Gold(III) porphyrins as a new class of anticancer drugs: cytotoxicity, DNA binding and induction of apoptosis in human cervix epitheloid cancer cells†
Chi-Ming
Che
*,
Raymond Wai-Yin
Sun
,
Wing-Yiu
Yu
,
Chi-Bun
Ko
,
Nianyong
Zhu
and
Hongzhe
Sun
*
Department of Chemistry and Open Laboratory of Chemical Biology of the Institute of Molecular Technology for Drug Discovery and Synthesis, The University of Hong Kong, Pokfulam Road, Hong Kong. E-mail: cmche@hku.hk; Fax: +(852)2857-1586
First published on 16th June 2003
Abstract
A series of gold(III) tetraarylporphyrins are stable in the presence of glutathione and exert much higher potency than cisplatin in killing human cancer cells, including the drug-resistant variants; the gold-induced cytotoxicity occurs through an apoptotic pathway according to laser confocal microscopy and flow cytometric studies.
Gold compounds are promising candidates for new anticancer drugs, few of them exhibit good stability under physiological conditions due to reduction of Au(III) to Au(I).1 The most successful example is a cyclometallated gold(III) complex2,3 denoted as [AuIII(dmamp)Cl2] [dmamp = 2-(dimethylaminomethyl)phenyl] which exhibits potent in vitro and in vivo antitumor activities against human carcinoma xenografts.2a
Herein we present that the gold(III) meso-tetraarylporphyrins 1a–e (Fig. 1), which are stable against demetallation in physiological conditions, exhibit 100-fold stronger cytotoxicity than cisplatin against a panel of human cancer cell lines, including multidrug- (KB-V1) and cisplatin-resistant cancer cells (CNE-1). This finding highlights the prospect of gold(III) porphyrins as a promising lead for anticancer drug development.
 | ||
| Fig. 1 Gold(III) tetraarylporphyrin complexes. | ||
The [AuIII(p-Y-TPP)]Cl complexes [Y = H (1a), Me (1b), OMe (1c), Br (1d) and Cl (1e)] (Fig. 1) were prepared by a reported method.4 The molecular structure of [AuIII(TPP)]ClO4 has been established by X-ray crystallography.†‡ The redox properties of 1a–e in CH3CN containing 0.1 M nBu4NPF6 have been studied by cyclic voltammetry. The negative potential (−0.96 to −1.02 V vs Cp2Fe+/0; see Table S1, ESI†) required for one-electron reduction of the gold(III) porphyrins is consistent with stabilization of Au(III) by the dianionic porphyrin ligand. Indeed, the reversibility of the redox couples indicates no demetallation of the gold(III) porphyrins upon electrochemical reduction.5
The gold(III) porphyrins exhibit excellent stability in a Tris buffer–MeCN (9 : 1) solution (pH 7.2); no siginficant spectral changes were observed for 1a over 48 h at room temperature based on UV-vis spectroscopy. We examined the stability of the gold(III) porphyrins against reduction by glutathione (GSH), since elevated cellular GSH levels have been implicated in cisplatin-resistance presumably through reduction or sequestration of the cisplatin.6 Treating 1a with GSH (2 mM) in Tris buffer–MeCN (19 : 1 v/v, pH 7.2) did not cause any spectral changes within the first several hours, although upon standing the solution for 48 h reduction of the Soret band intensity by ca. 27% was observed.† Yet, we found that adding acetone (ca. 0.4 mL) to the buffer solution of 1a would completely recover the Soret band to its original band shape and intensity. Demetallation can be ruled out since the characteristic absorption peaks of the free base porphyrins were not observed (e.g. H2TPP: λmax = 514, 548, 590 and 640 nm). Therefore, the observed spectral change is attributed to band broadening due to self-aggregation of the metalloporphyrin. Likewise, treatment of 1b–e with GSH also led to similar observations.
The interaction of 1a with GSH has been studied by 1H NMR spectroscopy. By monitoring the “1a + GSH” mixture in a D2O–CD3CN (9 : 1 v/v) solution at 25 °C for 72 h, we did not observe significant spectral changes in the pyrrolic region (δH 7.2–8.4 ppm). Accordingly, formation of glutathione disulfide (GSSG) by oxidation of GSH was also not observed by 1H NMR (δH 3.30 ppm assignable to the β-CH2 group of GSSG).7 The lack of spectral changes at the pyrrolic region and the absence of GSSG formation indicate that the gold(III) porphyrins are stable against GSH reduction.
By means of the MTT assay, cytotoxicities of 1a–e toward some established human cancer cell lines, including some drug resistant variants were determined. The results are listed in Table 1. All the gold(III) complexes show significant cytotoxicities against human cancer cells with IC50 = 0.1–1.5 µM, and 1a displays the most prominent activities (IC50 = 0.1–0.8 µM). Importantly, 1a was found to be equally cytotoxic to cisplatin-resistant nasopharyngeal carcinoma (CNE1) cell lines with the IC50 value being 0.17 µM, which corresponds to ca. 240-fold higher in potency than cisplatin. The resistance factor, IC50 (CNE1)/IC50 (SUNE1), for cisplatin is 3.2, whereas the corresponding values for 1a–e are close to unity. The lack of cross resistance suggests that the gold(III) porphyrins and cisplatin induce cytotoxicity via different mechanisms.
| Entry | Compound | IC50/µM | ||||||
|---|---|---|---|---|---|---|---|---|
| KB-3-1 | HL-60 | HepG2 | SUNE1 | HeLa | KB-V1 | CNE1 | ||
| a n.d. = not determined. KB-3-1 = human oral epidermoid carcinoma; HL-60 = human promyelocytic leukemia; HepG2 = human hepatocellular carcinoma; SUNE1 = human nasopharyngeal carcinoma; HeLa = human cervix epitheloid carcinoma; KB-V1 = human oral epidermoid carcinoma (multi-drug resistance); CNE1 = human nasopharyngeal carcinoma (cisplatin resistance). | ||||||||
| 1 | 1a | 0.20 ± 0.03 | 0.73 ± 0.02 | 0.34 ± 0.02 | 0.11 ± 0.02 | 0.28 ± 0.02 | 0.11 ± 0.02 | 0.17 ± 0.02 |
| 2 | 1b | 0.41 ± 0.01 | 1.53 ± 0.03 | 1.11 ± 0.03 | 1.09 ± 0.05 | 0.52 ± 0.02 | 1.09 ± 0.05 | 0.99 ± 0.03 |
| 3 | 1c | n.d.a | 0.88 ± 0.02 | 0.76 ± 0.05 | 0.68 ± 0.03 | 0.66 ± 0.04 | n.d.a | 0.77 ± 0.03 |
| 4 | 1d | 1.29 ± 0.02 | 1.12 ± 0.09 | 1.21 ± 0.12 | 0.74 ± 0.08 | 0.89 ± 0.11 | 0.74 ± 0.09 | 0.98 ± 0.04 |
| 5 | 1e | 0.50 ± 0.01 | 0.87 ± 0.03 | 0.92 ± 0.02 | 0.34 ± 0.02 | 0.69 ± 0.02 | 0.34 ± 0.07 | 0.41 ± 0.06 |
| 6 | nBu4N[AuIIICl4] | 10.3 ± 1.02 | 9.71 ± 0.96 | 11.2 ± 0.78 | 9.94 ± 0.88 | 8.41 ± 0.99 | n.d.a | 11.5 ± 0.99 |
| 7 | Cisplatin | 13.2 ± 1.24 | 16.8 ± 1.42 | 13.5 ± 1.82 | 12.6 ± 0.92 | 11.2 ± 0.96 | 12.6 ± 0.78 | 40.8 ± 2.17 |
| 8 | [ZnII(TPP)] | >50 | >50 | >50 | >50 | >50 | n.d.a | >50 |
Complexes 1a–e also show significant cytotoxicities (IC50 = 0.1–1.1 µM) against KB-3–1 and its multi-drug resistant (KB-V1) variant, which possess more membrane P-glycoproteins for expulsion of drugs such as vinblastine and doxorubicin.8 The lack of cross resistance suggests that the P-glycoproteins are ineffective for the gold(III) porphyrins.
Notably [ZnII(TPP)] is at least 100-fold (IC50 > 50 µM) less potent than the gold(III) porphyrins in killing cancer cells. Yet, the porphyrin ligand is essential for the anticancer activities, since nBu4N[AuIIICl4] is 30–90 times less cytotoxic than 1a (Table 1). We reason that the porphyrin ligand should stabilize the Au(III) center and carry the metal to its cellular target.
Using human lung fibroblast cells (CCD-19Lu), the cytotoxicity of 1a to normal cells has been evaluated the IC50 value being 0.91 µM as determined by the MTT assay. Comparing the IC50 values (ca. 0.2 µM) obtained from the cancer cell lines, 1a is about 4.6-fold more toxic to cancer cells.
DNA is one of the major targets for anticancer drugs,9 and binding of metalloporphyrins to DNA has been studied extensively.10 We have examined the interaction of 1a with duplex DNA by UV-vis absorption titrations. Isosbestic spectral changes (isosbestic points at 290 and 425 nm) and significant hypochromicity (44%) of the Soret band upon addition of calf thymus DNA (0–5 μM) to a solution of 1a were observed (Fig. S5 in ESI]. The intrinsic binding constant (Kb) toward calf thymus DNA was determined from the the linear plot (R = 0.99) of [ctDNA]/Δεapvs [ctDNA] with the Kb value = (2.79 ± 0.34) × 106 dm3mol−1 at 293 K.
