
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of synthesis process on the Li-ion conductivity of LiTa2PO8 solid electrolyte materials for all-solid-state batteries†
Hayami
Takeda
 *,
Miki
Shibasaki
,
Kento
Murakami
,
Miki
Tanaka
,
Keisuke
Makino
,
Naoto
Tanibata
,
Hirotaka
Maeda
and
Masanobu
Nakayama
*,
Miki
Shibasaki
,
Kento
Murakami
,
Miki
Tanaka
,
Keisuke
Makino
,
Naoto
Tanibata
,
Hirotaka
Maeda
and
Masanobu
Nakayama
Department of Advanced Ceramics, Nagoya Institute of Technology, Gokiso, Showa, Nagoya, Aichi 466-8555, Japan. E-mail: takeda.hayami@nitech.ac.jp
First published on 11th July 2024
Abstract
Inorganic solid electrolytes are essential for developing safe and non-flammable all-solid-state batteries, with oxide-based ones having attracted attention owing to their excellent chemical stability. Recently, a new solid electrolyte material LiTa2PO8 (LTPO) was reported to have a bulk lithium-ion conductivity of 1.6 mS cm−1 at room temperature, which is one of the highest among oxide solid electrolytes. However, oxide solid electrolytes tend to have a high grain boundary resistivity and must be formed into dense sintered pellets. In this study, different dense LTPO materials were synthesised by adjusting the size of the starting powder particles, and their ionic conductivities were systematically investigated. Counterintuitively, larger raw particles resulted in a lower grain boundary resistivity. This was attributed to the micromorphology of the sintered pellets. The grain boundary resistance varied by up to one order of magnitude under the investigated synthesis conditions, and the optimised total ionic conductivity (including the bulk and grain boundary contributions) of LTPO was 0.95 mS cm−1 at 30 °C.
Introduction
The demand for large and safe storage batteries for electric vehicles and smart grids has increased recently, with all-solid-state batteries using non-flammable inorganic solid electrolytes instead of flammable organic electrolytes being more attractive, offering increased safety. Moreover, all-solid batteries can be stacked, are expected to be more compact, and have a higher capacity. Current inorganic solid electrolytes are based on sulfides,1–3 chlorides,4–6 and oxides. In particular, oxide-based solid electrolytes exhibit excellent chemical stability and can be assembled under atmospheric conditions. Some well-known examples include perovskite-type (e.g., Li3xLa2/3−xTiO3),7–9 garnet-type (e.g., Li7La3Zr2O12),10–13 NASICON-type phosphates (e.g., Li1+xAlxTi2−x(PO4)3, LiZr2(PO4)3),14–21 and LISICON-type materials (e.g., Li2+2xZn1−xGeO4).22,23 A common feature of these excellent Li ion-conducting oxides is that their structures contain a framework composed of metal-centred octahedra or tetrahedra that share a vertex.24In 2018, Kim et al. conducted a systematic search through the vertex-sharing polyanion Lix(MO6/2)m(TO4/2)n phases (where M and T represent octahedral and tetrahedral cation sites, respectively). They developed a novel material, monoclinic LiTa2PO8 [Lix(TaO6/2)2(PO4/2)1, hereafter abbreviated as LTPO], with a bulk conductivity of 1.6 mS cm−1 and a total conductivity (including the bulk and grain-boundary contributions) of 0.25 mS cm−1 at 25 °C. The crystalline system of LTPO is reported to be monoclinic with a C2/c space group.24,25 LTPO exhibits excellent water and alkali tolerance.26 Although the P5+ and Ta5+ ions are reduced upon contact with the Li metal anode,26 stable charge/discharge cycles were possible even when using a Li metal anode by interposing a lithium ion-conductive polyethylene oxide buffer phase between the LTPO and Li metal anode.26
Considering the high theoretical lithium-ion conductivity of LTPO (35 mS cm−1) based on density functional theory calculations,23 there should be plenty of room for improving its conductivity. To enhance the bulk conductivity, researchers have considered the partial substitution of Ta and P with other elements.27–29 To reduce the grain boundary resistivity, which is larger than the bulk resistivity, studies have reported the addition of excess Li30,31 and B2O332,33 or sintering methods such as spark plasma sintering and hot pressing.26,33 However, the highest reported bulk and total conductivities of LTPO at room temperature are 2.8 and 0.7 mS cm−1, respectively,33 still more than one order of magnitude lower than the theoretical values. Even when similar preparation conditions are followed, such as equal Li excess and the same firing temperature, the reported lithium-ion conductivities of current LTPO materials differ from author to author.24,31 This suggests the existence of influencing factors other than raw material composition and heating conditions. Optimising the synthesis process is necessary for improving the conductivity, particularly the grain boundary resistivity. Therefore, in this study, we examined the solid-state reaction of LTPO synthesis to clarify the relationship between the particle size of the raw materials and the ionic conductivity, which influences the reaction rate of the synthetic procedure.
Experimental methods
Synthesis of LTPO pellet
LTPO was synthesised via a solid-state reaction using stoichiometric ratios of Ta2O5 (99.9%; Kojundo Chemical Laboratory, Japan), (NH4)2HPO4 (99%; Wako Pure Chemical Industry, Japan), and Li2CO3 (99.9%; Kojundo Chemical Laboratory, Japan). Excess Li2CO3 (10 wt%) was used to compensate for the evaporation of Li2O at high temperatures.All reagents were initially passed through a sieve of mesh size 500 μm. The reagents were further classified using another sieve and mixed according to conditions A–D listed in Table 1. For sample A, unclassified reagents were processed in a planetary ball mill (Pulverisette 7, Fritsch, Germany) with a ZrO2 pot and balls (5 mm diameter). For samples B–D, Ta2O5 and (NH4)2HPO4 were further passed through sieves of mesh sizes 100 and 250 μm. The third reagent (Li2CO3) was not classified, because its particles were reduced to below 100 μm after a few minutes of mortar grinding. Samples B–D were prepared by mixing the reagents for 20 min using an agate magnetic mortar and a mortar stirrer (VPMS-180BD; AS ONE, Japan). The particle size distribution of the mixed reagent powder was analysed in the dry state using a laser diffraction particle size analyser (Mastersizer 3000, Malvern Panalytical Ltd. UK). The specific surface area of the mixed reagent powder was determined using the Brunauer–Emmett–Teller method via nitrogen gas adsorption analysis (Bellsorp-miniII, Microtrac BEL Corp. Japan) at −196 °C. Before analysis, the samples were heated at 80 °C under vacuum for a minimum of 2 hours.
| Sample | Sieve sizes for reagents Ta2O5 and (NH4)2HPO4/μm | Mixing and grinding condition (method/rotation per minute) |
|---|---|---|
| A | <500 (unsieved samples) | Ball-mill/300 |
| B | <100 | Magnetic mortar/140 |
| C | 100–250 | Magnetic mortar/140 |
| D | 250–500 | Magnetic mortar/140 |
The mixed reagent powders for samples A–D were preheated at 600 °C for 8 h and then at 1000 °C for 8 h (collectively called the 1st heating). The 1st heating was conducted without pelletisation to avoid any differences in the results caused by the crushing and grinding steps after the formation of the sintered body. The use of powder during the 1st heating prevented the introduction of differences in conductivity measurements due to human-derived errors during crushing. Samples agglomerated after the 1st heated were processed at 400 rpm for 30 min using a planetary ball mill with a ZrO2 pot and balls (10 mm diameter). Thereafter, the samples were pelletised and sintered at 1050 °C for 12 h (2nd heating) to obtain pellets for conductivity measurements. The sintered pellets were approximately 9 mm in diameter and 1.5 mm in thickness.
Characterisation
The crystalline phase of the samples was determined via X-ray diffraction (XRD) using a MiniFlex 600 diffractometer (Rigaku, Japan) with CuKα radiation. Rietveld analysis was performed using RIETAN-FP software.34 Micromorphology and elemental analyses of the samples were performed via scanning electron microscopy and energy dispersive X-ray spectroscopy (SEM-EDS; JCM-7000 Neoscope, JEOL, Japan). Compositional analysis via X-ray photoelectron spectroscopy (XPS) was performed using an AlKα radiation source (PHI Quantes, ULVAC-PHI, Inc., USA). The bulk density of the sintered pellets (after the 2nd heating) was calculated using the weight, diameter, and thickness of the pellets. The diameter and thickness of the sample were measured at three points and the average values were used; the true density of the LTPO sample was assumed to be the theoretical density of LTPO (5.85 g cm−3).35 The relative density was calculated as the ratio of the density of the sample to the theoretical density of LTPO. The ionic conductivities of the pellets were measured after the 2nd heating using the AC impedance method. Both sides of the pellet were polished with 2000-grit abrasive paper, and electrodes were prepared on the sides by sputtering gold. The complex impedance was measured with an impedance analyser (VMP-300, BioLogic, France) at −10, 0, 10, and 30 °C, a frequency range of 1–106 Hz, and a voltage of 100 mV. The activation energy of lithium transport was calculated using the Arrhenius plot. The electronic conductivity was determined from DC potentiostatic polarisation measurements under a constant voltage of 100 mV for 3600 s, using the pellets after the AC impedance measurements.Results
Characterisation of starting materials after particle size adjustment
The particle size distribution of the adjusted starting materials is illustrated in Fig. S1 (ESI†). The median diameters of the prepared samples A, B, C, and D were 7.64, 11.1, 22.6, and 24.1 μm respectively. The nitrogen gas adsorption curves of the adjusted samples and the calculated specific surface area are illustrated in Fig. S2 (ESI†). Sample A exhibited the highest nitrogen adsorption and the highest specific surface area. The nitrogen gas adsorption curves of samples B, C, and D overlapped and no significant difference in their specific surface areas was observed. Fig. 1(a–d) shows the SEM images of mixed reagents used to prepare samples A–D (defined in Table 1), respectively. In the ball-milled reagents for sample A, agglomerated particles of about 5 μm were observed, while most of the primary particles were sub-micron or smaller. In the reagents for samples B, C, and D, which were mixed with an agate magnetic mortar, the larger the reagent particle size, the coarser the remaining particles. Fig. S3 (ESI†) shows the distributions of P and Ta elements and the EDS spectra of the starting mixture for sample D. EDS analysis detected Ta and O in the coarse particles, and a small amount of P. Such a finding indicates that Ta2O5 tends to remain as coarse particles without being pulverised. | ||
| Fig. 1 SEM images of the precursor particles for (a) sample A, (b) sample B, (c) sample C, and (d) sample D. | ||
Temperature dependence of the crystal phase
To investigate the crystal phases formed at different temperatures, the mixed precursor powders for samples A and D were heated at 750, 850, 950, 1050, and 1100 °C for 30 min. Their XRD patterns are shown in Fig. 2(a) and (b), and the identified phases are listed in Table S1 (ESI†). The generated phases depended on the heating temperature. The characteristic patterns of Ta2O5 inorganic crystal structure database (ICSD: #9112) and TaPO5 (ICSD: #202041) were identified in both samples A and D heated at 750 and 850 °C. The differences between A and D became more pronounced above 950 °C. The diffraction peaks were broad and low in intensity for sample D, which consisted of the coarsest particles, and the diffraction peaks of Ta2O5 remained unchanged. On the other hand, the characteristic peaks of Ta2O5 disappeared and the peaks indicated by red circles ( ) appeared after heating to 950 °C in sample A, which consisted of the smallest primary particles. The peaks indicated by red circles do not correspond to any phase listed in the ICDD database, however, they are in good agreement with a previously reported phase called β-LiTa2PO8.35 Unfortunately, no structural refinement has been performed for this β-LiTa2PO8 phase. Therefore, this phase is referred to as “LTPO-Precursor” in this study. In sample A, the diffraction peaks of LTPO-Crystal (ICSD: #110781) were observed at 1050 °C, whereas sample D did not exhibit any such peaks and only very weak peaks were observed at 1100 °C. The above results confirm that the larger the particle size, the more Ta2O5 remained at higher temperatures, and the higher the temperature at which LTPO-Crystal was synthesised.
) appeared after heating to 950 °C in sample A, which consisted of the smallest primary particles. The peaks indicated by red circles do not correspond to any phase listed in the ICDD database, however, they are in good agreement with a previously reported phase called β-LiTa2PO8.35 Unfortunately, no structural refinement has been performed for this β-LiTa2PO8 phase. Therefore, this phase is referred to as “LTPO-Precursor” in this study. In sample A, the diffraction peaks of LTPO-Crystal (ICSD: #110781) were observed at 1050 °C, whereas sample D did not exhibit any such peaks and only very weak peaks were observed at 1100 °C. The above results confirm that the larger the particle size, the more Ta2O5 remained at higher temperatures, and the higher the temperature at which LTPO-Crystal was synthesised.
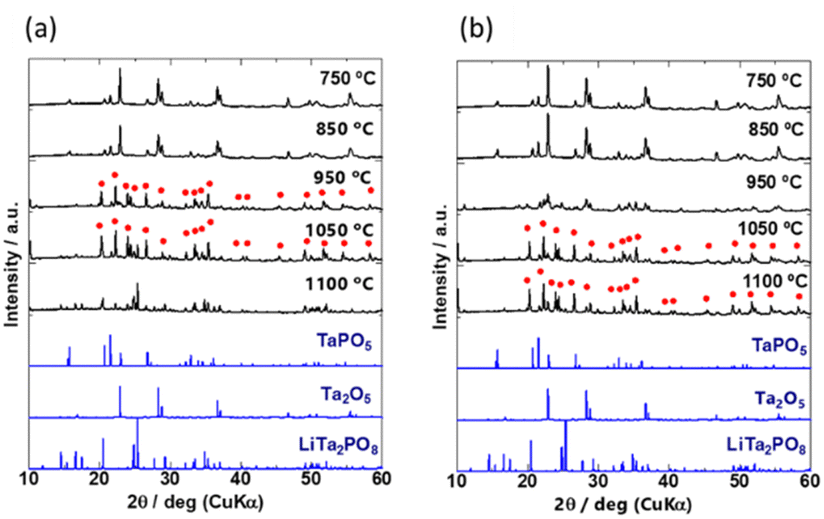 | ||
| Fig. 2 XRD patterns of (a) sample A and (b) sample D after heating for 30 min at 750, 850, 950, 1050, and 1100 °C. Bottom: Simulated reference patterns based on ICSD for TaPO5 (#202041), Ta2O5 (#9112), and LiTa2PO8 (#110781). The peaks indicated by red circles are attributed to LTPO-Precursor in this study. | ||
The XRD patterns of the four samples after the 1st heating (8 h at 600 °C and then 8 h at 1000 °C) and the 2nd heating (12 h at 1050 °C) are shown in Fig. 3(a) and (b), respectively. Due to extended exposure to high temperatures during the 2nd heating, the XRD patterns in Fig. 3(b) are different from those in Fig. 2. Prolonged heating promoted LTPO formation. As shown in Fig. 3(a), the XRD pattern of sample A after the 1st heating matched that of LTPO-Crystals. In contrast, the patterns of samples B, C, and D after the 1st heating only showed very weak characteristic peaks of LTPO-Crystals, matching with those of the LTPO-Precursor (red circles in Fig. 2). After the 1st heating, none of the samples showed diffraction peaks of the starting materials such as Ta2O5. The peak intensity of LTPO-Precursor increased in the order of B < C < D, and a few diffraction peaks of LTPO-Precursor were also observed in sample A after the 1st heating. Overall, the results in Fig. 3(a) indicate that LTPO synthesis is more likely to proceed at lower temperatures when the starting mixture consists of smaller particles. Although samples A–D formed different products after the 1st heating at 1000 °C, their XRD patterns after the 2nd heating at 1050 °C were very similar (Fig. 3(b)), confirming that the main phase of all sintered samples after the 2nd heating was LTPO-Crystal. In addition, slight diffraction peaks of LiTa3O8 (ICSD: #493) were observed in all four samples after the 2nd heating, which agreed well with previous reports.24 Rietveld analysis was performed on each sample (Fig. S4, ESI†) and the content of LiTa3O8 was calculated as A, 2.0 mass%; B, 3.3 mass%; C, 2.1 mass%; D, 2.2 mass%, exhibiting only minor differences in their content. The results of refinement are presented in Table S2-1-5 (ESI†).
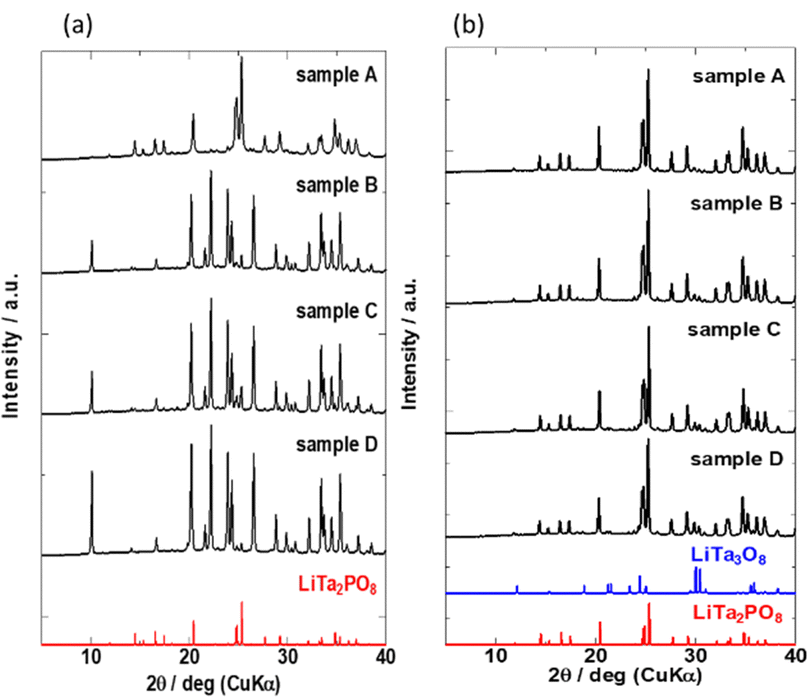 | ||
| Fig. 3 XRD patterns of samples (a) after the 1st heating and (b) after the 2nd heating. Bottom: Simulated reference patterns based on ICSD for LiTa3O8 (#493) and LiTa2PO8 (#110781). | ||
Micromorphology, density, and chemical composition
Fig. 4(a)–(d) illustrate SEM images of the sintered pellet surfaces obtained after the 2nd heating. All samples were ball-milled under identical conditions after the 1st heating, and hence their particle size was approximately the same before the 2nd heating. However, the microstructures of the samples after the 2nd heating were different. In sample A, where the main phase was LTPO-Crystal after the 1st heating, isotropic grains ranging from < 1 to 5 microns were observed. In contrast, samples B, C, and D, in which the formation of LTPO-Crystal was hardly observed after the 1st heating, were composed of micrometre-sized grains and uniaxially grown columnar grains, with almost no submicron grains visible. Table S3 (ESI†) shows the results of the XPS composition analysis for the atomic concentrations of Li and P when the atomic concentration of Ta is converted to 2. Li concentration was smaller with a smaller raw material grain size, however, the observed difference was considered to be an error. | ||
| Fig. 4 SEM images of after the 2nd heating for (a) sample A, (b) sample B, (c) sample C, and (d) sample D. | ||
Fig. 5(a)–(d) illustrate the distribution of P and Ta determined by EDS analysis. The distribution of P and Ta was more heterogeneous in samples B and C than in samples A and D. Small quantities of P were detected in areas with high Ta concentration in the SEM-EDS images (Fig. S5 and S6, ESI†). Based on the XRD profile, it was concluded that the regions rich in Ta and poor in P corresponded to the formation of LiTa3O8. Table S4 (ESI†) presents the relative densities of the samples after the 2nd heating. Sample A, which consisted of smaller grains, exhibited the highest relative density.
 | ||
| Fig. 5 P and Ta elemental distribution on the surface after the 2nd heating for (a) sample A, (b) sample B, (c) sample C, and (d) sample D. | ||
Lithium-ion conductivity
Fig. 6(a) and (b) show the AC impedance plots of the samples after the 2nd heating at two representative temperatures (30 and −10 °C, respectively). The AC impedance results measured at −10, 0, 10, and 30 °C were fitted with equivalent circuits (Rb-CPEb)(Rgb-CPEgb)(CPEel) using Z-view software, where R is the resistance, CPE is the constant phase element, and the subscripts b, gb, and el refer to the bulk, grain boundary, and electrode contributions, respectively. Only one semi-circle is observed in the impedance plot at 30 °C, attributed to the limit of the frequency range of the instrument. The bulk resistance appearing on the high-frequency side is considered to be the resistance represented by the R-CPE parallel circuit from the results of lower temperature measurements. Therefore, fitting with the R-CPE circuit was also employed on the 30 °C data. The obtained capacitances were 10−11–10−10 F for the semicircle on the high-frequency side and 10−9 F for that on the low-frequency side, corresponding to the bulk and grain boundary resistances, respectively.13,24,36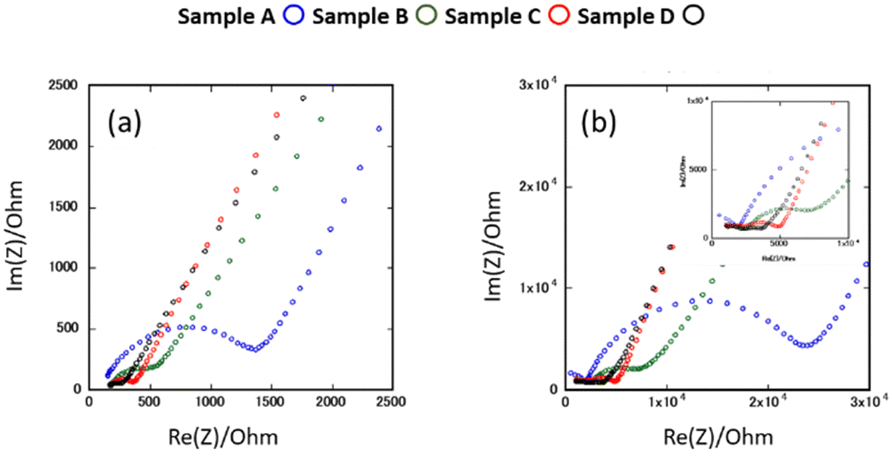 | ||
| Fig. 6 AC impedance plots for samples after the 2nd heating measured at (a) 30 °C and (b) −10 °C. | ||
Fig. 7 shows the Arrhenius plots of the Li-ion conductivity of samples after the 2nd heating. The Li-ion conductivities at each temperature and the activation energies calculated from the Arrhenius plots are listed in Table 2. The bulk and grain boundary resistances at 30 and −10 °C are shown in Fig. 8. The total ionic conductivity increased in the order of A < B < C < D. The total ionic conductivity of sample A at 30 °C was 0.19 mS cm−1 and that of sample D was 0.95 mS cm−1. Sample A exhibited the lowest total ionic conductivity and the highest density, as shown in Table S4 (ESI†). Fig. 8 also shows that samples A–D exhibit slight differences in their bulk resistance, whereas their grain boundary resistance is ranked as D < C < B < A. At −10 °C, the grain boundary resistance of sample A is 12 times higher than its bulk resistance, whereas it is about 2.5 times higher in sample B and almost equal in sample D. A similar trend was observed at 30 °C, where the grain boundary resistance of sample D was lower than the corresponding bulk resistance. In all samples, the bulk activation energy was lower than the grain boundary activation energy, and the total activation energy did not differ significantly, as shown in Table 2.
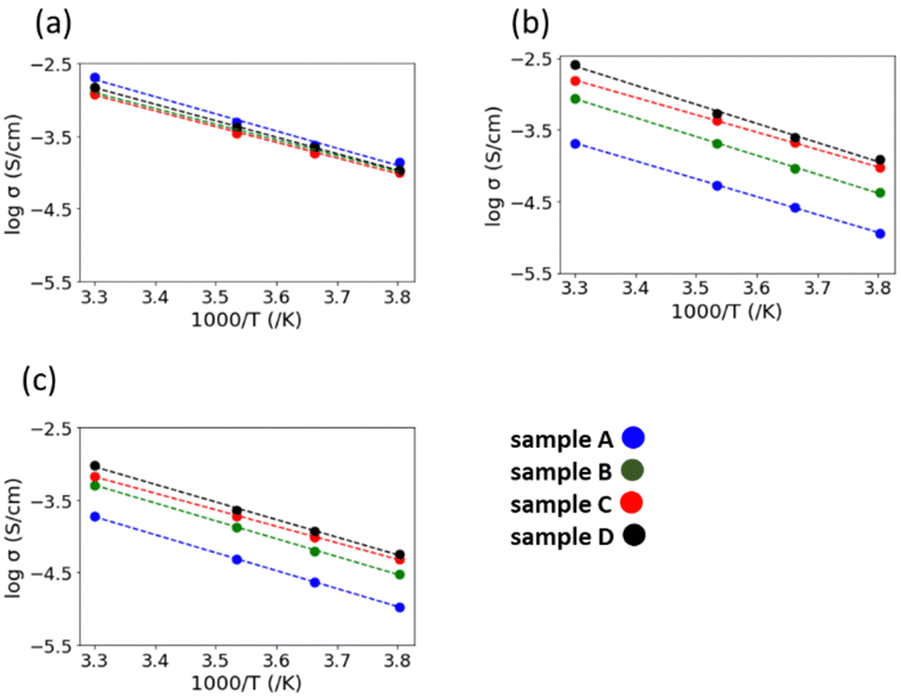 | ||
| Fig. 7 Arrhenius plots of the (a) bulk conductivity, (b) grain boundary conductivity, and (c) total ionic conductivity of the samples after the 2nd heating. | ||
| Sample | A | B | C | D | |
|---|---|---|---|---|---|
| Conductivity at 30 °C (mS cm−1) | Bulk | 2.04 | 1.28 | 1.20 | 1.49 |
| Grain boundary | 0.21 | 0.86 | 1.55 | 2.59 | |
| Total | 0.19 | 0.51 | 0.68 | 0.95 | |
| Conductivity at 10 °C (mS cm−1) | Bulk | 0.48 | 0.38 | 0.34 | 0.42 |
| Grain boundary | 0.05 | 0.21 | 0.42 | 0.53 | |
| Total | 0.05 | 0.13 | 0.19 | 0.23 | |
| Conductivity at 0 °C (mS cm−1) | Bulk | 0.24 | 0.20 | 0.18 | 0.22 |
| Grain boundary | 0.03 | 0.09 | 0.21 | 0.25 | |
| Total | 0.02 | 0.06 | 0.10 | 0.12 | |
| Conductivity at −10 °C (mS cm−1) | Bulk | 0.14 | 0.11 | 0.10 | 0.11 |
| Grain boundary | 0.01 | 0.04 | 0.09 | 0.12 | |
| Total | 0.01 | 0.03 | 0.05 | 0.06 | |
| Activation energy (eV) | Bulk | 0.47 | 0.43 | 0.42 | 0.45 |
| Grain boundary | 0.49 | 0.52 | 0.48 | 0.53 | |
| Total | 0.49 | 0.49 | 0.45 | 0.48 | |
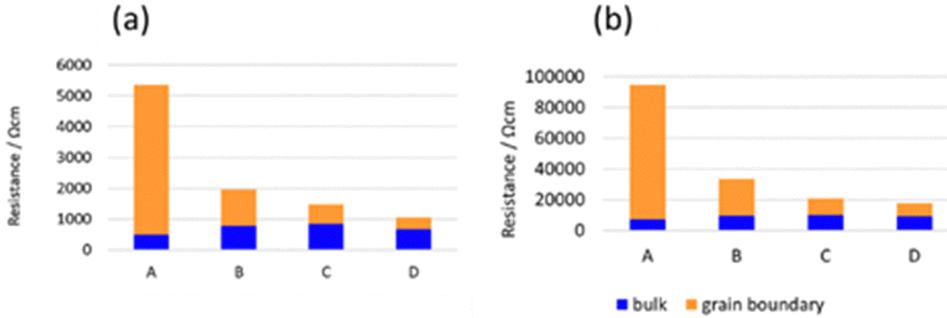 | ||
| Fig. 8 Bulk and grain boundary resistances of samples after the 2nd heating measured at (a) 30 and (b) −10 °C. | ||
We also investigated the total ionic conductivity of the sample D before and after heating at 200 °C for 1 h in order to remove possible impurities on the surfaces, such as LiOH-xH2O. Fig. S7 (ESI†) shows the Arrhenius plots of the total ionic conductivity before and after the heating treatment, and no change is indicated. Thus, the effect of impurity phase on the surface is negligible in the present study.
The DC polarisation results for sample D are shown in Fig. S8 (ESI†). Its electronic conductivity is 1.53 × 10−5 mS cm−1, which is four orders of magnitude smaller than its ionic conductivity and comparable to the reported value.24
Discussion
The synthesis of LTPO using various particle sizes ranging from submicron to hundreds of nanometres confirmed that particle size influenced the LTPO-Crystal formation temperature. The smaller the starting particle size, the lower the temperature at which LTPO-Crystal was formed. Specifically, heating at 1000 °C for 8 h produced LTPO-Crystal in sample A exhibiting the smallest particle size, whereas samples B, C, and D having larger particle sizes mainly generated LTPO-Precursor together with a trace amount of LTPO-Crystal. It was therefore concluded that the Ta2O5 particle size is an important factor for the kinetics of the LTPO-Precursor formation reaction, since the Ta2O5 particles remain as coarse particles in the starting powder mixture without being pulverised, and the coarse Ta2O5 particles remain unreacted until high temperatures due to the low diffusivity of high-valent Ta5+ ions. The difference in the phases formed during the 1st heating had a significant effect on grain growth during the 2nd heating. No growth of LTPO-Crystal particles was observed in sample A during the 2nd heating, resulting in sintered pellets comprising fine grains. Notably, sample A exhibited the highest relative density after the 2nd heating, indicating the formation of a dense sintered body by a fast-necking process among the particles. Conversely, the growth of LTPO-Crystal particles in samples B–D was clearly visible in a specific direction, and columnar grains were produced during the 2nd heating. Such results indicate that the size of Ta2O5 particles in the starting powder mixture affected the formation reaction of LTPO-Precursor and micromorphology of the sintered pellets.Sample A, which had the smallest particle size in the starting powder mixture, showed the lowest total lithium-ion conductivity, whereas sample D having the largest particle size showed the opposite trend. According to Fig. 8, the difference in the total conductivity among samples A–D was due to the grain boundary conductivity, since there was no large difference in their bulk conductivity. In contrast to the empirical knowledge that using a finer powder as the starting mixture produces homogeneous, dense sintered bodies37,38 and lowers the grain boundary resistance, our findings indicated the opposite: sample D having the largest particles initially showed the lowest grain boundary resistance after sintering.
Considering the micromorphology and grain boundary resistance, we propose that in the case of sample D, particle growth during the 2nd heating effectively reduced the grain boundary resistance, by shrinking the total grain boundary area. The presence of the LTPO-Precursor phase may control the kinetics of necking and particle growth during the 2nd heating. Furthermore, it has been reported that the presence of LiTa3O8 leads to a decrease in conductivity.33 However, Rietveld analysis demonstrated that sample A, which has the lowest conductivity, contains less LiTa3O8 than the other samples, while samples B, C, and D contain almost the same amount of LiTa3O8. Therefore, it is unlikely that the difference in conductivity is due to the LiTa3O8 content. It was concluded that the conductivity is influenced by the microstructure and elemental distribution; sample A has the lowest conductivity because it consists of the smallest particles and therefore has the largest grain boundary area (i.e. the highest grain boundary resistance). As illustrated in Fig. 5, the distribution of Ta and P in samples B and C is non-uniform compared to that in sample D, which is thought to have resulted in a higher intergranular resistance than in sample D.
Conclusion
In this study, we proved that the use of starting materials with a larger particle size can improve the lithium-ion conductivity of synthesised LTPO, primarily due to a reduction in its grain boundary resistance. Furthermore, increasing the raw material particle size inhibited the formation of LTPO crystals and promoted the formation of LTPO-precursor in the 1st heating. This finding contradicts the general trend that smaller particles are beneficial for producing homogeneous sintered samples with a low grain boundary resistance. Sample A, in which LTPO-Crystal was produced during the 1st heating, formed a sintered structure during the 2nd heating with a relatively high relative density but without visible particle growth. Furthermore, it exhibited the highest grain boundary resistance. In contrast, sample D containing coarser particles exhibited significant particle growth without the formation of the LiTa3O8 impurity phase, having the lowest grain boundary resistance after sintering. These findings suggest that the grain boundary resistance could be reduced by promoting particle growth, which lowers the proportion of grain boundaries. Such a conclusion is contrary to the general trend that smaller particles are beneficial for producing homogeneous sintered samples with a low grain boundary resistance. It was also found that the composition remained nearly constant irrespective of the synthesis conditions, however, the inhomogeneous concentration of the constituent elements in the sintered microstructure reduced the ionic conductivity. The conductivity was reduced in the samples that did not exhibit the formation of any intermediate products after the first sintering. This suggests that in the case of compounds such as LTPO, where intermediate products are formed during the synthesis process, the raw material particle size and sintering conditions affect the sintered body microstructure and significantly change the intergranular ionic conductivity properties. In this study, the same heating conditions were applied to the four samples. Optimising the firing conditions can lead to further improvements in conductivity.Author contributions
H. T. and M. N. conceived and designed the study. H.T., M. S., K. Mu., M. T., K. Ma., and H. M. performed the experiments and analysed the data. The manuscript was mainly written by H. T. and M. N. with contributions from all authors. All the authors approved the final version of the manuscript.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Grant-in-Aid for Scientific Research (grant numbers 19H05815, 20H02436, 21K04650, 21K14715, 21H01625, 22H02179, 24H02203, and 24K01157) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan; Data Creation and Utilization-Type Material Research and Development Project (grant number, JPMXP1122712807) from MEXT and JST CREST (grant number: JPMJCR21D3). English language editing was done by Editage (https://www.editage.com).References
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 109, 682–686 CrossRef PubMed.
- A. Kuhn, O. Gerbig, C. Zhu, F. Falkenberg, J. Maier and B. V. Lotsch, Phys. Chem. Chem. Phys., 2014, 16, 14669–14674 CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 14, 1–7 Search PubMed.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- N. Tanibata, S. Takimoto, S. Aizu, H. Takeda and M. Nakayama, J. Mater. Chem. A, 2022, 10, 20756–20760 RSC.
- N. Tanibata, S. Takimoto, K. Nakano, H. Takeda, M. Nakayama and H. Sumi, ACS Mater. Lett., 2020, 2, 880–886 CrossRef CAS.
- Y. Inaguma, L. Chen, M. Itoh and T. Nakamura, Solid State Ionics, 1994, 70–71, 196–202 CAS.
- O. Bohnke, C. Bohnke and J. L. Fourquet, Solid State Ionics, 1996, 91, 21–31 CrossRef CAS.
- Y. H. Cho, J. Wolfenstine, E. Rangasamy, H. Kim, H. Choe and J. Sakamoto, J. Mater. Sci., 2012, 47, 5970–5977 CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS.
- R. Iwasaki, K. Ishida, R. Yasuda, K. Nakano, N. Tanibata, H. Takeda, M. Nakayama and N. Watanabe, Phys. status solid, 2022, 2100546 CrossRef CAS.
- Z. Yang, S. Suzuki, N. Tanibata, H. Takeda, M. Nakayama, M. Karasuyama and I. Takeuchi, J. Phys. Chem. C, 2021, 125, 152–160 CrossRef CAS.
- I. N. David, T. Thompson, J. Wolfenstine, J. L. Allen and J. Sakamoto, J. Am. Ceram. Soc., 2015, 98, 1209–1214 CrossRef CAS.
- H. Xie, Y. Li and J. B. Goodenough, RSC Adv., 2011, 1, 1728 CAS.
- H. Fukuda, S. Kusakawa, K. Nakano, N. Tanibata, H. Takeda, M. Nakayama, M. Karasuyama, I. Takeuchi, T. Natori and Y. Ono, RSC Adv., 2022, 12, 30696–30703 RSC.
- M. Nakayama, K. Nakano, M. Harada, N. Tanibata, H. Takeda, Y. Noda, R. Kobayashi, M. Karasuyama, I. Takeuchi and M. Kotobuki, Chem. Commun., 2022, 58, 9328–9340 RSC.
- M. Harada, H. Takeda, S. Suzuki, K. Nakano, N. Tanibata, M. Nakayama, M. Karasuyama and I. Takeuchi, J. Mater. Chem. A, 2020, 8, 15103–15109 RSC.
- Y. Li, W. Zhou, X. Chen, X. Lü, Z. Cui, S. Xin, L. Xue, Q. Jia and J. B. Goodenough, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13313–13317 CrossRef CAS.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, J. Electrochem. Soc., 1989, 136, 590–591 CrossRef CAS.
- B. Lang, B. Ziebarth and C. Elsässer, Chem. Mater., 2015, 27, 5040–5048 CrossRef CAS.
- H. Takeda, H. Fukuda, K. Nakano, S. Hashimura, N. Tanibata, M. Nakayama, Y. Ono and T. Natori, Mater. Adv., 2022, 3, 8141–8148 RSC.
- R. Kanno, T. Hata, Y. Kawamoto and M. Irie, Solid State Ionics, 2000, 130, 97–104 CrossRef CAS.
- L. Zhang, M. Malys, J. Jamroz, F. Krok, W. Wrobel, S. Hull, H. Yan and I. Abrahams, Inorg. Chem., 2023, 62, 11876–11886 CAS.
- J. Kim, J. Kim, M. Avdeev, H. Yun and S. J. Kim, J. Mater. Chem. A, 2018, 6, 22478–22482 RSC.
- N. Ishigaki, K. Kataoka, D. Morikawa, M. Terauchi, K. Hayamizu and J. Akimoto, Solid State Ionics, 2020, 351, 115314 CAS.
- B. Huang, B. Xu, J. Zhang, Z. Li, Z. Huang, Y. Li and C. A. Wang, J. Mater. Sci., 2021, 56, 2425–2434 CrossRef.
- J. Lei, Z. Liu, H. Wang, Z. Li, R. Liao, S. Dmytro and Q. Zhang, Ceram. Int., 2023, 49, 1980–1986 CrossRef CAS.
- Z. L. Jingang Lei, Z. Q. L. Ziwei, H. Wang and D. Sydorov, J. Energy Strage, 2023, 72A, 108080 Search PubMed.
- S.-Y. Y. H. Kim, S. Choi and D.-H. Peck, Ceram. Int., 2023, 49, 8718–8724 CrossRef.
- Q. Zhang, F. Meng, R. Liao, L. Chen, M. Xu, S. Zhong, J. Chen and A. Lu, Front. Mater., 2021, 8, 670754 CrossRef.
- K. Kwatek, W. Ślubowska-Walkusz, E. Kwiatkowska, J. L. Nowiński, A. T. Krawczyńska, I. Sobrados, V. Diez-Gómez and J. Sanz, J. Eur. Ceram. Soc., 2023, 43, 5548–5556 CrossRef CAS.
- Z. Liu, J. Lei, W. Liu, B. Fang, L. Xie, S. Dmytro and Q. Zhang, J. Eur. Ceram. Soc., 2023, 43, 4437–4442 CrossRef CAS.
- R. Dai, M. Avdeev, S.-J. Kim, R. P. Rao and S. Adams, Chem. Mater., 2022, 34, 10571–10583 Search PubMed.
- F. Izumi and K. Momma, Three-dimensional visualization in powder diffraction, Solid State Phenom., 2007, 130, 15–20 CAS.
- R. Kim, L. J. Miara, J. H. Kim, J. S. Kim, D. Im and Y. Wang, Chem. Mater., 2021, 33, 6909–6917 CrossRef CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- E. Traversa, P. Nunziante, M. Sakamoto and Y. Sadaoka, Mater. Res. Bull., 1998, 33, 673–681 CrossRef CAS.
- M. Hirano and E. Kato, J. Am. Ceram. Soc., 1996, 579–816 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00180j |
| This journal is © The Royal Society of Chemistry 2024 |