- J. Kwak, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778 CrossRef CAS PubMed.
- X. Li, S. Yu, F. Wang, B. Wan and X. Yu, Angew. Chem., Int. Ed., 2013, 52, 2577 CrossRef CAS PubMed.
- C. Suzuki, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3990 CrossRef CAS PubMed.
- M. V. Pham, B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 10610 CrossRef CAS PubMed.
- Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 4698 CrossRef CAS PubMed.
- T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed.
- T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Chem. – Eur. J., 2013, 19, 9142 CrossRef CAS PubMed.
- Y. Li, B.-J. Li, W.-H. Wang, W.-P. Huang, X.-S. Zhang, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 2115 CrossRef CAS PubMed.
- A. S. Tsai, M. E. Tauchert, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2011, 133, 1248 CrossRef CAS PubMed.
- Y. Li, X. S. Zhang, K. Chen, K. H. He, F. Pan, B. J. Li and Z. J. Shi, Org. Lett., 2012, 14, 636 CrossRef CAS PubMed.
- Y. Li, X. S. Zhang, Q. L. Zhu and Z. J. Shi, Org. Lett., 2012, 14, 4498 CrossRef CAS PubMed.
- Y. Lian, R. G. Bergman and J. A. Ellman, Chem. Sci., 2012, 3, 3088 RSC.
- K. Gao and N. Yoshikai, Chem. Commun., 2012, 48, 4305 RSC.
-
(a) C.-H. Jun, J.-B. Hong, Y.-H. Kim and K.-Y. Chung, Angew. Chem., Int. Ed., 2000, 39, 3440 CrossRef CAS;
(b) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed.
- S. Chen, J. Yu, Y. Jiang, F. Chen and J. Cheng, Org. Lett., 2013, 15, 4754 CrossRef CAS PubMed.
- P. W. Tan, N. A. B. Juwaini and J. Seayad, Org. Lett., 2013, 15, 5166 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC.
- J. Y. Kim, S. H. Park, J. Ryu, S. H. Cho, S. H. Kim and S. Chang, J. Am. Chem. Soc., 2012, 134, 9110 CrossRef CAS PubMed.
- J. Ryu, K. Shin, S. H. Park, J. Y. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 9904 CrossRef CAS PubMed.
- K. Shin, Y. Baek and S. Chang, Angew. Chem., Int. Ed., 2013, 52, 8031 CrossRef CAS PubMed.
- S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS PubMed.
- H. Zhao, Y. Shang and W. Su, Org. Lett., 2013, 15, 5106 CrossRef CAS PubMed.
- C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed.
- Q. Jiang, D. Duan-Mu, W. Zhong, H. Chen and H. Yan, Chem. – Eur. J., 2013, 19, 1903 CrossRef CAS PubMed.
- W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362 CrossRef CAS PubMed.
- N. Chernyak, A. S. Dudnik, C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 8270 CrossRef CAS PubMed.
- A. V. Gulevich, F. S. Melkonyan, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 5528 CrossRef CAS PubMed.
- S. Bhadra, C. Matheis, D. Katayev and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 9279 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2009, 110, 890 CrossRef PubMed.
- A. J. Roering, L. V. A. Hale, P. A. Squier, M. A. Ringgold, E. R. Wiederspan and T. B. Clark, Org. Lett., 2012, 14, 3558 CrossRef CAS PubMed.
- K. M. Waltz and J. F. Hartwig, Science, 1997, 277, 211 CrossRef CAS.
- T. Mita, Y. Ikeda, K. Michigami and Y. Sato, Chem. Commun., 2013, 49, 5601 RSC.
- H. X. Dai and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 134 CrossRef CAS PubMed.
- Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431 CrossRef CAS PubMed.
- C.-G. Feng, M. Ye, K.-J. Xiao, S. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 9322 CrossRef CAS PubMed.
- T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455 CrossRef CAS PubMed.
- C. Li, T. Yano, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9801 CrossRef CAS PubMed.
-
(a) J. J. Li, T. S. Mei and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed;
(b) J.-J. Li, R. Giri and J.-Q. Yu, Tetrahedron, 2008, 64, 6979 CrossRef CAS PubMed.
-
(a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523 CrossRef CAS PubMed;
(b) S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142 CrossRef CAS PubMed;
(c) F. Kakiuchi, T. Kochi, H. Mutsutani, N. Kobayashi, S. Urano, M. Sato, S. Nishiyama and T. Tanabe, J. Am. Chem. Soc., 2009, 131, 11310 CrossRef CAS PubMed;
(d) B. Song, X. Zheng, J. Mo and B. Xu, Adv. Synth. Catal., 2010, 352, 329 CrossRef CAS;
(e) R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chem., Int. Ed., 2011, 50, 5524 CrossRef CAS PubMed.
- X.-T. Ma and S.-K. Tian, Adv. Synth. Catal., 2013, 355, 337 CAS.
- B. Du, X. Jiang and P. Sun, J. Org. Chem., 2013, 78, 2786 CrossRef CAS PubMed.
- K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed.
-
(a) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed;
(b) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed.
- S.-J. Lou, D.-Q. Xu, A.-B. Xia, Y.-F. Wang, Y.-K. Liu, X.-H. Du and Z.-Y. Xu, Chem. Commun., 2013, 49, 6218 RSC.
- K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094 CrossRef CAS PubMed.
-
(a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2001, 124, 390 CrossRef PubMed;
(b) J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr. and M. R. Smith, Science, 2002, 295, 305 CrossRef CAS PubMed.
- Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS PubMed.
-
(a) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS PubMed;
(b) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS PubMed;
(c) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed.
- H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed.
- L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056 CrossRef CAS PubMed.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778 CrossRef CAS PubMed.
- K. Sun, Y. Li, T. Xiong, J. Zhang and Q. Zhang, J. Am. Chem. Soc., 2011, 133, 1694 CrossRef CAS PubMed.
- J. P. Brand and J. Waser, Org. Lett., 2012, 14, 744 CrossRef CAS PubMed.
-
(a) M. Catellani, E. Motti and N. Della Ca’, Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS PubMed;
(b) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2010, 292, 1 CrossRef CAS.
- L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed.
- L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563 CrossRef CAS PubMed.
- L. Jiao and T. Bach, Angew. Chem., Int. Ed., 2013, 52, 6080 CrossRef CAS PubMed.
- Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350 CrossRef CAS PubMed.
- J. Kwak, M. Kim and S. Chang, J. Am. Chem. Soc., 2011, 133, 3780 CrossRef CAS PubMed.
- M.-L. Louillat and F. W. Patureau, Org. Lett., 2012, 15, 164 CrossRef PubMed.
- F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed.
- J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef CAS PubMed.
-
(a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed;
(b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed.
-
(a) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed;
(b) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed.
-
(a) T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed;
(b) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed;
(c) T. A. Davis, T. K. Hyster and T. Rovis, Angew. Chem., Int. Ed., 2013, 52, 14181 CrossRef CAS PubMed;
(d) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507 CrossRef CAS PubMed.
-
(a) S. Cui, Y. Zhang and Q. Wu, Chem. Sci., 2013, 4, 3421 RSC;
(b) M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, Org. Lett., 2013, 15, 1528 CrossRef CAS PubMed.
-
(a) B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573 CrossRef CAS PubMed;
(b) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548 CrossRef CAS PubMed;
(c) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS PubMed.
-
(a) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed;
(b) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592 CrossRef CAS PubMed.
- X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS PubMed.
- R. Zeng, S. Wu, C. Fu and S. Ma, J. Am. Chem. Soc., 2013, 135, 18284 CrossRef CAS PubMed.
- J. R. Huckins, E. A. Bercot, O. R. Thiel, T.-L. Hwang and M. M. Bio, J. Am. Chem. Soc., 2013, 135, 14492 CrossRef CAS PubMed.
- T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed.
-
(a) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204 CrossRef CAS PubMed;
(b) S. Cui, Y. Zhang, D. Wang and Q. Wu, Chem. Sci., 2013, 4, 3912 RSC.
-
(a) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed;
(b) P. C. Too, S. H. Chua, S. H. Wong and S. Chiba, J. Org. Chem., 2011, 76, 6159 CrossRef CAS PubMed.
-
(a) P. Too, T. Noji, Y. Lim, X. Li and S. Chiba, Synlett, 2011, 2789 CAS;
(b) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC;
(c) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS.
-
(a) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed;
(b) C. Kornhaass, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190 CrossRef CAS PubMed.
-
(a) K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2007, 10, 325 CrossRef PubMed;
(b) K. Parthasarathy and C.-H. Cheng, J. Org. Chem., 2009, 74, 9359 CrossRef CAS PubMed;
(c) K. Parthasarathy and C.-H. Cheng, Synthesis, 2009, 1400 CAS;
(d) R. M. Martin, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2012, 77, 2501 CrossRef CAS PubMed.
- G. Liu, Y. Shen, Z. Zhou and X. Lu, Angew. Chem., Int. Ed., 2013, 52, 6033 CrossRef CAS PubMed.
- Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366 CrossRef CAS PubMed.
- Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed.
- S.-C. Chuang, P. Gandeepan and C.-H. Cheng, Org. Lett., 2013, 15, 5750 CrossRef CAS PubMed.
-
(a) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed;
(b) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294 CrossRef CAS PubMed.
- D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed.
- X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970 CrossRef CAS PubMed.
- B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS PubMed.
-
(a) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740 CrossRef CAS;
(b) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed;
(c) H. A. Chiong, Q. N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS PubMed;
(d) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2009, 132, 468 CrossRef PubMed;
(e) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed;
(f) P. Gandeepan, K. Parthasarathy and C.-H. Cheng, J. Am. Chem. Soc., 2010, 132, 8569 CrossRef CAS PubMed;
(g) K. Morimoto, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 9548 CrossRef CAS PubMed;
(h) S. H. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372 CrossRef CAS PubMed;
(i) B. Xiao, T.-J. Gong, J. Xu, Z.-J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466 CrossRef CAS PubMed;
(j) A. Iglesias, R. Alvarez, A. R. de Lera and K. Muniz, Angew. Chem., Int. Ed., 2012, 51, 2225 CrossRef CAS PubMed;
(k) K. Graczyk, W. Ma and L. Ackermann, Org. Lett., 2012, 14, 4110 CrossRef CAS PubMed;
(l) F. Mo, L. J. Trzepkowski and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 13075 CrossRef CAS PubMed;
(m) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070 CrossRef CAS PubMed;
(n) Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574 CrossRef CAS PubMed;
(o) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed;
(p) X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed.
-
(a) P. Gandeepan and C.-H. Cheng, J. Am. Chem. Soc., 2012, 134, 5738 CrossRef CAS PubMed;
(b) P. Gandeepan and C.-H. Cheng, Org. Lett., 2013, 15, 2084 CrossRef CAS PubMed.
-
(a) N. Chernyak and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 5636 CrossRef CAS PubMed;
(b) Y. Minami, Y. Shiraishi, K. Yamada and T. Hiyama, J. Am. Chem. Soc., 2012, 134, 6124 CrossRef CAS PubMed;
(c) Y. Minami, K. Yamada and T. Hiyama, Angew. Chem., Int. Ed., 2013, 52, 10611 CrossRef CAS PubMed.
- X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163 CrossRef CAS PubMed.
-
(a) J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205 CrossRef CAS;
(b) M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164 CrossRef CAS PubMed;
(c) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898 CrossRef CAS PubMed.
-
(a) L. Y. Chan, L. Cheong and S. Kim, Org. Lett., 2013, 15, 2186 CrossRef CAS PubMed;
(b) X. Meng and S. Kim, Org. Lett., 2013, 15, 1910 CrossRef CAS PubMed;
(c) H.-L. Wang, R.-B. Hu, H. Zhang, A.-X. Zhou and S.-D. Yang, Org. Lett., 2013, 15, 5302 CrossRef CAS PubMed;
(d) D. Eom, Y. Jeong, Y. R. Kim, E. Lee, W. Choi and P. H. Lee, Org. Lett., 2013, 15, 5210 CrossRef CAS PubMed;
(e) J. Seo, Y. Park, I. Jeon, T. Ryu, S. Park and P. H. Lee, Org. Lett., 2013, 15, 3358 CrossRef CAS PubMed;
(f) T. Ryu, J. Kim, Y. Park, S. Kim and P. H. Lee, Org. Lett., 2013, 15, 3986 CrossRef CAS PubMed;
(g) D. Zhao, C. Nimphius, M. Lindale and F. Glorius, Org. Lett., 2013, 15, 4504 CrossRef CAS PubMed;
(h) Y. Unoh, Y. Hashimoto, D. Takeda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2013, 15, 3258 CrossRef CAS PubMed.
-
(a) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed;
(b) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC.
-
(a) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed;
(b) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed;
(c) M. Nakanishi, D. Katayev, C. Besnard and E. P. Kündig, Angew. Chem., Int. Ed., 2011, 50, 7438 CrossRef CAS PubMed;
(d) X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2014 |