By confocal microscopy, the gold(III) porphyrins were found to induce apoptosis of cancer cells. After treatment of HeLa cells with 1a (IC60 ∼ 0.5 µM at 37 °C) for 15 h, apoptotic cells were characterized by apoptotic body formation (Fig. 2). Notably, no necrotic cells were detected. In addition to morphological changes, fragmentation of genomic DNA was also evident after incubating the HeLa cells with 1a for 15 h.11
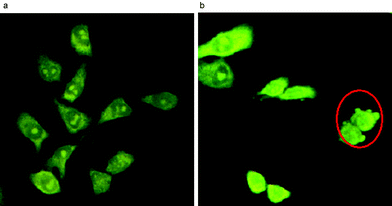 | ||
| Fig. 2 Laser confocal micrographs of the HeLa cells treated with 1a (0.5 µM) at time intervals of (a) 0 h and (b) 15 h. Apoptotic cells are marked by a red circle. | ||
Using fluorescein-labeled annexin V, we have quantified the apoptotic cells by flow cytometry.12 As shown in Fig. 3, upon treatment with 1a (0.5 µM) for 15 h, 57% of the HeLa cells were found to undergo apoptosis as visualized by the annexin V-FITC fluorescence. The gold(III)-induced apoptosis follows a dose-dependent manner: with 0.1 µM of 1a, only 8.6% and 32% of the HeLa cells became apoptotic after 6 and 15 h of incubation, respectively.†
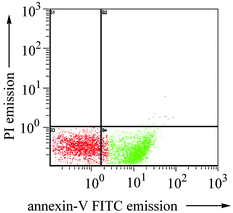 | ||
| Fig. 3 Flow cytometric study of 1a-induced apoptosis of HeLa cells. Plot shows the fluorescent data of propidium iodide (PI) and annexin V-fluorescein corresponding to 57.3% (t = 15 h) apoptotic cells. | ||
As a negative control, untreated HeLa cell shows >90% survival as revealed by the absence of the annexin V-FITC and PI signals. It is noteworthy that no apparent necrotic cells (< 1%) were detected by dual fluorescence from PI and annexin V-FITC due to rupture of the cell membrane thereby exposing both naked DNA and phosphatidylserine (Fig. S6).11
We acknowledge the financial support from The University of Hong Kong (Generic Drugs Research Program) and the Area of Excellence Scheme (AoE/P-10/01) administered by the University Grants Council (HKSAR, China).
Notes and references
- (a) Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512 CrossRef CAS; (b) C. F. Shaw III, Chem. Rev., 1999, 99, 2589 CrossRef CAS.
- (a) R. G. Buckley, A. M. Elsome, S. P. Fricker, G. R. Henderson, B. R. C. Theobald, R. V. Parish, B. P. Howe and L. R. Kelland, J. Med. Chem., 1996, 39, 5208 CrossRef CAS . See also; (b) G. Marcon, S. Carotti, M. Coronnello, L. Messori, E. Mini, P. Orioli, T. Mazzei, M. A. Cinellu and G. Minghetti, J. Med. Chem., 2002, 45, 1672 CrossRef CAS.
- (a) H.-Q. Liu, T.-C. Cheung, S.-M. Peng and C.-M. Che, J. Chem. Soc., Chem Commun., 1995, 1787 RSC; (b) C.-M. Che, M. Yang, K.-H. Wong, H.-L. Chan and W. Lam, Chem. Eur. J., 1999, 5, 3350 CrossRef CAS.
- E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373 Search PubMed.
- K. M. Kadish, W. E, Z. Ou, J. Shao, P. J. Sintic, K. Ohkubo, S. Fukuzumi and M. J. Crossley, Chem. Commun., 2002, 356 RSC.
- B. A. Teicher, S. A. Holden, T. S. Herman, E. A. Sotomayer, V. Khandekar, K. W. Rosbe, T. W. Brann, T. T. Korbut and E. Frei, Int. J. Cancer, 1991, 47, 252 CAS.
- H. Sun, S. C. Yan and W. S. Cheng, Eur. J. Biochem., 2000, 267, 5450 CrossRef CAS.
- D. W. Shen, C. Cardarelli, J. Hwang, M. Cornwell, N. Richert, S. Ishii, I. Pastan and M. M. Gottesman, J. Biol. Chem., 1986, 261, 7762 CAS.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188 CrossRef CAS.
- See for an example: L. A. Lipscomb, F. X. Zhou, S. R. Presnell, R. J. Woo, M. E. Peek, R. R. Plaskon and L. D. Williams, Biochemistry, 1996, 35, 2818 Search PubMed.
- (a) A. Hunt and G. Evan, Science, 2001, 293, 1784 CrossRef CAS; (b) M. O. Hengartner, Nature, 2000, 407, 770 CrossRef CAS.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental and crystallographic details. See http://www.rsc.org/suppdata/cc/b3/b303294a/ |
| ‡ Crystal data: [Au(TPP)]ClO4: C44H28AuClN4O4, M = 909.12, orthorhombic, Pnna, a = 8.1020(16), b = 20.964(4), c = 20.060(4) Å, a = 90, β = 90, γ = 90°, V = 3407.2(12) Å3, T = 253 K, Z = 4, μ = 4.451 mm−1, 13426 collected reflection, 3066 independent reflection, R indices (all data) R = 0.0645, wR = 0.1040. CCDC 201191. See http://www.rsc.org/suppdata/cc/b3/b303294a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |