DOI:
10.1039/C4AY01509F
(Paper)
Anal. Methods, 2014,
6, 8915-8923
Historical textile dyeing with Genista tinctoria L.: a comprehensive study by UPLC-MS/MS analysis†
Received
25th June 2014
, Accepted 22nd July 2014
First published on 22nd July 2014
Abstract
Polyphenolic components from Genista species have been well characterised because of their potential as antioxidants and as therapeutic leads; however, the identification of dyer's greenweed (Genista tinctoria L.) in historical textiles has been the subject of only limited studies. This paper presents a comprehensive UPLC-PDA MS/MS study of reference and historical yarns dyed with this species. Several so far unreported dye components that could assist with the identification of this dye source, were characterised by MS/MS. Furthermore, the effect of photo-degradation and textile preparation techniques (such as over-dyeing) on the dye fingerprint was investigated and the results correlated with those obtained from historical samples from the Burrell and Bodleian collections, UK.
1. Introduction
Polyphenolic components from Genista species have been well characterised because of their potential as antioxidants and as therapeutic leads;1,2 however, the identification of dyer's greenweed (Genista tinctoria L.) in historical textiles has been the subject of only limited studies.3,4 Although historically weld (Reseda luteola L.) was probably the most widely used European yellow dye plant, it is reported that other dye plants, including dyer's greenweed (Genista tinctoria L.) and sawwort (Serratula tinctoria L.), were used as substitutes.5 The flavones luteolin and apigenin, the isoflavone genistein, and the glycosides of these are known dye components of Genista species.6 But genistein, and its glycosides, are also the main dye components found in other varieties of broom; notwithstanding, the dye source for historical textiles is usually ascribed to dyer's greenweed.7,8 Whilst genistein is the principle component on which this attribution is made,4 our studies of the dye extracts of historical samples show the presence of additional dye components that could enhance this identification. This paper presents a comprehensive study of the structure of these additional dye components using UPLC-PDA‡ and ESI-MS/MS techniques. To achieve this, an analytical method was developed for a range of flavonoid and isoflavonoid dyes, allowing a more efficient separation of several isomeric dye components from textile samples. This method was then used to determine the relative amount of dyestuffs present in raw materials, modern yarns and historical yarns; examining differences between the plant extract and the dye components adsorbed onto textiles, and relating variations in component ratios to dyeing processes such as over-dyeing, and to the effects of photo-degradation. Data from a selection of historical yarns sampled from mid-sixteenth century English tapestries from the Burrell Collection in Glasgow, UK and the Bodleian Library in Oxford, UK were then placed in context using the results of this study.
2. Experimental
2.1 Materials and chemicals
2.1.1 Plant and textiles.
Dyer's broom (Genista tinctoria L.) from Fibrecrafts (George Weil & Sons Ltd.) was used to prepare the reference material. Alum mordanted wool (YW2) and silk (YS3a) yarns were dyed, and over-dyed (YS3b–d), as part of the Monitoring of Damage to Historic Tapestries project (FP5, EC contract number EVK4-CT-2001-00048).9
2.1.2 Flavone and isoflavone standards.
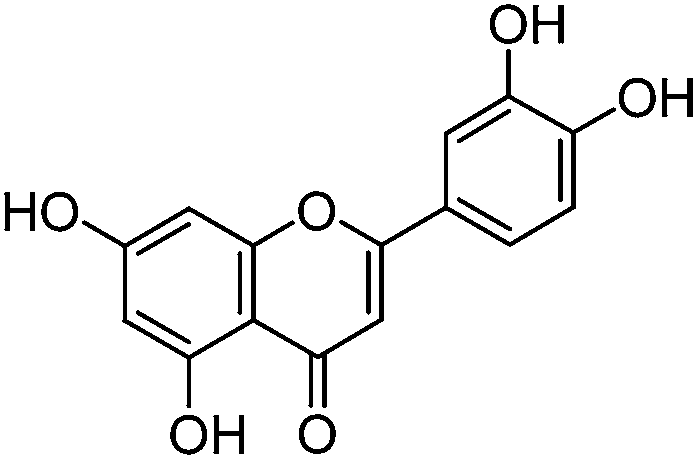
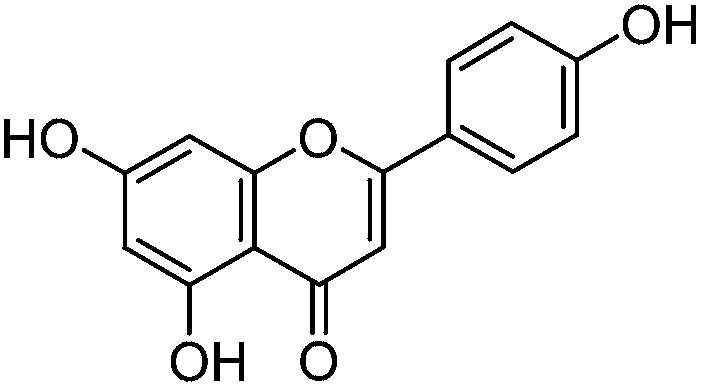
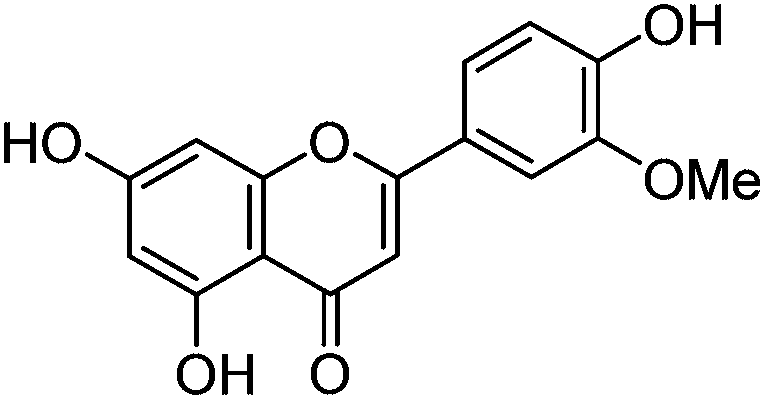
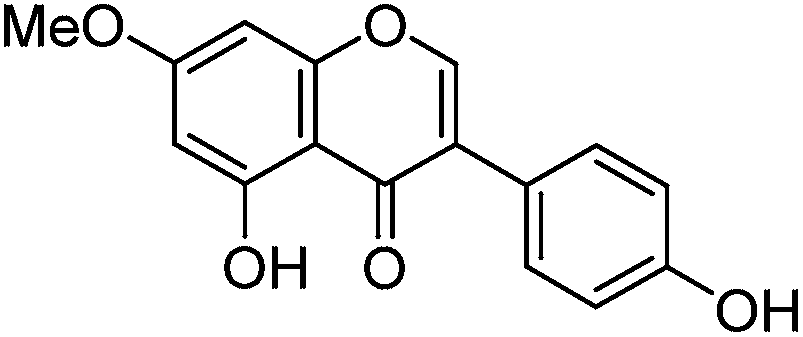
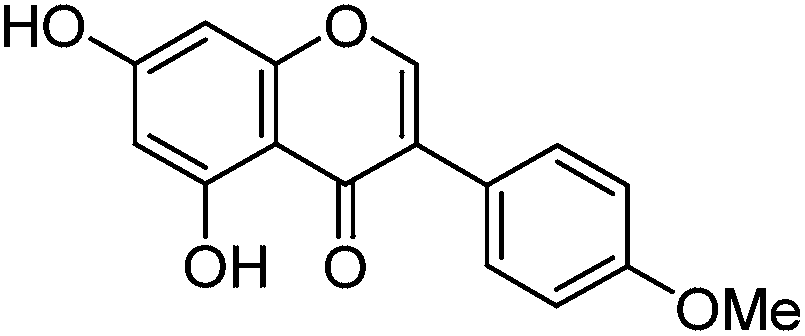
Luteolin (3′,4′,5,7-tetrahydroxyflavone), genistein (4′,5,7-trihydroxyisoflavone), apigenin (4′,5,7-trihydroxyflavone), prunetin (4′,5-dihydroxy-7-methoxyisoflavone), and biochanin A (5,7-dihydroxy-4′-methoxyisoflavone) standards were purchased from Sigma-Aldrich; glycitein (4′,7-dihydroxy-6-methoxyisoflavone) standard was purchased from Cayman Chemical Company; and chrysoeriol (4′,5,7-trihydroxy-3′-methoxyflavone) and diosmetin (3′,5,7-trihydroxy-4′-methoxyflavone) standards were purchased from ExtraSynthese. All commercial standards were >98% purity.
An authentic sample of isoprunetin (4′,7-dihydroxy-5-methoxyisoflavone) was prepared from genistein by selective acetylation (Ac2O, py) to give 7,4′-diacetoxy-5-hydroxyisoflavone,10 methylation of the remaining free hydroxyl [(MeO)2SO2 K2CO3, acetone] under high dilution conditions11 to give 7,4′-diacetoxy-5-methoxyisoflavone, followed by hydrolysis (NaHCO3 aq., MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF)10 of the acetate groups which allowed isolation of isoprunetin in >95% purity after chromatography (see ESI 1†).12
:THF)10 of the acetate groups which allowed isolation of isoprunetin in >95% purity after chromatography (see ESI 1†).12
2.1.3 Solutions of standards.
The UPLC® system was calibrated using stock solutions of flavonoid and isoflavonoid standards: (1) a solution containing luteolin and genistein (1.00 ± 0.01 mg of each standard) in H2O:MeOH [25 mL, 1:1 (v/v); equivalent to 40 μg mL−1]; (2) a solution containing apigenin, chrysoeriol (0.20 ± 0.01 mg of each standard) in H2O:MeOH [10 mL, 1:1 (v/v); equivalent to 20 μg mL−1]; (3) a solution of diosmetin (1.00 ± 0.01 mg) in H2O:MeOH [25 mL, 1:1 (v/v); equivalent to 40 μg mL−1]. Diluted solutions were then prepared with components at concentrations of 20, 10, 5, 1, 0.5, 0.1, 0.05, 0.02 and 0.01 μg mL−1, by dilution with H2O:MeOH [1:1 (v/v)] using calibrated micro-pipettes.
2.2 UPLC-PDA and ESI MS systems
2.2.1 UPLC-PDA.
The UPLC chromatographic method was developed using a Waters Acquity UPLC® system with sample detection using a Waters PDA detector (250 to 500 nm). Data were collected by Waters Empower 2 software and processed with Origin 8.5 (OriginLab, Northampton, MA, USA). Sample extracts were automatically injected via a Rheodyne injector with a 10 μL sample loop. The bandwidth (resolution) was 1.2 nm with a sampling rate of 5 points per s. The method used a PST BEH C18 reverse phase column, 1.7 μm particle size, 150 × 2.1 mm (length × i.d.), set-up with inline filter. The total run time was 37.33 min at a flow rate of 250 μL min−1 and the column was maintained at 55 ± 1 °C. A binary solvent system was used; A = 0.02% aqueous HCOOH (pH 3), B = MeOH. The elution program was isocratic for 3.33 min (77A:23B) then a linear gradient from 3.33 min to 29.33 min (10A:90B) before recovery of the initial conditions over 1 min and equilibration over 7 min.
2.2.2 UPLC-ESI MS.
UPLC-ESI MS analysis was performed using a Waters Acquity UPLC® system coupled with a Waters Synapt G2 (Waters Corporation, Manchester, UK) equipped with an electrospray ionization source, using the gradient described above. MS and tandem MS (MS/MS) analyses were conducted in negative ionization mode. Source conditions included a capillary voltage of 2.5 kV, a sample cone voltage of 35 V, a desolvation temperature of 300 °C and a source temperature of 120 °C. Argon was used as a collision gas. Precursor ions were selected on the basis of m/z and retention time. For MS/MS the collision energy was optimised to establish characteristic fragmentation patterns. Representative data using a collision energy of 25 eV to illustrate fragmentation patterns is presented. Data was collected using Masslynx 4.1 (Waters Corporation, Milford, MA, USA) and processed with Origin 8.5 (OriginLab, Northampton, MA, USA).
2.3 Plant and yarn extractions
2.3.1 Plant extraction.
1.5 g of dried leaves of dyer's greenweed were extracted in 100 mL (MeOH:H2O 1:1, v/v) in an ultrasonic bath for 2 hours at 40 °C. A fraction of the extract was centrifuged for 10 min at 10000 rpm and filtered using a PTFE Phenomenex syringe filter (0.2 μm, 4 mm) for UPLC-PDA analysis. A second fraction of the extract was subjected to the hydrolysis procedure described below for yarn analysis and again analysed by UPLC-PDA.
2.3.2 Yarn extraction.
The yarns (0.1–0.5 mg) were extracted with 37% HCl:H2O:MeOH [200 μL, 2:1:1 (v/v/v)], at 100 °C for 10 min.13 After ambient cooling to room temperature, the extract was centrifuged for 10 min at 10000 rpm and then filtered directly into Waters UPLC vials® (residual volume of 9 μL) using a PTFE Phenomenex syringe filter (0.2 μm, 4 mm). The extract was then cooled with liquid nitrogen and dried under vacuum using a freeze drier system. The dry residue was then reconstituted with H2O:MeOH [40 μL, 1:1 (v/v)] – allowing a single injection of 10 μL.
2.4 Accelerated light-ageing system
Accelerated light-ageing experiments were performed at National Museums Scotland using a Complete Lighting Systems light box (St Albans, UK), working at ambient temperature and relative humidity (RH). These levels were monitored hourly, together with the intensity of illuminance and UV levels using an Elsec IRLOG environmental Logger and the data periodically downloaded to computer. Wool yarns dyed with weld (Reseda luteola L.) and others with the single dye component genistein were removed over a period of 4571 hours and 1 mg of yarn was subsequently hydrolysed following the procedure described above and investigated by HPLC analysis following the method used by Peggie et al. in order to obtain the degradation profiles of luteolin and genistein components.14
3. Results and discussion
3.1 UPLC and MS study of flavone/isoflavone standards
3.1.1 UPLC method development.
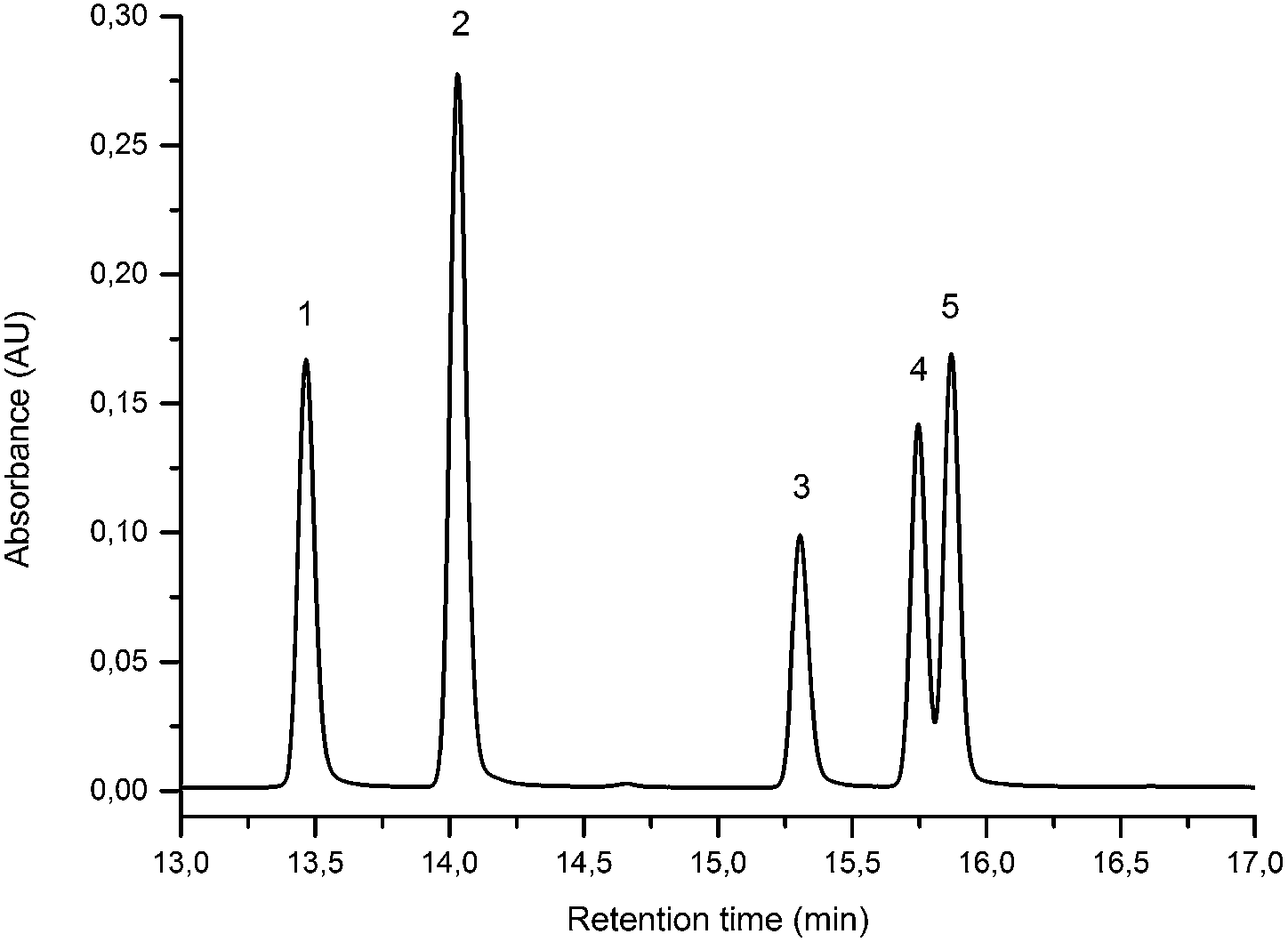
The UPLC method was evaluated based on several performance characteristics: the repeatability, the specificity, the limit of detection/quantification and the linearity.15 The method was developed to provide an improved separation and quantification of 5 dye standards: the flavones luteolin (1) and apigenin (3), the isoflavone genistein (2), and the two O-methylated flavones chrysoeriol (4) and diosmetin (5). For all the dye standards the average variation on 12 measurements ranged between 0.04–0.06%, and the method allowed a significantly greater separation of several dye components compared to that achieved in a recent UPLC study.16 The method allowed a complete separation of the flavone luteolin (1) and the isoflavone genistein (3) with an Rs factor of 3.13; while the separation of the flavone apigenin (3) and the O-methylated flavone chrysoeriol (4) was increased to a value of 2.88, compared to 0.78 obtained by conventional HPLC.17 Finally, the method also allowed the separation of the O-methylated regio-isomers chrysoeriol (4) and diosmetin (5) with an Rs factor of 1.32 (Fig. 1). The limits of detection (LoD) and limits of quantification (LoQ) were calculated based on the average value of the baseline noise Hnoise of several solvent blanks, considering all data points. The baseline of the UV detector at 254 nm averaged (9 ± 1) × 10−4 AU, resulting in detection limits ranging from 0.5 ng for genistein to 1.9 ng for apigenin for an injection volume of 5 μL (Table 1). These values equalled or improved upon the values published for luteolin (1), genistein (2) and apigenin (3) in other UPLC/UHPLC studies.16,18
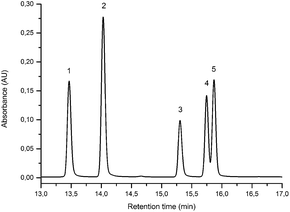 |
| | Fig. 1 UPLC Method using PST BEH C18, 1.7 μm, 2.1 × 150 mm column, showing the separation of luteolin (1), genistein (2), apigenin (3), chrysoeriol (4) and diosmetin (5). Standard solution at 10 μg mL−1 recorded at 254 nm (5 μL injection volume). | |
Table 1 Flavonoid and isoflavonoid standards used to calibrate the UPLC-PDA and ESI-MS systemsa
| Component |
Structure |
PDA Rt (min) |
LoD (LoQ) (ng) |
λ
max MeOH (nm) |
MS/MS Rt (min) |
[M − H]−m/z |
MS/MS (CE: 25 eV) m/z (% BPI) fragment |
|
The ions annotated with RDA correspond to retro-Diels–Alder fragments (1,3A−). The ions annotated with RDA* correspond to retro-Diels–Alder fragments where ionisation is in the B-ring (1,3B−). The ions annotated with iFF correspond to the isoflavonoid-specific fragmentation (0,3B−).
|
|
FLAVONES
|
| Luteolin (1) |
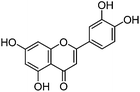
|
13.47 |
0.9 (2.6) |
|
13.56 |
285 |
285 (100) |
[M − H]− |
|
|
217 (12.5) |
[M − H − C3O2]− |
|
|
199 (17.8) |
|
| 252 |
175 (23.1) |
[M − H − C3O2 − C2H2O]− |
| 291 (sh) |
151 (45.2) |
RDA |
| 349 |
149 (20.2) |
(4′-OH) |
|
|
133 (63.5) |
RDA* |
|
|
121 (6.6) |
(4′-OH) − CO |
|
|
107 (12.1) |
RDA − CO2 |
| |
| Apigenin (3) |
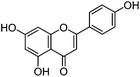
|
15.26 |
1.9 (5.7) |
|
15.58 |
269 |
269 (100) |
[M − H]− |
|
|
225 (28.5) |
[M − H − CO2]− |
|
|
201 (12.9) |
[M − H − C3O2]− |
| 267 |
183 (9.5) |
|
| 300 (sh) |
159 (12.6) |
[M − H − C3O2 − C2H2O]− |
| 338 |
151 (58.7) |
RDA |
|
|
149 (54.9) |
(4′-OH) |
|
|
121 (11.3) |
(4′-OH) − CO |
|
|
117 (79.1) |
RDA* |
|
|
107 (17.4) |
RDA − CO2 |
| |
| Chrysoeriol (4) |
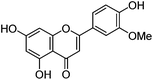
|
15.69 |
1.2 (3.4) |
250 |
15.80 |
299 |
299 (4.4) |
[M − H]− |
| 268 |
284 (100) |
[M − H − CH3]−˙ |
| 290 (sh) |
256 (30.1) |
[M − H − CH3 − CO]−˙ |
| 349 |
151 (1.3) |
|
| |
| Diosmetin (5) |
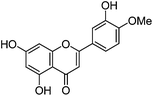
|
15.87 |
1.2 (3.7) |
250 |
15.93 |
299 |
299 (1.3) |
[M − H]− |
| 268 |
284 (100) |
[M − H − CH3]−˙ |
| 290 (sh) |
256 (4.7) |
[M − H − CH3 − CO]−˙ |
| 348 |
151 (1.0) |
RDA |
| |
|
ISOFLAVONES
|
| Genistein (2) |
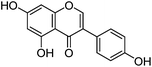
|
14.03 |
0.5 (1.6) |
|
14.16 |
269 |
269 (100) |
[M − H]− |
|
|
241 (2.0) |
[M − H − CO]− |
|
|
224 (6.4) |
[M − 2H − CO2]− |
| 260 |
213 (3.9) |
[M − H − 2CO]− |
| 332 (sh) |
201 (8.2) |
[M − H − C3O2]− |
|
|
197 (6.8) |
[M − H − CO − CO2]− |
|
|
159 (13.8) |
[M − H − C3O2 − C2H2O]− |
|
|
133 (20.4) |
iFF |
|
|
107 (9.1) |
RDA − CO2 |
| |
| Isoprunetin (6) |
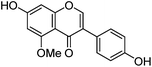
|
12.17 |
— |
|
12.16 |
283 |
283 (30.7) |
[M − H]− |
| 255 |
268 (63.0) |
[M − H − CH3]−˙ |
| 332 (sh) |
240 (100) |
[M − H − CH3 − CO]−˙ |
|
|
196 (6.7) |
[M − H − CH3 − CO − CO2]−˙ |
| |
| Glycitein (7) |
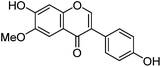
|
12.91 |
— |
235 (sh) |
12.98 |
283 |
283 (2.6) |
[M − H]− |
| 257 |
268 (100) |
[M − H − CH3]−˙ |
| 320 |
240 (58.2) |
[M − H − CH3 − CO]−˙ |
|
|
196 (4.3) |
[M − H − CH3 − CO − CO2]−˙ |
| |
| Prunetin (8) |
|
18.93 |
— |
|
19.04 |
283 |
283 (6.4) |
[M − H]− |
| 260 |
268 (100) |
[M − H − CH3]−˙ |
| 332 (sh) |
240 (7.7) |
[M − H − CH3 − CO]−˙ |
|
|
196 (1.2) |
[M − H − CH3 − CO − CO2]−˙ |
| |
| Biochanin A (9) |
|
19.30 |
— |
|
19.45 |
283 |
283 (9.5) |
[M − H]− |
| 260 |
268 (100) |
[M − H − CH3]−˙ |
| 332 (sh) |
240 (5.3) |
[M − H − CH3 − CO]−˙ |
|
|
239 (7.3) |
[M − 2H − CH3 − CO]− |
|
|
211 (3.5) |
[M − 2H − CH3 − 2CO]− |
3.1.2 UPLC-MS study.
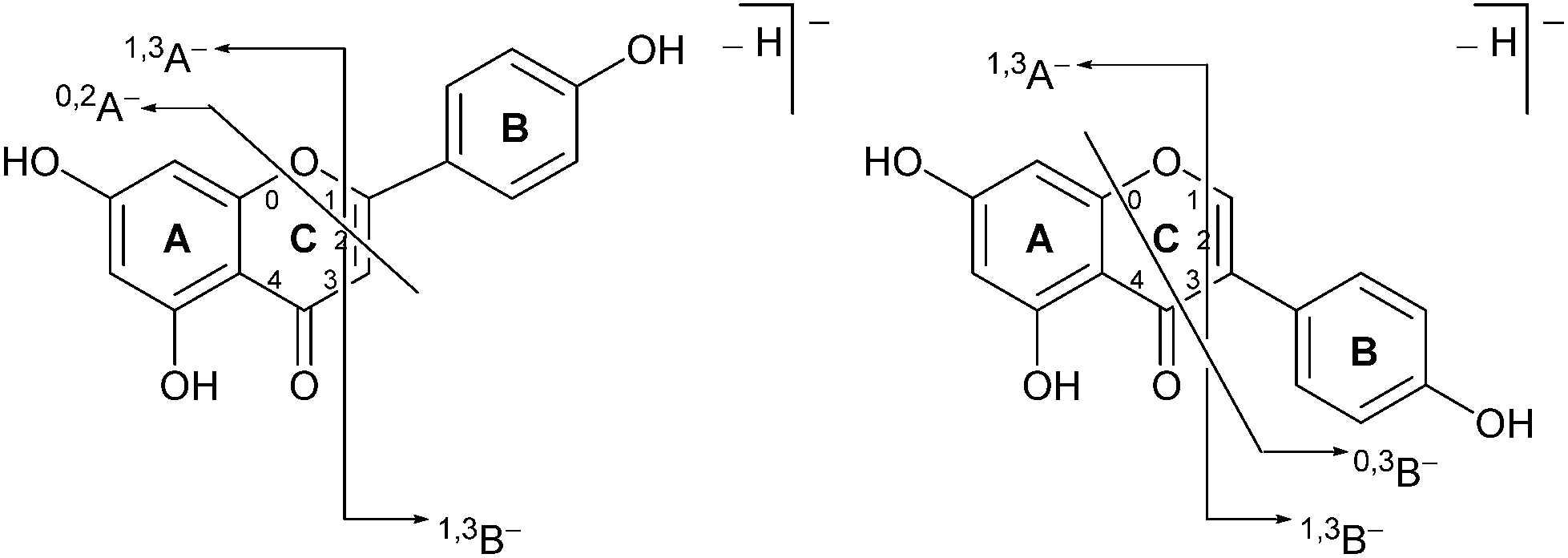
Previous studies have shown that mass spectrometric analysis of flavonoid and isoflavonoid dyes may be achieved effectively using electrospray ionisation (ESI) mass spectrometry in negative mode,3,19–21 and have allowed the characterisation of several fragmentation mechanisms using secondary and tertiary mass spectrometry ion fragmentation (MS2 and MS3).3,20 A formal notation which has been adopted for these fragmentation mechanisms is indicated in Fig. 2;22,23 it is based upon glycoside nomenclature with letters to indicate the ring carrying the charge and superscript numbers to indicate bond scission. Studies of isoflavonoids using negative ESI and MSn have identified detailed mechanisms for the loss of small neutral molecules including CO, CO2 and ketene (C2H2O),21 processes which are common across these classes. In addition, for methylated, or glycosylated, flavonoids and isoflavonoids the primary fragmentation mechanism has been shown to be radical cleavage resulting in the detection of [M − H − CH3]−˙ or [M − H − glycoside]−˙ ions.21
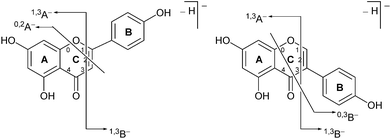 |
| | Fig. 2 Formal notation used for the retrocyclization fragmentation of the [M − H]− ions of (a) flavonoids and (b) isoflavonoids under negative ion electrospray ionisation conditions. | |
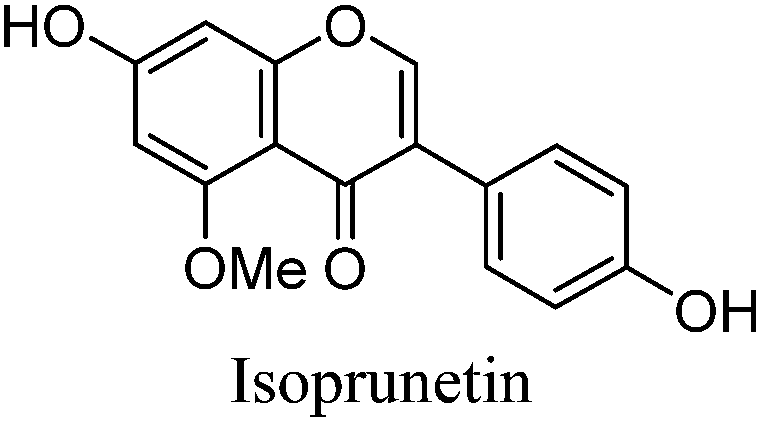
Alongside the standards used for optimisation of the UPLC conditions, several O-methylated isoflavones related to genistein (2) [isoprunetin (6), glycitein (7), prunetin (8) and biochanin A (9)], which were putative candidates for the minor components found in historical extracts, were selected for study by UPLC-MS. With this larger group of compounds some general trends in elution times could be deduced. Not surprisingly, compounds with an increased hydroxyl substitution pattern were shown to elute at shorter retention times under the UPLC conditions employed (cf. luteolin vs. apigenin, Table 1). Methylation generally gave rise to longer retention times (cf. genistein vs. prunetin, Table 1), except for where methylation disrupted the chelating motif formed by the 5-OH and the carbonyl of the C-ring (cf. genistein vs. isoprunetin, Table 1), in these cases shorter retention times were observed due to reduced binding to the stationary phase.
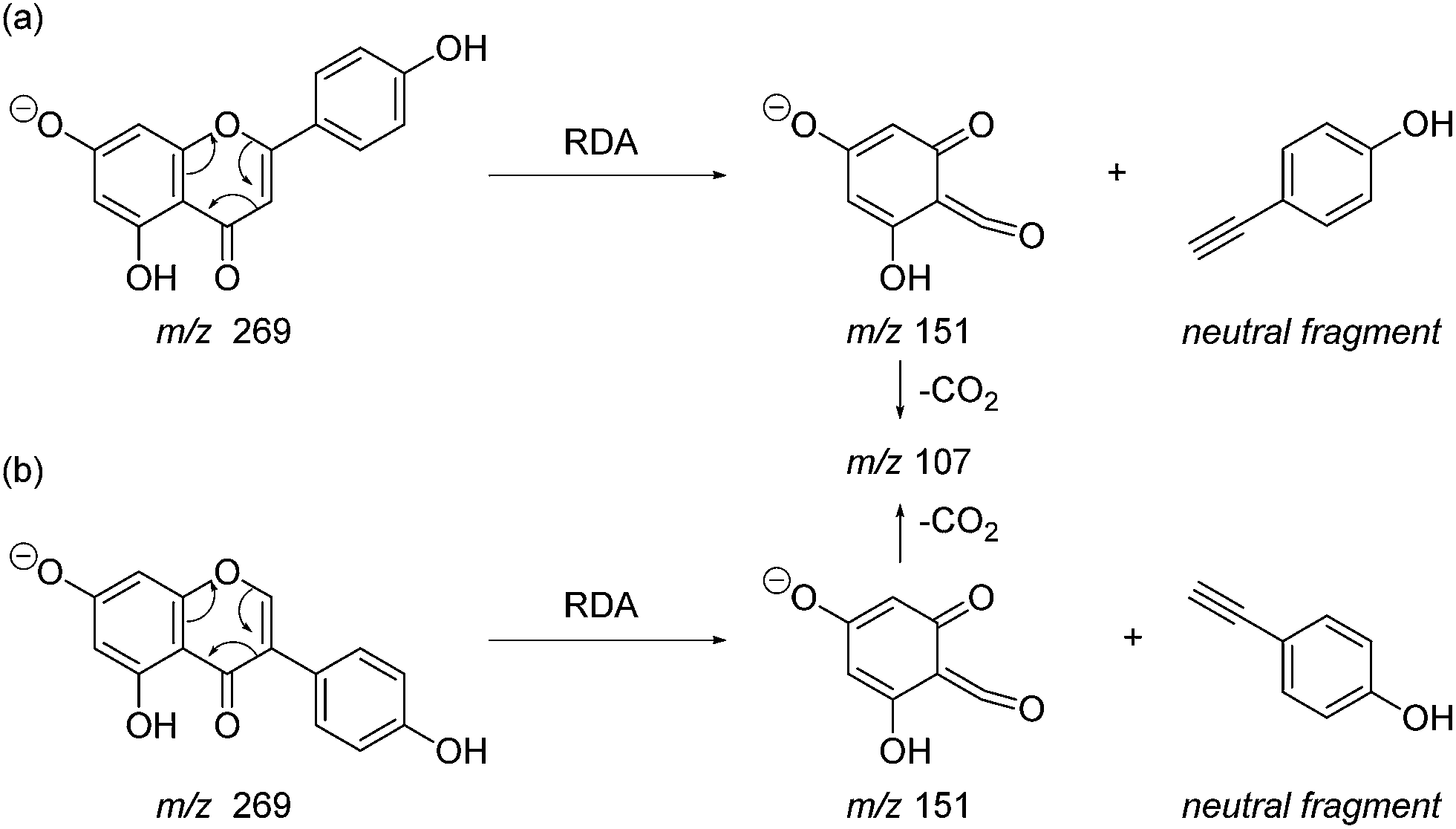
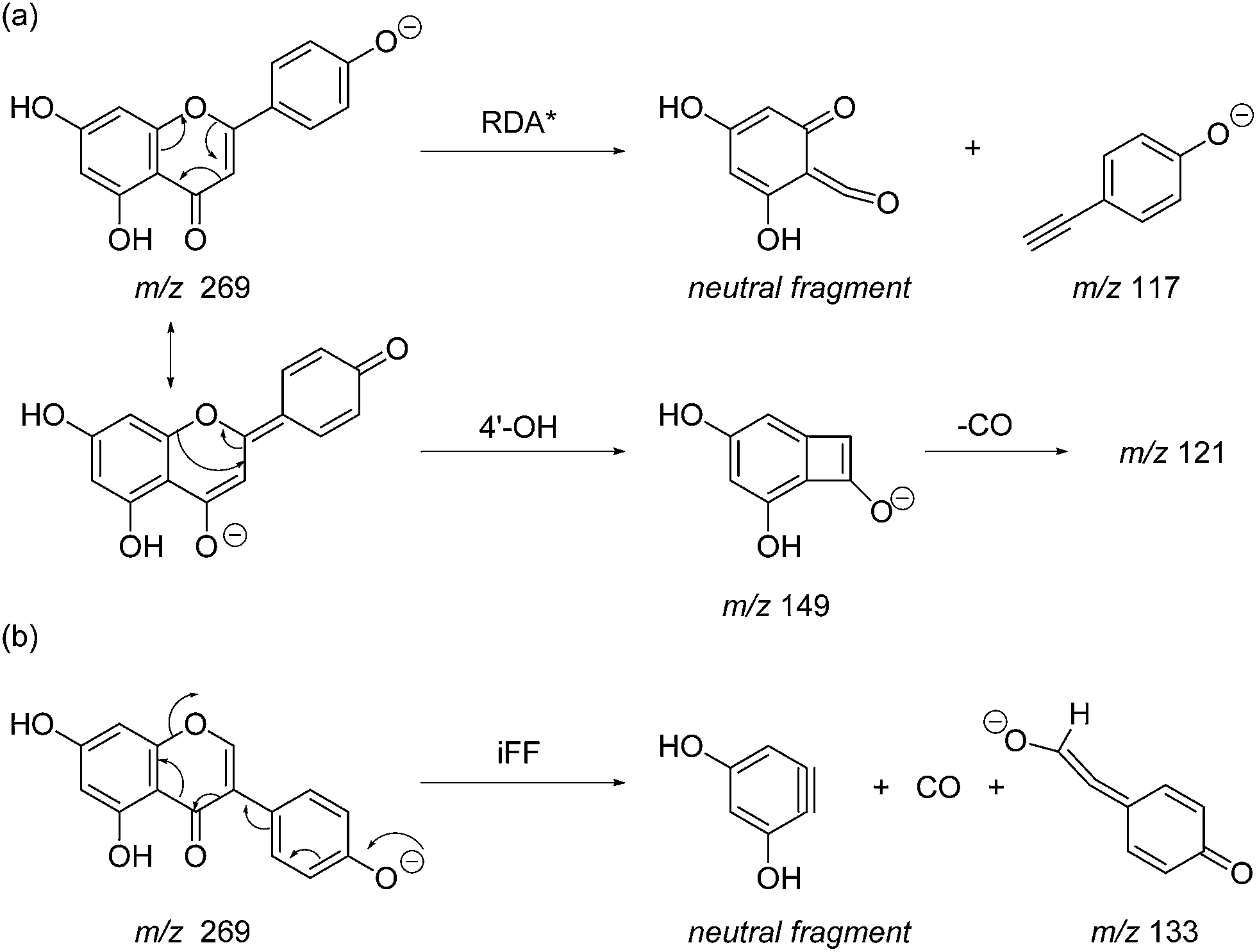
Prior to sample analysis, ESI conditions were optimised using a reference standard solution containing luteolin, genistein, apigenin, chrysoeriol and diosmetin at 10 μg mL−1 amongst others to maximise signal and minimise in-source fragmentation. Standard solutions were then used to characterise fragmentation patterns in the MS/MS experiments. For the MS/MS studies the deprotonated standards, [M − H]−, were subjected to fragmentation at collision energies ranging from 10–40 eV; with 25 eV determined to be optimal as it resulted in significant fragmentation across all species. The results of MS/MS fragmentation at this collision energy are presented in Table 1, with fragment ion intensities reported as a percentage of the base peak intensity (% BPI). Direct comparison of MS/MS fragmentation of deprotonated flavonoids and isoflavonoids under these conditions has allowed us to identify the following trends. When deprotonated on the A-ring, both flavonoids and isoflavonoids show cleavage due to a retro-Diels–Alder mechanism (RDA, 1,3A−, Scheme 1); this is more pronounced in the case of flavonoids and is frequently accompanied by the loss of CO2.20 However, when deprotonated on the B-ring, flavonoids show breakdown by the equivalent retro-Diels–Alder mechanism (RDA*, 1,3B−, Scheme 2), whereas there is little evidence for this in the isoflavonoids. Instead, an alternative 4′-OH isoflavonoid-specific fragmentation pathway predominates (iFF, 0,3B−, Scheme 2). This is in direct contrast to our previously reported 4′-OH flavonoid-specific pathway (4′-OH, 0,2A−, Scheme 2).20 As expected, upon methylation both flavonoids and isoflavonoids have as a principal fragment the [M − H − CH3]−˙ ion, which typically occurs as the base peak and is accompanied by neutral losses of CO and CO2; retrocyclization accounts for only a very small proportion of the fragmentation in these cases.
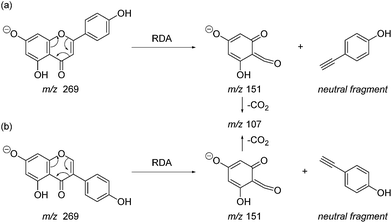 |
| | Scheme 1 Retro-Diels–Alder (RDA, 1,3A−) MS/MS fragmentation mechanisms for [M − H]− with ionisation at the 7-OH, illustrated for: (a) the flavonoid apigenin; and (b) the isoflavonoid genistein. | |
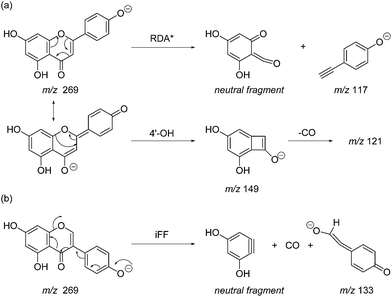 |
| | Scheme 2 MS/MS fragmentation pathways for [M − H]− with ionisation at the 4′-OH, illustrated for: (a) the flavonoid apigenin, with retro-Diels–Alder* (RDA*, 1,3B−) and 4′-OH flavonoid-specific (4′-OH, 0,2A−) mechanisms; and (b) the isoflavonoid genistein, with the 4′-OH isoflavonoid-specific fragmentation (iFF, 0,3B−) mechanism. | |
3.2 Dye components characterised in Genista tinctoria L.
3.2.1 UPLC-PDA study.
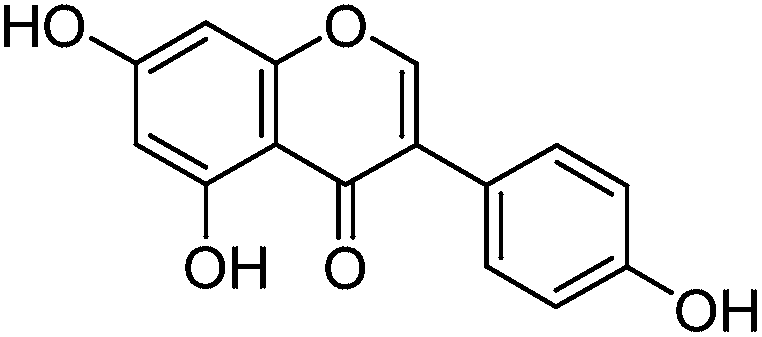
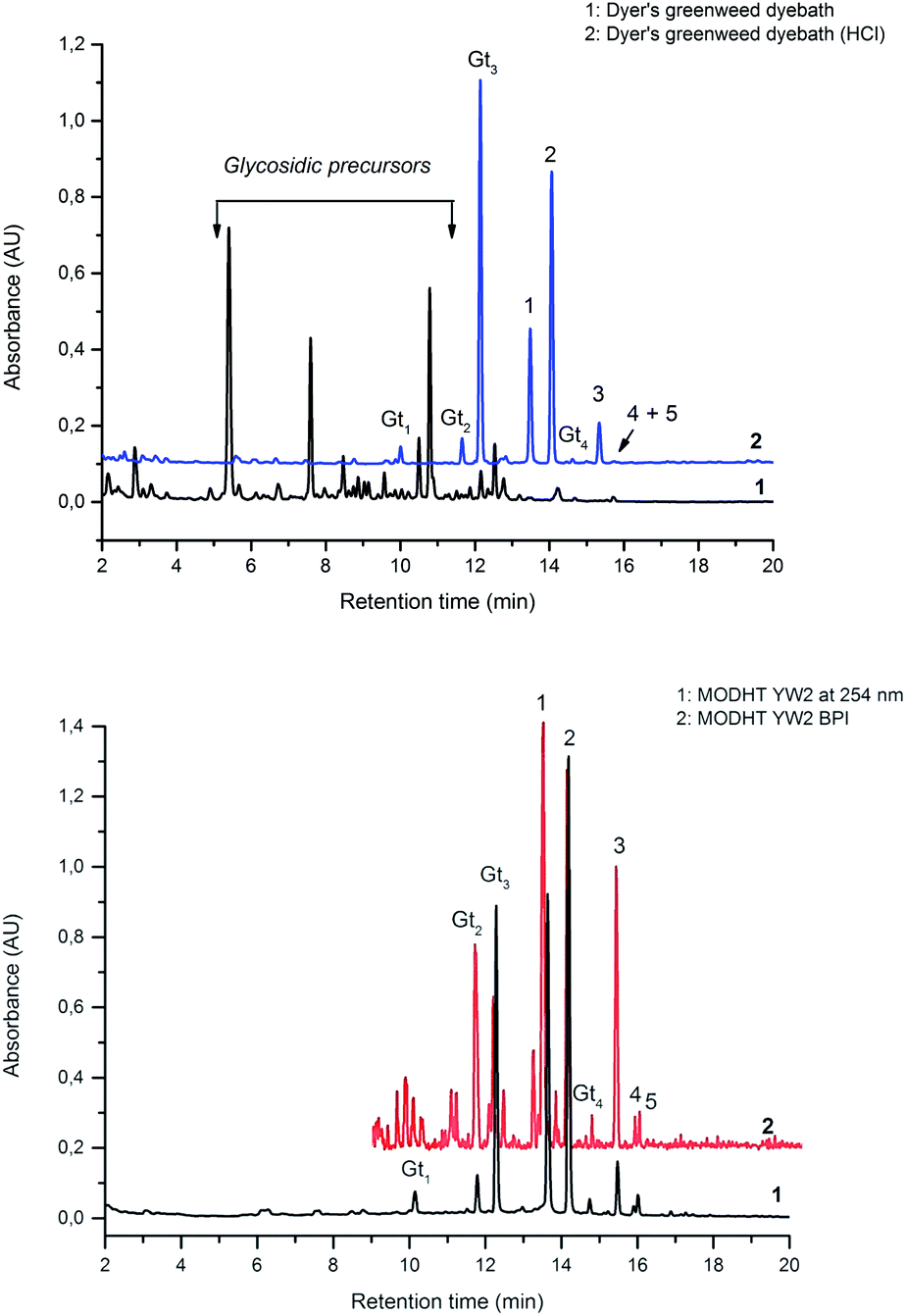
In common with other recent studies,24 UPLC-PDA investigation of the extract of dyer's greenweed leaves reveals a complex mixture of flavonoid glycosides and minor amounts of their aglycone equivalents; after acid hydrolysis the same extract exhibits a similar composition to the acid hydrolysed extracts from wool (YW2) and silk (YS3a) reference yarns (Fig. 3). Both fibre extracts present a very similar dye component profile; when analysed at 254 nm the main dye components luteolin (1), genistein (2), and apigenin (3) appear to be of quite similar intensity on each substrate (wool vs. silk; see ESI 4†). In addition, UPLC analysis shows that both luteolin methyl-ethers chrysoeriol (4) and diosmetin (5) are present at low levels in the acid hydrolysed extracts (Fig. 3B). Chrysoeriol (4) was found to average 1.3 and 1.2% (wool and silk, respectively), while diosmetin (5) was found to average 2.4 and 2.3% (wool and silk, respectively) of the total relative amount of the flavonoids present in the extracts (see ESI 4†). The presence of the methylated regioisomers chrysoeriol and diosmetin in the acid hydrolysed extracts of yarn dyed with Genista tinctoria L. has not been reported previously; but is not surprising given that chrysoeriol has been reported to occur in some Genista species.25 Four unknown components were also characterised in the acid hydrolysed extracts of both silk and wool yarns; some of these are thought to be aglycones as they were also found in the extract of dyer's greenweed leaves before acid hydrolysis (Fig. 3). Thus, these components might be useful markers for the identification of Genista tinctoria L. and were named Gt1 (Rt = 10.06 min), Gt2 (Rt = 11.67 min), Gt3 (Rt = 12.17 min) and Gt4 (Rt = 14.61 min) respectively; they all exhibit a maximum absorption between 255 and 261 nm, which indicates an isoflavonoid structure. Of these, Gt3 is often present at levels which are detectable in historical textile samples; hence we have further investigated the structure of these components by MS/MS, and examined the effect of the dyeing process on their uptake onto wool and silk fibres.
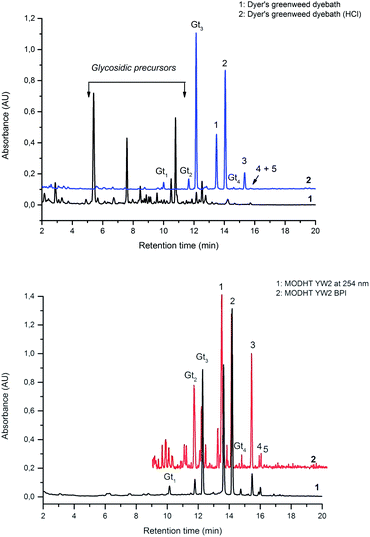 |
| | Fig. 3 (A – Upper): UPLC chromatogram of a solution of dyer's greenweed extract and its acid hydrolysed equivalent, both monitored at 254 nm. (B – Lower): UPLC chromatogram of the acid hydrolysed reference MODHT YW2 monitored at 254 nm and MS BPI trace. Indicated on (A) and (B) are the flavones luteolin (1) and apigenin (3), the isoflavone genistein (2), together with the four unidentified components Gt1, Gt2, Gt3 and Gt4 and the two luteolin methyl-ethers chrysoeriol (4) and diosmetin (5). | |
3.2.2 UPLC-MS study.
In an attempt to identify the Gt components unambiguously, the extract from the reference yarn YW2 was studied further by UPLC-ESI-MS (Fig. 3, lower). Although detection at 254 nm indicates the presence of a clear peak for Gt1, the ESI-MS BPI profile clearly shows the presence of several overlapping species with a very similar retention time, thus it was not possible to obtain further information about Gt1. However, the components Gt2–4 were readily isolated by ESI-MS analysis, allowing further study by MS/MS (ESI 3†).
Fragmentation of Gt2.
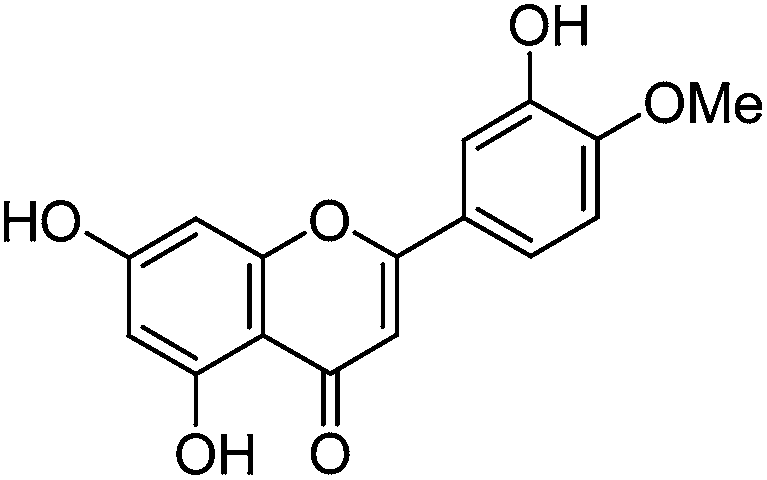
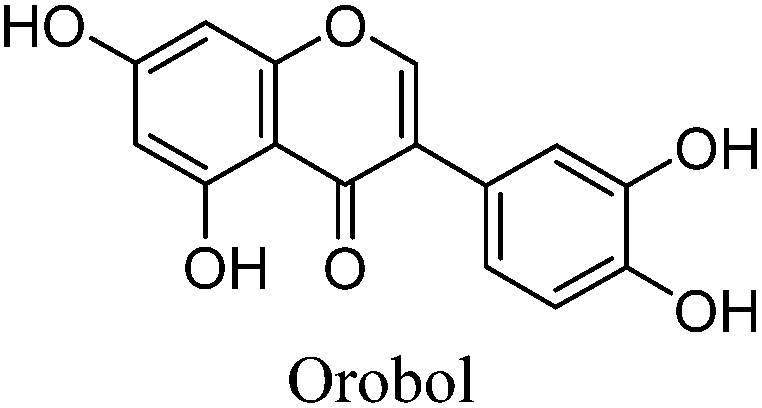
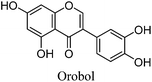
The minor component Gt2 exhibits a maximum absorption at 260 nm which indicates an isoflavonoid structure, with [M − H]− at m/z 285 that corresponds to the isoflavone genistein with an additional OH. Several isomeric isoflavone structures corresponding to this level of hydroxyl substitution have been reported in the literature of which 5,7,3′,4′-tetrahydroxyisoflavone (orobol); 6,7,3′,4′-tetrahydroxyisoflavone; 2,5,7,4′-tetrahydroxy-isoflavone and 7,8,3′,4′-tetrahydroxyisoflavone exhibit a maximum absorption between 258 and 262 nm.26 Detailed MS/MS studies showed a fragmentation pattern similar to genistein (Table 2); and the decrease in retention time under UPLC-MS conditions of 2.2 min relative to genistein compares favourably with the flavonoid pairing of luteolin and apigenin where an additional hydroxyl group in the B-ring decreases the retention time by 2.02 min (Table 1). These data, combined with the UV-vis spectra, suggest that Gt2 corresponds to 5,7,3′,4′-tetrahydroxyisoflavone (orobol), a hydroxylated isoflavone that has been identified in several Genista species growing in Italy and also in Genista tinctoria.27,28
Table 2 Investigation of Gt compoundsa
| Unknown component |
PDA Rt (min) |
λ
max MeOH (nm) |
MS/MS Rt (min) |
[M − H]−m/z |
MS/MS (CE: 25 eV) m/z (% BPI) fragment |
Proposed structure |
|
The ions annotated with iFF correspond to the isoflavonoid-specific fragmentation (0,3B−).
|
| Gt1 |
10.06 |
218 (sh) |
— |
— |
— |
|
Not identified |
| 255 |
|
| 290 (sh) |
|
| |
| Gt2 |
11.67 |
|
11.96 |
285 |
285 (100) |
[M − H]− |
|
|
|
268 (8.5) |
|
| 260 |
257 (13.2) |
[M − H − CO]− |
| 290 (sh) |
240 (6.4) |
[M − 2H − CO2]− |
| 330 (sh) |
229 (12.3) |
[M − H − 2CO]− |
|
|
217 (12.7) |
[M − H − C3O2]− |
|
|
198 (9.8) |
|
|
|
149 (14.3) |
iFF |
| |
| Gt3 |
12.17 |
|
12.38 |
283 |
283 (28.5) |
[M − H]− |
|
|
|
268 (64.8) |
[M − H − CH3]−˙ |
| 255 |
240 (100) |
[M − H − CH3 − CO]−˙ |
| 330 (sh) |
211 (3.4) |
[M − 2H − CH3 − 2CO]− |
|
|
196 (14.8) |
[M − H − CH3 − CO − CO2]−˙ |
|
|
184 (10.1) |
|
| |
| Gt4 |
14.61 |
|
14.89 |
299 |
299 (12.8) |
[M − H]− |
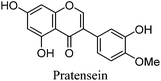
|
| 219 (sh) |
284 (100) |
[M − H − CH3]−˙ |
| 261 |
256 (7.3) |
[M − H − CH3 − CO]−˙ |
| 295 (sh) |
255 (6.0) |
[M − 2H − CH3 − CO]− |
| 340 (sh) |
227 (5.5) |
[M − 2H − CH3 −2CO]− |
|
|
200 (6.4) |
|
Fragmentation of Gt3.
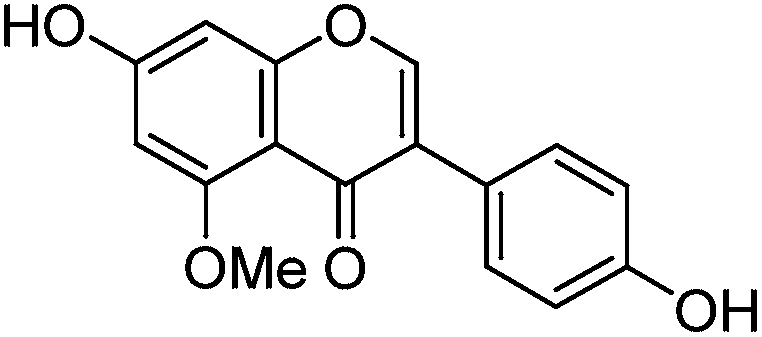
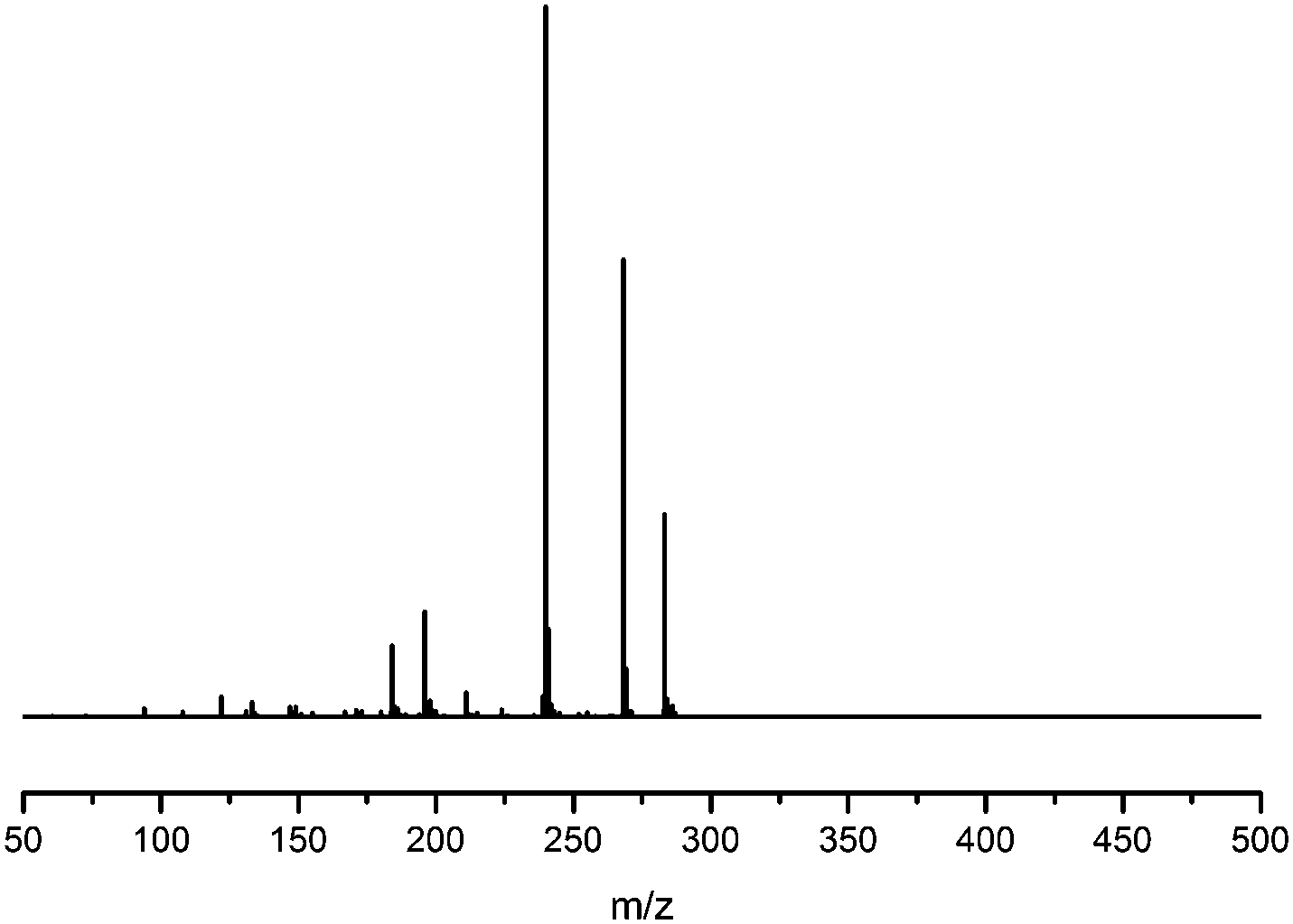
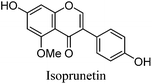
The minor component Gt3 exhibits a maximum absorption at 254 nm, with [M − H]− at m/z of 283 that corresponds to an O- or C-methylated isoflavonoid related to genistein. Detailed MS/MS studies (Fig. 4) showed the presence of a [M − H − CH3]−˙ fragment ion at m/z 268, characteristic of an O-methylated isoflavone. In addition, the base peak at CE 25 eV was found to be at m/z 240 corresponding to the fragment ion [M − H − CH3 − CO]−˙. The close match of the MS2 fragmentation pattern to that of the authentic standard, combined with the correlation of retention time and the UV-vis data allowed Gt3 to be unambiguously identified as 5-O-methyl genistein or isoprunetin (6). This was confirmed by a spiking experiment under the UPLC conditions in which authentic isoprunetin was added to the extract from reference yarn YW2 (ESI 4†). Although it has not previously been identified in historical textile extracts, the presence of high levels of isoprunetin and its associated glycoside in the Genista tinctoria L. textile extract is not surprising, as it is one of the main components reported in Genistea species,29 and has been previously characterised in several Genista species and also Genista tinctoria L. plant extracts.28,30
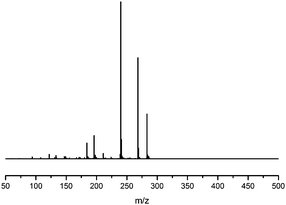 |
| | Fig. 4 MS/MS fragmentation of the [M − H]− ion (m/z 283) of unknown component Gt3 under negative ion electrospray ionisation conditions (CE: 25 eV). | |
3.3 Effect of textile preparation and ageing on the dye fingerprint
3.3.1 Over-dyeing process.
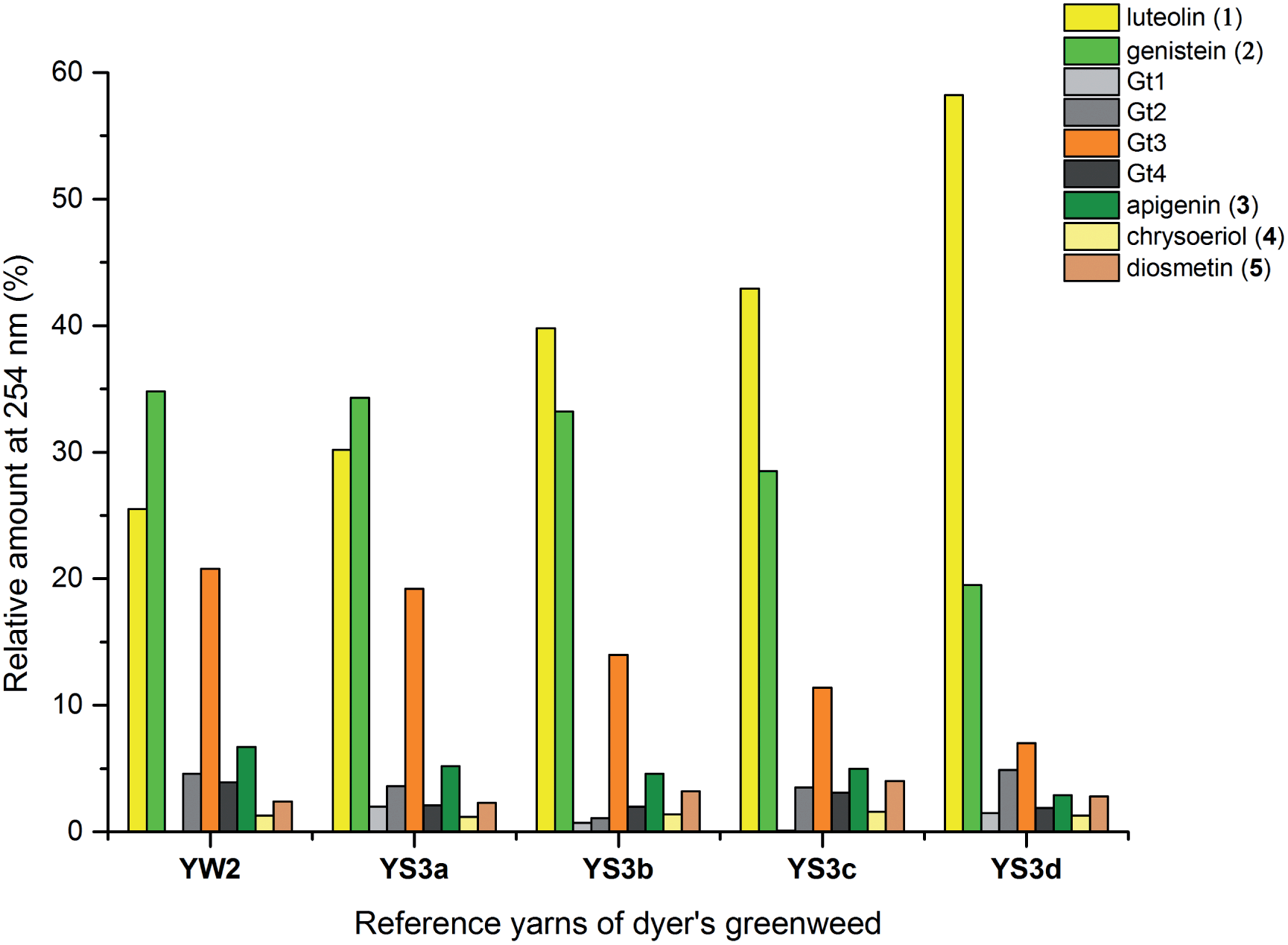
The composition of dyestuffs extracted from historical textile samples is known to vary due to the effects of ageing, but it is also thought to be dependent on workshop practices such as over-dyeing (where a yarn is treated with successive dyebaths in order to achieve the desired colour or hue).4,8 Silk reference samples prepared by over-dyeing with successive dyer's greenweed dye baths (YS3a–d, one to four baths respectively) were examined by UPLC (Fig. 5). While the silk reference YS3a shows the highest level of Gt3 and genistein (2) compared to luteolin (1), this is gradually reduced as the textile is over-dyed (YS3b–d). A number of explanations for this have been suggested including that luteolin (1) binds the mordant more efficiently than the isoflavone genistein (2),4 so that the latter is displaced with each additional dyeing, or that genistein (2), degrades preferentially under the dyebath conditions,4 or other reasons. The degradation of genistein (2), and Gt3, in the dyebath would be supported by several studies of soy products which showed that isoflavonoid compounds, especially genistein (2), degraded easily at elevated temperatures.34,35 However, in the context of dyeing a gradual equilibration to give the most stable dye/mordant/proteinaceous fibre complex is a more attractive explanation for this variation. Particularly as a displacement mechanism might also be expected to apply to Gt3, which should not bind as strongly due to the modified chelating motif provided by the 5-O-methylation, and in support of this the ratio of Gt3 to luteolin is the most noticeably reduced through subsequent overdyeing processes (YS3a–d). The presence of both colourless components, genistein (2) and Gt3, in high quantities on wool and silk yarns after a single dyebath would clearly affect the colour achieved and might explain why dyer's greenweed was judged to be of lower quality as a dyestuff than weld due to the need for multiple dyeing processes to obtain the desired yellow colour.8
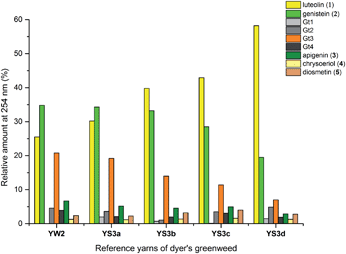 |
| | Fig. 5 Relative amounts of the flavonoid dyes present in the acid-hydrolysed extracts of wool (YW2) and silk (YS3a) MODHT reference samples and a series of over-dyed (YS3b–d) samples; monitored at 254 nm. | |
3.3.2 Light ageing.
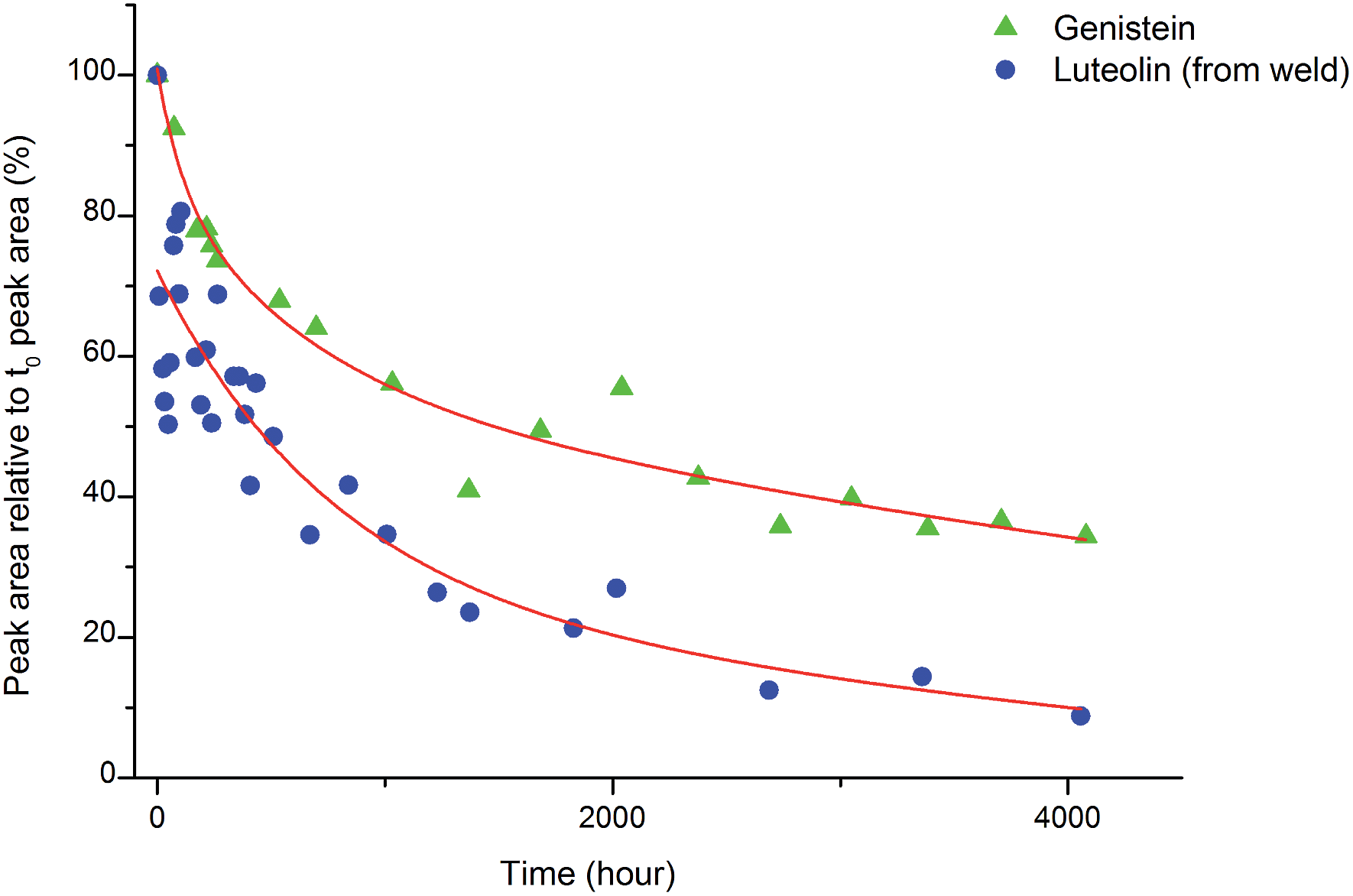
An investigation into the relative degradation rates of luteolin (1) and genistein (2) was carried out using reference wool yarn YW1 dyed with weld (Reseda luteola L.) and an alum mordanted wool yarn ‘dyed’ with pure genistein that were aged under accelerated conditions. The results were then compared with the degradation of luteolin in weld and have been plotted in Fig. 6, where the area of each component, per mg of yarn (monitored at 254 nm) has been expressed as a percentage of the area obtained in the un-aged sample extract. The degradation rate of both components can be fitted to a double exponential model, predicted by previous work.3 This is best explained, at least to a first approximation, by the two different degradation rates associated with the dye on the surface of the yarn and the dye in the bulk of the yarn. After 4000 h of exposure in the light box, the amount of luteolin observed in the extract (per mg of yarn) had fallen to ca. 10% of that observed in the original, un-aged, extract. In contrast, the amount of genistein observed in the extract after 4000 h of ageing had only fallen to ca. 35% of that observed in the original, un-aged, extract; thus confirming the observation that genistein (on an alum mordanted wool substrate) has a relatively slow photo-degradation rate compared to luteolin. These results suggest that although the relative amount of genistein initially present on the fibre is dependent on the dyeing process, its presence in the acid hydrolysed extract should act as a “marker” in historical samples.4
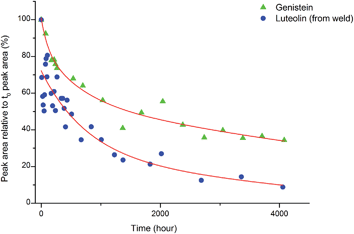 |
| | Fig. 6 The photo-degradation rates of genistein (2) and luteolin (1). | |
3.3.3 Historical tapestry samples.
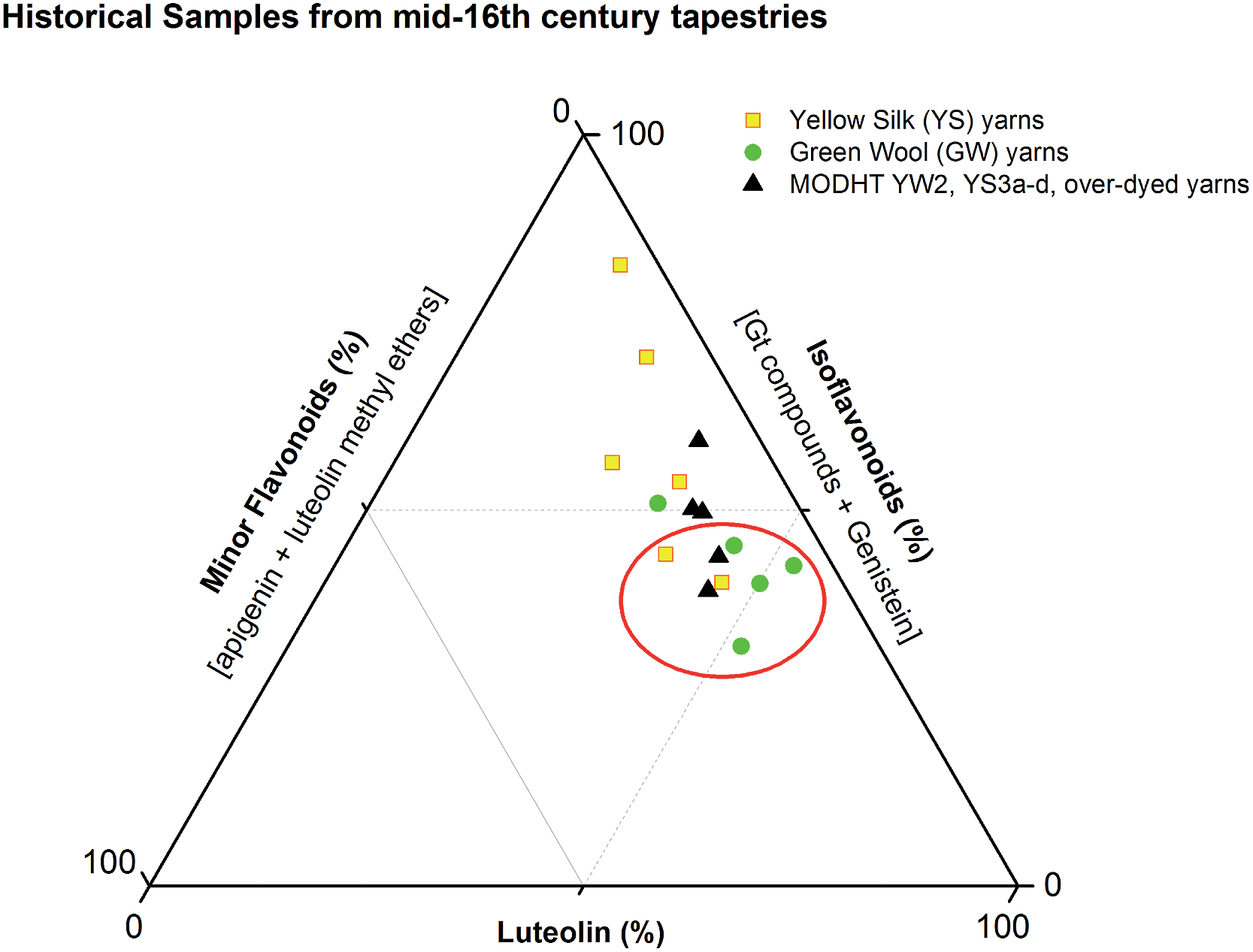
These studies have allowed us to contextualise the analytical data acquired from a small selection of historical yarns (yellow silk and green wool) sampled from mid-sixteenth century English tapestries from the Burrell Collection in Glasgow, UK and the Bodleian Library in Oxford, UK (ESI 4†). The dye profiles from these yarns are characterised by the presence of the flavones luteolin (1) and apigenin (3), the isoflavone genistein (2), the O-methylated flavone chrysoeriol (4) and the isoflavonoid compounds Gt3 and Gt4, along with indigotin in the green yarns. Interestingly, diosmetin (5) was only identified in two historical samples, highlighting the need for further investigation into the light fastness of both O-methylated flavone isomers. The presence of both the isoflavone genistein and the methylated isoflavonoid compound Gt3 in most of the historical yarns investigated would suggest that they exhibit similar photo-degradation rates. It was observed that the relative amounts of isoflavonoid dyes were very variable in historical yarns, which would suggest that these variations could be related to the textile preparation, as was observed in the reference yarns subjected to over-dyeing processes. A ternary representation of the relative amounts of the dyestuff components (Fig. 7) highlights the differences in composition that result from these differences in yarn preparation and allows the dye profile of the historical samples (ESI, Tables 2 and 3†) to be compared with those of over-dyed silk references. From these observations, it does appear that most of the yellow yarns were not over-dyed (with an isoflavonoid content of >50%), while in contrast several of the green yarns show a profile which more closely matches that of the over-dyed silk samples, i.e. with a higher ratio of luteolin:genistein. However, in light of the photo-degradation studies presented above, the scattering of the composition might also reflect variation in the levels of photo-degradation of the samples.
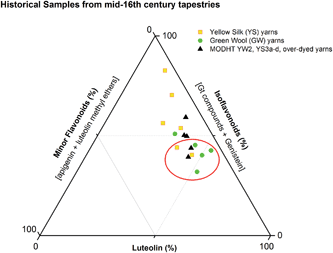 |
| | Fig. 7 Ternary representation of the relative amounts of dye components characterised in the acid hydrolysed extracts of historical silk and wool yarns and over-dyed silk reference yarns (MODHT YW2 and YS3a–d), monitored at 254 nm. Dyestuffs are grouped as follows: (i) luteolin (1); (ii) isoflavonoids [Gt3 and genistein (2), and minor components Gt1, Gt2 and Gt4 for reference samples]; and (iii) minor flavone components [apigenin (3), chrysoeriol (4), diosmetin (5)]. The red circle indicates the historical samples that it is thought were subjected to an over-dyeing process. | |
4. Conclusions
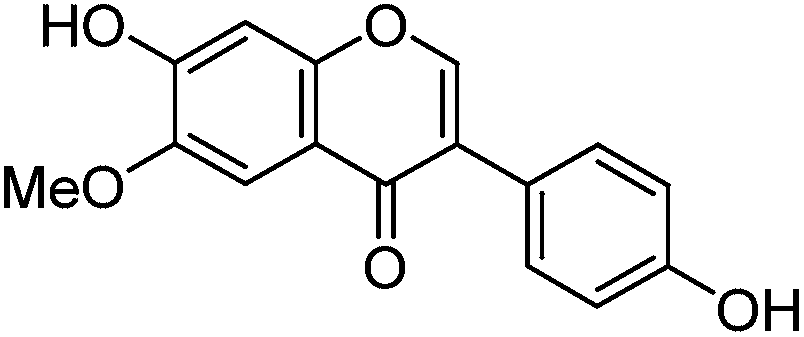
This UPLC study provides a greatly improved method for the identification of a range of flavonoid and isoflavonoid dyestuffs from historical textile samples, including the regio-isomeric compounds chrysoeriol and diosmetin. This method has allowed the unambiguous identification of additional dye components which occur in the plant extract of Genista tinctoria L. and also in the acid hydrolysed extracts of reference and historical yarns dyed with this species. Four unknowns were also identified in the extracts, designated Gt1–4 on the basis of their relative retention times, and further study of these by UPLC-MS/MS allows us to suggest a structural identification of Gt2 as orobol, Gt3 as isoprunetin and Gt4 as pratensein (or a related O-methylated isoflavone). We believe that these, in particular Gt3, might aid with the identification of dyer's greenweed as a plant dye source on historical textiles. This robust method was also used to study the effects of over-dyeing and photo-degradation on the relative compositions of dyestuff components on wool and silk substrates, allowing us to contextualise a selection of historical yarns sampled from mid-sixteenth century English tapestries. The application of UPLC and MS techniques to the study of historical textile samples opens up exciting possibilities for heritage applications, as objects from which sampling was previously not possible may now be examined due to the greatly reduced requirements for sample size and subsequent limit of detection obtained, enabling the identification of very minor components in heavily degraded samples. Given the clear benefits which this study demonstrates and the wealth of information which these methods provide, we anticipate that those working in the field of heritage science will rapidly adopt UPLC as the method of choice for natural product dyestuff analysis.
Acknowledgements
We thank Jane Rowlands and Patricia Collins (Burrell Collection, Glasgow, UK) and David Howell (Bodleian Library, Oxford, UK) for allowing sampling of the tapestries; Andrew Simpson from Waters UK for technical support on both HPLC and UPLC systems; Lorraine Gibson, Strathclyde University, Glasgow UK, for advice during method development; Logan Mackay, University of Edinburgh for assistance with UPLC-MS. Financial assistance was provided through the Science and Heritage programme (studentship to LGT, Grant ref. CDA08/411), the EC (contract number EVK4-CT-2001-00048, Monitoring of Damage to Historic Tapestries project), Glasgow Museums and National Museums Scotland. Tapestry detail in the graphical abstract reproduced courtesy of Glasgow Museums Collection.
Notes and references
- N. C. Veitch, Nat. Prod. Rep., 2013, 30, 988–1027 RSC.
-
V. Sharma and K. G. Ramawat, in Natural Products, ed. K. G. Ramawat and J. M. Mérillon, Springer-Verlag Berlin Heidelberg, 2013, pp. 1849–1865 Search PubMed.
-
E. S. B. Ferreira, PhD thesis, University of Edinburgh, 2002.
-
D. A. Peggie, PhD thesis, University of Edinburgh, 2006.
- D. Cardon, Dyes Hist. Archaeol., 1995, 13, 59–73 Search PubMed.
- E. S. B. Ferreira, H. McNab, A. N. Hulme and A. Quye, Chem. Soc. Rev., 2004, 33, 329–336 RSC.
-
D. Cardon, Le monde des teintures naturelles, Belin Paris, 2003 Search PubMed.
-
J. H. Hofenk de Graaf, The colourful past, Archetype Publications, 2005 Search PubMed.
-
A.-M. Hacke and A. Quye, Appendix 2 in Wroughte in Gold and Silk: Preserving the Art of Historic Tapestries, ed. A. Quye, K. Hallet and C. Herrero Carretero, NMS Enterprises Ltd. – Publishing, Edinburgh, UK , 2009 Search PubMed.
- N. Al-Maharik and N. P. Botting, Tetrahedron, 2003, 59, 4177–4181 CrossRef CAS.
- B. J. Compton, L. Larsen and R. T. Weavers, Tetrahedron, 2011, 67, 718–726 CrossRef CAS PubMed.
- S. A. Adensanya, M. J. O'Neill and M. F. Roberts, Phytochemistry, 1985, 24, 2699–2702 CrossRef.
- J. Wouters and A. Verhecken, Stud. Conserv., 1989, 34, 189–200 CrossRef CAS PubMed.
- D. A. Peggie, A. N. Hulme, H. McNab and A. Quye, Microchim. Acta, 2008, 162, 371–380 CrossRef CAS.
- C. Hartmann, J. Smeyers-Verbeke, D. L. Massart and R. D. McDowall, J. Pharm. Biomed. Anal., 1998, 17, 193–218 CrossRef CAS.
- A. Serrano, M. van Bommel and J. Hallett, J. Chromatogr. A, 2013, 29, 102–111 CrossRef PubMed.
-
L. G. Troalen, PhD thesis, The University of Edinburgh, 2013.
- A. Villela, E. J. C. van der Klift, E. S. G. M. Mattheussens, G. C. H. Derksen, H. Zuilhof and T. A. van Beek, J. Chromatogr. A, 2011, 1218, 8544–8550 CrossRef CAS PubMed.
- A. N. Hulme, H. McNab, D. A. Peggie and A. Quye, Phytochemistry, 2005, 66, 2766–2770 CrossRef CAS PubMed.
- H. McNab, E. S. B. Ferreira, A. N. Hulme and A. Quye, Int. J. Mass Spectrom., 2009, 284, 57–65 CAS.
- K. Ablajan, J. Mass Spectrom., 2011, 46, 77–84 CrossRef CAS PubMed.
- R. E. March, M. Xiu-Sheng, C. D. Metcalfe, M. Stobiecki and L. Marczak, Int. J. Mass Spectrom., 2004, 232, 171–183 CAS.
- J. Kang, L. A. Hick and W. E. Price, Rapid Commun. Mass Spectrom., 2007, 21, 857–868 CrossRef CAS PubMed.
- M. Łuczkiewicz, D. Głód, T. Bączek and A. Buciński, Chromatographia, 2004, 60, 179–185 Search PubMed.
- F. Tosum, C. Akyuz Kizilay and A. U. Tosun, Chem. Nat. Compd., 2009, 45, 83–84 CrossRef.
- S. E. Kulling, D. M. Honig, T. J. Simat and M. Metzler, J. Agric. Food Chem., 2000, 48, 4963–4972 CrossRef CAS PubMed.
- C. Noccioli, L. Luciardi, S. Barsellini, C. Favro, A. Bertoli, A. Bader, M. C. Loi and L. Pistelli, Chem. Nat. Compd., 2012, 48, 672–673 CrossRef CAS.
- D. Rigano, V. Cardile, C. Formisano, M. T. Maldini, S. Piacente, J. Bevilacqua, A. Russo and F. Senatore, Chem.-Biol. Interact., 2009, 180, 211–219 CrossRef CAS PubMed.
- J. B. Harborne, Phytochemistry, 1969, 8, 1449–1456 CrossRef CAS.
- O. Boumaza, R. Mekkiou, R. Seghiri, D. Sarri, S. Benayache, V. P. Garcia, J. Bermejo and F. Benayache, Chem. Nat. Compd., 2006, 42, 730–731 CrossRef CAS PubMed.
- M. Chen, L. Liu and X. Chen, Can. J. Chem., 2013, 11, 1147–1154 CrossRef.
- I. I. Ozimina and V. A. Bandyukova, Khim. Prir. Soedin., 1985, 507–510 CAS.
- K. Lech, K. Witkoś and M. Jarosz, Anal. Bioanal. Chem., 2014, 406, 3703–3708 CrossRef CAS PubMed.
- Y. Ungar, O. F. Osundahunsi and E. Shimoni, J. Agric. Food Chem., 2003, 51, 4394–4399 CrossRef CAS PubMed.
- B. Eisen, Y. Ungar and E. Shimoni, J. Agric. Food Chem., 2003, 51, 2212–2215 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of an authentic sample of isoprunetin; (ESI−) MS2 fragmentation of flavonoid and isoflavonoid standards at 25 eV; UPLC-PDA and (ESI−) MS2 fragmentation of Gt components at 25 eV; UPLC-PDA investigation of references and historical yarns. See DOI: 10.1039/c4ay01509f |
| ‡ As commonly adopted by the field, the abbreviation UPLC has been used for the technique of ultra-high pressure liquid chromatography (UHPLC). |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a