
Synthesis of guar gum-g-(acrylic acid-co-acrylamide-co-3-acrylamido propanoic acid) IPN via in situ attachment of acrylamido propanoic acid for analyzing superadsorption mechanism of Pb(II)/Cd(II)/Cu(II)/MB/MV†
Nayan Ranjan
Singha
 *a,
Manas
Mahapatra
a,
Mrinmoy
Karmakar
a,
Arnab
Dutta
a,
Himarati
Mondal
a and
Pijush Kanti
Chattopadhyay
b
*a,
Manas
Mahapatra
a,
Mrinmoy
Karmakar
a,
Arnab
Dutta
a,
Himarati
Mondal
a and
Pijush Kanti
Chattopadhyay
b
aAdvanced Polymer Laboratory, Department of Polymer Science and Technology, Government College of Engineering and Leather Technology (Post Graduate), Maulana Abul Kalam Azad University of Technology, Salt Lake, Kolkata – 700106, West Bengal, India. E-mail: drs.nrs@gmail.com
bDepartment of Leather Technology, Government College of Engineering and Leather Technology (Post Graduate), Maulana Abul Kalam Azad University of Technology, Salt Lake, Kolkata – 700106, West Bengal, India
First published on 13th October 2017
Abstract
For the first time, guar gum-g-(acrylic acid-co-acrylamide-co-3-acrylamido propanoic acid) (GGAAAMAPA), a smart environment-friendly and sustainable interpenetrating polymer network hydrogel having unprecedented thermomechanical/physicochemical properties and excellent recyclability, has been synthesized by the grafting of guar gum (GG) and in situ adjunct allocation of 3-acrylamido propanoic acid (APA) during solution polymerization of acrylic acid (AA) and acrylamide (AM), using N,N′-methylenebisacrylamide (MBA) and potassium persulfate (PPS)–sodium bisulfite (SBS) as a crosslinker and a redox pair of initiators, respectively, through systematic multistage optimization of the constituents and the temperature of the reaction, for a comprehensive understanding of the individual/synergistic superadsorption mechanism during recycling of hazardous dyes, like 2,8-dimethyl-3,7-diamino-phenazine (i.e. safranine, SF), 3,7-bis(dimethylamino)-phenothiazin-5-ium chloride (i.e. methylene blue, MB) and sodium 4{[4(dimethylamino)phenyl]diazenyl}benzene-1-sulfonate (i.e. methyl orange, MO), along with the adsorptive exclusion of metal ions, i.e. M(II), like Pb(II), Cd(II) and Cu(II). The unambiguous in situ addition of APA, the grafting of GG into the AA-co-AM-co-APA (AAAMAPA) matrix and the superadsorption mechanism have been systematically determined by extensive analyses of FTIR, 1H-/13C-NMR, O 1s-/N 1s-/C 1s-/Pb 4f7/2,5/2-/Cd 3d5/2,3/2- and Cu 2p3/2,1/2-XPS, TGA, DSC, XRD, FESEM, EDX, and storage/loss moduli and supported by computational measurements via density functional theory (DFT), along with the measurements of %gel content (%GC), pH at point of zero charge (pHPZC), %graft ratio (%GR) and network parameters of hydrogels. The prevalence of ionic (I) and variegated interactions, like monodentate (M), bidentate bridging (BB) and bidentate chelating (BC), between GGAAAMAPA and M(II), has also been rationalized by FTIR and fitting of kinetics data to the pseudosecond order model and by the measurement of activation energies of adsorption. The individual/interactive effects of cationic and/or anionic dyes have been rationally determined via extensive UV-Vis analyses at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2 and 2:1 mole ratios of dyes and pHi = 2/9, interpreted through the appearance of the metachromic effect, dye-aggregates, π–π stacking interaction, diprotonated MO etc. The BET and Langmuir isotherms have fitted the best to MB and SF/Cd(II)/Cu(II)/Pb(II), respectively. Thermodynamically spontaneous chemisorption processes have shown the maximum adsorption capacities (ACs) of 27.06, 39.35, 40.55, 39.42, and 41.98 mg g−1 for MB, SF, Cd(II), Cu(II) and Pb(II), respectively, at 303 K, adsorbent dose = 0.025 g and initial concentration of dyes/M(II) = 30 ppm.
:1, 1:2 and 2:1 mole ratios of dyes and pHi = 2/9, interpreted through the appearance of the metachromic effect, dye-aggregates, π–π stacking interaction, diprotonated MO etc. The BET and Langmuir isotherms have fitted the best to MB and SF/Cd(II)/Cu(II)/Pb(II), respectively. Thermodynamically spontaneous chemisorption processes have shown the maximum adsorption capacities (ACs) of 27.06, 39.35, 40.55, 39.42, and 41.98 mg g−1 for MB, SF, Cd(II), Cu(II) and Pb(II), respectively, at 303 K, adsorbent dose = 0.025 g and initial concentration of dyes/M(II) = 30 ppm.
Introduction
Hydrogels, 3D networks of polymeric chains, display variable swelling based on the relative ratios of hydrophilic to hydrophobic functional groups, introduced through the variation of synthesis conditions.1 Though H-bonding and electrostatic and/or covalent interactions make hydrogels insoluble in water at ambient temperature and pHi, these possess the unique ability to engulf larger amounts of water than their dry weight.1 In recent years, interpenetrating polymer network (IPN) superadsorbent hydrogels, possessing a perfect balance between biodegradability and mechanical strength by incorporating suitable ratios of synthetic to natural polymers, have been gaining great attention for the removal of toxic components. In fact, perfect blending of biodegradability and mechanical properties can only be achieved through the grafting of several biopolymers within a synthetic polymer network. Such natural polymer based hydrogels also possess stimuli-responsivity, necessary for their prospective use in drug delivery, adhesives and environmental monitoring. In this context, IPNs are designated crosslinked polymeric chains, in which monomer(s) are reacted to produce homo-/co-/ter-polymers, followed by the incorporation of another natural/synthetic polymer and crosslinking of the polymer(s) to produce semi-IPN or full-IPN.2–5In recent years, waste effluents of various industries usually contain several toxic substances, like dyes and M(II), imparting severe threat to the whole eco-system.6 Among the toxic organic dyes, MB, a cationic phenothiazin dye, has been immensely used for colouring paper, for dyeing clothes and wool, and as a hair colorant, a photosensitizing agent and a redox indicator in analytical chemistry. On the other hand, SF, a cationic phenazine dye, is mainly used for dyeing food, in flavouring and colouring candies and cookies, and in dyeing leather, cotton, bast fibers, wool, silk, leather and paper. These dyes cause several acute health hazards, including stomach pain and irritation, to the skin, mouth, throat, tongue, and lips and permanent damage to eyes. Again, Pb, one of the most toxic elements, causes dysfunction of brain, liver, kidney and bones due to the impairment of the central nervous system, reduction in haemoglobin formation, mental retardation, infertility and abnormalities in pregnant women, anaemia, headache, dizziness, irritability and weakness of muscles.7,8 In succession, Cu, extensively used in the manufacture of fungicides and anti-fouling paints and electrical industries, may cause acute vomiting, cramps, convulsions and even carcinogenicity.9 In addition, Cd causes high toxicity for the majority of living organisms by the unique ability to bind specifically with proteins, nucleic acids and small metabolites in living organisms, and thus, inhibits their functions.10 Therefore, considerable attention has been devoted to developing innovative, efficient, economical and fast treatment methods to reduce such toxic species from aqueous systems, prior to their discharge, using several methods, like biological treatment,11 ion exchange,11 coagulation/flocculation,11 chemical oxidation,12 photocatalytic degradation,12 adsorption6 and membrane separation.13–15 Of these, adsorption using proper biocompatible adsorbents is one of the most widely studied, facile and accepted techniques for decontaminating polluted waste water due to its flexible design, potential application prospects and easy regeneratibility of adsorbents.
Meanwhile, incorporation of natural polymers, like pectin,1 agarose,16 alginate, dextrin,17 chitosan18 and gum ghatti,19 in fabricating IPNs is gaining great attention owing to its ability for enhancing physicochemical characteristics, chemical stability and selectivity in separations, in addition to the natural abundance, renewability and biodegradability. However, GG, a biodegradable, non-toxic and renewable polysaccharide, has been modified through functionalization,20 grafting and network formation21 to find potentially high ACs and entrap drugs, like diclofenac sodium.22 Again, crosslinked hydrogels of modified and unmodified GG,23 GG-g-poly(AM),24 GG-g-poly(acrylate)25 and GG-g-poly(sodium acrylate)/rectorite26 hydrogels, have been reported for the removal of different dyes and M(II/VI), like Cu(II), Ni(II), Pb(II), Zn(II), Fe(II), Cr(VI) and Hg(II). In this context, though researchers have utilized a few acrylamido derivatives, like 2-acrylamido glycolic acid,27 2-acrylamido-2-methylpropane sulfonic acid,28,29 2-acrylamido-2-methyl-1-propanesulfonic acid30,31 and 3-acrylamido phenylboronic acid,32,33 for synthesizing copolymer hydrogels, the in situ incorporation of APA within the network of GG grafted AAAMAPA (i.e. GGAAAMAPA), via solution polymerization of AA and AM, is yet to be reported.
Meanwhile, several homo-/co-polymer, IPN and composite hydrogels have been reported for the individual removal of dyes and M(II). In fact, the synthesis of a GGAAAMAPA IPN superadsorbent, via in situ attachment of APA and grafting of GG, with very distinct chemical networks, using the optimized composition of AA:AM, GG and temperature, allows for the introduction of excellent thermomechanical properties and unprecedented performance potential in individual and/or synergistic removal of dyes/M(II). Rational systematic characterization of both the unadsorbed and adsorbed GGAAAMAPAs to determine the changes in several physicochemical properties of the optimized hydrogel is reported in the present study. In addition, the determination of the chemisorption mechanism through analyses of XPS, FTIR, DFT, EDX, FESEM, DSC and TGA and by measuring isotherm, kinetics, diffusion and thermodynamics parameters, is yet to be reported. Indeed, an important objective behind the selection of M(II), like Cu(II), Pb(II), and Cd(II), was to investigate the consequences of the Jahn–Teller effect in Cu(II), hemi/holo structures of Pb(II) complexes and d10 configuration of Cd(II), in diversified coordination modes of interaction, such as M, BB and BC, within adsorbed GGAAAMAPAs, on the adsorption and desorption behaviour and, hence, the recyclability of GGAAAMAPA. Finally, the systematic synthesis of GGAAAMAPA, via in situ addition of APA, for the attainment of excellent swellability with network integrity, comprehensive characterization of both unloaded and loaded GGAAAMAPAs and determination of the adsorption mechanism for adsorptive removal of chemically different dyes and M(II) have been thoroughly investigated in the present study.
Experimental section
Materials
Na2HPO4, Na2B4O7·10H2O, HCl, AA, AM, GG, MBA, PPS and SBS, of analytical grades, were purchased from Merck and used without any further modification. MB, SF, nitrate salts of Pb(II) and Cu(II) and chloride salt of Cd(II), used in adsorption studies, were purchased from Sigma Aldrich.Synthesis of hydrogels
Hydrogels were synthesized via free radical solution polymerization using PPS and SBS as a redox pair of initiators and MBA as a crosslinker under a nitrogen atmosphere (Scheme 1). However, the exact composition of ingredients and the reaction temperature were optimized by measuring the equilibrium swelling ratio (ESR) and gelation time (GT) of several hydrogels, synthesized at varying compositions and temperatures (Scheme 2). Finally, the most stable hydrogel, i.e. GGAAAMAPA, possessing the maximum ESR and GT, was preferably taken for further study. In brief, a three neck reactor was placed on a magnetic stirrer cum hot plate (SCHOTT Instruments GmbH, Germany). A homogeneous dispersion of 0.5 g GG powder in 30 mL H2O at 313 K, 0.632 mol of AA in 27 mL H2O at pH = 5.5 and 0.064 mol of AM in 25 mL of water were then successively incorporated under constant stirring at 300 rpm. In succession, 0.384 mmol of MBA in 8 mL of water was added and the resultant mixture was kept under a nitrogen atmosphere for 8 h. PPS and SBS of 0.462 and 1.202 mmol, respectively, taken in 10 mL distilled water, was added to initiate the polymerization reaction (Scheme 1). The as-prepared GGAAAMAPA was allowed to swell in an excess 1:3 CH3OH:H2O (v:v) solution and washed several times for removing unreacted components and water soluble oligomers. Finally, GGAAAMAPA was air dried for 100 h and then under vacuum for another three days at 323 K.
 | ||
| Scheme 1 Synthesis of GGAAAMAPA. | ||
 | ||
| Scheme 2 Systematic optimization. | ||
Characterization and methodology
GGAAAMAPAs, both unadsorbed and adsorbed, were characterized by Fourier transform infrared (FTIR) spectroscopy through Spectrum-2, Singapore in the range of 4000 to 400 cm−1; 1H-/13C-nuclear magnetic resonance (1H-/13C-NMR) spectroscopy using a Bruker-Advance Digital 300 MHz instrument in CDCl3 as a solvent with TMS as an internal reference/JEOL ECX400, at a proton frequency of 400 MHz; X-ray photoelectronic spectroscopy (XPS) via ESCA+, Omicron Nanotechnology, Oxford Instruments, Germany, equipped with an Al source (Kα radiation hν = 1486.7 eV) monochromator, operated at 15 kV and 20 mA; thermogravimetric analysis (TGA) using Pyris6 TGA, The Netherlands, under a N2 atmosphere with flow and scanning rates of 20.0 cm3 min−1 and 10 °C min−1, respectively, from 30 to 700 °C; differential scanning calorimetry (DSC) via Pyris6 DSC, The Netherlands, under a N2 atmosphere with a flow rate of 20.0 cm3 min−1, within 30–442 °C; X-ray diffraction (XRD) by X'Pert PRO, PANalytical B.V., The Netherlands, using Ni-filtered CuKα radiation (λ = 1.5406 Å) having an angle of diffraction within 5–100°; field emission scanning electron microscopy (FESEM) and energy dispersive X-ray (EDX) using a ZEISS EVO-MA 10 having a resolution of 3 nm with W filament and Sb as sources and rheological analyses through an Anton Paar MCR 102 rheometer. GGAAAMAPA was also characterized by measuring %GC, %–COOH, pHPZC and ESRs, at different pHi. DFT analyses were carried out by using Gaussian 09, Revision A.02, software. All graphics based analyses were carried out by using Origin 9.0 software. Dye and M(II) solutions of varying concentrations (i.e. 10–60 ppm) were prepared by exact dilution of 1000 ppm stock solutions. In the present study, 0.025 g of dry GGAAAMAPAs were added to 50 mL buffered solutions of dye and M(II), respectively, with constant stirring on a magnetic stirrer at 300 rpm. The progress of adsorption was monitored by withdrawing the supernatant solution after pre-determined time intervals and measuring the absorbance, at λmax, using a UV-Vis spectrophotometer (PerkinElmer Lambda 365) and an atomic absorption spectrometer (PerkinElmer A-ANALYST 100) for dyes and M(II), respectively. From the pre-calibrated equation, the dye concentration (Ct) was calculated, from which qt (mg g−1) was determined using eqn (1). | (1) |
Here, C0/Ct (ppm), V (mL) and ms (g) are feed dye concentrations at t = 0/t, volume of adsorbate solutions and mass of GGAMSAASPs, respectively.
Results and discussion
pHPZC, %GC, %GR, %–COOH and network parameters of GGAAAMAPA
pHPZC, %GC, %GR and %–COOH of GGAAAMAPA were calculated by the method reported elsewhere,1 and were found to be 6.72 (Fig. 1a), 94.31, 2.31 and 9.61, respectively. The network theory of Flory and Rehner was employed to determine several network parameters from the respective densities and ESR of hydrogels, composed of different amounts of MBA, initiators, GG, AA and AM (Table 1). The hydrogels showed inverse variation CDs and ESR with an increase in the amount of MBA.1 However, the gradual increase in the amounts of MBA from 0.1 to 0.4 wt% resulted in moderate enhancement of ρc from 4.42 × 10−6 to 4.88 × 10−4via the formation of a closely spaced tighter network of hydrogels.1 In fact, a relatively tighter network of hydrogels was related to the decrease in Mc from 16.28 × 106 to 1.48 × 105. Similar results were obtained through an enhancement of initiator amounts from 0.2 to 0.5 wt% that resulted in the decrease in Mc from 13.88 × 106 to 1.36 × 106 and the increase in ρc from 5.18 × 10−6 to 5.28 × 10−5. In this context, a good balance between ESR and ρc was obtained at 0.4 wt% of the initiator. Moreover, Mc of hydrogels was noted to increase from 1.26 × 105 to 8.06 × 106 as a consequence of the increase in the GG amount from 1 to 2 wt%, whereas further enhancement of the GG amount resulted in substantial increment in solution viscosity and, thus, restricted polymerization reaction due to lack of fluidity of the solution. However, the maximum amount of grafting was ensured at 2 wt% of GG in the network of hydrogels. In fact, ESR and Mc of hydrogels were found to increase within 7.21–16.26 and 4.19 × 105–8.06 × 106, respectively, via the increase in AA:AM content from 2:1 to 10:1, whereas ρc showed an inverse variation, in which ρc decreased from 1.73 × 10−4 to 8.94 × 10−6.
 | ||
| Fig. 1 Reversibility of (a) pHPZC of GGAAAMAPA, and (b) AAAMAPA/GGAAAMAPA, and swelling study of (c) AAAMAPA and (d) GGAAAMAPA at different pHi. | ||
| Hydrogels (AA:AM/MBA/PPS + SBS/GG) |
Density (g mL−1) | Swelling ratio in water (g g−1) | Volume fraction of swollen hydrogel (φp) | Polymer–water interaction parameter (χ) | Average molar mass between crosslinks (Mc) | Crosslink density (ρc) |
|---|---|---|---|---|---|---|
(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1/0.2/0.4/2) :1/0.2/0.4/2) |
1.2934 | 7.21 | 0.0960 | 0.5320 | 4.19 × 105 | 1.73 × 10−4 |
| (5:1/0.2/0.4/2) |
1.3049 | 10.22 | 0.0691 | 0.5230 | 1.48 × 106 | 4.87 × 10−5 |
| (10:1/0.2/0.4/2) |
1.3179 | 16.26 | 0.0441 | 0.5147 | 8.06 × 106 | 8.94 × 10−6 |
| (10:1/0.1/0.4/2) |
1.3065 | 20.00 | 0.0365 | 0.5122 | 16.28 × 106 | 4.42 × 10−6 |
| (10:1/0.2/0.4/2) |
1.3179 | 16.26 | 0.0442 | 0.5147 | 8.06 × 106 | 8.94 × 10−6 |
| (10:1/0.3/0.4/2) |
1.3282 | 11.25 | 0.0621 | 0.5207 | 2.25 × 106 | 3.20 × 10−5 |
| (10:1/0.4/0.4/2) |
1.3314 | 5.12 | 0.1268 | 0.5423 | 1.48 × 105 | 4.88 × 10−4 |
| (10:1/0.2/0.2/2) |
1.2846 | 19.55 | 0.0379 | 0.5126 | 13.88 × 106 | 5.18 × 10−6 |
| (10:1/0.2/0.3/2) |
1.3012 | 18.68 | 0.0391 | 0.5130 | 12.50 × 106 | 5.76 × 10−6 |
| (10:1/0.2/0.4/2) |
1.3179 | 16.26 | 0.0441 | 0.5147 | 8.06 × 106 | 8.94 × 10−6 |
| (10:1/0.2/0.5/2) |
1.2822 | 10.22 | 0.0702 | 0.5234 | 1.36 × 106 | 5.28 × 10−5 |
| (10:1/0.2/0.4/1) |
1.3072 | 5.00 | 0.1315 | 0.5438 | 1.26 × 105 | 5.72 × 10−4 |
| (10:1/0.2/0.4/2) |
1.3179 | 16.26 | 0.0441 | 0.5147 | 8.06 × 106 | 8.94 × 10−6 |
| (10:1/0.2/0.4/3) |
1.3249 | 10.00 | 0.0695 | 0.5232 | 1.47 × 106 | 4.91 × 10−5 |
Swelling study
The swelling properties of GGAAAMAPA were studied at 30 °C and pHi = 3.0, 5.0, 7.0, 9.0 and 12.0 (Fig. 1c and d). GGAAAMAPA of the given amount was immersed in excess buffer solution and then the swollen GGAAAMAPA was withdrawn from the medium after preselected time intervals. The excess solvent was wiped out with blotting paper and the increase in weight was measured. The measurement was continued till the weight of the swollen GGAAAMAPA attained the constant value. %SR (g/g) of GGAAAMAPA was calculated using eqn (2). | (2) |
Here, Wi and Wt (g) are the weights of GGAAAMAPA at t = 0 and t (min), respectively. In succession, ESR was also obtained by replacing Wt by Wf in eqn (2). Furthermore, swelling and deswelling studies of GGAAAMAPA were continued for a time period of 1 h at pHi = 2 and 12, respectively (Fig. 1b). Indeed, all the cycles were continued repetitively until the loss of the GGAAAMAPA network.
Stimulus responsive “smart” materials, like GGAAAMAPA, generally undergo conformational change via open to fully solvated, followed by coils, and finally, desolvated globular conformations over a moderate range of pHi.34 The pHi responsive swelling varied with several synthesis parameters, like the amounts of AA:AM, initiator (PPS + SBS), crosslinker (MBA) and natural polymer (GG) along with the pHi and temperature of experiment. Several intrinsic parameters, like the available free volume, chain relaxation and ionizable functional groups, i.e. –COOH, –CONH2, –CH2OH and O–H, can also affect the swelling properties of GGAAAMAPA. Swelling kinetics of AAAMAPA and GGAAAMAPA at different pHi are presented in Fig. 1c and d, in which the ESR increased from pHi = 3 to 10 and then decreased from pHi = 10 to 12. At pHi > pHPZC, the prevalent repulsive force between –COO− caused an enhancement of chain relaxation and, thus, macromolecular expansion resulted in the enhanced swelling and fully solvated open coil conformation (Scheme 3). However, at a very high pHi = 12, the GGAAAMAPA network was ionized rapidly causing higher counter ion concentration inside the matrix that reduced the electrostatic repulsion with a consequent reduction in ESR. At pHi < pHPZC, extensive protonation of the –COO− groups reasonably decreased the extent of hydrogen bonding with water molecules and thus resulted in a compact globular conformation. In this context, better relative population of water loving functional groups in GGAAAMAPA resulted in relatively higher ESR as compared to that of AAAMAPA. Again, the grafting of hydrophilic GG, within AAAMAPA, caused an enhanced ESR as well as chain flexibility, reflected by the retention of structural integrity even after the completion of several swelling–deswelling cycles. On the contrary, denser networks resulted from the presence of higher amounts of GG, leading to restricted water diffusion into the matrixes and relaxation of the polymer chains.
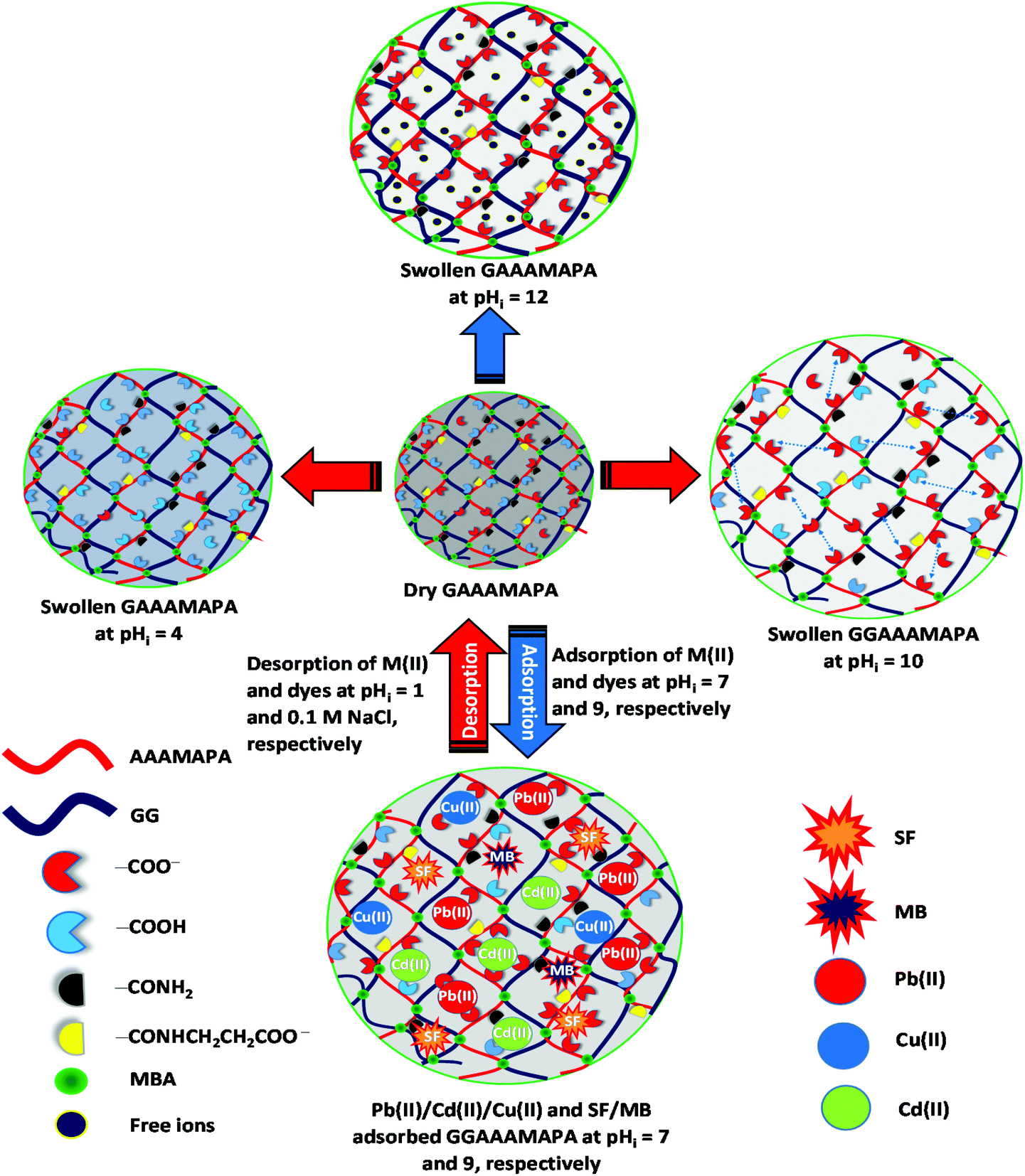 | ||
| Scheme 3 Swelling of GGAAAMAPA at different pHi and adsorption/desorption of M(II)/dyes. | ||
FTIR analyses
From the FTIR spectrum of GGAAAMAPA (Fig. 2a), intimate interactions between GG and AAAMAPA could be inferred from the disappearance of several characteristic peaks of GG and AAAMAPA within the moieties of GGAAAMAPA. For instance, the specific GG peak at 870 cm−1 (Fig. 2a), ascribed to the coupled C–C/C–O with anomeric C–H vibrations of β-conformers, was absent in GGAAAMAPA.35 Moreover, the characteristic GG peak at 768 cm−1, assigned to (1–6) linkage of galactose (gal) and mannose (mann), was slightly shifted to 770 cm−1 in GGAAAMAPA.36 Furthermore, the characteristic peaks at 1023 and 1075 cm−1, attributed to –CH2– twisting and –CH2OH of GG, respectively,36 did not appear in GGAAAMAPA, which indicated the possible conversion of primary –CH2OH into ether or ester. In this context, the appearance of a new small peak at 1121 cm−1 in GGAAAMAPA is usually attributed to the presence of –CH2–O–CH2– type of ether linkage within the network.37 As –CH2–O–CH2– moieties were absent in both individual GG or AAAMAPA, the formation of the –CH2–O–CH2– type of ether linkage, during GGAAAMAPA formation, could be rationalized, via interaction between –CH2–O˙, produced from the primary –CH2OH of GG, and AA/AM monomer.38 As AAAMAPA is devoid of any ether linkages and GG contains glycosidic types of >CH–O–CH2–/(1–6) and >CH–O–CH < /(1–4) linkages rather than the ether linkage of the –CH2–O–CH2– kind, the arrival of a new type of ether linkage reasonably confirmed the grafting of GG, also substantiated by the appearance of a new C–O–C def. peak at 425 cm−1 of aliphatic ether in GGAAAMAPA.37 Indeed, grafting related changes in the GGAAAMAPA matrix were manifested through the shift of vibrations for –CH2– and –O–CH2– groups. In addition, substantial alterations in the H-bonding environment, resulting from phase mixing within GGAAAMAPA, were comprehensively understood by the appearance of new H-bonded O–H str. peaks at 3616, 3626, 3646 and 3650 cm−1 in GGAAAMAPA along with a slight shift of the broad peak from 3435 of AAAMAPA to 3436 cm−1 in GGAAAMAPA, corresponding to combined O–H and N–H str. vibrations.39 In fact, significant changeover in the H-bonding environment was also realized from the arrival of new peaks at 2353 and 2331 cm−1.1 It seemed that the in-plane vibrations of both C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O def. and N–CO in primary amides were considerably altered as a result of grafting and changes in the H-bonding environment.37 In this regard, the C–CO def. peak at 481 cm−1 of AAAMAPA disappeared in GGAAAMAPA along with a marginal shift of the C–CO def. peak from 473 cm−1 of AAAMAPA to 472 cm−1 in GGAAAMAPA. In fact, changes in the H-bonding environment were also reflected through the significant shift of N–CO in plane bending vibration at 516 and 506 cm−1 of primary α-branched aliphatic amides (i.e. >CH–CO–NH2) to 519 and 511 cm−1, respectively.37 Similar to primary amides, vibrations of secondary amides were also affected by the change in the H-bonding environment, as reflected by the disappearance of several characteristic N–CO in plane bending peaks at 670, 657, 646 and 632 cm−1 with the simultaneous formation of a couple of peaks at 672 and 648 cm−1 in GGAAAMAPA. Indeed, grafting and the associated intermingling of the component chains produced a decrease in str. and bending vibrations of the C–N bond of amide III from 1323 cm−1 of AAAMAPA to 1320 cm−1 in GGAAAMAPA.40 Indeed, several characteristic peaks, corresponding to amide III, C–CO and N–CO of amides, indicated the transformation of primary amides into secondary ones via addition reaction between –CO–NH˙ and unsaturated chains to produce the –CO–NH–CH2– segment, which eventually altered the prevalent –CH2– and C–N peaks in GGAAAMAPA. In this regard, occurrence of both –COOH and –COO− in GGAAAMAPA was inferred from the prevalent peaks at 1743, 1738, 1718 and 1705 cm−1 corresponding to CO str. of –COOH along with νs(–COO−) and νas(–COO−) at 1405 and 1560 cm−1, respectively.41–43 In fact, the difference (Δν) between νas(–COO−) and νs(–COO−) in GGAAAMAPA was 155 cm−1 (Table 2), which was well within the prescribed limit of 136–164 cm−1 for ionic interaction (I).44 Indeed, peaks at 1705 and 1743 cm−1 may be assigned to CO str. modes of the cyclic H-bonded –COOH in dimeric form and free –COOH of GGAAAMAPA, respectively.43 In this regard, the presence of anhydrides could be neglected as the spectrum of GGAAAMAPA was devoid of any peak at 1030 and 1803 cm−1, corroborated by the fact that the formation of anhydride crosslinks within GGAAAMAPA would be very slow as the drying temperature (i.e. 70 °C) was well below 150 °C.43
O def. and N–CO in primary amides were considerably altered as a result of grafting and changes in the H-bonding environment.37 In this regard, the C–CO def. peak at 481 cm−1 of AAAMAPA disappeared in GGAAAMAPA along with a marginal shift of the C–CO def. peak from 473 cm−1 of AAAMAPA to 472 cm−1 in GGAAAMAPA. In fact, changes in the H-bonding environment were also reflected through the significant shift of N–CO in plane bending vibration at 516 and 506 cm−1 of primary α-branched aliphatic amides (i.e. >CH–CO–NH2) to 519 and 511 cm−1, respectively.37 Similar to primary amides, vibrations of secondary amides were also affected by the change in the H-bonding environment, as reflected by the disappearance of several characteristic N–CO in plane bending peaks at 670, 657, 646 and 632 cm−1 with the simultaneous formation of a couple of peaks at 672 and 648 cm−1 in GGAAAMAPA. Indeed, grafting and the associated intermingling of the component chains produced a decrease in str. and bending vibrations of the C–N bond of amide III from 1323 cm−1 of AAAMAPA to 1320 cm−1 in GGAAAMAPA.40 Indeed, several characteristic peaks, corresponding to amide III, C–CO and N–CO of amides, indicated the transformation of primary amides into secondary ones via addition reaction between –CO–NH˙ and unsaturated chains to produce the –CO–NH–CH2– segment, which eventually altered the prevalent –CH2– and C–N peaks in GGAAAMAPA. In this regard, occurrence of both –COOH and –COO− in GGAAAMAPA was inferred from the prevalent peaks at 1743, 1738, 1718 and 1705 cm−1 corresponding to CO str. of –COOH along with νs(–COO−) and νas(–COO−) at 1405 and 1560 cm−1, respectively.41–43 In fact, the difference (Δν) between νas(–COO−) and νs(–COO−) in GGAAAMAPA was 155 cm−1 (Table 2), which was well within the prescribed limit of 136–164 cm−1 for ionic interaction (I).44 Indeed, peaks at 1705 and 1743 cm−1 may be assigned to CO str. modes of the cyclic H-bonded –COOH in dimeric form and free –COOH of GGAAAMAPA, respectively.43 In this regard, the presence of anhydrides could be neglected as the spectrum of GGAAAMAPA was devoid of any peak at 1030 and 1803 cm−1, corroborated by the fact that the formation of anhydride crosslinks within GGAAAMAPA would be very slow as the drying temperature (i.e. 70 °C) was well below 150 °C.43
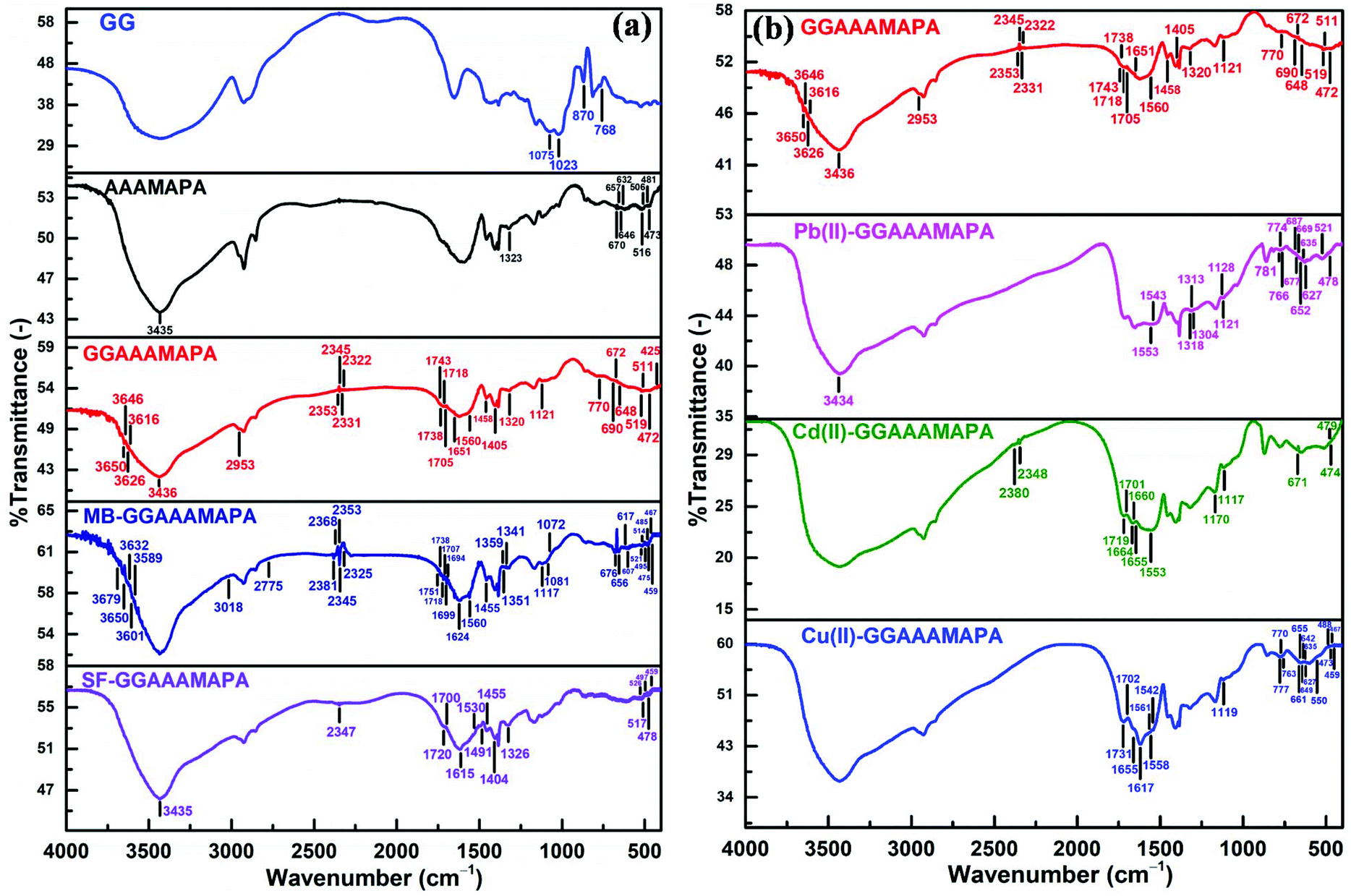 | ||
| Fig. 2 FTIR of (a) GG, AAAMAPA, GGAAAMAPA, MB-GGAAAMAPA and SF-GGAAAMAPA, and (b) GGAAAMAPA, Pb(II)-GGAAAMAPA, Cd(II)-GGAAAMAPA and Cu(II)-GGAAAMAPA. | ||
| Sample | ν as(–COO−) − νs(–COO−) = Δν (cm−1) | Mode(s) of interactiona |
|---|---|---|
| a I = ionic, M = monodentate, BB = bidentate bridging and BC = bidentate chelating. | ||
| GGAAAMAPA | 1560 − 1405 = 155 | I |
| Cu(II)-GGAAAMAPA | 1617 − 1407 = 210 | M |
| 1558 − (1407/1454) = 151/104 | I, BB | |
| 1561 − (1407/1454) = 154/107 | I, BB | |
| 1542 − (1407/1454) = 135/88 | BC | |
| 1617 − 1454 = 163 | I, BB | |
| Pb(II)-GGAAAMAPA | 1553 − (1398/1454) = 155/99 | I, BB |
| 1543 − (1398/1454) = 145/89 | I, BC | |
| Cd(II)-GGAAAMAPA | 1575 − (1404/1455) = 171/120 | I, BB |
| 1558 − (1404/1455) = 154/103 | I, BB | |
| 1553 − (1404/1455) = 149/98 | I, BB | |
| 1546 − (1404/1455) = 142/91 | I, BC | |
| 1539 − (1404/1455) = 135/84 | I, BC | |
Substantial and variegated shifts of characteristic peaks, corresponding to both νas(–COO−) and νs(–COO−), in Cu(II) adsorbed GGAAAMAPA (Cu(II)-GGAAAMAPA) were noted (Fig. 2b). Characteristic peaks appearing at 1558/1561 cm−1 and 1617 cm−1, corresponding to νas(–COO−), determined the availability of BB mode (Scheme 4) of –COO− coordination with Cu(II) in Cu(II)-GGAAAMAPA.41 In this regard, a new peak at 550 cm−1 could be related to the asymmetrical Cu–O vibration of the bridged Cu(II) complex within Cu(II)-GGAAAMAPA.45 Theoretically, an increase in νas(–COO−) of metal acetates than that of sodium salt is indicative of bridging, whereas a decrease in the same reflects the presence of chelation. Moreover, it is well known that monodentate complexes (M) possess much higher Δν, within 164–171 cm−1 (Table 2), in FTIR spectra than that of ionic (I), while for bidentate –COO− complexes, such Δν values are much smaller.46 In fact, monodentate coordination removes the equivalence of the two oxygen atoms of –COO− and a pseudo-ester configuration is obtained, in which the C–O bond orders are appreciably affected. In the present studies, the calculated value of Δν was 154 cm−1, which reemphasized the prevalent bidentate complex within Cu(II)-GGAAAMAPA (Table 2).
 | ||
| Scheme 4 Different binding modes of GGAAAMAPA. | ||
In this context, considerable lowering in νas(–COO−) at 1542 cm−1 in Cu(II)-GGAAAMAPA (Fig. 2b) than GGAAAMAPA indicated the presence of BC mode with Cu(II) in Cu(II)-GGAAAMAPA (Scheme 4). Thus, both BB and BC modes of interactions were prevalent within Cu(II)-GGAAAMAPA along with the smaller extent of M mode. Such BB and BC coordination modes were responsible for the formation of ring structures containing –CH2– bridges in Cu(II)-GGAAAMAPA, as inferred from the substantial increase in νs and νas of –CH2– groups.1 Moreover, all the strong H-bonded O–H str. peaks, within 2322–2353 cm−1, disappeared along with the appearance of new small peaks at 1702 and 1731 cm−1 (shoulder), indicating significant alteration in cyclic H-bonded –COOH in dimeric form and free –COOH of Cu(II)-GGAAAMAPA, respectively.43 Again, intrusion and positioning of Cu(II) in the hydrogel network brought about notable changes in the representative amide I peaks of GGAAAMAPA from 1651 to 1655 cm−1, (1–6) glycosidic linkage, –CH2–O–CH2– type of ether linkage from 1121 to 1119 cm−1, C–CO deformation and N–CO in plane bending vibration of primary α-branched (>CH–CO–NH2) amides and N–CO in plane bending vibration of amide II. For instance, the designated peak at 768 cm−1 for 1–6 glycosidic linkage of gal and mann36 was replaced by three small peaks at 777, 770 and 763 cm−1. Similarly, numerous C–CO def. peaks appeared at 488, 473, 467 and 459 cm−1 in Cu(II)-GGAAAMAPA along with the disappearance of N–CO in plane bending vibration of amide I at 519 and 511 cm−1.37 Moreover, a set of new small peaks at 661, 655, 649, 642, 635 and 627 cm−1, corresponding to N–CO in plane bending vibration of secondary amide,37 were produced in Cu(II)-GGAAAMAPA, which suggested significant changes in the GGAAAMAPA structure resulting from BC, BB and non-covalent interactions.
Similar to Cu(II)-GGAAAMAPA, several significant changes were observed within Pb(II)-GGAAAMAPA (Fig. 2b), resulting mostly from BB and BC types of coordination between Pb(II) and –COO−.44 In Pb(II)-GGAAAMAPA, both νas(–COO−) and νs(–COO−) were shifted considerably to lower frequencies than those of GGAAAMAPA (Table 2). Accordingly, the Δν values were considerably lower than those of the I and M modes, confirming the absence of M within Pb(II)-GGAAAMAPA unlike Cu(II)-GGAAAMAPA. In fact, the broad nature of the peaks at 1543 and 1553 cm−1 in Pb(II)-GGAAAMAPA indicated the introduction of various BB coordination modes, such as syn–syn, syn–anti or anti–anti configurations, possibly due to the variegated coordination abilities of Pb(II) forming hemi or holo type complexes.1 Such variations of coordination number, from 2 to 10, in Pb(II)-GGAAAMAPA were also responsible for the significantly broader nature of the peak at 3434 cm−1 than that of Cu(II)-GGAAAMAPA as a result of radical changes in mutual H-bonding interactions between O–H and N–H groups. However, similar to Cu(II)-GGAAAMAPA, stronger H-bonds among O–H groups were completely disrupted in Pb(II)-GGAAAMAPA, as envisaged from the complete disappearance of peaks at 2353, 2345, 2331 and 2322 cm−1. In fact, various binding modes of –COO− with Pb(II) resulting in different bond orders of C–O in –COO− were also envisaged from the shift of δs(–COO−) from 690 to 687/677 cm−1.44 In addition, significant changes were also noted in vibrations of different ether linkages including the glycosidic bonds. In this context, vibration of the (1–6) linkage of gal and mann, corresponding to the GG component, changed to 766, 774 and 781 cm−1, whereas –CH2–O–CH2– vibration introduced an extra peak at 1128 cm−1 in addition to the usual peak at 1121 cm−1.
Again, similar to Cu(II), incorporation of Pb(II) induced several changes in the various vibrational modes of amides (Fig. 2b). For instance, C–CO def. and N–CO in plane bending vibrations of primary amide, corresponding to AM side chains, were considerably shifted to 478 and 521 cm−1, respectively. Indeed, followed by the adsorption of Pb(II), N–CO in plane bending vibrations of α-branched aliphatic secondary amides underwent significant alterations to generate peaks at 669, 652, 635 and 627 cm−1 in Pb(II)-GGAAAMAPA.37 Hence, in addition to O-donor ligands, possible coordination of N-donor ligands, like –NH2 and –NH of amides, with Pb(II) was inferred from the change in amide III peak from 1320 to 1318 (shoulder), 1313 and 1304 cm−1.40
Similar to Pb(II), Cd(II) interacted with –COO− of GGAAAMAPA through various BB and BC modes, which were elucidated from the characteristic Δν values and the νas(–COO−), along with ionic type of interactions in Cd(II)-GGAAAMAPA. Interestingly, the FTIR spectrum of Cd(II)-GGAAAMAPA (Fig. 2b) showed three broad and intense peaks at 1719, 1660 and 1553 cm−1 due to the diversified CO str. of –COOH, amides and –COO−, respectively. The availability of free –NH2 in Cd(II)-GGAAAMAPA was less likely as the respective spectrum was devoid of the designated peak for free –NH2 groups at 1613 cm−1 and, hence, Cd(II) could penetrate deep inside the core of GGAAAMAPA to engage with the readily accessible –NH2 groups.47 Indeed, such easier penetration of relatively smaller Cd(II) was manifested in the powdery appearance of the deposited particles on the Cd(II)-GGAAAMAPA surface. In this context, though the adsorbed Cd(II), like the other M(II), actuated variegated alterations in the respective vibrations at 1719 and 1701 cm−1 of –COOH, 1664, 1655, 479 and 474 cm−1 of primary and 671 cm−1 for secondary amides, through influencing the surrounding H-bonding environment, the strong H-bonds among O–H groups remained almost unaffected, as evident from the appearance of peaks at 2348 and 2380 cm−1. Similar to other M(II)-GGAAAMAPAs, the characteristic vibrations of all the ether linkages, including the glycosidic bonds, were induced by the electron deficient Cd(II) as envisaged from the marginal shift of respective peaks to 1117 and 1170 cm−1.
In comparison with GGAAAMAPA, several new structural features were observed in MB-GGAAAMAPA (Fig. 2a), including the presence of water attached to the adsorbed MB molecules, realized from the arrival of new peaks at 3601, 3589, 3650, 3679 and 3632 cm−1 in MB-GGAAAMAPA. In fact, two weak peaks at 1694 and 1699 cm−1, in MB-GGAAAMAPA, were ascribed to ChetN+(CH3)2 str. of MB.48 In this regard, the appearance of the characteristic Chet–N(CH3)2 peak at 2775 cm−1 in MB-GGAAAMAPA suggested the availability of Chet–N(CH3)2 along with ChetN+(CH3)2 in MB-GGAAAMAPA.48 Though the peak at 1560 cm−1, corresponding to C–N vibration of MB, was noticed in the FTIR spectrum of MB-GGAAAMAPA, characteristic peaks at 1624 and 3018 cm−1 of MB for CN/CC and C–H of the heterocycle, respectively, receded from view in MB-GGAAAMAPA. The possible interaction of CS+ in the phenothiazin ring of MB with water or anions of GGAAAMAPA resulted in the shifting of the characteristic bending vibrations from 614 to 617/607 cm−1 of C–S–C. In addition, C–S–C vibrations of the MB heterocycles, appearing at 1081 and 1072 cm−1, could be attributed to ionic and H-bonding interactions between MB and GGAAAMAPA. In this context, new peaks at 1341, 1351 and 1359 cm−1 indicated divergent interactions via H-bonds of CS+ moieties in the phenothiazin ring of adsorbed MB with water molecules. Since two of these particular peaks prevailed within 1339–1356 cm−1, the presence of MB dimers and H-aggregates in MB-GGAAAMAPA should not be ruled out. Indeed, the dimer and H-aggregate formation in the MB adsorbed GGAAAMAPA was assisted by the water of crystallization via intermolecular H-bonds of the type Nhet⋯H–O⋯S+het.48 In fact, such types of reasonably strong intermolecular H-bonds might also be responsible for producing numerous peaks at 2381, 2368, 2353, 2345 and 2325 cm−1 of varying intensities. In this regard, the lengths of H-bonds of the types Nhet⋯H and O⋯S+het were 0.20 nm and 0.34 nm, respectively.48 In fact, the H-bond length of 0.20 nm for Nhet⋯H, comparable to the strong H-bond length of 2.55 ± 0.05 Å, was ascribed to the prevalence of such peaks at around 2350 cm−1.49
Intimate association of these crystalline water molecules within GGAAAMAPA, via reasonably stronger H-bonds was also realized from the restricted removal of these water molecules through evaporation at a considerably higher temperature range, within 150–200 °C, during thermal analyses. Furthermore, the adsorption of MB on GGAAAMAPA brought about diversified alterations in the H-bonding environment around primary and secondary amides, realized from the appearance of peaks at 459, 467, 475, 485 and 495 cm−1 for C–CO deformation, 514 and 521 cm−1 for N–CO in plane bending vibrations for primary and 676 and 656 cm−1 for the secondary amides. Moreover, appreciable changes in both non-bonded and H-bonded –COOH were reflected in the shift of characteristic peaks from 1743 and 1705 cm−1 of GGAAAMAPA to 1751 and 1707 cm−1 in MB-GGAAAMAPA, along with a couple of unaltered peaks at 1738 and 1718 cm−1. In this context, reduction in both νas(–COO−) and νs(–COO−) suggested restriction imposed on str. vibration of –COO−, which was involved in ionic interaction with MB. Moreover, the involvement of ether linkages, including –CH2–O–CH2–, in H-bonding interaction with MB was realized from the complete disappearance of the peak at 2953 cm−1, corresponding to the –O–CH2– of νas(–CH2–O–CH2–),37 along with the considerable shift of –O–CH2– bending and –CH2–O–CH2– vibration from 1458 to 1455 and 1121 to 1117 cm−1, respectively.
Different characteristic peaks of SF including peaks at 3144 and 1640 cm−1 (Fig. 2a), ascribed to aromatic ring C–H and CC str. vibrations, respectively,50 disappeared in SF-GGAAAMAPA. However, peaks at 1615, 1326 and 1530 (broad shoulder)/1491 cm−1, designated NC, N–C and CC str. of the phenazine ring in SF, respectively, were almost retained in SF-GGAAAMAPA. Indeed, unlike MB, SF-GGAAAMAPA produced substantial damage to the constitution and characteristics of both stronger and weaker type of H-bonds, as manifested in the conversion of numerous peaks within 2353–2322 cm−1 into a single peak at 2347 cm−1 and the simultaneous disappearance of several peaks within 3600–3650 cm−1, determining the absence of crystalline water held by intermolecular H-bonds within SF-GGAAAMAPA. In addition, usual alterations in mutual H-bonding among O–H and N–H was also evident in the respective spectra which showed marginal shift of the broad peak from 3436 of GGAAAMAPA to 3435 cm−1 in SF-GGAAAMAPA. Similar to MB, the H-bonding interaction between GGAAAMAPA and SF influenced νas(–CH2–) in –O–CH2– leading to the disappearance of the peak at 2953 cm−1.
Moreover, the prevalent ionic interaction between SF cations and –COO− was inferred from the lowering of νs(–COO−) to 1404 cm−1 and the presence of νas(–COO−) in the FTIR spectra of SF-GGAAAMAPA. However, simultaneous prevalence of both –COO− and –COOH, both H-bonded and non H-bonded, was inferred from the appearance of characteristic CO str. peaks at 1700 and 1720 cm−1, respectively. Finally, the influence of adsorbed SF on the H-bonding environment of GGAAAMAPA was reflected from the change in C–N str. and N–CO in plane bending vibrations of primary α-branched aliphatic amides (>CH–CO–NH2) to 1326 and 517/526 cm−1, respectively, along with appearance of numerous new C–CO def. peaks at 459, 469, 478, 488 and 497 cm−1.
NMR analyses
From the 1H NMR spectrum of AAAMAPA (Fig. 3), the appearance of a saturated backbone, originating through the free radical solution polymerization of monomers and crosslinkers, was determined from the prevalent characteristic peaks within the ranges of 0.82–1.54 ppm and 2.05–2.58 ppm for methylene (–CH2–) and methine (>CH–) protons, respectively. Indeed, such a type of alteration was also noted in GGAAAMAPA, reflected by the appearance of characteristic peaks within the ranges of 0.81–1.56 and 2.06–2.80 ppm for –CH2– and >CH– protons, respectively.51 Moreover, complete disappearance of characteristic peaks of vinyl protons within the ranges of 6.03–6.59, 5.70–6.30 and 5.59–6.13 ppm for AA, AM and MBA, respectively, (Fig. S1†) comprehensively indicated the inclusion of AA and AM monomers as well as the crosslinking by MBA within the GGAAAMAPA network (Fig. 3). In succession, the formation of the crosslinked spatial network was also ascertained by the prevalence of characteristic peaks at 4.24 and 4.66 ppm for –CH2–, connecting –NH– groups of MBA, in AAAMAPA and GGAAAMAPA, respectively.1 However, the arrival of new peaks from 3.45 to 3.61 ppm in the spectrum of both AAAMAPA and GGAAAMAPA determined the formation of –CH2– groups of –CH2–NH–(CO)–,52–56 appearing through free radical chain propagation between –C(O)NH˙ and SA/AA. However, the attainment of such reactions was emphasized by the use of a relatively lower amount of more reactive AM, resulting in the lower relative availability of reactive free radicals of AM, as compared to that of AA/SA. Furthermore, –NH– peaks of –NH–(CO)– and newly formed –CH2–NH–(CO)– moieties of the crosslinked MBA appeared at 6.83 and 7.71 ppm in AAAMAPA and 6.87, 7.56 and 8.04 ppm in GGAAAMAPA.1,57 Again, GG consists of α-(1,6) branched gal sugar units distributed randomly in the β-(1,4) mann backbone. Herein, the prevalence of mann and gal units was ascertained from the appearance of characteristic peaks of anomeric protons of gal (i.e. gal-1) and mann (i.e. mann-1) in 1H NMR at 4.75 and 5.05 ppm, respectively (Fig. 3).8 In addition, the chemical shifts of non-anomeric protons of gal-2, gal-5, gal-4, mann-4 and mann-2 were found at 3.83, 3.86, 4.01, 3.85 and 4.24 ppm, respectively.58,59 However, the weaker peak at 3.67 ppm was attributed to the gal-6 and mann-6 protons,60 whereas a set of emblematic peaks for non-anomeric protons of residual sugar units appeared within 4.34–4.64 ppm. In contrast to AAAMAPA, GGAAAMAPA consisted of some distinct peaks at 3.87 and 3.61 ppm, attributed to gal-5/mann-4 and gal-/mann-6 protons of GG.58–60 Again, grafting of –CH2–OH groups of GG into AAAMAPA was inferred from the significant shifting of the characteristic –CH2–O peak of GG from 3.67 to 3.61 ppm and prevalence of the anomeric GG peak at 5.33 and 5.40 ppm in GGAAAMAPA.
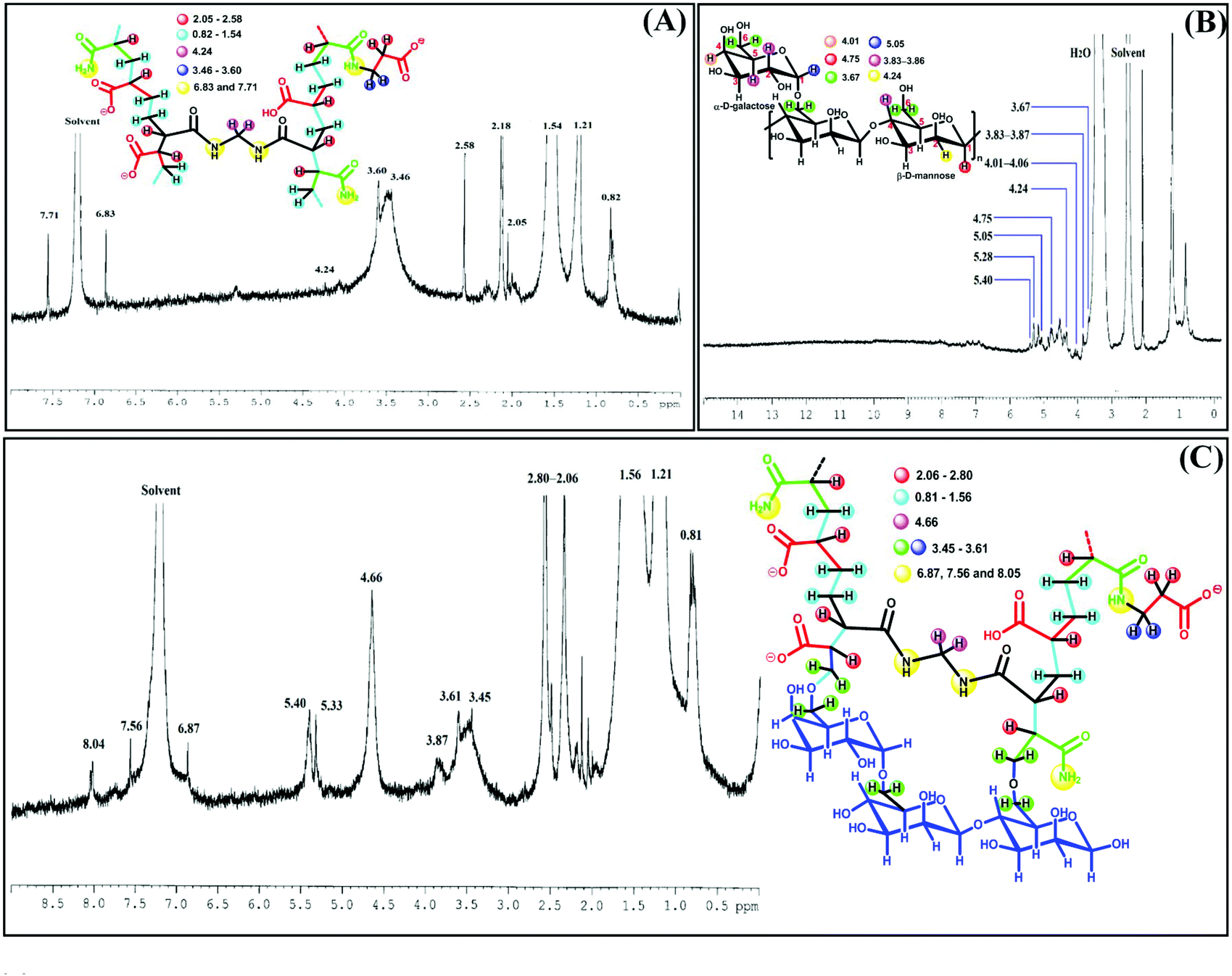 | ||
| Fig. 3 1H NMR of (A) AAAMAPA, (B) GG and (C) GGAAAMAPA. | ||
In addition, the intense and maximum downfield peaks at 185.28 and 185.88 ppm in 13C NMR (Fig. 4) were ascribed to the acrylic –COOH, –COO− and –CONH2 groups of AAAMAPA and GGAAAMAPA. However, the prevalent equivalent chemical environment of –CH2– of MBA and newly formed backbones, like –CH2–, –CH–COOH, –CH–COO−, –CH–CONH2 and –CH2NHCO– of GGAAAMAPAs, were determined through the appearance of several characteristic broad peaks within 40.27–46.50 and at 42.74 ppm for AAAMAPA and GGAAAMAPA, respectively. Moreover, several characteristic peaks at 64.27, 70.74, 72.22, 73.59, 79.31, 82.73, 85.89, 100.03 and 102.56 ppm of GG were found in 13C NMR of GGAAAMAPA.58 In fact, the prevalence of both β-(1,4) mann and α-(1,6) gal units of GG in GGAAAMAPA was confirmed by the prevalence of characteristic peaks of mann-1 and gal-1 at 102.56 and 100.03 ppm, respectively. In addition, though the characteristic peaks of gal-2, gal-3 and gal-4 units prevailed in close vicinity, which seemed to occur at around 70.74 ppm, mann-2, mann-3 and maan-4 units showed three distinct chemical shifts at 72.22, 73.59 and 79.31 ppm, respectively. In this context, both gal-6 and mann-6 peaks of GG appeared at 64.27 ppm, whereas the prevalent residual peaks of mann and gal units were found at 82.73, 85.89 ppm, respectively.58,61
 | ||
| Fig. 4 Solid state 13C NMR of AAAMAPA and GGAAAMAPA. | ||
XPS analyses
X-ray photoelectron spectroscopy (XPS) is an important characterization technique for analyzing the chemical composition and state of crosslinked polymeric solids. Herein, the relative compositional changes and mode of binding in both GGAAAMAPA and M(II)-GGAAAMAPA were examined by XPS analyses. The O 1s XPS spectrum for GGAAAMAPA (Fig. 5b) was deconvoluted into three peaks with binding energies (BEs) of 530.78, 536.42, 540.74 eV, assigned to OC,62O–H63 of –COOH and shake-up satellite band of the O atom in the –CO–NH–CH2– segment of GGAAAMAPA due to π → π* transition, noted earlier in polyurea.64 In this context, the presence of the –CO–NH–CH2– segment in GGAAAMAPA has already been inferred from NMR and FTIR analyses. However, the O 1s peak at 540.74 eV (Fig. 5b) indicated the possible presence of mutually H-bonded water clusters of low dimension around the GGAAAMAPA. Moreover, three deconvoluted C 1s peaks of GGAAAMAPA at 284.63, 285.21, 286.12 eV (Fig. 5a) were ascribed to C–C,62C–N62 and CO65 of amide/acid fragments, respectively. The possible modes of interactions between M(II) and GGAAAMAPA were ascertained from the relative changes in the respective XPS spectra for M(II)-GGAAAMAPAs, as compared to that of GGAAAMAPA. Indeed, variegated bindings between Pb(II) and GGAAAMAPA were inferred from the diversified shifting of O 1s peaks from 530.78 eV in GGAAAMAPA to 530.51, 532.10, 533.40, 534.12 and 536.61 eV in Pb(II)-GGAAAMAPA (Fig. 5d), along with the substantial lowering in BEs from 139.30 (Pb 4f7/2) and 143.8 eV (Pb 4f5/2) of pure Pb(II) to 136.54 eV (Pb 4f7/2) and 141.31 eV (Pb 4f5/2) in Pb(II)-GGAAAMAPA (Fig. 5e), reflecting significant increase in electronic density around Pb(II) by the formation of Pb(II)–O coordinate bonds.66 In this context, diversified interactions between Pb(II) and GGAAAMAPA, producing hemi or holo type complexes, were inferred earlier from the FTIR analyses and the arrival of the maximum number of peaks in the respective EDX spectrum (Fig. 7f). The stronger interaction between O-donor atom of GGAAAMAPA and Pb(II) was also envisaged via considerable increase in BEs for C 1s of both C–N of amide and CO of amide/acid fragments to 286.10 and 287.85 eV, respectively, in Pb(II)-GGAAAMAPA (Fig. 5c). Indeed, almost negligible increase in BE for C 1s at 284.66 eV (Fig. 5c) indicated little influence on C–C backbone, linked indirectly with Pb(II) in Pb(II)-GGAAAMAPA. In this context, the preferential ionic binding between Cd(II) and GGAAAMAPA was evident from the appearance of peak at 405.83 (Cd 3d5/2) and 412.63 eV (Cd 3d3/2) in Cd(II)-GGAAAMAPA (Fig. 5h) as compared to 405 eV (Cd 3d5/2) and 412 eV (Cd 3d3/2) for Cd(II) of CdS.67 In fact, the consequent alteration in the BEs of O 1s to 531.51, 532.27, 535.64 eV (Fig. 5g), designated to CO of –COOH, –COO− and O–H of –COOH, respectively, corroborated the formation of Cd–O ionic bond. Thus, Cd(II), possessing d10 configuration, preferably involved in ionic bonding with the –COO− of GGAAAMAPA in Cd-GGAAAMAPA.68 In fact, ionic interaction between GGAAAMAPA and Cd(II) also resulted in substantial shifting of all the C 1s peaks to 284.91, 285.53 and 288.70 eV (Fig. 5f).63 Interestingly, the O 1s spectra of Cu(II)-GGAAAMAPA demonstrated four distinct deconvoluted peaks at 531.52, 532.83,65 533.61 and 540.42 eV (Fig. 5j), of which the peak at 532.83 eV indicated the prevalence of BB and BC coordination modes producing Cu–O bidentate coordinate bonds. In this context, M and I type interactions produced nonequivalent O 1s peaks at 531.52 and 533.61 eV (Fig. 5j). The strong coordination of Cu(II) with GGAAAMAPA, owing to Jahn–Teller distortion of Cu(II),1 mainly through O-atom of AA moieties, was envisaged by the fluctuation of Cu 2p peaks of Cu-GGAAAMAPA. In fact, Cu–O bonding resulted in significantly shifted distinct peaks, at 933.17 eV (Cu 2p3/2) and 953.10 eV (Cu 2p1/2) (Fig. 5k) in Cu(II)-GGAAAMAPA, as compared to the characteristic peaks at 935.50 (2p3/2) and 953.70 eV (2p1/2) of pure Cu(II).1 In principle, Cu(II) gained electrons mainly from the O-donors leading to the lowering in BEs for Cu 2p.68 In this regard, the shake-up satellite band of O-atom at 540.74 eV for GGAAAMAPA (Fig. 5b) was slightly shifted to 540.42 eV in Cu-GGAAAMAPA (Fig. 5j), suggesting alteration of O 1s BE for –CO–NH–CH2– segment, mainly through the formation of stronger H-bonding, induced slightly bigger water clusters within Cu-GGAAAMAPA. However, stronger coordination also resulted prominent changes in C 1s spectra of Cu-GGAAAMAPA, which exhibited signals at 284.88, 285.34 and 287.42 eV (Fig. 5i) corresponding to –CH2–/–CH–, C–N69 and CO bonds of AM and AA moieties of GGAAAMAPA.
 | ||
| Fig. 5 XPS analyses of C 1s (a, c, f and i), O 1s (b, d, g and j) for GGAAAMAPA and Pb(II), Cd(II) and Cu(II) adsorbed GGAAAMAPA; Pb 4f7/2 and 5/2 (e), Cd 3d5/2 and 3/2 (h) and Cu 2p3/2 and 1/2 (k) of Pb(II), Cd(II) and Cu(II) adsorbed GGAAAMAPA, respectively. | ||
TGA analyses
From Fig. 6a, the initiation of major thermal degradation of GG was observed to begin at 230 °C, after the initial removal of loosely bound moistures and volatile matters within 50–130 °C.70,71 Such rapid degradation was continued till 320 °C, resulting in a net mass loss of 49 wt%. However, beyond 320 °C, this degradation was retarded gradually to register a mass loss of 71 wt% at 400 °C, comparable to the earlier findings by Srivastava et al.70 In fact, the overall thermal degradation of GG passed through random chain scission followed by molecular rearrangements, resulting in almost negligible residue of 0.06 wt% at 700 °C. | ||
| Fig. 6 TGA of (a) GG, AAAMAPA, GGAAAMAPA, MB- and SF-GGAAAMAPA, (b) GGAAAMAPA, Pb(II)-, Cd(II)- and Cu(II)-GGAAAMAPA, DSC of (c) GG, AAAMAPA, GGAAAMAPA, MB- and SF-GGAAAMAPA and (d) GGAAAMAPA, Pb(II)-, Cd(II)- and Cu(II)-GGAAAMAPA and XRD of (e) GG, AAAMAPA and GGAAAMAPA, (f) GGAAAMAPA, Pb(II)-, Cd(II)-, Cu(II)-, MB- and SF-GGAAAMAPA, (inset of a) TGA of MB and SF and (inset of f) XRD of MB and SF. | ||
Notably, the TGA thermogram of AAAMAPA was associated with three-stage decomposition pattern, i.e. 200–350, 350–420 and 420–500 °C (Fig. 6a), in addition to the usual moisture loss step completed within 200 °C. In fact, in the first stage, NH3 gas evolved via conversion of neighbouring amides of AAAMAPA chains to imides, whereas the degradation of –COO− and imides was initiated mostly in the second region. Again, the third stage was attributed to the degradation of polymer backbone along with the formation of paraffinic materials. However, beyond 500 °C, no further weight loss was observed, and a char-like material, having significant quantities of inorganic salts, might be generated to register significantly high residue content (40 wt%). In fact, the presence of a significant quantity of inorganic salts could be rationally correlated with the partial neutralization of AA during synthesis at pHi = 5.5.
Since grafting of GG on AAAMAPA could be possible with either AA or AM, the coupled thermal decomposition behavior of both GGAA and GGAM should be manifested during the thermal decomposition of GGAAAMAPA (Fig. 6a). In fact, grafting of GG on AAAMAPA imparted marginally enhanced thermal resistance up to 420 °C and thereafter, the degradation profiles of both GGAAAMAPA and AAAMAPA remained almost similar until 600 °C. Interestingly, within 600–700 °C, the thermal resistance of AAAMAPA was relatively better than that of GGAAAMAPA, producing a greater amount of residues or char for AAAMAPA. Such lower residue or char formation in GGAAAMAPA could be attributed to the remarkably lower residue content of 0.06 wt% for GG, as evident from the thermogram of pure GG (Fig. 6a). However, a slightly higher thermal resistance of GGAAAMAPA, especially in the initial two decomposition stages, might be attributed to the reduced conversion of amides into imides, owing to the lower availability of neighbouring amide side chains, since grafted GG chains might act as barriers between two closely spaced amide side chains. Similarly, the grafted GG chains might resist the anhydride formation via arresting the dehydration of neighbouring –COOH in the initial stage and the associated decarboxylation of anhydrides in later stages by partially affecting the closer approach between adjacent –COOH.
Among all the M(II)-GGAAAMAPAs, the highest thermal stability of Pb(II)-GGAAAMAPA, up to 380 °C (Fig. 6b), was mostly attributed to the unavailability of neighboring amides and –COOH, since, considerable portion of such groups were involved in bonding with Pb(II) via coordination bonds in the BB and BC modes (Scheme 4). Indeed, considerably higher thermal stability of Pb(II)-GGAAAMAPA over Cd(II)-GGAAAMAPA, especially up to 380 °C, could also be corroborated to the higher stability of Pb(C2O4)22− than Cd(C2O4)22− and comparatively higher decomposition temperature of Pb(OOCCH3)2·4H2O over Cd(OOCCH3)2·2H2O in vacuum. However, within 380–620 °C, Cd(II)-GGAAAMAPA superseded the thermal resistance of Pb(II)-GGAAAMAPA (Fig. 6b), indicating the prevalence of more reinforced polymeric backbones resulting in well organized microstructures in Cd(II)-GGAAAMAPA, possibly through the finer particle size distribution of Cd(II) salts within the hydrogel matrix. Moreover, since the adsorption of M(II) was conducted at pHi > pHpzc, a large portion of –COOH was expected to convert into –COO− that drastically reduced the possibility of anhydride formation from the neighbouring –COOH, via dehydration, leading to a lesser extent of moisture loss up to 200 °C in Cd(II)/Pb(II)-GGAAAMAPAs, with respect to GGAAAMAPA. In this context, the phenomenon of increased moisture loss up to 200 °C in Cu(II)-GGAAAMAPAs, as compared to GGAAAMAPA, was ascribed to the loss of water from the adsorbed Cu(II) crystals accumulated at the respective hydrogel.1 In fact, within 200–400 °C, relatively rapid thermal decomposition of Cu(II)-GGAAAMAPA was associated with the removal of coordinated water and ligand moieties from Cu(II) complex to produce thermally stable ash, constituting of mostly CuO, as envisaged from almost negligible mass loss beyond 430 °C (Fig. 6b). In succession, poor thermal stability of Cu(II)-GGAAAMAPA could be linked to the relatively poor thermal resistances of both Cu-acrylate and Cu-polyacrylate, among various metal acrylates and polyacrylates, since such structural moieties were expected to be present in Cu(II)-GGAAAMAPA. In this context, as compared to GGAAAMAPA, the excess residue of Cd(II)-GGAAAMAPA, at 700 °C, was ascribed mostly to the presence of metallic Cd whose evaporation or sublimation starts slowly above 800 °C.
From Fig. 6a, the initial thermal degradation of MB-GGAAAMAPA was observed to register marginally restricted mass loss than GGAAAMAPA, followed by slightly higher moisture loss beyond 150 °C due to the detachment and vaporization of water molecules sandwiched within MB multilayers attached to the surface of GGAAAMAPA via strong H-bonds, as also realized from the respective FTIR spectrum (Fig. 2a). However, unlike MB-GGAAAMAPA, the moisture loss from SF-GGAAAMAPA initiated at lower temperature in a more regular manner, confirming the lesser population of water molecules within the SF-GGAAAMAPA. In fact, the presence of strongly held crystalline water molecules in MB powder dyes was also realized from the relatively delayed moisture loss beyond 150 °C, as compared to the rapid moisture loss from SF powder dyes at the relatively lower temperature range (inset of Fig. 6a). Interestingly, within the intermediate temperature range of 200–500 °C, the overall decomposition of dyes, dye adsorbed hydrogels and GGAAAMAPA proceeded almost in a similar manner, except a slightly higher thermal resistance of dye loaded GGAAAMAPAs over the unloaded was observed within 400–500 °C (Fig. 6a). Such an improved thermal resistance of dyed GGAAAMAPAs, within 400–500 °C, could rationally be explained from the reversible binding of both SF and MB cations with the polyanions of GGAAAMAPA at pHi > pHPZC. In fact, in such experimental pHi, the possible decomposition pathway consisting of conversion of –COOH into ketone, via anhydride formation could be arrested initially by transformation of significant amount of –COOH into –COO− during adsorption. Moreover, the adsorbed dye molecules could strengthen the microstructural organization and architecture within the adsorbed GGAAAMAPA via diversified physical crosslinking with the polyanions of GGAAAMAPAs.
More importantly, in the final decomposition stage (i.e. 200–650 °C), superior thermal stabilities of dye-GGAAAMAPA assemblies over the individual powder dyes and unadsorbed GGAAAMAPA reemphasized the prevalence of crosslinking, within the dyed GGAAAMAPAs, stabilizing the GGAAAMAPAs skeleton.
DSC analyses
The DSC thermogram of GG showed endothermic transitions at 76 and 239 °C (Fig. 6c), which were ascribed to the loss of moisture and the cleavage of gal/mann units from GG, respectively.36 Interestingly, the wide decomposition peak at 239 °C was considerably lower than the peaks at 253 and 248 °C.36 The shorter chain length and hence, lesser average MW of GG, should be responsible for such decrease in decomposition temperature. Moreover, another shoulder appeared at 296 °C was also in agreement with the reported works.36 Indeed, the endothermic transitions at 239 and 296 °C were closely related to the rapid thermal degradation of GG within 230–320 °C (Fig. 6a). Though AAAMAPA was composed of both acid and salt forms, the endothermic peak at 181 °C was arrived mostly due to the presence of the acid form (Fig. 6c). In this context, Maurer and Harvey72 observed the existence of same endothermic peak corresponding to acid form of AA-co-AM rather than the salt form of AA-co-AM. In fact, arrival of such endothermic peak through the formation of anhydrides via dehydration of neighbouring –COOH was expected only in the acid form of AAAMAPA. In addition, the endothermic peak at 241 °C was attributed to conversion of amides and –COOH groups to imides and anhydrides, respectively, whereas the transition at 344 °C was related mostly to the decarboxylation of anhydrides. In this regard, the melting temperature of polyAM at 237 °C is very close to the endothermic peak at 241 °C.73 Finally, the endothermic peak at 433 °C indicated the breakdown of AAAMAPA chains along with complete decarboxylation of acrylate carboxyl groups.A new peak at 124 °C along with the appearance of broad shoulders at 80 and 238 °C in GGAAAMAPA indicated partial retention of characteristic GG peaks in GGAAAMAPA. In fact, broader nature of the shoulder, appeared at 238 °C, was resulted from almost simultaneous multiple thermal transformations, including the cleavage of gal/mann units of GG, coupled with the formation of imides/anhydrides. However, distinct degradation peak of anhydrides at 341 °C was considerably less intense in comparison to the same peak of AAAMAPA, indicating the formation of fewer anhydrides, formed by the limited availability of neighbouring –COOH, in GGAAAMAPA, envisaged earlier in the respective TGA plots (Fig. 6a). Among all the M(II)-GGAAAMAPAs, two new peaks at 254 and 261 °C were appeared in the DSC thermogram of Cu(II)-GGAAAMAPA in addition to the lowering of characteristic GGAAAMAPA peak from 124 to 121 °C (Fig. 6d). It appeared that the characteristic transition peak of GGAAAMAPA might be shifted from 241 °C to the higher temperature at 254 and 261 °C due to substantial lowering of chain flexibility and segmental mobility as a result of complexation between O-donor ligands of GGAAAMAPA and Cu(II) via different modes of coordination (Table 2). Again, Pb(II) interacted with GGAAAMAPA mostly via BB mode of interaction leading to the appearance of a single broad peak at 255 °C. In this regard, the same peak was shifted to 263 °C in Cd(II)-GGAAAMAPA as a consequence of greater immobilization of chains by smaller dimension and the greater interfacial area of contact between Cd(II) and GGAAAMAPA. Interestingly, appearance of peaks at 342 and 401 °C were assigned as the glass transition temperature (Tg) of Cu(II)-GGAAAMAPA.74 In this context, slightly elevated endothermic transitions at 354 and 401.5 °C of Pb(II)-GGAAAMAPA were appeared in consequence to the increased stability, inferred earlier in the TGA studies. Interestingly, sharp and intense exothermic peak at 332 °C for Pb(II)-GGAAAMAPA could be ascribed to the energy released due to transformation of GGAAAMAPA into a highly elastic state.75 Moreover, Tg of M(II)-GGAAAMAPAs should also be directly dependent on the relative extent of adsorbed M(II)s within the GGAAAMAPA. In this regard, the better adsorption of Pb(II) over Cu(II) and higher charge density of Pb(II) could result in slightly improved Tg of Pb(II)-GGAAAMAPA over Cu(II)-GGAAAMAPA. Though, Pb(II) possesses the higher charge density than Cd(II), Cd(II)-GGAAAMAPA showed a remarkably intense endothermic peak at a significantly higher temperature of 437 °C, corroborated to the comparatively smaller dimension and greater interfacial area of contact between Cd(II) and GGAAAMAPA as discussed later in the analysis of FESEM. In comparison to MB-GGAAAMAPA, relatively broad and intense endothermic transition of SF-GGAAAMAPA could be interpreted by the continuous moisture loss from SF-GGAAAMAPA, as realized earlier during TGA analyses (Fig. 6a). Accordingly, restricted moisture loss from MB-GGAAAMAPA was ascribed to the prevalence of strongly H-bonded water, allocated within MB multilayers in MB-GGAAAMAPA. Sharp endothermic peak at 345 °C in SF-GGAAAMAPA and relatively less intense peak at 356 °C in MB-GGAAAMAPA were corroborated to the significant destabilization of the GGAAAMAPA via reversible binding of the dyes within loaded GGAAAMAPAs, especially by SF, as SF possibly intruded deep interior to destruct GGAAAMAPA more actively as compared to MB that preferably accumulated at the surface.
XRD analyses
Loss of several characteristic peaks at 20.44°, 41.00° and 71.0° (Fig. 6e) of GG36 in GGAAAMAPA indicated substantial intermixing of the GG within AAAMAPA network. Indeed, the presence of a considerable extent of rigid and crystalline domains in pure GG resulted in high endothermic transition temperature of 77 °C (Fig. 6e). In fact, disintegration of such crystalline moieties of GG through grafting in GGAAAMAPA resulted in considerable lowering and broadening of the endothermic peak, already elucidated in the respective DSC thermogram (Fig. 6c). In this context, though the peak position of GGAAAMAPA was almost similar to that of the AAAMAPA, considerable enhancement of peak intensity in GGAAAMAPA was attributed to the formation of enhanced crystalline domains via grafting of more crystalline GG moieties within GGAAAMAPA (Fig. 6e).76,77 Among all the M(II)-GGAAAMAPAs, relative enhancement of crystalline nature in Cu(II)-GGAAAMAPA (Fig. 6f) was attributed to the deposition of small crystalline particulates at the adsorbent surface. In fact, water of crystallization and coordinated water molecules of such deposited crystals were eventually removed during the TGA analyses (Fig. 6b). Moreover, the interaction between Cu(II) with –COO− of GGAAAMAPA via various coordination modes, like BB, BC and M, resulted in the formation of ordered architecture in Cu(II)-GGAAAMAPA. On contrary, absence of any peak in the XRD spectrum of Pb(II)-GGAAAMAPA (Fig. 6f) determined the nonprevalent crystalline moieties at the GGAAAMAPA surface. In fact, this observation could also be corroborated from the absence of particulate crystalline deposits at the surface of Pb(II)-GGAAAMAPA (Fig. 7d). However, hemispherical protrusions or inflammations of amorphous nature were reflected by the absence of any characteristic peaks in the respective XRD spectrum (Fig. 6f). Almost similar amorphous structural organization was formed within Cd(II)-GGAAAMAPA, manifested via absence of the characteristic peak in the respective XRD spectrum. Numerous characteristic XRD peaks of MB and SF were passed from sight in dye absorbed GGAAAMAPA (Fig. 6f). For instance, characteristic peaks of SF at 7.20° and 18.02°, corresponding to length and thickness of 1.1 and 0.49 nm, respectively,78 became inexistent in the XRD spectrum of SF-GGAAAMAPA that suggested the positioning of higher extent of SF in the bulk of the GGAAAMAPA rather than at the surface (Fig. 6f). Furthermore, sharp and intense peaks at 25.99° and 27.00° of SF along with other prominent peaks were transformed into a broad peak at 25.45° in SF-GGAAAMAPA (inset of Fig. 6f). Notably, this peak was absent either in the unadsorbed GGAAAMAPA or in the AAAMAPA (Fig. 6f). Indeed, such a marginal improvement of crystallinity in SF-GGAAAMAPA over GGAAAMAPA was rationalized from the inclusion of partly ordered arrangement of flat phenazine rings of SF via stacking interaction promoted by the linear array of negative charges of polyanionic GGAAAMAPA template. Similarly, the characteristic peak of MB at 28.66°, designated to the interplanar distance of 3 Å between flat MB molecules of MB dimer,48 was disappeared in MB-GGAAAMAPA to produce broad peak at 25.52°, indicating simultaneous prevalence of MB dimers, both hydrated and dehydrated, of variegated thicknesses within 3–4 Å at the surface of MB-GGAAAMAPA. In this context, the presence of hydrated MB dimers and aggregates in MB-GGAAAMAPA was already determined from the respective FTIR (Fig. 2a) and TGA (Fig. 6a) analyses. | ||
| Fig. 7 FESEM microphotographs of (a) AAAMAPA, (b) GGAAAMAPA, (c) Cu(II)-GGAAAMAPA, (d) Pb(II)-GGAAAMAPA and (e) Cd(II)-GGAAAMAPA at high KX and their respective insets show microphotographs at low KX; EDX spectrum of (f) Pb(II)- (g) Cd(II)- and (h) Cu(II)-GGAAAMAPA. | ||
FESEM and EDX analyses
Distinct phase boundaries, manifested in the relatively heterogeneous surface of the AAAMAPA (Fig. 7a and inset), were not observed in the photomicrograph of GGAAAMAPA (Fig. 7b and inset) due to the finer intermixing of phases, leading to the destruction of phase boundaries along with incorporation of some minor cracks at the surface of GGAAAMAPA. It may so happen that the relatively loose structural organization of the AAAMAPA was reinforced via grafting with GG to generate a well organized and compact structure of GGAAAMAPA that produced such a finer break. Followed by adsorption of Cu(II), FESEM photomicrograph of Cu(II)-GGAAAMAPA (Fig. 7c and inset) envisaged tiny particulate deposition of Cu compounds at the surface of GGAAAMAPA, also inferred from the numerous Cu peaks of variable intensities in the respective EDX spectrum (Fig. 7h).On contrary, once the Pb(II) intruded in the GGAAAMAPA, discrete swollen state along with surface heterogeneities were developed at the surface of Pb(II)-GGAAAMAPA (Fig. 7d and inset), suggested massive alterations in the structural organization, via H-bonding and variegated coordinate bonding, within the adsorbent. The appearance of variegated interactions between Pb(II) and the GGAAAMAPA were also realized from the multiple peaks of varied intensities in the respective EDX spectrum (Fig. 7f). However, crystalline moieties of GGAAAMAPA was destructed by the adsorbed Cd(II), which preferably interacted with the GGAAAMAPA in a limited fashion (Fig. 7e/7e inset and g), ascribed to the comparatively smaller dimension and greater interfacial area of contact between Cd(II) and GGAAAMAPA and hence, resulted in the phaseless morphology.
DFT studies of AAAMAPA and GGAAAMAPA
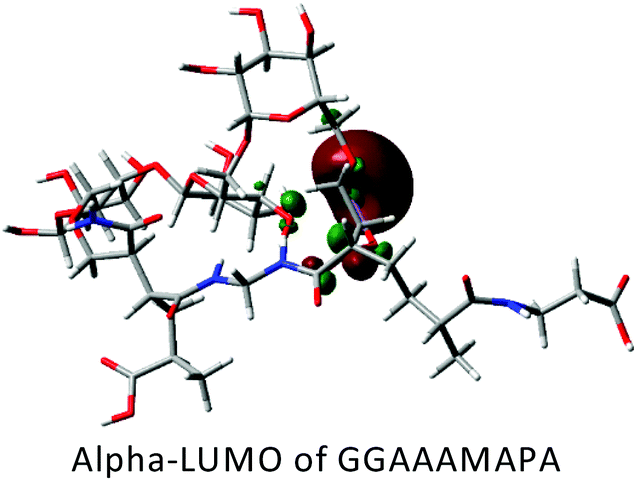
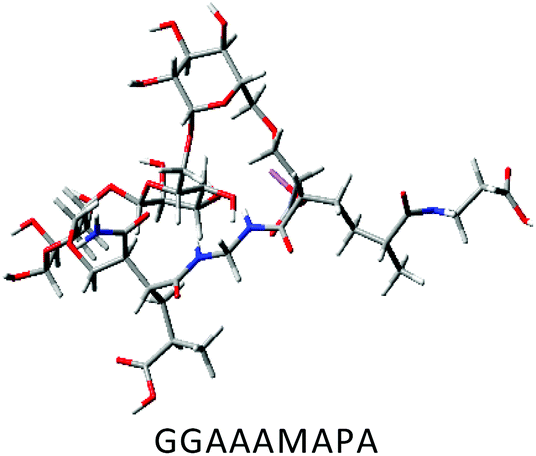
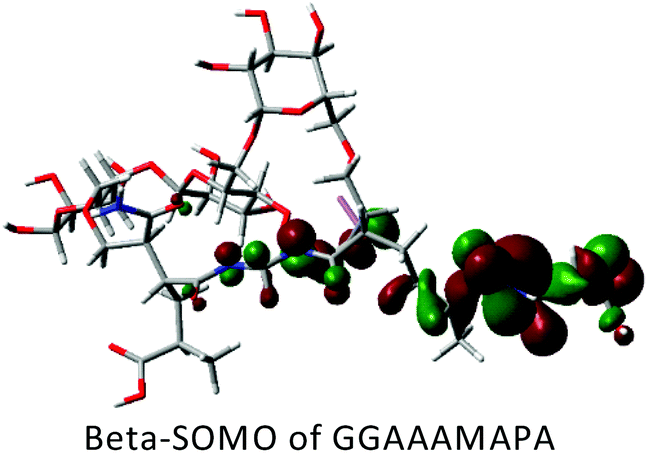
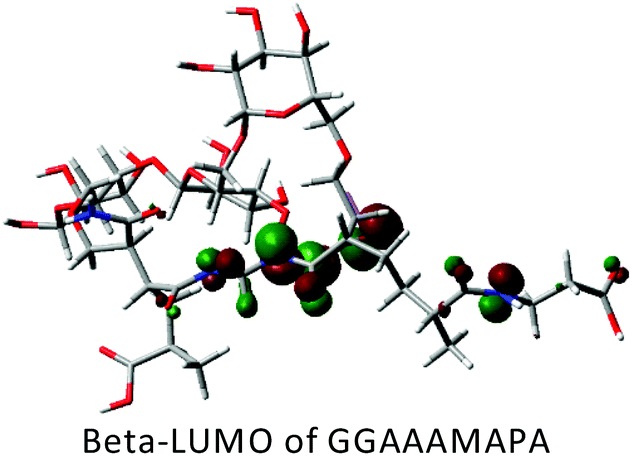
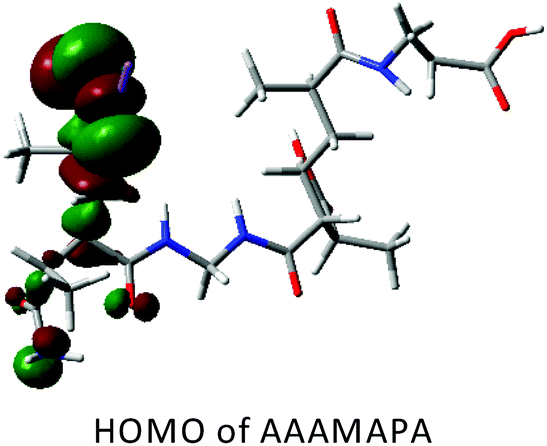
DFT calculations in ground state were performed using Lee–Yang–Parr correlation function (B3LYP) and the basis set 6-31G (s, p). The ground state energy (GSE), dipole moment (DP), optimized structure and (HOMO/SOMO/LUMO) of AAAMAPA and GGAAAMAPA were determined using Gaussian 09 software for comprehensive understanding of change in relative stability via grafting of GG within AAAMAPA network. The theoretically computed GSE and DP for the optimized AAAMAPA (Table 3) were found to be −2031.39 au and 15.75 D, respectively. Indeed, the better overall stability of GGAAAMAPA than AAAMAPA was ascertained from the attainment of relatively lower values of both GSE (i.e. −3936.97 au) and DP (i.e. 3.72 D) (Table 3), ascribed to substantial intramolecular H-bonding within GGAAAMAPA and grafting of GG within AAAMAPA network. Meanwhile, the prevalent grafting and H-bonding was also rationally determined through NMR and FTIR analyses.| Polymer | HOMO/SOMO | LUMO |
|---|---|---|
| Red: O, blue: N, gray: C, LT gray: H and purple: Na. | ||
|

|
|
|
|
|

|
|

|
Rheological studies of AAAMAPA and GGAAAMAPA
Significant increase in storage modulus with the change in frequency from 0 to 80 rad s−1 was observed in GGAAAMAPA (Fig. 8). In contrast, no such marginal change was noted in AAAMAPA within 0–42.3 rad s−1. However, the crossover frequencies of AAAMAPA and GGAAAMAPA were determined at 47.49 and 89.97 rad s−1, respectively. Indeed, above such crossover frequencies, lowering in storage modulus with respect to loss modulus was ascribed to the structural breakdown of hydrogel components, held mostly by weak physical forces.1 Higher values of crossover frequency at 89.97 rad s−1 in GGAAAMAPA than AAAMAPA (i.e. 47.49 rad s−1) demonstrated the overall structural stability and compactness, which were introduced through the grafting of GG in AAAMAPA. Furthermore, a decrease in loss moduli within 80.60–100.00 and 52.40–80.60 rad s−1 for AAAMAPA and GGAAAMAPA, respectively, might comprehensively be linked to the increasing resistance offered towards the laminar flow in stable viscous liquid form, produced through the breakdown of hydrogels at relatively higher frequencies. | ||
| Fig. 8 Variation of storage (G′) and loss (G′′) moduli with frequency in AAAMAPA and GGAAAMAPA. | ||
Adsorption isotherm study
The mutual interaction of adsorbate (i.e. MB, SF, Pb(II), Cd(II) and Cu(II)) and adsorbent (i.e. GGAAAMAPA), during isothermal adsorption, was rationally comprehended through fitting of experimental data with different adsorption isotherm models (eqn (S1)–(S3)†). Isotherm studies were executed by taking 0.025 g of xerogel and 50 mL buffered solutions of different initial concentrations from 5 to 25 ppm of dyes/M(II) within 296–317 K. In fact, equilibrium adsorption data of SF and all the M(II)s fitted the best with Langmuir isotherm model (Fig. S2a and b†), as evident from the highest R2 values, in the entire ranges of temperature and concentration (Table 4). Since, Langmuir model predominantly considers the prevalence of monolayer adsorption on structurally equivalent adsorbent surface, predominant interaction was existent between SF/M(II) and GGAAAMAPA than the interaction between unadsorbed and already adsorbed GGAAAMAPAs. In fact, the excellent performance potential of GGAAAMAPA for the adsorptive removal of SF and M(II) could be understood from high adsorption capacities and more than 90% removal proficiency. Again, the kL values of SF adsorption were observed to increase from 1.0149 to 1.4457 with the increase in temperature, indicated the prevalence of chemisorption. In fact, the existence of chemical interaction between SF and GGAAAMAPA was further justified from the enhancement of qmax from 37.61 to 41.85 mg g−1 with increasing temperature. From FTIR analyses of SF-GGAAAMAPA, the pervasiveness of strong ionic interactions between the available functional groups of SF, i.e.N+Me2/N+HMe and –COO− of GGAAAMAPA, at pHi > pHPZC, also supported the occurrence of chemisorption. Again, the qmax values followed the trend: Pb(II) (41.98 mg g−1) > Cd(II) (40.55 mg g−1) > Cu(II) (39.42 mg g−1). In this context, ionic and coordinative interactions between M(II) and GGAAAMAPA, at pHi > pHPZC, have already been established from FTIR analyses of M(II) loaded GGAAAMAPAs. Though all types of –COO− interactions, like I, M, BB and BC, appeared during Cu(II) adsorption, the lowest molecular weight of Cu(II) reduced the amount of adsorbed Cu(II) within GGAAAMAPA. In fact, the prevalence of small Cu(II) crystals on the surface of GGAAAMAPA was also confirmed from the respective FESEM image (Fig. 7c) and XRD analyses (Fig. 6f). However, the highest atomic weights of 207.20 and 112.41 amu for Pb and Cd, respectively, and the maximum coordination number of 10 for Pb(II) caused an enhancement in the extent of Pb(II) adsorption1 than Cd(II) and, hence, qmax was found to be the maximum for Pb(II). In fact, the order of qmax for M(II) was also found to be in accordance with EDX analyses (Fig. 7f–h) and the residues obtained during TGA analyses (Fig. 6b). In this regard, the dimensionless separation factor (RL), a quantity measuring the feasibility of adsorption (RL > 1, 0 < RL < 1, RL = 1 and RL = 0) indicate unfavourable, favourable, linear and irreversible adsorptions, respectively), was also estimated based on eqn (3). | (3) |
| Temperature (K) | ||||
|---|---|---|---|---|
| 296 | 303 | 310 | 317 | |
| Models and parameters | MB | |||
| BET | ||||
| q BET (mg g−1)/pHi/C0 (ppm) | 26.63/9/5–25 | 27.06/9/5–25 | 27.26/9/5–25 | 27.49/9/5–25 |
| k 1 (L mg−1) | 1.8698 | 1.8813 | 1.8951 | 1.9305 |
| k 2 (L mg−1) | 0.0689 | 0.0659 | 0.0640 | 0.0620 |
| R 2 | 0.9948 | 0.9957 | 0.9966 | 0.9974 |
| F | 1069.65 | 1294.97 | 1645.19 | 2158.77 |
| χ 2 | 1.1547 | 0.9517 | 0.7483 | 0.5695 |
| Pseudosecond order | ||||
| q e,cal (mg g−1)/pHi/C0 (ppm) | 33.06/9/20 | 32.47/9/20 | 32.93/9/20 | 32.79/9/20 |
| q e,exp (mg g−1) | 32.06 ± 0.96 | 32.07 ± 0.97 | 32.10 ± 0.99 | 32.12 ± 1.02 |
| k 2 × 103 (g mg−1 min−1) | 4.57 | 4.92 | 7.72 | 8.97 |
| R 2 | 0.9964 | 0.9911 | 0.9925 | 0.9955 |
| F | 16563.23 |
5759.02 | 9942.41 | 16541.49 |
| χ 2 | 0.3233 | 0.8572 | 0.5951 | 0.3579 |
| Intraparticle diffusion | ||||
| k ip (mg g−1 min−0.5)/pHi/C0 (ppm) | 0.1786/9/25 | |||
| Intercept | 7.33 | |||
| R 2 | 0.9988 | |||
| F | 826.97 | |||
| SF | ||||
|---|---|---|---|---|
| Langmuir | ||||
| q max (mg g−1)/pHi/C0 (ppm) | 37.61/9/5–25 | 39.35/9/5–25 | 40.57/9/5–25 | 41.85/9/5–25 |
| k L (L mg−1) | 1.0149 | 1.0823 | 1.2243 | 1.4457 |
| R 2 | 0.9727 | 0.9876 | 0.9935 | 0.9982 |
| F | 462.63 | 1004.09 | 1910.52 | 6768.73 |
| χ 2 | 4.5259 | 2.2289 | 1.2413 | 0.3732 |
| Pseudosecond order | ||||
| q e,cal (mg g−1)/pHi/C0 (ppm) | 32.75/9/25 | 34.81/9/25 | 36.67/9/25 | 38.34/9/25 |
| q e,exp (mg g−1) | 31.84 ± 0.98 | 33.94 ± 0.99 | 35.79 ± 1.02 | 37.85 ± 1.07 |
| k 2 × 103 (g mg−1 min−1) | 5.30 | 5.85 | 8.06 | 9.64 |
| R 2 | 0.9961 | 0.9891 | 0.9901 | 0.9981 |
| F | 17750.75 |
6918.87 | 7131.62 | 38282.51 |
| χ 2 | 0.3248 | 0.9692 | 1.0349 | 0.2111 |
| Intraparticle diffusion | ||||
| k ip (mg g−1 min−0.5)/pHi/C0 (ppm) | 0.3115/9/25 | |||
| Intercept | 28.20 | |||
| R 2 | 0.9991 | |||
| F | 3087.22 | |||
| Pb(II) | Cd(II) | Cu(II) | |
|---|---|---|---|
| Langmuir | |||
| q max (mg g−1)/pHi/C0 (ppm) | 41.98/7/5–25 | 40.55/7/5–25 | 39.42/7/5–25 |
| k L (L mg−1) | 1.6249 | 1.1446 | 1.3447 |
| R 2 | 0.9983 | 0.9986 | 0.9996 |
| F | 6931.72 | 9051.88 | 35059.32 |
| χ 2 | 0.3715 | 0.2591 | 0.0667 |
| Pseudosecond order | |||
| q e,cal (mg g−1)/pHi/C0 (ppm) | 38.95 | 37.08 | 36.61 |
| q e,exp (mg g−1) | 37.96 ± 1.15 | 36.36 ± 1.04 | 35.75 ± 0.96 |
| k 2 × 103 (g mg−1 min−1) | 7.27 | 7.11 | 5.31 |
| R 2 | 0.9911 | 0.9941 | 0.9925 |
| F | 7431.05 | 13120.77 |
10103.49 |
| χ 2 | 1.0656 | 0.5898 | 0.7357 |
| Intraparticle diffusion | |||
| k ip (mg g−1 min−0.5)/pHi/C0 (ppm) | 0.3531/7/25 | 1.0291/7/25 | 0.6786/7/25 |
| Intercept | 34.43 | 27.52 | 28.57 |
| R 2 | 0.9214 | 0.9726 | 0.9559 |
| F | 36.15 | 72.03 | 47.61 |
However, the spontaneity of adsorption for SF and M(II) was confirmed by the appearance of RL values within 0.05–0.95. On the other hand, MB adsorption data, at equilibrium, was found to fit the best with BET isotherms (Fig. S2c†) in the entire concentration and temperature ranges studied (Table 4). Like other adsorbates, the %adsorption of MB on GGAAAMAPA was also extraordinarily high (∼90%) under the experimental conditions. In this context, the BET isotherm states the prevalence of multilayer adsorption, via dye–adsorbent and dye–dye interactions, on the previously occupied sites of the adsorbent surface. Moreover, qBET and k1 values were found to increase with the rise in temperature, indicating more favourable adsorption at relatively higher temperature. In fact, the reverse variation of k2 with increasing temperature confirmed the decrease in dye–dye interaction at relatively higher temperature.
Adsorption kinetics study
In isothermal adsorption, a kinetics study was carried out to establish the adsorption mechanism via identifying the rate determining step (RDS), mass transport and diffusion processes. Here, kinetics studies were conducted by taking 0.025 g of xerogel at different initial concentrations of dyes/M(II)s, a definite pHi of 9/7 for dyes/M(II) (Fig. 9a–e) and different temperatures (Fig. 9f and g). In this context, the equilibrium adsorption data of SF, MB, Pb(II), Cd(II) and Cu(II) were observed to follow pseudosecond order kinetics (eqn (S4)†) better than pseudofirst order (eqn (S5)†) at the experimental temperature ranges (Table 4). In fact, the applicability of pseudosecond order kinetics confirmed the prevalence of chemisorption. In this context, ionic and variegated coordinate interactions (i.e. M, BB and BC) between GGAAAMAPA (i.e. –COO−) and M(II) had already been established, from XPS (Fig. 5) and FTIR analyses (Fig. 2b) of unloaded and M(II) loaded GGAAAMAPAs, as the key driving force of the adsorption process. Similarly, FTIR studies (Fig. 2a) of dyes adsorbed GGAAAMAPAs have already established that ionic interactions betweenN+Me2 and N+HMe of dyes and –COO− of GGAAAMAPAs were responsible for adsorption. Again, the pseudosecond order rate constants (i.e. k2) for both the dyes were observed to increase sharply with the increase in temperature, indicating faster rate of chemisorption at relatively higher temperature. Indeed, the higher k2 values of SF than those of MB could be explained by considering the formation of the MB dimer, i.e. (MB)2, causing an enhancement of the overall molecular dimension. Thus, (MB)2 might have felt restrictions in the smooth passage and was forced to adopt a tortuous motion, which affected the rate of adsorption (Table 4). However, the larger overall molecular weight of (MB)2 than that of SF might be the possible reason for the presence of a larger amount of MB within the polymer matrix (Table 4), and hence, qe, cal of MB was found to be higher than that of SF. Such a higher value of qe, cal for MB justified a larger amount of residues obtained from TGA analyses of MB-/SF-GGAAAMAPA. Again, k2 for M(II) adsorption at 303 K was varied in the order: Pb(II) > Cd(II) > Cu(II) (Table 4).
 | ||
| Fig. 9 Pseudosecond order kinetics plots for (a) Pb(II)-GGAAAMAPA, (b) Cd(II)-GGAAAMAPA and (c) Cu(II)-GGAAAMAPA (pHi = 7, T = 303 K and adsorbent dose = 0.05 g L−1), pseudosecond order kinetics plots for (d) MB-GGAAAMAPA and (e) SF-GGAAAMAPA (pHi = 10, T = 303 K and adsorbent dose = 0.05 g L−1), pseudosecond order kinetics plots for (f) SF-GGAAAMAPA and (g) MB-GGAAAMAPA (pHi = 7, Co = 30 ppm and adsorbent dose = 1 g L−1), (inset of (f), (g)) full scan kinetics data of SF and MB, respectively, (h) Weber and Morris fitting and (i) Boyd fitting of dyes and M(II). | ||
| qt = kipt0.5 + c | (4) |
Here, kip (mg g−1 min−0.5) and c indicate the intraparticle diffusion rate constant and the resistance of mass transfer, respectively. However, in the present study, the plots of qtvs. t0.5 (Fig. 9h) were observed to be multilinear, possessing three distinct linear segments, of which the slope and intercept of the second segment measured kip.
This observation signified the concurrent incidence of bulk, film and pore/intraparticle diffusions for all dyes/M(II). Additionally, the prevalence of intraparticle diffusion was also confirmed by considering the following Boyd equation (eqn (5)):
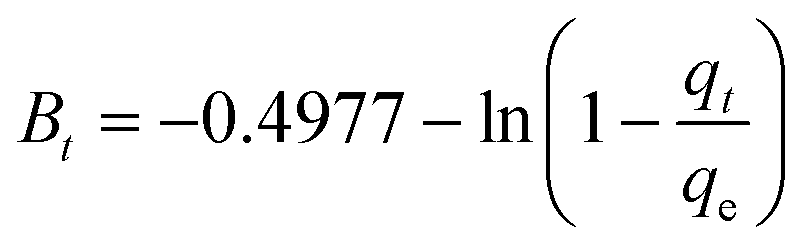 | (5) |
Here, qt and qe are ACs at time t and equilibrium, respectively. Bt is the Boyd parameter, obtained by measuring fractional adsorption (qt/qe) at various times (t). In fact, the nature of the Btvs. t plot can explain the diffusion characteristics of adsorption. However, in the present study, Boyd plots for all the dyes/M(II) were found to be almost linear with the origin as the starting point (Fig. 9i), confronting the existence of pore-diffusion through the micropores of GGAAAMAPA. Again, kip of the used dyes followed the similar order of k2, i.e. SF > MB (Table 4), indicating the tortuous passage of bulky (MB)2 through the micropores of GGAAAMAPA. Again, the order of kip for M(II) adsorption followed the trend: Cd(II) > Cu(II) > Pb(II) due to the smallest particle size of Cd(II) compounds (Fig. 7e).
 | (6) |
Here, k0 and Ea are the temperature independent factor (g mg−1 min−1) and activation energy of adsorption (kJ mol−1), respectively. In fact, from the slope of the linearized lnk2vs. 1/T plot, Ea of adsorption can be evaluated (Fig. S3a†). However, the Ea values were found to be 27.52 and 23.52 kJ mol−1 for MB and SF, respectively, indicating the prevalence of chemisorption for all dyes.6
Thermodynamics of adsorption
The spontaneity of the chemisorption processes was established by estimating the change in standard Gibbs free energies (ΔG0) of dyes using eqn (7).| ΔG0 = −RTlnkd | (7) |
Here, the distribution coefficient (kd) is defined as the ratio of dye concentrations in solid to liquid phases at equilibrium (eqn (8)).
 | (8) |
In fact, ΔG0 can alternatively be represented as the difference between changes in adsorption enthalpy and entropy (i.e. ΔH0 and ΔS0), at a constant temperature, through the van't Hoff equation (eqn (9)):
 | (9) |
Thus, the slope and intercept of the linear plot of lnkdvs. 1/T (Fig. S3b and c†) could be successfully used to determine the values of ΔH0 and ΔS0, respectively (Tables 5 and 6). In fact, the negativity of ΔG0 values for dyes/M(II) (Tables 5/6) suggested the prevalence of thermodynamic spontaneity. Indeed, the gradual increment of −ΔG0 values for the used dyes with increasing temperature confirmed the intense chemisorption at relatively higher temperature.
| Concentration (ppm)/temperature (K) | −ΔG0 (kJ mol−1) of MB/SF | ΔH0 (kJ mol−1) of MB/SF | ΔS0 (J mol−1 K−1) of MB/SF |
|---|---|---|---|
| 5/296 | 9.87/8.64 | −2.09/14.54 | 26.31/78.21 |
| 5/303 | 10.06/9.10 | ||
| 5/310 | 10.25/9.72 | ||
| 5/317 | 10.42/10.27 | ||
| 10/296 | 8.45/7.05 | 4.14/27.52 | 42.47/115.05 |
| 10/303 | 8.68/7.49 | ||
| 10/310 | 9.02/7.83 | ||
| 10/317 | 9.34/8.41 | ||
| 15/296 | 7.34/6.95 | 2.47/17.86 | 33.14/82.11 |
| 15/303 | 7.54/7.12 | ||
| 15/310 | 7.81/7.35 | ||
| 15/317 | 8.04/7.67 | ||
| 20/296 | 6.38/5.50 | 0.61/11.22 | 23.64/55.17 |
| 20/303 | 6.53/5.59 | ||
| 20/310 | 6.71/5.71 | ||
| 20/317 | 6.88/5.88 | ||
| 22/296 | 5.14/4.69 | 0.34/13.59 | 18.51/60.68 |
| 22/303 | 5.27/4.91 | ||
| 22/310 | 5.41/5.06 | ||
| 22/317 | 5.53/5.30 | ||
| 25/296 | 4.99/3.31 | −1.66/21.19 | 11.25/−56.43 |
| 25/303 | 5.07/3.72 | ||
| 25/310 | 5.15/4.07 | ||
| 25/317 | 5.23/4.50 | ||
| Concentration (ppm)/temperature (K) | −ΔG0 (kJ mol−1) of Pb(II)/Cd(II)/Cu(II) |
|---|---|
| 5/296 | 9.52/8.73/9.16 |
| 10/296 | 8.85/7.89/8.10 |
| 15/296 | 7.99/6.98/7.04 |
| 20/296 | 6.11/5.26/5.41 |
| 22/296 | 5.48/4.77/4.75 |
| 25/296 | 4.53/4.12/3.97 |
Synergistic adsorption of MO and MB/SF from alkaline and acidic media
In the alkaline environment, adsorption spectra of the binary mixtures containing equals amount of MO and MB exhibited characteristic peaks at 467.50, 615.64 and 664.10 nm of MO anions, (MB)2 and MB, respectively (Fig. 10a). Accordingly, retention of such peaks even in the binary mixtures indicated the absence of metachromic interactions between MO anions and MB cations. In this regard, the relatively poor adsorption of MO anions on anionic GGAAAMAPA, through weaker ionic interactions among oppositely charged species in the solution, was understandable, whereas appreciable adsorption of cationic MB was observed throughout the adsorption process (Scheme 5a). However, with the progress of adsorption, relative enhancement of the peak intensity indicated that the increase in the concentration of MB monomers compared to that of (MB)2, in the binary mixture (Fig. 10a), envisaged the gradual instability of hydrophobic interactions within (MB)2 as a result of continuously increased dilution and polarity of the aqueous medium resulting in the gradual dissociation of MB dimers into MB monomers. | ||
| Fig. 10 Synergistic removal of (a/b/c) MO:MB (1:1/1:2/2:1) and (d/e/f) MO:SF (1:1/1:2/2:1) at pHi = 9 and (g/h/i) MO:MB (1:1/1:2/2:1) and (j/k/l) MO:SF (1:1/1:2/2:1) at pHi = 2. | ||
 | ||
| Scheme 5 Synergistic removal of MO and MB at (a) pHi = 9 and (c) pHi = 2 and MO and SF at (b) pHi = 9 and (d) pHi = 2. | ||
Almost similar phenomena were observed when MO:MB ratios were changed to either 1:2 or 2:1 (Fig. 10b and c). In both the cases, throughout the adsorption process, the time dependent dilution effect became relatively more pronounced, especially at a 1:2 ratio, resulting in progressive enhancement of relative peak intensities or concentrations of MB monomer over the (MB)2. On the contrary, the visible region of the absorption spectra for a 1:1 mixture of MO and SF showed neither the characteristic individual peak at 465 nm of MO anions nor the sole absorption peak at 516.15 nm of SF cations (Fig. 10d). In fact, individual adsorption peaks of both the dyes (insets of Fig. 9f and g) were considerably shifted to produce a peak at 491.65 nm initially (t = 0) through the metachromic interactions as compared to the weaker type of ionic interactions between MO and MB. Moreover, as the adsorption proceeded, the sole adsorption peak at 491.65 nm was gradually blue shifted to 470.05 nm with a moderate time dependent hypochromic shift that indicated the relative lowering in adsorption of MO–SF in comparison with that of the MO–MB system. Such an inferior adsorbing tendency of the MO–SF mixture by the polyanionic GGAAAMAPA was attributed to the metachromic interaction driven stability of the MO–SF adduct in solution. As the adsorption proceeded, the metachromic interaction was substantially affected by the enhanced dilution and polarity of the solution, leading to the time dependent continuous breakdown of the MO–SF adduct to liberate SF cations. Thus, relatively better population of MO anions in solution, generated via cleavage of the MO–SF adduct (Scheme 5b), was evident from the final λmax at 468.75 nm, which was very close to the individual λmax at 465 nm of MO anions. As the MO:SF ratio was changed from 1:1 to 1:2, the initial λmax was red shifted to 507.65 nm (Fig. 10e and f). Indeed, the population driven shift of the initial λmax was also corroborated by the significant blue shift of the initial λmax from 491.65 nm for 1:1 to 475.25 nm for 2:1 composition that was mostly populated with MO. Moreover, similar to the 1:1 binary mixture, the time dependent dilution induced breakdown of the MO–SF adduct in a 1:2 mixture, followed by the attachment of liberated SF cations with GGAAAMAPA, was realized from the appearance of gradual blue shifts from 507.65 and 475.25 to 471.35 and 468.75 nm, respectively. Altogether, the preferential adsorption of SF cations over MO anions by the GGAAAMAPA was corroborated by the increasing hypochromic effect with the increased stoichiometric ratio of SF in the binary mixture.
In the acidic environment, for an equimolar binary mixture of MO and MB, the absence of metachromic interactions between MO tautomers, in ammonium and azonium forms, and MB cations was confirmed by the almost unaltered characteristic peaks for ammonium/azonium (λmax = 320.85/500–540 nm) tautomers of MO (Fig. S4†), MB (λmax = 664.10 nm) and (MB)2 (λmax = 615.64 nm) (Fig. 10g–i). However, the enhanced dilution effect of the adsorption process increased the dissociation of (MB)2 into MB, as observed from the time dependent increment of relative peak intensity of MB over the (MB)2. Interestingly, both the peaks, assigned to ammonium and azonium tautomers (Scheme 5c), showed an initial hypochromic shift up to 200 min followed by a hyperchromic effect up to 2120 min. In particular, the characteristic peaks for the azonium tautomer demonstrated an unusual hypsochromic shift from 507.55 to 480.40 nm, coupled with the hypochromic effect within 200 min, and thereafter, a bathochromic shift from 480.40 to 505.10 nm was observed up to 2120 min along with the hyperchromic effect. In fact, such phenomena were accompanied by the gradual appearance of a shoulder within 400–420 nm, followed by continuous disappearance of that particular shoulder in the later stage up to 2120 min. Such a combined hypsochromic and hypochromic effect along with the appearance of a shoulder within 400–420 nm indicated the possible protonation of a part of the azonium tautomers into a diprotonated form of MO79 due to the gradual numerical advantage of the available protons over the azonium tautomers in solution phase with gradual attachment of azonium tautomers by GGAAAMAPA. In this regard, such a contribution of MO anions (λmax = 320.85 nm) for producing the combined hypsochromic and hypochromic shift could be neglected at strongly acidic pH. In the later stage, the unstable diprotonated form of MO dissociated in the increasingly polar environment to produce azonium tautomers. Altogether, the time dependent change in the relative population of various MO zwitterions, like ammonium–azonium tautomer and diprotonated forms, in the solution phase caused such peculiar alterations in the visible spectrum. Similar types of more pronounced phenomena were observed in the case of the MO–MB binary mixture in the molar ratio of 1:2.
The characteristic peak for azonium tautomers reflected an unusual hypsochromic shift from 508.80 to 470.10 nm coupled with the hypochromic effect within the first 240 min, followed by a bathochromic shift within 470.10–505.10 nm up to 2060 min along with the hyperchromic effect. In contrast, such phenomena became less evident when the binary mixture was populated with relatively more hydrophilic MO that imparted more hydrophilic character in solution. It may so happen that a relatively polar environment might interrupt the formation of the diprotonated form as this was expected to be less stable in a relatively more polar environment. In this context, the visible region of the spectra for the MO–SF binary mixture envisaged a single peak at 512.45 nm corresponding to the characteristic peak of both MO azonium ions and SF monomers. As the simultaneous adsorption of an equimolar MO–SF mixture continued in the acidic environment, a combined hypsochromic and hypochromic effect was observed up to 200 min, suggesting the occurrence of similar events as demonstrated during adsorption of MO–MB. In contrast, such combined hypsochromic and hypochromic effects became almost irrelevant once the molar ratio was increased up to 2:1 in favour of SF (Fig. 10k). This observation was just the opposite of the earlier observations of adsorption from MO–MB having an identical molar ratio. Earlier it was mentioned that the formation of diprotonated MO was assisted in the case of the MO–MB binary mixture having a 1:2 molar ratio. Conversely, the production of diprotonated MO was arrested in a 1:2 molar ratio of MO–SF. Though MO was more hydrophilic than SF, the prevalent metachromatic interaction between MO and SF prevented the formation of diprotonated MO (Scheme 5d). In this context, most of the MO azonium ions, relatively lower than SF, were engaged in the metachromic interaction with SF cations. However, during the adsorption, the formation of diprotonated MO would be accentuated and even more probable in 2:1 than 1:1 mole ratio of MO–MB (Fig. 10j and l).
Reusability and recyclability
Desorption studies were conducted to determine the possible reusability/recyclability of the GGAAAMAPA superadsorbent via adsorption of 25 ppm of MB and SF/M(II) at pHi = 9/7 followed by desorption of MB and SF/M(II) loaded GGAAAMAPA at 0.1 N NaCl/pHi = 1 (Fig. 11a). The simpler mechanism for GGAAAMAPA recycling was ascribed to the pH switchability of M(II). In fact, desorption of M(II) at low pHi indicated the disruption of the prevalent electrostatic interactions of M(II)-GGAAAMAPAs. Again, the increased desorption of dyes from dyed GGAAAMAPA in the strongly electrolytic solution was due to the decreased dye-GGAAAMAPA ionic interaction, as the Cl− of NaCl competitively blocked the available cationic sites of dyes needed for sorption. In our study, ∼90% desorption was observed at 0.1 N NaCl/pHi = 1 that confirmed electrostatic attraction of cationic MB and SF/M(II) with the anionic –COO− during adsorption at pHi = 9/7. In fact, five consecutive adsorption–desorption cycles were carried out under the identical experimental conditions using the regenerated GGAAAMAPAs and %adsorption in the consecutive cycles was observed to be ∼95% (Fig. 11b). | ||
| Fig. 11 Desorption of (a) MB/SF at 0.1 N NaCl solution and (b) reusability plot for GGAAAMAPA. | ||
Comparison of the results
Several micro-/nano-material based adsorbents, blends, homo-/co-/ter-polymers, and IPN hydrogels have been employed for the adsorptive removal of MB, SF, Pb(II), Cd(II) and Cu(II) from aqueous solutions of varying initial concentrations (i.e. 10–1000 ppm), temperature (i.e. 293–333 K) and pHi (Table S1†). Although it is tantamount to mention that AC predominantly depends on the initial concentrations, the ACs of GGAAAMAPA, within initial concentration of 5–25 ppm, were found to be the highest among all the reported studies (Table S1†).Conclusions
The most exciting outcome of this work is that the green synthesis of a novel and sustainable GGAAAMAPA superadsorbent, via in situ attachment of non-toxic APA during solution polymerization of AA and AM and grafting of GG within AAAMAPA, has been confirmed through 1H-/13C-NMR, FTIR and XPS analyses and supported by DFT studies. The plausible chemical structure of GGAAAMAPA, intimate physicochemical interactions, superadsorption mechanism, variegated interactions and several associated microstructural changes have been rationally inferred via systematic characterization of both loaded and unloaded GGAAAMAPAs, through spectroscopic, microscopic, diffractometric, thermal, computational and rheological analyses, and by the measurements of %GC, pHPZC, %GR and network parameters. A series of hydrogels have been synthesized, using varying compositions and temperatures, of which GGAAAMAPA possessing the optimum ESR, GT and structural integrity has shown excellent ACs for cationic phenothiazine/phenazine dyes and Cu(II)/Pb(II)/Cd(II). The mechanism of M(II) adsorption onto the as-prepared GGAAAMAPA, via the appearance of I, M, BB and BC modes of interactions, has been rationally understood by FTIR and XPS analyses. The attachment of hydrated MB dimers on the GGAAAMAPA surface has been determined via FTIR, XRD, TGA and DSC analyses, supported by the BET isotherm model. The pervasiveness of chemical attachment onto the GGAAAMAPA, at pHi > pHPZC, has also been rationalized via fitting kinetics data to the pseudosecond order model as well as from the measurement of Ea. The individual/interactive effects of cationic and/or anionic dyes have been rationally realized, through the appearance of dye-aggregates, diprotonated MO, π–π interaction, a hydrophilic/hydrophobic environment etc. with varying pHi and mole ratios of dyes. This tailor-made hydrogel, with excellent thermomechanical properties, can also be attempted for targeted drug delivery, tissue engineering, self-healing materials, membrane based separation and sensors. Altogether, owing to the rationally optimized combination of synthetic and natural polymers, imparting an excellent balance between physicochemical properties, including recyclability, performance characteristics towards selective individual/synergistic chemisorption of M(II)/dyes, and extraordinary adsorption efficiency, systematic design of a sustainable GGAAAMAPA has shown novelty (Table S1†) in a kinetically fast, green and hazardous waste remediation and/or recycling process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The corresponding author gratefully acknowledges the Department of Science and Technology (DST), Government of India [YSS/2015/000886] and the DST, Government of West Bengal [773(Sanc.)/ST/P/S&T/15G-2/2015] for providing financial assistance and the Department of Higher Education, Government of West Bengal for giving an opportunity to participate in the inter-institutional collaboration with the University of Calcutta. Mr Mahapatra and Mr Karmakar are grateful to the University Grants Commission (UGC) and DST INSPIRE, Government of India, respectively, for providing fellowships.References
- N. R. Singha, M. Karmakar, M. Mahapatra, H. Mondal, A. Dutta, C. Roy and P. K. Chattopadhyay, Polym. Chem., 2017, 8, 3211–3237 RSC.
- N. R. Singha, S. B. Kuila, P. Das and S. K. Ray, Chem. Eng. Process., 2009, 48, 1560–1565 CrossRef CAS.
- N. R. Singha, S. Kar, S. Ray and S. K. Ray, Chem. Eng. Process., 2009, 48, 1020–1029 CrossRef CAS.
- N. R. Singha, S. Kar and S. K. Ray, Sep. Sci. Technol., 2009, 44, 422–446 CrossRef CAS.
- N. R. Singha and S. K. Ray, Sep. Sci. Technol., 2010, 45, 2298–2307 CrossRef CAS.
- M. Karmakar, M. Mahapatra, A. Dutta, P. K. Chattopadhyay and N. R. Singha, Int. J. Biol. Macromol., 2017, 102, 438–456 CrossRef CAS PubMed.
- L. Ling, W. Liu, S. Zhang and H. Jiang, J. Mater. Chem. A, 2016, 4, 10336–10344 CAS.
- Y. Lei, W. Chen, B. Lu, Q. Ke and Y. Guo, RSC Adv., 2015, 5, 98783–98795 RSC.
- N. Li and R. Bai, Sep. Purif. Technol., 2005, 42, 237–247 CrossRef CAS.
- F. Yuan, C. Song, X. Sun, L. Tan, Y. Wang and S. Wang, RSC Adv., 2016, 6, 15201–15209 RSC.
- O. A. Attallah, M. A. Al-Ghobashy, M. Nebsen and M. Y. Salem, RSC Adv., 2016, 6, 11461–11480 RSC.
- Y. Song, X. Song, C. Cheng and Z. Zhao, RSC Adv., 2015, 5, 87030–87042 RSC.
- N. R. Singha, P. Das and S. K. Ray, J. Ind. Eng. Chem., 2013, 19, 2034–2045 CrossRef CAS.
- S. Ray, N. R. Singha and S. K. Ray, Chem. Eng. J., 2009, 149, 153–161 CrossRef CAS.
- S. Roy and N. R. Singha, Membranes, 2017, 7 CrossRef PubMed , 53.
- G. K. Mehta, S. Kondaveeti and A. K. Siddhanta, Polym. Chem., 2011, 2, 2334–2340 RSC.
- D. Das, P. Patra, P. Ghosh, A. P. Rameshbabu, S. Dhara and S. Pal, Polym. Chem., 2016, 7, 2965–2975 RSC.
- L. Hu, P. Zhao, H. Deng, L. Xiao, C. Qin, Y. Du and X. Shi, RSC Adv., 2014, 4, 13477–13480 RSC.
- K. Sharma, V. Kumar, B. S. Kaith, V. Kumar, S. Som, A. Pandey, S. Kalia and H. C. Swart, New J. Chem., 2015, 39, 3021–3034 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- M. Sharma, D. Mondal, C. Mukesh and K. Prasad, RSC Adv., 2013, 3, 16509–16515 RSC.
- A. Giri, T. Bhunia, L. Goswami, A. B. Panda and A. Bandyopadhyay, RSC Adv., 2015, 5, 41736–41744 RSC.
- R. Barbucci, R. Giardino, M. D. Cagna, L. Golini and D. Pasqui, Soft Matter, 2010, 6, 3524–3532 RSC.
- M. P. Gowrav, U. Hani, H. G. Shivakumar, R. A. M. Osmani and A. Srivastava, RSC Adv., 2015, 5, 80005–80013 RSC.
- K. S. V. P. Chandrika, A. Singh, A. Rathore and A. Kumar, Carbohydr. Polym., 2016, 149, 175–185 CrossRef CAS PubMed.
- W. Wang and A. Wang, Carbohydr. Polym., 2009, 77, 891–897 CrossRef CAS.
- B. Urbano and B. L. Rivas, Polym. Int., 2012, 61, 23–29 CrossRef CAS.
- E. K. Yetimoğlu, M. V. Kahraman, Ö. Ercan, Z. S. Akdemir and N. K. Apohan, React. Funct. Polym., 2007, 67, 451–460 CrossRef.
- S. Durmaz and O. Okay, Polymer, 2000, 41, 3693–3704 CrossRef CAS.
- K. Kataoka, H. Miyazaki, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 1061–1062 CrossRef CAS.
- M. A. Jalali, A. D. Koohi and M. Sheykhan, Carbohydr. Polym., 2016, 142, 124–132 CrossRef PubMed.
- M. Yadav and K. Y. Rhee, Carbohydr. Polym., 2012, 90, 165–173 CrossRef CAS PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- S. Yuen, S. Choi, D. L. Phillips and C. Ma, Food Chem., 2009, 114, 1091–1098 CrossRef CAS.
- D. Mudgil, S. Barak and B. S. Khatkar, Int. J. Biol. Macromol., 2012, 50, 1035–1039 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons. Ltd, England, 2001 Search PubMed.
- A. G. Sullad, L. S. Manjeshwar and T. M. Aminabhavi, Ind. Eng. Chem. Res., 2010, 49, 7323–7329 CrossRef CAS.
- N. R. Singha, T. K. Parya and S. K. Ray, J. Membr. Sci., 2009, 340, 35–44 CrossRef CAS.
- D. Huang, W. Wang, Y. Kang and A. Wang, J. Macromol. Sci. Part. A :Pure Appl. Chem., 2012, 49, 971–979 CrossRef CAS.
- P. H. Mccluskey, R. L. Snyder and R. A. Condrate, J. Solid State Electrochem., 1989, 83, 332–339 CrossRef CAS.
- W. Li, H. Zhao, P. R. Teasdale, R. John and S. Zhang, React. Funct. Polym., 2002, 52, 31–41 CrossRef CAS.
- J. Dong, Y. Ozaki and K. Nakashima, Macromolecules, 1997, 30, 1111–1117 CrossRef CAS.
- M. D. Keersmaecker, T. Hauffman, O. V. D. Berg, S. Vandewalle, T. Muselle, K. Verbekend, A. Hubin, F. D. Prez and A. Adriaens, Electrochim. Acta, 2017, 229, 8–21 CrossRef.
- K. Chauhan, G. S. Chauhan and J. H. Ahn, Bioresour. Technol., 2009, 100, 3599–3603 CrossRef CAS PubMed.
- G. I. Dzhardimalieva and A. D. Pomogailo, Russ. Chem. Rev., 2008, 77, 259–301 CrossRef CAS.
- M. Eldamer-Rinaudo, J. C. Lassegues, J. Portier, J. Salardenne, J. Grondin and R. Cavagnat, J. Mater. Chem., 1997, 7, 1549–1552 RSC.
- O. V. Ovchinnikov, A. V. Evtukhova, T. S. Kondratenko, M. S. Smirnov, V. Y. Khokhlov and O. V. Erina, Vib. Spectrosc., 2016, 86, 181–189 CrossRef CAS.
- N. Zotov and H. Keppler, Am. Mineral., 1998, 83, 823–834 CrossRef CAS.
- H. B. Sonmez, B. F. Senkal and N. Bicak, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3068–3078 CrossRef CAS.
- D. Arunbabu, H. Shahsavan, W. Zhang and B. Zhao, J. Phys. Chem. B, 2013, 117, 441–449 CrossRef CAS PubMed.
- G. B. H. Chua, P. J. Roth, H. T. T. Duong, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 1362–1374 CrossRef CAS.
- B. L. Rivas and A. Maureira, Eur. Polym. J., 2008, 44, 523–533 CrossRef CAS.
- D. Roy and B. S. Sumerlin, ACS Macro Lett., 2012, 1, 529–532 CrossRef CAS.
- P. Ilgin and O. Ozay, Iran. Polym. J., 2017, 26, 391–404 CrossRef CAS.
- Y. Kaneko, S. Sato, J. Kadokawa and N. Iyi, J. Mater. Chem., 2006, 16, 1746–1750 RSC.
- D. Das, P. Ghosh, A. Ghosh, C. Haldar, S. Dhara, A. B. Panda and S. Pal, ACS Appl. Mater. Interfaces, 2015, 7, 14338–14351 CAS.
- V. Crescenzi, M. Dentini, D. Risica, S. Spadoni, G. Skjåk-Bræk, D. Capitani, L. Mannina and S. Viel, Biomacromolecules, 2004, 5, 537–546 CrossRef CAS PubMed.
- P. L. R. Cunha, I. G. P. Vieira, A. M. C. Arriaga, R. C. M. de Paula and J. P. A. Feitosa, Food Hydrocolloids, 2009, 23, 880–885 CrossRef CAS.
- A. Bahamdan and W. H. Daly, Polym. Adv. Technol., 2007, 18, 652–659 CrossRef CAS.
- H. Kono, F. Otaka and M. Ozaki, Carbohydr. Polym., 2014, 111, 830–840 CrossRef CAS PubMed.
- D. Pathania, D. Gupta, A. H. Al-Muhtaseb, G. Sharma, A. Kumar, M. Naushad, T. Ahamad and S. M. Alshehri, J. Photochem. Photobiol., A, 2016, 329, 61–68 CrossRef CAS.
- S. Celebi, A. K. Erdamar, A. Sennaroglu, A. Kurt and H. Y. Acar, J. Phys. Chem. B, 2007, 111, 12668–12675 CrossRef CAS PubMed.
- S. C. Yoon and B. D. Ratner, Macromolecules, 1986, 19, 1068–1079 CrossRef CAS.
- E. Orozco-Guareño, F. Santiago-Gutiérrez, J. L. Morán-Quiroz, S. L. Hernandez-Olmos, V. Soto, W. de la Cruz, R. Manríquez and S. Gomez-Salazar, J. Colloid Interface Sci., 2010, 349, 583–593 CrossRef PubMed.
- J. Gao, Y. Wang, W. Yang and Y. Li, Bull. Korean Chem. Soc., 2010, 31, 406–414 CrossRef CAS.
- X. Deng, C. Wang, H. Yang, M. Shao, S. Zhang, X. Wang, M. Ding, J. Huang and X. Xu, Sci. Rep., 2017, 7, 3877–3888 CrossRef PubMed.
- S. Deng and Y. P. Ting, Langmuir, 2005, 21, 5940–5948 CrossRef CAS PubMed.
- J. Fan, Z. Shi, M. Lian, H. Li and J. Yin, J. Mater. Chem. A, 2013, 1, 7433–7443 CAS.
- A. Srivastava, V. Mishra, P. Singh, A. Srivastava and R. Kumar, J. Therm. Anal. Calorim., 2012, 107, 211–223 CrossRef CAS.
- M. Mahapatra, M. Karmakar, B. Mondal and N. R. Singha, RSC Adv., 2016, 6, 69387–69403 RSC.
- J. J. Maurer and G. D. Harvey, Thermochim. Acta, 1987, 121, 295–306 CrossRef CAS.
- H. Dweik, W. Sultan, M. Sowwan and S. Makharza, Int. J. Polym. Mater., 2008, 57, 228–244 CrossRef CAS.
- N. Gupta and A. K. Srivastava, Macromolecules, 1995, 28, 827–832 CrossRef CAS.
- Z. Huang, S. Liu, G. Fang and B. Zhang, Carbohydr. Polym., 2013, 92, 2314–2320 CrossRef CAS PubMed.
- P. Das, S. K. Ray, S. B. Kuila, H. S. Samanta and N. R. Singha, Sep. Purif. Technol., 2011, 86, 159–173 CrossRef.
- M. Karmakar, M. Mahapatra and N. R. Singha, Korean J. Chem. Eng., 2017, 34, 1416–1434 CrossRef CAS.
- S. P. Sun, T. A. Hatton, S. Y. Chan and T. Chung, J. Membr. Sci., 2012, 401–402, 152–162 CrossRef CAS.
- K. M. Tawarah and H. M. Abu-Shamleh, Dyes Pigm., 1991, 16, 241–251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01564j |
| This journal is © The Royal Society of Chemistry 2017 |