
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-opening metathesis polymerization-induced self-assembly (ROMPISA)
Spyridon
Varlas
 ,
Jeffrey C.
Foster
* and
Rachel K.
O'Reilly
*
,
Jeffrey C.
Foster
* and
Rachel K.
O'Reilly
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: J.C.Foster@bham.ac.uk; R.OReilly@bham.ac.uk
First published on 15th July 2019
Abstract
Polymerization-induced self-assembly (PISA) has simplified the preparation of polymeric nanoparticles, expanding their commercial importance. Recently, PISA mediated via ring-opening metathesis polymerization (ROMPISA) has emerged as a powerful alternative to existing PISA methodologies. ROMPISA can be conducted under air in minutes, producing nano-object morphologies with unique characteristics. Herein, we highlight recent advances in this field.
 Spyridon Varlas | Spyridon Varlas received his BSc in Chemistry from the National and Kapodistrian University of Athens (Greece) in 2014. He then continued his studies at the same institution under the supervision of Prof. Hermis Iatrou, where he obtained a 1st class MSc in Polymer Science in 2016, studying the self-assembly behavior of stimuli-responsive polypeptide-based nanostructures for drug delivery applications. Currently, he is pursuing a PhD in Chemistry at the University of Birmingham (UK) under the supervision of Prof. Rachel K. O’Reilly focusing on the development of functional polymersome nanoreactors via RAFT- and ROMP-mediated polymerization-induced self-assembly (PISA). |
 Jeffrey C. Foster | Jeffrey C. Foster obtained his BS and MS degrees from California Polytechnic State University, San Luis Obispo in 2010 and 2012, respectively, and his PhD in Chemistry at Virginia Tech under the supervision of Prof. John Matson. After a postdoctoral fellowship at the University of Warwick (UK) with Prof. Rachel K. O’Reilly, he was promoted to the position of Group Leader at the University of Birmingham (UK). His research focuses on the use of metathesis chemistry for polymer-based applications in aqueous milieu. |
 Rachel K. O'Reilly | Rachel K. O’Reilly received her first degree from the University of Cambridge (UK), working with Professor Brian Johnson FRS for her Masters project. She then went on to complete her PhD at Imperial College London (UK) in 2003, working with Professor Vernon C. Gibson. Her PhD was in collaboration with BP and focused on organometallic catalysts for radical polymerization. She then moved to the US working as a postdoctoral fellow under the joint direction of Professors Craig J. Hawker (IBM Almaden) and Karen L. Wooley (Washington University in Saint Louis). In 2004, she was awarded a Royal Commission for the Exhibition of 1851 research fellowship which she held in the US for 1 year before returning to the UK in 2005. She then started her independent career in 2005 at the University of Cambridge (UK) as a Royal Society Dorothy Hodgkin Fellow. In 2009, she moved to the University of Warwick (UK) and in 2012 was promoted to full Professor. In January 2018, she took up a position of Chair in the School of Chemistry at the University of Birmingham (UK), and was appointed as Head of School later that year. |
In recent years, a rapidly expanding methodology for the facile and reproducible fabrication of block copolymer nano-objects has emerged – termed polymerization-induced self-assembly (PISA).1–8 Contrary to conventional block copolymer self-assembly processes, where multiple laborious post-polymerization steps under dilute conditions ([solids] ≤ 1% w/w) are generally required for the development of self-assembled nanostructures, the high efficiency and broad applicability of PISA lies in the fact that polymerization and self-assembly occur simultaneously in a single synthetic step at high solids concentration (typically 10–30% w/w).9 Fundamental studies on the PISA mechanism revealed that in situ self-assembly of amphiphilic block copolymers is achieved upon chain-extension of a solvophilic corona-forming polymer using appropriately selected core-forming monomers such that, after a critical point, the growing polymer chains become insoluble in the reaction medium, resulting in particle micellization (Scheme 1). Morphological evolution toward higher-order nanostructures, such as worm-like micelles and polymersomes, is commonly observed during PISA and is dictated by continuous alterations of the relative volume fractions (i.e., packing parameter) between the corona- and core-forming blocks upon increasing the degree of polymerization (DP) and/or total solids content.1,10 Importantly, the resulting PISA morphologies can also be tuned through a plethora of additional parameters, such as the length of the stabilizer block, core-block hydrophobicity, solvent composition, stimuli-responsiveness, and copolymer architecture.11,12
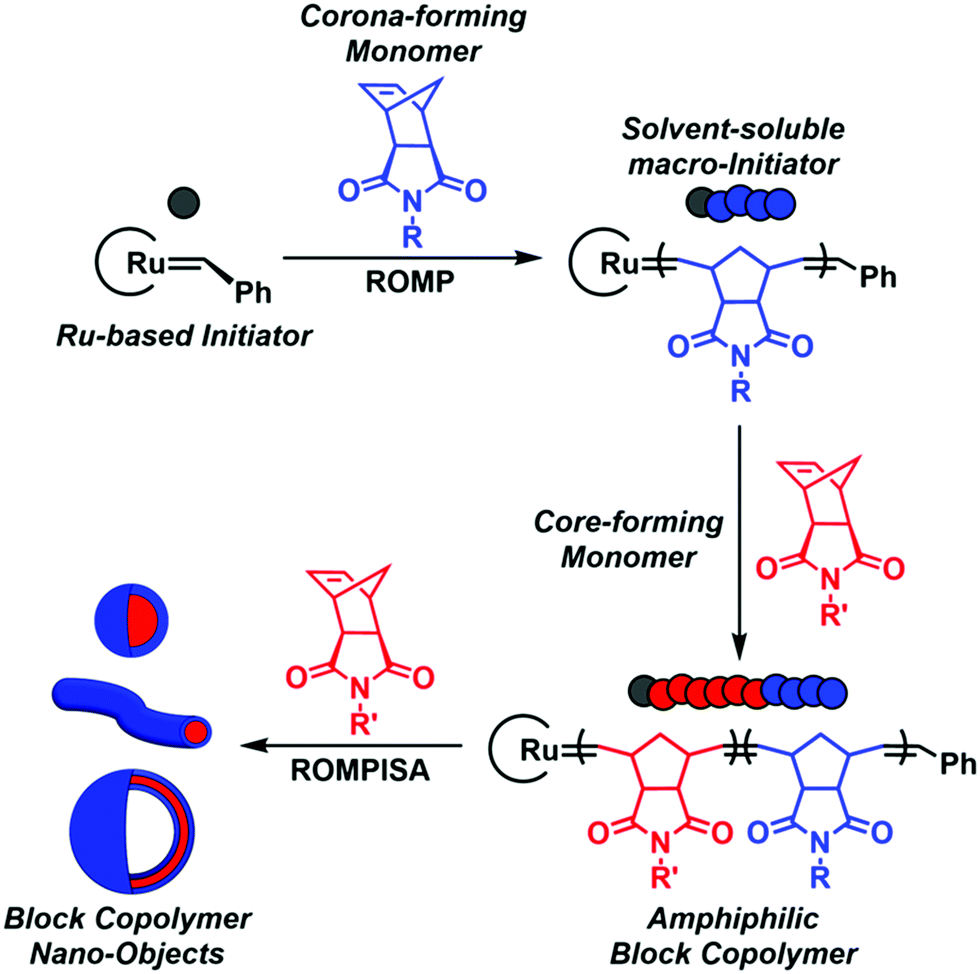 | ||
| Scheme 1 Schematic representation of ROMPISA mechanism mediated by a Ru-based initiator for in situ formation of block copolymer nano-objects of various morphologies. | ||
In the majority of PISA reports thus far, reversible-deactivation radical polymerization (RDRP) techniques including atom transfer radical polymerization (ATRP),13,14 reversible addition–fragmentation chain-transfer (RAFT) polymerization,3,15–17 nitroxide-mediated polymerization (NMP),18,19 and organometallic-mediated radical polymerization (OMRP)20 are utilized for the preparation of block copolymer nano-objects under dispersion or emulsion polymerization conditions.9,21 In addition to traditional thermally initiated RDRP approaches, a significant number of studies have focused on visible light-initiated PISA (photo-PISA) mediated by wavelength-specific photoinitiators and special photoredox catalysts or via the “photoiniferter” mechanism of chain transfer agents for the development of well-defined nano-objects at ambient reaction temperatures with biologically relevant applications.22–27
Recently, ring-opening metathesis polymerization-mediated PISA (ROMPISA) has been established as an alternative non-radical methodology for the synthesis of polynorbornene (PNB)-based nano-objects of various morphologies in both organic and aqueous milieu owing to the robustness and functional group tolerance (alcohols, ethers, carbonyl compounds, disulfides, thioethers, imines, tertiary amines, and others are tolerated) of Ru-based catalysts that also allow for ROMPISA under mild reaction conditions.28–30 ROMPISA can be carried out under air on the timescale of minutes, and interesting backbone chemistries (i.e., degradable linkages) can be more readily introduced through the use of functional cyclic monomers compared to radical polymerization strategies. In addition, the excellent molecular weight control afforded by ROMP can allow for the synthesis of PNB copolymers with high blocking efficiencies over a wide range of architectures, number of blocks (multi-block copolymers with ≥10 blocks have been reported), and molecular weight (up to 106 Da).31,32 However, study of aqueous ROMPISA has been limited due the lack of water-soluble and highly active metathesis catalysts as well as the small number of reported core-forming ROMPISA monomers. We believe that this field will greatly expand in the near future as new catalysts developed for aqueous metathesis of small molecules and the identification of new core-forming monomers have provided a roadmap toward overcoming these key challenges.33,34
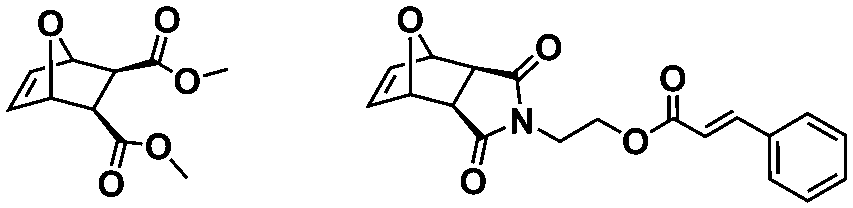
To the best of our knowledge, the first study introducing the concept of in situ preparation of block copolymer nano-objects via ROMPISA was reported by Xie and coworkers.35 In this early report, a solvent-soluble poly(2,3-bis(2-bromoisobutyryloxymethyl)-5-norbornene) (PBNBE) macroinitiator was first synthesized using (PCy3)2(Cl)2Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (G1) as the catalyst. Spherical micelles ranging from 140 to 200 nm were formed upon chain-extension of PBNBE macroinitiator in toluene using an oxanorbornene dicarboxylic acid dimethyl ester (ONBDM) monomer as the core-forming block. The same group extended this work by preparing micelles bearing photo-cross-linkable moieties within their cores via ROMPISA copolymerization of a cinnamyl-based ONB monomer and ONBDM in toluene, using a PBNBE macroinitiator.36 UV irradiation resulted in core-cross-linked micelles, whilst the retention of their morphology after dissolution in a good solvent for all blocks was also confirmed.
CHPh (G1) as the catalyst. Spherical micelles ranging from 140 to 200 nm were formed upon chain-extension of PBNBE macroinitiator in toluene using an oxanorbornene dicarboxylic acid dimethyl ester (ONBDM) monomer as the core-forming block. The same group extended this work by preparing micelles bearing photo-cross-linkable moieties within their cores via ROMPISA copolymerization of a cinnamyl-based ONB monomer and ONBDM in toluene, using a PBNBE macroinitiator.36 UV irradiation resulted in core-cross-linked micelles, whilst the retention of their morphology after dissolution in a good solvent for all blocks was also confirmed.
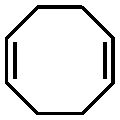
Significant advancements in the field of self-assembled nanostructures via ROMPISA in organic media have also been demonstrated by Choi and coworkers. In a seminal study, a solvophilic polynorbornene (PNB) macroinitiator was chain-extended using cyclooctatetraene (COT) as the solvent-miscible monomer for formation of a gradually insoluble polyacetylene (PA) block and (H2IMes)(py)2(Cl)2RuCHPh (G3) as the catalyst in various organic solvents under mild reaction conditions.37 Interestingly, spontaneous self-assembly of the growing PNB-b-PA diblock copolymer resulted in the development of morphologies ranging from small micelles to progressively larger populations of long “nanocaterpillars” as the DP of the PA block increased (Fig. 1). The formation of the latter intriguing morphology was attributed to strong π–π interactions between adjacent spheres to minimize the solvent-exposed area of PA cores, offering enhanced stability to the otherwise unstable (in air) PA segments. Furthermore, tuning of the reaction temperature and the stereochemistry of the PA block eliminated the nanosphere population and resulted in uniform single-phase “nanocaterpillars” from diblock copolymers with cis-major PA cores that were possible to be converted to trans-major ones upon thermal isomerization.38 Additionally, changing the shell of the assemblies from the more flexible coil-like PNB to a more rigid rod-like poly(tricyclo[4.2.2.0]deca-3,9-diene) (PTD) corona-forming block promoted stronger interactions between PA blocks and allowed for the 1D “nanocaterpillars” to undergo a further structural evolution to 3D nano- and microaggregates.39,40 In a different study, the Choi group developed polysulfane-rich diblock copolymer micelles of tunable size and refractive indices upon ROMPISA of a cyclic polysulfane norbornene-based monomer at high concentration in either DCM or THF, using a PNB-G3 macroinitiator.41
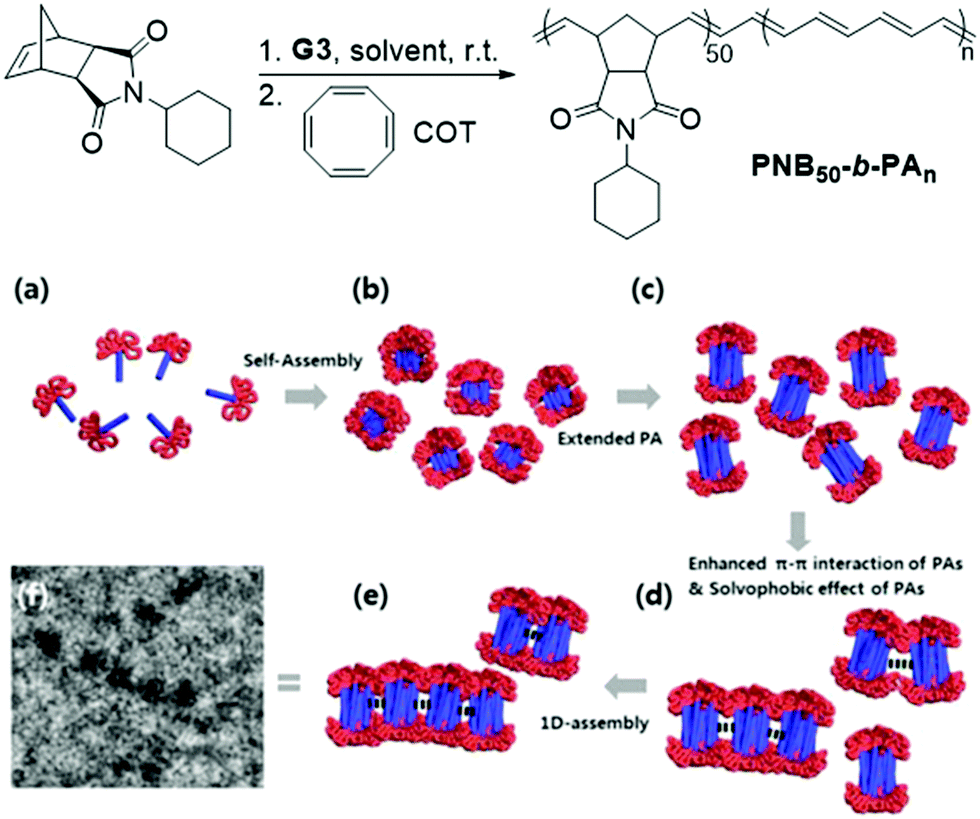 | ||
| Fig. 1 Preparation of PNB50-b-PAn diblock copolymer “nanocaterpillars” via a one-pot ROMPISA process and proposed 1D self-assembly mechanism. Adapted with permission from ref. 37. Copyright 2012 American Chemical Society. | ||
More recently, Gianneschi et al. reported a one-pot ROMPISA procedure utilizing an oligo(ethylene glycol) (OEG)-based NB monomer for formation of the steric stabilizer block and a NB monomer functionalized with a short protected peptide sequence for the solvophobic core-forming component, using G3 under inert atmosphere in DMF/MeOH solvent mixtures.42 Importantly, when a longer brush-like corona block was used, morphological evolution was limited and only spherical micelles of varying size were developed, whereas a shorter corona block allowed for distinct higher-order nano-object morphologies of worm-like micelles and vesicles to also be accessed. Moreover, Delaittre and coworkers reported the fabrication of spherical micelles with tunable size (10–100 nm) bearing TEMPO units in their outer shell via an ultra-fast two-step ROMPISA methodology in organic milieu.43 A solvent-soluble random copolymer from OEG-NB and TEMPO-NB was first synthesized via ROMP using G1 in THF. This was then used as the macroinitiator for chain-extension of PNB under inert atmosphere in EtOH/THF mixture leading to simultaneous amphiphilic block copolymer synthesis and self-assembly. The developed nitroxide-containing micelles could be readily transferred to aqueous media and were found to be both non-cytotoxic and efficient catalysts for a series of alcohol oxidation reactions with eminent recyclability.
Alternative approaches driven by other controlled metathesis processes (i.e., ring-closing metathesis) for in situ self-assembly of block copolymer nano-objects of different morphologies in organic media have also been investigated mainly by the Choi and Xie groups.44–48 In the majority of cases, they involve ROMP for the synthesis of a solvophilic stabilizer block using the highly efficient G3 catalyst and tandem metathesis cyclopolymerization (MCP) of 1,6-heptadiyne-derived monomers bearing various functional groups for the formation of conjugated core-forming polymers resulting in nanostructured materials with enhanced dielectric properties.
Progress has also been made towards implementing ROMPISA in aqueous systems, originating from initial studies on formation of PNB-based nanolatexes via aqueous emulsion ROMP.49,50 According to popular belief, water and oxygen must be rigorously excluded from Ru-catalyzed olefin metathesis. Indeed, the vast majority of metathesis reactions are conducted in dry organic solvent under inert atmosphere to avoid catalyst deactivation. This belief is deeply rooted in the metathesis community, originating from the strong nucleophilicity of early metal alkylidene complexes based on W or Mo metal centers. However, modern Ru-based metathesis catalysts are highly stable, opening up the possibility for conducting ROMP and other metathesis transformations in aqueous media. Catalysts for aqueous metathesis have been reviewed extensively elsewhere and will not be discussed here.34 It should be noted, however, that despite significant progress in the development of water-soluble Ru-based metathesis catalysts, few possess catalytic activity comparable to that of G3 in organic solvent. Water-soluble G3-derivatives have been reported, but these generally require the use of additional additives such as Brønsted or Lewis acids to promote phosphine or pyridine dissociation.51–54 As described above, the issues of catalyst solubility and activity must be addressed in order to conduct ROMP in aqueous media in a controlled manner. Hence, it is understood why aqueous ROMP-mediated PISA has only very recently emerged.
An increasing number of studies on aqueous ROMPISA have been reported in the past two years, expanding the scope of PISA in aqueous solutions beyond RAFT polymerization and building toward the establishment of ROMPISA as a powerful technique for development of block copolymer nano-objects. In particular, the rapid kinetics of ROMP coupled with the unique rigidity of PNBs are expected to provide specific advantages over RAFT formulations with respect to particle properties including: (1) ready access to out-of-equilibrium nano-objects via kinetic trapping; (2) enhanced particle stability resulting from high Tg values of PNBs; and (3) easier access to higher-order morphologies (e.g. worm-like micelles and vesicles) and in situ synthesis of multi-block copolymer nano-objects via sequential monomer additions.
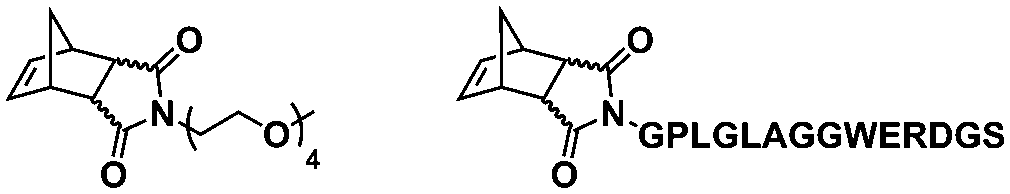
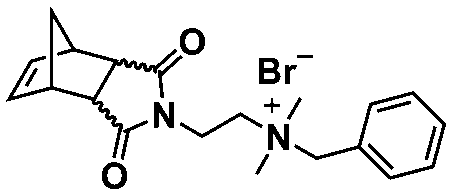
In an initial study, Gianneschi and coworkers utilized a water-soluble NHC-modified (H2IMes)(Cl)2RuC(H)(C6H4OR) (HG2) catalyst for the synthesis of a corona-forming OEG-based PNB block via solution ROMP under inert atmosphere in water. One-pot aqueous ROMPISA was achieved upon subsequent chain-extension of the resulting macroinitiator under identical polymerization conditions using a water-miscible quaternary amine phenyl NB dicarboximide (MX) monomer for synthesis of the insoluble core-forming block and in situ preparation of self-assembled nanostructures with morphologies evolving from spherical and worm-like micelles to large unilamellar vesicles, upon increasing the DP of the second block (Scheme 2A).55 Building on this concept, the same group prepared enzyme-responsive spherical micelles and phase-separated framboidal vesicles, following a similar one-pot aqueous-phase ROMPISA methodology.56 In particular, a hydrophilic PNB block bearing a 15-peptide sequence was first synthesized using the same water-soluble HG2 catalyst by open-to-air ROMP in water at ambient temperature. Nano-object formation was achieved via aqueous dispersion ROMPISA using PMX as the core-forming block, whilst enzymatic cleavage of the corona peptide pendant groups by the proteolytic enzyme thermolysin resulted in occurrence of morphological transitions toward larger aggregates induced by changes in the packing parameter of the assemblies, highlighting their potential use in biomedical applications. In a later study, Gianneschi et al. extended their original protocol for aqueous ROMPISA by directly incorporating a cis-platin NB derivative into the core of non- and pH-responsive spherical nano-objects.57 The effect of size and surface charge of Pt-loaded prodrug nanoparticles (NPs) on drug release behavior and in vitro cytotoxicity was investigated, revealing that smaller and positively-charged NPs showed enhanced cell uptake and increased cytotoxicity.
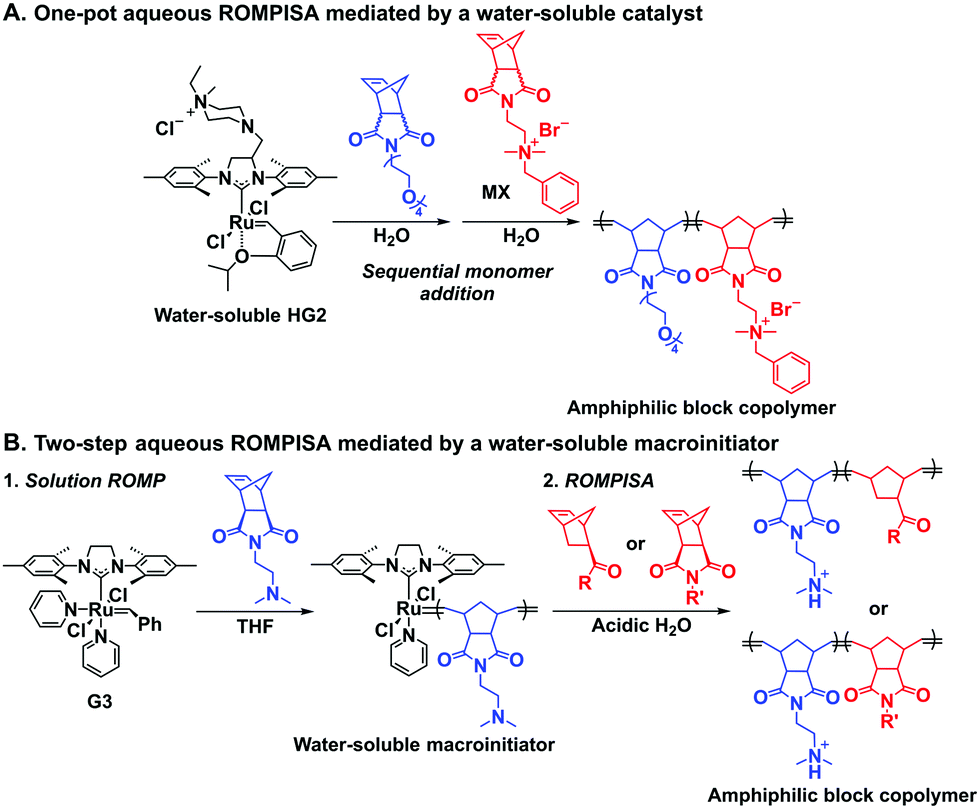 | ||
| Scheme 2 Reported synthetic strategies to conduct aqueous dispersion ROMPISA for fabrication of diblock copolymer nano-objects of various morphologies. (A) One-pot aqueous ROMPISA mediated by a water-soluble HG2 catalyst, and (B) two-step aqueous ROMPISA mediated via a water-soluble PNB macroinitiator prepared in organic solvent using G3. | ||
Recently, our group introduced an alternative two-step process for controlled open-to-air ROMP in aqueous media involving the synthesis of a hydrophilic PNB macroinitiator in a water-miscible organic solvent using commercially available G3 at ambient temperature.54 In this initial report, transfer of the prepared macroinitiator to an acidic solution of appropriately selected core-forming monomers allowed for ionization of the pendant tertiary amine groups of the macroinitiator to yield a steric stabilizer block and for in situ aqueous ROMPISA resulting in amphiphilic diblock copolymer nano-objects with common self-assembled morphologies (i.e., spheres, worms, and vesicles) (Scheme 2B). In a fundamental PISA study, O’Reilly and coworkers reported an in silico methodology for identification of monomers suitable for use as either corona- or core-forming blocks in aqueous RAFT-mediated PISA that involves theoretical calculations of surface area (SA)-normalized octanol–water partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Poct/SA) for their corresponding oligomeric models. The computational hydrophobicity evaluation method was also exploited for the reliable prediction of the resulting block copolymer nano-object morphology at a certain core block length.58 Very recently, our group combined this predictive methodology with the above described two-step approach for aqueous ROMP to expand the selection pool of core-forming monomers for use in aqueous dispersion ROMPISA.59 Through oligomer hydrophobicity calculations for a series of previously and newly reported NB-based monomers, a diverse set of core-forming ROMPISA monomers were able to be identified and validated experimentally. Moreover, important structure-hydrophobicity relationships were also determined that allowed for successful nano-object morphology prediction (Fig. 2).
Poct/SA) for their corresponding oligomeric models. The computational hydrophobicity evaluation method was also exploited for the reliable prediction of the resulting block copolymer nano-object morphology at a certain core block length.58 Very recently, our group combined this predictive methodology with the above described two-step approach for aqueous ROMP to expand the selection pool of core-forming monomers for use in aqueous dispersion ROMPISA.59 Through oligomer hydrophobicity calculations for a series of previously and newly reported NB-based monomers, a diverse set of core-forming ROMPISA monomers were able to be identified and validated experimentally. Moreover, important structure-hydrophobicity relationships were also determined that allowed for successful nano-object morphology prediction (Fig. 2).
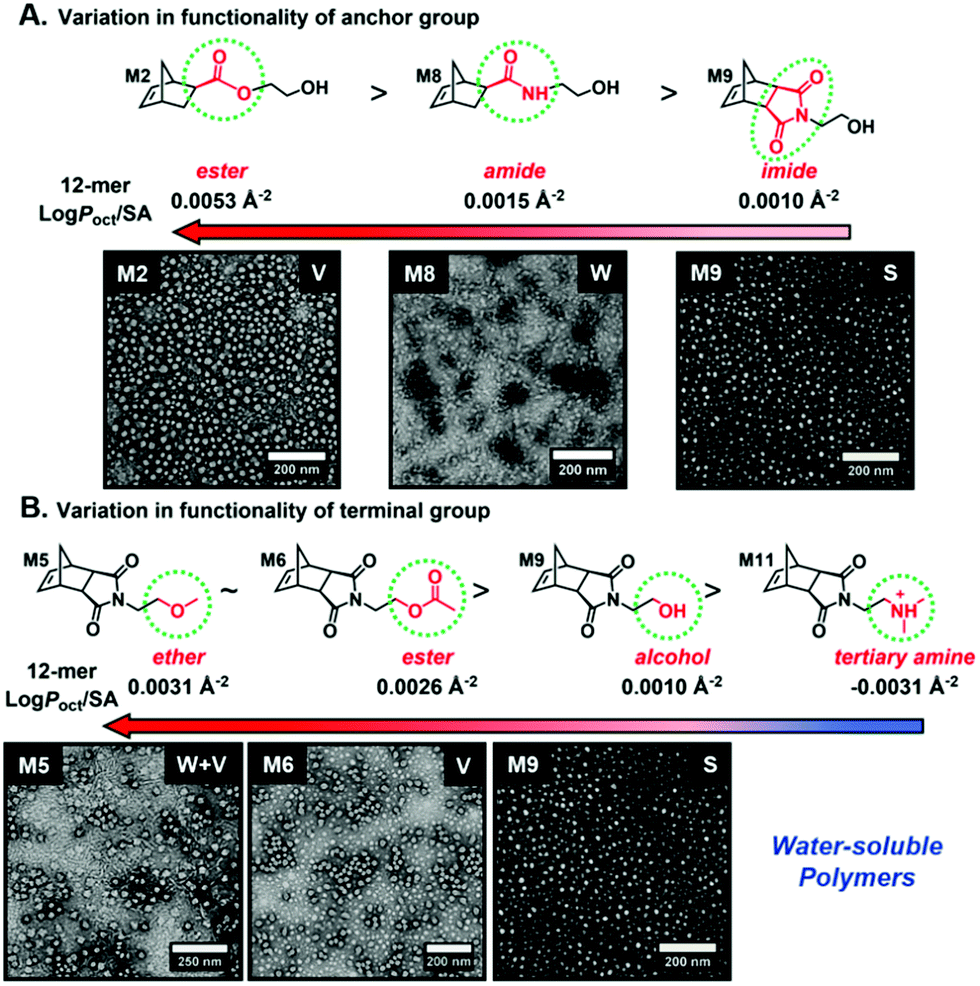 | ||
| Fig. 2 Oligomer logPoct/SA values as a function of (A) anchor and (B) terminal group variations for NB-based monomers suitable for use as core-forming blocks during aqueous ROMPISA, and representative nano-object TEM images for corresponding diblock copolymers of approximately the same length. Adapted with permission from ref. 59. Copyright 2019 American Chemical Society. | ||
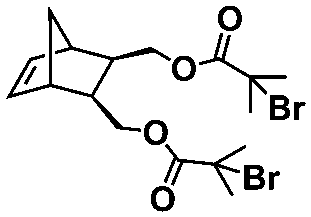
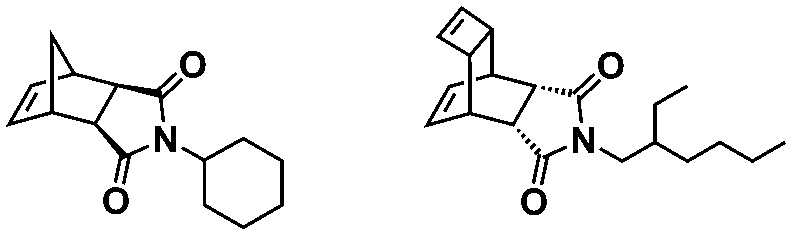
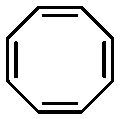
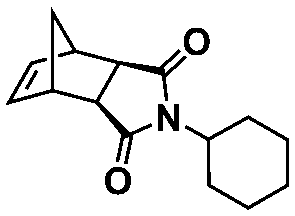
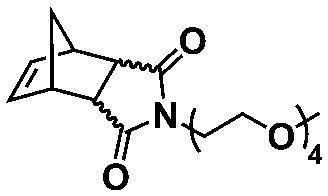
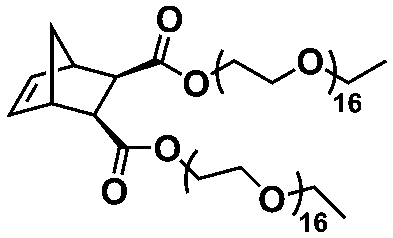
An alternative pathway toward the preparation of diblock copolymer nano-objects via ROMPISA in aqueous media has been reported by Cunningham and coworkers.60 In the first step, the synthesis of a hydrophilic steric stabilizer PNB-(PEG16)2 block was carried out via homogeneous ROMP initiated by a water-soluble G3 catalyst bearing PEGylated pyridine ligands under inert atmosphere at room temperature. Then, water-insoluble 1,5-cyclooctadiene (COD) monomer was added to the macroinitiator solution to allow for one-pot aqueous ROMPISA under emulsion polymerization conditions. Typical for heterogeneous emulsion PISA systems, stable (PNB-(PEG16)2)-b-PCOD diblock copolymer nanolatexes (i.e., kinetically trapped spherical micelles) of ∼200 nm size and low PD were obtained as judged by DLS and dry-state TEM analysis when a hydrophilic-to-hydrophobic molar ratio of 30:70 mol% was maintained. Currently identified corona- and core-forming monomer structures along with the catalysts and solvents utilized for ROMPISA in either organic or aqueous media are summarized in Table 1.
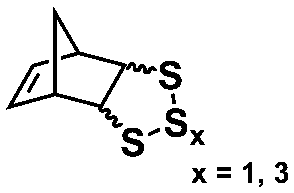
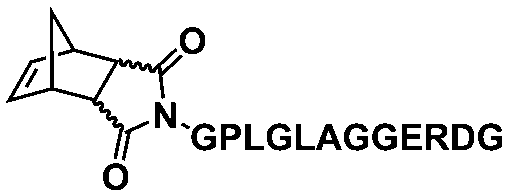
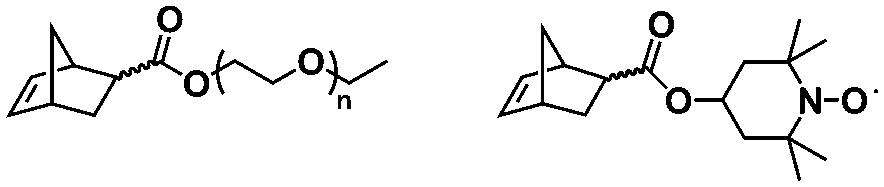
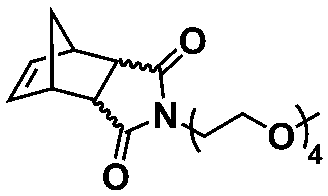
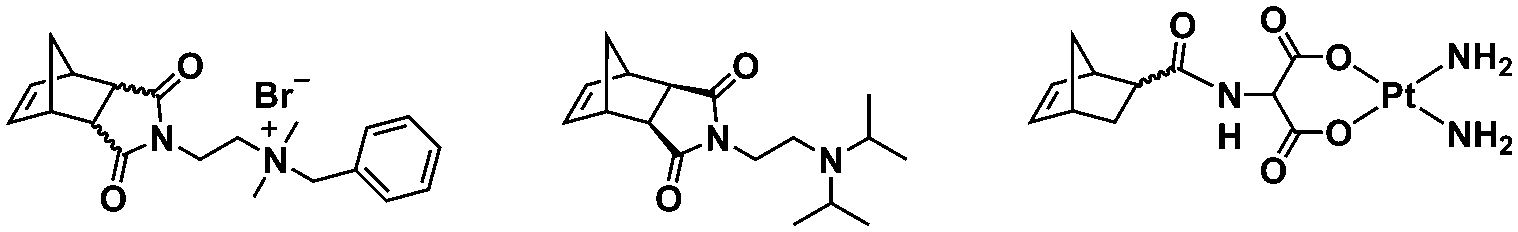
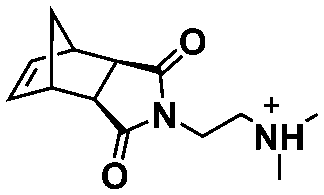
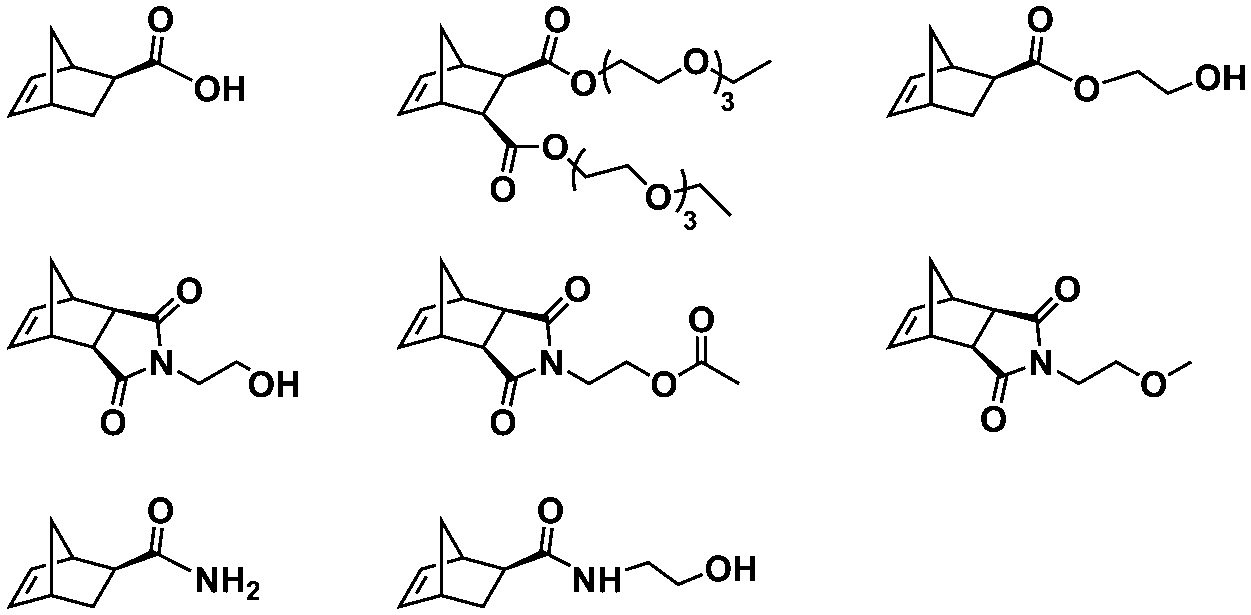
In summary, ROMPISA represents a complementary strategy for the in situ preparation of nano-objects with specific advantages over RAFT or other radical polymerization mechanisms. ROMPISA is fast, achieves quantitative conversions, is broadly tolerant to monomer functionality (or other functional groups in solution), can be carried out under air, and allows for facile access to common nano-object morphologies such as spherical or worm-like micelles and vesicles. Similar to RAFT-mediated PISA, formulation parameters such as initial solids concentration or block ratio can be tuned to access these morphologies. Future studies will likely reveal unique self-assembly behavior and/or novel applications which exploit the distinctive rigidity of polynorbornene-based assemblies as compared to their (meth)acrylate/acrylamide-based counterparts. Degradable functionalities can also be more readily incorporated within the backbone of polymers produced by ROMPISA compared with other water-compatible polymerization mechanisms. With the development of different formulation approaches (i.e., water-soluble catalysts vs. macroinitiator synthesis in organic solvent) and the recent identification of new core-forming monomers, ROMPISA has become more accessible, paving the way for research opportunities beyond the scope of radical polymerization. Still, this burgeoning field is currently limited by the small number of commercially available ROMPISA monomers and the relatively high cost, innately poor solubility and/or rapid decomposition of most Ru-based metathesis catalysts, as well as the existing difficulty of quantitatively removing catalysts from the nano-object cores following polymerization. In addition, acid additives are often required to achieve catalyst activity in aqueous media that is comparable to that of organic solvent. Thus, the development of highly soluble and active metathesis catalysts suitable for use in either organic or aqueous ROMPISA processes represents a grand challenge to be overcome to expand the usefulness of ROMPISA, especially with regards to its commercial implementation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (grant number 615142) and the University of Birmingham.Notes and references
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 RSC.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS.
- M. Chen, J.-W. Li, W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 CrossRef.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O’Reilly, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 CrossRef.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- G. Wang, Z. Wang, B. Lee, R. Yuan, Z. Lu, J. Yan, X. Pan, Y. Song, M. R. Bockstaller and K. Matyjaszewski, Polymer, 2017, 129, 57–67 CrossRef CAS.
- M. Williams, N. J. W. Penfold, J. R. Lovett, N. J. Warren, C. W. I. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 3864–3873 RSC.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef.
- G. Mellot, J.-M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., Int. Ed., 2019, 58, 3173–3177 CrossRef CAS.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 CrossRef CAS.
- X. G. Qiao, P. Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Polym. Chem., 2017, 8, 4014–4029 RSC.
- D. Cordella, F. Ouhib, A. Aqil, T. Defize, C. Jérôme, A. Serghei, E. Drockenmuller, K. Aissou, D. Taton and C. Detrembleur, ACS Macro Lett., 2017, 6, 121–126 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D’Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O’Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS.
- S. Varlas, L. D. Blackman, H. E. Findlay, E. Reading, P. J. Booth, M. I. Gibson and R. K. O’Reilly, Macromolecules, 2018, 51, 6190–6201 CrossRef CAS.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS.
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905 CrossRef CAS.
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927–2946 CrossRef CAS.
- S. C. Radzinski, J. C. Foster, S. J. Scannelli, J. R. Weaver, K. J. Arrington and J. B. Matson, ACS Macro Lett., 2017, 6, 1175–1179 CrossRef CAS.
- M. L. Gringolts, Y. I. Denisova, E. S. Finkelshtein and Y. V. Kudryavtsev, Beilstein J. Org. Chem., 2019, 15, 218–235 CrossRef CAS PubMed.
- J. Tomasek and J. Schatz, Green Chem., 2013, 15, 2317–2338 RSC.
- V. Sabatino and T. R. Ward, Beilstein J. Org. Chem., 2019, 15, 445–468 CrossRef CAS.
- L. Zhang, C. Song, J. Yu, D. Yang and M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5231–5238 CrossRef CAS.
- J. Liu, Y. Liao, X. He, J. Yu, L. Ding and M. Xie, Macromol. Chem. Phys., 2011, 212, 55–63 CrossRef CAS.
- K. Y. Yoon, I. H. Lee, K. O. Kim, J. Jang, E. Lee and T. L. Choi, J. Am. Chem. Soc., 2012, 134, 14291–14294 CrossRef CAS.
- K.-Y. Yoon, I.-H. Lee and T.-L. Choi, RSC Adv., 2014, 4, 49180–49185 RSC.
- K. Y. Yoon, S. Shin, Y. J. Kim, I. Kim, E. Lee and T. L. Choi, Macromol. Rapid Commun., 2015, 36, 1069–1074 CrossRef CAS.
- S. Shin, K.-Y. Yoon and T.-L. Choi, Macromolecules, 2015, 48, 1390–1397 CrossRef CAS.
- J. Lim, Y. Cho, E. H. Kang, S. Yang, J. Pyun, T. L. Choi and K. Char, Chem. Commun., 2016, 52, 2485–2488 RSC.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 CrossRef CAS.
- D. Le, M. Dilger, V. Pertici, S. Diabaté, D. Gigmes, C. Weiss and G. Delaittre, Angew. Chem., Int. Ed., 2019, 58, 4725–4731 CrossRef CAS.
- J. Kim, E.-H. Kang and T.-L. Choi, ACS Macro Lett., 2012, 1, 1090–1093 CrossRef CAS.
- E.-H. Kang, S. Yang, S. Y. Yu, J. Kim and T.-L. Choi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3058–3066 CrossRef CAS.
- W. Liu, X. Liao, Y. Li, Q. Zhao, M. Xie and R. Sun, Chem. Commun., 2015, 51, 15320–15323 RSC.
- J. Chen, Y. Wang, H. Li, H. Han, X. Liao, R. Sun, X. Huang and M. Xie, Chem. Mater., 2018, 30, 1102–1112 CrossRef CAS.
- J. Chen, R. Sun, X. Liao, H. Han, Y. Li and M. Xie, Macromolecules, 2018, 51, 10202–10213 CrossRef CAS.
- J. P. Claverie, S. Viala, V. Maurel and C. Novat, Macromolecules, 2001, 34, 382–388 CrossRef CAS.
- B. Ö. Öztürk, A. Kolberg and S. Karabulut Şehitoğlu, Macromol. Chem. Phys., 2017, 218, 1600594 CrossRef.
- D. M. Lynn, S. Kanaoka and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 784–790 CrossRef CAS.
- D. Samanta, K. Kratz, X. Zhang and T. Emrick, Macromolecules, 2008, 41, 530–532 CrossRef CAS.
- D. M. Lynn, B. Mohr and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 1627–1628 CrossRef CAS.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS.
- D. B. Wright, M. A. Touve, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2018, 7, 401–405 CrossRef CAS.
- D. B. Wright, M. P. Thompson, M. A. Touve, A. S. Carlini and N. C. Gianneschi, Macromol. Rapid Commun., 2019, 40, 1800467 CrossRef.
- D. B. Wright, M. T. Proetto, M. A. Touve and N. C. Gianneschi, Polym. Chem., 2019, 10, 2996–3000 RSC.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS.
- S. Varlas, J. C. Foster, L. A. Arkinstall, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O’Reilly, ACS Macro Lett., 2019, 8, 466–472 CrossRef CAS.
- O. L. Torres-Rocha, X. Wu, C. Zhu, C. M. Crudden and M. F. Cunningham, Macromol. Rapid Commun., 2019, 40, 1800326 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |