
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ evolution of bulk-active γ-CoOOH with immobilized Gd dopants enabling efficient oxygen evolution electrocatalysis†
Tianjue
Hou‡
a,
Ruotao
Yang‡
a,
Jiaxin
Xu
a,
Xiaodie
He
a,
Hongyuan
Yang
b,
Prashanth W.
Menezes
 *bc and
Ziliang
Chen
*ab
*bc and
Ziliang
Chen
*ab
aInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 199 Ren'ai Road, Suzhou, 215123, Jiangsu, China. E-mail: zlchen@suda.edu.cn
bMaterials Chemistry Group for Thin Film Catalysis–CatLab, Helmholtz-Zentrum Berlin für Materialien und Energie, Albert-Einstein-Str. 15, 12489 Berlin, Germany. E-mail: prashanth.menezes@helmholtz-berlin.de
cDepartment of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: prashanth.menezes@mailbox.tu-berlin.de
First published on 25th July 2024
Abstract
Promoting the in situ reconstruction of transition metal (TM)-based precatalysts into low-crystalline and well-modified TM (oxy)hydroxides (TMOxHy) during the alkaline oxygen evolution reaction (OER) is crucial for enhancing their catalytic performances. In this study, we incorporated gadolinium (Gd) into a cobalt hydroxide precatalyst, achieving a deep reconstruction into cobalt oxyhydroxide (γ-CoOOH) while retaining the incorporated Gd during the activation process of the alkaline OER. The unconventional non-leaching Gd dopants endow γ-CoOOH with reduced crystallinity, increasing the exposure of electrolyte-accessible Co atoms and enhancing its bulk activity. Furthermore, the modulation of the electronic structure of γ-CoOOH substantially boosts the intrinsic activity of the active Co sites. As a result, when supported on nickel foam, the catalyst exhibits remarkable alkaline OER performance, achieving a current density of 100 mA cm−2 at a low overpotential of approximately 327 mV. Notably, an ultrahigh current density of 1000 mA cm−2 is robustly maintained for 5 days, highlighting its immense potential for practical applications in large-scale hydrogen production.
Introduction
Hydrogen (H2) is widely recognized as a promising alternative to fossil fuels, the overconsumption of which has induced the current severe energy and environmental crisis.1,2 Sustainable energy-driven electrochemical water splitting, involving the anodic oxygen evolution reaction (OER) and the cathodic hydrogen evolution reaction (HER), has garnered significant attention because it can produce H2 in an environmentally benign and non-carbon-emitting manner.3,4 However, the OER involves a complex four-electron transfer step with sluggish kinetics, which reduces the overall efficiency of water electrolysis.3,5 Therefore, it is of high necessity to develop efficient electrocatalysts capable of accelerating the OER. Industrially, the anode of alkaline water electrolysis is usually made of catalysts based on nickel (Ni) or its alloy (e.g., RANEY® Ni), but their catalytic performance still needs further boosting.6,7 In this regard, numerous efforts have been devoted to designing and synthesizing a new generation of low-cost non-noble transition metal (TM)-based catalysts, particularly Fe, Co, Ni, and Mn-based compounds, with highly efficient alkaline OER capability to satisfy the requirements of practical applications.6–13When examining most TM-based compounds, including (oxyhydr)oxides, pnictides, chalcogenides, borophosphates, and intermetallics, in the alkaline OER in aqueous media, they tend to reconstruct into the associated higher-valence TM-based (oxyhydr)oxides (TMOxHy), which serve as the real active structures.10,14–17 Given the widespread observation of component oxidation to soluble oxyanions and subsequent leaching into electrolytes during the OER, it is crucial to consider several key points.14–22 Firstly, among these single-phase OER-active TMOxHy, CoOxHy exhibits excellent corrosion resistance and, notably, the highest electrical conductivity.23,24 This makes Co-based materials promising precursors for efficient alkaline OER catalysts. Secondly, the alkaline OER capabilities of pure CoOxHy are normally still far from satisfactory, likely due to the inadequate intrinsic activity of the redox-active Co sites.24 To address this issue, appropriate modifications are required for the in situ reconstructed CoOxHy to increase the number of accessible redox-active Co atoms with evidently improved inherent activity. Moreover, stabilizing the active Co sites with other components is of great significance to achieve the robust durability of CoOxHy, yet remains a challenge.25,26 Under these conditions, incorporating elements that do not significantly leach out during the alkaline OER in aqueous solutions into Co-based precatalysts may offer opportunities to effectively regulate the properties of in situ evolved CoOxHy, eventually resulting in an enhanced oxygen evolution performance.25,27
On the other hand, element doping has been widely proposed in previous works, as it can evidently promote the structural reconstruction of TM-based precatalysts into the associated TMOxHy by reducing the reconstruction energy barrier or increasing the surface porosity.8,28 However, the destabilization tendency of most doped atoms often results in their serving only as sacrificial agents, and the modulation effect cannot be maintained over prolonged OER electrochemistry. An intriguing observation is that OER-active TMs themselves can also be utilized as dopants, especially in the case of incorporating Fe into Ni- or Co-based catalyst systems where the resulting alkaline OER activity can be substantially improved.11,29–33 However, the dissolution phenomenon during the operational OER process persists8,34 and, therefore, the exact active sites responsible for the alkaline OER are still under debate.33 Based on these factors, one can envision that if a Co-based precatalyst can be incorporated into an OER-inactive but stable component, its in situ deep reconstruction into a distinctive active CoOxHy phase could be facilitated. Due to the regulation effect of the immobilized dopant, as well as the exclusion of interference from other potential active sites, such CoOxHy could likely exhibit significantly improved alkaline OER performance. More importantly, this improvement can be explicitly attributed to the increased availability of redox-active Co sites with optimized intrinsic activity.
Taking into account the aforementioned considerations and to determine the role of the non-leaching feature of f-block rare earth metals under alkaline OER conditions, we deliberately incorporated f-block rare earth metals into a cobalt hydroxide (α-CoGd(OH)2) precatalyst.27 Herein, gadolinium (Gd) was specifically chosen owing to its high chemical stability, strong corrosion resistance, and high conductivity.35,36 As expected, compared with pristine α-Co(OH)2 doping, the Gd doping induced a deep OER-driven reconstruction into low-crystallinity γ-CoOOH with increased structural defects, facilitating the participation of more redox-active Co atoms in OER catalysis with enhanced bulk activity. Besides, the Gd dopants remained intact, enabling the regulation of the electronic structure of the γ-CoOOH host, thus boosting the intrinsic activity of the active Co sites. Benefiting from these advancements, the fully activated α-CoGd(OH)2 exhibited strikingly superior OER performance compared to pristine α-Co(OH)2 when deposited on both fluorine-doped tin oxide glass (FTO) and nickel foam (NF). Particularly noteworthy is the performance of α-Co0.95Gd0.05(OH)2 on NF, requiring an overpotential of ∼327 mV to achieve a current density of 100 mA cm−2, which can then sustain an ultrahigh current density of 1000 mA cm−2 for five days without visible degradation, displaying promising potential for further larger-scale application.
Results and discussion
Synthesis and characterization of the as-prepared samples
The α-CoGd(OH)2 precatalyst was directly grown on an FTO substrate (α-CoGd(OH)2/FTO) through electrodeposition using a typical three-electrode system, in which the FTO glass, a saturated calomel electrode (SCE), and Pt foil acted as the working electrode, reference electrode, and counter electrode, respectively. Co(NO3)2·6H2O and Gd(NO3)3·H2O were dissolved in deionized water as the deposition solution with a molar ratio of 0.95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05. After applying a constant current of −1 mA for a duration of 400 s, the targeted α-CoGd(OH)2 supported on FTO was obtained. Subsequently, it was subjected to OER activation using cyclic voltammetry (CV) for 20 cycles, resulting in the in situ formation of Gd-doped γ-CoOOH. Pristine α-Co(OH)2/FTO was fabricated and activated using the same methods, except that no Gd sources were incorporated (details are given in Fig. 1 and the Experimental section).
:0.05. After applying a constant current of −1 mA for a duration of 400 s, the targeted α-CoGd(OH)2 supported on FTO was obtained. Subsequently, it was subjected to OER activation using cyclic voltammetry (CV) for 20 cycles, resulting in the in situ formation of Gd-doped γ-CoOOH. Pristine α-Co(OH)2/FTO was fabricated and activated using the same methods, except that no Gd sources were incorporated (details are given in Fig. 1 and the Experimental section).
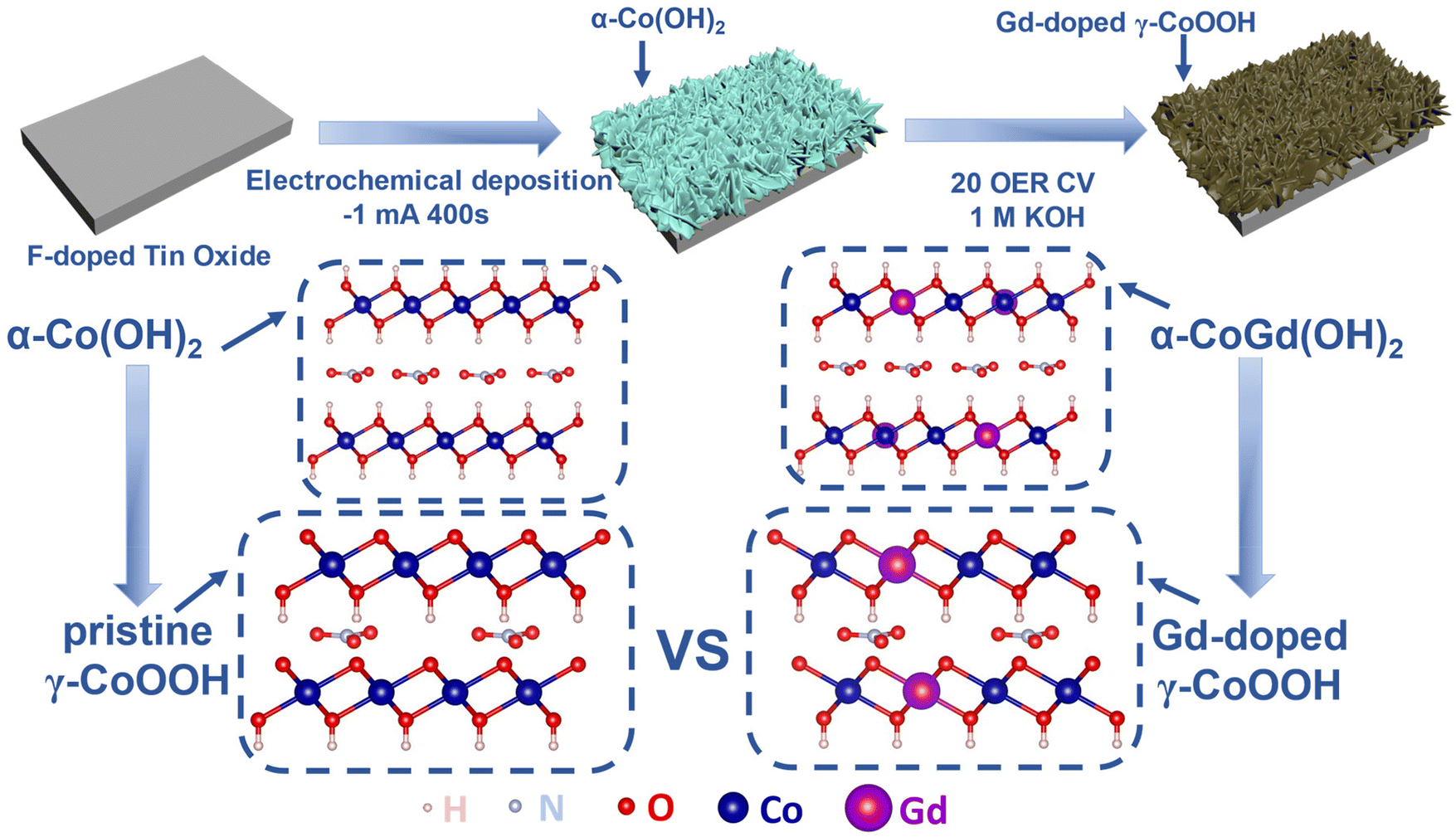 | ||
| Fig. 1 Schematic representation of the synthesis of FTO-supported α-CoGd(OH)2 and α-Co(OH)2 and the activation of α-CoGd(OH)2. | ||
Hereafter, the as-prepared α-CoGd(OH)2/FTO and pristine α-Co(OH)2/FTO, both before and after activation, were comprehensively characterized to acquire information about their morphology, phase, crystallinity, composition, and chemical state. Initially, field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) were employed to probe the morphology of the two as-prepared samples, revealing a typical nanosheet-like morphology (Fig. 2a–d and Fig. S1a–c†). High-resolution TEM (HRTEM) imaging exhibited lattice fringes with interplanar distances of ∼0.23 and ∼0.20 nm for fresh α-CoGd(OH)2, corresponding to the (105) and (108) planes of α-Co(OH)2, respectively. Note that the distance was slightly larger than that of the theoretical value, indicating lattice expansion induced by the incorporation of Gd atoms (Fig. 2e). Additionally, the selected area electron diffraction (SAED) pattern (Fig. 2f) uncovered the (105), (102), and (110) crystal facets of α-Co(OH)2 (PDF #46-0605), providing further evidence of the successful synthesis of the α-Co(OH)2 host phase. Similar observations were made for the pristine α-Co(OH)2 sample without Gd incorporation (Fig. S1d and e†). However, it is worth mentioning that the diffraction rings presented in the SAED pattern, as well as the lattice fringes shown in the HRTEM image, turned out to be more broken and indistinct for the α-CoGd(OH)2 sample compared with those of pristine α-Co(OH)2, signifying the low crystallinity feature of the host α-Co(OH)2 after Gd doping. The formation of more structural defects contributed to promoting the OER-driven reconstruction.28,37 Additionally, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) pattern and the energy-dispersive X-ray spectroscopy (EDS) elemental mapping images confirmed a uniform distribution of Co, Gd, and O within the selected region of the sample, directly indicating the successful Gd doping into the α-Co(OH)2 phase (Fig. 2g–j and Table S1†). Additionally, as shown in Table S2,† inductively coupled plasma (ICP) was used to measure the atomic ratio of Co and Gd, delivering a similar result. Similar observations were made for pristine α-Co(OH)2 (Fig. S1f–h†). The powder X-ray diffraction (XRD) technique was subsequently used to examine α-CoGd(OH)2/FTO and pristine α-Co(OH)2/FTO with bare FTO as a reference. As depicted in Fig. 2k, in addition to the XRD peaks originating from the FTO substrate, the α-Co(OH)2/FTO sample exhibited a discernible peak, which matched well with the most ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif)
![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif)
![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) (003) peak of a typical α-Co(OH)2 pattern (PDF #46-0605).37,38 However, such a peak in the case of α-CoGd(OH)2/FTO appeared non-evident and negatively shifted. This is because the Gd doping with a larger size compared with Co led to a slight lattice expansion, consequently reducing the crystallinity of the α-Co(OH)2 host, which is in agreement with the above HRTEM and SAED data. Subsequently, the XRD patterns with 2 theta values below 10° for α-Co(OH)2 and α-CoGd(OH)2 together with full width at half maximum (FWHM) analysis on their respective (003) peaks are also provided in Fig. S2,† from which the broadening and negative shift of the (003) peak can be easily identified, illustrating the crystallinity reduction and lattice expansion induced by Gd introduction. Additionally, by employing calculations based on the Scherrer formula, it was determined that the average crystal size of α-Co(OH)2 was ∼7.27 nm, whereas α-CoGd(OH)2 exhibited an average crystal size of 9.29 nm. These findings substantiated that successful Gd doping can induce the increase of the average crystal size.39 Afterward, we also used X-ray photoelectron spectroscopy (XPS) to gain more information on the chemical states and phases of fresh α-CoGd(OH)2/FTO and pristine α-Co(OH)2/FTO. In Fig. S3a,† the Co 2p3/2 spectrum of the latter sample can be fitted at ∼781.1 and ∼780.1 eV, corresponding to Co2+ and Co3+, respectively.26 However, the fitted peaks indicative of Co2+ and Co3+ in the Co 2p3/2 XPS spectrum of α-CoGd(OH)2/FTO both shifted toward higher binding energy, indicating an electronic interaction between the Gd dopants and the Co atoms in the host lattice.7,31 Moreover, the Gd 4d3/2 and 4d5/2 XPS peaks (Fig. S3b†) can be fitted at ∼148.1 and ∼142.3 eV, respectively, illustrating that Gd was doped in the form of Gd3+.40,41 In addition, the O 1s XPS spectrum of pristine α-Co(OH)2/FTO was located at ∼531.3 eV, similar to that previously reported for the α-Co(OH)2 phase.42 Besides, a positive shift of the O 1s XPS spectrum can be identified for α-CoGd(OH)2/FTO, further confirming the electronic modifications to α-Co(OH)2 induced by the introduction of Gd atoms (Fig. S3c†).
(003) peak of a typical α-Co(OH)2 pattern (PDF #46-0605).37,38 However, such a peak in the case of α-CoGd(OH)2/FTO appeared non-evident and negatively shifted. This is because the Gd doping with a larger size compared with Co led to a slight lattice expansion, consequently reducing the crystallinity of the α-Co(OH)2 host, which is in agreement with the above HRTEM and SAED data. Subsequently, the XRD patterns with 2 theta values below 10° for α-Co(OH)2 and α-CoGd(OH)2 together with full width at half maximum (FWHM) analysis on their respective (003) peaks are also provided in Fig. S2,† from which the broadening and negative shift of the (003) peak can be easily identified, illustrating the crystallinity reduction and lattice expansion induced by Gd introduction. Additionally, by employing calculations based on the Scherrer formula, it was determined that the average crystal size of α-Co(OH)2 was ∼7.27 nm, whereas α-CoGd(OH)2 exhibited an average crystal size of 9.29 nm. These findings substantiated that successful Gd doping can induce the increase of the average crystal size.39 Afterward, we also used X-ray photoelectron spectroscopy (XPS) to gain more information on the chemical states and phases of fresh α-CoGd(OH)2/FTO and pristine α-Co(OH)2/FTO. In Fig. S3a,† the Co 2p3/2 spectrum of the latter sample can be fitted at ∼781.1 and ∼780.1 eV, corresponding to Co2+ and Co3+, respectively.26 However, the fitted peaks indicative of Co2+ and Co3+ in the Co 2p3/2 XPS spectrum of α-CoGd(OH)2/FTO both shifted toward higher binding energy, indicating an electronic interaction between the Gd dopants and the Co atoms in the host lattice.7,31 Moreover, the Gd 4d3/2 and 4d5/2 XPS peaks (Fig. S3b†) can be fitted at ∼148.1 and ∼142.3 eV, respectively, illustrating that Gd was doped in the form of Gd3+.40,41 In addition, the O 1s XPS spectrum of pristine α-Co(OH)2/FTO was located at ∼531.3 eV, similar to that previously reported for the α-Co(OH)2 phase.42 Besides, a positive shift of the O 1s XPS spectrum can be identified for α-CoGd(OH)2/FTO, further confirming the electronic modifications to α-Co(OH)2 induced by the introduction of Gd atoms (Fig. S3c†).
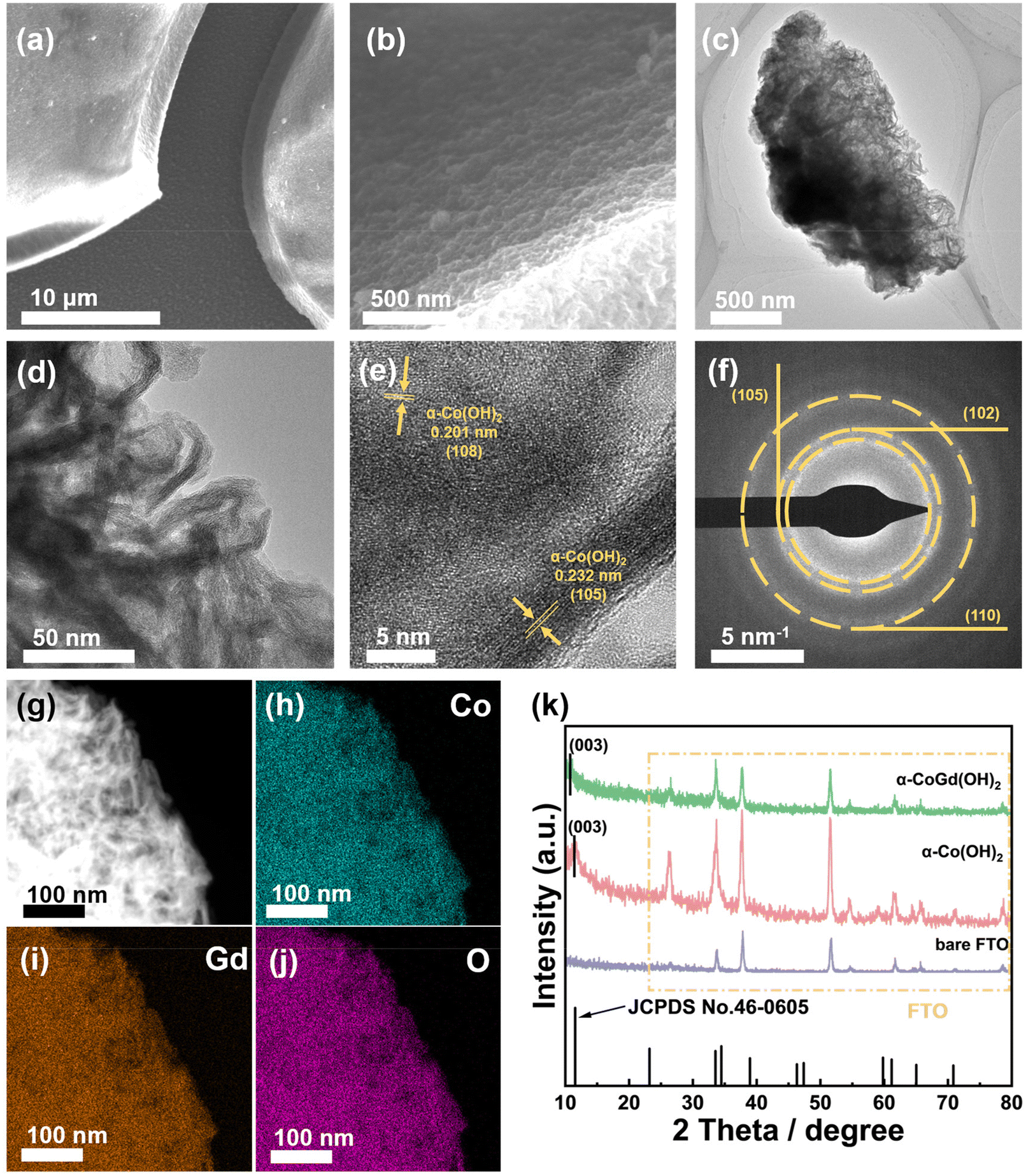 | ||
| Fig. 2 (a and b) The FESEM images of α-CoGd(OH)2/FTO. (c and d) The TEM images, (e) HRTEM image, and (f) SAED pattern together with the corresponding (g) HAADF pattern as well as the elemental mapping of (h) Co, (i) Gd, and (j) O for α-CoGd(OH)2 peeled from the FTO substrate. (k) The powder XRD patterns of bare FTO, α-Co(OH)2/FTO, and α-CoGd(OH)2/FTO. | ||
Characterization of the samples after OER activation
The well-defined α-CoGd(OH)2/FTO was fully activated using OER CV (details are provided in the Experimental section) to enable the in situ evolution of the active oxyhydroxide that is responsible for the OER electrocatalysis. Notably, the optical images in Fig. 3a disclosed that the color of α-CoGd(OH)2 completely changed after activation, indicating a deep structural reconstruction. This finding was further supported by the powder XRD patterns (Fig. 3b), where the broad (003) diffraction peaks of α-Co(OH)2 can be found (except the peaks corresponding to the FTO substrate). We have further performed GI-XRD characterization on both activated α-Co(OH)2 and α-CoGd(OH)2 while employing a beam knife to minimize air scattering in the low 2 theta region. In the former case free of Gd doping, the (003) diffraction peak of α-Co(OH)2 was still easily identified. Conversely, such a peak was absent in the XRD pattern of activated α-CoGd(OH)2 (Fig. S4†). This comparison clearly illustrated that the amorphization of α-CoGd(OH)2 after activation mainly stemmed from the Gd doping instead of the pure anodic activation. In situ Raman spectra were subsequently recorded to gain deeper insights into the phase reconstruction during activation. At the open circuit potential (OCP), the spectrum of the α-CoGd(OH)2 sample (Fig. 3c) exhibited two peaks at ∼455 and ∼518 cm−1, attributed to the Co–O vibrations of the α-Co(OH)2 phase,43 which remained intact until 1.0 V (vs. RHE). Notably, the transformation of this precatalyst was initiated at 1.1 V (vs. RHE), where a hydrous Co(OH)2 phase was formed, as confirmed by a large peak at ∼595 cm−1.44 Moreover, at 1.2 V (vs. RHE), the associated peaks at ∼454 and ∼518 cm−1 almost disappeared and two new bands at ∼462 and ∼571 cm−1 emerged. This indicated that the precatalysts reconstructed into the γ-CoOOH phase.43,44 Notably, the reconstructed γ-CoOOH stabilized with increasing applied potentials upon returning to the OCP. Moreover, for both α-CoGd(OH)2/FTO and α-Co(OH)2/FTO after OER activation, their Co 2p3/2 XPS spectra revealed an apparent increase in the Co3+ concentration, aligning with the conclusion of the in situ evolution of higher-valence γ-CoOOH (Fig. 3d). Remarkably, the fitted peaks indexed to both Co3+ and Co2+ in the XPS spectrum of the former sample were located at higher binding energy positions, which implied a significant modification of the incorporated Gd. Fig. 3e displays a clear Gd 4d XPS spectrum of α-CoGd(OH)2/FTO after activation, with the fitted peaks assigned to 4d3/2 and 4d5/2 still representing the existence of Gd3+, confirming that Gd was preserved during the structural reconstruction, resulting in a Gd3+-doped high-valence γ-Co3+δOOH active phase.40,41 Also, there was no signal of Gd in α-Co(OH)2 after activation. The O 1s XPS spectra in Fig. 3f further validated the evolution of oxyhydroxides for these two samples after OER activation.45 The peaks corresponding to absorbed water, metal–OH and metal–O were found at ∼532.9, ∼531.1 and ∼529.4 eV, respectively.46 Based on the above data, it can be concluded that α-CoGd(OH)2 deeply reconstructed into Gd-doped low-crystalline γ-CoOOH during OER activation, while pristine α-Co(OH)2 only achieved a lower degree of reconstruction, forming pure γ-CoOOH with higher crystallinity. Analogous to most cases of TM-based precatalysts, the reconstruction of pristine α-Co(OH)2 occurs only on its surface because the transformed layer is usually densely stacked during the OER process. This will hinder the penetration of OH− ions (reactants of the alkaline OER) into the inner bulk, thereby limiting further reconstruction. Upon incorporating Gd into the α-Co(OH)2 precatalyst to diminish crystallinity and create more structural defects, more exposed TM sites could be provided, thereby facilitating deeper penetration of OH− reactants. Consequently, a deep surface transformation was induced.25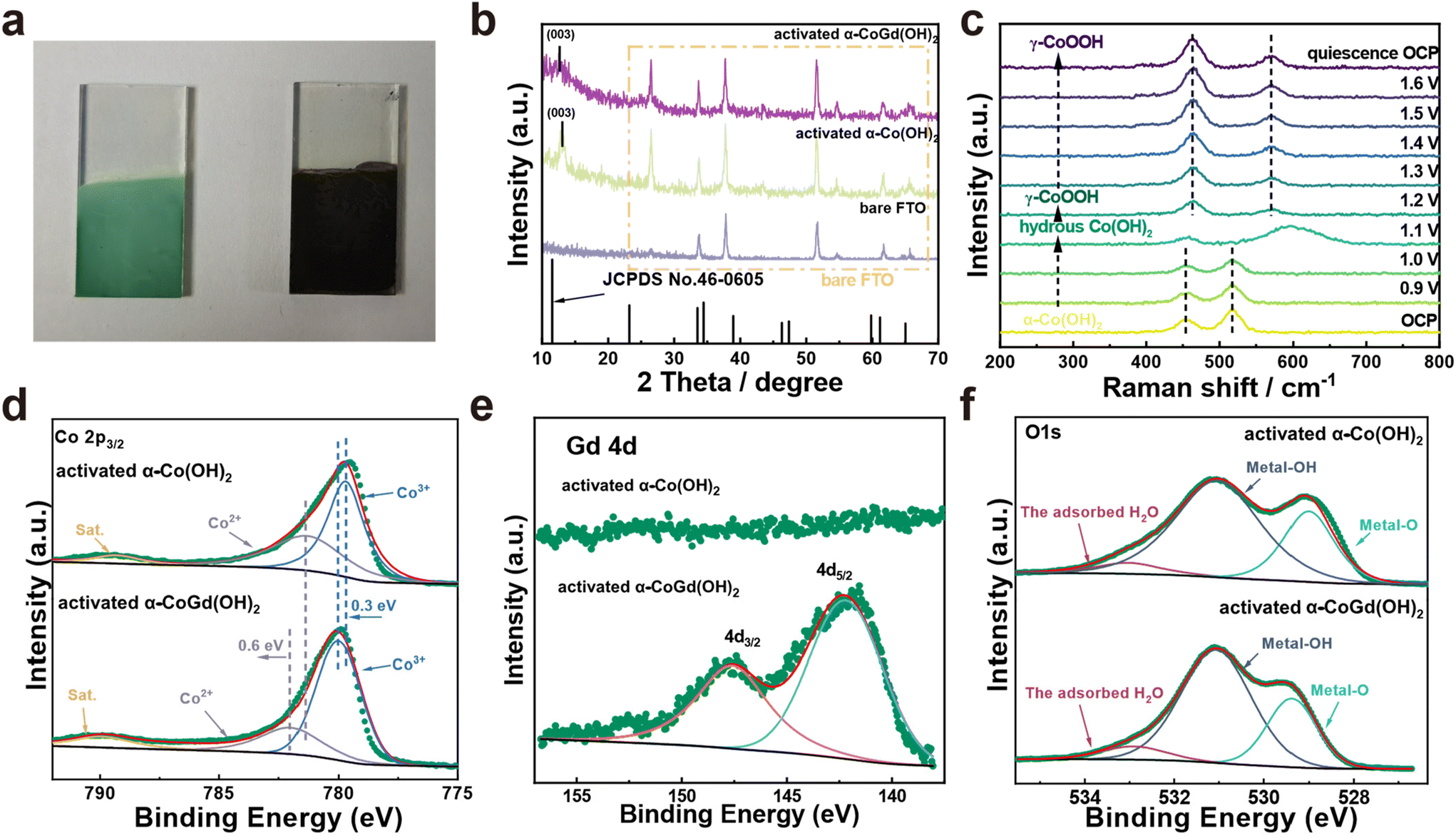 | ||
| Fig. 3 (a) The optical images of α-CoGd(OH)2 before and after activation. (b) The powder XRD patterns of activated α-CoGd(OH)2 and α-Co(OH)2. Herein, the XRD pattern of bare FTO was used as a reference. (c) In situ Raman spectra of activated α-CoGd(OH)2 at 0.9–1.6 V vs. RHE, as well as the ex situ ones at the OCP. (d) Co 2p3/2 XPS spectra of α-CoGd(OH)2 and α-Co(OH)2 after activation. (e) The Gd 4d XPS spectrum of α-CoGd(OH)2 after activation. (f) The O 1s XPS spectra of α-CoGd(OH)2 and α-Co(OH)2 after activation. | ||
The morphologies of the activated α-CoGd(OH)2/FTO and α-Co(OH)2/FTO were investigated through FESEM and TEM characterization studies, revealing that both maintained nanosheet structures characteristic of oxyhydroxides (Fig. S5a–d and S5a–c†). Furthermore, the HRTEM analysis in Fig. 4e confirmed that the lattice fringes of α-CoGd(OH)2 after OER activation were broken and indistinct, whereas within the short-range ordered region, the lattice fringes possessed an interplanar spacing distance of 0.24 nm, primarily attributed to the (101) plane of γ-CoOOH (PDF# 14-0673), suggesting the dominant presence of a low-crystalline γ-CoOOH phase within α-CoGd(OH)2 after activation. This conclusion was supported by the SAED pattern of this sample (Fig. 4f). It is noteworthy that the measured interplanar distance was slightly larger than the ideal value of γ-CoOOH, possibly due to steady Gd incorporation (Fig. 4e). Conversely, the activated pristine α-Co(OH)2 sample (Fig. S5d and e†) exhibited relatively unbroken lattice fringes in its HRTEM image. Meanwhile, its SAED pattern showed distinct diffraction rings, assigned to both reconstructed γ-CoOOH (PDF# 14-0673) and remaining α-Co(OH)2 (PDF #46-0605). These findings highlight the pivotal role of Gd doping in structural reconstruction during the alkaline OER. Furthermore, the HAADF-STEM pattern and the EDS elemental mapping images showed uniform distribution of Co, Gd, and O throughout the selected region of α-CoGd(OH)2 after activation, indicating the stable retention of incorporated Gd (Fig. 4g–j and Table S1†), which was also well corroborated by the findings of ICP in Table S2.† Note that the structure of CoOxHy materials is typically composed of nanodomains formed by edge-sharing [MO6] octahedral layers.47 The large-sized nanodomains tend to be orderly arranged for the directly synthesized CoOxHy (such as the ones obtained from the electrochemical deposition in this study), resulting in limited access for the electrolyte to the bulk active sites.48 As a result, only near-surface activity can be achieved. Herein, the in situ formation of low-crystallinity γ-CoOOH, facilitated by the stable incorporation of Gd, is believed to create more structural defects and voids. This enhances the penetration of the KOH electrolyte, thereby increasing the number of accessible redox-active Co atoms. Eventually, a higher bulk activity of the reconstructed active structure was realized. On the other hand, the stable incorporation of Gd dopants in the reconstructed γ-CoOOH could result in a distinctive gradient orbital coupling of Gd(4f)–O(2p)–Co(3d), increasing the electronic density and covalency of the Co−O bond. Such an electron structure could optimize the adsorption energies towards the OER intermediates.11,35 Consequently, the intrinsic activity of the transformed active phase is significantly enhanced. The enhancements in both bulk and intrinsic activities of γ-CoOOH resulting from Gd doping are substantiated by a series of electrochemical analyses detailed in the following sections.
 | ||
| Fig. 4 (a and b) The FESEM images of α-CoGd(OH)2/FTO after activation. (c and d) The TEM images, (e) HRTEM image, and (f) SAED pattern together with the corresponding (g) HAADF pattern as well as the elemental mapping of (h) Co, (i) Gd, and (j) O for α-CoGd(OH)2 after activation (peeled from the FTO substrate). | ||
Alkaline OER electrochemistry of the activated samples and characterization studies after stability tests
After activation using CV cycles to ensure deep reconstruction into Gd-doped γ-CoOOH, all evaluations of the OER catalytic performances of α-CoGd(OH)2/FTO were conducted in 1 M KOH (details are provided in the Experimental section) with pristine α-Co(OH)2/FTO after the same activation procedure used as a reference. The linear sweep voltammetry (LSV) tests were performed for α-CoGd(OH)2/FTO with different Gd doping concentrations. Fig. S6a† illustrates that among them, the sample prepared with a molar ratio of Co to Gd of 0.95:0.05 exhibited optimum activity (the one characterized throughout the above sections). Notably, as is depicted in Fig. 5a, the activated α-CoGd(OH)2/FTO required an overpotential of ∼371 mV to afford 10 mA cm−2 current density, substantially superior to that of the activated α-Co(OH)2/FTO. The negligible OER activity of the bare FTO substrate indicated that the observed activity mainly originated from the deposited catalysts. Subsequently, steady-state measurements were conducted to determine the Tafel slopes of α-CoGd(OH)2/FTO and α-Co(OH)2/FTO after activation.49 As shown in Fig. 5b, α-CoGd(OH)2/FTO exhibited a significantly lower Tafel slope, indicating that Gd incorporation promoted more favorable reaction kinetics of reconstructed γ-CoOOH. Additionally, CV was performed on both activated α-CoGd(OH)2/FTO and α-Co(OH)2/FTO and their respective reduction peaks were analyzed to determine the number of redox-active Co atoms that were involved in OER electrocatalysis.47 Specifically, the former showed ∼1.4 times more redox-active Co sites than the latter (Fig. 5c and S7†), which can be related to the lower crystallinity of the active γ-CoOOH induced by the immobilized Gd dopants. The lower crystallinity meant more structural defects, probably resulting in more disordered stacking of the edge-sharing [MO6] octahedra layers, allowing greater electrolyte penetration.26,50–52 As a result, more Co atoms can be accessed and an enhanced bulk activity was achieved.7,48 Based on this, we further normalized the current from the LSV data in Fig. 5a against such determined numbers of redox-active Co atoms to evaluate their intrinsic activity.46 As is shown in Fig. S8,† the Gd-doped γ-CoOOH in the activated α-CoGd(OH)2/FTO sample exhibited a distinctly higher inherent activity than that of the pure γ-CoOOH in the activated α-Co(OH)2/FTO sample, which can be further verified by the higher turnover frequency (TOF) and mass-normalized activity of the activated α-CoGd(OH)2/FTO (Fig. S11 and S12†).53,54 Combining these results with the lower Tafel slope values (Fig. 5b), it can be concluded that the introduction of Gd atoms profoundly enhanced the intrinsic activity of the active sites of γ-CoOOH for the alkaline OER. The electrochemical impedance spectroscopy (EIS) results (Fig. 5d and Table S3†) demonstrated an enhanced charge transfer ability of the reconstructed γ-CoOOH during the OER upon doping Gd. The stabilities of activated α-CoGd(OH)2/FTO and α-Co(OH)2/FTO were further assessed through a 24 h chronoamperometry (CA) measurement at a current density of ∼10 mA cm−2. As shown in Fig. 5e, negligible degradation was found for α-CoGd(OH)2/FTO, whereas α-Co(OH)2/FTO showed a visible activity decline, suggesting that Gd doping into γ-CoOOH might stabilize its alkaline OER activity. Considering that CoOxHy typically features good electric conductivity and resistance to alkaline OER corrosion, the improvements in the reconstructed γ-CoOOH through doping Gd should primarily be attributed to the elevated bulk activity owing to the increased accessible redox-active Co sites, as well as enhanced intrinsic activity of such catalytic atoms.24 Moreover, through a set of characterization methods, including (HR)TEM, SAED, HAADF-STEM, and EDS-elemental mapping, on the post-CA sample, it was found that the morphology, phase, crystallinity, composition content and distribution of the reconstructed Gd-doped γ-CoOOH were almost unchanged (Fig. S9†), verifying the applicability and durability of Gd incorporation.
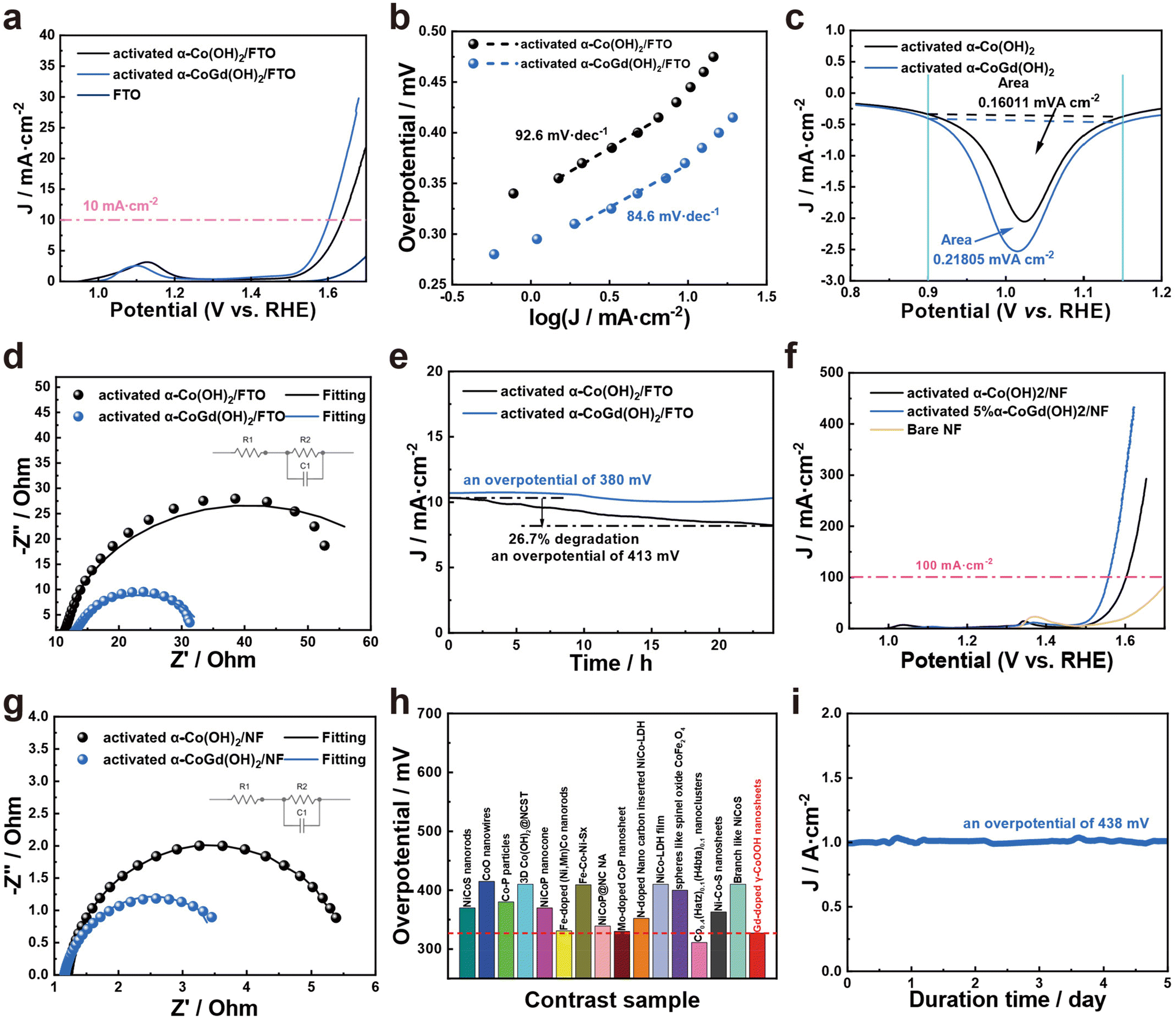 | ||
| Fig. 5 (a) The LSV curves (together with the bare FTO reference), (b) Tafel slope plots, (c) integration of reduction peaks, (d) Nyquist plots obtained from EIS fitting to an equivalent circuit (inset) at an anodic potential of 1.6 V vs. RHE, and (e) CA curves of α-CoGd(OH)2/FTO and α-Co(OH)2/FTO recorded for 24 h. (f) The LSV curves (together with the bare NF reference) and (g) Nyquist plots obtained from EIS fitting to an equivalent circuit (inset) at an anodic potential of 1.55 V vs. RHE for α-CoGd(OH)2/NF and α-Co(OH)2/NF. (h) The activity comparison between α-CoGd(OH)2/NF and other recently reported advanced Co-based catalysts supported on NF for the alkaline OER. (i) The CA curve of α-CoGd(OH)2/NF recorded for 5 d. All the above electrochemistry was conducted after activating the studied samples using 20 cycles of OER CV. | ||
Encouraged by the excellent OER catalytic performances of α-CoGd(OH)2/FTO after activation, α-CoGd(OH)2, together with the α-CoGd(OH)2 reference, was further deposited on NF. After activation by CV cycles, the α-CoGd(OH)2 peeled from the NF substrate exhibited similar features to the activated α-CoGd(OH)2 extracted from the FTO substrate (Fig. S10†). The LSV (Fig. 5f), Tafel slope (Fig. S13†), and EIS (Fig. 5g and Table S3†) results of activated α-CoGd(OH)2/NF and α-Co(OH)2/NF exhibited a trend similar to that observed when deposited on FTO. Especially, as depicted in Fig. 5f and Fig. S13,† the activated α-CoGd(OH)2/NF can deliver a high current density of ∼100 mA cm−2 at only ∼327 mV with a Tafel slope value of merely ∼79.5 mV dec−1. Furthermore, Fig. 5h and Table S4† demonstrate that its activity was superior or comparable to most of the recently reported advanced Co-based catalysts supported on NF for alkaline OER electrocatalysis. Finally, the possible industrial potential of the activated α-CoGd(OH)2/NF was confirmed by subjecting it to an ultrahigh current density (∼1000 mA cm−2) for 5 days without any obvious decline (Fig. 5i). The material exhibited remarkable stability during this period, indicating its enormous promise for practical applications in industry.
Conclusions
In summary, our innovative Gd doping strategy has facilitated a significant transformation of the α-Co(OH)2 precatalyst into γ-CoOOH, incorporating well-immobilized Gd during alkaline OER activation. The presence of stable Gd dopants led to a low crystallinity of the reconstructed γ-CoOOH, thereby enhancing accessibility and increasing the number of redox-active Co atoms. Consequently, a substantial increase in bulk activity was observed. Moreover, the inherent activity of these active Co atoms was largely enhanced due to profound modifications in the electronic structure of γ-CoOOH. As a result of these advantages, the activated α-CoGd(OH)2/NF achieved a remarkable current density of ∼100 mA cm−2 at only ∼327 mV and impressively sustained an ultrahigh current density of ∼1000 mA cm−2 for 5 days without noticeable activity degradation. Therefore, we believe that this newly developed α-CoGd(OH)2 holds great potential for applications in large-scale green hydrogen production.Experimental
Chemicals and materials
Deionized water was used to carry out all the experiments. Potassium hydroxide (KOH, 95%) and cobalt nitrate (Co(NO3)2·6H2O, 99.99%) were obtained from Sinopharm Group Co. Ltd. Gadolinium nitrate (Gd(NO3)3·6H2O, 99.9%) was obtained from Macklin Biochemical Technology Co., Ltd. The electrode substrates NF and FTO were purchased from Merck & Co., Inc., respectively.Synthesis of α-CoGd(OH)2 with different Gd doping concentrations and their activation
In order to prepare α-CoGd(OH)2, electrodeposition was conducted in a standard three-electrode system, where the working electrode, counter electrode, and reference electrode were bare FTO, Pt foil, and a SCE, respectively. Co(NO3)2·6H2O and Gd(NO3)3·6H2O were dissolved in 50 mL of deionized water with a molar ratio of 0.95:0.05. Using a CHI760e electrochemical workstation, a constant applied current of −1 mA was maintained for a duration of 400 s. The resulting loading mass obtained was ∼0.22 mg cm−2. Furthermore, NF was employed as an alternative working electrode while keeping the remaining deposition process consistent with that described above, eventually yielding a loading mass of ∼0.68 mg cm−2. Before all electrochemistry tests, the as-prepared α-CoGd(OH)2 was activated using OER CV for 20 cycles in another three-electrode system (CHI760e electrochemical workstation). Note that the FTO- or NF-supported α-CoGd(OH)2 was used as the working electrode, while Pt foil and an Hg/HgO electrode acted as the counter and reference electrodes, respectively. The electrolyte was 1 M KOH aqueous solution. Besides, the utilized potential window of CV activation ranged from 0.9 to 1.5 V vs. RHE. Based on the same procedures mentioned above in the case of α-CoGd(OH)2 (except that no Gd source was introduced), pristine α-Co(OH)2 deposited on FTO or NF was prepared and subsequently activated. In addition, α-CoGd(OH)2 samples with different Gd doping concentrations were fabricated by adjusting the molar ratio between Co(NO3)2·6H2O and Gd(NO3)3·6H2O to 0.975:0.025, 0.925:0.075, and 0.9:0.1. These samples were denoted as 2.5% α-CoGd(OH)2, 7.5% α-CoGd(OH)2, and 10% α-CoGd(OH)2, respectively.
Characterization
The phase information of the investigated samples was obtained using powder XRD on a Bruker D8 ADVANCE X-ray diffractometer with Cu Kα radiation (λ = 1.5406 A). Powder XRD in GI mode with a beam knife was performed using a PANalytical X-ray diffractometer with Cu Kα radiation. The calculation of the average crystal size was performed using the Scherrer formula: D = K λ/β cosθ, where D is the crystallite size, K is the Scherrer constant (0.9), λ is the wavelength of the X-rays used (0.15406 nm), β is the FWHM, and θ is the peak position.39 FESEM (Hitachi SU800 electron microscope, Tokyo, Japan) was utilized to acquire the morphologies and structures of the target samples at an accelerating voltage of 10 kV. Furthermore, TEM (FEI Talos F200X and JEM F200) was used to further analyze the existence and distribution of their microstructures, phases, and components. ICP (PerkinElmer, USA) was utilized to measure the compositions. XPS measurements were performed using a Thermo Scientific ESCALAB 250Xi spectrometer (USA) with a power of 100 W (10 kV and 10 mA) and a monochromatic α X-ray source (hν = 1486.65 eV). All samples were analyzed under vacuum conditions of less than 1.0 × 10−9 Pa and the spectra were obtained with a 0.05 eV step size using Advantage software (version 5.979). Raman spectra were recorded using a Horiba LabRam HR Evolution confocal microscope with a laser excitation wavelength of 473 nm, a laser power of 1.25 mW, and an objective magnification of 100× (NA = 0.95). The Raman spectrum was initially recorded at the OCP using a Renishaw Raman system (inVia Qontor, 532 nm laser), followed by in situ Raman testing from 0.9 V to 1.6 V, and finally the catalyst was re-measured after being placed at the OCP for an hour. In situ Raman spectroscopy was performed using a Renishaw Raman system integrated with an electrochemical workstation (CHI, 760E).
Electrochemical measurements
The electrochemical experiments were performed using CHi760e and EC-Lab electrochemical workstations in a typical three-electrode configuration. The working electrodes consisted of catalysts electrodeposited on different substrates, while a counter electrode made of Pt foil (1 × 1 cm) and a reference electrode composed of Hg/HgO were employed. The LSV, CV, CP, and CA experiments were carried out with an iR compensation of 90% on the CHi760e instrument. Furthermore, the CV and LSV tests were conducted at a scan rate of 5 mV s−1. All measured potentials were calibrated with respect to the RHE based on the following equation: E (vs. RHE) = E (vs. Hg/HgO) + 0.098 V + (0.059 × pH) V, where the pH value of 1 M KOH was adopted from our previous report.55 The Tafel slope analysis and EIS were conducted using the EC-Lab electrochemical workstation. The Tafel slope was determined through a steady-state measurement, i.e., the CA was performed at each fixed potential for 3 minutes, and the applied potentials gradually increased with an interval of 15 mV.49 The average current density obtained at each potential was used for the calculation of Tafel plots. The Tafel slope was determined according to the Tafel equation: η = blogj + a, where η, b, and j represent the overpotential (V), Tafel slope (mV dec−1), and current density (mA cm−2), respectively. In order to obtain the effective catalytic metal atoms of the catalyst, we determined the number of atoms capable of catalytic conversion using the formula: n = A/s q, which was achieved by analyzing the reduction peak area of a catalyst supported on FTO with an equivalent mass at a sweep rate of 5 mV s−1. Here, n represents the number of transferred electrons, A denotes the reduction peak area, s signifies the sweep rate, and q represents the charge carried by a single electron.19 The EIS spectrum was recorded at 1.60 V (vs. RHE) for the catalysts deposited on FTO and at 1.55 V (vs. RHE) for those on NF. The amplitude of the sinusoidal wave was determined within a frequency range from 100 kHz to 1 mHz. The charge transfer resistance (Rct) was reflected by the diameter of the semicircle in the Nyquist plots after fitting using the EC-Lab electrochemical workstation. The turnover frequency (TOF) was calculated using the formula: TOF = j NA/F n Ί, where j is the current density, NA is the Avogadro number, n is the number of electrons transferred for the evolution of a single O2 molecule, F is the Faraday constant, and Γ is the surface concentration or the number of active Co sites.53
Author contributions
T. H.: experimental data, writing – editing, characterization, and analysis. R. Y.: characterization, experimental data, and writing – editing. J. X.: experimental data. H. Y.: writing – editing. P. M.: conceptualization, writing – editing, and funding acquisition. Z. C.: conceptualization, methodology, writing – editing, supervision, and funding acquisition. The manuscript was written with the contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52201269), the Natural Science Foundation of Jiangsu Province (BK20210735), the Collaborative Innovation Center of Suzhou Nano Science & Technology, the 111 Project, the Suzhou Key Laboratory of Functional Nano & Soft Materials and the Jiangsu Key Laboratory for Advanced Negative Carbon Technologies. Z. C. gratefully acknowledges the funding from the Gusu leading talent plan for scientific and technological innovation and entrepreneurship (ZXL2022487). H. Yang thanks the China Scholarship Council (CSC) for the Ph.D. fellowship. P. W. Menezes greatly acknowledges support from the German Federal Ministry of Education and Research in the framework of the project Catlab (03EW0015A/B) and the project PrometH2eus (03HY105C).References
- T. T. Le, P. Sharma, B. J. Bora, V. D. Tran, T. H. Truong, H. C. Le and P. Q. P. Nguyen, Int. J. Hydrogen Energy, 2024, 54, 791 CrossRef CAS.
- J. G. Love, A. P. O'Mullane, F. A. Boulaire and I. D. R. Mackinnon, Sustainable Energy Fuels, 2022, 6, 4008–4023 RSC.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. I. Jamesh and X. Sun, J. Power Sources, 2018, 400, 31–68 CrossRef CAS.
- H. Yang, G. Vijaykumar, Z. Chen, J. N. Hausmann, I. Mondal, S. Ghosh, V. C. J. Nicolaus, K. Laun, I. Zebger, M. Driess and P. W. Menezes, Adv. Funct. Mater., 2023, 33, 2303702 CrossRef CAS.
- H. Yang, P. V. Menezes, G. Dai, G. Vijaykumar, Z. Chen, M. Al-Shakran, T. Jacob, M. Driess and P. W. Menezes, Appl. Catal., B, 2023, 324, 122249 CrossRef CAS.
- D. Yao, L. Gu, B. Zuo, S. C. Weng, S. Deng and W. Hao, Nanoscale, 2021, 13, 10624–10648 RSC.
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, Adv. Mater., 2023, 35, 2302462 CrossRef CAS PubMed.
- S. Lee, Y. Chu, L. Bai, H. M. Chen and X. Hu, Chem. Catal., 2022, 3, 100475 CrossRef.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- I. Mondal, P. V. Menezes, K. Laun, T. Diemant, M. Al-Shakran, I. Zebger, T. Jacob, M. Driess and P. W. Menezes, ACS Nano, 2023, 17, 14043–14052 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- H. Yang, J. N. Hausmann, V. Hlukhyy, T. Braun, K. Laun, I. Zebger, M. Driess and P. W. Menezes, ChemCatChem, 2022, 14, e202200293 CrossRef CAS.
- B. Dasgupta, J. N. Hausmann, R. Beltrán-Suito, S. Kalra, K. Laun, I. Zebger, M. Driess and P. W. Menezes, Small, 2023, 19, 2301258 CrossRef CAS PubMed.
- S. Ghosh, B. Dasgupta, S. Kalra, M. L. P. Ashton, R. Yang, C. J. Kueppers, S. Gok, E. G. Alonso, J. Schmidt, K. Laun, I. Zebger, C. Walter, M. Driess and P. W. Menezes, Small, 2023, 19, 2206679 CrossRef CAS PubMed.
- P. W. Menezes, C. Walter, B. Chakraborty, J. N. Hausmann, I. Zaharieva, A. Frick, E. von Hauff, H. Dau and M. Driess, Adv. Mater., 2021, 33, 2004098 CrossRef CAS PubMed.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- H. Yang, G. Dai, Z. Chen, J. Wu, H. Huang, Y. Liu, M. Shao and Z. Kang, Small, 2021, 17, 2101727 CrossRef CAS PubMed.
- Z. Chen, H. Yang, Z. Kang, M. Driess and P. W. Menezes, Adv. Mater., 2022, 34, 2108432–2108432 CrossRef CAS PubMed.
- H. Lei, L. Ma, Q. Wan, S. Tan, B. Yang, Z. Wang, W. Mai and H. J. Fan, Adv. Energy Mater., 2022, 12, 2202522 CrossRef CAS.
- S. Zou, M. S. Burke, M. G. Kast, J. Fan, N. Danilovic and S. W. Boettcher, ACS Nano, 2015, 27, 8011–8020 CAS.
- Q. Xu, H. Jiang, X. Duan, Z. Jiang, Y. Hu, S. W. Boettcher, W. Zhang, S. Guo and C. Li, Nano Lett., 2020, 21, 492–499 CrossRef PubMed.
- H. Liu, S. Zhang, Y. Zhou, W. Yu, Y. Ma, S. Wang, Y. Chai and B. Dong, Small, 2023, 19, 2301255 CrossRef CAS PubMed.
- H. Yang, Z. Chen, W. Hao, H. Xu, Y. Guo and R. Wu, Appl. Catal., B, 2019, 252, 214–221 CrossRef CAS.
- Z. Ye, T. Li, G. Ma, Y. Dong and X. Zhou, Adv. Funct. Mater., 2017, 27, 1704083 CrossRef.
- L. Reith, J. N. Hausmann, S. Mebs, I. Mondal, H. Dau, M. Driess and P. W. Menezes, Adv. Energy Mater., 2023, 13, 2203886 CrossRef CAS.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C. Sun, L. Li, D. Sokaras, C. Dong, S. Qiao, X. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- K. Yu, H. Yang, H. Zhang, H. Huang, Z. Wang, Z. Kang, Y. Liu, P. W. Menezes and Z. Chen, Nano-Micro Lett., 2023, 15, 186 CrossRef CAS PubMed.
- Z. Chen, H. Yang, S. Mebs, H. Dau, M. Driess, Z. Wang, Z. Kang and P. W. Menezes, Adv. Mater., 2023, 35, 2208337 CrossRef CAS PubMed.
- U. Shamraiz, A. Badshah and B. Raza, Langmuir, 2020, 36, 2223–2230 CrossRef CAS PubMed.
- T. Shinagawa, N. Kotobuki and A. Ohtaka, Nanoscale Adv., 2023, 5, 96–105 RSC.
- S. A. Hassanzadeh-Tabrizi, J. Alloys Compd., 2023, 968, 171914 CrossRef CAS.
- C. Yu, T. Xuan, Y. Chen, Z. Zhao, X. Liu, G. Lian and H. Li, J. Alloys Compd., 2016, 688, 611–619 CrossRef CAS.
- R. Li, J. Yang, S. Xu, Y. Zhou, X. Wang, H. Peng and J. Du, Catalysts, 2020, 10, 298 CrossRef CAS.
- H. Jin, S. Mao, G. Zhan, F. Xu, X. Bao and Y. Wang, J. Mater. Chem. A, 2017, 5, 1078–1084 RSC.
- L. Aguilera, P. C. M. Aguiar, Y. L. Ruiz, A. Almeida, J. A. Moreira, R. R. Passos and L. A. Pocrifka, J. Mater. Sci.: Mater. Electron., 2020, 31, 3084–3091 CrossRef CAS.
- C. Jing, T. Yuan, L. Li, J. Li, Z. Qian, J. Zhou, Y. Wang, S. Xi, N. Zhang, H. Lin, C. Chen, Z. Hu, D. Li, L. Zhang and J. Wang, ACS Catal., 2022, 12, 10276–10284 CrossRef CAS.
- P. W. Menezes, C. Panda, S. Garai, C. Walter, A. Guiet and M. Drieß, Angew. Chem., Int. Ed., 2018, 57, 15237–15242 CrossRef CAS PubMed.
- J. N. Hausmann, P. V. Menezes, G. Vijaykumar, K. Laun, T. Diemant, I. Zebger, T. Jacob, M. Driess and P. W. Menezes, Adv. Energy Mater., 2022, 12, 2202098 CrossRef CAS.
- J. N. Hausmann, S. Mebs, K. Laun, I. Zebger, H. Dau, P. W. Menezes and M. Driess, Energy Environ. Sci., 2020, 13, 3607–3619 RSC.
- J. N. Hausmann, M. L. P. Ashton, S. Mebs, C. Walter, S. Selve, M. Haumann, T. Sontheimer, H. Dau, M. Driess and P. W. Menezes, Small, 2024, 2309749 CrossRef CAS PubMed.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- Y. Sun, J. Wu, Z. Zhang, Q. Liao, S. Zhang, X. Wang, Y. Xie, K. Ma, Z. Kang and Y. Zhang, Energy Environ. Sci., 2022, 15, 633–644 RSC.
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Adv. Energy Mater., 2021, 2003755 CrossRef CAS.
- Y. Sun, J. Wu, Y. Xie, X. Wang, K. Ma, Z. Tian, Z. Zhang, Q. Liao, W. Zheng, Z. Kang and Y. Zhang, Adv. Funct. Mater., 2022, 202207116 Search PubMed.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., Int. Ed., 2021, 60, 23051–23067 CrossRef CAS PubMed.
- L. Yu, S. Sun, H. Li and Z. J. Xu, Fundam. Res., 2021, 1, 448–452 CrossRef CAS.
- J. N. Hausmann, B. Traynor, R. J. Myers, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 3567–3571 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01743a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |