
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirectional asymmetry over multiple length scales in reticular porous materials
Alexandre
Legrand
 a,
Zaoming
Wang
ab,
Javier
Troyano
a and
Shuhei
Furukawa
*ab
a,
Zaoming
Wang
ab,
Javier
Troyano
a and
Shuhei
Furukawa
*ab
aInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: shuhei.furukawa@icems.kyoto-u.ac.jp
bDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 19th October 2020
Abstract
In nature and synthetic materials, asymmetry is a useful tool to create complex and functional systems constructed from a limited number of building blocks. Reticular chemistry has allowed the synthesis of a wide range of discrete and extended structures, from which modularity permits the controlled assembly of their constituents to generate asymmetric configurations of pores or architectures. In this perspective, we present the different strategies to impart directional asymmetry over nano/meso/macroscopic length scales in porous materials and the resulting novel properties and applications.
1. Introduction
Asymmetry is a fundamental concept in nature and science spanning from the molecular level to the cosmic scale. In nature, a broad range of systems exists with various degrees of imperfection or asymmetry, which allows generation of complexity and new properties from a limited number of building blocks. In living systems, asymmetry is present at many levels of organization ranging from individual cells, through organs, to entire body-shapes and plays a key role in the evolution of organisms and species.1 The benefits of asymmetry extend from improved spatial arrangements to different functions. At the molecular level, asymmetry can be expressed through the chirality of a molecule. Enantiomers can show distinct properties such as taste, smell or biological actions due to different interactions with their receptors.2–4 At the mesoscale, biological membranes exhibit a marked structural asymmetry in the distribution of their components (phospholipids) constituting the inside or the outside of the lipid bilayer and thus present different affinity of interactions with analytes.5 At the macroscale, nature provides several examples of living-organisms showing preferential handedness, which affect their behavior. Indeed, in fiddler crabs, males develop asymmetry in the size of their claws, with one being larger than the other, which may favor adaptive responses in the walking legs for signaling and reproduction purposes.6Asymmetric features, which generate an uneven spatial distribution of building blocks in one system leading to new functions and properties in a wide range of length scales, can be applied to design synthetic materials.7–10 In particular, we focus on reticular porous materials, in which molecular building blocks are connected by strong interactions that maintain the structural integrity. The connection point can be (i) covalent bonds between organic molecules that form finite porous organic cages (POCs) and extended covalent–organic frameworks (COFs) or (ii) coordination bonds between organic ligands and metal ions leading to discrete metal–organic cages (MOCs) and extended metal–organic frameworks (MOFs). Because of the modularity of reticular frameworks, asymmetric configurations can be induced through the controlled assembly of their constituents over multiple length scales. Indeed, in reticular chemistry, the separation between connection and function allows tuning, with atomic precision, of the chemical composition, structure, porosity and chemical functionality while maintaining the connectivity.11 In particular, MOFs have emerged as suitable materials for designing asymmetric systems thanks to the possibility of inducing heterogeneity within a material without losing crystallinity.12 The fabrication of hierarchical or heterogeneous MOFs has already been discussed in many reviews including strategies to control the pore structure, composition and defects within the porous system, as well as their distribution at the mesoscale.13–15 Benefiting from these studies, multi-component hierarchical porous materials have been created with tunable structures and properties, including multivariate MOFs,16 MOFs with micropores transformed into mesopores,17 core–shell MOFs18 and Janus-type MOFs.19 Depending on how these components are spatially integrated, the heterogeneity of the MOF materials could be well controlled. However, asymmetric tailoring in such heterogeneous porous materials has rarely been discussed although some of them already display asymmetric microporous structures. In most of the cases, researchers focused on the hierarchical aspects of MOF structures rather than asymmetric ones, even though both have been suggested by Kitagawa as attractive features for the development of future porous materials.20
In this perspective, we aim to provide an overview of the different strategies that have been reported to break the inherited symmetrical nature of porous materials. To further expand the diversity and properties of porous materials, herein we focus on the challenging design of directional asymmetry in porous materials, in which parts of the architecture or the porosity configuration of a system that differ consistently from each other. Thus, point chirality arising from asymmetric atoms introduced in the backbone of the ligands will not be discussed here. The different approaches to impart directional asymmetry in porous materials will be divided into three parts corresponding to the different length scales where asymmetry can be introduced, i.e. nano-, meso- and macroscopic regimes which correspond to the asymmetry at the molecular component, the networked structure and the material assembly, respectively (Fig. 1). Indeed, the modularity of these systems allows for building blocks to be spatially positioned in the structure at different length scales. The applications of asymmetric porous materials in various fields including gas separation and artificial ion channels to name a few are also discussed. We expect this review to inspire more efforts towards the future development of hierarchical structures with higher complexity, where the asymmetry of molecular building blocks at the molecular level could be transmitted over multiple length scales through their supramolecular assembly, providing advanced functions.
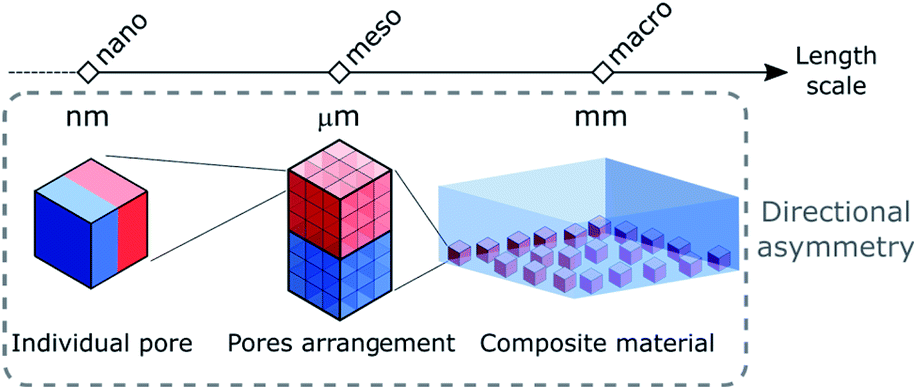 | ||
| Fig. 1 Schematic representation of the directional asymmetry concept starting from (left) the individual pore system at the nanoscale to (middle) their arrangement in a crystal at the mesoscale and (right) the creation of a composite material at the macroscale. | ||
2. Asymmetry at the nanoscale
The molecular scale is defined here as a molecular cage system, which represents the smallest porous unit. Its finite nature makes it an ideal model to demonstrate how asymmetry can be introduced not from the viewpoint of strict symmetry operations, but rather from the perspective of architecture or pore configuration. We define here directional asymmetry as a molecular system that can be divided into two distinct parts similar to what is observed with Janus particles.Creating asymmetry in metal–organic cages (MOCs) is challenging due to the difficulty of controlling the reactivity and conformation of the initial building blocks. Indeed, their self-assembly often leads to the formation of isotropic and spherical structures as the most stable thermodynamic product. In this regard, the use of multiple components in the assembly process not only allows the formation of heterometallic or heteroleptic cages, but also leads to the increase of complexity, novel geometries, and directional asymmetry in their structures. Nevertheless, the multiple component strategy does not always lead to directional asymmetry in the resulting cage. Recently, Omoto et al. reported the formation of a heteroleptic bipolar MOC, [Cu12(ipR)6(L)6], via a self-sorting strategy using two ligands with different bending angles (ipR = 5-substituted isophthalic acid, R = carbazole dendron functional group and L = 3,3′-(ethyne-1,2-diyl)dibenzoic acid).21 The ligands self-sorted into a uniform anisotropic cage thanks to the steric repulsion of the bulky dendritic macromolecular moiety on one of the ligands forcing it to arrange at the axial positions of the MOC core. However, this strategy cannot break the symmetry of the cage obtained due to the high dissimilarity between the backbone angles of the ligands used.
In this section, the different strategies developed for the controlled formation of structures with directional asymmetry are presented. This can be achieved either via (i) the rational design of complementary ligands and metal complexes (Fig. 2a) or (ii) the replacement of one of the constituents (metal or ligand) in a preformed cage (Fig. 2b).
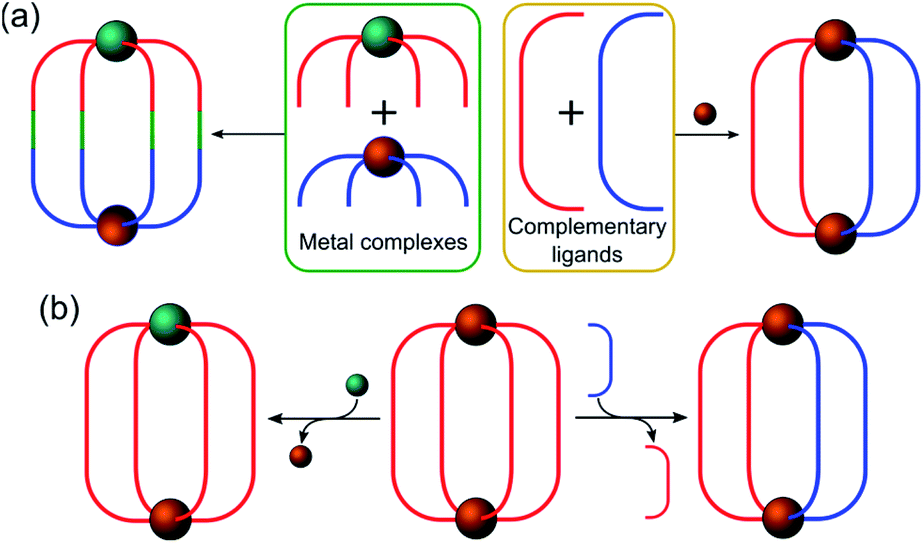 | ||
| Fig. 2 Schematic representation of the different approaches to control the positioning of cage constituents in order to obtain directional asymmetry at the nanoscale via (a) the rational design of metal complexes (left) and complementary ligands (right) or (b) the partial replacement of one of the metals (left) or the ligands (right). | ||
2.1. Heterometallic cages
Heterometallic cages can be synthesized using predesigned metalloligand complexes serving as building blocks, where the primary metal ion influences the directional bonding of the reactive sites in the organic part.22,23 Lisboa et al. reported the synthesis of the [PdPt(Lab)4]4+ cage by combining a platinum(II) tetrapyridylaldehyde complex ([Pt(La)4]2+) with an amino substituted pyridylamine derivative (Lb) and a palladium(II) metal precursor (Fig. 3a).24 The metal ions have the same coordination environment but the cage formation is concomitant with the imine bond formation (Lab) and coordination of the pyridinyl moieties to the Pd(II) ion. The molecular cage is considered asymmetric because of the different polarities created by the metal centers.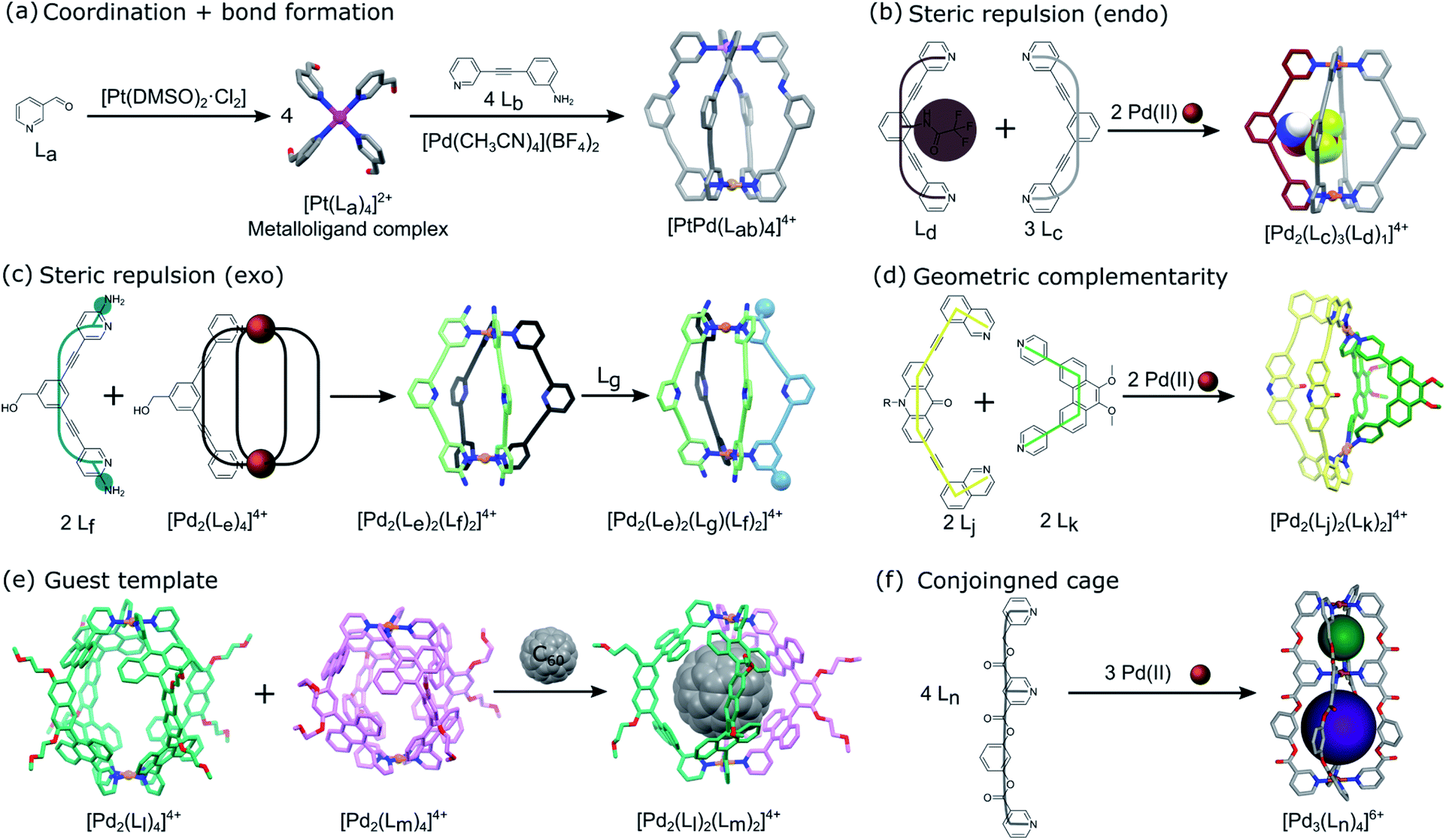 | ||
| Fig. 3 Schematic representation of the different techniques reported in the literature to impart directional asymmetry at the nanoscale in metal–organic cages using (a) similar coordination environments of metal complex and imine bond formations,24 (b and c) steric repulsion via endo25 and exo26 functionalized ligands, respectively, (d) geometric complementarity between ligands with different bend angles,28 (e) guest template induced reorganization of homoleptic cages,32 and (f) a conjoined cage based on tridentate ligands with asymmetric coordination sites in the backbone ligands.39 The asymmetric pore configuration is shown via spheres of different colours occupying the void spaces of the structure. | ||
2.2. Heteroleptic cages
For heteroleptic cages, self-assembly occurs through the self-sorting of the ligands thanks to intermolecular interactions (H-bonding or steric hindrance), the templating effect or rational design of complementary linkers. Different approaches have been developed to control the heterogeneous distribution of ligands to induce directional asymmetry in the cage architecture.The control of the position of substituted functionalities in a linker affects the resulting self-sorting and symmetry of the cage obtained. Johnson et al. substituted a 2,6-bis(pyridin-3-ylethynyl)benzene ligand (Lc) with a trifluoroacetamide functionality at the endo position (Ld). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [Pd2(Lc)4]4+ and [Pd2(Lc)3(Ld)1]4+ is obtained when the endohedral functionalized tripyridyl ligand is combined with the non-modified one and a palladium precursor (Fig. 3b).25 The selective formation of the heteroleptic cage is due to the steric constraints of the internal substituent. Indeed, molecular modelling shows that the bulky functional group occupies half the space inside disfavoring other outcomes of the self-sorting.
:1 mixture of [Pd2(Lc)4]4+ and [Pd2(Lc)3(Ld)1]4+ is obtained when the endohedral functionalized tripyridyl ligand is combined with the non-modified one and a palladium precursor (Fig. 3b).25 The selective formation of the heteroleptic cage is due to the steric constraints of the internal substituent. Indeed, molecular modelling shows that the bulky functional group occupies half the space inside disfavoring other outcomes of the self-sorting.
Similarly, Preston et al. performed ligand exchange on a homoleptic cage based on a tripyridyl ligand ([Pd2(Le)4]4+) to form the kinetically metastable mixed-ligand [Pd2(Le)2(Lf)2] cage.26 The addition of an electron-rich amino side-chain in the ortho position of the terminal ligating pyridyl units of the tripyridyl ligand (Lf) created steric repulsion (protection of the metal) that directed the formation of the heteroleptic cage. The asymmetric cis isomer formed was found to be more stable than the trans one according to density functional theory (DFT) calculation due to hydrogen-bonding interactions between the amino groups of Lf and acidic α-hydrogens of the adjacent Le ligands (Fig. 3c). Furthermore, adding a meta PEG-substituted ligand (Lg) to the previously formed cage led to a triple-ligand cage architecture [Pd2(Le)(Lg)(Lf)2]4+. NMR experiments provided strong evidence that the statistical mixture of heteroleptic cages formed adopted the cis geometry. More recently, Zhu et al. combined endo and exo steric repulsion to directly form an asymmetric heteroleptic [Pd2(Lh)2(Li)2]4+ cage.27 Combining tripyridyl ligands with the ortho methyl group of the terminal ligating pyridyl units pointing either inside (Lh) or outside (Li) led to a preferred cis 2in/2out configuration of the ligands in the cage as observed by ESI-MS, 2D NMR and DFT calculation.
Instead of using steric repulsion to control the outcome in a metal–organic cage made up of different ligands, Bloch et al. used geometric complementarity based on the directional bonding approach to build an asymmetric heteroleptic coordination cage (Fig. 3d).28 The careful design of symmetric ditopic N-donor linkers with different backbone angles but mutually compatible geometries (Lj = 10-hexyl-2,7-bis(isoquinolin-8-ylethynyl)acridin-9(10H)-one and Lk = 9,10-dimethoxy-3,6-bis(4-pyridyl)phenanthrene) leads to the formation of the most energetically favorable cis-[Pd2(Lj)2(Lk)2]4+ due to the complementary arrangement of the linkers with respect to the Pd(II) coordination sphere. The same strategy was used with three or four complementary ligands providing a mixture of asymmetric cages.29 Sun et al. previously reported the selective formation of a [Pd12(DPB)12(BPPEB)12]24+ (DPB = 1,3-di(pyridine-4-yl)benzene and BPPEB = 1,3-bis((4-(pyridine-4-yl)phenyl)ethynyl)benzene) 3/4 cantellated cuboctahedron and its pseudoisomer.30 The use of two geometrically complementary ligands, with similar shapes but different lengths, led to intramolecular self-sorting that formed a well-defined but mixed asymmetric cage. Similarly, Li et al. used a ligand exchange strategy with the same cuboctahedral MOP as Fujita but based on Cu2 paddlewheel metal nodes and bidentate dicarboxylate ligands ([Cu24(t-Bu-bdc)24] with t-Bu-bdc = 5-t-butyl-1,3-benzenedicarboxylic acid).31 The partial ligand exchange with 2,7-naphthalenedicarboxylic acid (2,7-ndc) led to the formation of a directionally asymmetric cage with 3/4 cantellated cuboctahedral topology, [Cu24(t-Bu-bdc)12(2,7-ndc)12].
The use of a guest molecule as a template is another strategy that allows the formation of an otherwise non-accessible multicomponent cage system. Yamashina et al. reported that the mixing of homoleptic cages ([Pd2(Ll)4]4+ and [Pd2(Lm)4]4+), bearing similar bispyridine derivative ligands but having different lengths, with a C60 template leads to the selective formation of the [Pd2(Ll)2(Lm)2]4+ heteroleptic cage instead of a complex mixture.32 Indeed, the guest molecule induced reorganization of the initial cages into a more stable thermodynamic asymmetric product due to efficient host–guest aromatic–aromatic interactions (Fig. 3e). The asymmetry of the cage arises from the favored cis-isomer geometry giving a Janus-like structure to the cage. Previously, Hiraoka et al. reported the direct assembly of a Pd(II)-based cage by combining a Pd(II) complex with two different sized tridentate pyridine derivative ligands, P (PL larger than PS, PL = 1,3,5-tris(pyridin-4-ylmethyl)benzene and PS = 4,4′-((5-(pyridin-4-yl)-1,3-phenylene)bis(methylene))dipyridine).33 The use of large (spherical) or small (flat) guests selectively stabilized the [Pd3(PL)2]6+ or [Pd3(PS)2]6+ homoleptic cages, respectively, while medium-sized guest preferentially formed the [Pd3(PL)(PS) ]6+ heteroleptic cage.
Lal et al. used a different strategy based on mixed-linkers with different properties (hydrophobic as opposed to hydrophilic) to obtain an asymmetric cuboctahedral MOP, [Cu24(OH-bdc)12(C8-bdc)12] (OH-bdc = 5-hydroxy-1,3-benzenedicarboxylic acid and C8-bdc = 5-octyloxy-1,3-benzenedicarboxylic acid).34 Indeed, the use of equimolar isophthalic acid ligands bearing polar (OH) and non-polar (alkene chain) groups at the 5-position can form MOPs with both ligands evenly and symmetrically distributed over the polyhedron surface or segregated to different faces to give a Janus-like MOP. Only the latter shows solvent-dependent self-assembly behavior while the homogeneous distribution of functional groups over the MOP core prevents the former from aggregating. Nonetheless, this strategy offers limited control over the position of the functional moiety and is solely restricted to the formation of mixed-phase products.
On the other hand, porous organic cages (POCs) are inherently heteroleptic assemblies prepared from dynamic covalent bond formation between at least two different organic ligands leading to a large variety of supramolecular cage structures.35,36 Even though chiral POCs were reported,37,38 to the best of our knowledge, no directional asymmetric POCs have been described so far. This may be due to the difficulty in controlling the ligands' respective positions within the structure as one of the linkers will serve as the vertex while the other as edges.
2.3. Conjoined cage
In the previous examples, asymmetry arises from the ligands or metal distribution in the multicomponent cage architectures. In contrast, Samantray et al. designed a tridentate ligand (Ln = 5-((pyridin-3-ylmethoxy)carbonyl)nicotinic acid) with coordination sites placed non-symmetrically in the linker backbone.39 This allows the construction of a discrete molecular cage with multiple 3D cavities where the directional asymmetry originates from the difference in the resulting cavity sizes. The combination of a Pd(II) precursor with the tridentate ligand leads to the formation of [Pd3(Ln)4]6+ and [Pd6(Ln)8]12+ homoleptic conjoined-cages with only the former showing asymmetry (Fig. 3f). When di (Lo = pyridin-3-ylmethyl nicotinate) and tridentate (Ln) ligand mixtures are used, self-sorting occurs leading to a heteroleptic [Pd4(Ln)4(Lo)2]8+ cage with asymmetric cavity distribution.3. Asymmetry at the mesoscale
At the mesoscale, porous materials (MOFs, COFs, MOCs, and POCs) exist in the form of crystals. Therefore, the appearance of directional asymmetry could be derived from the asymmetric arrangement of multiple components inside individual crystals via the controlled positioning of multiple linkers or metal nodes and the generation of defects.12,13,16–19 This can be achieved via the introduction of spatial heterogeneity or gradient, which is the gradual change of structures or properties along one specific direction of the crystals. However, these strategies remain challenging due to the isotropic nature of crystal growth and the difficulty of characterizing such systems (Fig. 4 left).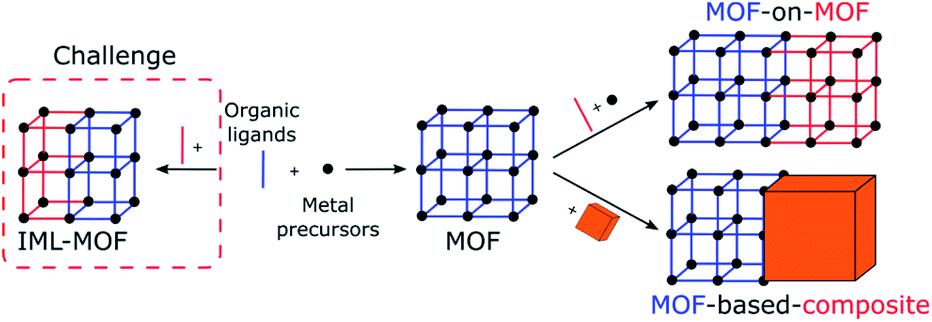 | ||
| Fig. 4 Schematic representation of the strategies leading to directional asymmetry at the mesoscale which can be achieved (left) through controlling the position of the building blocks within the crystal structure leading to an isostructural mixed ligand (IML) MOF or (right) through the integration of a MOF with other materials or MOFs. | ||
The versatility of MOF synthesis in developing MOF composites offers an alternative way to create directional asymmetry. At this scale, this could be achieved via the integration of a MOF with other materials or another MOF with noticeably different properties (Fig. 4 right). In such a case, directional asymmetric systems will be achieved through the control over the spatial distribution of MOFs relative to their counterparts. For instance, in the simplest binary system, two different arrangements are known: centrosymmetric core–shell and non-centrosymmetric Janus materials.19 Therefore, this becomes an ideal method to break symmetry by forming Janus-like MOF composites at the mesoscopic level. In this section, two main types of MOF composites are classified: MOFs integrated with other MOFs (MOF-on-MOF) and MOFs connected with another material (MOF-based composites). Note that many other materials have been reported to be integrated with MOFs but without showing directional asymmetry.40–43
3.1. MOF-on-MOF
Instead of random copolymerization of multiple linkers to form MOFs with mixed composition, one MOF can be sequentially grown on the surface of another MOF. In 2009, Furukawa et al. first demonstrated this MOF-on-MOF concept and succeeded in the fabrication of hybridized MOF heterocrystals.44 In this work, two similar tetragonal porous frameworks built from dicarboxylate layer ligands and N-donor pillar ligands, [M2(dicarboxylate)2(pillar)] (M = metal ions), were selected based on the criterion of lattice matching that promises pore connection at the interface between crystals. The epitaxial growth of a secondary MOF, [Cu2(1,4-ndc)2(dabco)] (1,4-ndc = 1,4-naphthalene dicarboxylate and dabco = diazabicyclo[2.2.2]octane), could be directed on the faces of the core MOF crystals [Zn2(1,4-ndc)2(dabco)] under careful selection of linkers and isostructural MOF systems with similar unit cell parameters. However, this strategy led to a core–shell structure without directional asymmetry. Based on this consideration, the same group further developed their strategy to realize face-selective epitaxial growth by targeting the {001} surfaces that possess square lattices based only on layer ligands. Thus, the secondary growth no longer requires lattice matching along the 〈001〉 direction, allowing different pillar ligands to be used. This leads to an anisotropic block MOF-on-MOF crystal with distinct pore surface functionalities, as shown in Fig. 5a, where [Zn2(1,4-ndc)2(dabco)] was grown on the core crystal of [Zn2(1,4-ndc)2(dpndi)] (dpndi = N,N′-di(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide).45 Though this MOF hybrid crystal does not have a directed asymmetry configuration, one can generate it simply by cutting the core crystal in the middle. In 2016, a similar strategy was used by Choi et al. to demonstrate the anisotropic growth of a MIL-68 analogue ([In(OH)(1,4-ndc)], MOF-NDC) only on the six rectangular facets (ac plane) of the MIL-68 template, [In(OH)(bdc)] (bdc = 1,4-benzenedicarboxylic acid), due to well-matched lattices in the direction of the a and c axes. However, due to the lattice mismatch in the direction of the b axis, the MOF-NDC cannot grow on the two hexagonal facets (ab plane) thus leading to the formation of semi-tubular particles (Fig. 5a bottom right).46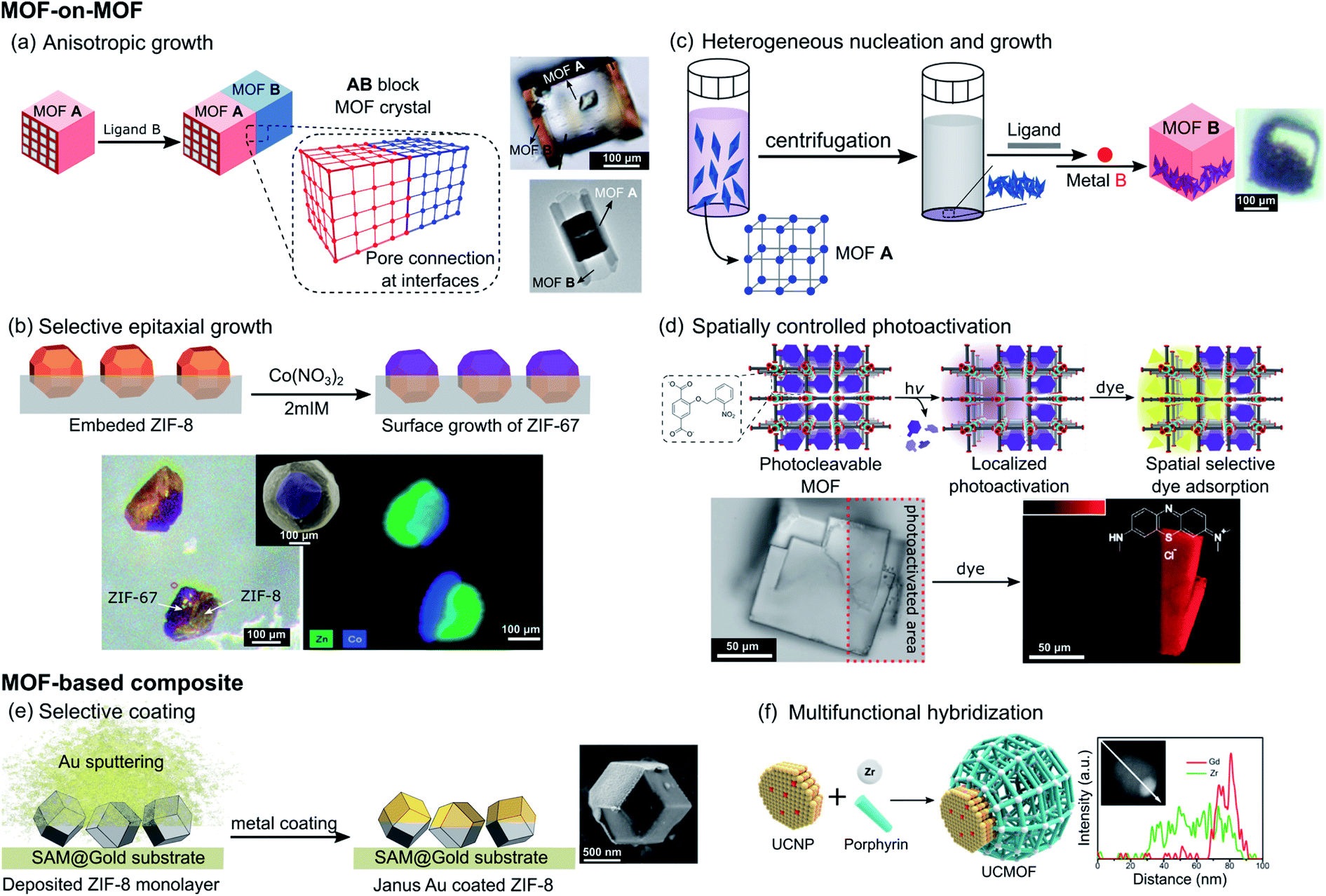 | ||
| Fig. 5 Schematic representation of the different strategies reported in the literature to impart directional asymmetry at the mesoscale in MOF crystals using (a) anisotropic growth of MOF A onto MOF B through lattice matching with the corresponding SEM images of the crystals obtained via face selective epitaxial growth (top right, adapted from ref. 45 with permission from the Royal Society of Chemistry) and semi-tubular crystals via lattice mismatch strategies (bottom right, adapted with permission from ref. 46, Copyright 2016, American Chemical Society), (b) selective epitaxial coating of ZIF-8 with ZIF-67 by surface blockage and the corresponding SEM images and elemental analysis of the isolated Janus particles (adapted from ref. 48 with permission from the Royal Society of Chemistry), (c) synthesis-directed control of the apportionment of core MOFs inside the shell MOFs thanks to preliminary centrifugation of the core crystals and the corresponding SEM image (adapted with permission from ref. 49, Copyright 2018, American Chemical Society), (d) spatially controlled photoactivation of MOF pores with the SEM images showing the spatially controlled adsorption of the fluorescent guest (adapted from ref. 51 with permission from the Royal Society of Chemistry), (e) gold selectively coated on ZIF-8 particles immobilized on a surface-assembled monolayer on the gold surfaces (SAM@Gold) with the corresponding SEM image of the Janus particle obtained (adapted from ref. 53 with permission from the Royal Society of Chemistry), and (f) anisotropic growth of MOFs on a preformed metal nanoparticle with the corresponding TEM image and elemental analysis along the composite particle (adapted with permission from ref. 57, Copyright 2017, American Chemical Society). | ||
When any exposed face of the initially synthesized crystals does not share crystallographic parameters with the newly added reactants, selective and partial coating of MOFs is an alternative strategy for the creation of directional asymmetry. In 2013, Szilágyi et al. reported the formation of Janus particles MOF-5@IRMOF-2 by the static route, where the IRMOF-2 shell of [Zn4O(Br-bdc)3] (Br-bdc = 2-bromo-1,4-benzenedicarboxylate) was directly grown onto the surface of a freshly prepared MOF-5 core crystal, [Zn4O(bdc)3], without any disturbance.47 In this case the shell MOF partially coated the core crystals since the latter formed on the vial wall, which prevented the complete formation of the core–shell structure. For more controlled surface blockage of MOFs, Tan et al. reported the synthesis of a bimetallic Zn/Co Janus MOF by selective epitaxial growth of ZIF-67, [Co(MeIm)2] (MeIm = 2-methylimidazolate), on the surface of ZIF-8 ([Zn(MeIm)2]) particles, with the latter partially embedded in a poly(methyl methacrylate) (PMMA) film. The polymer film was removed using ethyl acetate after secondary growth to isolate the ZIF-67/ZIF-8 Janus particles (Fig. 5b).48 Apart from the synthesis of Janus MOF-on-MOF crystals, this strategy is also applicable in the formation of Janus composites integrating MOFs with other materials, which will be discussed later.
Another effective approach to control the apportionment of MOF components in a composite system can be implemented through the tuning of the synthetic conditions in order to decrease the energy barrier associated with heterogeneous nucleation. Under the guidance of stability consideration and surface functionalization, Feng et al. recently demonstrated the controlled formation of hierarchical MOF-on-MOF composites, in which the spatial arrangement of different MOFs can be tuned, to obtain well-mixed, core–shell, or half–half asymmetric Janus distributions.49 PCN-222, [Zr6O8(TCPP)2] (TCPP = tetrakis(4-carboxyphenyl)porphyrin), was first synthesized and the surface of the core nanocrystals was functionalized with an excess of bdc linkers. Then addition of Zn(NO3)2 metal precursors led to the formation of the PCN-222@MOF-5 composite. The increase of the concentration of MOF-5 precursors added to the solution of the core MOF changed the mutual positions of the MOF-on-MOF composite from well-mixed to core–shell distributions. In contrast, when centrifugation was used to precipitate the core PCN-222 at the bottom of the reaction vessels, followed by the addition of MOF-5 precursors, the heterogeneous nucleation and growth of MOF-5 gave Janus-like crystals, where PCN-222 crystals are incorporated within one side of the PCN-222@MOF-5 composite (Fig. 5c). In addition, the same group modified their strategy to form Janus-like composites of MOFs with COFs, where the latter served as a core scaffold.50
Instead of controlling the growth of MOFs in an anisotropic way, post-synthetic modification can be implemented to locally change the MOF compositions. For instance, Stassen et al. reported the spatial control of the pore accessibility of isostructural MOF-5 single crystals containing linkers with photo-cleavable and pore-blocking o-nitrobenzyl pendant groups, [Zn4O(bdc-NB)3] (bdc-NB = 2-((2-nitrobenzyl)oxy)-1,4-benzenedicarboxylic acid).51 The spatially controlled photo-irradiation led to the localized and selective removal of o-nitrobenzyl moieties, which makes the MOF porosity accessible for fluorescent guests (Fig. 5d). This strategy allows introduction of complexity and asymmetry in the pore size distribution within the MOF single crystal, in which the distribution of MOFs with the photo-cleaved large pore can be tuned within a single crystal of the parent MOF with the pendant group.
3.2. MOF-based composites
It is widely accepted that the hybridization of different materials is a strategy to improve the properties and functionalities of materials and plenty of MOF composites have been studied.52 Based on this strategy, many asymmetric materials were created at the macroscale, which will be discussed in the next section, whereas only a few examples have been reported at the mesoscale.In 2016, Ayala et al. reported the formation of Janus MOF particles by the partial coating of colloidal MOF particles with metal vapor.53 In this work, the pre-synthesized single ZIF-8 crystals were immobilized on a self-assembled monolayer on Au surfaces, followed by coating with a metal using an electron-beam evaporator. Because only the top (exposed) surfaces of these crystals were coated with the metal layer, Janus particles were successfully generated after detaching the composite from the planar surfaces through ultrasonication in MeOH (Fig. 5e). Different metals (Au, Co or Pt) were used to coat different MOFs (UiO-66 [Zr6(OH)4O4(bdc)6] or UiO-66-SH [Zr6(OH)4O4(bdc-SH)6] with bdc-SH = 2,5-dimercapto-1,4-benzenedicarboxylic acid), indicating the versatility of this method.
Instead of partial coating on preformed MOF crystals, the anisotropic growth of MOFs on preformed templates is a similar approach for the formation of Janus-like composite materials. For example, HKUST-1, [Cu3(btc)2] (btc = 1,3,5-tricarboxylic acid), has been selectively grown on Cu beads54 and polymer spheres55via electrodeposition and layer-by-layer crystal growth, respectively. In the former case, indirect bipolar electrodeposition was applied to trigger the accumulation of metal ions upon polarization on one specific surface of the Cu beads, which can be subsequently reacted with btc ligands in solution to selectively grow HKUST-1 and to yield an asymmetric composite. For the latter case, Janus-like polymer spheres were first synthesized by the electrohydrodynamic co-jetting process from solutions of poly(n-butyl methacrylate-co-cinnamoylethyl methacrylate) (P(nBMA-co-CEMA)) and poly(t-butyl methacrylate-co-cinnamoylethyl methacrylate) (P(tBMA-co-CEMA). This process allows the compartmentalization of the two polymers in opposite hemispheres. Once crosslinked via the photoreaction of the cinnamoyl group to maintain the spherical shape of polymer particles, the tBMA groups were selectively deprotected with trifluoroacetic acid (TFA) for further spatially controlled growth (layer-by-layer) of HKUST-1 in one of the hemispheres.
Zhang et al. reported the synthesis of multifunctional hybrid materials based on the asymmetric combination of MOFs (ZIF-8), Pd nanosheets (PdNS) and cyclodextrin (CD).56 First, polyacrylic acid (PAA) was preferentially coated on one side of the hexagonal PdNS thanks to the difference in the interfacial energy. Then, ZIF-8 was selectively grown only on the surface of the PAA side, while the other exposed side was modified with mono(6-mercapto-6-deoxy)-beta-CD, leading to the formation of three component Janus nanoparticles.
Li et al. succeeded in the preparation of asymmetric compounds composed of porphyrinic MOFs and lanthanide-doped upconversion nanoparticles (UCNPs).57 The UCNP is composed of a NaGdF4 core layer doped with Yb3+ sensitizer and Er3+ activator ions. The seed cores were then reacted with a solution containing the precursors of the shell to produce, by epitaxial growth, the NaGdF4:Yb,Er@NaGdF4 composite. The core–shell UCNP nanoplates were further coated with polyvinylpyrrolidone (PVP). The DFT calculation suggested that PVP preferentially binds to the {001} facet of the UCNPs. The hydrophobic polymer layer facilitated the preferential anisotropic growth of the Zr-based porphyrinic MOFs (PCN-222) on one side of the nanoplates. Indeed, the carbonyl pendant group from the pyrrolidone repeating unit can bind the Zr metal ions at the UCNP surface via coordination interactions, followed by heterogeneous nucleation and MOF growth leading to the formation of Janus UCNP-MOF particles in a controllable fashion (Fig. 5f).
Most of the work on COF-based composites has so far led to the synthesis of core–shell COF composites, in which COF crystals were grown as the shell on the surface of MOF crystals and metal-oxide nanoparticles but without displaying any directional asymmetry in their structures.58–61 Additional strategies should be proposed to obtain a directional asymmetric system based on COF composites by taking inspiration from the work already reported by Feng et al.49,50
4. Asymmetry at the macroscale
In this section we show how asymmetric systems at the macroscale can be generated by controlling the spatial location of mesoscopic porous materials within macroscopic objects. Such materials are typically constructed by combining two or more different materials with significantly different physical/chemical properties to form composites. Note here that asymmetric composite materials comprise systems, in which at least one material is arranged spatially to give vertical/lateral heterogeneity or programmed gradient patterns, and at least one material is a continuous phase. Thus, systems built-up from distinct domains of materials with different properties, joined at an interface, such as supported MOF films,62–64 are not included in this category.Only a few exceptional examples of macroscopic single component asymmetric systems have been reported,65–67 whereas the number of studies dealing with the generation of asymmetric composite materials is much larger as we will discuss later. This difference can be attributed to the fact that in single component systems, each part displays identical physicochemical properties, leading to a homogeneous distribution at the macroscale. In contrast, the use of different components displaying at least one different physicochemical property (e.g. solubility, density, and reactivity) favors the generation of gradients. In general, macroscale asymmetric porous materials are found in the form of composite films, in which particles of porous materials act as fillers, distributed asymmetrically in a polymeric continuous matrix.
In this section, we present relevant and illustrative examples of methods that have been developed to achieve directional asymmetric porous materials at the macroscale. The reported strategies for constructing such asymmetric systems can be divided into two categories: one-step methods, in which the appearance of asymmetry is concomitant with the formation of the macroscopic object, and multi-step methods, in which the porous asymmetric structures are prepared sequentially (Fig. 6).
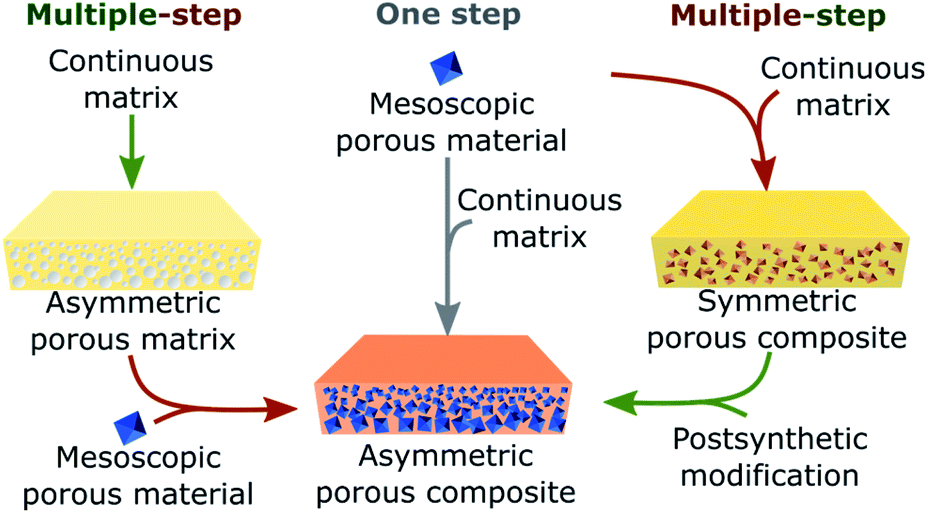 | ||
| Fig. 6 Schematic representation showing the different strategies to obtain asymmetric porous composites at the macroscale. These materials can be prepared in one step (middle) by mixing preformed mesoscopic porous materials with a continuous matrix or via a multiple-step approach with (left) first preparing an asymmetric porous matrix in which the porous material is then incorporated or (right) through the initial formation of a symmetric porous composite formed by mixing the continuous matrix and porous materials which are then post modified. | ||
4.1. One-step methods
One of the most common strategies used for fabricating porous membranes with directional asymmetry is the phase inversion method. Although this strategy was typically developed for the fabrication of polymeric asymmetric membranes, several examples of asymmetric MOF/polymer membranes have been reported during the last decade. In this method, the MOF filler particles are initially dispersed in a polymer solution, which is cast onto a support and then immersed in a solvent, causing the precipitation of the polymer. Factors such as choice of polymer and solvents, temperature or time, among others, could have a major effect upon the membrane structure.68 In 2010, Basu et al. prepared Matrimid®/Cu3(btc)2 and Matrimid®/PSf/Cu3(btc)2 membranes (Matrimid® = polyimide, PSf = polysulfone) with variable MOF content, in which MOF crystals are embedded and well distributed in the polymer matrix. The directional asymmetric structure arises from the difference in the pore sizes between the dense layer and the layer with macro voids.69 Similarly, by following this method, Zhu et al. fabricated directional asymmetric membranes by embedding a post-functionalized MIL-53(Al), [Al(OH)(bdc)], into a polyetherimide Ultem® 1000 polymer matrix.70 In this case, a post-modification of MIL-53(Al) with aminosilane was implemented to improve the particle-polymer interfacial interaction. Although the phase inversion approach has been mostly applied for the synthesis of asymmetric composite membranes with MOFs, this method was also explored for the production of functionally graded COF-based composites. Recently, Yang et al. reported the synthesis of an asymmetric COF/polymer membrane via phase inversion using a TpHZ COF and poly(ether sulfone) (PES) polymer (TpHZ = co-condensation of 1,3,5-triformylphloroglucinol (Tp) and hydrazine hydrate (HZ)).71 TpHZ nanosheets with a lamellar morphology were first mixed with PES in a DMF/THF solvent mixture and cast on a glass plate (Fig. 7a). Next, the solvent was allowed to evaporate for a short period of time (15 s) and then the glass plate was immersed into a coagulation bath. The resulting membrane showed a graded distribution of COF nanosheets, with a thin uniform COF-rich layer and a thick porous bottom layer. The generation of this asymmetric arrangement is attributed to the presence of a denser upper layer of PES, favored by the volatile THF evaporation prior to immersion, which reduces the migration capacity of COF nanosheets and entraps them. Remarkably, this methodology also allows tuning of the gradient distribution of COFs by changing the COF/polymer ratio and phase inversion temperature. Another effective strategy to induce asymmetry in materials is interfacial synthesis, also called the interfacial polymerization method. The basic principle relies on the confinement of the reaction at the interface between two immiscible phases, which is governed by the reactant diffusion toward the interface. Thus, this method can be conveniently exploited for the construction of macroscopic asymmetric systems, if a unidirectional diffusion gradient of reactive species is attained. The feasibility of this strategy was demonstrated by Lu et al. by synthesizing a free-standing MOF-based membrane with different composition and morphology in the top and bottom layer.65 These MOF membranes were synthesized through a liquid–liquid interfacial coordination mechanism by layering a hexane solution containing triethylamine (TEA) over a DMF solution of MOF precursors (zinc nitrate and terephthalic acid). The resulting asymmetric membrane is composed of a top layer of small MOF-5 particles and a bottom layer of larger MOF-2 crystals with sheet-like morphology (Fig. 7b). This asymmetric distribution is a consequence of the difference in the precursor and TEA diffusion rate across the interface formed by the two immiscible solvents during the synthesis, which generates a vertical pH gradient. In the higher pH region (upper part) the formation of octahedral Zn4O(CO2)6 clusters is favored and consequently 3D MOF-5 is obtained. In contrast, in the lower pH region (bottom part) the construction of dinuclear Zn2(CO2)4 paddlewheels is promoted, resulting in the formation of 2D MOF-2. Recently, Wang et al. demonstrated that interfacial synthesis can be directly performed on polymeric substrates for the synthesis of COF-based asymmetric composite membranes.72 In order to achieve this, a p-phenylenediamine (Pa) aqueous solution and 1,3,5-triformylphloroglucinol (Tp) in an n-hexane solution were separately charged into a diffusion cell, in which a polyvinylidene fluoride (PVDF) membrane was placed between both liquid phases. Under these conditions, only Pa molecules can diffuse across the PVDF membrane and, consequently, the formation of the TpPa COF is limited to one side of the PVDF substrate.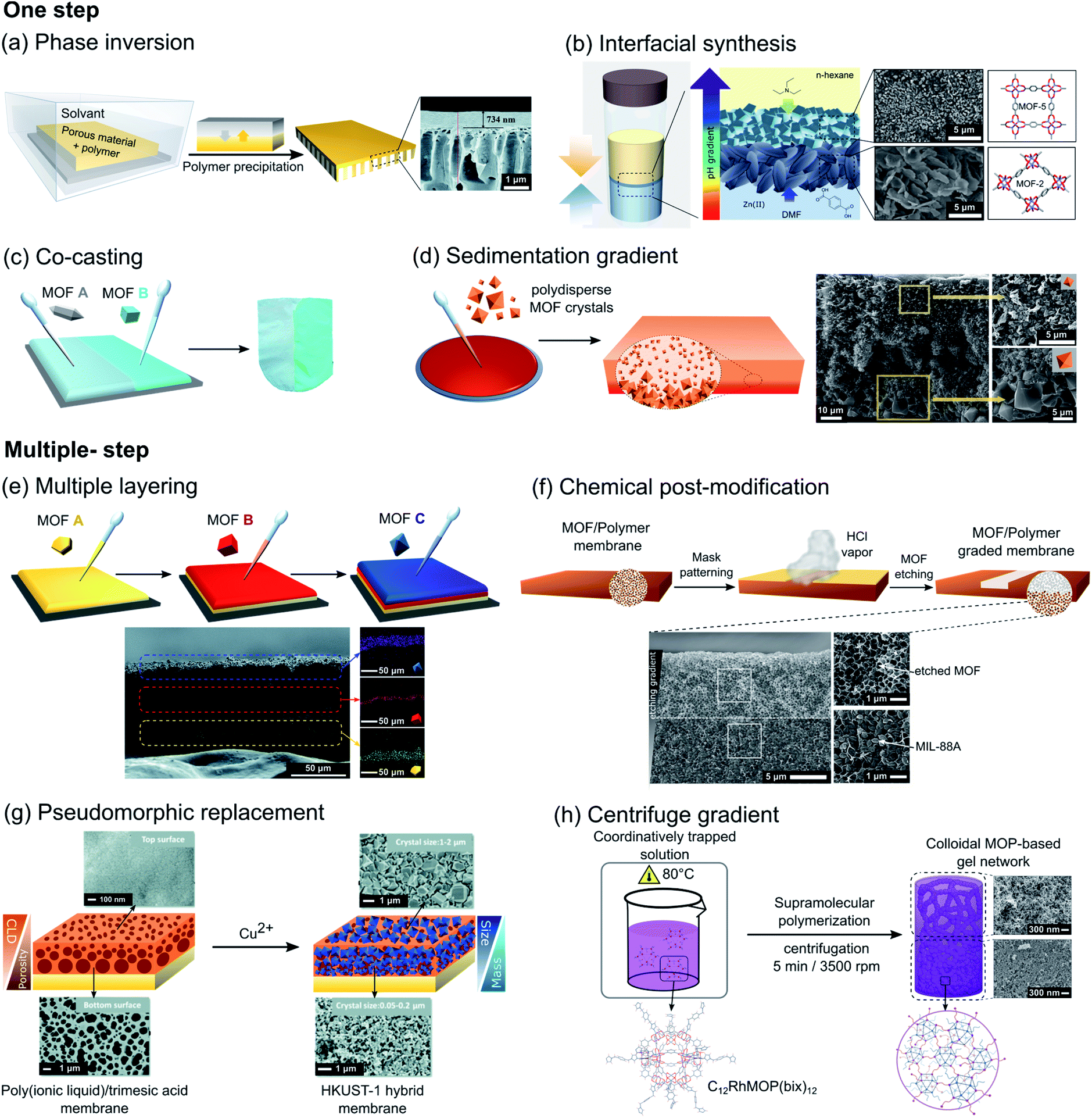 | ||
| Fig. 7 Overall schematic representation of different reported strategies for the fabrication of asymmetric systems at the macroscale divided into one-step (a–d) and multiple-step (e–h) methods. (a) Schematic diagram of the phase inversion method used for the fabrication of asymmetric PES/TpHZ-8 membranes. Adapted from ref. 71 with permission from the Royal Society of Chemistry. (b) Schematic of the liquid–liquid interfacial coordination mechanism used for the fabrication of free-standing asymmetric MOF-2/MOF-5 membranes. Adapted with permission from ref. 65, Copyright 2013, Wiley. (c) Schematic representation of the co-casting method using different MOF inks to form asymmetric MOF@PVDF membranes with different discrete regions. Adapted from ref. 73 with permission from the Royal Society of Chemistry. (d) Schematic of the formation of asymmetric MIL-88A@PVDF membranes showing the distribution of bigger (bottom) and smaller (top) MOF crystals across the membrane. Adapted with permission from ref. 74, Copyright 2018, Wiley. (e) Schematic of the repeated casting process with subsequent MOF layers to form layered MOF@PVDF layers with different MOF species. Adapted from ref. 73 with permission from the Royal Society of Chemistry. (f) Schematic representation of the patterning of MIL-88A@PVDF films by chemical etching with HCl to produce asymmetric graded membranes. Adapted with permission from ref. 78, Copyright 2019, Wiley. (g) Schematic of the pseudomorphic replacement approach for the preparation of an asymmetric HKUST-1 membrane from a porous PIL membrane template. Adapted from ref. 79 with permission from the Royal Society of Chemistry. (h) Creation of a graded C12RhMOP-bix gel network by applying centrifugal force at the specific moment of colloidal aggregation, yielding a less dense network of colloidal particles at the top compared to the gel formed at the bottom. Adapted from ref. 66 with permission from the Royal Society of Chemistry. | ||
In the last few years, some straightforward drop-casting methods have also been investigated. These strategies, while simple in concept, allow easy generation of asymmetric macroscopic systems with a broad compositional versatility. These methods have been used for the fabrication of composite membranes with asymmetric distribution of MOF crystals. For instance, Denny et al. reported a co-casting strategy for preparing composite materials by casting MOF/PVDF mixtures of different MOFs into discrete regions, allowing them to come into contact with each other to yield a monolithic film after solvent evaporation (Fig. 7c).73 Although this method can involve some mixing of the species at the interface, the degree of mixing of the MOF types at the interface is very low due to the high viscosity of MOF/polymer mixtures, which favors better control over the spatial distribution of the different components. Another example of an asymmetric MOF-based composite film prepared by drop-casting was recently reported by Troyano et al.74 In this work, a MIL-88A/PVDF composite film (MIL-88A = [Fe3OH(DMF)2(fumarate)3]) is synthesized by following a conventional drop-casting method. But in this case, the use of MOF crystals with a broad size distribution (ranging from 0.5 μm to 9.0 μm) is responsible of the directional asymmetry created. During solvent evaporation, the inhomogeneity in the size of the crystals generates a vertical gradient of MOF crystal distribution across the thickness, in which the smaller crystals are homogeneously distributed across the polymer matrix, but the larger crystals accumulate at the bottom (Fig. 7d).
A different method to achieve a gradient distribution of MOFs inside a composite membrane, via gravity-assisted solvent evaporation, was recently proposed by Peng et al.75 In their work, a commercial Celgard membrane (polypropylene, PP) was immersed into a Cu(NO3)2 and H3btc solution in DMF at 100 °C for a certain period of time to allow the diffusion of the precursors into the membrane. Then, the membrane was transferred to a glass plate and kept at 80 °C for solvent evaporation and growth of HKUST-1 seeds. In this step, the solution in the membrane tends to move down due to gravity, and consequently MOF seeds concentrate at the bottom between the membrane and the glass plate. Then, secondary growth of HKUST-1 crystals was performed by immersing the membrane again in a fresh precursor solution at 100 °C.
4.2. Multi-step methods
The fabrication of microporous asymmetric structures at the macroscale can be also achieved sequentially. For example, by repeating a drop casting process, asymmetric MOF/polymer membranes with a vertical distribution of different MOFs can be obtained. This approach, first reported by Peterson et al., formed the first MOF/polymer layer by casting and, in the second step, an additional layer by drawing a MOF/polymer casting mixture across the first layer.76 Under these conditions, the solvent partially dissolves the polymer of the bottom layer and fuses the layers together, resulting in a monolithic multi-layered system that cannot be mechanically separated. By following this methodology repeatedly, the authors created multi-layered MOF/polymer membranes with a different set of MOF fillers and polymeric matrices. More recently, a straightforward but effective modification of this strategy was reported by Denny et al.73 This approach is based on the reinforcement of the PVDF polymer matrix via diamine crosslinking (Fig. 7e). Thus, after the formation of the first MOF/PVDF layer, the second layer was cast on top without destruction of the underlying layer, creating a vertical distribution of MOFs within the same PVDF polymer matrix. A different strategy for the fabrication of multi-layered COF membranes was recently reported by Fan et al.67 In this work, on a previously functionalized surface, aldehyde and complementary diamine/hydrazine were mixed and heated sequentially at two different temperatures. Under these conditions, the first COF layer was formed on the surface via aldehyde–imine condensation during the first heating step, followed by the synthesis of the second COF layer via aldehyde–hydrazine condensation at higher temperature. This temperature-swing approach resulted in a layered-stacking structure without boundary defects between the two COF layers.A different strategy was demonstrated to convert a pre-existing symmetric system into an asymmetric one by spatially locating a chemical transformation. In particular, in the case of MOF/polymer membranes, chemical transformations selectively affect the polymeric matrix or the filler particles, leading to different asymmetric conformations of the resulting composite film. For instance, Wijenayake et al. reported the controlled cross-linking of the polymer matrix of a previously formed homogeneous MOF-based composite membrane.77 This membrane, composed of a 6FDA-durene polyimide matrix with ZIF-8 crystals as the filler, was further modified via surface cross-linking with ethylenediamine (EDA) vapor. In this case, the diffusion of EDA vapor into the membrane causes a gradual crosslinking of the polymer matrix from the surface towards the center. Remarkably, this methodology not only allows creation of vertical gradients of the cross-linked polymer, but also provides control of the thickness of the cross-linked layer by changing the exposure time of the homogeneous films to EDA vapor. On the other hand, the creation of an asymmetric MOF/polymer membrane from a symmetric one via controlled chemical transformation can also be achieved by post-synthetically modifying the embedded MOF particles. This method was recently reported by Troyano et al.78 Homogeneous MIL-88A/PVDF membranes were transformed into asymmetric systems by selectively etching the MOF crystals with HCl vapor. The excellent chemical resistance of the PVDF matrix allows the selective etching of more reactive MIL-88A MOF fillers (Fig. 7f). Thus, when one side of a homogeneous MIL-88A@PVDF film was exposed to HCl gas, it diffuses into the polymer film generating a vertical gradient by etching the MOF crystals. As shown in the previous example, this gradient can be tuned by controlling the exposure time. Remarkably, the authors extended this method for the construction of more complex structures by using a predesigned mask, allowing the control of the vertical and horizontal distribution of the MOF crystals within the polymeric film.
Another effective sequential approach for the generation of microporous asymmetric systems is based on the incorporation of microporous materials into a previously formed asymmetric structure. This strategy was demonstrated by Sun et al. who reported the fabrication of an asymmetric MOF/polymer membrane through coordination-driven pseudomorphic replacement.79 In the first step, a membrane was formed by casting a mixture of poly (ionic liquid) and H3btc on a glass substrate. Then, this film was immersed in an aqueous NH3 solution to deprotonate H3btc, which generated an ionic complex with the cationic chains of the polymer. As a consequence of the gradual diffusion of NH3 into the polymer, the resulting membrane presented a crosslinking gradient: as more ammonia penetrated into the polymer, the higher concentration of btc3− resulted in a higher degree of electrostatic crosslinking and consequently a denser layer. Once the asymmetric membrane was formed, its structure was replicated by immersion into a Cu(NO3)2 solution, resulting in the formation of HKUST-1 crystals with an asymmetric distribution (Fig. 7g).
More recently, Legrand et al. reported an example of macroscopic asymmetric MOP-based materials, showing the potential of MOPs as building blocks for constructing complex porous hierarchical architectures. In this case, the intrinsic porosity and the excellent stability of robust Rh-MOPs are exploited to form an asymmetric macroscopic gel with gradients of stiffness and porosity across its structure (Fig. 7h).66 In this case, pre-synthesized MOP molecules C12RhMOP, [Rh2(C12-bdc)2]12 (bdc-C12 = 5-dodecoxybenzene-1,3-dicarboxylate) are assembled with a ditopic linker, 1,4-bis(imidazole-1-ylmethyl)benzene (bix), by coordination-driven supramolecular polymerization.80 This process involves several stages, in which after initial formation of nuclei from the linking of a few MOPs, they are further fused into colloidal particles. Finally, these colloids percolate to give a gel network. Time-resolved dynamic light scattering (TRDLS) was used to investigate the assembly process, allowing the determination of the duration of the distinct stages of polymerization. Understanding the mechanism of gel formation permits creation of a gradient by imposing gravitational stress on the system once the colloids are formed but before the gelation stage starts. Thus, a graded gel structure could be obtained by heating a solution containing C12RhMOP and bix at 80 °C for a certain period of time, after which the mixture was centrifuged for 5 min in order to create an asymmetric distribution of colloids, followed by further heating to continue the gelation.
5. Applications
As has been shown, the construction of asymmetric architectures, based on reticular porous materials, across different length scales can be achieved by means of diverse chemical and physical methods that allow achievement of a further level of control over their structures and properties. In this sense, the introduction of directional asymmetry is not only an effective way to increase the complexity of porous systems but also a powerful strategy to improve their performances or even to achieve new functions.At the nanoscale, even if the introduction of asymmetry through multi-component assembly is supposed to generate more elaborate cage structures with multiple functions, applications directly derived from asymmetric cages are scarce.
The heterometallic cage ([PdPt(Lab)4]4+) developed by Lisboa et al. reversibly forms the open-cage [PtLab]2+ in the presence of N,N′-dimethylaminopyridine (DMAP) and a [Pd(DMAP)4]2+ complex.24 This stimuli-responsive system is expected to be used in drug delivery or switchable catalysis by taking advantage of metals with different biological and photophysical properties.
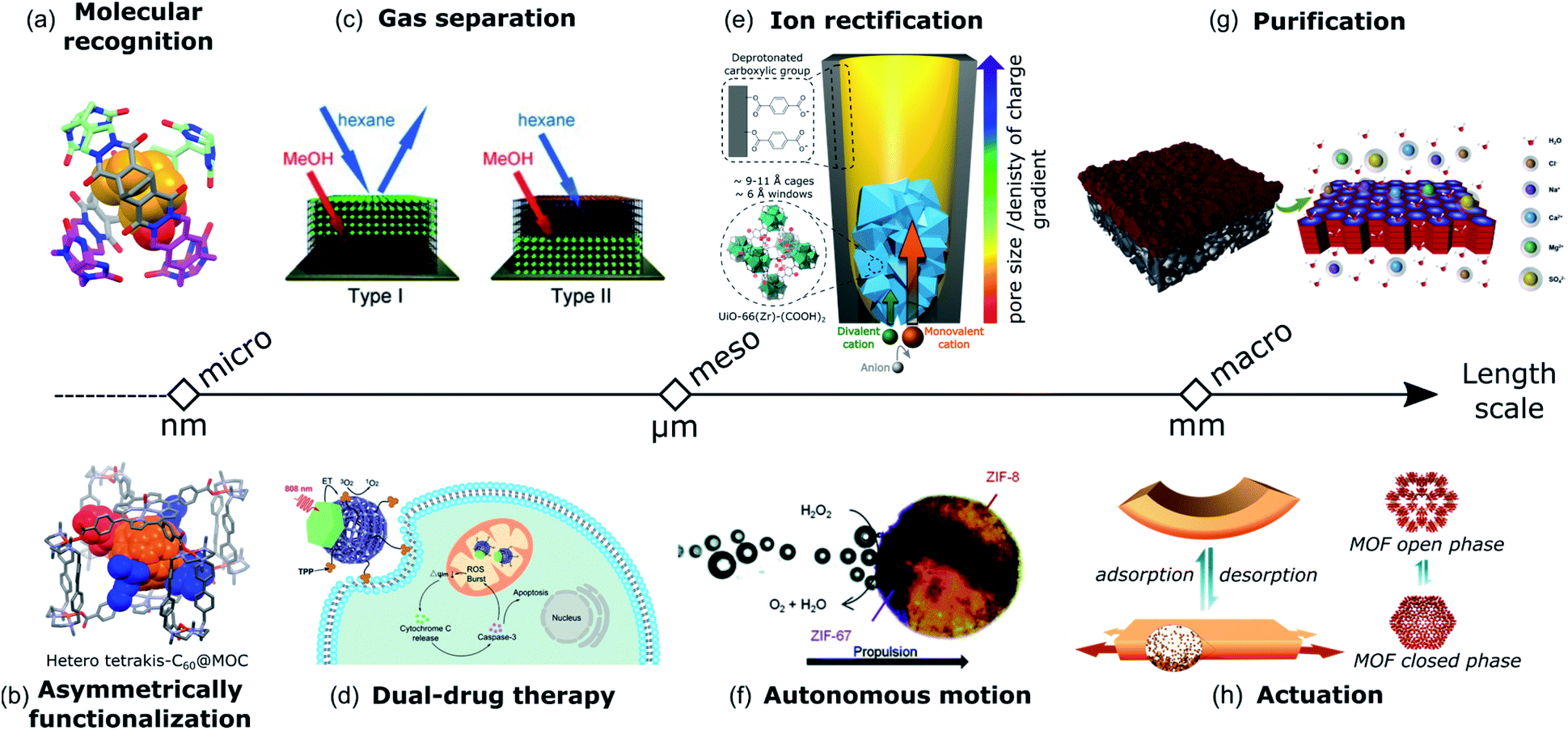
Heteroleptic cages also benefit from their Janus-like architectures to develop complementary pairs that can be used as advanced host-structures for sensing, drug delivery, or catalytic applications. For example, the asymmetric environment of the cis [Pd2(Lj)2(Lk)2]4+ coordination cage reported by Bloch et al. preferentially recognized stereoisomeric guests through shape complementary binding mode within the cavity.28 The conjoined cages with asymmetric pore configuration developed by Samantray et al. demonstrated selective binding of certain anions in the smaller cavity while the larger one contained solvent molecules.39 Rivera et al. reported the formation of dissymmetric diastereoisomer capsules from the self-assembly of symmetric organic molecules and the chiral template through hydrogen bonding complexation.81 The “softball” demonstrated molecular recognition properties for camphor derivative guests by preferentially forming one of the enantiomeric capsules in 32% excess (Fig. 8a).
 | ||
| Fig. 8 Different applications based on directional asymmetric systems over multiple length scales from the microscale (left) to the macroscale (right). At the molecular scale the possible applications arising from directional asymmetry are (a) molecular recognition based on dissymmetric diastereoisomer capsules81 and (b) asymmetric functionalization of fullerene using a metal–organic cage as a mask template.82 At the mesoscale, the applications are (c) gas separation thanks to the different polarity of MOF-on-MOF structures (adapted with permission from ref. 19, Copyright 2013, Wiley), (d) triphenylphosphine functionalized MOFs grown on upconversion nanoparticles for photodynamic therapy (adapted with permission from ref. 86, Copyright 2019, Wiley), (e) artificial ion channels based on a hybrid polymeric/UiO-66-(COOH)2 membrane with directional asymmetry arising from the gradient of pore size distribution and the gradient of charge created by the COO− functional group decorating the MOF pores and the PET membrane87 (adapted with permission from ref. 88, Copyright 2020, Springer Nature), and (f) self-propelling Janus-like MOF particles due to the specific side catalytic decomposition of H2O2. Adapted from ref. 48 with permission from the Royal Society of Chemistry. At the macroscale, the applications are (g) water purification using a COF/polymer-based membrane (adapted with permission from ref. 90, Copyright 2020, Elsevier) and (h) an actuator responsive to humidity based on a gradient of MOF crystals across a membrane. Adapted with permission from ref. 74, Copyright 2018, Wiley. | ||
A porous cage can also be used as a supramolecular mask for the asymmetric functionalization of guest molecules. In this regard, the approach reported by Fuertes-Espinosa et al. is elegant as it offers the possibility of regioselective functionalization of fullerenes.82 The metal–organic cage [(Pd2Me2pp)4(porphyrin)2]8+ (Pd2Me2pp = dipalladium(II) polyazamacrocycle and porphyrin = 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin-ZnII) serves as a host for the encapsulation and stabilization of fullerene in its void. The tetragonal prismatic geometry of the cage with four lateral apertures allows access to and control of the reactivity of the equatorial position of the confined C60 guest for Bingel–Hirsch cyclopropanation. Therefore, equatorial bis-, tris- and tetrakis-C60 hetero-adducts with asymmetric distribution were synthesized (Fig. 8b). The isomer-pure poly-functionalized fullerene was easily released from the host cage, offering alternatives to the costly and time-consuming chromatographic separation/purification of multi-adduct mixtures.
At the mesoscale, in order to increase the efficiency of porous materials for gas separation applications, besides the specific control of the pore size and functionality, a critical point is how these pores are spatially arranged over the whole structure. Thus, by imparting directional asymmetry on porous materials, it is possible to create structures with anisotropic porosity, leading to the enhancement of gas selectivity. For example, Meilikhov et al. fabricated binary Janus MOFs where polar and nonpolar framework types were combined spatially by layer-by-layer growth. Due to the existence of MOFs with different properties (i.e. polarity and pore size), different affinities for analytes and thus gas separation were obtained (Fig. 8c).19 Also, MOF/polymer composite membranes with asymmetric configuration have been demonstrated for gas separation applications such as selective removal of CO269,70,83,84 or hydrogen recovery.85 Typically, these membranes consist in a porous polymer layer and a thin layer of a polymer with MOF crystals as fillers. The effectiveness of these membranes relies on their asymmetric design, which combines an increase of the permeability, as the porous structure increases gas flux, and an excellent selectivity, as the selective skin increases the gas separation factor. MOF-based Janus NPs have emerged as promising materials for therapeutic treatment. Zhang et al. developed a hybrid material where PAA-ZIF-8 was selectively grown on one surface of PdNS while the opposite surface was modified with CD.56 The distinct surfaces (PAA-ZIF-8 and CD) of PdNS allow the loading, preservation and sequential delivery of two drugs with different properties. In addition, the PdNS provide near-infrared surface plasmon resonance properties making the hybrid system useful for synergistic dual-drug chemotherapy and photothermal therapy in the NIR-II bio-window (1000–1350 nm). Following the same direction, Li et al. demonstrated the importance of the Janus structure in NIR-induced photodynamic therapy (PDT) for cancer treatment using porphyrinic nanoscale MOFs anisotropically grown on the surface of Nd3+-sensitized UCNPs.57 Effective luminescence resonance energy transfer (LRET) from the UCNP side into the MOF domain enabled the activation of the MOF to produce cytotoxic reactive oxygen species (ROS) with an 808 nm laser. Such a strategy was not able be realized by the solution mixture of MOF NPs and UCNPs and was only achieved with the synergistically enhanced functions of their dual optical properties. Later, the same group functionalized the MOF surface of the system described above with triphenylphosphine, a ligand capable of targeting mitochondria, the organelle responsible for initiating the apoptotic pathway.86 This organelle-targeting strategy was able to amplify the photodynamic therapy efficacy through the in situ generation of ROS in mitochondria which activated cancer cell death (Fig. 8d).
In biology, asymmetry in cell membranes is a prerequisite for the selective, fast and preferential transportation direction of ions with similar properties between the extracellular space and cell interior. Recently, Lu et al. reported the design of artificial ion channels based on a hybrid polymeric/UiO-66-(COOH)2 membrane.87,88 Bullet-shaped channels were created in a PET membrane decorated with deprotonated carboxylic acid groups that were used to grow the MOF at the tip of the channel. This strategy allowed creating a gradient of pore size distribution (Å to nm) and a gradient of charge created by the COO− functional group decorating the MOF pores and the PET membrane (Fig. 8e). Indeed, the asymmetry in the pore geometry along the diffusion direction and the electrochemical potential gradient on the membrane surface are key parameters for ion rectification properties and high transport rates. In addition, the angstrom-sized pores of the MOF allow ion dehydration at the pore entrance and increase interactions with the surface wall, which provide high ion selectivity.
The introduction of asymmetry not only results in the improvement of the performance, but also endows the porous materials with new functionalities, which were not found in their symmetric counterparts, like self-propulsion or self-shaping properties. For example, to induce directional autonomous motion, it is necessary to combine a propulsion mechanism, such as surface tension gradients, electrochemical potential or bubble-ejection, with an asymmetric structure.89 Using MOFs as active materials, several examples of self-propulsion asymmetric systems were reported. For instance, Tan et al. reported the fabrication of Janus ZIF-8/ZIF-67 crystals by the polymer blockage method (Fig. 8f).48 Since the Janus crystals only catalyze the decomposition of H2O2 to H2O and O2 on the ZIF-67 surface, the directional propulsion of the crystals was realized by oxygen bubbles. In a similar manner, Ayala et al. constructed Janus MOF/metal composites, in which the surface of ZIF-8 crystals was partially coated with platinum as the catalyst. Directional bubble propulsion was triggered by the asymmetrically distributed Pt coating, driving the autonomous motion of these crystals.53
At the macroscale, asymmetric composite membranes based on COFs have found applications in liquid separation. In these materials, a COF thin film is predominantly concentrated on one side of the membrane, acting as the selective layer. These asymmetric COF composite membranes showed increased hydrophilicity and higher water permeance, making them ideal for different liquid separation applications. For instance, a COF-based asymmetric membrane constructed from TpHZ and PES exhibited high water/ethanol separation efficiency.71 The enhancement of separation performance is attributed to the gradient distribution of TpHZ across the membrane. Here, a gradual transition from the solution-governed zone (low concentration of COFs) to the diffusion-governed zone (high concentration of COFs) allows the formation of water-selective channels while favoring water transport. The excellent separation characteristics of the asymmetric TpHz/PES membrane were also exploited for water desalination, showing high salt rejection capacity (Fig. 8g).90 Further, similar asymmetric COF-based membranes with TpPa as the COF were prepared in combination with PSF or PVDF polymers, demonstrating their efficiency for rejection of dyes from water.72,91
Another practical application that was successfully exploited to gain better performance is pollutant removal. For example, the versatility of the multilayer casting method allowed the fabrication of layered MOF/polymer membranes with enhanced removal of chemical warfare agents (CWAs).76 These membranes can act as reactive barriers to CWAs while maintaining high moisture vapor transport through the composite, making them breathable while maintaining their protective barrier function.
In the field of water remediation one of the major drawbacks of using MOFs as adsorbents is the high cost derived from their recovery after use. One effective strategy to overcome this problem consists of introducing magnetism into MOFs by their hybridization with magnetic particles, resulting in composites whose movement could be well guided by applying a magnetic field. Ayala et al. reported the fabrication of metallic Janus MOF particles partially coated with a metal (Co). Apart from the magnetic properties, the asymmetric nature provides the materials with higher extraction efficiency, given the higher exposure of MOFs to target pollutants at the uncovered face compared to their corresponding core–shell composites.53
In the case of self-shaping systems that are capable of changing their shape as a consequence of internal stress, an asymmetric structure is crucial. In these materials, the responsive behavior is derived from heterogeneous expansion/shrinking phenomena, which force the material to minimize its elastic energy by changing its shape. Thus, by achieving asymmetric distributions of MOFs with swelling properties, materials with self-shaping properties were successfully produced. For instance, the aforementioned coordination-driven pseudomorphic replacement used by Sun et al. allowed the fabrication of asymmetric HKUST-1/polymer membranes, which showed self-folding behavior toward NH3 vapors as a result of the expansion of the MOF crystal upon adsorption.79 By using MIL-88A crystals, Troyano et al. reported the fabrication of humidity responsive composite film actuators by forming a vertical gradient of MOF crystals across the membrane (Fig. 8h).74 Later, the same group expanded this approach by patterning the MOF crystals using chemical etching with HCl.78 This strategy allowed control of the location of MOF crystals in both the vertical and lateral directions thus leading to novel actuations such as lifting cargo, mechanical gripping, and unidirectional walking.
6. Conclusions and outlook
We showed the diversity of strategies to introduce directional asymmetry in reticular porous materials thanks to the modularity of these systems. At the nanoscale, the rational design of the molecular building blocks and their molecular interactions (inter or intra) allow control of their positioning in the molecular cationic cages based on N-donor ligands and square planar metal nodes. On the other hand, examples of neutral cages with directional asymmetry based on carboxylate ligands are scarcer and mostly rely on post synthetic ligand exchange on a preformed cage or on the surface functionalization of the ligands imparting opposite properties (hydrophobic/hydrophilic). Similar strategies used for cationic cages (steric repulsion, geometric complementarity, and guest template) could be applied to neutral cages in order to extend the number of cage systems with directional asymmetry. In addition, cages built from carboxylate ligands offer increased stability due to their stronger coordination bonds, which is necessary to fabricate solid-state porous materials by assembling cages with directional asymmetry with transferring the asymmetric information to higher length scales.At the mesoscale, most examples of directional asymmetry rely on MOF structures. Indeed, their modularity allows them to be directionally integrated with other MOFs thanks to the controlled orientation of the epitaxial growth through (i) the careful selection of ligands and framework systems with similar unit cell parameters or (ii) the partial blockage of core surfaces. Alternatively, the control of the synthetic conditions and application of centrifugation can also lead to the formation of Janus-like MOF composites. A different approach is the selective and spatially controlled post-synthetic modification on symmetric porous materials using external stimuli in order to form directional asymmetric systems. MOF composites can also be obtained by hybridizing MOFs with other functional materials by preferential coating or selective growth on a template. However, the challenge remains in the introduction of directional asymmetry through multiplicity and variation of the components in a single MOF system. In addition, advanced characterization techniques are required to obtain a full understanding of the spatial localization of the components.92,93
Finally, at the macroscopic level, asymmetric structures have been typically achieved by controlling the spatial location of preformed mesoscopic components inside a polymeric matrix. In general, the reported strategies for the synthesis of such asymmetric composite films involve the mixing of the components to yield a homogeneous casting solution. Then, solvent diffusion, gravitational sedimentation, or even the manual control of the casting can be used to attain an asymmetric configuration. Considering that at the macroscale the transport phenomena can be easily controlled, these methodologies could be efficiently extended to other types of porous materials. Nevertheless, other strategies involving the in situ formation or the post-synthetic chemical modification of microporous materials have also been proven to be successful. In this sense, the use of different external stimuli, such as electromagnetic fields94 or laser guided patterning95 could be considered useful tools to achieve precise spatial positioning.
One of the possible research directions is the transfer of the directional asymmetry of molecular building blocks to meso and macro hierarchical structures through the controlled assembly of asymmetric units across multiple length scales. This could be directly inspired by nature, which is able to in vivo transform molecular chirality into macroscopic chirality as observed from the relation of multilevel asymmetry in Drosophila larvae. Indeed, at the molecular level, the chirality of myosin 1D gene induces preferential twisting of cells and single organs, which turn-out to modify the whole-body symmetry and motion behavior.96 This demonstrates that the breaking of the symmetry at all biological scales is determined by a single asymmetry at the molecular scale that propagates to higher levels.
Based on a bottom-up approach, directional asymmetry across multiple length scales could be achieved through the generation of a gradient of the structure or porosity within a continuous system. Using the microfluidic technique, it has already been demonstrated that through the fine tuning of the reactant diffusion, control of the nonequilibrium shape of crystals at the mesoscale has been achieved.97 Though more time consuming, the layer-by-layer growth method also takes advantage of the MOFs' epitaxial growth modularity and controlled crystal orientation to create asymmetric structures or pore configurations. With this in mind, the formation of a continuous asymmetric structure at the macroscopic level can be achieved with the right chiral inducer during the assembly of porous materials at the mesoscale. In all cases, this could lead to materials with unidirectional transport properties of guests due to the asymmetric environment or ferroelectric properties when subjected to external stimuli like polarization.
Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
Z. W. acknowledges the China Scholarship Council (CSC) for the scholarship support. J. T. is grateful to the JSPS Postdoctoral Fellowship Program for Foreign Researchers. This study was supported by JSPS KAKENHI Grant Number 20K15366 (Wakate) for A. L. and 19H04575 (Coordination Asymmetry) and 18H01995 (Kiban B) for S.F.References
- A. R. Palmer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14279–14286 CrossRef CAS
.
- J. C. Brookes, A. P. Horsfield and A. M. Stoneham, J. R. Soc., Interface, 2009, 6, 75–86 CrossRef CAS
- A. Mosandl, J. Chromatogr. Sci., 2004, 42, 440–449 CAS
- W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760–770 CrossRef CAS
- J. Rothman and J. Lenard, Science, 1977, 195, 743–753 CrossRef CAS
- B. J. Allen and J. S. Levi nton, Functional Ecology, 2007, 21, 154–161 CrossRef
- Z. Zhang, L. Wen and L. Jiang, Chem. Soc. Rev., 2018, 47, 322–356 RSC
- T. Pakizeh, A. Dmitriev, M. S. Abrishamian, N. Granpayeh and M. Käll, J. Opt. Soc. Am. B, 2008, 25, 659–667 CrossRef CAS
- A. K. Chew and R. C. Van Lehn, J. Phys. Chem. C, 2018, 122, 26288–26297 CrossRef CAS
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS
- L. Feng, K.-Y. Wang, G. S. Day and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC
- Y. Luo, M. Ahmad, A. Schug and M. Tsotsalas, Adv. Mater., 2019, 31, 1901744 CrossRef
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS
- V. Guillerm, H. Xu, J. Albalad, I. Imaz and D. Maspoch, J. Am. Chem. Soc., 2018, 140, 15022–15030 CrossRef CAS
- O. Kwon, J. Y. Kim, S. Park, J. H. Lee, J. Ha, H. Park, H. R. Moon and J. Kim, Nat. Commun., 2019, 10, 3620 CrossRef
- M. Meilikhov, S. Furukawa, K. Hirai, R. A. Fischer and S. Kitagawa, Angew. Chem., Int. Ed., 2013, 52, 341–345 CrossRef CAS
- S. Kitagawa, Acc. Chem. Res., 2017, 50, 514–516 CrossRef CAS
- K. Omoto, N. Hosono, M. Gochomori, K. Albrecht, K. Yamamoto and S. Kitagawa, Chem. Commun., 2018, 54, 5209–5212 RSC
- H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS
- F. Li and L. F. Lindoy, Aust. J. Chem., 2019, 72, 731–741 CrossRef CAS
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101 CrossRef CAS
- A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS
- R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V. Schäfer and G. H. Clever, Chem.–Eur. J., 2018, 24, 12976–12982 CrossRef CAS
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS
- K. E. Ebbert, L. Schneider, A. Platzek, C. Drechsler, B. Chen, R. Rudolf and G. H. Clever, Dalton Trans., 2019, 48, 11070–11075 RSC
- Q.-F. Sun, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2014, 53, 13510–13513 CrossRef CAS
- J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2015, 21, 4200–4204 CrossRef CAS
- S. Hiraoka, Y. Kubota and M. Fujita, Chem. Commun., 2000, 1509–1510 RSC
- G. Lal, S. J. Lee, D. M. Spasyuk and G. K. H. Shimizu, Chem. Commun., 2018, 54, 1722–1725 RSC
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS
- R. L. Greenaway, V. Santolini, A. Pulido, M. A. Little, B. M. Alston, M. E. Briggs, G. M. Day, A. I. Cooper and K. E. Jelfs, Angew. Chem., Int. Ed., 2019, 58, 16275–16281 CrossRef CAS
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS
- E. Berardo, R. L. Greenaway, L. Turcani, B. M. Alston, M. J. Bennison, M. Miklitz, R. Clowes, M. E. Briggs, A. I. Cooper and K. E. Jelfs, Nanoscale, 2018, 10, 22381–22388 RSC
- S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 880 CrossRef CAS
- K. Koh, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2009, 6162–6164 RSC
- Y. Ikezoe, G. Washino, T. Uemura, S. Kitagawa and H. Matsui, Nat. Mater., 2012, 11, 1081–1085 CrossRef CAS
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS
- J. Liang, A. Nuhnen, S. Millan, H. Breitzke, V. Gvilava, G. Buntkowsky and C. Janiak, Angew. Chem., Int. Ed., 2020, 59, 6068–6073 CrossRef CAS
- S. Furukawa, K. Hirai, K. Nakagawa, Y. Takashima, R. Matsuda, T. Tsuruoka, M. Kondo, R. Haruki, D. Tanaka, H. Sakamoto, S. Shimomura, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 1766–1770 CrossRef CAS
- S. Furukawa, K. Hirai, Y. Takashima, K. Nakagawa, M. Kondo, T. Tsuruoka, O. Sakata and S. Kitagawa, Chem. Commun., 2009, 5097–5099 RSC
- S. Choi, T. Kim, H. Ji, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS
- P. Á. Szilágyi, M. Lutz, J. Gascon, J. Juan-Alcañiz, J. van Esch, F. Kapteijn, H. Geerlings, B. Dam and R. van de Krol, CrystEngComm, 2013, 15, 6003–6008 RSC
- T. T. Y. Tan, J. T. M. Cham, M. R. Reithofer, T. S. Andy Hor and J. M. Chin, Chem. Commun., 2014, 50, 15175–15178 RSC
- L. Feng, S. Yuan, J.-L. Li, K.-Y. Wang, G. S. Day, P. Zhang, Y. Wang and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 1719–1726 CrossRef CAS
- L. Feng, K.-Y. Wang, X.-L. Lv, T.-H. Yan, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 3069–3076 CrossRef CAS
- I. Stassen, I. Boldog, C. Steuwe, D. De Vos, M. Roeffaers, S. Furukawa and R. Ameloot, Chem. Commun., 2017, 53, 7222–7225 RSC
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- A. Ayala, C. Carbonell, I. Imaz and D. Maspoch, Chem. Commun., 2016, 52, 5096–5099 RSC
- S. Yadnum, J. Roche, E. Lebraud, P. Négrier, P. Garrigue, D. Bradshaw, C. Warakulwit, J. Limtrakul and A. Kuhn, Angew. Chem., Int. Ed., 2014, 53, 4001–4005 CrossRef CAS
- T.-H. Park, K. J. Lee, S. Hwang, J. Yoon, C. Woell and J. Lahann, Adv. Mater., 2014, 26, 2883–2888 CrossRef CAS
- L. Zhang, S. Li, X. Chen, T. Wang, L. Li, Z. Su and C. Wang, Adv. Funct. Mater., 2018, 28, 1803815 CrossRef
- Y. Li, Z. Di, J. Gao, P. Cheng, C. Di, G. Zhang, B. Liu, X. Shi, L.-D. Sun, L. Li and C.-H. Yan, J. Am. Chem. Soc., 2017, 139, 13804–13810 CrossRef CAS
- Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 CrossRef
- F.-M. Zhang, J.-L. Sheng, Z.-D. Yang, X.-J. Sun, H.-L. Tang, M. Lu, H. Dong, F.-C. Shen, J. Liu and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 12106–12110 CrossRef CAS
- L. Garzón-Tovar, J. Pérez-Carvajal, A. Yazdi, J. Hernández-Muñoz, P. Tarazona, I. Imaz, F. Zamora and D. Maspoch, Angew. Chem., Int. Ed., 2019, 58, 9512–9516 CrossRef
- J. Tan, S. Namuangruk, W. Kong, N. Kungwan, J. Guo and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 13979–13984 CrossRef CAS
- O. Shekhah, J. Liu, R. A. Fischer and C. Wöll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC
- Y. Zhang, X. Feng, S. Yuan, J. Zhou and B. Wang, Inorg. Chem. Front., 2016, 3, 896–909 RSC
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC
- H. Lu and S. Zhu, Eur. J. Inorg. Chem., 2013, 2013, 1294–1300 CrossRef CAS
- A. Legrand, G. A. Craig, M. Bonneau, S. Minami, K. Urayama and S. Furukawa, Chem. Sci., 2019, 10, 10833–10842 RSC
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS
-
M. Mulder, in Encyclopedia of Separation Science, ed. I. D. Wilson, Academic Press, Oxford, 2000, pp. 3331–3346 Search PubMed
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, J. Membr. Sci., 2010, 362, 478–487 CrossRef CAS
- H. Zhu, L. Wang, X. Jie, D. Liu and Y. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 22696–22704 CrossRef CAS
- H. Yang, H. Wu, Z. Yao, B. Shi, Z. Xu, X. Cheng, F. Pan, G. Liu, Z. Jiang and X. Cao, J. Mater. Chem. A, 2018, 6, 583–591 RSC
- R. Wang, X. Shi, Z. Zhang, A. Xiao, S.-P. Sun, Z. Cui and Y. Wang, J. Membr. Sci., 2019, 586, 274–280 CrossRef CAS
- M. S. Denny, M. Kalaj, K. C. Bentz and S. M. Cohen, Chem. Sci., 2018, 9, 8842–8849 RSC
- J. Troyano, A. Carné-Sánchez, J. Pérez-Carvajal, L. León-Reina, I. Imaz, A. Cabeza and D. Maspoch, Angew. Chem., Int. Ed., 2018, 57, 15420–15424 CrossRef CAS
- S. Peng, L. Zhang, C. Zhang, Y. Ding, X. Guo, G. He and G. Yu, Adv. Energy Mater., 2018, 8, 1802533 CrossRef
- G. W. Peterson, A. X. Lu, M. G. Hall, M. A. Browe, T. Tovar and T. H. Epps, ACS Appl. Mater. Interfaces, 2018, 10, 6820–6824 CrossRef CAS
- S. N. Wijenayake, N. P. Panapitiya, S. H. Versteeg, C. N. Nguyen, S. Goel, K. J. Balkus, I. H. Musselman and J. P. Ferraris, Ind. Eng. Chem. Res., 2013, 52, 6991–7001 CrossRef CAS
- J. Troyano, A. Carné-Sánchez and D. Maspoch, Adv. Mater., 2019, 31, 1808235 CrossRef
- J.-K. Sun, H.-J. Lin, W.-Y. Zhang, M.-R. Gao, M. Antonietti and J. Yuan, Mater. Horiz., 2017, 4, 681–687 RSC
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 CrossRef
- J. M. Rivera, T. Martín and J. Rebek, Science, 1998, 279, 1021–1023 CrossRef CAS
- C. Fuertes-Espinosa, C. García-Simón, M. Pujals, M. Garcia-Borràs, L. Gómez, T. Parella, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Chem, 2020, 6, 169–186 CAS
- Y. Dai, J. R. Johnson, O. Karvan, D. S. Sholl and W. J. Koros, J. Membr. Sci., 2012, 401–402, 76–82 CrossRef CAS
- S. Basu, A. Cano-Odena and I. F. J. Vankelecom, Sep. Purif. Technol., 2011, 81, 31–40 CrossRef CAS
- J. Hu, H. Cai, H. Ren, Y. Wei, Z. Xu, H. Liu and Y. Hu, Ind. Eng. Chem. Res., 2010, 49, 12605–12612 CrossRef CAS
- C. Liu, B. Liu, J. Zhao, Z. Di, D. Chen, Z. Gu, L. Li and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 2634–2638 CrossRef CAS
- J. Lu, H. Zhang, J. Hou, X. Li, X. Hu, Y. Hu, C. D. Easton, Q. Li, C. Sun, A. W. Thornton, M. R. Hill, X. Zhang, G. Jiang, J. Z. Liu, A. J. Hill, B. D. Freeman, L. Jiang and H. Wang, Nat. Mater., 2020, 19, 767–774 CrossRef CAS
- A. Legrand and S. Furukawa, Nat. Mater., 2020, 19, 701–702 CrossRef CAS
- K. K. Dey and A. Sen, J. Am. Chem. Soc., 2017, 139, 7666–7676 CrossRef CAS
- R. Wang, M. Wei and Y. Wang, J. Membr. Sci., 2020, 604, 118090 CrossRef CAS
- R. Wang, X. Shi, A. Xiao, W. Zhou and Y. Wang, J. Membr. Sci., 2018, 566, 197–204 CrossRef CAS
- A. M. Katzenmeyer, J. Canivet, G. Holland, D. Farrusseng and A. Centrone, Angew. Chem., Int. Ed., 2014, 53, 2852–2856 CrossRef CAS
- Z. Ji, T. Li and O. M. Yaghi, Science, 2020, 369, 674–680 CrossRef CAS
- F. Cheng, E. S. Marshall, A. J. Young, P. J. Robinson, J.-S. G. Bouillard, A. M. Adawi, N. A. Vermeulen, O. K. Farha, M. R. Reithofer and J. M. Chin, Chem.–Eur. J., 2017, 23, 15578–15582 CrossRef CAS
- R. Li, S. Yuan, W. Zhang, H. Zheng, W. Zhu, B. Li, M. Zhou, A. Wing-Keung Law and K. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 40564–40574 CrossRef CAS
- G. Lebreton, C. Géminard, F. Lapraz, S. Pyrpassopoulos, D. Cerezo, P. Spéder, E. M. Ostap and S. Noselli, Science, 2018, 362, 949–952 CrossRef CAS
- A. Sorrenti, L. Jones, S. Sevim, X. Cao, A. J. deMello, C. Martí-Gastaldo and J. Puigmartí-Luis, J. Am. Chem. Soc., 2020, 142, 9372–9381 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |