
Vapor phase synthesis of metal–organic frameworks on a nanofibrous aerogel creates enhanced functionality†
Vahid
Rahmanian
 ,
Muhammed Ziauddin
Ahmad Ebrahim
,
Seyedamin
Razavi
,
Mai
Abdelmigeed
,
Eduardo
Barbieri
,
Stefano
Menegatti
,
Gregory N.
Parsons
,
Fanxing
Li
,
Tahira
Pirzada
* and
Saad A.
Khan
*
,
Muhammed Ziauddin
Ahmad Ebrahim
,
Seyedamin
Razavi
,
Mai
Abdelmigeed
,
Eduardo
Barbieri
,
Stefano
Menegatti
,
Gregory N.
Parsons
,
Fanxing
Li
,
Tahira
Pirzada
* and
Saad A.
Khan
*
Department of Chemical & Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695-7905, USA. E-mail: tpirzad@ncsu.edu; khan@eos.ncsu.edu
First published on 6th November 2023
Abstract
This study introduces an innovative approach for synthesizing metal–organic frameworks (MOFs) on 3D-structured hierarchically porous nanofibrous aerogels (NFAs). The NFA was initially fabricated by solid-templating cellulose diacetate (CDA)–silica electrospun nanofibers and subsequently coated with ZIF-8 MOF via vapor phase synthesis. The ZIF-8 MOF – known for its CO2 adsorption, heavy metal removal, and antibacterial properties – was synthesized directly on the NFA, producing a uniform layer of densely packed ZIF-8 crystals on the CDA–silica nanofibers. The integrated MOF–NFA features a rich functional diversity. First, measurements of mechanical compressibility demonstrated the robustness and strain recovery of the CDA–silica@ZIF-8 NFAs, and thus their applicability in environments that entail exposure to mechanical stress. Furthermore, the ZIF-8-coated aerogel, with its high CO2 adsorption rate and capacity (4.04 mmol g−1), holds promise as a substrate for CO2 separation or sequestration. The aerogel also exhibits excellent performance in removing heavy metals, particularly Cu(II) ions (>99% removal). The high adsorption capacity is rooted in the thin and uniform ZIF-8 coating of the NFA, and grants applicability for water purification. The antibacterial properties of the hybrid aerogel demonstrate efficacy against both Gram-negative and Gram-positive bacteria. In summary, the hybrid NFA shows promise for various applications, particularly in tackling challenges in environmental and biomedical fields, encompassing sorption, catalysis, filtration, water purification, and antimicrobial applications.
1. Introduction
Metal–organic frameworks (MOFs) are a class of crystalline porous coordination polymers composed of metal ions and organic linkers. These materials exhibit high crystallinity and thermal stability,1 and their large specific surface area makes them attractive substrates for water purification,2 drug delivery,3 gas storage,4 separation,5 catalysis,5 sensing6 and energy storage.7,8 In their native form of powders or colloidal crystals, however, MOFs display limited processability.9,10 Additionally, their intrinsic micro/meso-porosity can limit the access and diffusion kinetics of large molecules and nanomaterials.11 The integration of MOFs with various substrates represents a versatile and cost-effective route to increase their formability and enhance their adsorptive behavior. In this context, recent approaches focused on depositing layers of MOF crystals on fibers, inorganic particles, and nanosheets via layer-by-layer,12 spray13 and in situ growth methods.10,14,15 These methods, however, are often limited by the roughness, surface wettability, and functionality of the matrix, and require surface modification to increase the compatibility between the MOF layer and the substrate.16–18 MOF crystals were also embedded into hollow fibers,19 electrospun micro and nanofiber mats,20 and polymer films21,22 to prepare composite membranes. However, particle aggregation and the poor interaction between the MOF particles and the polymer matrix resulted in low MOF contents and thus low specific surface areas.93D self-supported aerogels have recently emerged as excellent candidates for the synthesis of MOF composite materials owing to their innate hierarchical porosity and surface area.23,24 Hybrid MOF-aerogels have been fabricated by growing or aggregating MOF particles on aerogel networks via either simple mixing, solvothermal synthesis, or high-internal-phase emulsion.23–28 Early MOF-loaded aerogels, prepared by mixing MOF nanoparticles in a dispersion of (bio)polymers, followed by gelation and freeze-drying, relied on relatively weak interactions (e.g., physical entanglement and van der Waals forces) and suffered from limited stability.24,25 Other MOF-aerogels were fabricated via in situ synthesis of MOFs on the surface of aerogel precursors, followed by gelation, solvent exchange, and freeze-drying – a process known as solvothermal approach, which is considerably more laborious and provides networks with inadequate structural integrity.9,26,29,30 Hybrid MOF aerogels have also been fabricated via high-internal-phase emulsion (HIPE), which consists of synthesizing MOFs on the interface of Pickering particles followed by freeze-drying.27 This procedure, however, is challenging due to the instability of 3D Pickering emulsion architectures during MOF synthesis. Notably, these methods relied on MOF integration into conventional aerogels, which requires precise tuning of fabrication conditions to achieve gelation over crystallization and precipitation, thus limiting their applicability to a narrow range of pore apertures, porosities, and densities.31 Furthermore, irregular agglomeration of MOF particles decreases the specific surface area of the coat, thus limiting the functionality of the hybrids.32 These limitations highlight the need for a robust and sustainable fabrication method that affords durable integration of MOF particles on aerogels' interconnected networks.
Nanofibrous aerogels (NFAs) are a young family of highly porous and ultra-light self-supported materials composed of a 3D network of interconnected nanofibers.33–35 These fibrous networks provide a mechanically robust substrate with a higher surface area compared to conventional aerogels.35–38 NFA fabrication via freeze-drying of a dispersion of short electrospun nanofibers in a non-solvent produces a fibrous entanglement featuring a bimodal distribution of primary (4–7 μm wide) and secondary pores (40–60 μm diameter).34,39 Compared to the laborious and expensive synthesis of conventional aerogels, this efficient method has allowed researchers to broaden the landscape of NFAs' functionality. Among hybrid NFAs, thermally treated cellulose diacetate (CDA)–silica has received considerable attention for its excellent thermal and mechanical stability, sustainability, flexibility, formability, and hierarchical porosity.35 The addition of a small amount of silica to CDA chains followed by thermal treatment affords a robust 3D structure, with strong bonding between fibers. Notably, CDA–silica NFAs can sustain significant deformations under high compressive strains (>80%) without collapse, which has been attributed to the hierarchical cellular structure of the aerogel, consisting of flexible walls made of densely entangled nanofibers.35 Furthermore, the CDA–silica nanofibers feature high specific surface area, ideal for the formation, growth, or attachment of MOF crystals.
In this study, we synthesized MOF crystals directly on 3D self-supported, ultralight CDA–silica NFA. Unlike conventional aerogels, our NFAs are prepared via a straightforward solid templating approach without involving time-consuming processing steps and large amounts of organic solvents. We adopted ZIF-8, a member of the zeolitic imidazolate framework family, as model MOF to deposit on the NFA. Our fabrication approach consists of depositing thin layers of zinc oxide (ZnO) on the CDA–silica NFA substrate via atomic layer deposition (ALD) followed by converting ZnO to ZIF-8 by treatment with organic vapors of 2-methylimidazole (2-MeIm) linker. Vapor phase synthesis of MOFs has several advantages over traditional solution-phase solvothermal synthesis, such as higher yield, lower cost and environmental impact, and broader functional outcomes.40–42 Recent efforts in developing solvent-free MOF synthesis – including chemical vapor deposition (CVD), ALD, molecular layer deposition (MLD), and combined physical vapor deposition-CVD, have focused on depositing MOF thin films on 2D substrates.43
Studies combining vapor phase MOF synthesis with 3D substrates remain largely unexplored, with the beauty of our approach being to preserve the high micro/meso-porosity of ZIF-8 crystals while combining it with the hierarchical porosities, flexibility, formability, and low density of NFAs. The distinctive combination of sol–gel electrospinning, aerogel fabrication and vapor phase MOF synthesis creates a sustainable and potentially scalable platform for superior performance of ZIF-8 powders in a multitude of applications. The uniqueness of this study lies in (a) the fabrication MOF–nanofibrous aerogels, in particular the use of a solvent-free approach to create MOFs on the nanofibrous surface of the aerogels. Such surface decoration results in firmly attached MOF particles and enhances access to the MOF, (b) the use of nanofibrous instead of conventional aerogels in MOF incorporation wherein the NFA requires fewer steps and provides enhanced functionalities, (c) the mechanical robustness of our systems, possibly resulting from the nanofibrous structure of the aerogels and fabrication protocols; and, (d) multifunctional performance the hybrid NFAs offer. Our proof of concept results on CO2 capture, antibacterial properties and heavy metal removal highlight the versatility of our system. Nevertheless, they are by no means comprehensive and future studies need to be undertaken to delve into each of these further.
2. Result and discussion
2.1 Construction, morphology and characterization
We synthesized NFAs by freeze-drying a dispersion of hybrid CDA–silica nanofibers following our previously established protocol.35 Briefly, the initial step involves sol–gel electrospinning of bead-free hybrid CDA–silica nanofibers under controlled process conditions and precursor mixture composition. The resulting 3D self-supported CDA–silica NFA is generated by chopping the nonwoven electrospun nanofibers into short fibers of specific aspect ratios in tertiary butanol (t-BuOH, a nonsolvent), followed by freeze-drying and thermal treatment. Freeze-drying leads to the creation of a hierarchical cellular structure due to the concentration of entangled fiber fragments between growing solvent crystals. Then, the NFA is conformally coated with thin layers of ZnO using an ALD process. Fig. S2c† shows the scanning electron microscope (SEM) of the ZnO coated NFAs with the hierarchical porosity. Finally, the ZnO is converted to ZIF-8 to create CDA–silica@ZIF-8, by hanging ZnO-decorated CDA–silica NFA in a closed glass jar containing a powder bed of 2-MeIm for 6 hours at 135 °C under vapor phase conversion conditions (schematic in Fig. 1). This process results in a densely packed, uniformly distributed layer of ZIF-8 crystals to the NFA substrate. The hierarchical porous structure of the NFA facilitates diffusion and access to the active sites located within ZIF-8 micro/mesopores.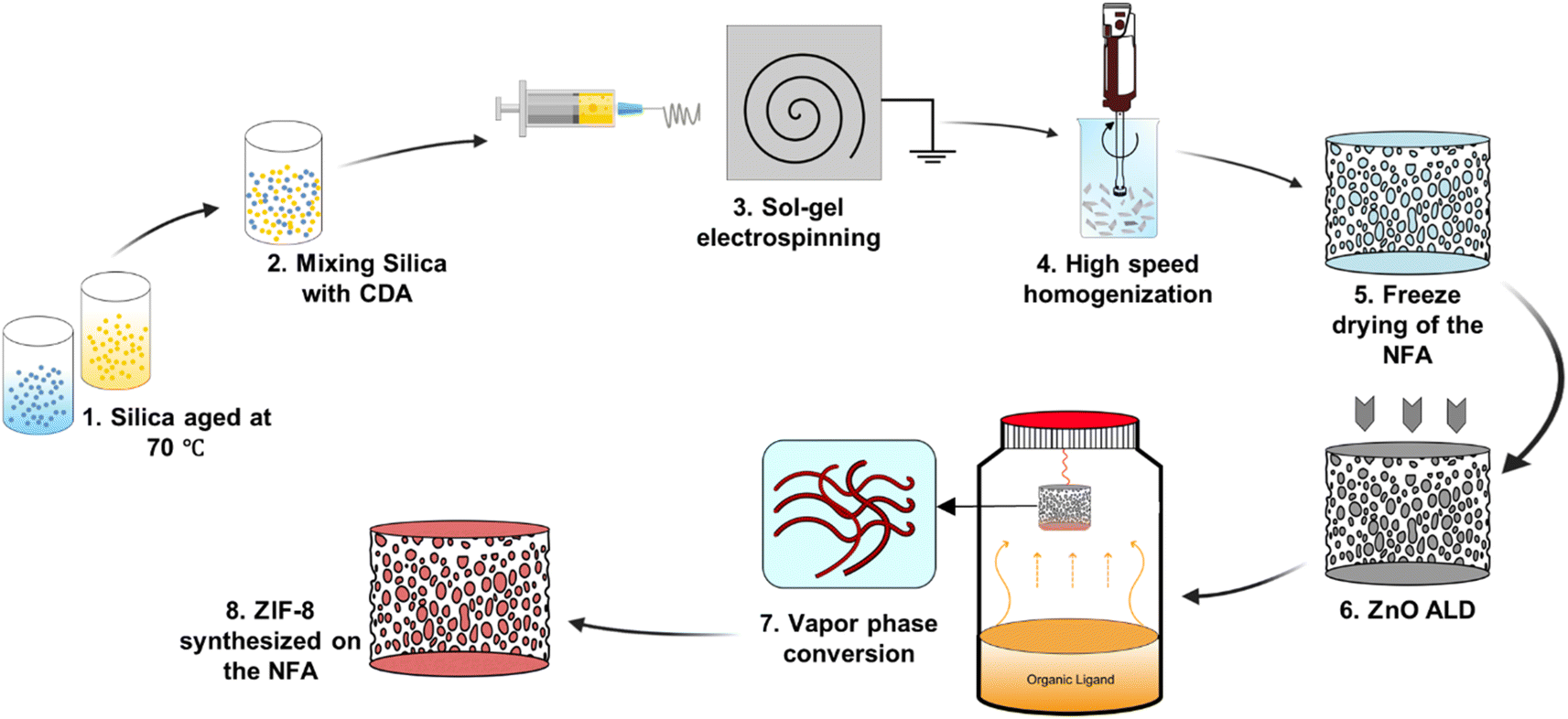 | ||
| Fig. 1 Pathway toward fabrication of CDA–silica@ZIF-8 NFA. | ||
As anticipated, the ZnO-coated CDA–silica NFA substrate enables the creation of a uniform and conformal layer of the metal oxide for ZnO-to-ZIF-8 conversion, while also serving as an adhesion layer between ZIF-8 and the NFA substrate. The morphology of CDA–silica nanofibers and NFA coated with ZIF-8 crystals is presented in Fig. 2. As shown by SEM images, the ZIF-8 crystals are tightly packed, uniformly distributed, and appear to be strongly attached to the fiber surface coated with 60 (Fig. 2a and b) and 120 (Fig. 2d and e) cycles of ZnO ALD. The morphology of pristine CDA–silica NFA demonstrated in Fig. 2c shows the hierarchical porous structure consisting of secondary and primary pores and is retained after the growth of ZIF-8 (Fig. 2f). Video S1† demonstrates qualitatively that the ZIF-8 crystals remain firmly attached to the NFA substrate without detachment following mechanical deformation. Additionally, Energy Dispersive X-ray (EDX) mapping displayed in Fig. S1† confirms the uniform distribution of zinc and nitrogen in the ZIF-8 decorated CDA–silica nanofibers. This level of homogeneity indicates a high conversion rate of ZnO to ZIF-8 crystals, which is desirable in our approach. As demonstrated in SEM images shown in Fig. 2, coating nanofibers with 60 and 120 ZnO ALD cycles resulted in crystallization of ZIF-8 during vapor phase synthesis. In order to investigate the impact of different numbers of ZnO ALD cycles on the crystallization of ZIF-8, we investigated the vapor phase synthesis on nanofibers coated with 200 and 300 ZnO ALD cycles. The SEM images in Fig. S2† illustrate the agglomeration of ZIF-8 crystals on the fiber surface for both the 200 (Fig. S2a†) and 300 (Fig. S2b†) cycles of ZnO ALD. This resulted in a decrease in mechanical properties, rendering the nanofibers brittle.
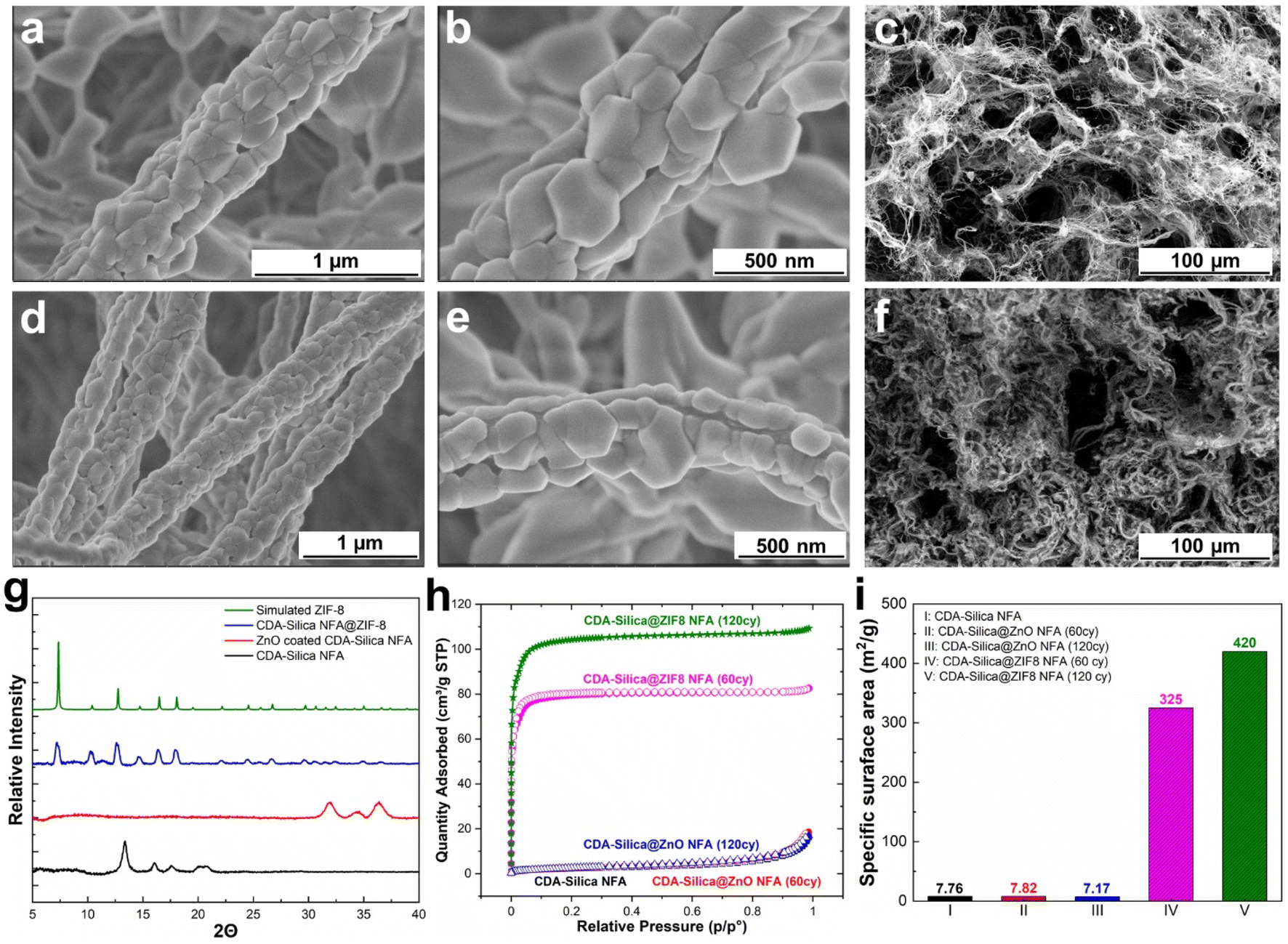 | ||
| Fig. 2 Morphology and characterization of CDA–silica@ZIF8 hybrid: (a and b) SEM images of CDA–silica@ZIF8-60cy at two magnifications. (c) SEM image of pristine CDA–silica NFA. (d and e) SEM image of CDA–silica@ZIF8-120cy at two magnifications. (f) SEM image showing the porous structure of CDA–silica@ZIF8-120cy. (g) XRD patterns of CDA–silica, CDA–silica@ZnO, CDA–silica@ZIF8-60cy and simulated ZIF-8 powder. (h) N2 physisorption isotherms of CDA–silica, CDA–silica@ZnO-60cy, CDA–silica@ZnO-120cy, CDA–silica@ZIF8-60cy and CDA–silica@ZIF8-120cy NFAs at 77 K. (i) BET specific surface area calculated form N2 physisorption isotherms. | ||
To further evaluate the presence of ZIF-8 crystals on CDA–silica NFA, we conducted a comparison of XRD between CDA–silica, ZnO coated CDA–silica and CDA–silica@ZIF-8 NFAs. As shown in Fig. 2g, the XRD of the CDA–silica@ZIF-8 NFA with 60 cycles of ZnO ALD and 6 hours of conversion displayed characteristic diffraction peaks at 2θ values of 7.3°, 10.4°, 12.8°, 14.7°, 16.5° and 18.1°, which correspond to the (110), (200), (211), (220), (013) and (222) crystal faces of ZIF-8, indicating successful conversion of the ZnO to ZIF-8 crystals during vapor phase conversion. The peak broadening observed in the sample XRD pattern suggests the formation of nanosized crystals. The crystallite size was calculated to be approximately ∼80–120 nm using the Scherrer equation.44 This observation supports the role of hierarchical morphology of the NFA as well as the nanoscale diameter of the nanofibers in facilitating the formation of smaller crystals on the substrate and promoting the distribution of ZIF-8 crystals. FTIR spectroscopy was also performed and revealed characteristic IR peaks at 1574 cm−1, 1146 cm−1, 758 cm−1, and 420 cm−1, corresponding to aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–N, out-of-plane bending of the 2-MeIm ring, and Zn–N stretching modes of ZIF-8, respectively (Fig. S3a†).
