
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcimer evolution hampers symmetry-broken charge-separated states†
Ebin
Sebastian‡
 ,
Jeswin
Sunny‡
and
Mahesh
Hariharan
*
,
Jeswin
Sunny‡
and
Mahesh
Hariharan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala P.O., Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: mahesh@iisertvm.ac.in
First published on 23rd August 2022
Abstract
Achieving long-lived symmetry-broken charge-separated states in chromophoric assemblies is quintessential for enhanced performance of artificial photosynthetic mimics. However, the occurrence of energy trap states hinders exciton and charge transport across photovoltaic devices, diminishing power conversion efficiency. Herein, we demonstrate unprecedented excimer formation in the relaxed excited-state geometry of bichromophoric systems impeding the lifetime of symmetry-broken charge-separated states. Core-annulated perylenediimide dimers (SC-SPDI2 and SC-NPDI2) prefer a near-orthogonal arrangement in the ground state and a π-stacked foldamer structure in the excited state. The prospect of an excimer-like state in the foldameric arrangement of SC-SPDI2 and SC-NPDI2 has been rationalized by fragment-based excited state analysis and temperature-dependent photoluminescence measurements. Effective electronic coupling matrix elements in the Franck–Condon geometry of SC-SPDI2 and SC-NPDI2 facilitate solvation-assisted ultrafast symmetry-breaking charge-separation (SB-CS) in a high dielectric environment, in contrast to unrelaxed excimer formation (Ex*) in a low dielectric environment. Subsequently, the SB-CS state dissociates into an undesired relaxed excimer state (Ex) due to configuration mixing of a Frenkel exciton (FE) and charge-separated state in the foldamer structure, downgrading the efficacy of the charge-separated state. The decay rate constant of the FE to SB-CS (kFE→SB–CS) in polar solvents is 8–17 fold faster than that of direct Ex* formation (kFE→Ex*) in non-polar solvent (kFE→SB–CS≫kFE→Ex*), characterized by femtosecond transient absorption (fsTA) spectroscopy. The present investigation establishes the impact of detrimental excimer formation on the persistence of the SB-CS state in chromophoric dimers and offers the requisite of conformational rigidity as one of the potential design principles for developing advanced molecular photovoltaics.
Introduction
Symmetry-breaking charge separation (SB-CS) is a photoexcited-state process by which a pair of identical chromophores forms a charge-separated state with the electron and hole localized on different chromophoric units.1–7 Since discovering their existence in photosynthetic reaction centers, SB-CS processes have received tremendous attention.8–13 Accomplishing SB-CS in a multichromophoric system is analogous to radical pair formation in silicon semiconductors, where exciton binding energy between a hole and an electron is overcome by thermal energy in silicon semiconductors.3 The fundamental understanding of structure–property correlation and related excited-state dynamics among the molecular architectures has enabled researchers to rationalize the design of organic photovoltaics (OPVs).14–18 Conventional donor–acceptor based OPVs generally exhibit low open-circuit voltages (VOC) due to the significant energy difference between the optical band gap and charge-separated state at the interface.16,19,20 However, the energy loss during SB-CS is low compared to that during charge separation (CS) in donor–acceptor systems.3,21 Moreover, SB-CS processes in solid-states, transition metal complexes, and polymers are a progressing topic of interest and have been widely explored in weakly coupled multichromophoric systems.2–4,22–24 Developing robust organic materials exhibiting optimized SB-CS and recombination dynamics is hence pivotal for the advancement of organic photovoltaics.The factors determining the excited-state dynamics of SB-CS in multichromophoric architectures are the relative spatial orientation, distance and strength of electronic interaction between the monomeric units and the surrounding dielectric environment.3,25–30 Among the various weakly coupled chromophoric assemblies investigated for SB-CS, multichromophoric perylenediimide (PDI) architectures are widely explored due to their exciting optoelectronic properties.3,24,27,31 Of several interesting PDI molecular constructs reported to date, the spiro-conjugated orthogonal arranged PDI dimer (Sp-PDI2) reported by our group exhibited a prolonged SB-CS state (kSB–CS/kCR = 2647 in acetonitrile).32 Ultrafast SB-CS in PDI dimers was first experimentally demonstrated by Wasielewski and coworkers in cofacial and head-to-tail stacked PDI dimers.33 Würthner, Kim and coworkers recognized excimer as an intermediate state to SB-CS in a cofacially arranged PDI dimer.26 In excimer-mediated excited state processes, the proficiency of the desired state is limited by the intermediate parasitic excimer formation.26,34 The superposition of Frenkel exciton and the charge-transfer states in a chromophoric assembly populates the excimer state, which is generally considered an exciton trap state that diminishes the efficacy of OPVs.35–39 Structural flexibility in the form of rotational or translational degrees of freedom in multichromophoric systems profoundly affects the population of distinct excited states.40,41 The interchromophoric torsional motion in a bianthryl system facilitates the transition from the local excited state to the SB-CS state in accordance with solvent polarity.42,43 Planarization of chromophores in the excited state reported in flexible molecular systems opens up diversified excited state dynamics.44,45 Hence controlling the conformational flexibility and the concomitant manifestation of undesired radiative and non-radiative deactivation pathways of the SB-CS state is vital for a long-lived CS state, thereby enhancing photovoltaic efficiency.3,46 Core-annulated near-orthogonal arranged perylenediimide dimers have found extensive applications as non-fullerene acceptors in organic solar cells owing to their greater solubility, reduced aggregation and excimer formation.47,48 Though vital for photovoltaic applications, the excited-state dynamics of heteroatom annulated PDI dimers remain elusive. Our continuous efforts to understand the excited-state dynamics of orthogonal/near-orthogonal arranged multichromophoric architectures motivated us to dwell in the realm of excited-state structural rearrangement and the associated deactivation pathways.32,44,49–53 Herein, we showcase the transformation of an initially populated SB-CS state to a detrimental excimer state due to excited-state conformational changes. Near-orthogonal stacked SC-SPDI2 and SC-NPDI2 undergo ultrafast structural relaxation to a foldamer structure, leading to a direct population of the excimer state in toluene. However, ultrafast SB-CS is favored over excimer formation in a polar environment due to the effective electronic coupling in Franck–Condon geometry (FC) and the thermodynamic feasibility of charge separation. The initially populated SB-CS state decays to an excimer state as the chromophore rearranges to a foldamer structure due to torsional flexibility, which is in contrast with SB-CS being facilitated by the torsional motion in bianthryl systems. Rigidification of the near-orthogonal arranged dimers emerges as an ideal strategy for achieving a long-lived charge-separated state and diminishing the unwanted deactivation pathway.
Results and discussion
Synthesis, characterization and geometry optimizations
In this work, sulfur and nitrogen annulated perylenediimide dimers, SC-SPDI2 and SC-NPDI2, respectively, were synthesized and characterized following the reported and modified procedures (Fig. 1 and Scheme S1†).47,54,55SPDI and NPDI are the monomer units of SC-SPDI2 and SC-NPDI2, respectively. In SC-SPDI2 and SC-NPDI2, the monomeric units are covalently connected via a carbon–carbon single bond in the bay region of the perylenediimide core. The ground-state optimized geometries of SC-SPDI2 and SC-NPDI2 in vacuum were found to have an edge-to-edge arrangement between the monomeric units with a dihedral angle (φ) of 86.5° in SC-SPDI2 and 83.6° in SC-NPDI2 (Fig. 1c and d). The single-crystal X-ray diffraction structure of SC-NPDI2 reported by Welch and coworkers exhibited a similar edge-to-edge arrangement with a dihedral angle of 66°, which is significantly lower than that observed in the optimized structure due to the crystal packing forces in the solid-state.47 The incorporation of S and N atoms on the perylene core of PDI imposed a curved or bowed molecular geometry as a consequence of the ring strain of the fused heteroring (Fig. S1†).56,57 The computed carbon-heteroatom bond lengths in the annulated heteroring of SC-SPDI2 and SC-NPDI2 indicate that the ring strain induced by the thiophene ring (C–S bond length: 1.77 Å, Fig. S2†) on the PDI core is weaker than that induced by the pyrrole ring (C–N bond length: 1.41 Å, Fig. S2†).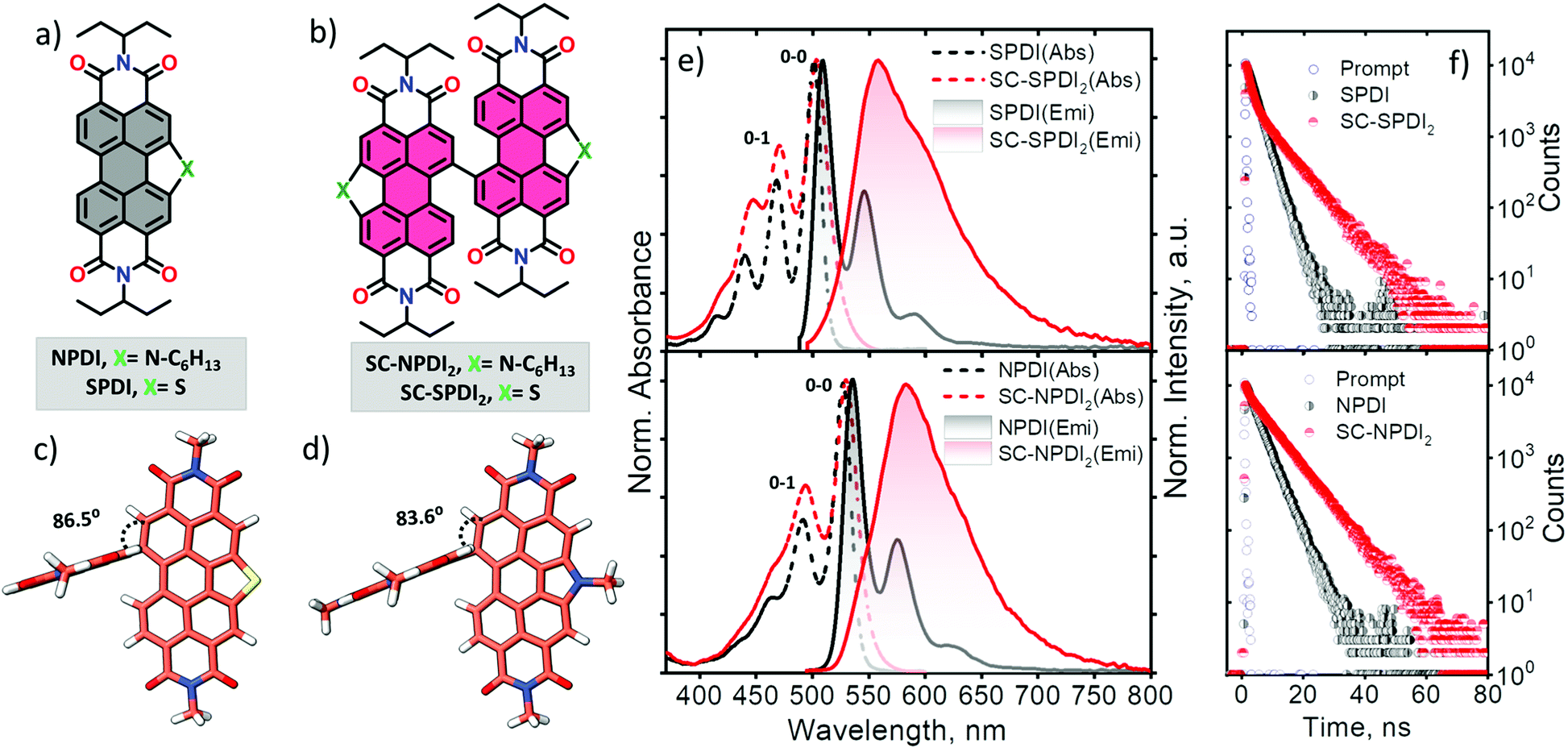 | ||
| Fig. 1 Molecular structures of monomeric SPDI and NPDI (a) and dimeric SC-SPDI2 and SC-NPDI2 (b). Optimized geometries of SC-SPDI2 (c) and SC-NPDI2 (d) with the corresponding dihedral angles between the monomeric planes. Normalized UV-vis absorption and emission spectra (e) and fluorescence decay profile (f) of SC-SPDI2 and SC-NPDI2 in toluene at room temperature. | ||
Optical properties
The steady-state electronic properties of SC-SPDI2 and SC-NPDI2 along with the monomeric units were explored by electronic absorption and photoluminescence spectroscopy measurements in toluene (TOL, c0 = 0.1–0.3 μM) at room temperature (Fig. 1e). The UV-visible absorption spectrum of NPDI shows characteristic spectral signatures of perylenediimide (PDI) dye with an absorption maximum (λAbsmax) at 527 nm, while SPDI (λAbsmax = 501 nm) shows a 26 nm hypsochromic shift compared to PDI and NPDI (Fig. S3†). As a result of the near-orthogonal arrangement, SC-SPDI2 and SC-NPDI2 show negligible red-shifted electronic absorption spectra with absorption maxima centered at 503 and 530 nm (Tables S1–S4†).32,58 The ratio of the intensity of the first and second vibronic bands, I0–0/I0–1, of SC-SPDI2 (1.42) and SC-NPDI2 (1.55) decreased from that of monomeric SPDI (1.70) and NPDI (1.92), indicating weak H-type excitonic coupling between the monomeric units (Fig. 1e).59,60 In addition, the slight broadening of the 0–0 vibronic absorption band of SC-SPDI2 and SC-NPDI2 as compared to that of the monomeric unit reiterates the weak excitonic coupling in near-orthogonal dimers.As shown in Fig. 1e, photoexcitation of SPDI and NPDI at 470 nm displays a characteristic PDI photoluminescence spectrum with an emission maximum (λEmmax) observed at 509 nm and 535 nm, respectively (Stokes shift of ∼8 nm). The photoluminescence quantum yield (ϕFl) of SPDI and NPDI is quantified as 0.58 and 0.68 in toluene, respectively (Table 1). Fascinatingly, SC-SPDI2 and SC-NPDI2 reveal a significantly red-shifted (Stokes shift of ∼54 nm) and broadened photoluminescence spectrum relative to the monomeric SPDI and NPDI in TOL (λEmmax = 558 nm for SC-SPDI2 and 584 nm for SC-NPDI2). Broad, featureless, and extensive Stokes shifted photoluminescence spectra are the characteristic spectral features of an excimer (Ex) state.29,35,61,62 Ex states, an admixture of Frenkel exciton (FE) and charge transfer (CT) states, are generally considered as energy trap states with increased non-radiative decay rates.27,54,55 However, the photoluminescence quantum yields of dimers are not drastically quenched, ϕFl = 0.60 for SC-SPDI2 and ϕFl = 0.71 for SC-NPDI2, as compared to that of the reference molecules (SPDI and NPDI), suggesting an excimer-like state having a large FE contribution (vide infra, Table 1).4,63,64
| Solvents | SPDI | NPDI | SC-SPDI2 | SC-NPDI2 | ||||
|---|---|---|---|---|---|---|---|---|
| ϕ Fl | τ Fl (ns) | ϕ Fl | τ Fl (ns) | ϕ Fl | τ Fl (ns) | ϕ Fl | τ Fl (ns) | |
| a Photoluminescence quantum yield (±5%). | ||||||||
| TOL (ε = 2.38) | 0.58 | 2.73 | 0.68 | 3.34 | 0.60 | 1.42 (42), 8.35 (58) | 0.71 | 0.84 (15), 7.86 (85) |
| ACE (ε = 20.70) | 0.53 | 2.21 | 0.61 | 3.39 | 0.09 | 5.89 | 0.08 | 3.82 |
| ACN (ε = 37.50) | 0.55 | 2.15 | 0.62 | 3.31 | 0.08 | 5.81 | <0.01 | 2.23 |
To further scrutinize the nature of the emissive state of SC-SPDI2 and SC-NPDI2, fluorescence lifetime measurements were performed in TOL. SC-SPDI2 and SC-NPDI2 exhibit biexponential decay of emission with lifetimes of τFl1 = 1.42 ns and τFl2 = 8.15 ns for SC-SPDI2 and τFl1 = 0.84 ns and τFl2 = 7.86 ns for SC-NPDI2 (Fig. 1f and Table 1), suggesting the presence of dual emissive states in the dimers. Monomeric SPDI and NPDI in TOL display a monoexponential decay profile with a lifetime of τFl = 2.73 ns and τFl = 3.34 ns, respectively. Since the fluorescence lifetime of the Ex state is reported to be longer than that of the monomer unit, the long-lived emissive component of SC-SPDI2 (τFl2 = 8.15 ns) and SC-NPDI2 (τFl2 = 7.86 ns) might stem from the Ex states.26,35 The dual emissive nature of SC-SPDI2 and SC-NPDI2 indicates that the dimers in the FC geometry may undergo a structural change in the excited state and form a different state, possibly an unrelaxed excimer state (Ex*), which can emit photons before completely relaxing to the relaxed excimer state.36,40,65 However, the most intriguing aspect of the emissive excited-state of SC-SPDI2 and SC-NPDI2 is that the population of Ex states does not quench photoluminescence quantum yield. What could be the excited-state structure that promotes the aforementioned optical properties? To address this, theoretical and experimental techniques have been employed to unravel the underlying excited-state phenomena.
Excited-state geometry optimizations
In order to obtain an in-depth insight into the excited-state relaxed structure, density functional theory-based geometry optimization has been performed in the excited-state by applying the dispersion corrected functional (TD-B3LYP-D3/def2-svp level of theory).66–69 The theoretical calculations propose that the ground state (S0) near-orthogonal arrangement of SC-SPDI2 and SC-NPDI2 gets transformed into the nearly π-stacked foldamer conformation in the excited state (S1) (Fig. 2 and S4†). Due to excited-state structural relaxation, the electronic interaction between the monomeric units increases as the dihedral angle (φ) between monomeric units decreases from 86.5° to 57.6° in SC-SPDI2 and from 83.6° to 62.5° in SC-NPDI2. Furthermore, the centroid-to-centroid distance (d) between annulated PDIs decreases from 5.94 Å to 4.47 Å for SC-SPDI2 and 5.73 Å to 4.46 Å for SC-NPDI2 upon excited-state structural relaxation (Fig. S4†). Several bichromophores and multichromophores have also been identified to exhibit foldamer arrangements in distinct environments.70–73 Schwartz and coworkers reported the existence of two conformers (near-orthogonal and nearly π-stacked arrangement) in the ground state of a non-annulated bay-linked perylenediimide dimer (di-PDI, Fig. S5†).74 The absence of two minima in the ground state potential energy surfaces of SC-SPDI2 and SC-NPDI2 when the dihedral angle changes from 45° to 125°, in contrast to di-PDI, could be ascribed to the effect core-annulation has in altering the energy landscapes (Fig. S6†).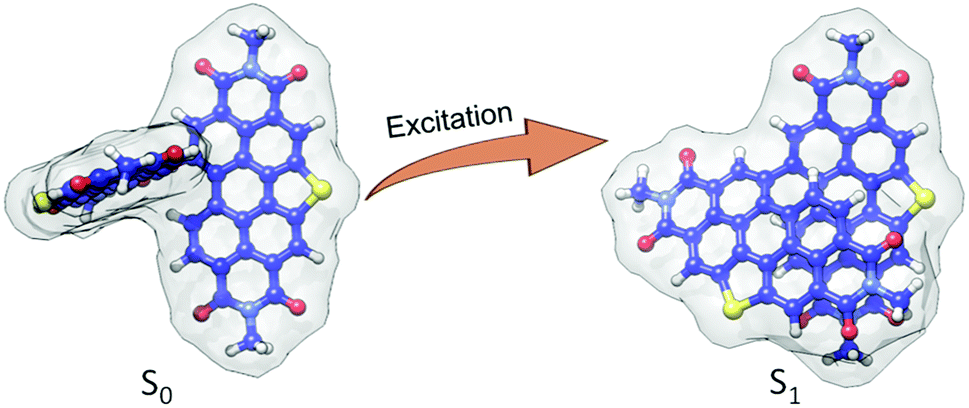 | ||
| Fig. 2 Excited-state structural relaxation of SC-SPDI2 dimer upon photoexcitation. | ||
Time-resolved emission spectroscopy measurements
To obtain deep insights into the energy landscape of excited-state structural relaxation and excimer formation of SC-SPDI2 and SC-NPDI2 in TOL, we have performed picosecond time-resolved emission spectroscopy (TRES) measurements (pulse width <60 ps, Fig. S7 and S8,† ESI, Section 1.2†). The global analysis of TRES data of SC-SPDI2 and SC-NPDI2 using sequential model A→B indicate the presence of two dependent emissive states. Evolution associated spectra of the first species (EASA) show an intense emission band that resembles the steady-state emission spectra of SC-SPDI2 with λEmmax at 559 nm, which is assigned to the unrelaxed excimer (Ex*, Fig. S7 and S9†).75 The Ex* state decays to a weakly emissive stable excimer state (Ex) with a time constant of τEx*→Ex = 1.37 ns. Weakly emissive, broadened, and red-shifted (λEmmax = 564 nm) photoluminescence features are characteristics of an excimer state (EASB) with a lifetime of 8.30 ns (Fig. S7†). Similarly, SC-NPDI2 in TOL exhibits emissive unrelaxed and relaxed excimer states (EASA and EASB), which decay with time constants of 0.74 ns and 7.74 ns (Fig. S8 and S9†). The relaxed excimer state (Ex) observed is weakly emissive in nature with red-shifted (λEmmax = 584 nm) and broader spectral properties than those of the strongly emissive unrelaxed excimer state (λEmmax = 579 nm). TRES measurement unambiguously confirms that the structural relaxation energetically stabilizes the excimer state and gradually reduces the photoluminescence properties.Temperature-dependent photoluminescence measurements
Further insights into the excited-state structural relaxation of SC-SPDI2 and SC-NPDI2 dimers in TOL were provided by steady-state temperature-dependent photoluminescence (PL) measurements from 90 K to 250 K (Fig. 3).75–77 Interestingly, at 90 K, SC-SPDI2 and SC-NPDI2 show PL spectra with well-resolved vibronic progression and a hypsochromic shifted emission maximum (λEmmax = 540 nm for SC-SPDI2 and λEmmax = 557 nm SC-NPDI2) as compared to the PL characteristics of dimers observed at room temperature (Fig. S10†). Moreover, the 0–0 transition approximately matches the corresponding absorption spectrum onset and is assigned to emission from the Frenkel exciton (FE) state. The observed PL spectra of SC-SPDI2 and SC-NPDI2 at 90 K resemble emission features of a monomeric unit with a decrease in the 0–0 to 0–1 PL line strength ratio, which confirms the delocalization of excitation energy among both monomeric units in the Franck–Condon geometry (Fig. S11†).78 As the temperature increases, a gradual reduction in the PL intensity and an inversion in the vibronic peak ratio are observed, indicating the transformation of FC geometry to an H-type foldamer conformation (Fig. 3).79,80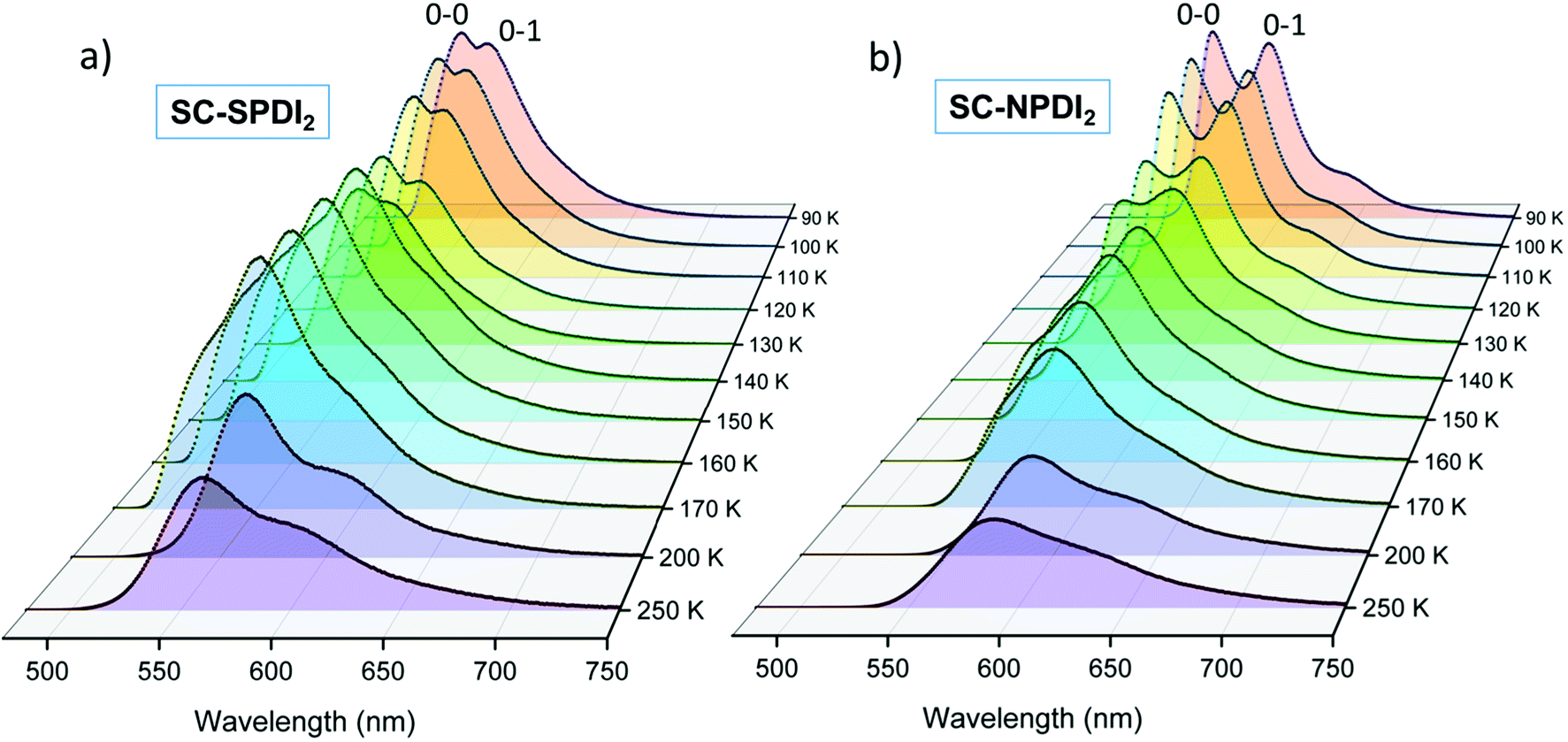 | ||
| Fig. 3 The temperature-dependent photoluminescence spectra of (a) SC-SPDI2 and (b) SC-NPDI2 in toluene. | ||
At the warming temperature of TOL between 140 K and 170 K, vibronically resolved PL features from the FE disappear and broader, less featured, and red-shifted PL spectra evolve (λEmmax = 552 nm and 582 nm for SC-SPDI2 and SC-NPDI2, respectively). The observed PL spectra (140 K–170 K) resemble the time-resolved emission spectral signatures of the unrelaxed excimer (Ex*) state and emission from the Ex* state is more predominant with an overlapping component of a minor amount of emission from the FE state (Fig. S12†). The PL intensity of SC-SPDI2 at warming temperature is slightly higher than that at 130 K, indicating the higher transition strength of the Ex* state than the FE state. At the same time, the PL intensity of SC-NPDI2 decreases gradually as the temperature increases from 90 K to 170 K. Finally, at high temperatures (>170 K), PL spectra are further broadened and red-shifted (λEmmax = 560 and 587 nm for SC-SPDI2 and SC-NPDI2, respectively) and PL intensity decreases dramatically, representing the formation of a relaxed excimer state (Ex), which resembles the room temperature PL spectra. The overlapping emissions of the unrelaxed excimer state (Ex*) and energy stabilized excimer state (Ex) define the PL emission spectra in this region (>170 K), and FE state emission is virtually absent, which could be due to the ultrafast structural reorganization (vide infra). The temperature-dependent PL measurement demonstrates the transformation of the FC geometry of SC-SPDI2 and SC-NPDI2 to a relaxed foldamer structure by excited-state structural relaxation at room temperature. Similarly, PL measurements of SC-SPDI2 and SC-NPDI2 in a polystyrene matrix support the near-orthogonal arrangement in the solid state (Fig. S13†).81
Solvent-dependent optical properties
To understand the surrounding dielectric environment moderated excited-state properties of SC-SPDI2 and SC-NPDI2, we carried out absorption and fluorescence measurements in solvents of different dielectric constants, i.e., toluene (TOL, ε = 2.38), acetone (ACE, ε = 20.70), and acetonitrile (ACN, ε = 37.50). The solvent-dependent UV-vis absorption spectra of SC-SPDI2 and SC-NPDI2 exhibit virtually unperturbed absorption line shapes, which is also the case for monomeric SPDI and NPDI (Fig. S14–S16† and Table 1). On the other hand, normalized solvent-dependent emission spectra of SC-SPDI2 and SC-NPDI2 show emission band broadening and a red-shift in the emission maximum as the polarity of the solvent increases from TOL to ACE (Fig. S17†). This solvent dependency of SC-SPDI2 and SC-NPDI2 implies the stabilization of the emissive excimer state as the solvent polarity increases. As the solvent polarity increases from ACE to ACN, SC-NPDI2 reflects gradual peak broadening and an increase in Stokes shifts in normalized emission spectra. This observation reveals the stronger charge-transfer character of the excimer state in SC-NPDI2.29,82 However, the normalized emission spectra of SC-SPDI2 exhibit minor changes as the polarity changes from ACE to ACN, indicating the weaker charge transfer characteristics of the excimer state of SC-SPDI2 in polar solvents.The photoluminescence quantum yield (ϕFl) of SC-SPDI2 and SC-NPDI2 drastically decreases with the increase in solvent polarity from TOL to ACE, presumably due to competitive non-radiative decay channels and the increased CT character of the excimer state (ϕFl = 0.60 in TOL, 0.09 in ACE for SC-SPDI2 and ϕFl = 0.71 in TOL, 0.08 in ACE for SC-NPDI2, Table 1). SC-NPDI2 exhibits further quenching of photoluminescence as solvent polarity changes from ACE to ACN; in contrast, SC-SPDI2 exhibits negligible effects as the solvent polarity changes from ACE to ACN. The unique dielectric environment-dependent ϕFl of SC-SPDI2 and SC-NPDI2 indicates the variation in the CT nature of the emissive excimer state as the solvent and molecular structure change (ϕFl = 0.09 in ACE, 0.08 in ACN for SC-SPDI2 and ϕFl = 0.08 in ACE, <0.01 in ACN for SC-NPDI2).
The solvent-dependent fluorescence lifetime measurements of SC-SPDI2 and SC-NPDI2 show the disappearance of a shorter-lived fluorescence decay component and a decrease in the fluorescence lifetime of the second component as the solvent polarity increases from TOL to ACE (Table 1 and Fig. S18†). Additionally, as the dielectric medium changes from ACE to ACN, a negligible difference in the fluorescence lifetime was observed for SC-SPDI2 (τFl = 5.89 ns in ACE and 5.81 ns in ACN). However, SC-NPDI2 displays a slight decrease in the fluorescence lifetime as the polarity of the solvent increases (τFl = 3.82 ns in ACE and 2.23 ns in ACN; Table 1 and Fig. S18†). This discrepancy implies the dielectric dependency of the excimer state of SC-NPDI2. The solvent-dependent photoluminescence quenching and decrease in the fluorescence lifetime of SC-SPDI2 and SC-NPDI2 in more polar solvents (ACE and ACN) implies the existence of an additional non-radiative deactivation channel such as SB-CS and an increase in the CT character of the excimer.62 Photoluminescence quantum yield and fluorescence lifetimes of monomers SPDI/NPDI are nearly insensitive to solvent polarity (Fig. S19† and Table 1).
Energetics
In order to determine the feasibility of the photoinduced SB-CS in SC-SPDI2 and SC-NPDI2 in polar solvents, the thermodynamic driving force (ΔGCS) for the SB-CS process has been analysed using the Weller formulation (Fig. S20,†Tables 2 and S9,† ESI, Section 1.4).83 The Weller analysis gives ΔGSB–CS ≈ −0.42 and −0.46 eV for SC-SPDI2 and ΔGSB–CS ≈ −0.40 and −0.43 eV for SC-NPDI2 in ACE and ACN respectively, indicating that SB-CS is thermodynamically favored in ACE and ACN (exergonic process) as compared to non-polar TOL (ΔGSB–CS ≈ +0.20 and +0.25 eV respectively, endergonic process).84 The electronic interaction between the frontier molecular orbitals of the adjacent monomeric units in the FC geometry of SC-SPDI2 and SC-NPDI2 provides the required electronic coupling for charge separation (Fig. S21 and S22†).3,32| Solvents | ΔGCS (eV) | τ CS (ps) | k CS × 1011 (s−1) | |
|---|---|---|---|---|
| SC-SPDI2 | TOL | +0.20 | — | — |
| ACE | −0.42 | 1.5 ± 0.3 | 6.67 | |
| ACN | −0.46 | 0.9 ± 0.2 | 11.1 | |
| SC-NPDI2 | TOL | +0.25 | — | — |
| ACE | −0.40 | 2.7 ± 0.2 | 3.70 | |
| ACN | −0.43 | 1.4 ± 0.1 | 7.14 |
Excited-state population dynamics
To shed light on excited-state dynamics accountable for the excimer formation, solvent-dependent fluorescence quenching and SB-CS in SC-SPDI2 and SC-NPDI2, femtosecond transient absorption (fsTA) measurements of the annulated PDI dimers were performed (ESI, Sections 1.5 and 1.6†). The solvent-dependent fsTA spectra of reference molecules, SPDI and NPDI, exhibit the characteristic spectral features of a singlet excited-state (S1 to Sn transition) of a PDI chromophore in the initial time, which decays with the evolution of a triplet state of the monomeric unit with a weak spectral signature (Fig. S23 and S24†).24,35,57,85,86Upon photoexcitation of SC-SPDI2 in TOL at 470 nm, fsTA spectra exhibit a strong negative ground state bleach (GSB) and weak stimulated emission (SE) between 450 and 563 nm, and excited-state absorption (ESA) maxima at 657 nm in the initial few picoseconds, assigned to the singlet excited state of SC-SPDI2 (1*SC-SPDI2, Fig. 4 and S25†). The rapid broadening of the ESA features between 570 and 760 nm and the evolution of a small negative SE signal in the 538–578 nm region within several hundred picoseconds are indicative of the structural relaxation to the unrelaxed excimer state (Ex*; vide infra). The SE signal maximum centered at 558 nm is comparable with the steady-state emission maximum (λEmmax = 558 nm), suggesting the emissive nature of the corresponding component. Later, singlet ESA disappears with the rise of a new broad and structureless ESA feature between 530 and 760 nm with an isosbestic point at ∼601 nm, representing the relaxed excimer state (Ex). The presence of two excimer states arises from the structural rearrangement of the initially formed high-energy unrelaxed excimer state (Ex*) to a stable excimer state.25,36,82 Global fitting of the fsTA data shows that the singlet excited-state/FE (A) decays to an unrelaxed excimer state (B) with a time constant of τA→B = 11.3 ± 0.1 ps, followed by the rise of a structurally relaxed excimer state (C) occurring with a time constant of τB→C = 1.12 ± 0.02 ns. Furthermore, the long-lived relaxed excimer state (C) does not decay completely within the experimental time window which is fitted with a time constant of 8.10 ± 0.01 ns.
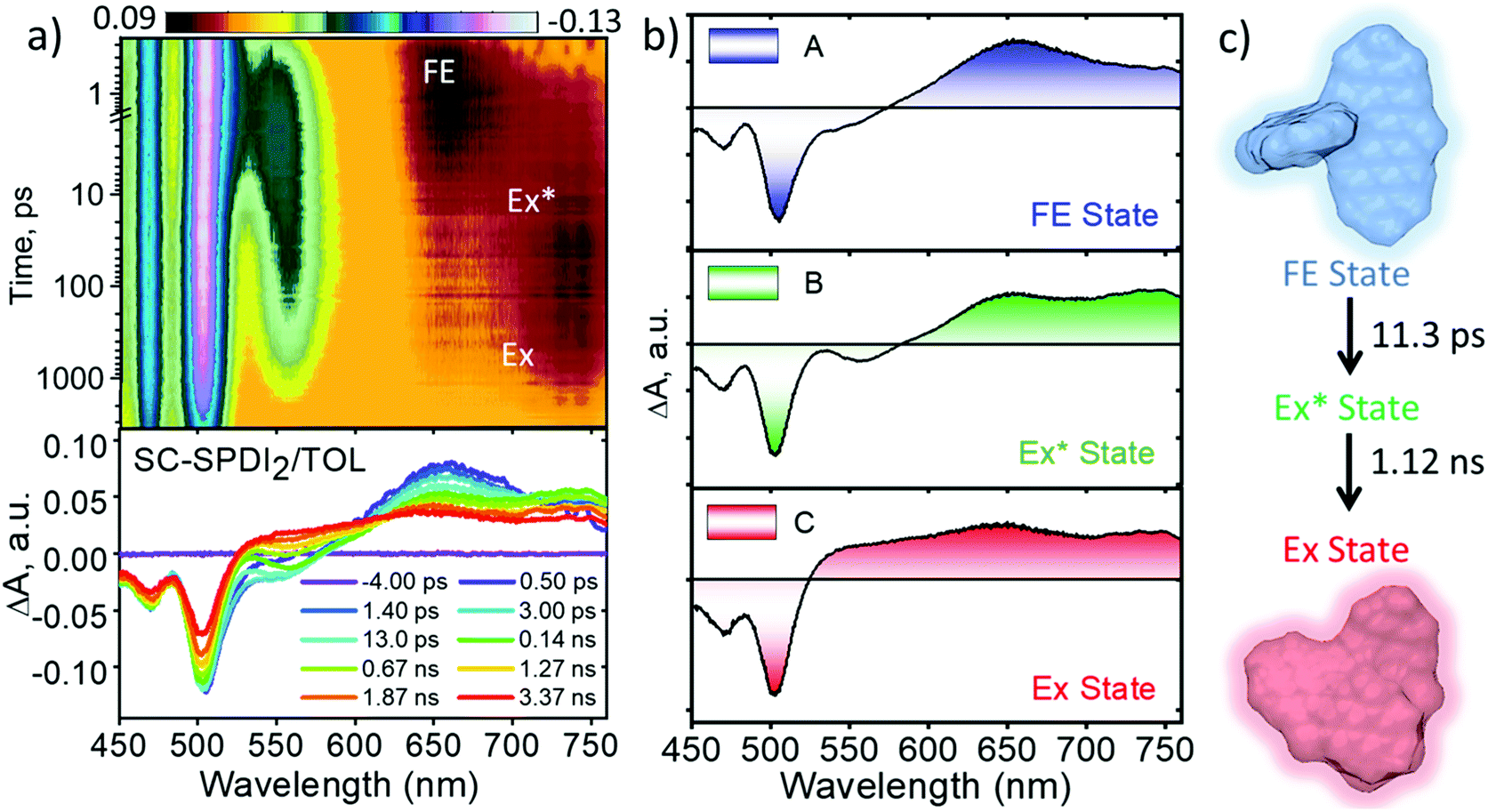 | ||
| Fig. 4 (a) Femtosecond transient absorption contour maps (top) and spectra (bottom) of SC-SPDI2 in toluene (TOL) showing the excited-state dynamics after photoexcitation at 470 nm. (b) Evolution associated difference spectra reconstructed from global analysis of the A → B → C → D model, where FE is the Frenkel exciton/singlet excited state; Ex* is the unrelaxed excimer state; Ex is the relaxed excimer state. (c) Schematic representation of excited-state dynamics of SC-SPDI2 in toluene at room temperature. | ||
Similarly, the fsTA spectra of SC-NPDI2 in TOL (Fig. S26 and S27†) display singlet excited or FE state features in early times (GSB/SE at 450–560 nm and ESA maxima at 620 nm), which decays to an intermediate unrelaxed excimer state (τA→B = 23.6 ± 0.2 ps) characterized by the formation of a new SE band centered at 583 nm (λEmmax = 584 nm) and broadening of ESA spectral features in the 620–760 nm region (the isosbestic point at ∼580 nm). Furthermore, the unrelaxed high-energy excimer (B) relaxes to form a stable, relaxed excimer state (C, τB→C = 0.79 ± 0.02 ns) characterized by featureless and broad ESA, which does not decay within the experimental time window and is fitted with a time constant of 7.87 ± 0.02 ns. The two different excimer decay time constants agree with the fluorescence lifetime of SC-SPDI2 and SC-NPDI2 in TOL, indicating the emissive nature of the components B and C (Tables S6† and 1). The evolution of the new SE band maximum of SC-SPDI2 and SC-NPDI2 matches the steady-state emission maximum observed in TOL (Fig. S28†). The difference in the ESA signature of unrelaxed (B) and relaxed excimer (C) states is due to the higher photoluminescence behavior of the unrelaxed excimer state (Ex*) as compared to the stable excimer state (Ex). The approximate two-fold increase in the time constant for structural relaxation of SC-SPDI2 compared to SC-NPDI2 in TOL could have resulted from the difference in the excited-state energy landscapes.
In polar solvents (ACE and ACN), SC-SPDI2 shows GSB between 450 and 538 nm and ESA maxima at ∼662 nm in the initial time delays (Fig. 5, S29–S31† and Table 2). Subsequently, the synchronized evolution of the two transient species is characterized by positive ESA features in the visible region at ∼575 and 669 nm. The newly evolving ESA signature is attributed to the radical cation (ESA ∼ 575 nm) and radical anion (EAS ∼ 669 nm) of SC-SPDI2, i.e., the SB-CS state (Fig. S32–S34†). The concurrent growth of radical pairs of chromophores is the characteristic signature of intramolecular charge separation occurring between identical chromophores due to the solvent or structural vibration-induced symmetry breaking.7,65,87,88 Furthermore, the radical pair of SC-SPDI2 decays to form a new transient state with broad spectral signatures resembling the characteristic excimer state features.35,62,89 The sequential fitting of the fsTA data shows the ultrafast decay of the singlet excited state/Frenkel exciton (A) of SC-SPDI2 with the evolution of the SB-CS state (B) with a time constant of 1.5 ± 0.1 ps and 0.9 ± 0.1 ps in ACE and ACN, respectively, which is ∼8–13 fold faster than excimer formation in TOL. Afterward, the SB-CS state (B) decays to the relaxed excimer (C) with a time constant of approximately 2.01 ± 0.05 ns in ACE and 0.84 ± 0.02 ns in ACN (Table 2). The long-lived excimer state (C) is fitted with a time constant of 5.89 ± 0.01 ns in ACE and 5.81 ± 0.03 ns in ACN, assuming that photoluminescence of SC-SPDI2 in ACE (τFl=5.89 ns) and ACN (τFl=5.81 ns) originated from the relaxed excimer state (Ex).
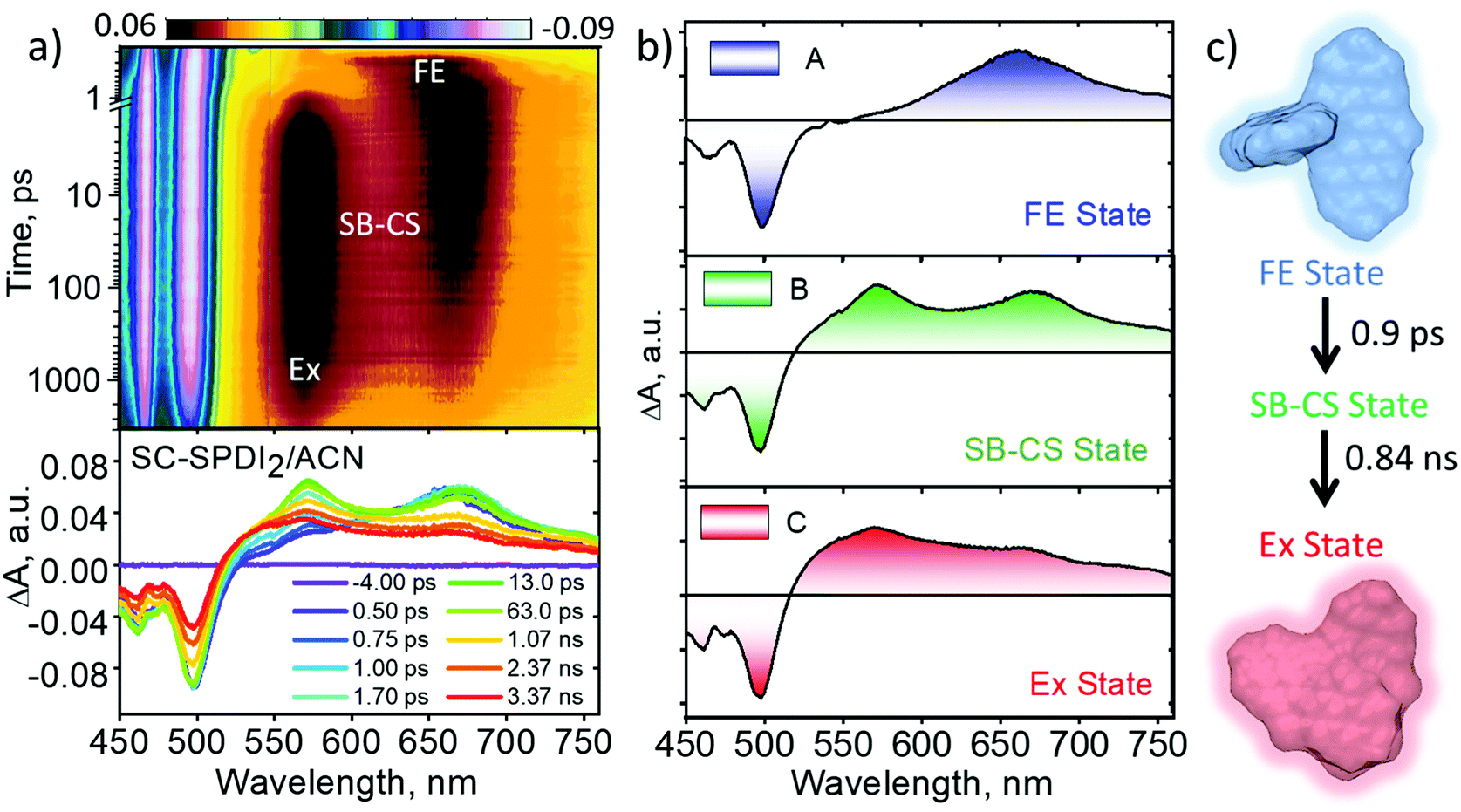 | ||
| Fig. 5 (a) Femtosecond transient absorption contour maps (top) and spectra (bottom) of SC-SPDI2 in acetonitrile (ACN) showing the excited-state dynamics after photoexcitation at 470 nm. (b) Evolution associated difference spectra reconstructed from global analysis of the A → B → C → D model, where FE is the Frenkel exciton/singlet excited state; SB-CS is the symmetry-breaking charge-separated state; Ex is the excimer state. (c) Schematic representative excited-state dynamics of SC-SPDI2 in acetonitrile at room temperature. | ||
The excited-state dynamics of SC-NPDI2 in ACE and ACN are similar to those of SC-SPDI2 in ACE and ACN, as shown in Fig. S35–S37.† The fsTA spectra at 0.5 ps show singlet excited-state/Frenkel exciton (A) features of SC-NPDI2 (ESA = 555−760 nm/GSB = <555 nm), which decays to the SB-CS state (B, τA→B = 2.7 ± 0.02 ps for ACE and 1.4 ± 0.01 ps for ACN). The ESA bands centered at ∼561 nm (radical cation of SC-NPDI2) and 673 nm (radical anion of SC-NPDI2) appear in the initial few picoseconds, indicating the ultrafast SB-CS state formation (Fig. S35†).24,57 Later, the SB-CS state (B) decays with the formation of a new excimer state (C) having a charge-transfer character (τB→C = 1.66 ± 0.02 ns for ACE and 0.71 ± 0.02 ns for ACN). Energy level diagrams showing the solvent-dependent distinct excited-state decay processes occurring in SC-SPDI2 and SC-NPDI2 along with the geometries of the dimers in the ground state and relaxed excited-state are shown in Fig. 6. The excimer state of SC-SPDI2 and SC-NPDI2 in polar solvents resembles a SB-CS state with spectral broadening. As observed by Kim, Würthner and coworkers in bay-substituted perylenediimide cyclophane, this broad spectral feature can be assigned to an excimer state with a charge-transfer (CT) resonance character.62 The lifetime of the long-lived CT resonance excimer state of SC-SPDI2 in ACE and ACN is fitted with (τC→D = 3.82 ± 0.04 ns in ACE and 2.3 ± 0.04 ns in ACN) the observed fluorescence lifetime (Table 1). The decrease in the ratio of 0–0 and 0–1 vibronic band intensities in the GSB may be an indication of excited-state structural relaxation of SC-SPDI2 and SC-NPDI2 dimers from near-orthogonal arrangement to a relaxed foldamer structure (Fig. S38†).36 Ultrafast spectroscopic techniques including time-resolved impulsive stimulated Raman spectroscopy (TR-ISRS), time-resolved IR spectroscopy (TR-IR) and femtosecond stimulated Raman spectroscopy (FSRS) have been previously employed to elucidate the structural dynamics of excimer evolution.90–93
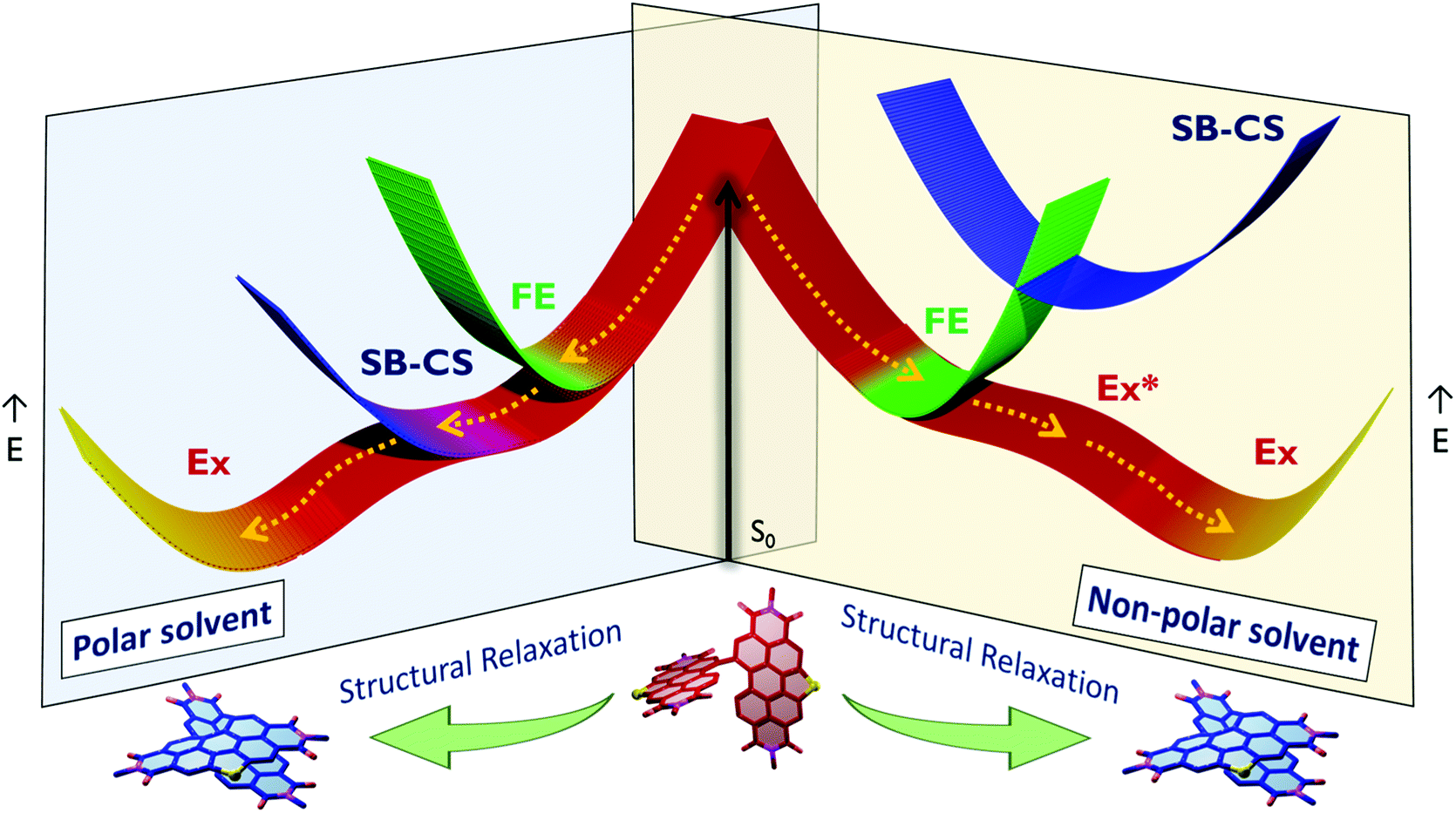 | ||
| Fig. 6 The potential energy diagram summarizes the excited-state dynamics of SC-SPDI2 and SC-NPDI2 undergoing structural relaxation in polar solvents (ACE and ACN) and non-polar solvent (TOL), where FE is the Frenkel exciton, Ex* is the unrelaxed excimer state, Ex is the stable excimer state and SB-CS is the symmetry-breaking charge-separated state. The x-axis represents the reaction coordinate and E on the y-axis indicates the potential energy. | ||
To obtain further evidence on the late events of SC-SPDI2 and SC-NPDI2, nanosecond transient absorption (nsTA) spectroscopy measurements were performed in different solvents (Fig. S39–S42†). In TOL, nsTA spectra of SC-SPDI2 show a positive ESA band at around 550 nm corresponding to the T1 → Tn transition and negative GSB below 515 nm, which decay with a lifetime of 6.62 ± 0.02 μs. At the same time, SC-NPDI2 in TOL does not show any spectral signature of the triplet state, which might be a consequence of the low spin–orbit-induced intersystem crossing (SO-ISC).94 Conversely, SC-SPDI2 and SC-NPDI2 in polar solvents (ACE and ACN) show the characteristic of a triplet manifold (GSB/ESA = 515 nm/550 nm for SC-SPDI2 and GSB/ESA = 535 nm/585 nm for SC-NPDI2), which decay to the ground state with a lifetime of 3.15 ± 0.02 μs and 3.16 ± 0.03 μs for SC-SPDI2 and 2.21 ± 0.01 μs and 2.18 ± 0.02 μs for SC-NPDI2, respectively. The overall excited state dynamics of SC-SPDI2 and SC-NPDI2 in different solvents are shown in Fig. S43–S46.† The population of excimer states through two competing pathways as a result of the varied interchromophoric coupling was reported by Wasielewski and co-workers in a cofacially arranged perylene dimer.25 Due to cofacial arrangement, the perylene dimer exhibits partial excimer formation through an SB-CS intermediate state and direct excimer population (major pathway) in polar solvents. In the present case, effective electronic coupling between the FE state and SB-CS state in the near-orthogonal FC geometry of SC-SPDI2 and SC-NPDI2 facilitates the ultrafast SB-CS and suppresses the excimer formation in the initial time. This is further supported by the rate constant of SB-CS in polar solvents being 8–17 fold greater than the rate constant of Ex* formation or excited state structural relaxation in toluene (Table S10†). However, when the chromophores rearrange into a foldamer conformation, the initially populated SB-CS dissociates to the excimer state.
Excited-state fragment-based analysis
The nature of different excited-states of SC-SPDI2 and SC-NPDI2, in their Franck–Condon (FC) near-orthogonal geometry and relaxed excited-state foldamer geometry, were evaluated by fragment-based excited-state analysis (Fig. 7, ESI, Section 1.7†).95,96 The participation ratio (PR) defines the extent of delocalization of the excitation energy in different fragments (each annulated PDI monomer unit of dimers is considered as a fragment) and its magnitude will range from 2 (delocalized on 2 fragments) to 1 (localized on one fragment). The charge-transfer (CT) value describes the nature of the molecule's different excited-states. The CT value can vary from 1 to 0, where the excited-state having CT < 0.2 indicates the Frenkel exciton (FE) state and CT > 0.8 describes the charge-transfer state. Finally, the excited-state defined by CT values ranging from 0.2 to 0.8 (0.2 < CT < 0.8, PR > 1.25) is assigned to an excimer state.95,97 In the FC geometry, SC-SPDI2 and SC-NPDI2 have four delocalized Frenkel exciton (FE) states, S1, S2, S3, and S4 (CT ≅ 0 and PR = 2) and two near-degenerate charge-transfer/resonance states, S5 and S6 (CT ≅ 0.94 and PR = 2, Fig. S47–S49,† Tables S7 and S8†). The charge resonance states (S5 and S6, CT ≈ 1) and delocalized Frenkel exciton states (S1–S4, CT ≈ 0) are energetically well-separated in the FC geometry.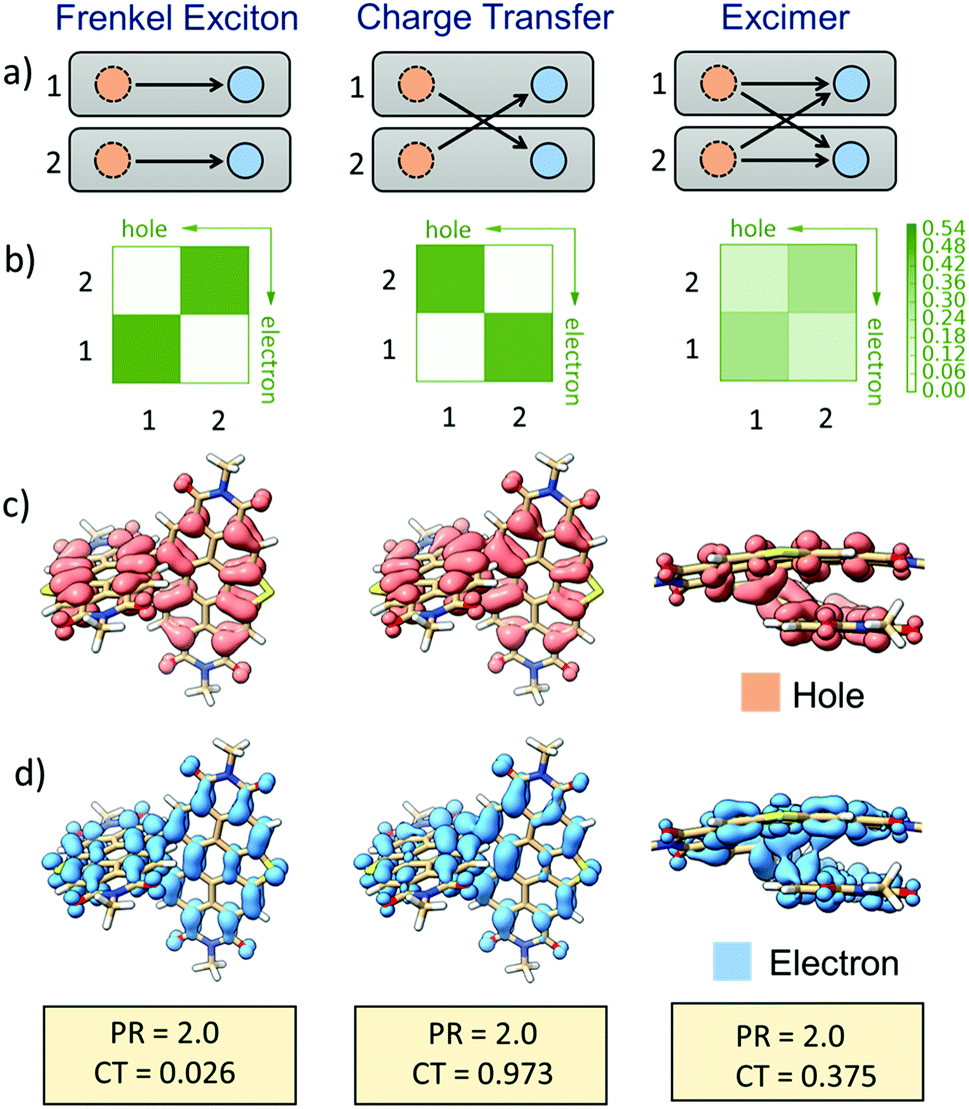 | ||
| Fig. 7 Pictorial representation of the delocalized Frenkel exciton (left), charge resonance states (middle) of FC geometry, and the excimer state (right) of the relaxed foldamer structure of SC-SPDI2. (a) Schematic representation of hole–electron distribution in the dimer, the rectangular box represents each fragment, dashed orange circles depict holes, and solid blue circles depict excited electrons. Hole–electron correlation plots (b) and the corresponding isosurface of the hole (c, orange) and electron (d, blue) distribution of different excited-states of SC-SPDI2. The CT and PR values are given at the bottom to define the nature of excitations. | ||
Fascinatingly, configuration mixing between the Frenkel exciton state and charge-resonance state occurs when the FC geometry (near-orthogonal arrangement) of SC-SPDI2 and SC-NPDI2 is transformed into an excited-state equilibrium geometry (π-stacked foldamer arrangement). The CT value of the first singlet excited-state (S1) of the relaxed foldamer geometry shows a magnitude of 0.37 for SC-SPDI2 and 0.39 for SC-NPDI2, along with a PR value of 2, indicating that the monomer units interact strongly to form the stabilized excimer state.98i.e., the S1 state of SC-SPDI2 and SC-NPDI2 in the foldamer arrangement does not remain as a pure Frenkel exciton but instead gains a partial CT character. The description is schematically and pictorially represented in Fig. 7 and S47† using electron–hole correlation plots and isosurfaces of SC-SPDI2 and SC-NPDI2. The different excited states of SC-SPDI2 and SC-NPDI2 in the foldamer configuration are given in Tables S9 and S10† and Fig. S50 and S51.† In the case of the delocalized Frenkel exciton, both electron–hole (hole) and excited-electron densities are localized on the same molecular fragment and these local excitations on monomeric units get coupled to form a delocalized state (Fig. 7, left side). In the charge resonance state, the electron–hole and excited-electron densities are completely localized on different monomeric units and a linear combination of two charge-transfer transitions opposing each other with no net charge transfer is observed (Fig. 7, middle). Finally, the excimer state implies that electron–hole and excited-electron densities are delocalized over two monomeric units. i.e., both the electron–hole and excited-electron of a monomeric unit can be found in the same or different monomeric units (Fig. 7, right side).95–97
Interplay of FE and CT contribution to the excimer state
The relative contributions of the FE and the CT states to an excimer state depend on the distance and relative orientation between the chromophore and dielectric medium around the chromophore.4,97,99,100 In a non-polar solvent, TOL, the energy of the CS/CT state of SC-SPDI2 and SC-NPDI2 is higher than that of the singlet state/FE state; as a result, efficient mixing of the FE state with CT/CS is less probable. Consequently, the excimer having a more prominent FE character than CT character is observed in the fsTA spectra of SC-SPDI2 and SC-NPDI2 in TOL, which could be the reason for high ϕFl of dimers in TOL (Fig. 4 and S26†). However, the excimer state spectral signatures of SC-SPDI2 and SC-NPDI2 in polar solvents nearly resemble the SB-CS state, with the positions of the radical pair bands being the same with broader ESA. This observation can be rationalized by the fact that the energy of the CS state is more stabilized than the FE state in polar solvents, facilitating the efficient mixing of the FE and CS states in the relaxed excited-state foldamer structure. Charge-transfer contribution to the excimer state of SC-NPDI2 is higher than that of SC-SPDI2 in polar solvents, which could stem from the reduced centroid to centroid distance (d) between monomers in the excited-state foldamer structure and the reduced energy difference between the FE and the CS states of SC-NPDI2 compared to those of SC-SPDI2 (Fig. S4† and Table 2). Photoluminescence quenching and a decrease in the fluorescence lifetime of SC-NPDI2 as the dielectric medium changes from ACE to ACN is attributed to the enhanced charge-transfer (CT) resonance character of the emissive excimer state. Due to the weak CT resonance nature of the emissive excimer state in SC-SPDI2, the photoluminescence quantum yield and fluorescence lifetimes are nearly unperturbed by a change in solvent polarity, i.e., ACE to ACN (Table 1).Conclusions
In conclusion, we report unequivocal evidence for the excited-state structural relaxation of SC-SPDI2 and SC-NPDI2 hampering the efficiency of the SB-CS state. Conjoint spectroscopic and theoretical investigations have rationalized the structural dynamics in the excited state and the manifestation of an excimer-like state in the π-stacked foldamer structure. The solvent polarity-dependent fsTA measurements of SC-SPDI2 and SC-NPDI2 reveal ultrafast SB-CS in polar solvents, in contrast to direct excimer formation observed in non-polar TOL. Molecular rearrangement of SC-SPDI2 and SC-NPDI2 in the excited-state transforms the initially populated SB-CS state into a CT resonance-enhanced excimer state, limiting the SB-CS state lifetime. These observations reinstate the importance of rigid molecular architectures, reminiscent of conformationally rigid special pairs of photosynthetic reaction centers embedded in protein scaffolds, exhibiting efficient SB-CS as non-fullerene acceptors in OPVs.Data availability
All experimental/computational data and procedures are available in the ESI.†Author contributions
E. S., J. S. and M. H. conceived the project; E. S and J. S. carried out the measurements. E. S., J. S. and M. H. analysed the results; E. S., J. S., and M. H. wrote the manuscript; M. H. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. H. acknowledges the Nanomission project (DST/NM/TUE/EE-01/2019) of the Department of Science and Technology (DST), Government of India, for financial support. E. S. acknowledges UGC for financial assistance. We thank Dr Sooraj Kunnikuruvan for fruitful discussions. We greatly acknowledge the support for high-performance computing time at the Padmanabha cluster, IISER Thiruvananthapuram, India.References
- M. R. Wasielewski, Chem. Rev., 2002, 92, 435–461 CrossRef.
- E. Vauthey, ChemPhysChem, 2012, 13, 2001–2011 CrossRef CAS PubMed.
- E. Sebastian and M. Hariharan, ACS Energy Lett., 2022, 7, 696–711 CrossRef CAS.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- H. Halperin, K. Dejong, R. E. W Adams, C. C. Coggins, N. Hammond, D. S. Gotufried, M. A. Steffen and S. G. Boxer, Science, 1991, 251, 662–665 CrossRef PubMed.
- B. Dereka, M. Koch and E. Vauthey, Acc. Chem. Res., 2017, 50, 426–434 CrossRef CAS PubMed.
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2022, 7, 786–793 CrossRef PubMed.
- D. A. Cherepanov, I. V. Shelaev, F. E. Gostev, A. Petrova, A. V. Aybush, V. A. Nadtochenko, W. Xu, J. H. Golbeck and A. Y. Semenov, J. Photochem. Photobiol., B, 2021, 217, 112154 CrossRef CAS PubMed.
- H. Tamura, K. Saito and H. Ishikita, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16373–16382 CrossRef CAS PubMed.
- B. A. Diner and F. Rappaport, Annu. Rev. Plant Biol., 2003, 53, 551–580 CrossRef PubMed.
- S. Vasil’ev, J. R. Shen, N. Kamiya and D. Bruce, FEBS Lett., 2004, 561, 111–116 CrossRef.
- H. Tamura, K. Saito and H. Ishikita, Chem. Sci., 2021, 12, 8131–8140 RSC.
- D. J. Lockhart and S. G. Boxer, Biochemistry, 2002, 26, 664–668 CrossRef.
- J. Hong, M. J. Sung, H. Cha, C. E. Park, J. R. Durrant, T. K. An, Y. H. Kim and S. K. Kwon, ACS Appl. Mater. Interfaces, 2018, 10, 36037–36046 CrossRef CAS PubMed.
- J. L. Brédas, E. H. Sargent and G. D. Scholes, Nat. Mater., 2016, 16, 35–44 CrossRef PubMed.
- V. Coropceanu, X. K. Chen, T. Wang, Z. Zheng and J. L. Brédas, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S. J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T. Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS PubMed.
- M. Lv, X. Lu, Y. Jiang, M. E. Sandoval-Salinas, D. Casanova, H. Sun, Z. Sun, J. Xu, Y. Yang and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202113190 CAS.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy, 2017, 2, 1–6 Search PubMed.
- A. N. Bartynski, M. Gruber, S. Das, S. Rangan, S. Mollinger, C. Trinh, S. E. Bradforth, K. Vandewal, A. Salleo, R. A. Bartynski, W. Bruetting and M. E. Thompson, J. Am. Chem. Soc., 2015, 137, 5397–5405 CrossRef CAS PubMed.
- N. Kaul and R. Lomoth, J. Am. Chem. Soc., 2021, 143, 10816–10821 CrossRef CAS PubMed.
- C. E. Ramirez, S. Chen, N. E. Powers-Riggs, I. Schlesinger, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2020, 142, 18243–18250 CrossRef CAS PubMed.
- Y. Guo, Z. Ma, X. Niu, W. Zhang, M. Tao, Q. Guo, Z. Wang and A. Xia, J. Am. Chem. Soc., 2019, 141, 12789–12796 CrossRef CAS PubMed.
- R. E. Cook, B. T. Phelan, R. J. Kamire, M. B. Majewski, R. M. Young and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 1607–1615 CrossRef CAS PubMed.
- J. Sung, A. Nowak-Król, F. Schlosser, B. Fimmel, W. Kim, D. Kim and F. Würthner, J. Am. Chem. Soc., 2016, 138, 9029–9032 CrossRef CAS PubMed.
- P. Spenst, R. M. Young, M. R. Wasielewski and F. Würthner, Chem. Sci., 2016, 7, 5428–5434 RSC.
- P. Roy, G. Bressan, J. Gretton, A. N. Cammidge and S. R. Meech, Angew. Chem., Int. Ed., 2021, 60, 10568–10572 CrossRef CAS PubMed.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329–10339 CrossRef CAS PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- Y. Wu, R. M. Young, M. Frasconi, S. T. Schneebeli, P. Spenst, D. M. Gardner, K. E. Brown, F. Würthner, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2015, 137, 13236–13239 CrossRef CAS PubMed.
- E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2021, 143, 13769–13781 CrossRef CAS PubMed.
- J. M. Giaimo, A. V. Gusev and M. R. Wasielewski*, J. Am. Chem. Soc., 2002, 124, 8530–8531 CrossRef CAS PubMed.
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305–310 CrossRef CAS PubMed.
- E. A. Margulies, L. E. Shoer, S. W. Eaton and M. R. Wasielewski, Phys. Chem. Chem. Phys., 2014, 16, 23735–23742 RSC.
- Y. J. Bae, D. Shimizu, J. D. Schultz, G. Kang, J. Zhou, G. C. Schatz, A. Osuka and M. R. Wasielewski, J. Phys. Chem. A, 2020, 124, 8478–8487 CrossRef CAS PubMed.
- R. Singh, M. Kim, J. J. Lee, T. Ye, P. E. Keivanidis and K. Cho, J. Mater. Chem. C, 2020, 8, 1686–1696 RSC.
- R. Wang, C. Zhang, Q. Li, Z. Zhang, X. Wang and M. Xiao, J. Am. Chem. Soc., 2020, 142, 12751–12759 CrossRef CAS PubMed.
- J. Hoche, M. Flock, X. Miao, L. N. Philipp, M. Wenzel, I. Fischer and R. Mitric, Chem. Sci., 2021, 12, 11965–11975 RSC.
- T. Yamakado, S. Takahashi, K. Watanabe, Y. Matsumoto, A. Osuka, S. Saito, T. Yamakado, S. Takahashi, K. W. Atanabe, Y. M. Atsumoto, P. R. A. Osuka and S. Saito, Angew. Chem., Int. Ed., 2018, 57, 5438–5443 CrossRef CAS PubMed.
- J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, D. M. R. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, ACS Appl. Energy Mater., 2018, 1, 1083–1095 CrossRef CAS.
- M. Jurczok, P. Plaza, M. M. Martin, Y. H. Meyer and W. Rettig, Chem. Phys., 2000, 253, 339–349 CrossRef CAS.
- J. J. Piet, W. Schuddeboom, B. R. Wegewijs, F. C. Grozema and J. M. Warman, J. Am. Chem. Soc., 2001, 123, 5337–5347 CrossRef CAS PubMed.
- H. Song, H. Zhao, Y. Guo, A. M. Philip, Q. Guo, M. Hariharan and A. Xia, J. Phys. Chem. C, 2020, 124, 237–245 CrossRef CAS.
- Z. Piontkowski and D. W. McCamant, J. Am. Chem. Soc., 2018, 140, 11046–11057 CrossRef CAS PubMed.
- L. Estergreen, A. R. Mencke, D. E. Cotton, N. V. Korovina, J. Michl, S. T. Roberts, M. E. Thompson and S. E. Bradforth, Acc. Chem. Res., 2022, 55, 1561–1572 CrossRef CAS PubMed.
- A. D. Hendsbee, J. P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098–7109 CrossRef CAS.
- R. Xin, J. Feng, C. Zeng, W. Jiang, L. Zhang, D. Meng, Z. Ren, Z. Wang and S. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 2739–2746 CrossRef CAS PubMed.
- M. P. Lijina, A. Benny, R. Ramakrishnan, N. G. Nair and M. Hariharan, J. Am. Chem. Soc., 2020, 142, 17393–17402 CrossRef CAS PubMed.
- A. M. Philip, A. R. Mallia and M. Hariharan, J. Phys. Chem. Lett., 2016, 7, 4751–4756 CrossRef CAS PubMed.
- M. Madhu, R. Ramakrishnan, V. Vijay and M. Hariharan, Chem. Rev., 2021, 121, 8234–8284 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- A. Benny, R. Ramakrishnan and M. Hariharan, Chem. Sci., 2021, 12, 5064–5072 RSC.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- H. Qian, W. Yue, Y. Zhen, S. Di Motta, E. Di Donato, F. Negri, J. Qu, W. Xu, D. Zhu and Z. Wang, J. Org. Chem., 2009, 74, 6275–6282 CrossRef CAS PubMed.
- I. K. Madu, H. Jiang, A. Laventure, P. M. Zimmerman, G. C. Welch and T. Goodson, J. Phys. Chem. C, 2021, 125, 10500–10515 CrossRef CAS.
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS PubMed.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed.
- B. S. Basel, C. Hetzer, J. Zirzlmeier, D. Thiel, R. Guldi, F. Hampel, A. Kahnt, T. Clark, D. M. Guldi and R. R. Tykwinski, Chem. Sci., 2019, 10, 3854–3863 RSC.
- S. A. Bagnich, S. Athanasopoulos, A. Rudnick, P. Schroegel, I. Bauer, N. C. Greenham, P. Strohriegl and A. Köhler, J. Phys. Chem. C, 2015, 119, 2380–2387 CrossRef CAS.
- W. Kim, A. Nowak-Król, Y. Hong, F. Schlosser, F. Würthner and D. Kim, J. Phys. Chem. Lett., 2019, 10, 1919–1927 CrossRef CAS PubMed.
- M. Nazari, E. Cieplechowicz, T. A. Welsh and G. C. Welch, New J. Chem., 2019, 43, 5187–5195 RSC.
- R. J. Lindquist, K. M. Lefler, K. E. Brown, S. M. Dyar, E. A. Margulies, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2014, 136, 14912–14923 CrossRef CAS PubMed.
- T. Kim, J. Kim, H. Mori, S. Park, M. Lim, A. Osuka and D. Kim, Phys. Chem. Chem. Phys., 2017, 19, 13970–13977 RSC.
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H. M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. B. Bornhof, A. Bauzá, A. Aster, M. Pupier, A. Frontera, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2018, 140, 4884–4892 CrossRef CAS PubMed.
- B. Fimmel, M. Son, Y. M. Sung, M. Grüne, B. Engels, D. Kim and F. Würthner, Chem. - Eur. J., 2015, 21, 615–630 CrossRef CAS PubMed.
- J. Chen, A. W. Ziegler, B. Zhao, W. Wan and A. D. Q. Li, Chem. Commun., 2017, 53, 4993–4996 RSC.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- E. P. Farr, M. T. Fontana, C. C. Zho, P. Wu, Y. L. Li, N. Knutson, Y. Rubin and B. J. Schwartz, J. Phys. Chem. C, 2019, 123, 2127–2138 CrossRef CAS.
- H. Yongseok, W. Kim, T. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, Angew. Chem., Int. Ed., 2021, e202114474 Search PubMed.
- H. Osaki, C.-M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 128, 7247–7251 CrossRef.
- D. Veldman, S. M. A. Chopin, S. C. J. Meskers, M. M. Groeneveld, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. A, 2008, 112, 5846–5857 CrossRef CAS PubMed.
- F. C. Spano and H. Yamagata, J. Phys. Chem. B, 2010, 115, 5133–5143 CrossRef PubMed.
- L. Van Dijk, P. A. Bobbert and F. C. Spano, J. Phys. Chem. B, 2009, 113, 9708–9717 CrossRef CAS PubMed.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, D. M. R. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, ACS Appl. Energy Mater., 2018, 1, 1083–1095 CrossRef CAS.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- A. Weller, Z. Phys. Chem., 1982, 133, 93–98 CrossRef CAS.
- H. Imahori, K. Hagiwara, M. Aoki, T. Akiyama, S. Taniguchi, T. Okada, M. Shirakawa and Y. Sakata, J. Am. Chem. Soc., 1996, 118, 11771–11782 CrossRef CAS.
- A. Mohan, E. Sebastian, M. Gudem and M. Hariharan, J. Phys. Chem. B, 2020, 124, 6867–6874 CrossRef CAS PubMed.
- I. Solymosi, S. Krishna, E. Nuin, H. Maid, B. Scholz, D. M. Guldi, M. E. Pérez-Ojeda and A. Hirsch, Chem. Sci., 2021, 12, 15491–15502 RSC.
- V. Markovic, D. Villamaina, I. Barabanov, L. M. Lawson Daku and E. Vauthey, Angew. Chem., Int. Ed., 2011, 50, 7596–7598 CrossRef CAS PubMed.
- S. Amthor, C. Lambert, S. Dümmler, I. Fischer and J. Schelter, J. Phys. Chem. A, 2006, 110, 5204–5214 CrossRef CAS PubMed.
- K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588–2593 CrossRef CAS PubMed.
- Y. Wu, J. Zhou, B. T. Phelan, C. M. Mauck, J. F. Stoddart, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 14265–14276 CrossRef CAS PubMed.
- C. M. Mauck, R. M. Young and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 784–792 CrossRef CAS PubMed.
- W. Kim, T. Kim, S. Kang, Y. Hong, F. Würthner and D. Kim, Angew. Chem., Int. Ed., 2020, 59, 8571–8578 CrossRef CAS PubMed.
- E. R. Kennehan, C. Grieco, A. N. Brigeman, G. S. Doucette, A. Rimshaw, K. Bisgaier, N. C. Giebink and J. B. Asbury, Phys. Chem. Chem. Phys., 2017, 19, 24829–24839 RSC.
- K. M. Lefler, K. E. Brown, W. A. Salamant, S. M. Dyar, K. E. Knowles and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 10333–10345 CrossRef CAS PubMed.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- L. M. Ibele, P. A. Sánchez-Murcia, S. Mai, J. J. Nogueira and L. González, J. Phys. Chem. Lett., 2020, 11, 7483–7488 CrossRef CAS PubMed.
- A. L. L. East and E. C. Lim, J. Chem. Phys., 2000, 113, 8981 CrossRef CAS.
- J. Sung, A. Nowak-Król, F. Schlosser, B. Fimmel, W. Kim, D. Kim and F. Würthner, J. Am. Chem. Soc., 2016, 138, 9029–9032 CrossRef CAS PubMed.
- D. Casanova, Int. J. Quantum Chem., 2015, 115, 442–452 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04387d |
| ‡ E. S. and J. S. contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |