
Direct formation of nano-objects via in situ self-assembly of conjugated polymers
Gregory I.
Peterson†
*,
Sanghee
Yang†
and
Tae-Lim
Choi
 *
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: gpeterson@snu.ac.kr; tlc@snu.ac.kr
First published on 9th December 2020
Abstract
Polymer self-assembly is a widely utilized method to prepare a wide range of nano-objects in solution. Typically, preparation of such objects relies on the use of block copolymers and challenging post-polymerization treatment steps. While polymerization induced self-assembly (PISA) can simplify their preparation, the resulting nano-objects typically have poor stability (including sensitivity to solvent, temperature, and mechanical stimuli). An alternative approach is to use conjugated polymers, with a strong driving force for self-assembly, to achieve semiconducting nano-objects. This process is termed in situ nanoparticlization of conjugated polymers (INCP) or PISA using conjugated polymers. With INCP, self-assembled nano-objects can be obtained (without any post-polymerization treatment steps) from block copolymers, using one-pot or one-shot methods, or even homopolymers. Due to the use of conjugated polymers, the nano-objects from INCP have the potential for use in various optoelectronic applications. In this Perspective, we summarize the development of INCP by discussing synthetic methods, accessible nano-objects morphologies, and mechanisms of nano-object formation.
Introduction
Block copolymers (BCPs) can self-assemble into various nanostructures with a broad range of morphologies and nearly unlimited combinations of functional groups.1–4 Self-assembly procedures generally require post-polymerization treatment steps, such as addition of selective solvents or additives, changing temperature or pH, aging, or dialysis. An alternative approach, which has garnered significant attention in recent years, is to form nanostructures during polymerization with a process termed polymerization-induced self-assembly,5–9 which was coined as PISA by Armes and coworkers in 2012.10 With this method, an insoluble “core” forming block is polymerized from a soluble “shell” block, leading to the formation of micelles, worms, lamellae, and vesicles in high concentrations without post-polymerization treatment steps (Fig. 1). PISA has been applied to prepare functional nanostructures for catalysis,11 drug delivery,12 and various other biomedical applications.13,14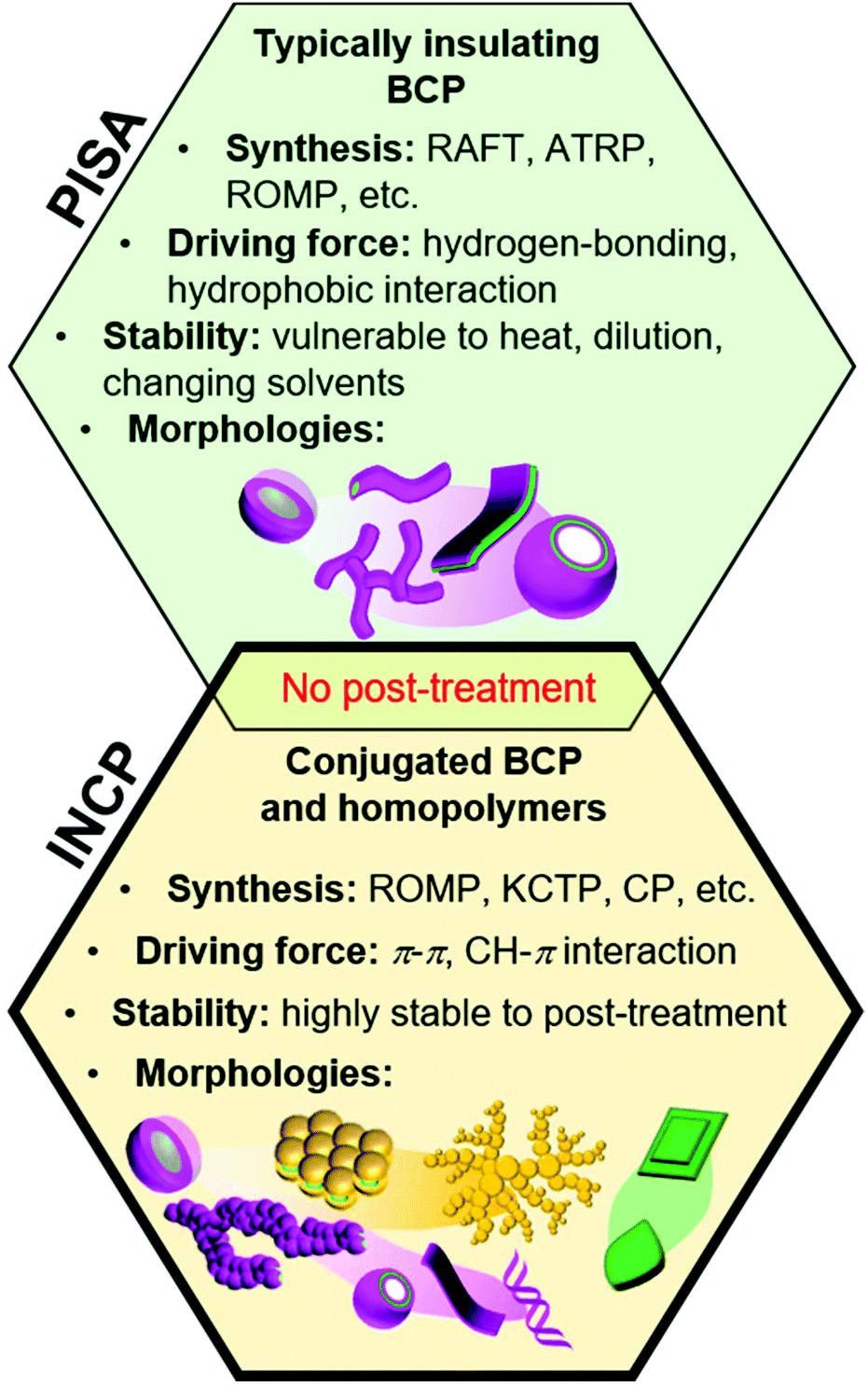 | ||
| Fig. 1 Preparation of nano-objects during polymerization. Similarities and differences between conventional PISA (with non-conjugated polymers) and INCP. | ||
The core-forming blocks of nanostructures used in PISA are generally sensitive to solvent, temperature, and concentration. Even small changes in these conditions can alter or break-up the nanostructures. While the stability can be greatly enhanced with cross-linking, this requires additional post-polymerization treatment steps and can make obtaining non-spherical nano-objects challenging (in the case of in situ cross-linking).15 In 2012, we developed another PISA strategy to achieve highly stable nano-objects, which was termed in situ nanoparticlization of conjugated polymers (INCP, Fig. 1).16,17 In regard to terminology, “INCP” is analogous to “PISA of conjugated polymers”, although we use the former term in this Perspective as only a few papers that conduct self-assembly of conjugated polymers use the PISA term. With INCP, strong π–π or CH–π interactions drive self-assembly, resulting in nano-objects that are highly stable toward different temperatures, solvents, and mechanical stimuli. The features that further differentiate conventional PISA (with non-conjugated polymers) and INCP is that the latter is not limited to BCPs (i.e., homopolymers can undergo INCP), the resulting nano-objects have semiconducting properties, and more diverse nanostructures can be achieved (such as nanocaterpillars, nanostars, fractals, etc.).
Conjugated polymer nanoparticles are generally prepared by post-polymerization treatment and have been targeted for various applications including sensors, imaging agents, photovoltaics, light-emitting diodes, and catalysts.18–21In situ nanoparticlization can also be achieved with the aid of surfactants or stabilizers using miniemulsion or dispersion polymerizations (techniques that are mostly limited to producing spherical nanoparticles),22–25 but these methods are beyond the scope of this Perspective. In contrast, INCP does not require any stabilizers and various 1D, 2D, or 3D nano-objects can be obtained. With a few exceptions, INCP has been performed using ring-opening metathesis polymerization (ROMP),26–28 cyclopolymerization (CP),29–31 and catalyst-transfer polycondensation (CTP).32–34 In this Perspective, we will discuss the development of INCP, including synthetic methods, nano-object morphologies, and mechanisms of nano-object formation (organized by the polymerization type).
INCP with ROMP
Polyacetylene (PA) is a highly insoluble conjugated polymer that has attracted much attention due to its interesting electronic properties.35 One of the simplest ways to synthesize PA is via the ROMP of 1,3,5,7-cyclooctatetraene (COT).36 We envisioned achieving INCP via standard block copolymerization of a norbornene monomer (NB1), to give a soluble poly(norbornene) (PNB) block with a living chain end, followed by addition and polymerization of COT, which would induce the self-assembly process.16,37 Polymerizations were optimized to suppress side reactions (e.g., benzene formation) and achieve high conversion of COT into PA. Using low temperature, high monomer concentration, and a highly active Grubbs third-generation catalyst (G3-Cl) enabled the synthesis of PNB150-b-PAn (the subscripts indicate the degree of polymerization, DP) with PA DPs up to 208 (Fig. 2A). In situ1H NMR and dynamic light scattering (DLS) experiments were conducted to determine if nanoparticles had formed during the polymerization.37 When the block copolymerization was conducted in deuterated solvent, only PNB signals were observed throughout the experiment, supporting the aggregated state of the PA blocks. Furthermore, aliquots were removed during the conventional block copolymerization and analyzed by DLS, without termination or precipitation. Large diameter assemblies were observed in as little as 15 min, which were differentiable from a smaller diameter population (which we attributed to single BCP chains). These large assemblies continued to grow in size with increasing polymerization time (Fig. 2B). Thus, we concluded that nanoparticles were formed during the polymerization. | ||
| Fig. 2 (A) Synthesis of PA-based BCPs. (B) DLS measurements of aliquots from the polymerization solution of PNB150-b-PAn. Times are after COT addition. Numbers above the peaks correspond to the average hydrodynamic diameter (Dh, with units of nm). Adapted from ref. 37 with permission from the Royal Society of Chemistry, copyright 2014. (C) DLS measurements of aliquots from the polymerization solution of PTD150-b-PAn. Adapted from ref. 38 with permission from John Wiley and Sons, copyright 2015. (D) AFM (on mica) images of nanospheres from PNB150-b-PA40. Adapted from ref. 16 with permission from American Chemical Society, copyright 2012. (E) AFM (on mica) and TEM (on a carbon-coated copper grid) images of nanocaterpillars from PNB150-b-PA208. Adapted from ref. 37 with permission from the Royal Society of Chemistry, copyright 2014. (F) AFM (on mica) images of the nano/micro-object morphologies for PTD150-b-PAn. Adapted from ref. 38 with permission from John Wiley and Sons, copyright 2015. | ||
To determine the morphology of the nanoparticles, dilute solutions of BCPs were spin-coated on mica surfaces and imaged with atomic force microscopy (AFM). For PNB150-b-PA40, nanospheres were observed (Fig. 2D).16 As the length of the PA block was increased, a morphological transition from nanospheres to 1D nano-objects was observed (Fig. 2E). High resolution AFM and transmission electron microscopy (TEM) images revealed undulated linear nanostructures, termed nanocaterpillars, which were also confirmed to exist in solution using cryogenic TEM (cryo-TEM). Defect- (i.e., nanosphere) free nanocaterpillars were obtained by enhancing the stereo-purity of the PA block (cis-PA was favored when polymerizations were conducted at 0 °C) and decreasing the molecular weight dispersity.37 Importantly, the highly stable nature of the nano-objects was supported by DLS experiments, as nanoparticle size distributions remained constant in solution after heating at 90 °C or sonicating for 30 min.16
We envisioned that we might be able to obtain higher-order nanostructures (i.e., 3D nano-objects) by using a more rigid shell-block, which would promote stronger PA block interactions.38 Thus, we switched from NB1 to an endo-tricyclo[4.2.2.0]deca-3,9-diene (TD) monomer (TD1) to achieve BCPs with a rigid poly(TD) (PTD) shell-block (Fig. 2A). While polymerizations were slower with TD1, PTD150-b-PAn with PA DPs up to 156 were still obtainable. The formation of nanoparticles during the polymerization was again supported by DLS measurements (Fig. 2C). The structural evolution from nanospheres to nanocaterpillars, with increasing PA block length, was also still observed (Fig. 2F). This transition, however, appeared to occur at much shorter PA block lengths for PTD150-b-PAn (nanocaterpillars were observed with a PA DP of 34) than for PNB150-b-PAn (nanocaterpillars were observed with PA DPs of 80 or higher). Further increasing the PA block length for PTD150-b-PAn resulted in the formation of 3D nano- and then micro-objects. DLS and cryo-TEM were used to confirm that the nanostructures did not form as a result of aggregation during drying for AFM/TEM imaging. Of note, the INCP process could also be achieved in a one-shot manner (without sequential monomer addition) due to the disparity between the polymerization rates of NB1 or TD1 and COT.39 The use of the one-shot process did not disrupt or alter the nano-object formation processes, thereby making the one-shot INCP one of the simplest method for preparing nano-objects.
Based on these results, we proposed a model (Fig. 3) for the self-assembly and morphological transition of nanospheres to nanocaterpillars and 3D micro-objects.38 As the PA block grows during the polymerization, the BCPs spontaneously self-assemble into micelle-like nanospheres due to strong π–π interactions of the PA blocks (the propagating carbenes remain active in the core). COT diffuses into the PA core and propagation continues. Eventually, the PA core-blocks become elongated enough that they are exposed to solvent. Solvophobic and π–π interactions promote the spontaneous assembly of the nanospheres into nanocaterpillar morphologies. Due to the relatively flexible nature of PNB, the PA core is well-shielded from solvent and other nanospheres or nanocaterpillars, such that they do not assemble into higher-order morphologies (even as the PA core continues to grow). The rigid nature of PTD,40 however, prohibits adequate shielding of the PA core. Thus, as the PA blocks of the PTD-based nanocaterpillars grow, the PA core-blocks become exposed, leading to aggregation of the nanocaterpillars into 3D nano- and then micro-objects.
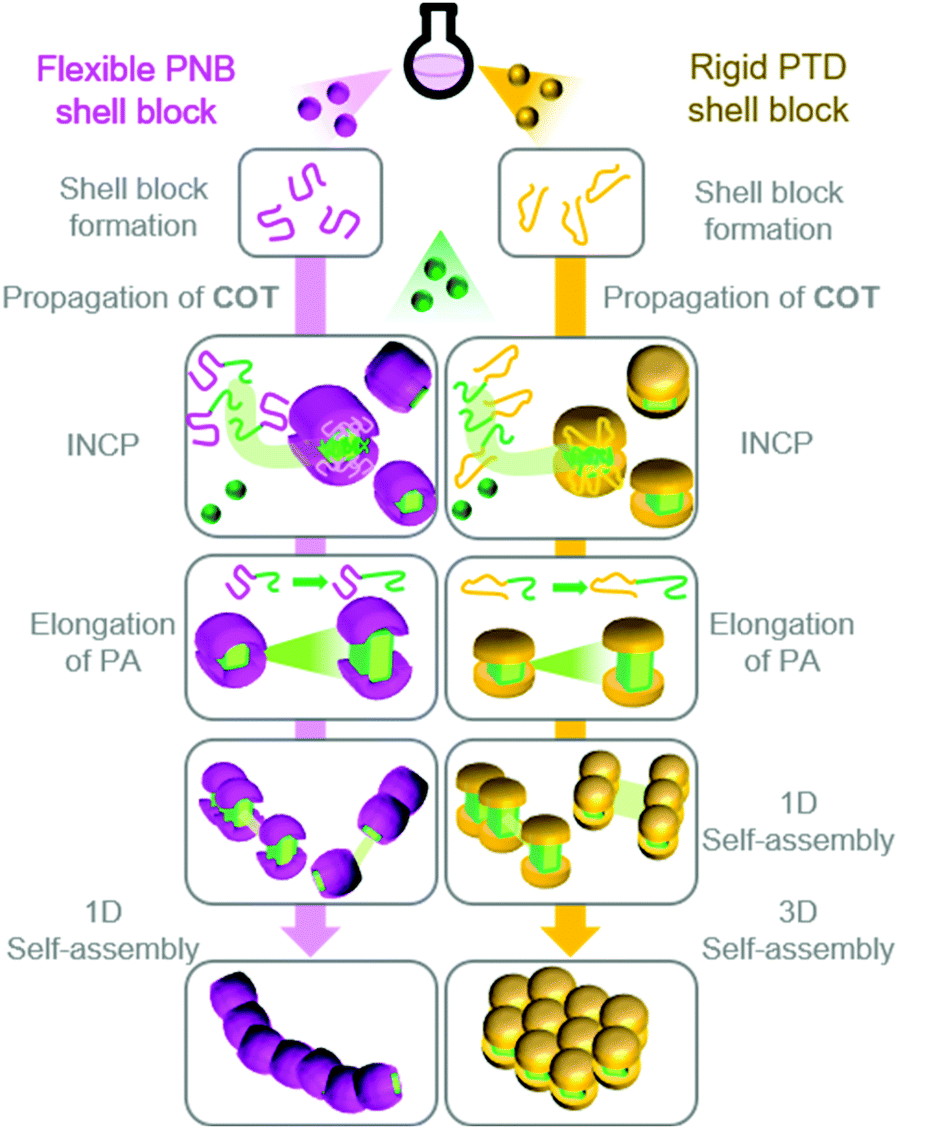 | ||
| Fig. 3 Proposed model for the formation of nanocaterpillars from PNB-based BCPs and 3D nano-objects from PTD-based BCPs via INCP. | ||
Poly(p-phenylenevinylene)s (PPVs) are highly fluorescent semiconducting polymers used in various optoelectronic applications and devices.41 A facile method of preparing PPVs is the ROMP of [2.2]paracyclophane-1,9-diene (PD). We envisioned conducting INCP with BCPs having PNB shell- and PVP core-blocks.42 Due to the disparate reactivity between NB1 and PD, one-shot polymerizations were achieved with G3-Cl, yielding PNB150-b-PPVn with PPV DPs up to 60 (Fig. 4A). Aliquots were removed from the polymerization and were characterized by DLS, AFM, and TEM, without termination or purification, to support in situ nanoparticlization. AFM and TEM images revealed a morphological transition from nanospheres to nanocaterpillars with increasing PPV block length (analogous to that described above for PA), followed by a transition from nanocaterpillar to nanostar morphologies (Fig. 4B). Water-dispersible nanocaterpillars were also prepared using NB2 (Fig. 4A). The nanocaterpillars were stable in tetrahydrofuran (THF), chloroform, and water, which highlights one of the advantages of preparing nanoparticles with INCP. The nanocaterpillars were also shown to undergo fluorescence quenching in the presence of trace amounts of nitroaromatics, including 2,4,6-trinitrotoluene (TNT), supporting their potential use in explosive sensing applications.
 | ||
| Fig. 4 (A) Synthesis of PPV-based BCPs. (B) AFM (on mica) and TEM (on a carbon-coated copper grid) images of the nano-object morphologies for PNB150-b-PPVn. Adapted from ref. 42 with permission from the Royal Society of Chemistry, copyright 2017. (C) AFM (on mica) images of the nanoparticle morphologies for PTD1200-b-PPVn. Adapted ref. 43 with permission from American Chemical Society, copyright 2018. (D) Proposed mechanism for fractal nano-object formation via INCP. (E) TEM (on a carbon-coated copper grid) images of PTD2200-b-PPV15-based nanostructures prepared with or without 3,5-DCP as an additive to tune the polymerization kinetics. Adapted from ref. 43 with permission from American Chemical Society, copyright 2018. | ||
We also explored the INCP of PPV-based BCPs having a PTD shell-block.43 The self-assembly behavior of PTD-b-PPVs was quite different from their PNB-based counterparts. In situ formation of nanospheres was still observed during the one-shot polymerization of PTD1200-b-PVPn. However, instead of forming uniform nanocaterpillars, rod-like and branched morphologies were formed with increasing polymerization time, then unexpected and unique fractal nano-objects were produced (Fig. 4C). To explain the origin of these unprecedented nano-fractal structures, kinetics studies were conducted. We observed slow initiation of PD from the living PTD blocks, followed by fast propagation, and thus, low initiation-to-propagation rate ratios (ki/kp). This implied that the polymerization was uncontrolled, leading to the gradual generation of BCPs and then micellar nanostructures (instead of simultaneous self-assembly). We proposed a model in which this gradual generation of nanospheres would maintain a sufficiently low nanoparticle concentration such that a diffusion limited aggregation mechanism would be operative (Fig. 4D).44–46 We imagined that if we could tune the polymerization kinetics to improve control of the polymerization, we would achieve uniform generation of nanospheres and observe the formation of nanocaterpillars (following the mechanism shown in Fig. 3). Switching to TD2 (which forms a shell-block with decreased steric bulk, enabling faster initiation of PD), fractal nano-objects were still observed (Fig. 4E). With the use of 3,5-dichloropyridine (3,5-DCP) as an additive (which reversibly binds to the propagating Ru species and slows PD propagation), a 13.5 times larger ki/kp value was obtained for PD (compared to polymerizations using TD1 without an additive). As expected, this resulted in a well-controlled block copolymerization, more uniform nanosphere formation, and the formation of nanocaterpillars (Fig. 4E). Therefore, we demonstrated that tuning the polymerization kinetics was a useful strategy to prepare unique nano-objects that would otherwise be challenging to accomplish via conventional equilibrium-driven self-assembly processes.
INCP with CP
A popular method to prepare functional PA derivatives is with the CP of 1,6-heptadiyne monomers.29 The resulting poly(cyclopentenylene-vinylene) (PCPV) polymers are fully conjugated and have functionalized 5-membered rings in their backbone. We found the combination of ROMP (for the first block) and CP (for the second block) to be effective for forming BCPs that could undergo the INCP process (Fig. 5A). Specifically, nanospheres were formed during the block copolymerization of NB3 and a Meldrum's acid functionalized 1,6-heptadienye (HD1).17 Thermolysis of the Meldrum's acid moieties generated ketene functional groups in the core-block, which underwent cycloaddition to afford cross-linked cores and improved the stability of the nanospheres. Nanospheres were also obtained during the block copolymerization of NB4 or TD1 and HD1.47 Higher-order morphologies (1D or 3D nanostructures) were not obtained from the INCP of PCPV-based BCPs. However, higher-order morphologies could be obtained by aging or photo-isomerization of the alkene bonds in the polymer backbone. The cis-to-trans isomerization of the PCPV core increased its rigidity, and therefore, its exposure to solvent. This enabled the self-assembly (with a mechanism analogous to that in Fig. 3) of PNB450-b-PCPV194-based nanospheres into nanocaterpillar and branched nanocaterpillar morphologies (Fig. 5B) and PTD50-b-PCPV130-based nanospheres into 3D micro-objects (Fig. 5C).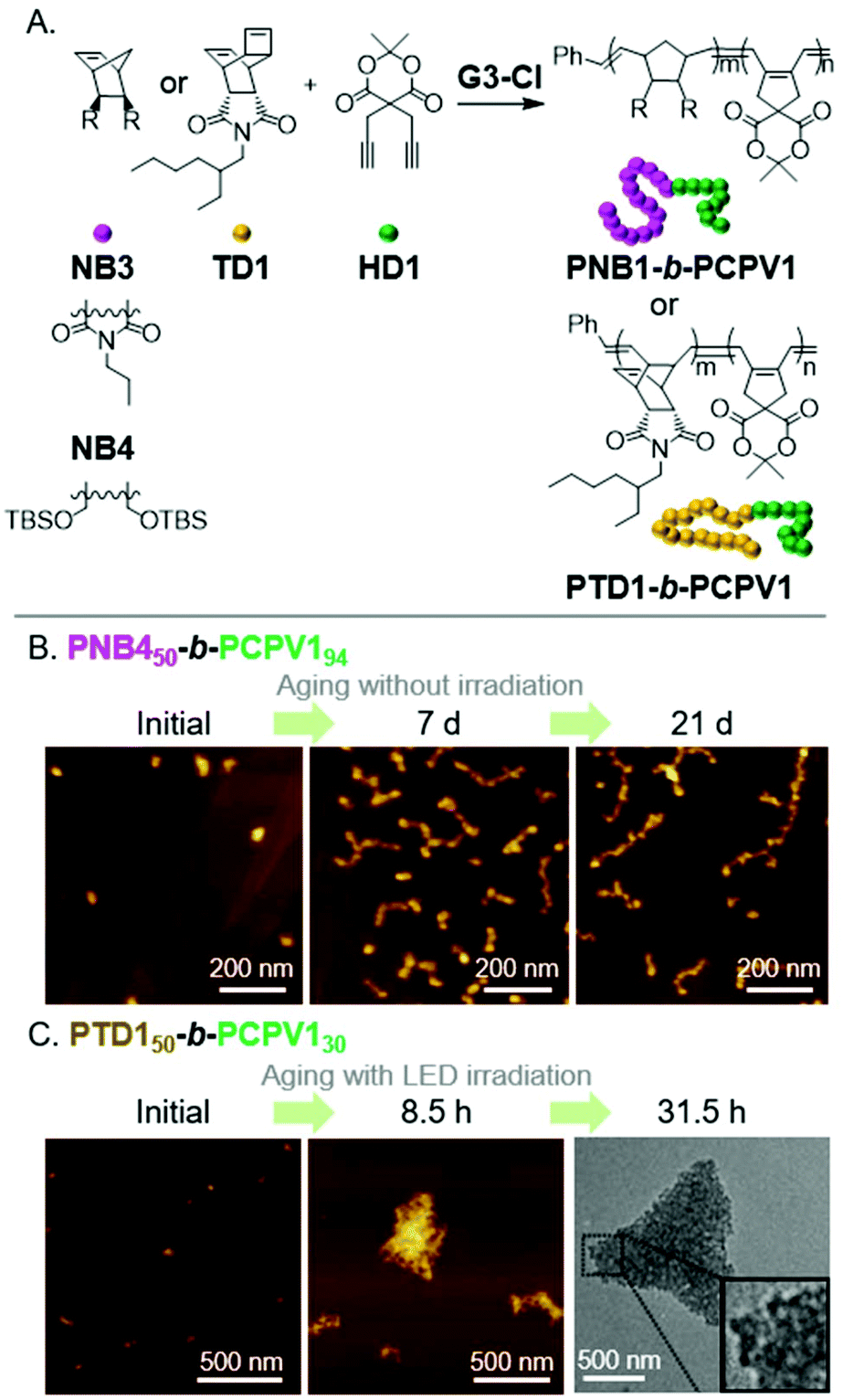 | ||
| Fig. 5 (A) Synthesis of PCPV-based BCPs. (B) Structural evolution of PNB450-b-PCPV194-based nanospheres (which were prepared via INCP) to nanocaterpillars and branched nanocaterpillars with aging (a non-INCP process), and (C) structural evolution of PTD150-b-PCPV130-based nanospheres (which were prepared via INCP) to 3D micro-objects with aging under light irradiation (another non-INCP process). Adapted from ref. 47 with permission from John Wiley and Sons, copyright 2017. | ||
To produce fully conjugated nano-objects without insulating shell blocks, we prepared BCPs using a soluble 1,6-heptadiyne monomer (HD2) and a fluorene-based monomer (HD3) (Fig. 6A).48 We found that the resulting PCPV250-b-PCPV3n underwent INCP, and self-assembled into long nanofibers (Fig. 6B) without going through an intermediate nanostructure (such as nanospheres). We attributed this difference in self-assembly behavior to the higher solubility of the PCPV3 block, compared to unfunctionalized PA blocks which underwent rapid nanoparticlization (as they were completely insoluble). Instead, the PCPV3 block underwent crystallization, forming well-defined cores for the nanofibers, with the PCPV2 block acting as a solubilizing shell (Fig. 6B). Remarkably, we also observed direct formation of higher-order nano-objects via INCP for PCPV3 homopolymers.49 Specifically, 2D leaf structures (Fig. 6C and D) were observed with in situ sampling during the polymerization of HD3 in THF. This result highlights one of the primary features that differentiates INCP from conventional PISA; that INCP is not limited to BCPs. Interestingly, when the leaf nano-objects were dispersed in chloroform, a morphological transition from leaf to rectangle morphologies was observed (Fig. 6D). Comparison of the diffraction patterns by fast Fourier transform (FFT) analysis of high-resolution TEM images for the two different morphologies provided insight into the self-assembly mechanism (Fig. 6E). The diffraction patterns suggested the presence of defects in chain packing for the leaf nano-objects and more ordered packing for the rectangular nano-objects. Specifically, the latter supported an orthorhombic crystal lattice and an interdigitated slip-stack packing of the polymer chains, without chain folding, leading to strong van der Waals and CH–π interactions serving as the driving force for self-assembly.
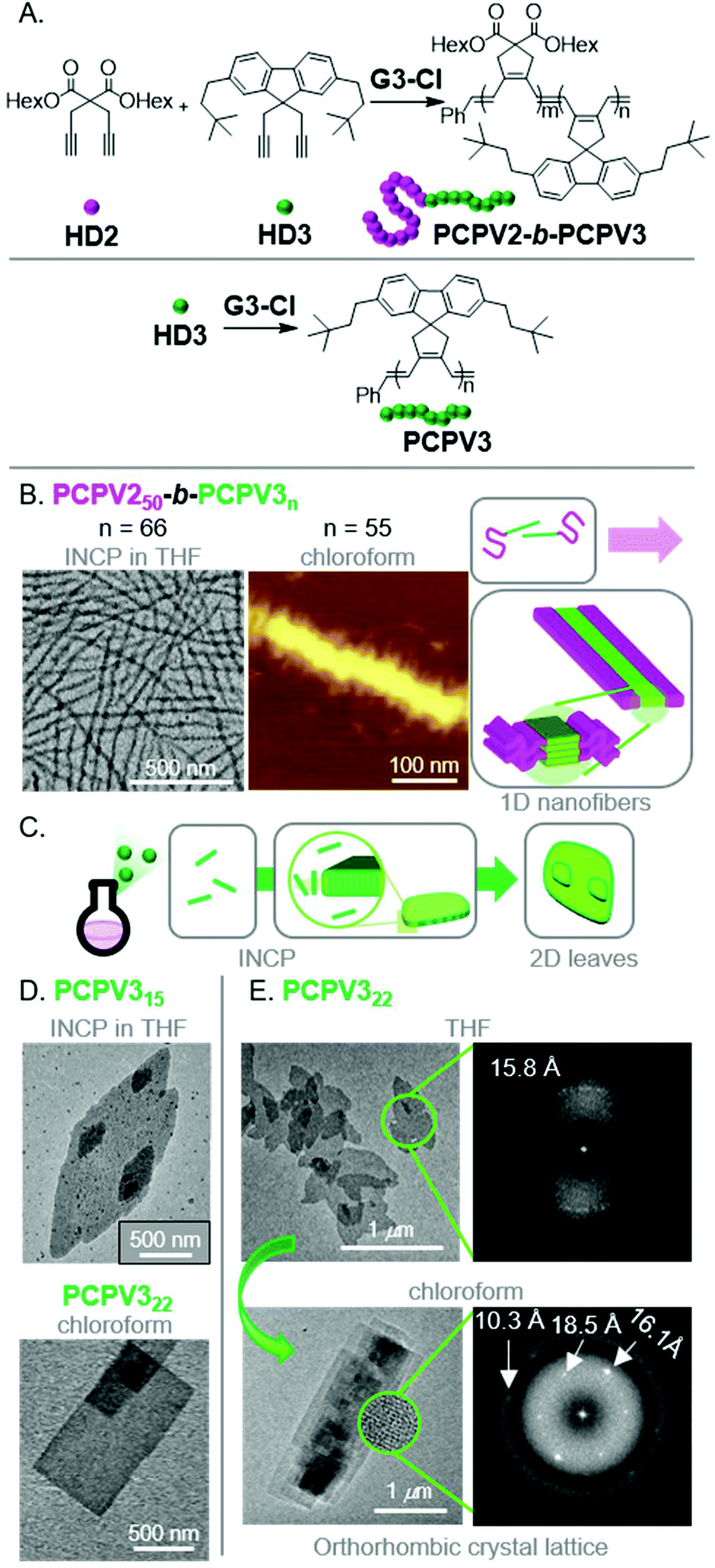 | ||
| Fig. 6 (A) Synthesis of PCPV-based BCPs and homopolymers. (B) TEM (on a carbon-coated copper grid) and AFM (on mica) images of the nanofibers for PCPV250-b-PCPV3n. Adapted from ref. 48 with permission from the Royal Society of Chemistry, copyright 2020. (C) Proposed mechanism for 2D leaf nano-object formation via INCP. (D) TEM (on a carbon-coated copper grid) images of PCPV3n-based 2D leaf nano-objects (via INCP) and rectangular nano-objects (after a non-INCP process), and (E) corresponding FFT electron diffraction patterns of each morphology. The highly ordered pattern from 2D rectangles in chloroform corresponded to the orthorhombic crystalline lattice with three main d-spacing values of 10.3, 16.1, and 18.5 Å. Adapted from ref. 49 with permission from American Chemical Society, copyright 2017. | ||
Xie, Sun, and coworkers observed micellular or aggregated structures from various PNB-b-PCPVs via INCP (Fig. 7A).50,51 These polymers showed good potential for obtaining unique, higher-order morphologies. For instance, PNB575-b-PCPV450 formed vesicles upon evaporating the polymerization solvent (increasing the nano-object concentration in THF, Fig. 7B). It was unclear if vesicles were formed during the polymerization as well, or only after the post-treatment. PNB675-b-PCPV425 formed nanotubes after changing from the polymerization solvent (THF) to chloroform (Fig. 7C). Direct formation of nanotubes, by conducting the polymerization in chloroform, was not explored. Li, Xie, and coworkers also demonstrated the formation of irregularly aggregated micellular structures from PNB6-b-PCPV5via INCP in chloroform.52 More uniform structures could be achieved by re-dispersing the nanostructures in THF, suggesting that more uniform nanospheres might have been achieved if THF was used as a polymerization solvent. Xie, Liao, and coworkers also observed self-assembly of PCPV homopolymers into unique structures.53PCPV7316 formed (concentration-dependent) nanospheres and hollow cylindrical structures in THF. PCPV629 formed (concentration-dependent) solid or hollow cylindrical structures in THF or chloroform, respectively (Fig. 7D). It was unclear if the cylindrical nano-objects were generated by INCP or were only a result of the solvent evaporation process in preparation for imaging. Regardless, these polymers represent promising candidates for further study.
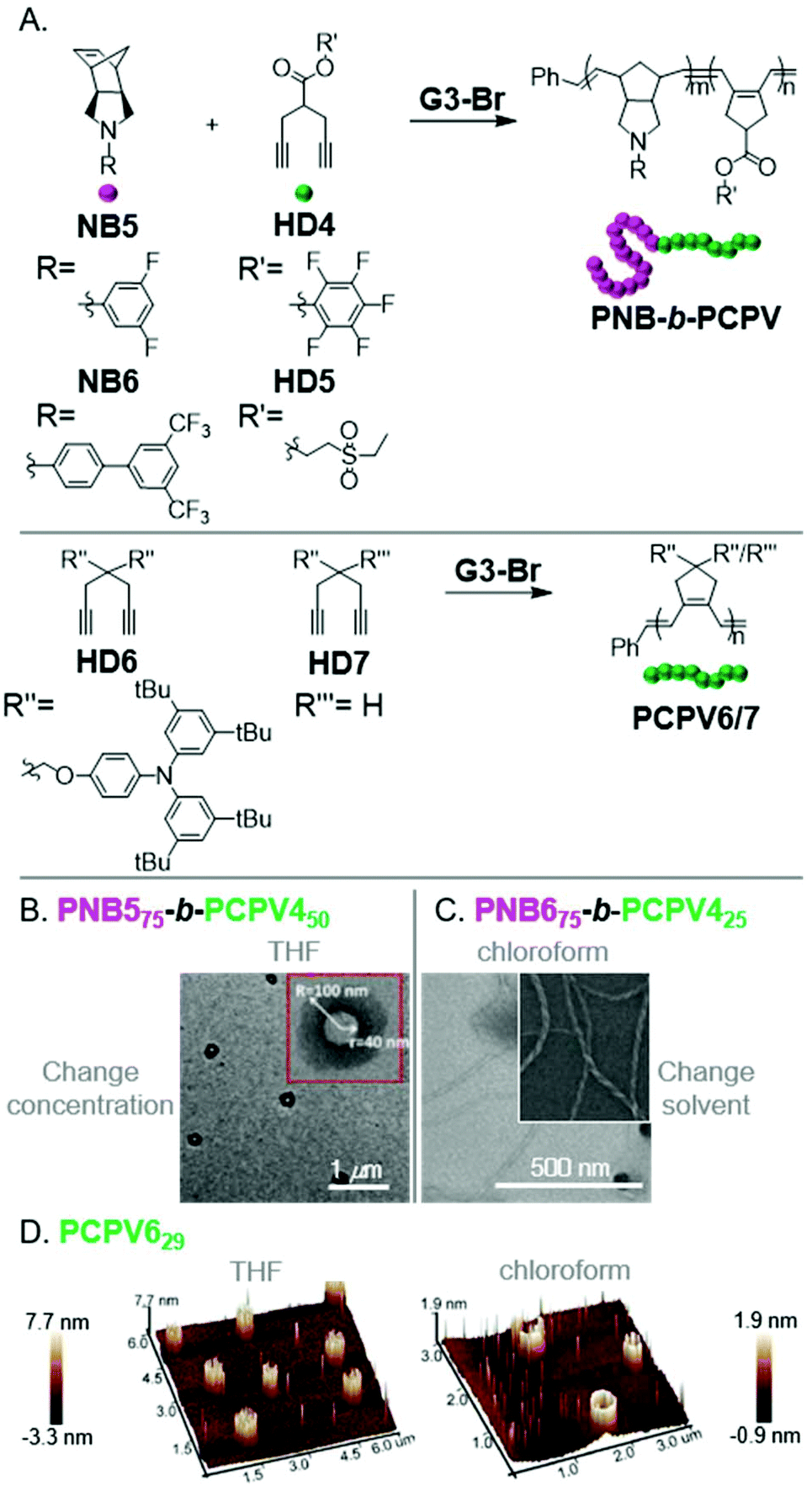 | ||
| Fig. 7 (A) Synthesis of PCPV-based BCPs and homopolymers. (B) TEM (on a carbon-coated copper grid) image of PNB575-b-PCPV450-based vesicles formed by changing solvent concentration. Adapted from ref. 50 with permission from the Royal Society of Chemistry, copyright 2015. (C) TEM (on a carbon-coated copper grid) image of PNB675-b-PCPV425-based nanotubes formed by changing the dispersion solvent. Adapted from ref. 51 with permission from the Royal Society of Chemistry, copyright 2017. (D) 3D AFM (on mica) images of cylindrical nano-objects from PCPV6. Adapted from ref. 53 with permission from American Chemical Society, copyright 2015. | ||
INCP with CTP
Polythiophene (PT) and its derivatives are among the most extensively studied conjugated polymers, having found use in a wide range of optoelectronic applications.54,55 With the use of CTP, we envisioned preparing fully conjugated BCPs that could undergo INCP.56 Toward that aim, we first prepared BCPs from T1 and T2 monomers using a Grignard metathesis (GRIM) polymerization, also known as a Kumada CTP (KCTP, Fig. 8A).57–60 We observed the formation and structural evolution of nano-objects analogous to that observed for PNB150-b-PPVn (Fig. 4B). That is, for PT1100-b-PT2n, mixtures of nanospheres and nanocaterpillers were first observed. As the PT2 block length increased, nanostars and then network morphologies were observed (Fig. 8B). Support for the nano-objects being formed by INCP was obtained by DLS measurements. Importantly, dispersions of the nano-objects were shown to be stable at elevated temperatures and under sonication conditions. These results can be contrasted with PT-based nanoparticles formed by crystallization-driven self-assembly (CDSA). For instance, poly(3-hexylthiophene)-b-poly(dimethylsiloxane)61 or poly(3-hexylthiophene)-b-poly(2-vinylpyridene)62 were shown to undergo self-assembly in a selective solvent (or mixed solvent system). Sonication of the resulting structures led to a drastic reduction in nanoparticle size (which was utilized for seeded-growth). These results further highlight the highly stable nature of nano-objects from INCP.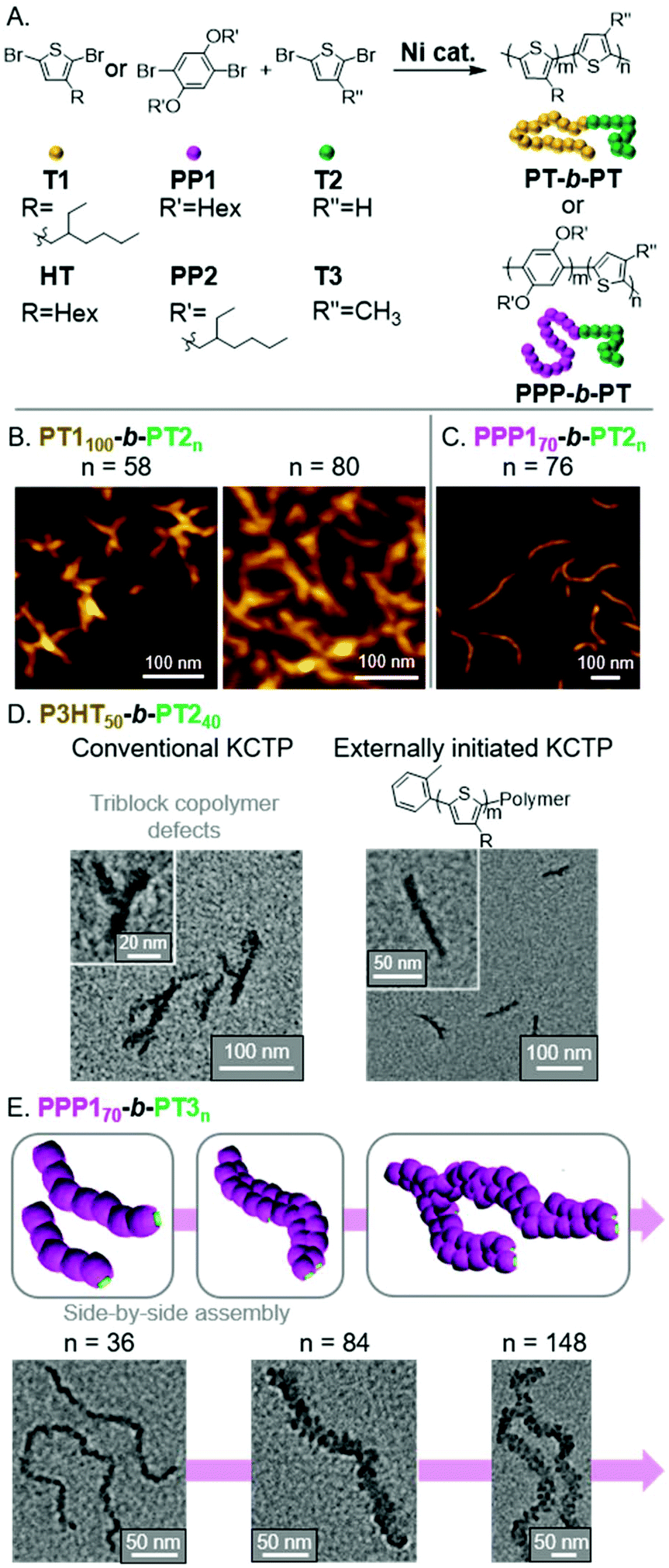 | ||
| Fig. 8 (A) Synthesis of PT-based BCPs. (B) AFM images of the nanostar to branched network evolution for PT1100-b-PT2n with increasing PT2 block length. Adapted from ref. 56 with permission from American Chemical Society, copyright 2013. (C) AFM image of nanocaterpillars from PPP170-b-PT2n. Adapted from ref. 63 with permission from the Royal Society of Chemistry, copyright 2014. (D) TEM images of P3HT50-b-PT240-based nano-objects using conventional or externally initiated KCTP. Adapted from ref. 64 with permission from the Royal Society of Chemistry, copyright 2016. (E) Schematic and TEM images for the generation of multi-line nanocaterpillars from PPP170-b-PT3n. Adapted from ref. 65 with permission from the Royal Society of Chemistry, copyright 2016. | ||
We next explored the influence of two different shell blocks on the PT-based nanoparticle morphology. We first looked at switching the first block to a dihexyloxy substituted poly(p-phenylene) (PPP1), which was expected to have a larger hydrodynamic radius than PT1 and would enable better shielding of the PT2 core-block.63 The resulting PPP170-b-PT2n formed well-defined nanocaterpillar structures (Fig. 8C), which still showed good stability to elevated temperatures and sonication. The second shell block we studied was poly(3-hexylthiophene) (P3HT), which we initially found to generate irregular aggregated nanostructures using conventional KCTP conditions (Fig. 8D).64 To improve the control of the nanoparticle morphology, we implemented an externally initiated KCTP method.66–69 The conventional KCTP leads to the production of triblock-impurities, whereas externally initiated KCTP gives clean diblock structures due to unidirectional polymerization from the o-tolyl-based Ni initiator/catalyst.
Specifically, defect-free o-tolyl-P3HT50-b-PT2n generated nanocaterpillars (with a PT2 DP of 40) and branched nanocaterpillars at higher PT2 block lengths (Fig. 8D).
Unique nano-object morphologies were observed by switching the core-block (from PT2 to PT3) for PPP1-based BCPs.65PT3 and PT2 undergo different types of crystalline packing. Specifically, PT3 and PT2 undergo staggered π-stacking (forming a face-centered lattice) and edge-to-face (herringbone) stacking, respectively.70 A consequence of the differences in packing is that PT3 has weaker π–π interactions. Interestingly, this led to the formation of multi-line nanostructures at higher PT3 DPs. Specifically, we observed the morphological evolution of nanospheres to nanocaterpillars to multi-line nanocaterpillars to branched multi-line nanocaterpillar structures (Fig. 8E). However, the weaker interactions came at the cost of decreased nano-object stability. While multi-line nanocaterpillars were robust to elevated temperatures, sonication resulted in their fragmentation into shorter multi-line structures. Very long nanocaterpillars (over 2 μm) could also be achieved by changing the PPP1 block to the more soluble PPP2, with branched 2-ethylhexyl side chains, without disrupting the ability to form multi-line nanocaterpillars.
INCP with other methods
The use of different polymerization methods enables a broader scope of polymer structures compatible with INCP, as well the potential to generate nano-objects with novel morphologies. Unfortunately, reports of using other polymerization methods for INCP (other than ROMP, CP, and CTP) are rare. Two recent examples describe the polymerization of acetylene-based monomers, specifically phenyl acetylenes (PA1 and PA2)71 and propargyl alcohol (PGA),72 to give substituted-PA backbones (Fig. 9A). The resulting polymers were able to form complex nano-objects via INCP. For instance, PPA180-b-PPA2n formed vesicles (with a PPA2 DP of 136).71 As the PPA2 block length increased, the vesicles transitioned to ribbons/lamellae (DP of 190), then helical ribbons (DP of 390, Fig. 9B). The morphological transition (Fig. 9C) was driven by the PPA2 block's solubility, which decreases with increasing length due to the formation of a contracted helical conformation stabilized by intramolecular hydrogen bonds. In the other example, PPGA homopolymers were shown to generate various solvent-dependent nano-object morphologies.72 Specifically, polymerizations in chloroform, dimethylformamide (DMF), or water led to the formation of micellular nanospheres, nanostars, or nanonetwork structures, respectively (Fig. 9D). This free radical polymerization also induced differing extents of oxygenation (in DMF and water) or chlorination (with chloroform) of the polymer backbones, making it difficult to elucidate why the various nano-object morphologies were observed. This work, however, is significant as it demonstrates that INCP is also feasible using non-controlled polymerization conditions.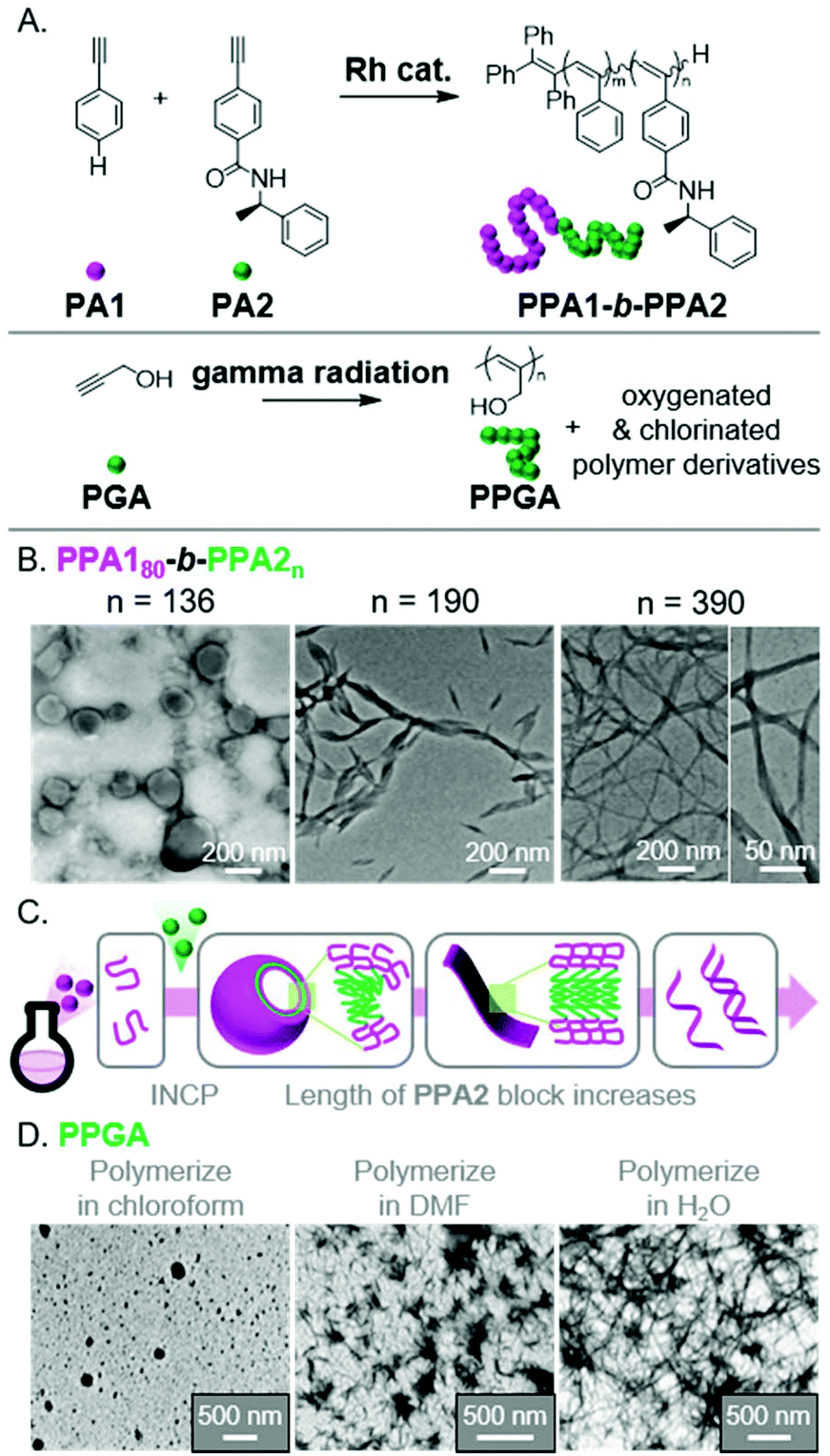 | ||
| Fig. 9 (A) Synthesis of functional PA BCPs (PPAX) and homopolymers (PPGA). (B) TEM images of the vesicle to helical ribbon evolution for PPA180-b-PPA2n with increasing PPA2 block length. Adapted from ref. 71 with permission from American Chemical Society, copyright 2020. (C) Proposed mechanism for nano-object formation from PPAX-based BCPs. (D) TEM images of the micellular, nanostar, and network nanostructures from PPGAvia INCP in different solvents. Adapted from ref. 72 with permission from Springer Nature, copyright 2018. | ||
Outlook
In comparison to the broad scope of known conjugated polymers, only a small fraction has been utilized for INCP. This is in part due to the requirements of chain-growth and living polymerizations to achieve BCP formation. Additional controlled polymerization methodologies can be explored to further expand the scope of applicable conjugated polymer structures. For instance, there are a broad range of CTPs that have not been explored in INCP (e.g., Suzuki–Miyaura, Stille, Negishi, etc.).32 Monomer deactivation polymerizations may also be useful, as they have been applied to the synthesis of block copolymers.73 Cascade polymerizations can also generate conjugated polymers with more diverse structures (e.g., the metathesis and metallotropy polymerization).74,75 For homopolymers, however, less controlled polymerizations might be utilized (e.g., the free radical polymerization of PGA, see Fig. 9), which could significantly increase the polymer scope. As previously described, in situ nanoparticlization can be achieved with dispersion polymerizations employing various step-growth polymerizations (based on Suzuki–Miyaura,23,76,77 Heck,22 and Sonogashira,78 coupling reactions). With careful monomer design, it might be possible to eliminate the stabilizers and achieve INCP using step-growth polymerizations. Achieving transition metal-free synthesis of conjugated polymers, using methods such as the Horner–Wadsworth–Emmons polymerization,79 would also provide a notable advance given that trace metals can have deleterious effects on a polymer's optoelectronic performance.80 The use of catalyst-free photo-controlled polymerizations could also be advantageous in that regard.81 Expanding the available polymerization methodologies for INCP should greatly increase the scope of conjugated polymer structures, which may lead to a greater breadth of optoelectronic properties and nano-object morphologies.We can also look to other self-assembly methods for inspiration. Various conjugated polymers (homopolymers or within BCPs) have been shown to undergo self-assembly using methods such as CDSA or self-seeding. While some of those polymers have already been explored in INCP, they have been shown to produce different nano-objects morphologies (than INCP), such as nanowires from oligo(p-phenylenevinylene)82,83 and 2D squares from PPV.84 Some of these polymers may be well suited for INCP, such as oligo(p-phenylene ethynylene) (which forms nanowires)85 and poly(9,9-dihexylfluorene) (which forms spherical nanoparticles and nanowires).86–89 Other polymers may be more challenging to adapt to INCP, such as P3HT (which was previously utilized as the soluble block in INCP), despite its ability to form nanofibers and 2D-rectangles using other methods.61,62,90–95 Similarly, poly(3-octylthiophene) (which forms nanowires)96 and poly[3-(2,5,8,11-tetraoxatridecanyl)thiophene] (which forms nanoribbons)97 would also be challenging due to their side chains which provide good solubility.
Other self-assembly methods can also provide insight into new nano-object morphologies that might be obtained with INCP (if proper polymers and polymerization conditions were used). For instance, higher-order morphological structures such as “octopi” or “jellyfish” were obtained by conventional PISA with the addition of a small amount of solvophilic comonomer into the insoluble block of a BCP.98 The use of copolymer core-blocks in PISA has also enabled inverse morphologies such as compound vesicles, sponges, and cubosomes.99 Specific examples, based on CDSA, which might be adaptable to INCP include the formation of diamond or hexagonal 2D nano-objects.100,101 One distinct advantage of CDSA over INCP is the ability to precisely control the structural dimensions of nano-objects.101,102 Further exploration of such control should done for INCP. Furthermore, even more complex structures can be achieved with CDSA due to the use of multi-polymer systems.103,104 Conducting INCP in the presence of other polymers could be an interesting area of exploration to generate novel nano-objects.
To date, the majority of INCP examples have focused on expanding the scope of polymers and nano-object morphologies, as well as understanding the mechanisms of self-assembly. More thorough characterization of nano-object properties (and perhaps morphology-dependent properties) will be important to identifying potential applications for which they would be best suited. Furthermore, despite the use of semiconducting polymers, the optoelectronic properties of the nano-objects have not been well characterized. As described above, semiconducting nanoparticles generally have a broad range of applications (sensors, imaging agents, photovoltaics, light-emitting diodes, and catalysts),105 and given that unique morphologies can be obtained with INCP, there is good reason to explore these applications in detail.
Conclusions
INCP is a useful method to prepare nano-objects directly from polymerizations, without post-treatment steps. Only a few polymerization types have been demonstrated with INCP, however, the polymerization kinetics, polymer dispersity, defects, and impurities can all influence the self-assembly process, leading to a wide range of nano-object morphologies. Advantages of INCP (compared to conventional PISA) include more stable nano-objects, alternative nano-object morphologies, and the ability to use homopolymers. The use of conjugated polymers also provides semiconducting properties, which may yield promising applications, although this remains a topic which has been little explored. With the adoption of new polymerization techniques and polymer structures, the scope of INCP can be expanded further and will ideally lead to new functional self-assembled nano-objects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the NRF of Korea through the following grants: Nano-Material Technology Development Program, Postdoctoral Fellowship Program (Nurturing Next-generation Researchers, Grant 2020R1A6A3A01096563), and Young Investigator Grant (Grant 2018R1C1B6003054).Notes and references
- J. Lequieu and A. J. D. Magenau, Polym. Chem., 2020 10.1039/D0PY00722F.
- J. C. Brendel and F. H. Schacher, Chem. – Asian J., 2018, 13, 230–239 CrossRef CAS.
- J. J. Shin, E. J. Kim, K. H. Ku, Y. J. Lee, C. J. Hawker and B. J. Kim, ACS Macro Lett., 2020, 9, 306–317 CrossRef CAS.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, Macromolecules, 2017, 50, 3439–3463 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- S. R. Mane, New J. Chem., 2020, 44, 6690–6698 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- S. Varlas, J. C. Foster and R. K. O'Reilly, Chem. Commun., 2019, 55, 9066–9071 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 CrossRef.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 CrossRef.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 CrossRef.
- W.-J. Zhang, J. Kadirkhanov, C.-H. Wang, S.-G. Ding, C.-Y. Hong, F. Wang and Y.-Z. You, Polym. Chem., 2020, 11, 3654–3672 RSC.
- K.-Y. Yoon, I.-H. Lee, K. O. Kim, J. Jang, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 14291–14294 CrossRef CAS.
- J. Kim, E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2012, 1, 1090–1093 CrossRef CAS.
- D. Tuncel, Nanoscale Adv., 2019, 1, 19–33 RSC.
- J. Li, J. Rao and K. Pu, Biomaterials, 2018, 155, 217–235 CrossRef CAS.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS.
- S. Ciftci and A. J. C. Kuehne, Macromolecules, 2015, 48, 8389–8393 CrossRef CAS.
- A. J. C. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1088 CrossRef.
- J. Huber, C. Jung and S. Mecking, Macromolecules, 2012, 45, 7799–7805 CrossRef CAS.
- J. Pecher, J. Huber, M. Winterhalder, A. Zumbusch and S. Mecking, Biomacromolecules, 2010, 11, 2776–2780 CrossRef CAS.
- O. Nuyken and S. D. Pask, Polymer, 2013, 5, 361–403 Search PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1990, 23, 158–165 CrossRef CAS.
- G. I. Peterson, S. Yang and T.-L. Choi, Acc. Chem. Res., 2019, 52, 994–1005 CrossRef CAS.
- D. Pasini and D. Takeuchi, Chem. Rev., 2018, 118, 8983–9057 CrossRef CAS.
- M. R. Buchmeiser, Polym. Rev., 2017, 57, 15–30 CrossRef CAS.
- M. A. Baker, C.-H. Tsai and K. J. T. Noonan, Chem. – Eur. J., 2018, 24, 13078–13088 CrossRef CAS.
- R. Grisorio and G. P. Suranna, Polym. Chem., 2015, 6, 7781–7795 RSC.
- Z. J. Bryan and A. J. McNeil, Macromolecules, 2013, 46, 8395–8405 CrossRef CAS.
- H. Shirakawa, Angew. Chem., Int. Ed., 2001, 40, 2574–2580 CrossRef.
- F. L. Klavetter and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 7807–7813 CrossRef CAS.
- K.-Y. Yoon, I.-H. Lee and T.-L. Choi, RSC Adv., 2014, 4, 49180–49185 RSC.
- K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, Macromol. Rapid Commun., 2015, 36, 1069–1074 CrossRef CAS.
- S. Shin, K.-Y. Yoon and T.-L. Choi, Macromolecules, 2015, 48, 1390–1397 CrossRef CAS.
- F. Dutertre, K.-T. Bang, E. Vereroudakis, B. Loppinet, S. Yang, S.-Y. Kang, G. Fytas and T.-L. Choi, Macromolecules, 2019, 52, 3342–3350 CrossRef CAS.
- N. Zaquen, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2016, 7, 1355–1367 RSC.
- S. Shin, J. Lim, M.-L. Gu, C.-Y. Yu, M. Hong, K. Char and T.-L. Choi, Polym. Chem., 2017, 8, 7507–7514 RSC.
- S. Shin, M.-L. Gu, C.-Y. Yu, J. Jeon, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2018, 140, 475–482 CrossRef CAS.
- J. R. Nicolás-Carlock, J. L. Carrillo-Estrada and V. Dossetti, Sci. Rep., 2016, 6, 19505 CrossRef.
- L. M. Sander, Contemp. Phys., 2000, 41, 203–218 CrossRef.
- T. A. Witten and L. M. Sander, Phys. Rev. Lett., 1981, 47, 1400–1403 CrossRef CAS.
- E.-H. Kang, S. Yang, S. Y. Yu, J. Kim and T.-L. Choi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3058–3066 CrossRef CAS.
- S. Yang and T.-L. Choi, Chem. Sci., 2020, 11, 8416–8424 RSC.
- S. Yang, S. Shin, I. Choi, J. Lee and T.-L. Choi, J. Am. Chem. Soc., 2017, 139, 3082–3088 CrossRef CAS.
- W. Liu, X. Liao, Y. Li, Q. Zhao, M. Xie and R. Sun, Chem. Commun., 2015, 51, 15320–15323 RSC.
- W. Liu, J. Chen, D. Zhou, X. Liao, M. Xie and R. Sun, Polym. Chem., 2017, 8, 725–734 RSC.
- J. Chen, R. Sun, X. Liao, H. Han, Y. Li and M. Xie, Macromolecules, 2018, 51, 10202–10213 CrossRef CAS.
- M. Guo, R. Sun, H. Han, J. Wu, M. Xie and X. Liao, Macromolecules, 2015, 48, 2378–2387 CrossRef CAS.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- T. M. S. K. Pathiranage, D. S. Dissanayake, C. N. Niermann, Y. Ren, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3327–3346 CrossRef CAS.
- I.-H. Lee, P. Amaladass, K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 17695–17698 CrossRef CAS.
- R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012–16013 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- S. K. Patra, R. Ahmed, G. R. Whittell, D. J. Lunn, E. L. Dunphy, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2011, 133, 8842–8845 CrossRef CAS.
- J. Gwyther, J. B. Gilroy, P. A. Rupar, D. J. Lunn, E. Kynaston, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Chem. – Eur. J., 2013, 19, 9186–9197 CrossRef CAS.
- I.-H. Lee, P. Amaladass and T.-L. Choi, Chem. Commun., 2014, 50, 7945–7948 RSC.
- I.-H. Lee and T.-L. Choi, Polym. Chem., 2016, 7, 7135–7141 RSC.
- I.-H. Lee, P. Amaladass, I. Choi, V. W. Bergmann, S. A. L. Weber and T.-L. Choi, Polym. Chem., 2016, 7, 1422–1428 RSC.
- H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS.
- M. Verswyvel, F. Monnaie and G. Koeckelberghs, Macromolecules, 2011, 44, 9489–9498 CrossRef CAS.
- E. F. Palermo and A. J. McNeil, Macromolecules, 2012, 45, 5948–5955 CrossRef CAS.
- T. Yamamoto, D. Komarudin, M. Arai, B.-L. Lee, H. Suganuma, N. Asakawa, Y. Inoue, K. Kubota, S. Sasaki, T. Fukuda and H. Matsuda, J. Am. Chem. Soc., 1998, 120, 2047–2058 CrossRef CAS.
- J. Chen, S. Cai, R. Wang, S. Wang, J. Zhang and X. Wan, Macromolecules, 2020, 53, 1638–1644 CrossRef CAS.
- A. A. Abdel-Fattah, Y. S. Soliman and M. M. Ghobashy, J. Polym. Res., 2018, 25, 106 CrossRef.
- L. Verheyen, P. Leysen, M.-P. Van Den Eede, W. Ceunen, T. Hardeman and G. Koeckelberghs, Polymer, 2017, 108, 521–546 CrossRef CAS.
- G. I. Peterson and T.-L. Choi, Chem. Sci., 2020, 11, 4843–4854 RSC.
- J. Yuan, W. Wang, Z. Zhou and J. Niu, Macromolecules, 2020, 53, 5655–5673 CrossRef CAS.
- J. B. ten Hove, J. Appel, J. M. van den Broek, A. J. C. Kuehne and J. Sprakel, Small, 2014, 10, 957–963 CrossRef CAS.
- N. Anwar, T. Willms, B. Grimme and A. J. C. Kuehne, ACS Macro Lett., 2013, 2, 766–769 CrossRef CAS.
- N. Anwar, A. Rix, W. Lederle and A. J. C. Kuehne, Chem. Commun., 2015, 51, 9358–9361 RSC.
- S. Ciftci, F. Jansen, V. Chimisso, J. Kler, K. Rahimi and A. J. C. Kuehne, Polym. Chem., 2018, 9, 2428–2433 RSC.
- F. C. Krebs, R. B. Nyberg and M. Jørgensen, Chem. Mater., 2004, 16, 1313–1318 CrossRef CAS.
- E. F. Woods, A. J. Berl and J. A. Kalow, Angew. Chem., Int. Ed., 2020, 59, 6062–6067 CrossRef CAS.
- D. Tao, C. Feng, Y. Lu, Y. Cui, X. Yang, I. Manners, M. A. Winnik and X. Huang, Macromolecules, 2018, 51, 2065–2075 CrossRef CAS.
- D. Tao, Y. Cui, X. Huang, G. Lu, I. Manners, M. A. Winnik and C. Feng, J. Colloid Interface Sci., 2020, 560, 50–58 CrossRef CAS.
- L. Han, M. Wang, X. Jia, W. Chen, H. Qian and F. He, Nat. Commun., 2018, 9, 865 CrossRef.
- J. Nie, Z. Wang, X. Huang, G. Lu and C. Feng, Macromolecules, 2020, 53, 6299–6313 CrossRef CAS.
- X. Xiao, W. Bai and J. Lin, Polym. Bull., 2014, 71, 2103–2112 CrossRef CAS.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Science, 2018, 360, 897–900 CrossRef CAS.
- H. Shaikh, X.-H. Jin, R. L. Harniman, R. M. Richardson, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2020, 142, 13469–13480 CrossRef CAS.
- S. T. G. Street, Y. He, X.-H. Jin, L. Hodgson, P. Verkade and I. Manners, Chem. Sci., 2020, 11, 8394–8408 RSC.
- S. Pan, M. Zhu, L. He, H. Zhang, F. Qiu, Z. Lin and J. Peng, Macromol. Rapid Commun., 2018, 39, 1800048 CrossRef.
- J. Qian, X. Li, D. J. Lunn, J. Gwyther, Z. M. Hudson, E. Kynaston, P. A. Rupar, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 4121–4124 CrossRef CAS.
- X. Li, P. J. Wolanin, L. R. MacFarlane, R. L. Harniman, J. Qian, O. E. C. Gould, T. G. Dane, J. Rudin, M. J. Cryan, T. Schmaltz, H. Frauenrath, M. A. Winnik, C. F. J. Faul and I. Manners, Nat. Commun., 2017, 8, 15909 CrossRef CAS.
- Y. Guo, Y. Han and Z. Su, J. Phys. Chem. B, 2013, 117, 14842–14848 CrossRef CAS.
- R. Qi, Y. Zhu, L. Han, M. Wang and F. He, Macromolecules, 2020, 53, 6555–6565 CrossRef CAS.
- T. Fukui, J. D. Garcia-Hernandez, L. R. MacFarlane, S. Lei, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2020, 142, 15038–15048 CrossRef CAS.
- U. Tritschler, J. Gwyther, R. L. Harniman, G. R. Whittell, M. A. Winnik and I. Manners, Macromolecules, 2018, 51, 5101–5113 CrossRef CAS.
- A. C. Kamps, M. H. M. Cativo, M. Fryd and S.-J. Park, Macromolecules, 2014, 47, 161–164 CrossRef CAS.
- D. Li, M. Huo, L. Liu, M. Zeng, X. Chen, X. Wang and J. Yuan, Macromol. Rapid Commun., 2019, 40, 1900202 CrossRef.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 CrossRef.
- X. He, M.-S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481–488 CrossRef CAS.
- M. Inam, G. Cambridge, A. Pitto-Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223–4230 RSC.
- M. E. Robinson, A. Nazemi, D. J. Lunn, D. W. Hayward, C. E. Boott, M.-S. Hsiao, R. L. Harniman, S. A. Davis, G. R. Whittell, R. M. Richardson, L. De Cola and I. Manners, ACS Nano, 2017, 11, 9162–9175 CrossRef CAS.
- X. Li, Y. Gao, C. E. Boott, M. A. Winnik and I. Manners, Nat. Commun., 2015, 6, 8127 CrossRef.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2020 DOI:10.1038/s41578-020-00233-4.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |