 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-(Aryl-sulfonyl)oxetanes as designer 3-dimensional fragments for fragment screening: synthesis and strategies for functionalisation†
Kate F.
Morgan
a,
Ian A.
Hollingsworth
b and
James A.
Bull
*a
aDepartment of Chemistry Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: j.bull@imperial.ac.uk; Tel: +44 (0)207 594 5811
bAstraZeneca Mereside, Alderley Park, Cheshire, SK10 4TG, UK
First published on 25th October 2013
Abstract
2-Sulfonyl-oxetanes have been prepared, affording non-planar structures with desirable physicochemical properties for fragment based drug discovery. The oxetane motif was formed by an intramolecular C–C bond formation. The fragments were further functionalised via organometallic intermediates at the intact oxetane and aromatic rings.
Fragment based drug discovery (FBDD) is now a well-established approach in the development of new drugs and lead compounds.1 Fragments provide desirable starting points for discovery chemistry that allow increases in MW and lipophilicity during the optimisation of potency and selectivity,2,3 whilst remaining in drug-like chemical space.4 The design of fragment libraries is a crucial element to the success of screening. Although desirable criteria for properties of fragments can vary by biological target, guidelines for ‘fragment space’ proposed by Astex are widely cited (Rule-of-3: MW < 300, c
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP ≤ 3, number of H-bond donors–acceptors ≤3).5 It has recently been suggested that the incorporation of more H-bond acceptors, up to 6, is advantageous in affording additional binding elements and further points for derivatisation during optimisation.6 Fragment screening samples a larger portion of available chemical space and as such FBDD is also a promising approach for complex and challenging targets such as protein–protein interactions.7 Fragment libraries dominated by sp2 rich molecules have been less successful in generating hits for these targets, suggesting more ‘3-dimensional’ fragments are likely to be required.8 Indeed, more sp3-rich molecules and aliphatic heterocycles can offer improved levels of success through development, relative to highly aromatic compounds.9,10
logP ≤ 3, number of H-bond donors–acceptors ≤3).5 It has recently been suggested that the incorporation of more H-bond acceptors, up to 6, is advantageous in affording additional binding elements and further points for derivatisation during optimisation.6 Fragment screening samples a larger portion of available chemical space and as such FBDD is also a promising approach for complex and challenging targets such as protein–protein interactions.7 Fragment libraries dominated by sp2 rich molecules have been less successful in generating hits for these targets, suggesting more ‘3-dimensional’ fragments are likely to be required.8 Indeed, more sp3-rich molecules and aliphatic heterocycles can offer improved levels of success through development, relative to highly aromatic compounds.9,10
Oxetanes have recently been highlighted as desirable low molecular weight motifs for drug discovery.11 Carreira, Rogers-Evans, Müller, and coworkers have shown an oxetane motif can act as an isosteric, polar replacement for a gem-dimethyl group or a carbonyl, which can result in improved physicochemical and biochemical properties relative to the parent molecule.12 Enhanced solubility, reduced lipophilicity, reduced hERG liability as well as improved metabolic stability were observed as potential beneficial effects. The oxetane motif is also found in several biologically active natural products, such as oxetanocin, taxol and mitrophorone.13 As a result, oxetanes have recently received significant interest in medicinal chemistry.14
We are interested in the preparation of novel non-planar fragments, with desirable properties, which contain biologically important motifs, and access new areas of chemical space.15 Consequently we designed 2-sulfonyl oxetanes (Fig. 1), as non-planar fragments to comply with the rule-of-3, modified for the number of hydrogen bond acceptors (HBA).16 We envisaged that these small and functional group rich molecules, with the potential to make interesting interactions, would be desirable fragments for screening in drug discovery or chemical biology programmes. In addition they would allow the structure to be ‘grown’ or ‘linked’ in several directions for optimisation, were they to be a hit. Here we report the synthesis of sulfonyl oxetanes as well as their further functionalization.
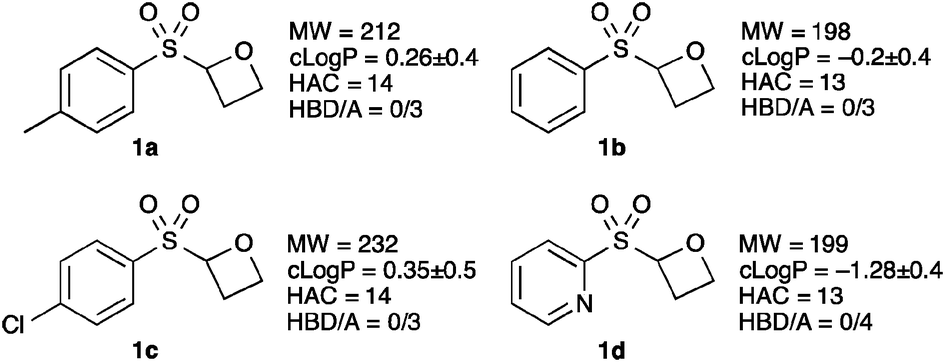 | ||
| Fig. 1 Sulfonyl-oxetane targets. MW = molecular weight; clogP = calculated logP (lipophilicity);‡ HAC = heavy atom count (number of non-H atoms); HBD/A = number of hydrogen bond donors–acceptors. | ||
The synthesis of oxetanes remains a challenge. Methods for the synthesis of oxetanes are mostly limited to two general approaches: photochemical Paternò–Büchi [2+2] reactions of carbonyl compounds with alkenes,17 or intramolecular Williamson etherification (Fig. 2).11,18 Recently epoxide ring opening/ring closing has been exploited as a facile route to the activated intermediate that undergoes etherification.19 Substituted oxetanes have also been generated from exo-methylene oxetanes.20 However, these approaches are unsuitable for our targets, particularly due to the instability of the often required α-sulfonyl oxy-anion intermediate.21
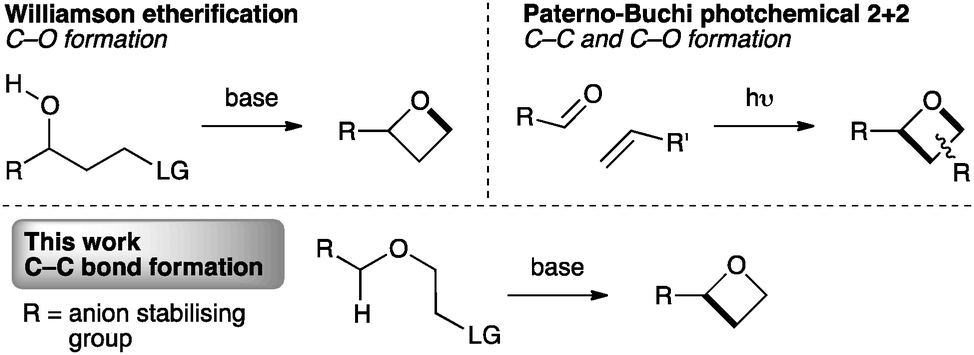 | ||
| Fig. 2 Approaches to 2-substituted oxetanes. | ||
To incorporate the 2-sulfonyl group, we envisaged a strategy involving C–C bond formation, as opposed to the usual C–O bond formation (Fig. 2, lower). While this approach is necessitated in ring closure to form cyclobutanes and is known in the synthesis of azetidines,22 it has not been exploited for oxetane synthesis. For oxetanes, a comparable mode of cyclisation has only been observed in intramolecular epoxide opening of lithiated benzyl ethers, using LDA-KOtBu at −78 °C.23,24
The route developed to sulfonyl-oxetanes 1a–d is shown in Scheme 1. The cyclisation precursors were accessed by alkylation of ethylene glycol with chloromethyl aryl-sulfide to afford S,O-acetal 3, using ethylene glycol as solvent to avoid double alkylation. Tosylation25 followed by oxidation with mCPBA afforded sulfone 4. The crucial step, the 4-exo-tet cyclisation, was optimised with 4a, varying reaction temperature, time, base and equivalents, solvent and concentration.26 Excellent yields were obtained using LiHMDS (1.1 equiv.) at 0 °C in THF. Under these conditions the reaction was complete in 1 h, with the carbenoid-like organolithium intermediate stable to decomposition. Cyclisation did not occur at lower temperatures and a larger excess of base led to decomposition of the product. The route could be readily scaled and the cyclisation was performed on 6.5 mmol affording >1 g of oxetane 1a in 93% yield. By this method the synthesis of oxetanes 1b–d, were similarly successful, modifying the size and electronics of the aromatic, including the incorporation of a pyridine ring.
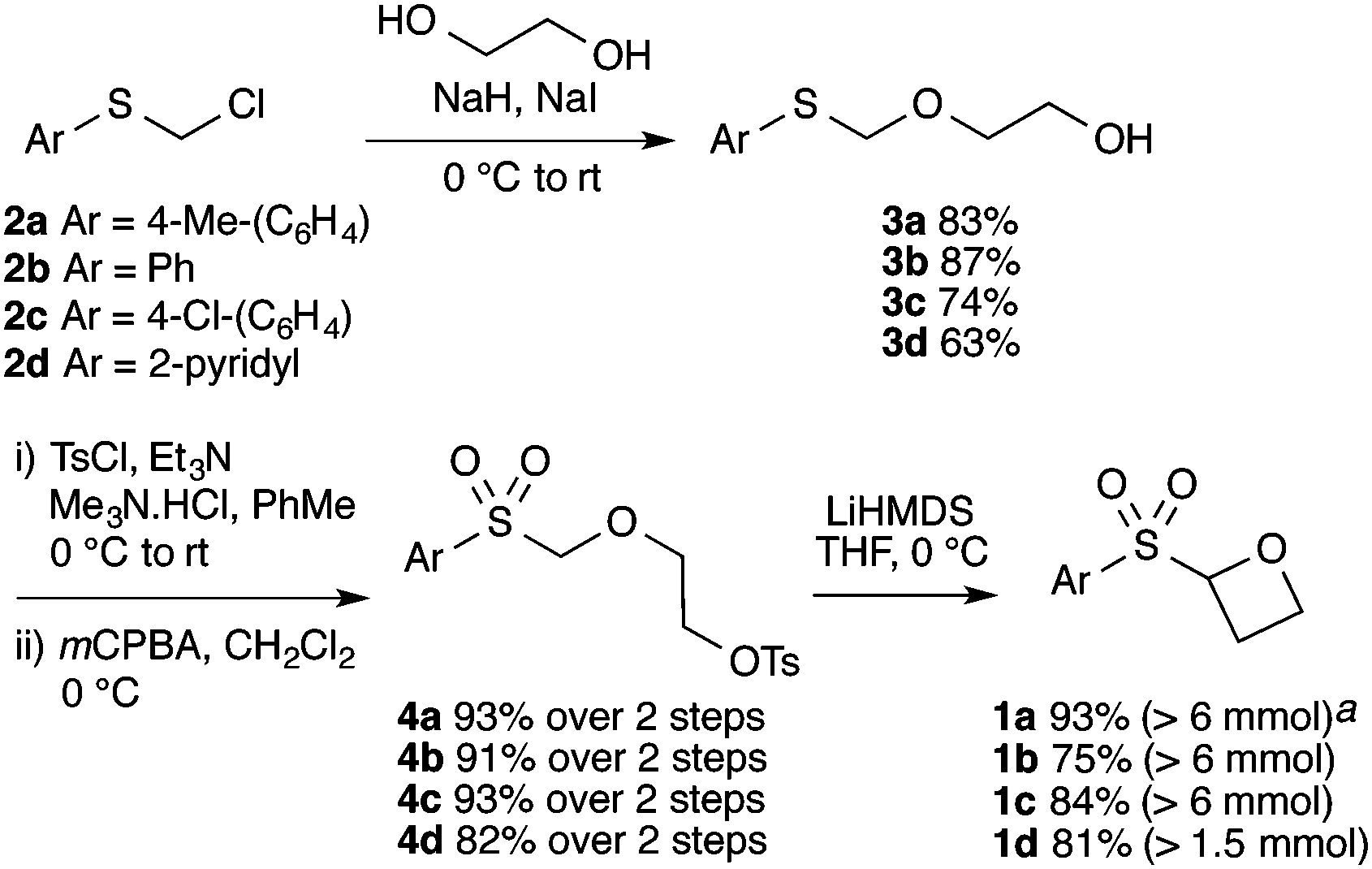 | ||
| Scheme 1 Synthetic route to sulfonyl oxetanes 1a–dvia C–C bond formation. aReaction scale in parentheses: mmol of 4 employed. | ||
An important part of our design was the ability to ‘grow’ the fragments by subsequent C–C bond formation in various directions. Therefore, having validated the route to the sulfonyl oxetanes we focused on the functionalisation of this molecule as a way to access a variety of fragment-like, and larger lead-like compounds. Sulfones can facilitate a wide array of transformations and here we envisaged they would allow functionalization of the intact oxetane ring by metallation. Capriati and co-workers recently reported an efficient route to 2-substituted phenyloxetanes via 2-lithio-2-phenyloxetane, formed by deprotonation with sBuLi, which was reacted with electrophiles.27 We envisaged that the sulfonyl oxetane would undergo regioselective deprotonation on the oxetane ring and this was examined first. We investigated several bases to successfully perform this deprotonation and identified 2 sets of reaction conditions that were appropriate for different electrophiles.26 The use of LiHMDS (1.2 equiv.) at −78 °C in THF afforded clean reactions with compatible electrophiles (Table 1, conditions A). Under these conditions methyl, ethyl and allyl groups were introduced in excellent yield (entries 1, 3 and 4).16
|
|
|||||
|---|---|---|---|---|---|
| Entry | Electrophile (E+) | Conditionsa | Oxetane | Yield (%) | |
|
a Conditions A: LiHMDS (1.2 equiv.), electrophile (2 equiv.) THF, −78 °C, 90 min; conditions B: nBuLi (1.3 equiv.), electrophile (2 equiv.) THF, −78 °C, 30 min.
b Isolated as a mixture of diastereoisomers, d.r. = 1:0.9.
|
|||||
| 1 | MeI | A |

|
5 | 93 |
| 2 | MeI | B | 71 | ||
| 3 | EtI | A |
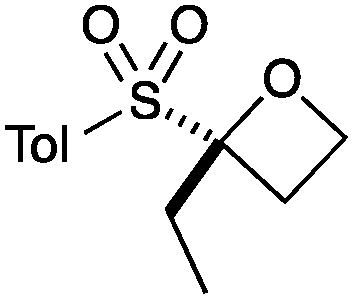
|
6 | 85 |
| 4 | Allyl bromide | A |
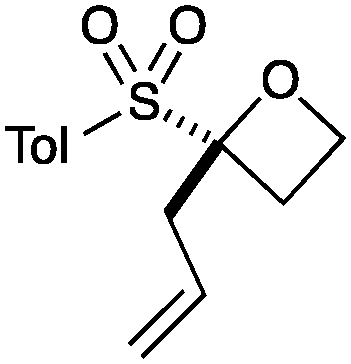
|
7 | 91 |
| 5 |
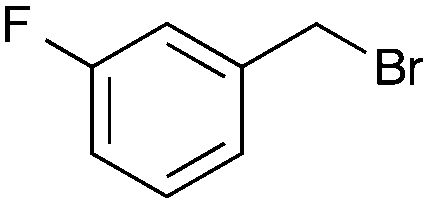
|
B |
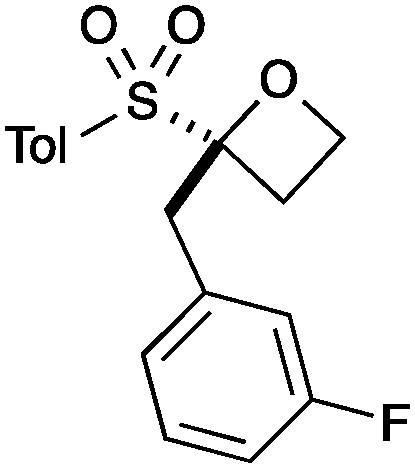
|
8 | 90 |
| 6 |

|
B |
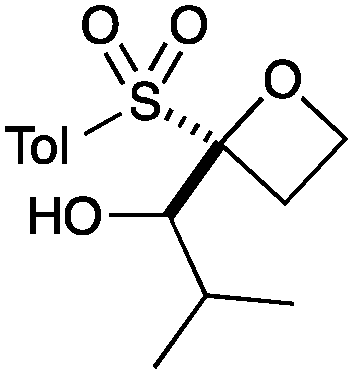
|
9 | 86b |
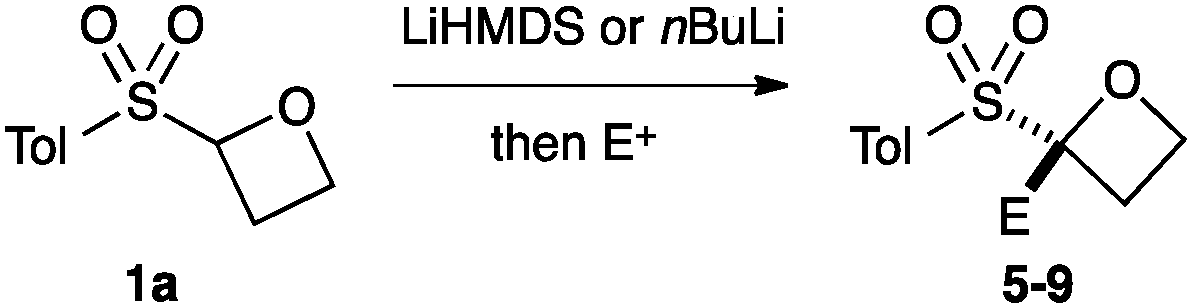
A second set of conditions using nBuLi (Table 1, conditions B) was more suitable for some electrophiles. These conditions were successful using methyl iodide and 3-fluorobenzyl bromide (entries 2 and 5). In addition, these conditions were successful with iso-butyraldehyde, which was unsuccessful using the amide base, to give oxetane 9 in high yield (entry 6).
Oxetane 5 was then derivatised at the aryl group by directed ortho-metallation (Scheme 2). Snieckus has demonstrated that tBu-aryl sulfones are powerful directing groups for ortho metallation.28,29 Employing the Snieckus conditions using MeI successfully afforded ortho-methylated oxetane 10.
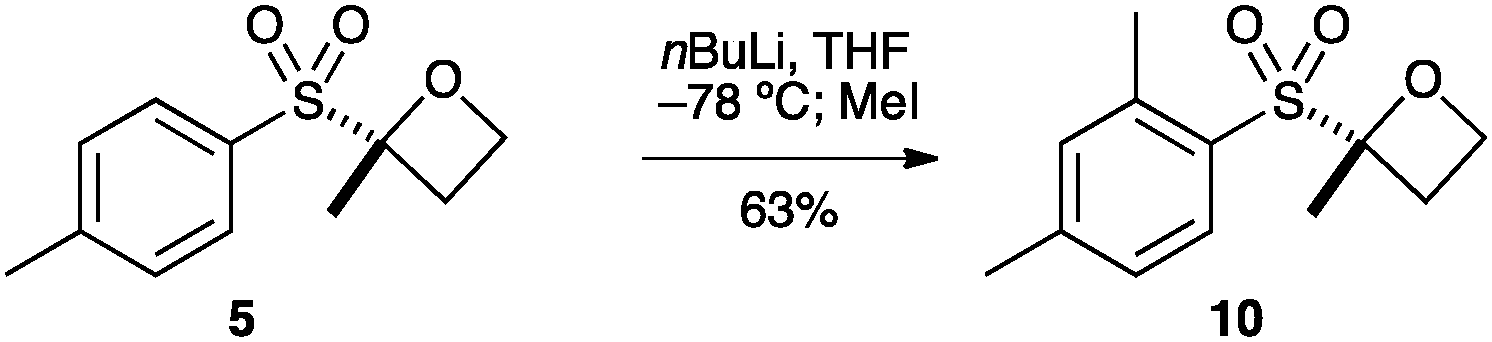 | ||
| Scheme 2 Directed ortho-metallation and reaction with MeI. | ||
Finally we examined the cross-coupling of sulfonyl oxetane 1c from the aryl chloride. The chloride substituent provides an interesting potential binding element and also provides a route to further derivatisation to access alkyl and aryl derivatives. Fürstner recently reported an iron-catalysed cross-coupling of Grignard reagents with aryl chlorides.30 Employing Fürstner's conditions with 1c afforded oxetanes 11 and 12 in good yields. Hexylmagnesium bromide and propylmagnesium chloride were successfully cross-coupled, in the presence of the acidic α-sulfonyl oxetane proton, and without noticeable ring opening of the oxetane (Scheme 3, conditions a).
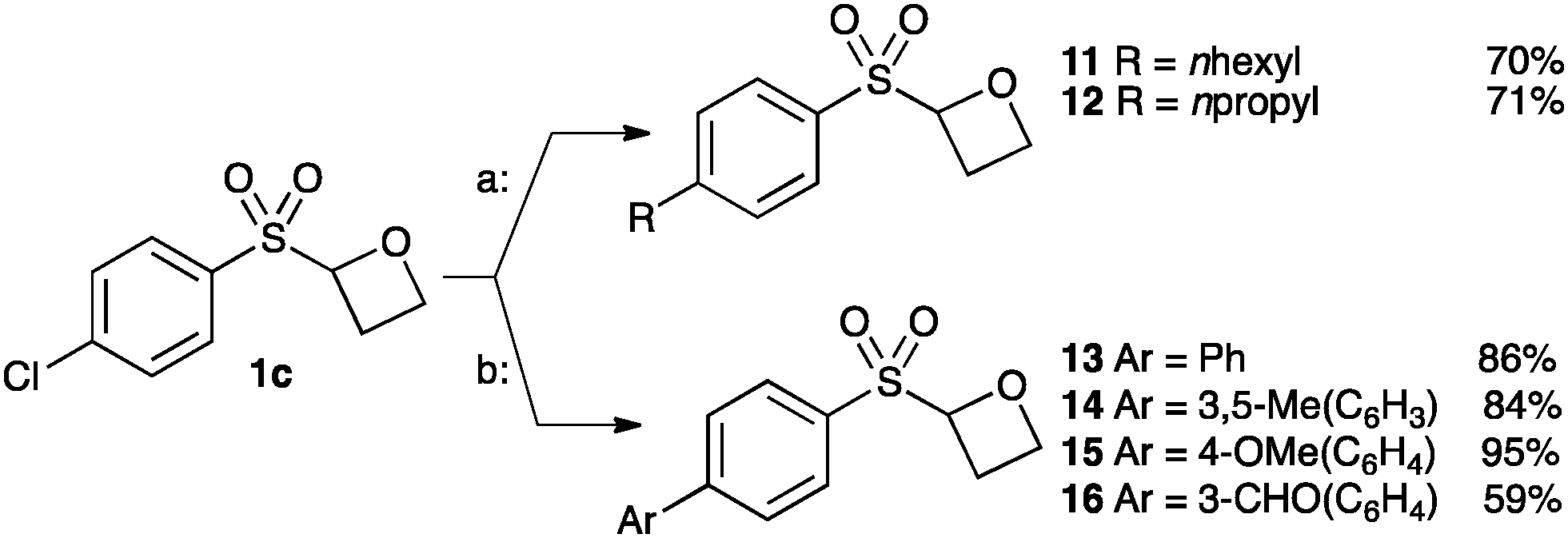 | ||
| Scheme 3 Catalytic cross-coupling of chloro-oxetane 1c. Conditions; a: Fe(acac)3 (5 mol%), RMgX (1.2 equiv.), THF/NMP, 0 °C to rt. b: SPhos (10 mol%), Pd(OAc)2 (5 mol%), K2CO3, dioxane:H2O, 65 °C. | ||
Suzuki cross-couplings of chloride 1c with boronic acids were also successful using Buchwald's SPhos ligand with Pd-catalysis (Scheme 3, b).31 Electron-rich and electron-poor aromatic boronic acids were successful, affording oxetanes 13–16.
In summary, here we report a new approach to the synthesis of 2-functionalised oxetanes that provides novel fragment-like compounds. The initial fragments can be further elaborated through lithiation on the oxetane ring itself, by directed ortho-metallation on the aromatic, as well as by iron and palladium catalysed cross-couplings of the aryl chloride. We are currently expanding the diversity of small ring fragments that can be obtained by this approach and developing enantioselective routes.
For financial support we gratefully acknowledge the EPSRC (Career Acceleration Fellowship to J.A.B., EP/J001538/1), Imperial College London, and AstraZeneca for CASE funding. Thank you to Prof Alan Armstrong for generous support and advice. We thank EPSRC National Mass Spectrometry Facility, Swansea.
Notes and reference
- (a) M. Baker, Nat. Rev. Drug Discovery, 2013, 12, 5 CrossRef CAS PubMed; (b) C. W. Murray, M. L. Verdonk and D. C. Rees, Trends Pharmacol. Sci., 2012, 33, 224 CrossRef CAS PubMed; (c) D. E. Scott, A. G. Coyne, S. A Hudson and C. Abell, Biochemistry, 2012, 51, 4990 CrossRef CAS PubMed.
- M. M. Hann, MedChemComm, 2011, 2, 349 RSC.
- A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed.
- M. M. Hann and G. M. Keserü, Nat. Rev. Drug Discovery, 2012, 11, 355 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, Drug Discovery Today, 2003, 8, 876 CrossRef.
- H. Köster, T. Craan, S. Brass, C. Herhaus, M. Zentgraf, L. Neumann, A. Heine and G. Klebe, J. Med. Chem., 2011, 54, 7784 CrossRef PubMed.
- J. Bower and A. Pannifer, Curr. Pharm. Des., 2012, 18, 4685 CrossRef CAS.
- A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- (a) T. J. Ritchie, S. J. F. Macdonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS PubMed; (b) T. J. Ritchie, S. J. F. Macdonald, S. Peace, S. D. Pickett and C. N. Luscombe, MedChemComm, 2013, 4, 673 RSC.
- J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed.
- (a) G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M. Rogers-Evans and K. Müller, J. Med. Chem., 2010, 53, 3227 CrossRef CAS PubMed; (b) G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736 CrossRef CAS PubMed; (c) G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512 CrossRef CAS PubMed.
- (a) H. Shimada, S. Hasegawa, T. Harada, T. Tomisawa, A. Fujii and T. Takita, J. Antibiot., 1986, 39, 1623 CrossRef; (b) M. C. Wani, H. L. Taylor, M. E. Wall, P. Caggon and A. T. McPhall, J. Am. Chem. Soc., 1971, 93, 2325 CrossRef CAS; (c) C. Li, D. Lee, T. N. Graf, S. S. Phifer, Y. Nakanishi, J. P. Burgess, S. Riswan, F. M. Setyowati, A. M. Saribi, D. D. Soejarto, N. R. Farnsworth, J. O. F. Iii, D. J. Kroll, A. D. Kinghorn, M. C. Wani and N. H. Oberlies, Org. Lett., 2005, 7, 5709 CrossRef CAS PubMed.
- For example, see: A. F. Stepan, K. Karki, W. S. McDonald, P. H. Dorff, J. K. Dutra, K. J. DiRico, A. Won, C. Subramanyam, I. V. Efremov, C. J. O'Donnell, C. E. Nolan, S. L. Becker, L. R. Pustilnik, B. Sneed, H. Sun, Y. Lu, A. E. Robshaw, D. Riddell, T. J. O'Sullivan, E. Sibley, S. Capetta, K. Atchison, A. J. Hallgren, E. Miller, A. Wood and R. S. Obach, J. Med. Chem., 2011, 54, 7772 CrossRef CAS PubMed.
- O. A. Davis, M. Hughes and J. A. Bull, J. Org. Chem., 2013, 78, 3470 CrossRef CAS PubMed.
- See ESI† for calculated molecular properties of all oxetane derivatives and energy-minimised structures of oxetanes 1a and 5 to indicate 3-D shape.
- M. Abe, J. Chin. Chem. Soc., 2008, 55, 479 CAS.
- For example see: (a) T. Aftab, C. Carter, M. Christlieb, J. Hart and A. Nelson, J. Chem. Soc., Perkin Trans. 1, 2000, 711 RSC; (b) S. F. Jenkinson and G. W. J. Fleet, Chimia, 2011, 65, 71 CrossRef CAS.
- (a) E. D. Butova, A. V. Barabash, A. A. Petrova, C. M. Kleiner, P. R. Schreiner and A. A. Fokin, J. Org. Chem., 2010, 75, 6229 CrossRef CAS PubMed; (b) T. Sone, G. Lu, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2009, 48, 1677 CrossRef CAS PubMed; (c) K. Okuma, Y. Tanaka, S. Kaji and H. Ohta, J. Org. Chem., 1983, 48, 5133 CrossRef CAS.
- For example see: (a) Y. Liang, N. Hnatiuk, J. M. Rowley, B. T. Whiting, G. W. Coates, P. R. Rablen, M. Morton and A. R. Howell, J. Org. Chem., 2011, 76, 9962 CrossRef CAS PubMed; (b) Also see: Y. Fang and C. Li, J. Am. Chem. Soc., 2007, 129, 8092 CrossRef CAS PubMed.
- F. Chemla, J. Chem. Soc., Perkin Trans. 1, 2002, 275 RSC.
- F. Couty, B. Drouillat, G. Evano and O. David, Eur. J. Org. Chem., 2013, 2045 CrossRef CAS.
- (a) A. Mordini, S. Bindi, A. Capperucci, D. Nistri, G. Reginato and M. Valacchi, J. Org. Chem., 2001, 66, 3201 CrossRef CAS; (b) A. Mordini, M. Valacchi, C. Nardi, S. Bindi, G. Poli and G. Reginato, J. Org. Chem., 1997, 62, 8557 CrossRef CAS.
- For an example of an alternative approach also see: S. P. Fritz, J. F. Moya, M. G. Unthank, E. M. McGarrigle and V. K. Aggarwal, Synthesis, 2012, 1584 CAS.
- Y. Yoshida, Y. Sakakura, N. Aso, S. Okada and Y. Tanabe, Tetrahedron, 1999, 55, 2183 CrossRef CAS.
- See ESI† for details of reaction optimisation.
- D. I. Coppi, A. Salomone, F. M. Perna and V. Capriati, Chem. Commun., 2011, 47, 9918 RSC.
- (a) M. Iwao, T. Iihama, K. K. Mahalanabis, H. Perrier and V. Snieckus, J. Org. Chem., 1989, 54, 24 CrossRef CAS; (b) S. L. MacNeil, O. B. Familoni and V. Snieckus, J. Org. Chem., 2001, 66, 3662 CrossRef CAS PubMed.
- For the use of oxetanes as an ortho-directing group, see; D. I. Coppi, A. Salomone, F. M. Perna and V. Capriati, Angew. Chem., Int. Ed., 2012, 51, 7532 CrossRef CAS PubMed.
- A. Fürstner and A. Leitner, Angew. Chem., Int. Ed., 2002, 41, 609 CrossRef.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details of reaction optimisation, calculated molecular properties, experimental, characterization data and NMR spectra (1H and 13C) for all novel compounds. See DOI: 10.1039/c3cc46450d |
| ‡ clogP values were determined using ACDlabs logP calculator http://www.acdlabs.com/resources/freeware/chemsketch/logp. |
| This journal is © The Royal Society of Chemistry 2014 |