N, C–N, out-of-plane bending of the 2-MeIm ring, and Zn–N stretching modes of ZIF-8, respectively (Fig. S3a†).
Later we determined the surface area of pristine CDA–silica, CDA–silica@ZnO, and CDA–silica@ZIF-8 under different conditions, using the Brunauer–Emmett–Teller (BET) method (Fig. 2h). The results further prove that the vapor phase synthesis of ZIF-8 was successful, as the surface area of the CDA–silica@ZIF-8 composites increases to 325 and 420 m2 g−1 for 60 and 120 ZnO ALD cycles respectively compared to 7.8 m2 g−1 for bare CDA–silica NFA. Furthermore, the pore size distribution of the CDA–silica@ZIF-8 composites was determined using the BET method, as depicted in Fig. S2b.† The pore sizes of the ZIF-8 component of the composites range from 8 to 11 Å, aligning closely with values reported in the literature.45,46 The micro/mesoporosity of the ZIF-8 crystals is responsible for the high specific surface area observed in the CDA–silica@ZIF-8 composites. However, when the number of ZnO ALD cycles is further increased to 200 and 300, the surface area decreases to 264 and 160 m2 g−1, respectively. This indicates that pore blockage, agglomeration (as illustrated in the SEM images in Fig. S2a and b†) or degradation of the MOF crystals may have occurred due to the presence of clustered granules contributed by 2-MeIm. Based on all the findings from the characterization analysis, we have determined that maintaining the number of ZnO ALD cycles at 60 and 120 is the most suitable approach for vapor phase conversion of ZnO to ZIF-8. These specific ZnO ALD cycle numbers consistently yield uniformly distributed ZIF-8 crystals with a significantly high specific surface area.
2.2 Mechanical properties
Mechanical properties of the NFAs are attributed to their robust hierarchical cellular structure, which allows for significant deformations without fracture. Unlike conventional aerogels, the NFAs can withstand a compressive strain (ε) of up to 80% and recover their original volume after stress release.34,35 To assess the compressibility, strength, and shape recovery of the NFAs, we conducted a series of compressive stress–strain (σ–ε) tests on CDA–silica and CDA–silica@ZIF-8 NFAs samples (details in the Experimental section). Fig. 3 shows compression–relaxation curves for the NFAs at set ε maxima of 10, 20, 40, 60, and 80%, revealing three characteristic deformation regimes: a Hookean or linear elastic regime at low strains, a plateau region representing elastic buckling of the cell walls, and a densification regime at higher strains with steeply increasing σ and dσ/dε. Both CDA–silica (Fig. 3a) and CDA–silica@ZIF-8 NFAs (Fig. 3b) exhibit a small Hookean region with an elastic modulus of 3.0 kPa. The similar Young's moduli of the two NFAs indicates that MOF synthesis has a small effect on their mechanical strength. CDA–silica@ZIF-8 NFA displays robust mechanical performance, sustaining large deformations without collapse at compressive strains up to 80% and exhibiting fatigue resistance after 50 stress–strain cycles at 60% strain (Fig. 3b and c). This resilience is attributed to the hierarchical cellular structure of flexible walls made of densely entangled nanofibers, which act as a cushion when compressed and bounce back to their original position after load removal.34,35 The retention of 3D geometry after repeated compressive cycles in both cases indicates the role played by strong interactions developed during thermal treatment in making the 3D structure more robust and flexible to support multiple cycles of heavy loads with shape recovery. The strain is absorbed by the deformation of primary pores and beyond 60% strain by the densification of secondary pores. Strain-dependent hysteresis loops in most of the loading–unloading curves indicate energy dissipation due to the fibrous network. The small quantity of silica (∼6 wt%) in the NFAs helps glue the CDA chains strong enough to hold the fibers together and develop a robust 3D structure upon heating.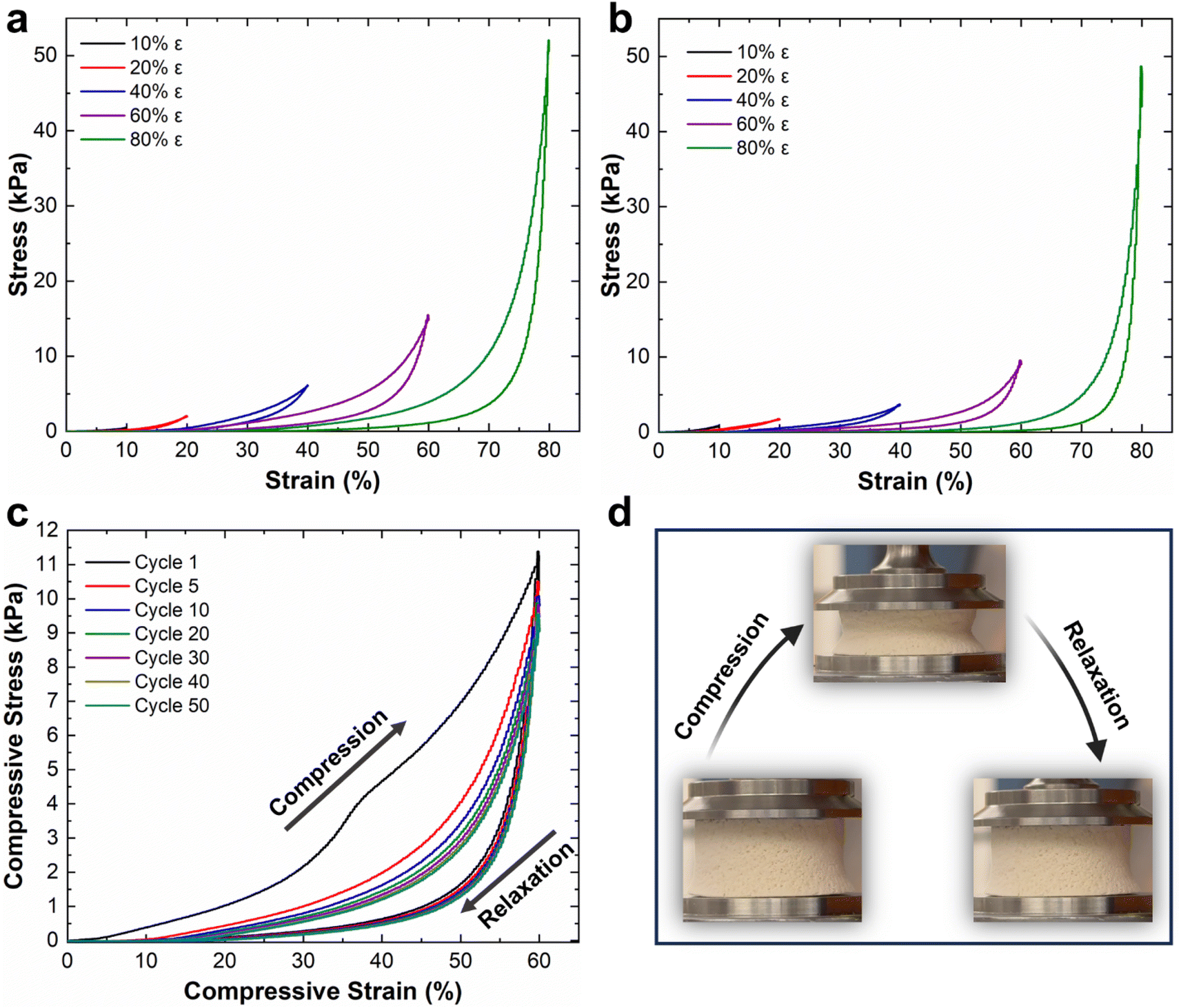 | ||
| Fig. 3 Compressive stress–strain experiments of (a) CDA–silica NFA, (b) CDA–silica@ZIF-8 NFA, (c) cyclic fatigue test of CDA–silica@ZIF-8 NFA at 60% strain for 50 cycles and (d) digital photograph of how the CDA–silica@ZIF-8 NFA changes under compressive deformation. | ||
The CDA–silica@ZIF-8 NFA exhibits a combination of unique properties, including ultralight weight, hierarchically porous structure including micro, meso and macroporosities, high specific surface area, and mechanical robustness. These distinctive characteristics make the CDA–silica@ZIF-8 NFAs highly versatile and suitable for an extensive array of applications. Their ultralight nature enables efficient transport and handling, while the hierarchically porous structure facilitates enhanced mass transfer and adsorption capabilities. The high surface area of the NFAs provides ample active sites for various reactions and sorption processes. Additionally, their mechanical robustness ensures long-lasting performance under challenging conditions.
2.3 CO2 adsorption
Thus far, our research has focused on the ZIF-8 vapor phase synthesis, morphology, characterization, and mechanical properties of CDA–silica@ZIF-8 NFA. In this and the following sections, we examine the functional attributes of our NFAs. We begin by investigating the ability of our hybrid NFA for application in carbon dioxide (CO2) adsorption.CO2 adsorption is recognized as an efficient approach for mitigating excessive CO2 emissions, which contribute to a range of ecological crises, including climate change, declining biodiversity, and glacial retreat.47–50 ZIF-8 is a promising material for the separation and adsorption of CO2 owing to its exceptional porous structure.51 Loading it on the aerogel structure provides significant surface area and exposes active sites in the micropores of ZIF-8, enabling the efficient adsorption of CO2 molecules.43 The thermal and chemical stability of ZIF-8 also makes it desirable for CO2 adsorption and separation processes.51 Finally, the pores within ZIF-8 can accommodate CO2 gas by providing Langmuir sites, which can replace other gas molecules with CO2 molecules (Fig. 4a).52
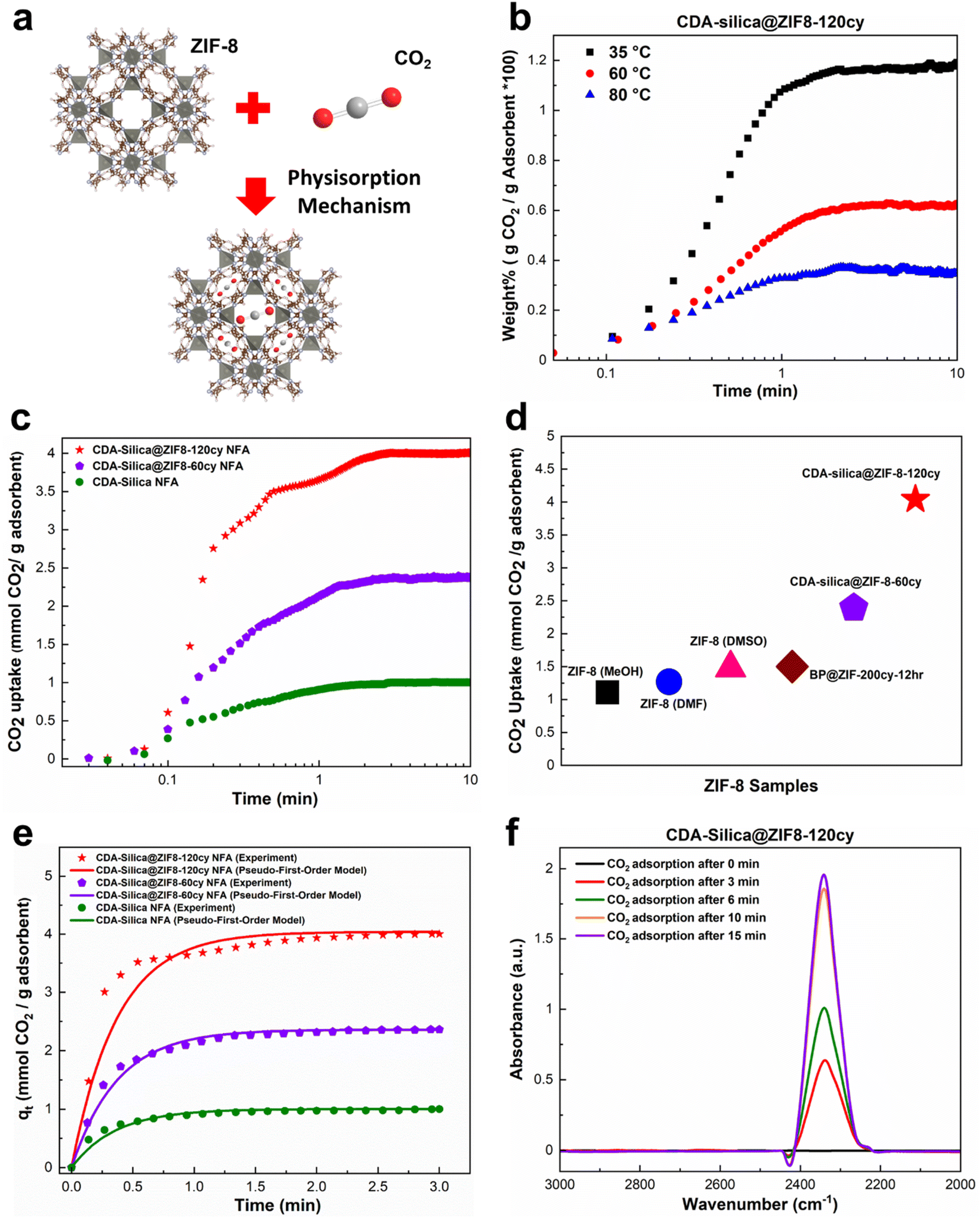 | ||
| Fig. 4 CO2 adsorption capacity of CDA–silica@ZIF-8 NFA: (a) schematic illustration of CO2 adsorption in ZIF-8 through physisorption mechanism, (b) CO2 uptake of CDA–silica@ZIF8-120cy at 35, 60 and 80 °C from a mixture of 15% CO2 and 85% Ar, (c) CO2 uptake of CDA–silica@ZIF-8 NFA at 35 °C from a mixture of 90% CO2 and 10% Ar, (d) CO2 adsorption capacity of our CDA–silica@ZIF-8 NFA compared to other works,43,53 (e) a comparison between the experimental and predicted CO2 adsorption curves using the pseudo-first-order model for CDA–silica@ZIF8-120cy, CDA–silica@ZIF8-60cy and CDA–silica NFAs at an adsorption temperature of 35 °C. (f) In situ FTIR of CO2 adsorbed on CDA–silica@ZIF8-120cy monitored as a function of time at 35 °C and 1 bar. | ||
To evaluate the CO2 adsorption capacity of CDA–silica@ZIF8-120cy and CDA–silica@ZIF8-60cy, a gravimetric approach was employed at 35, 60, and 80 °C. The TGA equipment was supplied with a mixture of CO2/Ar (15/85%) as the inlet gas. The results showed that the amount of CO2 adsorbed by both the NFAs, decreased with increasing temperature (Fig. 4b), as the adsorption process was exothermic. These materials exhibit fast adsorption rates, as evident from Fig. 4b and S4.† Both substances attained their maximum adsorption capacity within the first few minutes and reached equilibrium within ten minutes. Fig. 4b indicates that at a temperature of 35 °C, CDA–silica@ZIF8-120cy achieved 93.4% of its total adsorption within the first minute. The corresponding values at 60 °C and 80 °C were 86.7% and 90.9%, respectively. Similarly, for CDA–silica@ZIF8-60cy, the material achieved 82.6% of total adsorption at a temperature of 35 °C. The values for temperatures of 60 °C and 80 °C were 84.6% and 85.4%, respectively (Fig. S4†). Additionally, the cyclic stability of CDA–silica@ZIF8-120cy NFA was found to be consistent over 50 cycles as shown in Fig. S5,† suggesting the capability of these NFAs to remove CO2 in a cyclic adsorption/desorption procedure without loss in sorption capacity. The experiments conducted with high concentrations of CO2 (90% CO2 and 10% Ar at 35 °C and 1 bar) showed that the materials being tested exhibited excellent performance in terms of adsorption capacity and rate of adsorption, i.e., CDA–silica@ZIF8-120cy and CDA–silica@ZIF8-60cy were able to adsorb 4.04 mmol g−1 and 2.39 mmol g−1, respectively (Fig. 4c). The amount of CO2 adsorbed on CDA–silica@ZIF8-120cy and CDA–silica@ZIF8-60cy was also found to be significantly higher than similar materials, such as ZIF-8 (methanol, MeOH) (1.11 mmol g−1), ZIF-8 (dimethylformamide, DMF) (1.27 mmol g−1), ZIF-8 (dimethylsulfoxide, DMSO) (1.48 mmol g−1),53 and BP@ZIF8-200cy-12 h (1.5 mmol g−1),43 at a pressure of 1 atm at 35 °C (Fig. 4d).
A pseudo-first order model was adopted to fit the experimental results conducted with high concentrations of CO2. Such a model type is frequently utilized to predict the adsorption behaviors of physical adsorbents such as zeolites, silica, and activated carbon. Details of the model development and rationale discussed in the ESI† ultimately lead to the following equation:
| qt = qe(1 − e(−kft)) | (1) |
The saturated adsorption capacity observed in the experiments, as well as the anticipated adsorption capacity derived from the model, along with the calculated kinetic constants, are presented in Table S1.† The experimental results as well as the fitting model are illustrated in Fig. 4e, demonstrating a strong alignment between the data and the pseudo-first order model. This congruence strongly implies the physisorption mechanism since the pseudo-first-order model is frequently utilized to predict the adsorption behaviors of physical adsorbents. The interaction between CO2 molecules and NFA adsorption sites was investigated using in situ FTIR. The IR spectrum of ZIF-8 loaded with CO2 provided evidence for guest–host interactions between the ZIF-8 framework and CO2.54 In the case of CDA–silica@ZIF8-120cy and CDA–silica@ZIF8-60cy, the adsorption band caused by the asymmetric stretching vibration (V3 vibration) of CO2 molecules, was observed in the range of 2335–2350 cm−1 (Fig. 4f).55 The results showed that the adsorbent sites were filled with increasing time, and the peaks became stable after about 15 minutes.
2.4 Heavy metal removal
We also assessed the water purification performance of our hybrid aerogels, to illustrate the pivotal and functional role of ZIF-8 when combined with the CDA–silica aerogel matrix. Cu2+ stands out as a major heavy metal ion, known for its toxic effects on water bodies and biological systems. Batch adsorption experiments were conducted to measure the hybrid aerogels' maximum adsorption capacity for Cu2+ ions. A pH of 5 was maintained in the stock solution containing Cu2+ ions (100 mg L−1) to minimize electrostatic repulsion of the aerogel with Cu2+ ions before carrying out the experiments. Such a pH was selected to reduce its influence on the surface charge of the aerogel matrix. At lower pH levels, the increased concentration of H+ ions in the solution, along with a high degree of protonation of the aerogel surface, leads to competition with Cu2+ ions for adsorption into active sites. Conversely, higher pH or alkaline conditions cause the precipitation of cupric hydroxide due to larger content of OH− ions in the solution.Fig. 5a depicts the equilibrium adsorption capacity (Qe) for 20 mg of the adsorbent (aerogel) load after 24 hours. The control CDA–silica aerogel showed a maximum Qe of 189.7 mg g−1 with a removal efficiency of 75%. Upon the integration of ZIF-8, the Qe increases to 245.9 mg g−1 and 248.4 mg g−1 for CDA–silica@ZIF8-60cy and CDA–silica@ZIF8-120cy respectively. The removal efficiency, evaluated for ZIF-8 grown using both ALD cycles, consistently exceeded 95%. The sample treated with ZIF-8 grown using 120 cycles of ALD reached a maximum removal efficiency of 99%, giving us a final Cu concentration of 0.62 ± 0.16 mg L−1, which is within the standard set by both Environmental Protection Agency (EPA) at 1.3 mg L−1 and 2.0 mg L−1 by World Health Organization (WHO).56 Table S2† presents a comparison between our study and other recent research that focuses on the removal of copper (Cu) using composites based on ZIF-8. Our work demonstrates a high and comparable maximum adsorption capacity (Qe) when compared to these studies and it's important to note that we achieved this without even optimizing our approach. Moreover, these studies employed a solvothermal technique for ZIF-8 synthesis which is not environmentally desirable.
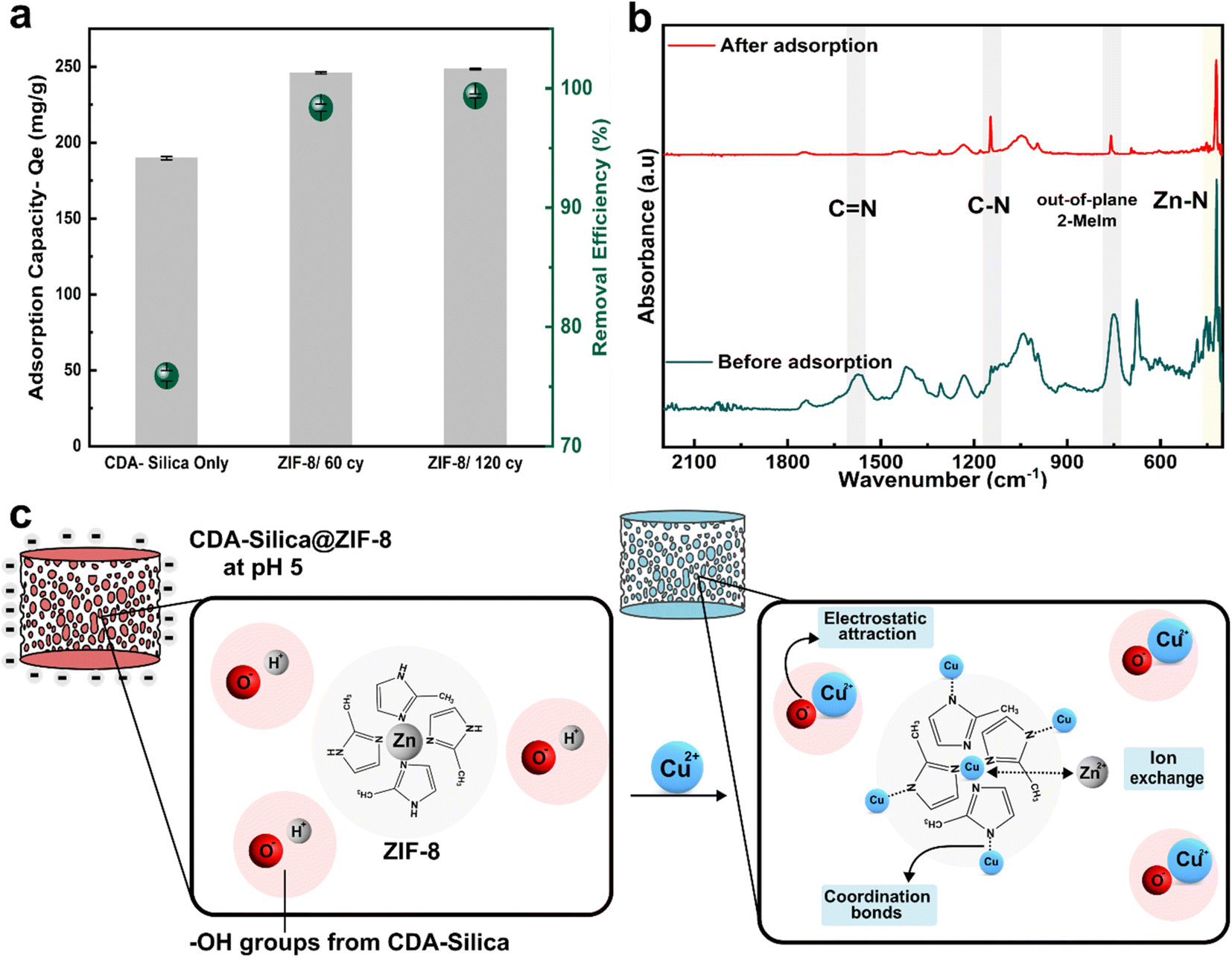 | ||
| Fig. 5 Heavy metal removal: (a) adsorption capacity (mg g−1) and removal efficiency (green circles, %) for 20 mg of the adsorbent (aerogel) load in 100 mg L−1 of stock solution of Cu2+ ions and (b) FTIR spectra of CDA–silica@ZIF-8/120cy before and after adsorption of Cu2+. Note that all three samples in (a) are aerogels. (c) Adsorption mechanism depicting ion-exchange with Zn, formation of coordination complex of Cu with N atoms of ZIF-8, and electrostatic interactions between hydroxyl groups of CDA-silica and Cu. | ||
Zeta potential measurements reveal that the surface of the CDA–silica (ζ-potential of −19.9 ± 0.72 mV) and CDA–silica@ZIF-8 (ζ-potential of −4.32 ± 0.22 mV) is negatively charged at pH 5. This facilitates an increased electrostatic attraction with Cu2+ ions. The presence of a large number of hydroxyl groups in CDA–silica offers many active sites and thus helps in the adsorption of Cu2+.35 To probe further, the adsorption mechanism was studied through FTIR analysis. As inferred from Fig. 5b, the FTIR spectra before adsorption show the characteristic peaks of CN, C–N, out-of-plane bending of 2-MeIm in ZIF-8 at 1580, 1146, 755, and 420 cm−1 respectively. However, after adsorption, the peaks corresponding to C–H, C–N, and the out-of-plane bending of 2-MeIm decreased in intensity. This indicates the formation of coordination bonds between Cu and N atoms on 2-MeIm. The Zn–N peak intensity also decreased after adsorption, revealing possible ion-exchange between Zn2+ and Cu2+. Based on all the evidence, we postulate that the adsorption of Cu2+ on the hybrid NFA occurs via a combination of electrostatic attraction, ion exchange, and the formation of a coordination complex with N atoms as depicted in Fig. 5c.56 The ZIF-8 works in concert with the aerogels to provide a framework with tunable pore size, hierarchical porosity, and abundant functional groups, facilitating strong binding sites for Cu2+ and enhanced the adsorption capacity. The mechanically stable and resilient nature for CDA–silica@ZIF-8 aerogel would further facilitate heavy metal ion adsorption without compromising structural integrity.
2.5 Antibacterial properties
Infectious diseases are a significant public health concern, causing global social and economic disruption. Pathogenic bacteria such as Escherichia coli (E. coli), Salmonella enterica, and others can cause severe illness.57,58 ZIF-8 has been recently recognized as an antimicrobial agent due to its capability to release zinc ions, which have been shown to have potential antimicrobial activity.43,59–61 Metal ions exert their microorganism (bacteria and fungi) growth inhibition through the generation of hydroxyl and superoxide anion free radicals through oxidation.62 Alternatively, a series of redox reactions can lead to cell death by impacting critical cell structures (cell wall, cell membrane, DNA, etc.) and metabolic pathways.63 The high porosity and uniform synthesis of ZIF-8 crystals on CDA–silica nanofibers in the hierarchical porous structure of the NFA enable CDA–silica@ZIF-8 hybrid NFA to exhibit efficient antimicrobial properties with increased accessibility to bacteria. We evaluated the antibacterial performance of CDA–silica@ZIF8-120cy NFA using bioassays with both Gram-negative (E. coli) and Gram-positive (Staphylococcus aureus, S. aureus) bacteria. The results illustrated in Fig. 6 revealed that after 30, 60, and 120 min of incubation with both bacteria, the pristine CDA–silica NFA displayed poor antibacterial activity. In contrast, CDA–silica@ZIF8-120cy NFA showed an antibacterial rate of 100% after 1 and 2 h of incubation with E. coli (Fig. 6a and b), and S. aureus (Fig. 6c and d). Alternatively, CDA–silica@ZIF8-120cy NFA showed a minimum of 4.47 log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 reduction of E. coli and S. aureus after 1 h of incubation, while CDA–silica NFA displayed no microbial reduction as listed in Table 1. It is worth mentioning that the detection limit of the antibacterial contact assay is 103 cfu mL−1, which explains the similar values for log10 reduction observed after 60 and 120 min as they are below this limit. Details on the analysis are presented in the Antibacterial contact assays section. The antibacterial efficiency of ZIF-8 is believed to be a result of the combination of the surface effect of zinc ions and physical contact with the bacteria. Additionally, the enhanced efficiency of our CDA–silica@ZIF8-120cy composite can be attributed to the uniformly coated and more accessible ZIF-8 from various sites of the hierarchically porous NFA. While these initial studies demonstrate the ability of CDA–silica@ZIF8-120cy NFA to inhibit bacterial growth, a detailed investigation of the functional attributes of the MOF hybrid NFAs can generate functional products that can be utilized in various applications, such as the development of membranes for removal of waterborne pathogens or for creation of antimicrobial membranes to interrupt the spread of waterborne pathogens, which has become a global concern.
10 reduction of E. coli and S. aureus after 1 h of incubation, while CDA–silica NFA displayed no microbial reduction as listed in Table 1. It is worth mentioning that the detection limit of the antibacterial contact assay is 103 cfu mL−1, which explains the similar values for log10 reduction observed after 60 and 120 min as they are below this limit. Details on the analysis are presented in the Antibacterial contact assays section. The antibacterial efficiency of ZIF-8 is believed to be a result of the combination of the surface effect of zinc ions and physical contact with the bacteria. Additionally, the enhanced efficiency of our CDA–silica@ZIF8-120cy composite can be attributed to the uniformly coated and more accessible ZIF-8 from various sites of the hierarchically porous NFA. While these initial studies demonstrate the ability of CDA–silica@ZIF8-120cy NFA to inhibit bacterial growth, a detailed investigation of the functional attributes of the MOF hybrid NFAs can generate functional products that can be utilized in various applications, such as the development of membranes for removal of waterborne pathogens or for creation of antimicrobial membranes to interrupt the spread of waterborne pathogens, which has become a global concern.
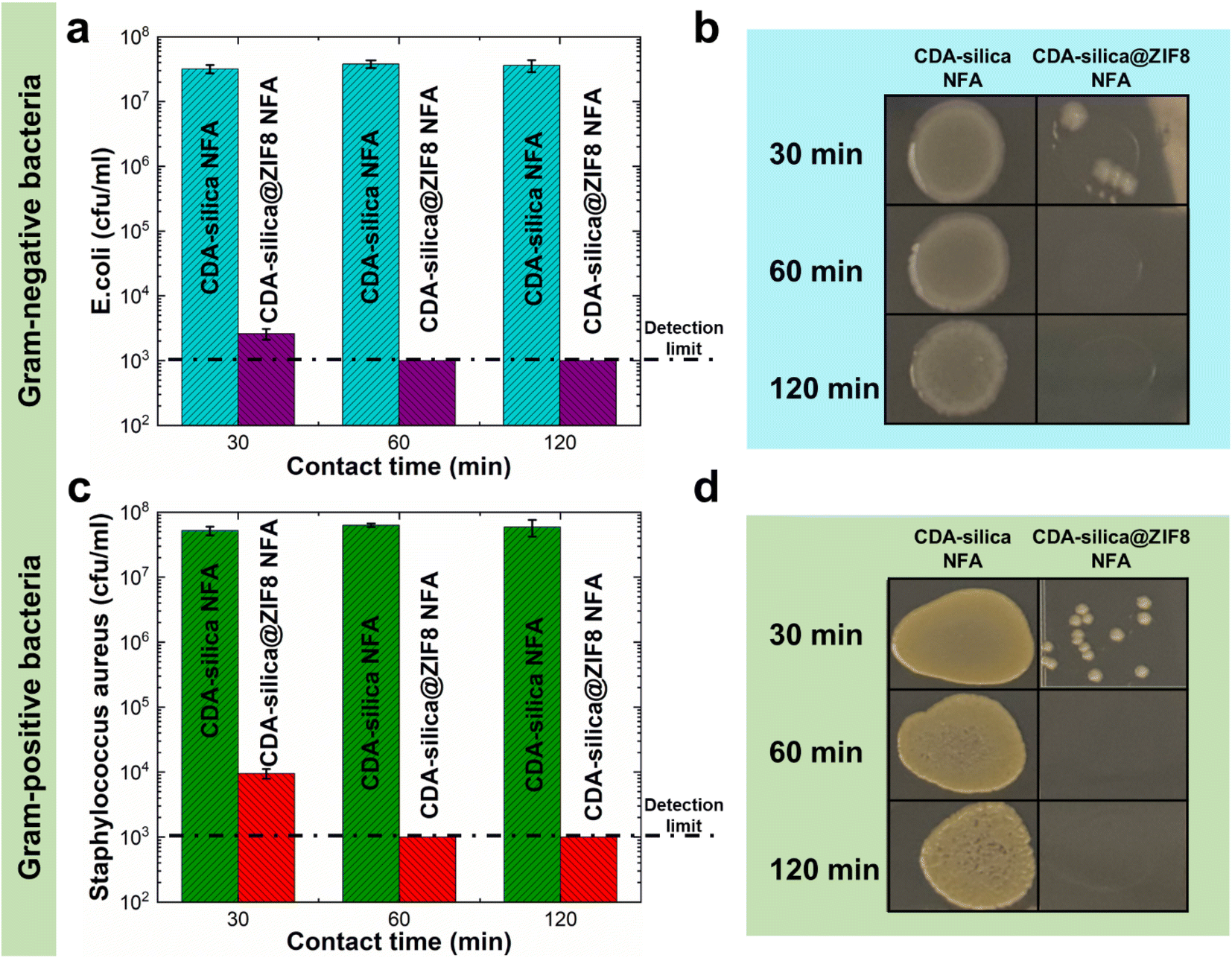 | ||
| Fig. 6 Antimicrobial properties of CDA–silica@ZIF8-120cy NFA against (a and b) Gram-negative (E. coli) and (c and d) Gram-positive (S. aureus) bacteria. | ||
10) reduction values of microbial loading measured with E. coli and S. aureus contacted with CDA–silica@ZIF8-120cy NFA
| 30 min | 60 min | 120 min | |
|---|---|---|---|
| E. coli log10 reduction | 4.06 | >4.47 | >4.47 |
| S. aureus log10 reduction | 3.50 | >4.47 | >4.47 |
3. Conclusions
This study presents a straightforward approach to synthesize MOF-based hybrid NFAs with enhanced functionality. We successfully integrated ZIF-8, a subclass of MOFs, onto a 3D structured hierarchically porous CDA–silica nanofibrous aerogel (NFA). The synthesis involved depositing thin layers of ZnO via ALD on the NFA substrate, followed by vapor phase conversion of ZnO to ZIF-8 using 2-MeIm as an organic linker vapor. The resulting CDA–silica@ZIF-8 NFA exhibited remarkable properties suitable for diverse applications. The hierarchical cellular structure and high specific surface area provided by the nanofibrous architecture, combined with the micro/mesoporosity of ZIF-8 crystals, led to efficient CO2 adsorption with rapid kinetics. The hybrid NFA also demonstrated excellent antibacterial properties against both Gram-negative and Gram-positive bacteria due to the presence of ZIF-8 and its antimicrobial zinc ions. Moreover, the material exhibited exceptional performance in the removal of heavy metal ions, particularly copper (Cu(II)), making it a potential candidate for water purification applications. The mechanically robust nature of the aerogel, withstanding large deformations without collapse, further enhances its applications in various fields. The unique combination of properties, including ultralight weight, hierarchical porosity, and mechanical resilience, makes the CDA–silica@ZIF-8 NFA a promising and versatile platform for applications in sorption, catalysis, filtration, and antimicrobial air filters. Our study highlights the potential of MOF-based hybrid nanofibrous aerogels as sustainable and scalable materials with superior functionality in various domains.4. Experimental section
4.1 Materials
2-MeIm (99%) and zinc nitrate hexahydrate (98%) were obtained from Acros Organics. Cellulose diacetate (CDA, acetyl: 39.8% and hydroxyl: 3.5%) was supplied by Eastman Chemicals. Tetraethyl orthosilicate (TEOS, 99.8%), N,N-dimethylformamide (DMF, 99.9%), dimethylacetamide (DMAC), acetone (99%), hydrochloric acid (HCl, 37%) and isopropanol (>99.5%) were obtained from Sigma-Aldrich. Anhydrous tertiary butanol (t-BuOH, 99+%) was purchased from Alfa Aesar. Diethylzinc (DEZ, 95%) was obtained from STREM Chemicals, Inc.4.2 Sol–gel electrospinning of CDA–silica nanofibers
Cellulose diacetate (CDA) solution (11 wt%) was prepared by stirring CDA in a cosolvent of DMAC/acetone (1:2 weight ratio) for an extended period. Silica precursor mixture was prepared by mixing TEOS, DMF, H2O, and HCl in a molar ratio of 1:10:18.75:0.021 and the resulting solution was subjected to magnetic stirring at 70 °C for one hour. The silica precursor mixture was then gradually added to the CDA solution in a weight ratio of 1:4 and the mixture was further stirred for an additional hour at 70 °C. Electrospinning was subsequently performed by loading the mixture of silica precursor and CDA solution into a 10 mL syringe fitted with a 22 G metal needle and spinning at a controlled feed rate of 0.5 mL h−1. A high voltage of 20 kV was applied, with a constant tip-to-collector distance of 15 cm. The resultant fibers were collected in the form of a nonwoven membrane on an aluminum foil wrapped around a metallic collector connected to a grounded electrode. The temperature and humidity during electrospinning were maintained at 21 ± 4 °C and 21 ± 2%, respectively.35
4.3 NFA fabrication
CDA–silica NFA was synthesized via freeze-drying of short nanofiber dispersions in t-BuOH. 135 mg of CDA–silica nanofiber mats were cut into 1 × 1 cm2 pieces. To achieve a stable dispersion, the nanofiber mats were homogenized with 20 mL of t-BuOH for 15 minutes at 12000 rpm using an IKA T25 homogenizer. The resulting dispersion was then poured into a glass beaker and frozen in a mixture of dry ice and 2-propanol for 20 minutes. The frozen mixture was then freeze-dried in a Labconco FreeZone 6L freeze-drying system at ∼0.003 mbar and −85 °C for 48 hours to obtain CDA–silica hybrid nanofibrous aerogel. Finally, the NFA were thermally treated at 180 °C for 2 hours, followed by an additional hour at 240 °C in a vacuum oven.35
4.4 ZnO ALD on CDA–silica nanofibers and NFA substrates
Thin layers of zinc oxide (ZnO) were coated on CDA–silica nanofibers and NFA substrate using a home-built hot-wall viscous-flow ALD reactor.15,43,64 Nitrogen (N2) was utilized as the purge and carrier gas during the deposition process, which was conducted at a temperature of 90 °C and a pressure of 1.1–1.2 torr. The deposition recipe consisted of four steps: 1s/40s/1s/40s corresponding to DEZ dose/N2 purge/H2O dose/N2 purge. The growth rate of ZnO was monitored by measuring the film thickness on silicon with native oxide (∼2 nm) using J. A. Woollam alpha-spectroscopic ellipsometer (SE) at an incidence angle of 70°. The results revealed a growth rate of approximately 1.7 Å per cycle (10 nm after 60 cycles) was obtained.4.5 Vapor phase synthesis of ZIF-8 on ZnO coated CDA–silica NFA
The conversion of ZnO to ZIF-8 was performed by utilizing ZnO coated CDA–silica NFA substrates. The NFA substrates were suspended using a string in a closed glass jar containing a powder bed of 2-MeIm (1 g) to avoid direct contact between the substrate and the powder bed. The substrates were suspended 1 cm above the 2-MeIm powder bed. The conversion process was conducted at a temperature of 135 °C for 6 hours. The samples are labeled as CDA–silica@ZIF8-Xcy, indicating that they are CDA–silica@ZIF-8 hybrid NFA that have undergone X cycles of ZnO ALD treatment. Note that details on the vapor phase synthesis of MOFs have been previously studied and well-documented in the literature.40–42 This has not been our focus as our primary interest is on using the vapor phase synthesis to create MOF on NFA for enhanced performance of MOF, something that has not been attempted before, to the best of our knowledge.4.6 CO2 adsorption
TA instruments SDT 650 was used to investigate the CO2 adsorption capacity of the CDA–silica@ZIF-8 NFA at various temperatures (35 °C, 60 °C, and 80 °C). A sample weighing between 5–10 mg was heated to 200 °C at a rate of 20 °C min−1 under Ar (99.999%) with a flow rate of 150 mL min−1 and left for 120 min to eliminate any adsorbed species. The sample was then held at this temperature for 120 minutes to remove any previously adsorbed species. After cooling to the desired analysis temperature, the samples were allowed to equilibrate for 60 minutes before switching the gas flow to a mixture of 15% CO2 and 85% Ar, with a flow rate of 150 mL min−1, for 10 minutes. The cyclic stability experiments were performed using a similar procedure for 50 cycles. Additional experiments were conducted to assess the adsorption of CO2 at higher concentrations (90% CO2 and 10% Ar) at 35 °C and 1 bar. The testing procedure for this experiment was similar to that of the CO2 adsorption experiment performed at different temperatures.The interaction between CO2 molecules and adsorption sites in the CDA–silica@ZIF-8 NFA was investigated using in situ FTIR. Thermo Fisher Nicolet iS50 FTIR, which was equipped with a DiffusIR sample chamber from Pike Technologies was employed for this analysis. The procedure was conducted by loading the sample into the in situ cell and subjecting it to Ar gas purging at 200 °C for 30 minutes. Subsequently, the sample was cooled down to a temperature of 25 °C. In order to acquire the background spectrum, the instrument recorded the spectrum within an Ar stream at 25 °C, which was subsequently automatically subtracted from the sample spectrum. CO2 gas was then introduced into the reaction cell at a flow rate of 40 mL min−1, and spectra were collected iteratively until the spectral curve reached a stabilized state.
4.7 Heavy metal removal
The NFAs were subjected to testing to study their adsorption performance for heavy metal ions. Copper ions (Cu2+) were specifically chosen as the element of interest to investigate the adsorption characteristics. First, a stock solution of 100 mg L−1 containing Cu2+ ions was prepared by dissolving copper sulfate in DI water and the pH was adjusted to 5 by using 0.1 M NaOH. Different quantities of aerogels were then placed into 50 mL of Cu2+ ions aqueous solution which was then subjected to agitation in an orbital shaker at 150 rpm for 24 hours at 25 °C. The resulting supernatant was passed through a 0.45 μm syringe filter. Finally, Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) was used to measure the concentration of Cu2+ in the filtrate. The obtained data was used to calculate the adsorption capacity (Qe, mg g−1) of the aerogels. All experiments were carried out in triplicates.The adsorption capacity of the aerogels per unit mass and removal efficiency were calculated using the following formulae.
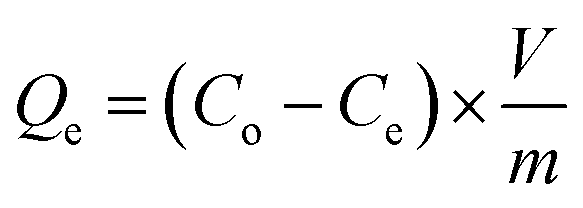 | (2) |
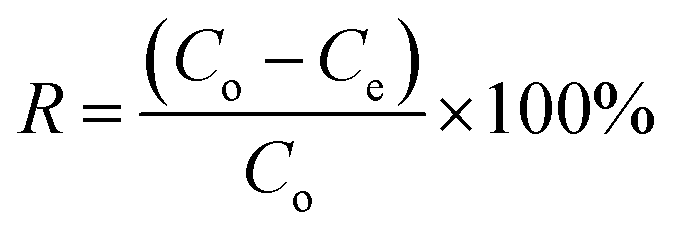 | (3) |
4.8 Antibacterial contact assays
The antibacterial activity of CDA–silica and CDA–silica@ZIF-8 NFA were investigated using Escherichia coli (E. coli, ATCC 8739) and Staphylococcus aureus (S. aureus, ATCC 6538P) cells as a model microorganism. The E. coli cells were grown in a growth media composed of 3.0 grams of beef extract, 5.0 grams of peptone, and 1 liter of deionized water (DI) for 4 hours. The cell suspension was then centrifuged at 4000 × g for 5 minutes and resuspended in phosphate-buffered saline (PBS). The optical density of the E. coli suspension was quantified using a Shimadzu UV-1800 spectrophotometer and adjusted to a final OD600 nm equal to 0.4. 20 mg of CDA–silica and CDA–silica@ZIF-8 NFA were placed in well plates and wetted with 10 μL of the E. coli suspension. A piece of polyethylene was gently placed on top of the samples. The samples were incubated at 37 °C in the dark for 1, 3, 8 and 24 hours. They were then removed, washed with 90 μL of neutralizing buffer, and serially diluted. Dilutions were deposited on top of agar plates and incubated for 18 hours at 37 °C. The final bacteria concentration was determined as the number of colonies per mL. A similar procedure was conducted to estimate the number of bacterial colonies after storing the control samples at 37 °C for 1, 3, 8 and 24 hours without CDA–silica and CDA–silica@ZIF-8 NFA. All experiments were conducted in triplicate. The antibacterial rate (% AR) of the samples was determined by using the following formula: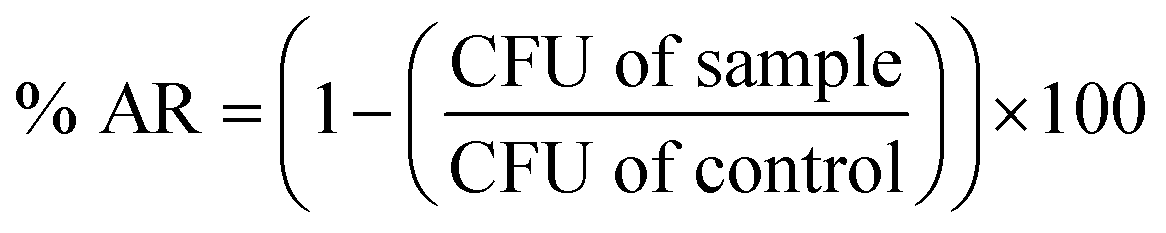 | (4) |
10 of initial bacteria concentration minus log10 after x minutes. The initial concentration for E. coli and S. aureus are 3 × 107 and 6 × 107 respectively.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported in part through the Kenan Institute for Engineering, Technology and Science (KIETS) at NC State University. Some experimental work was performed at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-1542015). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI). The authors gratefully acknowledge Dr Felix Castellano for letting us use his lab instruments and Austin Pyrch for his help with collecting the FTIR spectra. Part of this research was sponsored by the Army Research Office and was accomplished under Cooperative Agreement Number W911NF-19-2-0154. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Office or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341(6149), 1230444 CrossRef
.
- X. Jia, M. Peydayesh, Q. Huang and R. Mezzenga, Small, 2022, 18, 2105502 CrossRef CAS PubMed
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC
- M. S. Denny, J. C. Moreton, L. Benz and S. M. Cohen, Nat. Rev. Mater., 2016, 1, 16078 CrossRef CAS
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC
- C. Liu, Y. Bai, W. Li, F. Yang, G. Zhang and H. Pang, Angew. Chem. Int. Ed., 2022, 61, e2021162 Search PubMed
- G. Liu, V. Chernikova, Y. Liu, K. Zhang, Y. Belmabkhout, O. Shekhah, C. Zhang, S. Yi, M. Eddaoudi and W. J. Koros, Nat. Mater., 2018, 17, 283–289 CrossRef CAS PubMed
- L. Zhu, L. Zong, X. Wu, M. Li, H. Wang, J. You and C. Li, ACS Nano, 2018, 12, 4462–4468 CrossRef CAS
- G. W. Peterson, D. T. Lee, H. F. Barton, T. H. Epps and G. N. Parsons, Nat. Rev. Mater., 2021, 6, 605–621 CrossRef CAS
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC
- Y. Ma, Z. Dong, M. You, Y. Zhang, X. Feng, X. Ma and J. Meng, Chem. Commun., 2019, 55, 10146–10149 RSC
- M. Kalaj and S. M. Cohen, Angew. Chem., Int. Ed., 2020, 59, 13984–13989 CrossRef CAS
- J. Zhao, D. T. Lee, R. W. Yaga, M. G. Hall, H. F. Barton, I. R. Woodward, C. J. Oldham, H. J. Walls, G. W. Peterson and G. N. Parsons, Angew. Chem., 2016, 128, 13418–13422 CrossRef
- D. T. Lee, J. Zhao, C. J. Oldham, G. W. Peterson and G. N. Parsons, ACS Appl. Mater. Interfaces, 2017, 9, 44847–44855 CrossRef CAS
- M. Kalaj, M. S. Denny, K. C. Bentz, J. M. Palomba and S. M. Cohen, Angew. Chem., 2019, 131, 2358–2362 CrossRef
- Y. Deng, Y. Wang, X. Xiao, B. J. Saucedo, Z. Zhu, M. Xie, X. Xu, K. Yao, Y. Zhai, Z. Zhang and J. Chen, Small, 2022, 18, 2202928 CrossRef CAS PubMed
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC
- A. J. Brown, N. A. Brunelli, K. Eum, F. Rashidi, J. R. Johnson, W. J. Koros, C. W. Jones and S. Nair, Science, 2014, 345, 72–75 CrossRef CAS PubMed
- Y. Dou, W. Zhang and A. Kaiser, Adv. Sci., 2020, 7, 1902590 CrossRef CAS
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. M. Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed
- M. Matsumoto and T. Kitaoka, Adv. Mater., 2016, 28, 1765–1769 CrossRef CAS
- V. Rahmanian, T. Pirzada, S. Wang and S. A. Khan, Adv. Mater., 2021, 33, 2102892 CrossRef CAS PubMed
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 28, 7652–7657 CrossRef CAS PubMed
- J. Rostami, T. Benselfelt, L. Maddalena, C. Avci, F. A. Sellman, G. C. Ciftci, P. A. Larsson, F. Carosio, F. Akhtar, W. Tian and L. Wågberg, Adv. Mater., 2022, 34, 2204800 CrossRef CAS PubMed
- C. Tan, M. C. Lee, M. Arshadi, M. Azizi and A. Abbaspourrad, Angew. Chem., Int. Ed., 2020, 59, 9506–9513 CrossRef CAS
- J. Wang, J. Yang, H. Zhu, B.-G. Li and S. Zhu, Chem. Eng. J., 2023, 456, 141026 CrossRef CAS
- J. Rostami, K. Gordeyeva, T. Benselfelt, E. Lahchaichi, S. A. Hall, A. V. Riazanova, P. A. Larsson, G. C. Ciftci and L. Wågberg, Mater. Today, 2021, 48, 47–58 CrossRef CAS
- X. Ma, Y. Lou, X.-B. Chen, Z. Shi and Y. Xu, Chem. Eng. J., 2019, 356, 227–235 CrossRef CAS
- Y. Fei, M. Liang, L. Yan, Y. Chen and H. Zou, Chem. Eng. J., 2020, 392, 124815 CrossRef CAS
- P. K. Chattopadhyay and N. R. Singha, Coord. Chem. Rev., 2021, 446, 214125 CrossRef
- L. Wang, H. Xu, J. Gao, J. Yao and Q. Zhang, Coord. Chem. Rev., 2019, 398, 213016 CrossRef
- F. Deuber and C. Adlhart, Chimia, 2017, 71, 236 CrossRef CAS PubMed
- Y. Si, J. Yu, X. Tang, J. Ge and B. Ding, Nat. Commun., 2014, 5, 5802 CrossRef PubMed
- T. Pirzada, Z. Ashrafi, W. Xie and S. A. Khan, Adv. Funct. Mater., 2020, 30, 1907359 CrossRef CAS
- Y. Si, Q. Fu, X. Wang, J. Zhu, J. Yu, G. Sun and B. Ding, ACS Nano, 2015, 9, 3791–3799 CrossRef CAS PubMed
- V. Rahmanian, T. Pirzada, E. Barbieri, S Iftikhar, F. Li and S. A. Khan, Appl. Mater. Today, 2023, 32, 101784 CrossRef
- F. Deuber, S. Mousavi, L. Federer, M. Hofer and C. Adlhart, ACS Appl. Mater. Interfaces, 2018, 10, 9069–9076 CrossRef CAS PubMed
-
T. Pirzada, V. Rahmanian and S. A. Khan, in Applications of Polymer Nanofibers, Wiley, 2022, pp. 345–371 Search PubMed
- S. Han and C. B. Mullins, ChemSusChem, 2020, 13, 5433–5442 CrossRef CAS
- I. Stassen, N. Campagnol, J. Fransaer, P. Vereecken, D. De Vos and R. Ameloot, CrystEngComm, 2013, 15, 9308 RSC
- I. Stassen, D. De Vos and R. Ameloot, Chem.–Eur. J., 2016, 22, 14452–14460 CrossRef CAS
- S. Wang, T. Pirzada, W. Xie, E. Barbieri, O. Hossain, C. H. Opperman, L. Pal, Q. Wei, G. N. Parsons and S. A. Khan, Appl. Mater. Today, 2022, 28, 101517 CrossRef
- K. He, N. Chen, C. Wang, L. Wei and J. Chen, Cryst. Res. Technol., 2018, 53, 1700157 CrossRef
- S. Zhong, Q. Wang and D. Cao, Sci. Rep., 2016, 6, 21295 CrossRef CAS
- A. Gu, J. Chen, Q. Gao, M. M. Khan, P. Wang, Y. Jiao, Z. Zhang, Y. Liu and Y. Yang, Appl. Surf. Sci., 2020, 516, 146160 CrossRef CAS
- Y.-C. Lou, S.-C. Qi, D.-M. Xue, C. Gu, R. Zhou, X.-Q. Liu and L.-B. Sun, Chem. Eng. J., 2020, 399, 125845 CrossRef CAS
- S.-C. Qi, X.-J. Lu, Y.-C. Lou, R. Zhou, D.-M. Xue, X.-Q. Liu and L.-B. Sun, Engineering, 2022, 16, 154–161 CrossRef CAS
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. M. Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC
- X. Gong, Y. Wang and T. Kuang, ACS Sustain. Chem. Eng., 2017, 5, 11204–11214 CrossRef CAS
- K. Díaz, L. Garrido, M. López-González, L. F. del Castillo and E. Riande, Macromolecules, 2010, 43, 316–325 CrossRef
- Y. Guan, J. Shi, M. Xia, J. Zhang, Z. Pang, A. Marchetti, X. Wang, J. Cai and X. Kong, Appl. Surf. Sci., 2017, 423, 349–353 CrossRef CAS
- Y. Hu, Z. Liu, J. Xu, Y. Huang and Y. Song, J. Am. Chem. Soc., 2013, 135, 9287–9290 CrossRef CAS PubMed
- S. Bordiga, L. Regli, F. Bonino, E. Groppo, C. Lamberti, B. Xiao, P. S. Wheatley, R. E. Morris and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 2676 RSC
- N. Bakhtiari, S. Azizian, S. M. Alshehri, N. L. Torad, V. Malgras and Y. Yamauchi, Microporous Mesoporous Mater., 2015, 217, 173–177 CrossRef CAS
- A. K. Jha and K. Prasad, Biotechnol. J., 2010, 5, 285–291 CrossRef CAS PubMed
- F. Wang, Y. Si, J. Yu and B. Ding, Adv. Funct. Mater., 2021, 31, 2107223 CrossRef CAS
- Z. Liang, H. Wang, K. Zhang, G. Ma, L. Zhu, L. Zhou and B. Yan, Chem. Eng. J., 2022, 428, 131349 CrossRef CAS
- R. Li, T. Chen and X. Pan, ACS Nano, 2021, 15, 3808–3848 CrossRef CAS PubMed
- M. Taheri, D. Ashok, T. Sen, T. G. Enge, N. K. Verma, A. Tricoli, A. Lowe, D. R. Nisbet and T. Tsuzuki, Chem. Eng. J., 2021, 413, 127511 CrossRef CAS
- W.-J. Chen, P.-J. Tsai and Y.-C. Chen, Small, 2008, 4, 485–491 CrossRef CAS PubMed
-
C. López de Dicastillo, M. G. Correa, F. B. Martínez, C. Streitt and M. J. Galotto, in Antimicrobial Resistance - A One Health Perspective, IntechOpen, 2021 Search PubMed
- J. Zhao, B. Gong, W. T. Nunn, P. C. Lemaire, E. C. Stevens, F. I. Sidi, P. S. Williams, C. J. Oldham, H. J. Walls, S. D. Shepherd, M. A. Browe, G. W. Peterson, M. D. Losego and G. N. Parsons, J. Mater. Chem. A, 2015, 3, 1458–1464 RSC
- M. Su, R. Zhang, J. Li, X. Jin, X. Zhang and D. Qin, Cellulose, 2021, 28, 11713–11727 CrossRef CAS
- C. L. de Dicastillo, C. Patiño, M. J. Galotto, Y. Vásquez-Martínez, C. Torrent, D. Alburquenque, A. Pereira and J. Escrig, Beilstein J. Nanotechnol., 2019, 10, 1716–1725 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05299k |
| This journal is © The Royal Society of Chemistry 2024 |