
Nanomaterial combined engineered bacteria for intelligent tumor immunotherapy
Shurong
Qin†
a,
Guanzhong
He†
a and
Jingjing
Yang
 *b
*b
aCollege of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, Jiangsu Key Laboratory of Artificial Functional Materials, Nanjing University, Nanjing 210023, China
bDepartment of Biochemistry and Molecular Biology, School of Medicine, Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: jingjingyang@njucm.edu.cn
First published on 14th August 2024
Abstract
Cancer remains the leading cause of human death worldwide. Compared to traditional therapies, tumor immunotherapy has received a lot of attention and research focus due to its potential to activate both innate and adaptive immunity, low toxicity to normal tissue, and long-term immune activity. However, its clinical effectiveness and large-scale application are limited due to the immunosuppression microenvironment, lack of spatiotemporal control, expensive cost, and long manufacturing time. Recently, nanomaterial combined engineered bacteria have emerged as a promising solution to the challenges of tumor immunotherapy, which offers spatiotemporal control, reversal of immunosuppression, and scalable production. Therefore, we summarize the latest research on nanomaterial-assisted engineered bacteria for precise tumor immunotherapies, including the cross-talk of nanomaterials and bacteria as well as their application in different immunotherapies. In addition, we further discuss the advantages and challenges of nanomaterial-engineered bacteria and their future prospects, inspiring more novel and intelligent tumor immunotherapy.
 Jingjing Yang | Jingjing Yang is currently an associate professor at the School of Medicine, Nanjing University of Chinese Medicine. She received her PhD degree from the College of Engineering and Applied Sciences, Nanjing University, in 2021. In the same year, she joined Nanjing University of Chinese Medicine. Her current research focuses on biofunctional materials and microfluidics for tumor diagnosis and treatment. |
1. Introduction
In 2020, the World Health Organization reported that cancer was the leading cause of human fatality under the age of 70 in 112 countries.1 The most common methods for treating tumors are surgery, chemotherapy, and radiotherapy (RT), but their effectiveness is not satisfactory due to limited tumor targeting, insufficient tumor penetration, unfavorable prognosis, and a high probability of recurrence and metastasis.2 Surgical resection of tumors is a simple and direct method of treating tumors, but due to the low rate of local surgical resection and incomplete tumor resection, it is easy to cause tumor recurrence and metastasis.3 Chemotherapeutic drugs are susceptible to abnormal accumulation in normal tissues such as the liver, kidneys, and lungs due to poor targeting and selectivity, resulting in toxicity to healthy tissues and the immune system.4 Moreover, the poor tumor penetration and early multi-drug resistance of chemotherapy drugs further limit their clinical effectiveness.5 Similarly, although 77% of lung cancer patients have indications for RT, radiation toxicity due to accidental irradiation of adjacent tissues still exists, which would limit the application of RT in cancer treatment.6 Tumor immunotherapy is now considered a milestone in cancer treatment, due to its safety in normal tissues, potential to activate innate and specific immunity, and long-term immune memory.7 However, many difficulties and challenges still limit the effectiveness of immunotherapy in clinical treatment. For example, PD-L1 inhibitors are ineffective in most patients (more than 70%) due to the tumor immunosuppression microenvironment and immune escape mechanisms.8 Although chimeric antigen receptor T cell (CAR-T) therapy has achieved breakthrough success in treating hematological malignancies, its efficacy in solid tumors remains limited and requires further improvement.9 Therefore, further understanding of tumor immunology and novel approaches such as engineered bacterial oncotherapy are needed to improve cancer immunotherapy.The use of bacteria as an anti-tumor therapy has a long history.10 As early as 1863, W. Busch and his colleagues first discovered that infection with Streptococcus pyogenes could cause a patient's tumor mass regression. However, due to the inability to control the further infection of bacteria, the patient ultimately died.11 In 1898, William Coley and his colleagues further explored the reasons behind the bacteria's ability to shrink tumors, as well as the cause of the patient's death. Coley administered a mixture of heat-inactivated Serratia marcescens and Streptococcus pyogenes bacteria (now known as “Coley's toxin”) to many patients with unresectable soft tissue sarcomas.12,13 This treatment was surprisingly effective, but it caused a strong fever reaction, with patients' temperatures reaching 38–40 °C.14 After this discovery, the use of microorganisms, such as Clostridium, Listeria, Escherichia, and other bacteria, as a new tumor immunotherapy ignited the enthusiasm of researchers in the early 20th century.15 However, this first-generation bacterial therapy, mainly relying on natural or inactivated bacteria, did not continue to be researched due to its tendency to cause toxic shock and infection risks.16 With the development of molecular biology technology, second-generation bacterial therapy can use DNA recombinant technology to enable precise modification of bacterial DNA, thereby achieving precise regulation of bacterial behavior and function.17 Some genetically engineered bacteria can achieve reduced systemic toxicity by selectively knocking out virulence factor genes, such as Salmonella VNP20009.18 The others can be genetically programmed to express genes encoding antibodies or cytokines to regulate the tumor microenvironment (TME), such as programmed cell death 1 ligand 1 (PD-L1),19 cytotoxic T lymphocyte associate protein-4 (CTLA-4),20 and tumor necrosis factor-α (TNF-α)21 nanobodies. There are also some engineered bacteria that can kill tumor cells by disrupting tumor metabolism and producing their own metabolites. For example, engineered bacteria producing 5-ALA can convert it into protoporphyrin in tumor cells, which can then be ablated by near-infrared light irradiation.22 However due to the serious biosafety concerns and lack of spatiotemporal control, second-generation bacterial therapy is still not an ideal and intelligent therapy.
The third generation bacterial tumor therapy uses nanoparticle-assisted engineered bacteria, and has solved the dilemma of previous bacterial therapy and provided a new avenue for intelligent tumor immunotherapy.23 Recently, the development of nanotechnology has advanced bacterial therapy to a higher level by integrating multifunctional nanomaterials on or inside the bacteria. Yu and his colleagues encapsulated hematoporphyrin monomethyl ether (HMME) in ZIF-8, and then constructed MHS nanoparticles by adsorbing Cas9/sgRNA. These nanoparticles were subsequently combined on the surface of Lactobacillus plantarum, which in response to the acidic TME releases the CRISPR/Cas9 system and induces the enhancement of immunogenic cell death (ICD) and the maturation of dendritic cells (DCs), finally reversing the tumor immune microenvironment to ablate the tumor.24 Polydopamine-coated Escherichia coli Nissle 1917 (EcN) bearing CRISPR-Cas9 plasmid-loaded liposomes (Lipo-P), which could improve therapeutic agent penetration in tumor centers and reduce the thermal resistance of cancer cells, is utilized to enhance immunotherapy in deep tumors by activating both innate and adaptive immunity.25 A metal-based photosensitizer-bacteria hybrid Ir-HEcN, obtained by covalently labeling an iridium(III) photosensitizer to the surface of genetically engineered bacteria, was capable of inducing pyroptosis and ICD in tumor cells under a 525 nm laser.26 Interesting, bioluminescent bacteria transformed with the Luc-S.T.ΔppGpp plasmid serve as an internal light source, enabling effective photodynamic therapy (PDT) due to continuous light generation to excite photosensitizer chlorine6, enhancing antitumor immunity and effective suppression in opaque melanoma and large rabbit tumors.27 In summary, the nanoparticle-assisted engineered bacteria play a crucial role in cancer immunotherapy by enabling spatiotemporal control, enhancing tumor targeting, expanding the functionality of bacteria, and enabling real-time detection.
Hence in this review, we have focused on the cross-talk between nanomaterials and engineered bacteria, then summarized the latest research on nanoparticle-assisted engineered bacteria in different types of cancer immunotherapy, including immune checkpoint therapy, tumor vaccination, CAR-T, and nonspecific immunity (Scheme 1). Moreover, we have discussed the advantages and limitations of the current nanoparticle combined engineered bacteria hybrid system. In the end, we have proposed a possible future perspective on the third bacterial therapy, hoping to inspire novel ideas and methods to modify the engineered bacteria to enhance the efficacy of immunotherapy.
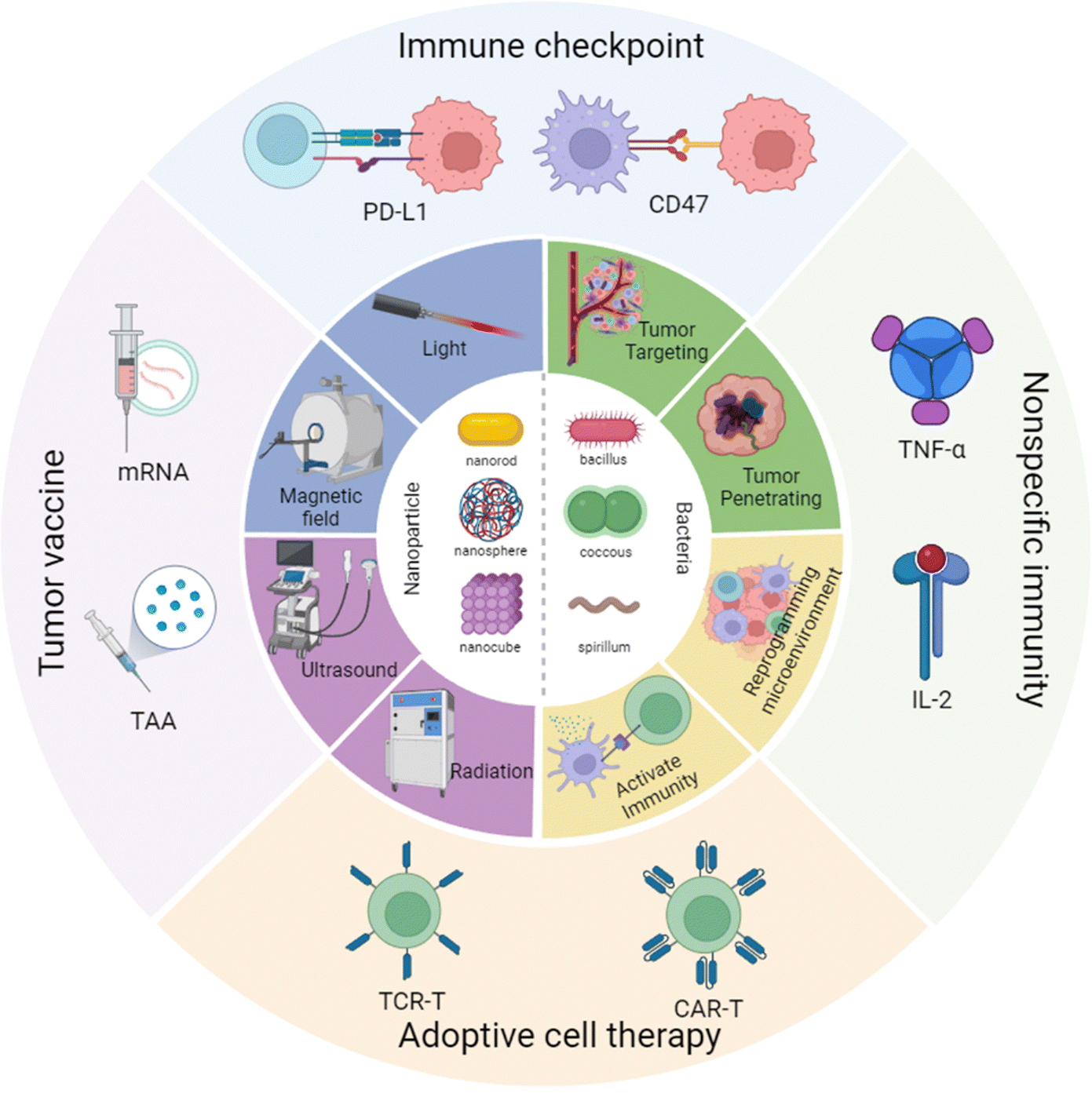 | ||
| Scheme 1 Nanoparticle combined engineered bacteria applied in cancer immunotherapy. Created with BioRender.com. | ||
2. The cross-talk roles of nanomaterials and bacteria
2.1 The effect of nanomaterials on bacteria
Engineering bacteria have achieved significant progress, but clinical translation remains unsatisfactory, primarily due to the challenge of effectively controlling bacterial behavior and function within a biological organism. Nanomaterial modification on bacterial surfaces or within bacteria via different reactions could allow bacteria to respond to external signals, such as light, magnetic, electric, acoustic, and radiation signals for imaging, activation of promoters, and precise spatiotemporal control of drug release (Table 1).| External signal | Nanomaterial | Bacteria | Tumor type | Cargo | Anti-tumor mechanism | Ref. |
|---|---|---|---|---|---|---|
| Light | INP | Salmonella typhimurium (YB1) | MB49 | ICG | PPT/PDT | 28 |
| ZIF-90 | Shewanella oneidensis | CT26 | Prussian blue | PPT and downregulate HSP90 | 29 | |
| PDA-liposome | EcN | 4T1 | HSP90 plasmid | HSP90 knockdown and enhanced PPT | 25 | |
| Au NPs | E. coli MG1655 | CT26 | ClyA plasmid | PPT and ClyA expression | 30 | |
| Polydopamine | EcN | CT26-OVA | OVA and α-PD-1 | Enhanced ICD and activation of T cells | 31 | |
| MC38-OVA | ||||||
| Au nanodot | EcN | 4T1 | 5-FU and ZOL | PPT, chemotherapy and polarization of macrophages | 32 | |
| HAS/ICG | Synchronous 7942 | 4T1 | ICG | Oxygen generation and enhanced PDT | 33 | |
| UCNP | EcN | H22 and 4T1 | FlaB plasmid | Activation of TLR5 and polarization of macrophages | 34 | |
| Magnetic field | Fe3O4@lipid nanocomposite | E. coli BL21 | CT26 and 4T1 | CD47 Nb | Release of CD47 Nb and reactivation of macrophages | 35 |
| Magnetic nanoparticle | EcN | CT26 | NDH-2 | Magnetothermal ablation and NDH-2-induced ROS | 36 | |
| Fe3O4 NPs | S. platensis | 4T1 | Chlorophyll | Chlorophyll-induced ROS enhanced RT | 37 | |
| INP | Magneto spirillum magnetism | MCF-7 | ICG | PTT/PDT | 38 | |
| Ultrasound | — | E. coli MG1655 | 4T1 and H22-Luc | IFN-γ | Apoptosis of cancer cells and polarization of macrophages | 39 |
| DOX-PFP-PLGA nanodroplets | E. coli MG1655 | 4T1 | — | Ultrasound-trigger drug release and enhanced chemotherapy | 40 | |
| Gas vesicle | E. coli BL21 | 4T1 | Gas vesicle | Mechanical damage of cancer cells | 41 | |
| IPA-CLs | E. coli BL21 | 4T1 | Gas vesicle | Hypoxia-activated chemotherapy | 42 | |
| Radiation | Bi2S3 NPs | E. coli MG1655 | 4T1-Luc | ClyA plasmid | Arrest cancer cell cycle and enhanced RT | 43 |
| GTe | Fusion membrane | 4T1 | GTe | Sensitizing RT and enhanced ICD | 44 | |
| — | EcN | 4T1 | Catalase | Oxygen sensitive RT with improved ROS generation | 45 | |
| Nanovesicles | Bacterial membrane | CT26 | Catalase | Long-term tumor hypoxia relief and enhanced RT | 46 | |
| Electricity | γ-Fe2O3 | S. putrefaciens CN32 | — | — | Improved extracellular electron transfer and bioelectricity generation | 47 |
| Carbon dots | S. oneidensis MR-1 | — | — | Boosted bacterial metabolism, electron transfer, and bioelectricity generation | 48 | |
| Polydopamine | Synechocystis sp. PCC6803 | — | — | Enhanced charge transfer | 49 | |
However, the inherent heat resistance of tumor cells and energy dissipation in deep tumor tissues may lead to the adaptation of certain resilient tumor cells to high-temperature thermal treatments.52 To overcome this limitation, Chen et al. developed a self-mineralized photothermal bacterium (PTB) using Shewanella oneidensis MR-1 to biomineralize palladium nanoparticles on its surfaces, and it was found to exhibit superior photothermal properties under NIR irradiation. They further constructed a hybrid bacterial system by conjugating ZIF-90-encapsulating MB to the bacterial surface (PTB@ZIF-90/MB). Under NIR irradiation, MB generated reactive oxygen species (ROS) to inhibit adenosine triphosphate (ATP) production and downregulate heat shock proteins by interfering with mitochondrial redox equilibrium. This hybrid system combined the precise tumor-targeting ability of the self-mineralized bacteria with the mitochondrial dysfunction-inducing capability of MB, providing a new approach for enhanced PTT.29 By overcoming tumor heat endurance and achieving precise tumor targeting, this strategy could potentially improve the efficacy of PTT. Similarly, Luo et al. employed the polydopamine-coated EcN bearing CRISPR-Cas9 plasmid-loaded liposomes (Fig. 1A), which could improve therapeutic agent penetration in tumor centers and reduce the thermal resistance of cancer cells, thus enhancing the PTT effect and immunotherapy in deep tumors.53 Moreover, Wang et al. developed a photothermal hybrid bacterial system (EM@Au) by using E. coli MG1655 to self-mineralize gold nanoparticles (Au NPs) on the bacterial surface. The E. coli was transformed with plasmids containing thermally sensitive promoters and the gene for ClyA, enabling precise control over the expression of therapeutic proteins. Upon NIR irradiation, the heat generated by Au NPs triggered the expression of ClyA, resulting in the targeted killing of tumor cells.30
 | ||
| Fig. 1 The nanomaterial-assisted engineered bacteria responded to external signals due to extracellular or intracellular nanoparticles. (A) A polydopamine-coated EcN hybrid system bearing CRISPR-Cas9 plasmid-loaded liposomes (Lipo-P) responded to NIR.53 Reproduced from ref. 53 with permission from ACS Publication, copyright 2023. (B) An engineered Escherichia coli with Fe3O4@lipid nanocomposites responded to the magnetic field.35 Reproduced from ref. 35 with permission from Springer, copyright 2023. (C) E. coli BL21 was genetically engineered by introducing a gene cluster encoding GVs and it responded to acoustic tweezers.41 Reproduced from ref. 41 with permission from Springer, copyright 2023. (D) An integrated nanosystem (Bac@BNP) composed of engineered bacteria (Bac) and Bi2S3 nanoparticles (BNPs) responded to radiation.43 Reproduced from ref. 43 with permission from ACS Publication, copyright 2022. | ||
Many studies have demonstrated that the complete elimination of residual tumor cells post-PTT primarily hinges on the stimulation of antitumor immune responses. However, elevated local inflammatory reactions, exacerbated by inflammatory factors and immune cells like tumor-associated macrophages (TAMs) recruited due to high temperatures, accelerate the development of an immunosuppressive microenvironment and immune evasion processes.54 Therefore, combining PTT with other modalities to activate immune systems and remodel the post-PTT microenvironment offers a viable strategy to enhance therapeutic outcomes. Bacteria with superior tumor site tracing and colonization capabilities are valuable for efficient multidrug loading and precise release regulation, further improving PPT treatment efficacy. Li et al. have developed a method to enhance tumor-specific immunotherapy using bacteria decorated with tumor-specific antigens and checkpoint blocking antibodies via polydopamine nanoparticles, which not only serve as a linker but also have a photothermal agent. When administered to tumor-bearing animals, the decorated bacteria show long tumor retention and demonstrate potent antitumor effects in tumor models. This approach presents a versatile platform for delivering tumor antigens, activating DCs, and reversing the immunosuppressive microenvironment through immune checkpoint inhibitors in a synergistic PPT–immunotherapy.31 Xie et al. developed a bacterial navigation hybrid system EcNZ/F@Au by incorporating 5-fluorouracil (5-Fu) and zoledronic acid (ZOL) into EcN, followed by Au nanodot modification on the EcN surface. EcNZ/F@Au autonomously navigated to tumor sites, and in situ transformed into biofilm-like granules (BGs) under the photothermal effect of Au nanodots triggered by NIR irradiation. In addition to the chemotherapeutic effect of FU, ZOL released from BGs promoted the polarization of TAMs from the M2 phenotype towards the antitumor M1 phenotype. And then these M1 TAMs secreted proinflammatory cytokines, reversing the immunosuppressive microenvironment and enhancing the synergistic efficacy of PTT, chemotherapy, and immunotherapy.32
PDT is a promising emerging technique due to its minimal invasiveness, low systemic toxicity, and limited drug resistance, making it a viable option for cancer treatment. It relies on the generation of ROS from photosensitizers (PSs) under laser exposure, which subsequently induces oxidative damage, apoptosis, and necrosis in tumor cells.55 However, the severe hypoxia in the TME poses a significant challenge in ROS generation, limiting the therapeutic effectiveness of PDT. Thus, numerous studies have attempted to alleviate tumor hypoxia to achieve efficient ROS generation in PDT. These approaches include using oxygen-evolving nanomaterials (e.g., MnO2 NPs),56 oxygen-carrying vectors (e.g., perfluorocarbon),57 and biocatalyzers (e.g., CAT).58 Moreover, Liu et al. developed an engineered Synchronous for tumor-targeted delivery of ICG and in situ oxygen generation to facilitate photosynthesis-assisted PDT. In this system, ICG and human serum albumin (HSA) were first assembled into HSA/ICG NPs via disulfide bonds. Then, these NPs were coupled onto the Syne cell wall via amide bonds, creating the biomimetic system S/HAS/ICG. This system could effectively target tumor sites and increase local O2 levels through photosynthetic reactions, significantly enhancing the therapeutic efficacy of PDT and immune stimulation under 660 nm laser irradiation.33
In addition, optogenetics has emerged as a novel method to control molecular and cellular behaviors by combining optical and genetic technologies.59 Zhang et al. constructed a light-controlled engineered bacteria system using upconversion nanoparticles (UCNPs) for accurate tumor diagnosis and therapy in colorectal cancer. The system achieves co-localization of engineered bacteria and UCNPs in tumor tissues, improving diagnostic accuracy, and triggers tumor cell death by releasing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) upon blue light irradiation. In vitro and in vivo experiments demonstrate the effectiveness of the system in accurate tumor diagnosis and light-controlled cancer therapy.60 Moreover, Wu et al. developed a two-step therapy platform for treating primary and distant tumors using UCNPs and engineered bacteria. Firstly, erbium ion (Er3+)-doped UCNPs activate the photosensitizer ZnPc to target primary tumors by generating 1O2. Secondly, thulium ion (Tm3+)-doped UCNPs emit blue-violet light upon NIR excitation, triggering engineered bacteria to produce interferon (INF-γ) in the intestine. This dual approach not only directly treats tumors but also enhances the immune response, synergistically inhibiting the growth of distant tumors through joint immunotherapy.61
Furthermore, it is possible to engineer bacteria to form nano-scale gas vesicles inside their bodies, enabling them to undergo real-time ultrasound imaging and spatiotemporally controlled lysis. Mikhail et al. have creatively developed a non-invasive, real-time “ear” for monitoring bacterial descent within the body using ultrasound imaging technology. By using genetic engineering, the “acoustic reporter genes” were introduced into E. coli and Salmonella, enabling the engineered bacteria to form a protein nanostructure called “gas vesicles.” In these engineered bacteria, these gas vesicles can reflect ultrasound waves, helping to detect and locate these bacteria deep within the tumor. The detection depth exceeds 10 cm, with a resolution smaller than 100 μm, allowing for the detection of engineered bacteria with a volume density of less than 0.01%.66 Yang et al. have developed genetically engineered bacteria that produce numerous sub-micron gas vesicles in the cytoplasm via the heterologous expression of specific gene clusters (Fig. 1C). These gas vesicles significantly enhance the acoustic sensitivity of the bacteria, enabling their manipulation by ultrasound. Using phased-array-based acoustic tweezers, these bacteria can be trapped and manipulated in vitro and in vivo using electronically steered acoustic beams, enabling controlled flow in the vasculature of live mice. This technology improves the aggregation efficiency of the bacteria in tumors, providing a platform for the in vivo manipulation of live cells and advancing cell-based biomedical applications.41 In addition, Wang et al. developed a biological targeting system using genetically engineered bacteria and multi-functional nanoparticles to treat malignant tumor. E. coli, which can target the tumor hypoxic microenvironment, was genetically engineered to carry an acoustic reporter gene responsible for gas vesicle (GV) formation. After the GV-producing E. coli (GVs-E. coli) colonized the tumor tissue, ultrasound imaging and focused ultrasound-acoustic synergy (FUAS) were performed. Subsequently, cationic lipid nanoparticles containing IR780 (a NIR dye), perfluoro hexane (a phase-change contrast agent), and banoxantrone dihydrochloride (AQ4N, a hypoxia-activated prodrug) were enriched in the tumor tissues via electrostatic adsorption, achieving targeted multimodal imaging and enhancing the curative effect of FUAS.42
In addition to cell cycle regulation, various strategies have been devised to enhance radiotherapeutic efficacy, particularly by remodeling the unfavorable TME, which differs from the normal internal microenvironment in terms of weak acidity, hypoxia, and high interstitial pressure.68 To address this issue, Huang et al. engineered a strain of EcN to express catalase with an NSP4 signal peptide under the tac promoter. The engineered bacteria specifically secreted catalase within the TME due to their hypoxic tropism, increasing oxygen availability and tumor susceptibility to RT. In vivo, these engineered bacteria combined with RT could inhibit tumor growth by up to 90%.45 Furthermore, Zai et al. developed catalase-containing E. coli membrane nanovesicles (EMs) with outstanding protease resistance, providing long-term relief from tumor hypoxia. Even when treated with a 100-fold excess of protease, EMs exhibited higher catalase activity than free catalase. When injected into tumors after 12 hours, EMs maintained their ability to relieve hypoxia, whereas free catalase lost its activity. Meanwhile, benefiting from their immune-stimulating properties, EMs could effectively enhance RT and induce antitumor immune memory.46
Although there are few studies on using electric fields to regulate nanomaterial-modified engineered bacteria for cancer therapy, numerous articles demonstrate that nanomaterial-modified bacteria can enhance electron transfer efficiency and bioelectricity. For example, Wang et al. developed the hybrid bacteria CN32@γ-Fe2O3, formed by self-assembling magnetic nanomaterials γ-Fe2O3 with S. putrefaciens CN32, showing significantly improved extracellular electron transfer and bioelectricity generation, achieving a more negative anodic peak potential and lower charge transfer resistance compared to controls.47 Yang et al. used water-soluble carbon dots (CDs) synthesized from sodium naphthalene reagent and triethylamine to enhance the bioelectricity generation of S. oneidensis MR-1.48 The CDs significantly boosted bacterial metabolism, electron transfer, and bioelectricity generation, achieving increases of approximately 7.34-fold in maximum current, 5.63-fold in total charge, 3.78-fold in maximum cell voltage, and 6.46-fold in power output compared to the control group. Moreover, Reggente et al. engineered a polydopamine (PDA) coating on the outer membrane of Synechocystis sp. PCC6803 to enhance charge transfer, leading to a three-fold increase in photocurrent compared to non-coated bacteria.49 These findings may suggest that placing nanomaterial-modified bacteria in electric fields for tumor treatment could potentially increase reactive oxygen species (ROS) production through enhanced electron transfer efficiency, possibly improving tumor-killing effects. Moreover, the introduction of bacteria might stimulate tumor immune activation, further coordinating and enhancing antitumor activity. Although this idea has not been reported in the literature, it presents a novel research direction to improve the antitumor efficacy of electrodynamic therapy.
2.2 The effect of bacteria on nanomaterials
In 1989, Matsumura and Maeda discovered the enhanced permeability and retention (EPR) effect, which triggered the development of nanomedicine.70 Nanomaterials exhibit unique characteristics that allow them to be used in cancer treatment. For instance, nanoparticles show enhanced accumulation in tumors and have extended half-lives in the body compared to traditional small molecule drugs.71 However, despite these advantages, the aggregation, penetration, and retention of nanomaterials in the deep regions of tumor tissues remain suboptimal, thereby hindering their full therapeutic potential.72 To overcome these limitations, researchers have explored the combination of nanomaterials with engineered bacteria, leveraging the unique properties of both components into promising combinatorial strategies for cancer therapy (Table 2).| Characteristic | Engineered methods | Bacteria | Mechanism | Ref. |
|---|---|---|---|---|
| Tumor targeting | — | EcN | Facultative anaerobic and the aid of flagella | 25 |
| — | Magnetostatic bacteria | Enhanced magnetic sensitivity | 73 | |
| Naturally secreted | EcN OMVs | EPR effect and ligand modification | 74 | |
| Coextrusion | Fusion membrane structure | EPR effect and tumor homing effect | 44 | |
| Tumor penetration | Expressing ClyA-Hy fusion protein | Hypervesiculating Escherichia coli Nissle | Break down the dense extracellular matrix | 75 |
| Expressing Hase | Attenuated S. typhimurium | Degrade extracellular matrix and decrease fluid pressure | 76 | |
| Naturally secreted | Clostridium butyricum spores | Penetrate the extracellular matrix | 77 | |
| TME reprogramming | Secrete tumstain | Bifidobacterium longum | Induces apoptosis of tumorous vascular endothelial cells | 78 |
| Secretes tLLO-CD105 | Listeria | Combats angiogenesis | 79 | |
| Deliver STAT3 siRNA endostatin | Salmonella typhimurium | Inhibit angiogenesis | 80 | |
| Expressing HlyE | Salmonella typhimurium | Induction of tumor cell apoptosis | 81 | |
| Expressing α-hemolysin | Escherichia coli strain χ6212 | Kill cancer cells | 82 | |
| Express oxidoreductase azurin | Pseudomonas aeruginosa | Triggering tumor cell apoptosis | 83 | |
| Immune activation | Express ADP-heptose | Yersinia pseudotuberculosis | Activate the NF-κB signal pathway and induce Alpk1-dependent inflammation | 84 |
| With flagellin | Salmonella | Activate the TLR5 signal pathway and the innate immunity | 85 | |
| Accumulate L-Arg | EcN | Activate T cells | 19 | |
| Express melanoma tumor antigens | Staphylococcus aureus | Activate CD4+ and CD8+ T cells | 86 | |
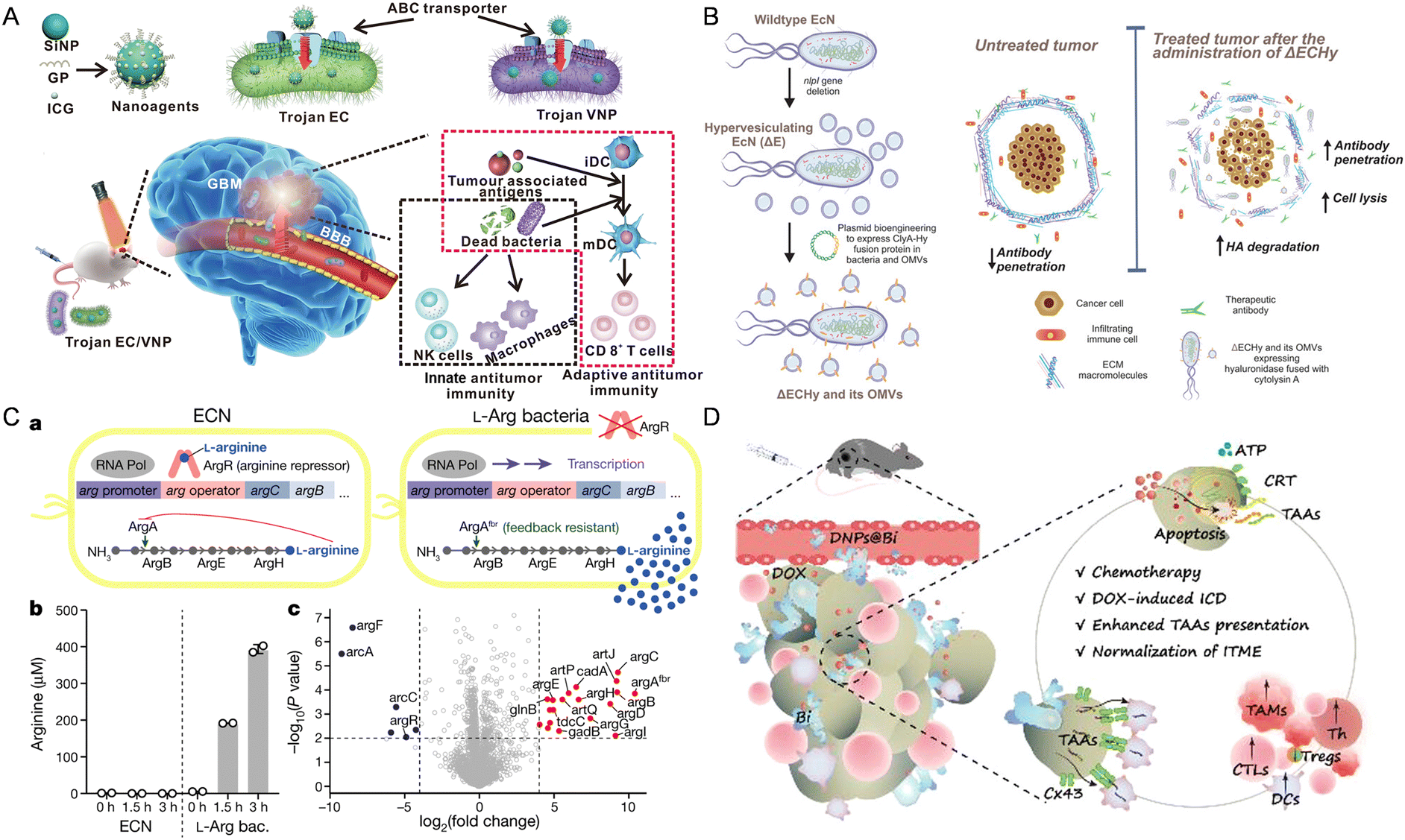 | ||
| Fig. 2 The nanomaterial-assisted engineered bacteria exhibited favorable features compared to the nanoparticles. (A) Bacteria loaded with the glucose polymer and photosensitive ICG silicon-nanoparticles exhibited tumor targeting ability in GBM.93 Reproduced from ref. 93 with permission from Springer, copyright 2022. (B) A hypervesiculating Escherichia coli Nissle (ΔECHy) and its OMVs capable of distributing cytolysin A (ClyA)–hyaluronidase (Hy) exhibited tumor penetrating ability via degrading tumor extracellular matrix macromolecules.75 Reproduced from ref. 75 with permission from John Wiley & Sons, Inc, copyright 2022. (C) An engineered probiotic EcN continuously converting ammonia into L-Arg exhibited the ability of reprogramming the TME.95 Reproduced from ref. 95 with permission from Springer, copyright 2021. (D) The probiotic Bifidobacterium bifidum modified with DOX-loaded CaP/SiO2 nanoparticles exhibited the ability of enhancing tumor immunity.96 Reproduced from ref. 96 with permission from ACS Publication, copyright 2023. | ||
In addition to hypoxia and nutrient targeting, recent studies have demonstrated immune cell-mediated bacteria delivery to enhance tumor targeting. Neutrophils and macrophages, as key components of the immune system, can be harnessed to transport nanomaterial-engineered bacteria to tumor sites. For instance, inspired by the targeting properties of inflammatory neutrophils, Liu et al. constructed a VNP delivery system. In this system, neutrophils first uptake the VNPs and subsequently release them into the tumor core, balancing safety and therapeutic efficacy.97 With a PD-1 nanobody-secreting VNP strain, the system significantly remodeled the tumor microenvironment, increasing activated dendritic cells, cytotoxic T cells, and antitumor macrophages. Similarly, Li et al. reported a nano-pathogenoid (NPN) system coated by bacteria-secreted outer membrane vesicles to hitchhike circulating neutrophils to supplement photothermal therapy (PTT).98 These hitchhiked neutrophils migrate to inflamed tumors and release NPNs, which respond to inflammation and are taken up by tumor cells, ultimately completely eradicating tumors in all treated mice.
Macrophages, particularly tumor-associated macrophages (TAMs), are abundant in the tumor microenvironment and can be reprogrammed to deliver therapeutic bacteria. For instance, Zhang et al. proposed a bacteria-based backpack (Mø@bac) that attaches bacteria to macrophages to guide cellular polarization in vivo by leveraging bacterial immunogenicity.99Escherichia coli, coated with polysaccharides to form an adhesive nanocoating, promote macrophage integration and sustainable M1 phenotype activation, repolarizing endogenous TAMs and remodeling the immunosuppressive TME. Additionally, Wu et al. developed “dead” yet “functional” Salmonella-loaded macrophages using liquid nitrogen cold shock, achieving an average loading of approximately 257 live bacteria per 100 cells. This “Trojan horse” strategy enhances bacterial tumor enrichment and activates the tumor microenvironment by increasing M1-like macrophages and decreasing M2-like macrophages while maintaining biosafety.33 These research studies demonstrate innovative approaches to leveraging immune cell properties for improved tumor targeting and therapeutic delivery of nanomaterial-engineered bacteria.
Using bacteria to produce cytotoxic proteins at tumor sites is also a way to reprogram the TME to treat cancer. This design primarily exploits the ability of bacteria to migrate to hypoxic foci within tumors and then release or secrete specific anti-tumor drugs through lysis, achieving targeted drug delivery to the tumor and inhibiting tumor growth. Jean et al. designed hemolysin E (HlyE)-expressing Salmonella typhimurium, which increased the necrosis rate of mouse mammary tumors and reduced tumor growth.81 St et al. showed that engineering the Escherichia coli strain χ6212 (Δasd) with Staphylococcus aureus-derived α-hemolysin (SAH) could cause tumor regression in 4T1 tumor-bearing mice and promote tumor necrosis.82 Yamada et al. used Pseudomonas aeruginosa to express low-toxicity oxidoreductase azurin, an anticancer protein whose intracellular translocation to the nucleus depends on p53. Azurin forms a complex with p53, stabilizing p53 and increasing the levels of intracellular p53 and Bax, leading to the release of mitochondrial cytochrome c into the cytoplasm and triggering tumor apoptosis.83 In particular, Canale et al. utilized synthetic biology to create a modified EcN strain, which resides in tumors and converts ammonia into L-arginine continuously (Fig. 2C). This bacterial colonization elevates intertumoral L-arginine levels, boosts tumor-infiltrating T cells, and enhances the effectiveness of PD-L1 blocking antibodies in tumor elimination, demonstrating the potential of engineered microbial therapies for improving immunotherapy efficacy by altering the TME metabolically.95
As for specific immunity, Fernando et al. have developed an engineered probiotic EcN strain using synthetic biology methods, which can colonize tumors and continuously convert accumulated nitrogenous metabolic waste in tumors into L-arginine. The specific accumulation of L-arginine at the tumor site can promote the activation of tumor-infiltrating T cells, and thereby in combination with PD-L1 inhibitors have a powerful anti-tumor effect.19 Moreover, Chen et al. have engineered Staphylococcus aureus to stimulate CD4 and CD8 positive cells both in vitro and on the skin to kill melanoma without damaging the skin barrier or causing skin inflammation.86 He et al. engineered Bifidobacterium bifidum (Bi) by covalently attaching DOX-loaded CaP/SiO2 nanoparticles (DNPs@Bi) for tumor therapy, inducing chemotherapy and ICD within the TME via pH-responsive drug release (Fig. 2D). Additionally, tumor-targeting Bi enhances antigen presentation to DCs via Cx43-dependent gap junctions, promoting DC maturation and CTL infiltration. In vivo antitumor experiments demonstrated that this approach leads to prolonged survival, inhibited tumor progression, and reduced metastasis, highlighting the potential of this bacterial-driven, hypoxia-targeting delivery system for tumor chemo-immunotherapy.96
2.3 The binding methods of engineered bacteria and nanoparticles
For effective synergistic therapy, both nanoparticles and engineered bacteria must accumulate within the tumor region. The binding methods of combining nanomedicines with engineered bacteria, such as physical, chemical, biological, and indirect methods, form the core of the synergistic anti-tumor therapy.Physical methods such as electroporation, electrostatic adsorption, mechanical extrusion, and so on are extensively utilized to achieve effective modification of engineered bacteria with nanoparticles. Electroporation, which temporarily increases cell membrane permeability via an electrical field, demonstrates high efficiency in incorporating drugs and nanoparticles into bacteria. For instance, Xie et al. employed electroporation to integrate 5-fluorouracil and zoledronic acid into E. coli Nissle 1917, followed by surface decoration with gold nanorods to create a hybrid system (EcNZ/F@Au), enhancing drug delivery and tumor targeting capabilities.32 Electrostatic adsorption is a widely used method for bacterial modification, exploiting the charge interactions between negatively charged bacterial surfaces and positively charged nanoparticles.103,104 Furthermore, Zhong et al. illustrated the application of this method by modifying photosynthetic bacteria with Fe3O4 nanoparticles, resulting in improved tumor imaging and therapeutic efficacy.105 Furthermore, mechanical extrusion with cell membranes, as demonstrated by Cao et al., significantly enhances the circulation time and tumor-targeting potential of the engineered bacteria by coating with red blood cell membranes.106 Collectively, these physical modification methods significantly enhance the interaction between engineered bacteria and nanoparticles, thereby augmenting the efficacy of targeted cancer therapy.
Owing to the diverse array of functional groups present on bacterial surfaces, various chemical methods, such as Schiff base reaction, carbodiimide chemistry, in situ growth techniques, and so on, are employed to functionalize bacteria with nanoparticles. For instance, Luo et al. conjugated upconversion nanorods and gold nanorods to Bifidobacterium breve and Clostridium difficile through Schiff base reaction, facilitating hypoxia-specific delivery for tumor imaging and therapy.107 Similarly, carbodiimide chemistry enables the formation of stable amide bonds between carboxyl and amine groups on bacterial surfaces and nanoparticles. Xu et al. demonstrated the binding of nano-photosensitizers to Salmonella bacteria via carbodiimide chemistry, enhancing their hypoxia-targeting and photothermal properties for solid tumor therapy.108 Additionally, Chen et al. reported in situ biomineralization of palladium nanoparticles on Shewanella oneidensis MR-1, creating a photothermal bacterium that hybridizes with mitochondria-targeted metal–organic frameworks, significantly augmenting photothermal tumor therapy.109
Furthermore, engineered bacteria can be effectively modified with nanoparticles using a range of biological methods, including exploiting biotin–streptavidin interactions, utilizing antigen–antibody reactions, and harnessing biofilm secretion processes. For instance, Suh et al. conjugated PLGA nanoparticles to Salmonella VNP20009 using biotin–streptavidin interaction, creating nanoscale bacteria-enabled autonomous drug delivery systems (NanoBEADS), which significantly enhanced the intratumoral transport of nanomedicine.110 Similarly, Chowdhury et al. used antigen–antibody reaction to program Escherichia coli with anti-PD-1 antibodies, inducing durable tumor regression and systemic antitumor immunity.111 Additionally, Wang et al. described the development of carcinoma cell-mimetic bacteria (CCMB), a set of multimodal oncolytic living agents, by coating them with tumor cell-derived biofilms.112 Under external stimulations, intracellularly pre-harbored bacteria are spontaneously coated with an extra membrane by forming apoptotic bodies from invaded tumor cells. This strategy significantly improves resistance, adhesion capacity, and targeting, which can be applied to cancer therapy for better targeting and retention.
Another effective modification of engineered bacteria with nanoparticles can be achieved using various indirect methods, including nanoparticle encapsulation and polymer cross-linking. For example, Wang et al. developed biohybrid bacterial microswimmers by encapsulating bacteria with metal–organic framework (MOF) exoskeletons.113 This method provided cytoprotection and enabled active drug delivery in harsh environments, significantly improving therapeutic efficacy in tumor treatment. Similarly, Xiong et al. demonstrated that a triple-layered nanogel cross-linked to bacterial surface accumulated in tumors. This nanogel, loaded with doxorubicin, selectively degraded in the bacteria-accumulated tumor environment, resulting in targeted drug release and enhanced cancer therapy.114 These indirect methods enhance the stability and functionality of engineered bacteria, thereby improving the efficiency and specificity of targeted cancer therapy.
3. Nanomaterial combined engineered bacteria and cancer immunotherapy
Compared to the second-generation bacterial therapy (engineered bacteria alone) for tumor therapy, combining nanomaterials with engineered bacteria offers enhanced tumor targeting, improved imaging capabilities, and better spatiotemporal control, making it a promising approach for intelligent tumor immunotherapy. Currently, cancer immunotherapy can be broadly categorized into four major classes: immune checkpoint inhibitors, tumor vaccines, cell-based immunotherapy, and nonspecific immunomodulators (Table 3).115| Type | Nanoparticles | Bacteria | Cargo | Tumor type | Therapeutic strategy | Ref. |
|---|---|---|---|---|---|---|
| Immune checkpoint therapy | F. nucleatum-mimetic nanomedicine | F. nucleatum | Colistin-loaded liposomes | CT26 and MC38 | Killing tumor-colonizing F. nucleatum and alleviating ICB inhibition | 116 |
| Polydopamine | E. coli | Melanin and α-PD-1 | 4T1 | Spatiotemporal release of α-PD-1 through photothermal effect and reverses the immunosuppressive TME | 117 | |
| Polydopamine | EcN | OVA and α-PD-1 | CT26-OVA and MC38-OVA | Enhancing ICD and reversing the immunosuppressive TME | 31 | |
| Fe3O4@lipid nanocomposites | E. coli BL21 | CD47 Nb | CT26 and 4T1 | Spatiotemporal release of CD47 Nb and reactivation of macrophages | 35 | |
| OMVs | E. coli | LyP1 peptide and PD-1 plasmid | B16-F10 and CT26 | Self-expressing α-PD-1 and self-blocking PD-L1 | 118 | |
| Tumor vaccine | Cationic nanoparticles | Attenuated Salmonella | VEGFR2 plasmid | B16-F10 | Inhibiting angiogenesis and activating T cells | 119 |
| HA-IR780 NPs | Vibrio vulnificus | Flagellin B | TC-1 | Suppressing immune-suppressive regulatory T cells (Tregs) and increasing the fraction of CD103+ migratory DCs | 120 | |
| Artificial bacterial nanoparticle | E. coli | Monophosphoryl lipid A and CpG | — | Inducing greater pro-inflammatory via co-presentation of CpG and MPLA | 121 | |
| OMVs | Bacillus | CpG and OVA | E. G7 | Enhancing antigen uptake and resulting in stronger immune activation for cancer vaccines | 122 | |
| Hybrid membrane nanocarriers | E. coli | — | CT26, 4T1, B16, and EMT6 | Inducing DC maturation and activating splenic T cells | 123 | |
| Bacterial membrane-coated nanoparticle | M. smegmatis | PC7A/CpG | B78 and NXS2 | Capturing cancer neoantigens and enhancing the uptake of DCs | 124 | |
| Eukaryotic–prokaryotic vesicle nanoplatform | Attenuated Salmonella | Poly(lactic-co-glycolic acid)–ICG | B16F10 | Stimulate the immune system and trigger the antitumor immune response | 125 | |
| Adoptive cell therapy | — | Brucella melitensis 16M ΔvjbR | Combined with CAR-T | MC38 | Polarize tumor macrophages and elevate CD8+ T cells | 126 |
| — | EcN | Heparin binding domain and CXCL16 | CT26, A20 and MC38 | Release ProCAR-Target by EcN and recruit ProCAR-T cell into the tumor site | 127 | |
| Nonspecific immunity | — | Staphylococcus epidermidis | Melanoma non-active antigens | B16-F10-OVA | Generate tumor-specific T cells | 86 |
| GP-ICG-SiNPs | Attenuated Salmonella strain VNP20009 and Escherichia coli 25922 | ICG | U87MG | Bypass the BBB, targeting, penetrating and thermally ablating GBM tissues | 93 | |
| OMVs | Salmonella | DOX | C6 glioma | Bypass the BBB, selectively recognized by neutrophils and enhance chemo/bacterial therapy | 128 | |
| OMVs | E. coli | Tumor antigen | MC38 and CT26 | Cross the intestinal barrier into the lamina propria and activate DC cells | 129 | |
| Au nanorod | EcN | 5-FU and macrophage phenotype regulator zoledronic acid | 4T1 | Enhance M1 polarization of macrophages and produce pro-inflammatory cytokines | 32 | |
| PLGA NPs | E. coli MG1655 | DOX and R848 | 4T1 | Polarizing macrophages and enhancing ICD | 130 | |
| Polydopamine | EcN | STING agonists | B16F10 | Activate the STING pathway and elevate the levels of IFN-I | 131 | |
| OMVs | E. coli BL21 (DE3) | IL-10 and E7 | CT26 and B16F10 | Increase systemic and intratumoral proportions of CD8+ T cells | 104 | |
| UCNPs-YS | L. lactis MG5267. | IFN-γ | B16F10 CT26 | Release IFN-γ, enhanced ICD and activation of T cells | 61 | |
| — | Salmonella enterica | TNF-α | — | Increase TNF-α in blood and promote the tumor-colonization of bacteria | 132 | |
| — | Salmonella VNP | TNF-α Nb | B16F10 | Secrete TNF-α Nb, stimulate DC maturation and activate CD8+ T cells | 133 | |
3.1 Nanomaterial combined engineered bacteria for immune checkpoint therapy
Immune checkpoint therapy, which has emerged as a pivotal immunotherapy, targets molecules involved in coinhibitory signaling pathways, which typically help maintain immune tolerance but are frequently exploited by cancer cells to escape immune surveillance.134 The presence of bacteria may either sensitize or inhibit the effectiveness of immune checkpoint inhibitors. In preclinical models, the effectiveness of anti-CTLA-4 therapy was shown to depend on Bacteroides species, as germ-free mice or mice treated with antibiotics did not respond to anti-CTLA-4 antibody; however, the response was restored by administering Bacteroides fragilis through gavage.135 Similarly, the gut microbiota can modulate the effectiveness of PD-L1 inhibitors against melanoma and cutaneous tumors.136,137To address these issues, immune checkpoint inhibitors, antibodies, or plasmids can also be encapsulated in nanoparticles or modified on the surface of bacteria or OMVs, thereby achieving enhanced efficacy of immune checkpoint therapy. Abedi et al. developed an ultrasound-thermal responsive promoter TcI42 and a GFP reporter gene construct in EcN (Fig. 3A), which could express and release the immune checkpoints CTLA-4 and PD-L1 nanobody to suppress tumor growth.65 Liu et al. proposed a dual engineered approach to modify live bacteria through genetic engineering and surface chemistry. The genetically engineered E. coli expressed melanin and used in situ polymerization of dopamine to modify the surface with anti-programmed death-1 antibodies (α-PD-1). This dual immunomodulatory approach, mediated by photothermal stimulation and checkpoint blockade, achieves a spatiotemporal release of anti-PD-1 antibodies and synergistic reprogramming of the immunosuppressive TME (Fig. 3B).117
 | ||
| Fig. 3 The nanomaterial-assisted engineered bacteria and immune checkpoints. (A) FUS-activated therapeutic bacteria could express immune checkpoint inhibitors such as CTLA-4 and PD-L1 nanobodies.65 Reproduced from ref. 65 with permission from Springer, copyright 2022. (B) An engineered E. coli was dually modified to concurrently carry photothermal melanin and immune checkpoint inhibitor αPD-1 by combining genetic manipulation and interfacial polymerization.117 Reproduced from ref. 117 with permission from John Wiley & Sons, Inc, copyright 2022. (C) The bacteria with triple immune nanoactivators are described for the creation of a tumor-resident living immunotherapeutic agent, wherein polydopamine nanoparticles, tumor-specific antigens and checkpoint-blocking antibodies are conjugated onto the bacterial surface.31 Reproduced from ref. 31 with permission from John Wiley & Sons, Inc, copyright 2022. (D) A novel LyP1 polypeptide-modified outer-membrane vesicle (LOMV) carrying a PD-1 plasmid is engineered to accomplish self-inhibition of PD-L1 in tumor cells.118 Reproduced from ref. 118 with permission from John Wiley & Sons, Inc, copyright 2022. | ||
Similarly, Liu et al. modified model antigen ovalbumin (OVA) and α-PD-1 antibodies simultaneously on the surface of bacteria using the same method. After the live bacteria reach the tumor site, the photothermal properties of polydopamine convert TAMs from the pro-tumor M2 phenotype to the anti-tumor M1 phenotype (Fig. 3C). The OVA antigen promotes the maturation of DCs, and activates antigen-specific T cell responses, while α-PD-1 antibodies block immune checkpoints to trigger anti-tumor immunity. This synergistic immunomodulation results in an 80% survival rate in animals within 40 days, whereas mice in other groups did not survive beyond 26 days.31 Nie et al. developed a tumor-homing, magnetically controlled bacterial system for precise ICB immunotherapy. Upon in situ accumulation of the Fe3O4@lipid nanocomposite engineered E. coli in colon tumors, the superparamagnetic Fe3O4 nanoparticles allow the engineered bacteria to receive magnetic signals and convert them into heat, thereby initiating the expression of lytic proteins under the control of a heat-sensitive promoter. Subsequently, the pre-expressed anti-CD47 nanobody within the bacteria is released. The potent immunogenicity of bacterial lysates, combined with the anti-CD47 nanobody, activates innate and adaptive immune responses. The gene expression and drug release behaviors of tumor-homing bacteria can be spatiotemporally controlled in vivo by magnetic fields, achieving tumor-specific CD47 blockade and precise tumor immunotherapy.35 Pan et al. designed and engineered E. coli expressing the target peptide LyP1 and then extracted LyP1 modified OMVs. By encapsulating the PD-1 plasmids inside, the LyP1-OMVs@PD-1 nanocarriers were obtained (Fig. 3D). Upon intravenous injection, these nanocarriers accumulate at tumor sites through the targeting ability of OMVs and are internalized into tumor cells via LyP1 mediation. Subsequently, the PD-1 plasmids are delivered to the cell nucleus and induce the expression of PD-1 in tumor cells. The PD-1-expressing tumor cells bind to the overexpressed PD-L1 on the surface of neighboring tumor cells, achieving self-blockade and relieving immune suppression. Meanwhile, the extracellular protein components of OMVs recruit CTL and NK cells to the tumor tissue and stimulate their secretion of IFN-γ, further enhancing the anti-tumor activity of the PD-1/PD-L1 pathway blockade. Additionally, OMVs promote the differentiation of CTL into Tcm, forming a persistent immune response.118
3.2 Nanomaterial combined engineered bacteria and tumor vaccine
Cancer vaccines have garnered significant attention due to their potential to counteract the immune system's tolerance to tumor-specific antigens.138 Due to the efficient secretion or presentation of tumor-associated antigens (TAAs) on bacterial cell surfaces, bacteria have been harnessed as potential vaccine vectors to deliver antigens in diverse forms for activating antitumor immunity.139 Furthermore, nanomaterial-modified bacteria, as a kind of new tumor vaccine, can facilitate tumor-targeted delivery of anti-tumor genes or drugs, enhance immunogenicity, enable real-time imaging, potentiate photothermal/photodynamic therapies and so on. For instance, Hu et al. developed an oral cancer vaccine (NP/SAL) comprising live attenuated Salmonella coated with VEGFR2 plasmid-loaded cationic nanoparticles through electrostatic interactions. The orally administered Salmonella facilitated the delivery of the nanoparticles into immune cells, which were readily engulfed by macrophages due to their pathogen-like characteristics. At the same time, the cationic nanoparticles facilitated the endo/lysosome escape of the live bacterial vector. Experimental results demonstrated that the oral vaccines exhibited remarkable antitumor efficacy, characterized by potent T-cell activation, cytokine production, and suppression of angiogenesis in the tumor vasculature, ultimately leading to tumor necrosis.119 Similarly, Nie et al. engineered E. coli to establish an orally administered tumor vaccine derived from genetically engineered bacteria-based OMVs. Following oral administration (Fig. 4A), these OMVs can be produced in situ in the intestinal tract, carrying tumor antigens, which effectively traverse the intestinal epithelial barrier and are recognized by immune cells in the gut-associated lymphoid tissue.128 Uthaman and colleagues combined PTT with immunotherapy by conjugating Vibrio vulnificus flagellin B with photoabsorber (HIF) loaded micelles. This approach demonstrated the vaccine-like properties of flagellin B, which concurrently suppressed Tregs and increased the secretion of antitumor cytokines, thereby achieving significant immunostimulatory effects in combination with PTT-produced TAAs.120 Siefert et al. proposed a biomimetic strategy using antigen-loaded NPs displaying monophosphoryl lipid A (MPLA) and encapsulating CpG, functioning as an artificial bacterial vaccine platform. In vitro and in vivo experiments of this approach demonstrated enhanced CpG potency and synergistic effects with MPLA, leading to robust pro-inflammatory, Th1-skewed cellular, and antibody-mediated responses, particularly critical for CD8+ T cell immunity.121 Ni et al. developed a demi-bacteria cancer vaccine, retaining bacterial PAMP molecules and pathogen-like physical properties inherited from its parental bacteria, essential for immune activation. By modifying Bacillus into a porous structure to load CpG and OVA, this optimal engineered nanoarchitecture allows multiple immunostimulatory elements to be integrated in a pattern closely resembling that of bacterial pathogens and elicits robust cellular and humoral immune responses for cancer immunotherapy.122 Moreover, Chen et al. developed a personalized cancer vaccine by modifying inactivated EcN with tumor antigens and β-glucan, an inducer of trained immunity (Fig. 4B). Upon subcutaneous injection, the vaccine (BG/OVA@EcN) accumulates at the injection sites, promoting phagocytosis by macrophages to induce trained immunity, facilitating DC maturation, T cell activation, and differentiation of M1-like macrophages in tumor tissues, ultimately demonstrating strong prophylactic and therapeutic efficacy against tumor growth through potent adaptive antitumor immunity and long-term immune memory.129 | ||
| Fig. 4 The nanomaterial-assisted engineered bacteria and tumor vaccines. (A) The genetically engineered bacteria produced antigen-bearing outer membrane vesicles as tumor vaccines.128 Reproduced from ref. 128 with permission from Springer, copyright 2022. (B) A personalized cancer vaccine is developed by engineering the inactivated probiotic EcN to load tumor antigens and β-glucan.129 Reproduced from ref. 129 with permission from John Wiley & Sons, Inc, copyright 2024. | ||
In addition, a variety of cell membranes, including those from erythrocytes, platelets, macrophages, neutrophils, and tumor cells, have been suggested to enhance the therapeutic effectiveness and safety of hybrid bacterial systems. Chen et al. developed a novel approach to create a hybrid membrane-based tumor vaccine using E. coli cytoplasmic membranes (EMs) and tumor cell membranes (TMs). TMs, obtained from surgically resected autologous tumor tissues, carried tumor neoantigens, which could trigger tumor-specific immune responses. Meanwhile, EMs acted as immune adjuvants, stimulating innate immune responses. This personalized tumor vaccine, combining antigens and adjuvants in a co-delivery hybrid membrane, showed superior efficacy in enhancing antitumor immune responses.123 Navi et al. have constructed a multifunctional bacterial membrane-coated nanoparticle (BNP) comprised of an immune activating PC7A/CpG polyplex core coated with the bacterial membrane and imide groups enhancing antigen retrieval. This BNP can capture cancer neoantigens following RT, enhance their uptake by DCs, and facilitate their cross-presentation to stimulate an anti-tumor T cell response. In mice bearing syngeneic melanoma or neuroblastoma, BNP + RT treatment results in activation of DCs and effector T cells, marked tumor regression, and induction of tumor-specific anti-tumor immune memory. This BNP facilitates in situ immune recognition of a radiated tumor, enabling a novel personalized approach to cancer immunotherapy using off-the-shelf therapeutics.124 Chen et al. engineered an eukaryotic–prokaryotic vesicle (EPV) nanoplatform by merging melanoma cytomembrane vesicles (CMVs) and attenuated Salmonella outer membrane vesicles (OMVs) together. The EPV inherits the beneficial properties of its parent components, integrating melanoma antigens with natural adjuvants to provide robust immunotherapy while allowing for easy functionalization with complementary therapeutics. In the melanoma model, the poly(lactic-co-glycolic acid)-ICG moiety (PI)-implanted EPV (PI@EPV) in combination with localized PTT exhibits synergistic antitumor effects as a therapeutic vaccine. This eukaryotic–prokaryotic fusion strategy offers novel insights for designing tumor-immunogenic, self-adjuvating, and expandable vaccine platforms.125
3.3 Nanomaterial combined engineered bacteria and adoptive cell therapy
Despite the success of chimeric antigen receptor (CAR)-T cells in treating hematologic malignancies, their effectiveness in targeting solid tumors has been limited. The challenge lies in the expression patterns of the tumor antigen, which makes identifying optimal targets for solid tumor therapies difficult and hinders the development of new CAR-T therapy.140,141 The majority of TAAs identified in solid tumors are not exclusive to tumors, increasing the risk of severe on-target, off-tumor toxicity due to cross-reactivity with proteins present in vital tissues.142,143 Furthermore, even if a safe target can be identified, TAAs are often expressed heterogeneously. Consequently, the application of targeted therapies exerts selection pressures, which can ultimately result in antigen-negative relapse.144,145Bacteria can be effectively integrated into chimeric antigen receptor (CAR)-T cell therapies. For instance, an attenuated bacterial strain, Brucella melitenis, was found to enhance proinflammatory M1 polarization of tumor macrophages and elevate the presence of CD8+ T cells within tumors. When administered alongside CAR-T cells in a murine model of colon cancer, this approach led to a decrease in tumor burden and a notable improvement in survival rates for the treated mice.126 Moreover, Vincent et al. developed probiotic-guided CAR-T cells (ProCARs) with engineered bacteria encoding a synthetic antigen recognized by CAR-T cells (Fig. 5). The synthetic antigen facilitated communication between the bacteria and ProCARs, enabling spatiotemporal CAR activation within tumors after bacterial release. In mouse models with human tumor xenografts, ProCARs significantly slowed tumor growth, outperforming a combination of bacterial vehicles and CAR-T cells. Increased ProCAR activation was observed due to synthetic antigen presence and TLR stimulation by bacteria, highlighting the potential of programmable medicine communities.127
 | ||
| Fig. 5 The engineered bacteria and adoptive cell therapy. A platform of probiotic-guided CAR-T cells (ProCARs) releasing synthetic targets, which can label tumor tissue for CAR-mediated lysis in situ.127 Reproduced from ref. 127 with permission from AAAS, copyright 2023. | ||
Additionally, Stein-Thoeringer and colleagues have revealed the role of gut microbiota in influencing the effectiveness of CD19 CAR-T cell therapy for patients with B-cell leukemia and lymphoma. Their findings suggest that prior treatment with broad-spectrum antibiotics before administering CD19-targeted CAR-T cell therapy could lead to suboptimal efficacy and other unfavorable outcomes. In this prospective study, the largest of its kind to date, researchers tracked 172 lymphoma patients who had previously failed multiple chemotherapy treatments, from before they began CAR-T cell therapy to two years after treatment. The 20% of patients who received broad-spectrum antibiotics (high-risk antibiotics) before CAR-T cell therapy showed poor clinical prognosis compared to patients who did not receive antibiotic treatment. Patients who received broad-spectrum antibiotics before CAR-T cell therapy tended to have higher tumor burden and systemic inflammation, and these adverse pre-treatment conditions resulted in poorer outcomes of subsequent CAR-T cell therapy. Several key gut microbiotas were further identified that played a curial role, including Bacteroides, Ruminococcus, Eubacterium, and Akkermansia, with Akkermansia being associated with higher baseline peripheral T cell levels before patients received CAR-T cell infusion.146 Therefore, it is expected that developing these identified gut microbiotas into nanohybrid systems or bacterial outer-membrane vesicle nanocarriers may help to predict and enhance the effectiveness of CAR-T therapy.
3.4 Nanomaterial combined engineered bacteria and nonspecific immunity
Nonspecific immunity encompasses tissue barriers, innate immune cells, and molecules. These tissue barriers comprise the skin, mucosal system, BBB, placental barrier, and intestinal barrier. The skin, functioning as the body's largest immune organ, acts as a protective barrier against pathogens and initiates immune responses, while also orchestrating a series of processes, including inflammation and tissue regeneration, to facilitate wound healing.147 As it is known that CD8+ T cells induced by Staphylococcus epidermidis promote skin homeostasis and expedite wound closure, Chen et al. constructed a strain of Staphylococcus epidermidis capable of expressing melanoma tumor antigens, which could stimulate antigen-specific CD8+ and CD4+ T cells both in vitro and on the skin, without disrupting the skin barrier or inducing skin inflammation.86 Bacteria have the capability to bypass the BBB, suggesting their potential in addressing central nervous system diseases like glioma. Hence, Sun et al. introduced a bacteria-based drug delivery system, named 'Trojan bacteria', loaded with glucose polymers and photosensitive ICG silicon-nanoparticles for GBM photothermal immunotherapy. In an orthotopic GBM mouse model, the constructed Trojan bacteria could effectively penetrate GBM tissues by bypassing the BBB. Upon 808 nm laser irradiation, the photothermal effects of ICG destroy bacterial and tumor cells, while the resulting bacterial debris and TAAs trigger antitumor immune responses, leading to prolonged survival in GBM-bearing mice.93 Similarly, Mi et al. described a bacteria-based drug delivery approach for BBB transportation, glioma targeting, and chemo-sensitization. The Salmonella first colonizes selectively in hypoxic tumor regions, modulating the TME and then facilitating targeted delivery of DOX via neutrophil-mediated transport of bacterial outer membrane vesicles (OMVs/DOX), resulting in enhanced drug accumulation in gliomas and complete tumor eradication with 100% survival in treated mice.148 Furthermore, Zhu et al. developed the first NIR optogenetic activation platform, combining EcN with UCNPs for cancer immunotherapy (Fig. 6A). The engineered EcN, transfected with a blue-light-activated plasmid encoding flagellin B (flaB), a potent immunoactivator activating Toll-like receptor 5 (TLR5) signaling, preferentially colonizes tumors and secretes flaB upon NIR photoirradiation, leading to tumor-associated macrophage repolarization and cytotoxic T cell infiltration.34 | ||
| Fig. 6 The nanomaterial-assisted engineered bacteria and nonspecific immunity. (A) An EcN was engineered to sense blue light and release the encoded flaB, then conjugated with lanthanide UCNPs for NIR nano-optogenetic cancer immunotherapy.34 Reproduced from ref. 34 with permission from John Wiley & Sons, Inc, copyright 2023. (B) The nonpathogenic EcN for tumor-targeted delivery of the STING agonist by coating biocompatible polydopamine on the bacterial surface.131 Reproduced from ref. 131 with permission from John Wiley & Sons, Inc, copyright 2023. (C) IL10-modified bacteria to extrude through the gap and self-assemble into bacterial biomimetic vesicles exposing IL10 (IL10-BBVs) on the surface with high efficiency.104 Reproduced from ref. 104 with permission from John Wiley & Sons, Inc, copyright 2021. (D) A novel VNP delivery system expressing the anti-TNF-α nanobody (VNPαTNF-α) in a hypoxic tumor environment.133 Reproduced from ref. 133 with permission from Springer, copyright 2023. | ||
Natural immune cells include macrophages, natural killer cells, and DCs, which would be activated through some signal pathway such as TLR5, TLR7/8, STING, and so on. For instance, Chen et al. demonstrated that the tumor-suppressive effects of Salmonella were significantly reduced in flagellum-deficient strains, even with higher bacterial doses, highlighting the crucial role of flagella in activating immune cells via the flagellin/Toll-like receptor 5 (TLR5) signaling pathway. The exogenous activation of TLR5 signaling enhanced the therapeutic efficacy of flagellum-deficient Salmonella against melanoma, suggesting the potential application of TLR5 agonists in cancer immunotherapy.149 Wei et al. loaded E. coli MG1655 with PLGA-R848 nanoparticles and PLGA-DOX NPs through electrostatic absorption. Leveraging their hypoxic tumor-targeting ability, the bacteria deliver these two nanoparticles to tumor sites. Afterward, the released TLR7/8 agonist R848 could reprogram TAMs into M1 macrophages alongside E. coli, and Dox could function as a chemotherapeutic to kill cancer cells while inducing ICD simultaneously.130 He et al. used non-pathogenic EcN to efficiently load the STING agonist MSA-2 through the oxidative polymerization reaction of dopamine to construct a new biological agent PDMN for targeted immunotherapy of tumors (Fig. 6B). This study also combined anti-PD-1 therapy to induce long-term protective immune responses in the body, thereby effectively preventing tumor recurrence and metastasis.131 More recently, Daniel et al. utilized non-pathogenic E. coli as a multifunctional platform (SYNB1891) to develop an immunotherapy for cancer treatment. This engineered bacterium can target and activate the STING pathway of APCs in tumors and activate the innate immune pathway, which can generate effective anti-tumor immunity and form immunological memory in mouse tumor models.150
Innate immune molecules include complement, cytokines, cytotoxins, and enzymes. The commonly used cytokines in anti-tumor immunotherapy include interleukin-2 (IL-2), interleukin-12 (IL-12), interferon-gamma (IFN-γ), and tumor necrosis factor (TNF).151 These cytokines play important regulatory roles in the immune system, activating, enhancing, or inhibiting the activity of immune cells, thereby enhancing anti-tumor immune responses. Bacteria possess a diverse array of pathogen-associated molecular patterns (PAMPs), such as LPS and flagella, which act as natural immune adjuvants to stimulate antitumor immunity.152 LPS primarily engages with TLR4 on DCs and macrophages, leading to the release of IL-2, tumor necrosis factor-alpha (TNF-α), and type I interferon (IFN-I), thereby triggering a T-cell-mediated antitumor response.153 Wei et al. engineered a glycol chitosan-coated E. coli MG1655, a nonpathogenic bacterium, by electrostatically absorbing resiquimod (R848)-loaded PLGA (PR848) onto its surface (Fig. 6C). This construct, known as Ec-PR848, was designed to actively target hypoxic tumor sites and induce polarization of TAMs. The hybrid system also led to a significant increase in the levels of TNF-α and IL-6, further enhancing immune responses.104 Yan et al. developed a novel tumor immunotherapy technique that leverages focused ultrasound to regulate bacterial gene expression by incorporating an ultrasound-responsive gene expression circuit into tumor-targeting bacteria. This approach takes advantage of ultrasound's non-invasiveness, focusability, high tissue penetration, and acoustic-thermal conversion to regulate gene expression at the tumor site. In preclinical models, this system effectively controlled IFN-γ expression, demonstrating potential for effective immunotherapy in subcutaneous breast cancer and orthotopic liver cancer.39
In addition, Leschner et al. found that there was a rapid surge of TNF-α in the bloodstream following intravenous injection of Salmonella enterica serovar typhimurium in an ectopic transplantable tumor model. This led to vascular disruption, causing a significant influx of blood into the tumors, which might be crucial in the initial stages of bacterial tumor colonization.132 Furthermore, Hua et al. developed a novel Salmonella-based delivery system expressing an anti-TNF-α nanobody (VNPαTNF-α), which significantly enhanced delivery efficiency by continuously releasing the nanobody within the hypoxic tumor environment (Fig. 6D). VNPαTNF-α promotes antitumor immune responses by mobilizing the tumor immune response in several ways, reducing tumor angiogenesis, and stimulating the maturation of DCs, leading to upregulation of granzyme-B and Ki67 and subsequent induction of tumor apoptosis.133
3.5 Nanomaterial combined engineered bacteria and chemotherapeutic agents
In addition to the aforementioned nanomaterial-modified engineered bacteria combined with immune checkpoint inhibitors, tumor vaccines, and adoptive cell therapy, nanomaterial-modified engineered bacteria can also be synergistically combined with chemotherapeutic drugs that elicit direct or indirect antitumor immune responses. Such a therapeutic strategy not only enhances the tumor-targeting capabilities and bioavailability of chemotherapeutic drugs but also augments immune responses within the tumor microenvironment, synergistically improving antitumor efficacy.Certain chemotherapeutic agents directly induce antitumor immune responses mainly by interacting with immune cells or modulating immune signaling pathways. When these agents are combined with nanomaterial-modified bacteria, a multi-pronged strategy is established that enhances tumor-specific targeting, improves drug bioavailability, and amplifies immune-mediated antitumor effects. For instance, Leventhal et al. utilized non-pathogenic E. coli Nissle 1917 to develop a STING agonist-producing live therapeutic (SYNB1891) using synthetic biology, which targets STING activation in phagocytic APCs and activates complementary innate immune pathways.154 Similarly, Chen et al. improved therapeutic efficacy by using nonpathogenic E. coli Nissle 1917 for tumor-targeted delivery of the STING agonist MSA-2, enhancing type I interferon levels and proinflammatory cytokines, inducing DC maturation, and showing profound tumor regression in mice, especially when combined with anti-PD-1 therapy.131 Moreover, Wei et al. developed a cancer treatment by loading PLGA-R848 (PR848), a toll-like receptor 7/8 (TLR7/8) agonist, onto the surface of MG1655b (Ec-PR848) to induce tumor-associated macrophage polarization therapy.155 The PR848/bacteria complex can effectively target tumors and polarize M2 macrophages to M1 macrophages. When supplemented with PLGA-DOX (PDOX)-induced ICD, this approach can reduce tumor immunosuppression and significantly enhance immunotherapy efficacy.
Nanomaterial-modified engineered bacteria can also be combined with chemotherapeutic agents that indirectly elicit antitumor immune responses. These agents include those that induce ICD of tumor cells, thereby activating DCs and subsequently indirectly triggering antitumor immunity. This approach harnesses the potential of engineered bacteria to enhance chemotherapeutic delivery and efficacy, while the ICD mechanism facilitates the activation of the immune system, leading to a synergistic enhancement of antitumor immunity and therapeutic outcomes. For example, Zhang et al. co-loaded an intelligent autophagy inhibitor hydroxychloroquine (HCQ) and chemotherapeutic drug doxorubicin (DOX) into “bulldozer” (HD@HH/EcN) driven by anoxic bacteria for PDAC chemo-immunotherapy.156 The probiotic E. coli Nissle 1917 carries hyaluronidase-hybridized albumin nanoparticles (HD@HH) to the PDAC tumor tissue, where the high GSH levels induce precise release of HCQ and DOX, enhancing DOX-induced ICD and improving the immunosuppressive tumor microenvironment by increasing MHC-I expression and CD8+ T cell recruitment. Du et al. proposed the ultrasound-responsive SonoBacteriaBot (DOX-PFP-PLGA@EcM), which encapsulates DOX and perfluoro-n-pentane (PFP) in PLGA nanodroplets, amide-bonded to the surface of E. coli MG1655.157 This system demonstrated high tumor-targeting efficiency, controlled drug release, increased tumor ICD, and enhanced ultrasound imaging, showing significant potential for real-time monitoring and chemotherapeutic agents’ delivery in clinical settings. Similarly, Liu et al. used a “biotin–streptavidin strategy” to combine live BCG with drug nanoparticles (PLGA-DOX) for improved bladder cancer treatment.158 This system precisely targets tumor cells, enhances drug transport, and inhibits cancer progression through the synergistic effects of BCG-immunotherapy, DOX-chemotherapy, and DOX-induced ICD, showing potential for clinical translation with improved tolerance and antitumor immunity.
4. Challenges and perspectives
Immunotherapy has significantly improved cancer survival rates by reactivating the immune system to eliminate tumors. Bacteria and their derivative-based hybrid nanocarriers have shown promise due to their ability to target tumors specifically and modulate the immune system. However, despite the potential advantages of this third-generation bacterial oncology approach, there are still challenges that need to be addressed. In this section, we will discuss the strengths and limitations of utilizing bacteria and their derivative-based hybrid nanocarriers for cancer immunotherapy, as well as explore future prospects in this field.4.1 Advantages
Engineered bacteria, benefiting from their inherent anaerobic and facultative anaerobic characteristics, can efficiently infiltrate the TME with minimal impact on healthy tissues. For instance, Hua et al. developed an engineered Salmonella strain that exhibits a 1000-fold increase in tumor targeting compared to the WT strain, with minimal distribution observed in the liver and kidneys.21 Moreover, under UV stimulation, bacteria within the cells are enclosed within apoptotic bodies released by tumor cells. Utilizing the homing ability of apoptotic bodies, a tenfold increase in the accumulation of bacteria within tumor cells has been achieved.164 By leveraging the tumor-targeting and infiltrating capabilities of engineered bacteria, more efficient and specific delivery of therapeutic agents can be achieved, potentially enhancing the efficacy and safety of cancer treatment.
In addition, nanomaterial-modified bacteria can enhance the immunostimulatory effects of bacteria by delivering additional immunomodulatory agents or by protecting the bacteria from the harsh tumor microenvironment. For example, nanoparticle-coated bacteria can deliver immune checkpoint inhibitors or other immunostimulatory molecules directly to the tumor site, thereby enhancing the activation of antitumor immunity.35,65,117,166
Furthermore, in conventional cancer therapy, metastasis is the predominant cause of cancer-related morbidity and mortality, responsible for up to 90% of cancer-associated deaths,167 while poor prognosis and tumor recurrence significantly contribute to diminishing cure rates.168 Therefore, it is crucial to consider the optimal prognostic outcomes of effectively inhibiting tumor metastasis and recurrence when evaluating the value of a therapy. Conventional therapeutics' reliance on cytotoxic effects often leads to incomplete tumor eradication, posing a hidden risk for recurrence and metastasis,169 whereas biomaterial-derived drug carriers have been reported to educate the immune system, establishing long-lasting immune memory through fusion with tumor antigens or adjuvant molecules, especially nanoparticle-assisted engineered bacteria playing crucial roles in supporting memory cell function.129,170–172 Additionally, nanomaterials can improve the stability and persistence of bacteria within the tumor, allowing for prolonged immune stimulation and more effective tumor eradication.
Furthermore, the cost of CAR-T therapy is much higher than that of bacterial therapy due to the complex preparation process and strict transportation and preservation conditions, rendering it unaffordable for most patients. The CAR-T therapy currently costs $373![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per patient, significantly higher than conventional chemotherapy agents. It is important to note that additional costs like hospitalization and supplemental drugs, not included in this figure, may add another $150000 to $200000 to the overall expense.176 For bacterial tumor treatment, in situ synthesis and delivery of anti-tumor agents via bacteria offer advantages over engineered immune cells. Bacteria can produce different drugs for synergistic treatment, minimizing the need for multiple dosing and eliminating the necessity for drug purification and storage, thereby significantly reducing treatment costs.177,178
000 per patient, significantly higher than conventional chemotherapy agents. It is important to note that additional costs like hospitalization and supplemental drugs, not included in this figure, may add another $150000 to $200000 to the overall expense.176 For bacterial tumor treatment, in situ synthesis and delivery of anti-tumor agents via bacteria offer advantages over engineered immune cells. Bacteria can produce different drugs for synergistic treatment, minimizing the need for multiple dosing and eliminating the necessity for drug purification and storage, thereby significantly reducing treatment costs.177,178
4.2 Shortcomings
Even in laboratory research, stability and quality concerns such as inconsistent drug loading, therapeutic concentration, and antitumor efficacy are the common challenges faced by scientists. In cancer therapies, most of the bacterial vectors are considered drug carriers for delivering small molecules or nanomaterials through surface modification, gene technology, membrane coating, etc. Regardless of the method used for drug loading, there is no assurance that the loading efficiency of the resulting product is consistent across each preparation. This results in medication being cumbersome and unpredictable. However, the excessive injection of empty bacterial vectors, lacking successful drug loading, may result in side effects, while isolating and purifying vectors with successful drug loading remains challenging. Furthermore, the proliferative capacity of live bacteria enables large-scale expansion but also introduces uncertainty regarding product quality, as the density of mechanically combined therapeutics may decrease with bacteria division.188 Hence, after achieving consistent production, overcoming the challenge of maintaining an effective concentration of drugs both in vitro and in vivo is a hurdle that requires research breakthroughs in the future. Finally, apart from the loaded cargos, the nanomaterial-assisted engineered bacteria also play a role in the tumoricidal capacity of these antitumor therapies. However, tumors have a tendency to develop mechanisms to block the biological activity of these live carriers. The vectors differ in their capacity to penetrate tumors, with physical barriers primarily formed by the dense extracellular matrix in certain solid tumors potentially hindering the penetration of carriers with limited infiltration ability.189
5. Conclusion
In conclusion, we have provided an overview of recent advancements in nanomaterial combined engineered bacteria for intelligent cancer immunotherapy. The hybrid system's superior immunogenicity, tumor-targeting capabilities, and spatiotemporal control offer promising strategies for future cancer treatments. However, it is crucial to address challenges such as immune overactivation, cytotoxicity, production stability, and cost-effectiveness of them. By combining with other therapies such as PTT, PDT, chemotherapy, and RT, these nanomaterial combined engineered bacterial systems have the potential to counteract the immunosuppressive TME and play a pivotal and indispensable role in cancer immunotherapy in the future.Author contributions
S. Qin and Prof. J. Yang designed the topic and outline of the review; S. Qin wrote the original draft; G. He and Prof. J. Yang reviewed and edited the manuscript.Data availability
No new data were generated or analyzed in support of this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the National Natural Science Foundation of China (82202347) and the Supporting project of Nanjing University of Chinese Medicine to NSFC (XPT82202347). The Priority Academic Program Development of Jiangsu Higher Education Institutions (Integration of Chinese and Western Medicine), Jiangsu Key Discipline Construction Fund of the 14th Five-Year Plan (Biology).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- L. Galluzzi, J. M. Bravo-San Pedro, S. Demaria, S. C. Formenti and G. Kroemer, Nat. Rev. Clin. Oncol., 2017, 14, 247–258 CrossRef CAS PubMed.
- O. Strobel, J. Neoptolemos, D. Jager and M. W. Buchler, Nat. Rev. Clin. Oncol., 2019, 16, 11–26 CrossRef CAS PubMed.
- I. F. Tannock, Lancet, 1998, 351(Suppl 2), SII9-16 Search PubMed.
- M. O. Palumbo, P. Kavan, W. H. Miller, Jr., L. Panasci, S. Assouline, N. Johnson, V. Cohen, F. Patenaude, M. Pollak, R. T. Jagoe and G. Batist, Front. Pharmacol., 2013, 4, 57 Search PubMed.
- S. K. Vinod and E. Hau, Respirology, 2020, 25(Suppl 2), 61–71 CrossRef PubMed.
- E. Pylaeva, S. Lang and J. Jablonska, Front. Immunol., 2016, 7, 629 Search PubMed.
- W. J. Lesterhuis, A. Bosco, M. J. Millward, M. Small, A. K. Nowak and R. A. Lake, Nat. Rev. Drug Discovery, 2017, 16, 264–272 CrossRef CAS PubMed.
- L. Chen, T. Xie, B. Wei and D. L. Di, Oncol. Lett., 2022, 24, 358 CrossRef CAS PubMed.
- S. Felgner, D. Kocijancic, M. Frahm and S. Weiss, Int. J. Microbiol., 2016, 2016, 8451728 Search PubMed.
- C. Jimenez-Jimenez, V. M. Moreno and M. Vallet-Regi, Nanomaterials, 2022, 12, 288 CrossRef CAS PubMed.
- H. C. Nauts, W. E. Swift and B. L. Coley, Cancer Res., 1946, 6, 205–216 CAS.
- E. F. McCarthy, Iowa Orthop. J., 2006, 26, 154–158 Search PubMed.
- G. S. Kienle, Glob Adv. Healthcare Med., 2012, 1, 92–100 CrossRef PubMed.
- N. S. Forbes, Nat. Rev. Cancer, 2010, 10, 785–794 CrossRef CAS PubMed.
- V. D. Joshi, D. V. Kalvakolanu and A. S. Cross, FEBS Lett., 2003, 555, 180–184 CrossRef CAS PubMed.
- B. F. Sieow, K. S. Wun, W. P. Yong, I. Y. Hwang and M. W. Chang, Trends Cancer, 2021, 7, 447–464 CrossRef CAS PubMed.
- M. G. Kramer, M. Masner, F. A. Ferreira and R. M. Hoffman, Front. Microbiol., 2018, 9, 16 CrossRef PubMed.
- F. P. Canale, C. Basso, G. Antonini, M. Perotti, N. Li, A. Sokolovska, J. Neumann, M. J. James, S. Geiger, W. Jin, J. P. Theurillat, K. A. West, D. S. Leventhal, J. M. Lora, F. Sallusto and R. Geiger, Nature, 2021, 598, 662–666 CrossRef CAS PubMed.
- C. R. Gurbatri, I. Lia, R. Vincent, C. Coker, S. Castro, P. M. Treuting, T. E. Hinchliffe, N. Arpaia and T. Danino, Sci. Transl. Med., 2020, 12, eaax0876 CrossRef CAS PubMed.
- L. Liu, X. Liu, W. Xin, L. Zhou, B. Huang, C. Han, Z. Cao and Z. Hua, Signal Transduction Targeted Ther., 2023, 8, 134 CrossRef CAS PubMed.
- J. Chen, X. Li, Y. Liu, T. Su, C. Lin, L. Shao, L. Li, W. Li, G. Niu, J. Yu, L. Liu, M. Li, X. Yu and Q. Wang, Microb. Biotechnol., 2021, 14, 2130–2139 CrossRef CAS PubMed.
- Q. W. Chen, J. Y. Qiao, X. H. Liu, C. Zhang and X. Z. Zhang, Chem. Soc. Rev., 2021, 50, 12576–12615 RSC.
- J. Yu, B. Zhou, S. Zhang, H. Yin, L. Sun, Y. Pu, B. Zhou, Y. Sun, X. Li, Y. Fang, L. Wang, C. Zhao, D. Du, Y. Zhang and H. Xu, Nat. Commun., 2022, 13, 7903 CrossRef CAS PubMed.
- W. Luo, Z. Zhang, D. Zhou, Y. Jiang, J. Yang, B. He, H. Yu and Y. Song, Nano Lett., 2023, 23, 8081–8090 CrossRef CAS PubMed.
- W. Lin, Y. Liu, J. Wang, Z. Zhao, K. Lu, H. Meng, R. Luoliu, X. He, J. Shen, Z. W. Mao and W. Xia, Angew. Chem., Int. Ed., 2023, 62, e202310158 CrossRef CAS PubMed.
- Z. Yang, Y. Zhu, Z. Dong, Y. Hao, C. Wang, Q. Li, Y. Wu, L. Feng and Z. Liu, Biomaterials, 2022, 281, 121332 CrossRef CAS PubMed.
- F. M. Chen, Z. S. Zang, Z. Chen, L. Cui, Z. G. Chang, A. Q. Ma, T. Yin, R. J. Liang, Y. T. Han, Z. H. Wu, M. B. Zheng, C. L. Liu and L. T. Cai, Biomaterials, 2019, 214, 119226 CrossRef CAS PubMed.
- Q. W. Chen, X. H. Liu, J. X. Fan, S. Y. Peng, J. W. Wang, X. N. Wang, C. Zhang, C. J. Liu and X. Z. Zhang, Adv. Funct. Mater., 2020, 30, 1909806 CrossRef CAS.
- L. Y. Wang, W. J. Qin, W. X. Xu, F. Huang, X. C. Xie, F. Wang, L. X. Ma and C. Zhang, Small, 2021, 17, 2102932 CrossRef CAS PubMed.
- J. J. Li, Q. Xia, H. Y. Guo, Z. Z. Fu, Y. Liu, S. S. Lin and J. Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202202409 CrossRef CAS PubMed.
- S. Z. Xie, P. Zhang, Z. L. Zhang, Y. Liu, M. H. Chen, S. Li and X. H. Li, Acta Biomater., 2021, 131, 172–184 CrossRef CAS PubMed.
- L. L. Liu, H. M. He, Z. Y. Luo, H. M. Zhou, R. J. Liang, H. Pan, Y. F. Ma and L. T. Cai, Adv. Funct. Mater., 2020, 30, 1910176 CrossRef CAS.
- X. Q. Zhu, S. H. Chen, X. W. Hu, L. J. Zhao, Y. Q. Wang, J. Z. Huang, J. W. Chen, Y. Z. Qiu, X. F. Zhang, M. D. Wang, X. L. Yang, Y. Zhang and Y. H. Zhu, Adv. Mater., 2023, 35, 2207198 CrossRef CAS PubMed.
- X. T. Ma, X. L. Liang, Y. Li, Q. Q. Feng, K. M. Cheng, N. N. Ma, F. Zhu, X. J. Guo, Y. L. Yue, G. N. Liu, T. J. Zhang, J. Liang, L. Ren, X. Zhao and G. J. Nie, Nat. Commun., 2023, 14, 1606 CrossRef CAS PubMed.
- H. T. Chen, Y. Z. Li, Y. J. Wang, P. Ning, Y. J. Shen, X. Y. Wei, Q. S. Feng, Y. L. Liu, Z. G. Li, C. Xu, S. Y. Huang, C. J. Deng, P. Wang and Y. Cheng, ACS Nano, 2022, 16, 6118–6133 CrossRef CAS PubMed.
- D. N. Zhong, W. L. Li, Y. C. Qi, J. He and M. Zhou, Adv. Funct. Mater., 2020, 30, 1910395 CrossRef CAS.
- J. H. Xing, T. Yin, S. M. Li, T. T. Xu, A. Q. Ma, Z. Chen, Y. M. Luo, Z. Y. Lai, Y. N. Lv, H. Pan, R. J. Liang, X. Y. Wu, M. B. Zheng and L. T. Cai, Adv. Funct. Mater., 2021, 31, 2008262 CrossRef CAS.
- Y. H. Chen, M. Du, Z. Yuan, Z. Y. Chen and F. Yan, Nat. Commun., 2022, 13, 4468 CrossRef CAS PubMed.
- M. Du, T. Wang, R. J. Feng, P. H. Zeng and Z. Y. Chen, Front. Bioeng. Biotechnol., 2023, 11, 1144963 CrossRef PubMed.
- Y. Yang, Y. Z. Yang, D. Y. Liu, Y. Y. Wang, M. Q. Lu, Q. Zhang, J. Q. Huang, Y. C. Li, T. Ma, F. Yan and H. R. Zheng, Nat. Commun., 2023, 14, 3297 CrossRef CAS PubMed.
- Y. T. Wang, Y. Tang, Y. Du, L. Lin, Z. Zhang, X. Ou, S. Chen, Q. Wang and J. Z. Zou, Acta Biomater., 2022, 150, 337–352 CrossRef CAS PubMed.
- P. Pan, X. Dong, Y. Chen, X. Zeng and X. Z. Zhang, ACS Nano, 2022, 16, 801–812 CrossRef CAS PubMed.
- P. Pan, X. Dong, Y. Chen, J. J. Ye, Y. X. Sun and X. Z. Zhang, Biomaterials, 2022, 289, 121810 CrossRef CAS PubMed.
- C. Y. Huang, F. B. Wang, L. Liu, W. Jiang, W. Liu, W. Ma and H. Zhao, Adv. Healthcare Mater., 2021, 10, 2002207 CrossRef CAS PubMed.
- W. J. Zai, L. Kang, T. J. Dong, H. R. Wang, L. N. Yin, S. J. Gan, W. J. Lai, Y. B. Ding, Y. Q. Hu and J. H. Wu, ACS Nano, 2021, 15, 15381–15394 CrossRef CAS PubMed.
- X. Wang, Z. Shi, Z. Wang and X. Wu, Materials, 2024, 17, 1501 CrossRef CAS PubMed.
- C. Yang, H. Aslan, P. Zhang, S. Zhu, Y. Xiao, L. Chen, N. Khan, T. Boesen, Y. Wang, Y. Liu, L. Wang, Y. Sun, Y. Feng, F. Besenbacher, F. Zhao and M. Yu, Nat. Commun., 2020, 11, 1379 CrossRef CAS PubMed.
- M. Reggente, C. Roullier, M. Mouhib, P. Brandl, H. X. Wang, S. Tacconi, F. Mura, L. Dini, R. Labarile, M. Trotta, F. Fischer and A. A. Boghossian, Nano Res., 2024, 17, 866–874 CrossRef CAS.
- H. P. Lee and A. K. Gaharwar, Adv. Sci., 2020, 7, 2000863 CrossRef CAS PubMed.
- M. Overchuk, R. A. Weersink, B. C. Wilson and G. Zheng, ACS Nano, 2023, 17, 7979–8003 CrossRef CAS PubMed.
- G. Hannon, F. L. Tansi, I. Hilger and A. Prina-Mello, Adv. Therap., 2021, 4, 2000267 CrossRef.
- W. Luo, Z. B. Zhang, D. T. Zhou, Y. T. Jiang, J. J. Yang, B. S. He, H. J. Yu and Y. J. Song, Nano Lett., 2023, 23, 8081–8090 CrossRef CAS PubMed.
- J. Li, L. S. Xie, B. Li, C. Yin, G. H. Wang, W. Sang, W. X. Li, H. Tian, Z. Zhang, X. J. Zhang, Q. L. Fan and Y. L. Dai, Adv. Mater., 2021, 33, 2008481 CrossRef CAS PubMed.
- M. Przygoda, D. Bartusik-Aebisher, K. Dynarowicz, G. Cieslar, A. Kawczyk-Krupka and D. Aebisher, Int. J. Mol. Sci., 2023, 24, 16890 CrossRef CAS PubMed.
- M. L. Song, T. Liu, C. R. Shi, X. Z. Zhang and X. Y. Chen, ACS Nano, 2016, 10, 633–647 CrossRef CAS PubMed.
- M. Gao, C. Liang, X. J. Song, Q. Chen, Q. T. Jin, C. Wang and Z. Liu, Adv. Mater., 2017, 29, 1701429 CrossRef PubMed.
- H. R. Wang, Y. Chao, J. J. Liu, W. W. Zhu, G. L. Wang, L. G. Xu and Z. G. Liu, Biomaterials, 2018, 181, 310–317 CrossRef CAS PubMed.
- H. J. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. S. Zhou, Adv. Mater., 2020, 32, 2004240 CrossRef CAS PubMed.
- Y. Y. Zhang, X. Xue, M. X. Fang, G. J. Pang, Y. J. Xing, X. Y. Zhang, L. Y. Li, Q. Chen, Y. X. Wang, J. Chang, P. Q. Zhao and H. J. Wang, ACS Appl. Mater. Interfaces, 2022, 41, 46351–46361 CrossRef PubMed.
- C. S. Wu, M. H. Cui, L. Cai, C. Chen, X. H. Zhu, Y. H. Wu, J. L. Liu, H. J. Wang and Y. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 13094–13106 CrossRef CAS PubMed.
- N. A. Valdez-Cruz, L. Caspeta, N. O. Pérez, O. T. Ramírez and M. A. Trujillo-Roldán, Microb. Cell Fact., 2010, 9, 1–16 CrossRef PubMed.
- M. H. Cui, T. Sun, S. B. Li, H. Z. Pan, J. Liu, X. Y. Zhang, L. Y. Li, S. S. Li, C. Y. Wei, C. Z. Yu, C. Yang, N. Ma, B. L. Ma, S. J. Lu, J. Chang, W. W. Zhang and H. J. Wang, Cell Rep., 2021, 36, 567 Search PubMed.
- Z. Izadifar, Z. Izadifar, D. Chapman and P. Babyn, J. Clin. Med., 2020, 9, 460 CrossRef PubMed.
- M. H. Abedi, M. S. Yao, D. R. Mittelstein, A. Bar-Zion, M. B. Swift, A. Lee-Gosselin, P. Barturen-Larrea, M. T. Buss and M. G. Shapiro, Nat. Commun., 2022, 13, 1585 CrossRef CAS PubMed.
- R. W. Bourdeau, A. Lee-Gosselin, A. Lakshmanan, A. Farhadi, S. R. Kumar, S. P. Nety and M. G. Shapiro, Nature, 2018, 553, 86–206 CrossRef CAS PubMed.
- M. Baumann, M. Krause, J. Overgaard, J. Debus, S. M. Bentzen, J. Daartz, C. Richter, D. Zips and T. Bortfeld, Nat. Rev. Cancer, 2016, 16, 234–249 CrossRef CAS PubMed.
- S. Gao, X. Y. Yang, J. K. Xu, N. Qiu and G. X. Zhai, ACS Nano, 2021, 15, 12567–12603 CrossRef CAS PubMed.
- E. D. Kirson, Z. Gurvich, R. Schneiderman, E. Dekel, A. Itzhaki, Y. Wasserman, R. Schatzberger and Y. Palti, Cancer Res., 2004, 64, 3288–3295 CrossRef CAS PubMed.
- H. Maeda, J. Perinat. Med., 2021, 11, 229 Search PubMed.
- V. Ejigah, O. Owoseni, P. Bataille-Backer, O. D. Ogundipe, F. A. Fisusi and S. K. Adesina, Polymers, 2022, 14, 2601 CrossRef CAS PubMed.
- W. Q. Yu, C. Hu and H. L. Gao, ACS Appl. Bio. Mater., 2020, 3, 5455–5462 CrossRef CAS PubMed.
- Q. L. Li, H. T. Chen, X. Y. Feng, C. C. Yu, F. Feng, Y. H. Chai, P. Lu, T. Song, X. Y. Wang and L. Yao, Small, 2019, 15, 1902626 CrossRef PubMed.
- J. Q. Qin, T. Yang, J. Y. Li, G. T. Zhan, X. Li, Z. H. Wei, Z. X. Chen, W. X. Zheng, H. B. Chen, X. L. Yang and L. Gan, Nano Today, 2022, 460, 101591 CrossRef.
- S. C. Thomas, T. Madaan, N. S. Kamble, N. A. Siddiqui, G. M. Pauletti and N. Kotagiri, Adv. Healthcare Mater., 2022, 11, 2101487 CrossRef CAS PubMed.
- J. S. Kim, J. E. Park, S. H. Choi, S. W. Kang, J. H. Lee, J. S. Lee, M. Shin and S. H. Park, J. Controlled Release, 2023, 355, 199–210 CrossRef CAS PubMed.
- Z. Y. Han, Q. W. Chen, Z. J. Fu, S. X. Cheng and X. Z. Zhang, Nano Lett., 2022, 21, 8608–8617 CrossRef PubMed.
- C. Wei, A. Y. Xun, X. X. Wei, J. Yao, J. Y. Wang, R. Y. Shi, G. H. Yang, Y. X. Li, Z. L. Xu, M. G. Lai, R. Zhang, L. S. Wang and W. S. Zeng, Technol. Cancer Res. Treat., 2016, 15, 498–508 CrossRef CAS PubMed.
- L. M. Wood, Z. K. Pan, P. Guirnalda, P. Tsai, M. Seavey and Y. Paterson, Cancer Immunol. Immunother., 2011, 60, 931–942 CrossRef CAS PubMed.
- H. J. Jia, Y. Li, T. S. Zhao, X. Li, J. D. Hu, D. Yin, B. F. Guo, D. J. Kopecko, X. J. Zhao, L. Zhang and D. Xu, Cancer Immunol. Immunother., 2012, 61, 1977–1987 CrossRef CAS PubMed.
- R. M. Ryan, J. Green, P. J. Williams, S. Tazzyman, S. Hunt, J. H. Harmey, S. C. Kehoe and C. E. Lewis, Gene Ther., 2009, 16, 329–339 CrossRef CAS PubMed.
- A. T. St Jean, C. A. Swofford, J. T. Panteli, Z. J. Brentzel and N. S. Forbes, Mol. Therapy, 2014, 22, 1266–1274 CrossRef CAS PubMed.
- T. Yamada, M. Goto, V. Punj, O. Zaborina, M. L. Chen, K. Kimbara, D. Majumdar, E. Cunningham, T. K. D. Gupta and A. M. Chakrabarty, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14098–14103 CrossRef CAS PubMed.
- P. Zhou, Y. She, N. Dong, P. Li, H. B. He, A. Borio, Q. C. Wu, S. Lu, X. J. Ding, Y. Cao, Y. Xu, W. Q. Gao, M. Q. Dong, J. J. Ding, D. Wang, A. Zamyatina and F. Shao, Nature, 2018, 561, 122–126 CrossRef CAS PubMed.
- J. X. Chen, Y. T. Qiao, G. Chen, C. J. Chang, H. Dong, B. Tang, X. W. Cheng, X. F. Liu and Z. C. Hua, Acta Pharm. Sin. B, 2021, 11, 3165–3177 CrossRef CAS PubMed.
- Y. E. Chen, D. Bousbaine, A. Veinbachs, K. Atabakhsh, A. Dimas, V. K. Yu, A. S. Zhao, N. J. Enright, K. Nagashima, Y. Belkaid and M. A. Fischbach, Science, 2023, 380, 203–210 CrossRef CAS PubMed.
- C. R. Gurbatri, N. Arpaia and T. Danino, Science, 2022, 378, 858–863 CrossRef CAS PubMed.
- Y. Ding, Y. Wang and Q. Hu, Exploration, 2022, 2, 20210106 CrossRef CAS PubMed.
- C. Yi, Y. Huang, Z. Y. Guo and S. R. Wang, Acta Pharm. Sin., 2005, 26, 629–634 CrossRef CAS PubMed.
- L. Van Mellaert, S. Barbé and J. Anné, Trends Microbiol., 2006, 14, 190–196 CrossRef CAS PubMed.
- D. H. Thamm, I. D. Kurzman, I. King, Z. J. Li, M. Sznol, R. R. Dubielzig, D. M. Vail and E. G. MacEwen, Clin. Cancer Res., 2005, 11, 4827–4834 CrossRef CAS PubMed.
- J. Stritzker, S. Weibel, P. J. Hlll, T. A. Oelschlaeger, W. Goebel and A. A. Szalay, Int. J. Med. Microbiol., 2007, 297, 151–162 CrossRef CAS PubMed.
- R. Sun, M. Z. Liu, J. P. Lu, B. B. Chu, Y. M. Yang, B. Song, H. Y. Wang and Y. He, Nat. Commun., 2022, 13, 5127 CrossRef CAS PubMed.
- K. Kuerban, X. W. Gao, H. Zhang, J. Y. Liu, M. X. Dong, L. N. Wu, R. H. Ye, M. Q. Feng and L. Ye, Acta Pharm. Sin. B, 2020, 10, 1534–1548 CrossRef CAS PubMed.
- F. P. Canale, C. Basso, G. Antonini, M. Perotti, N. Li, A. Sokolovska, J. Neumann, M. J. James, S. Geiger, W. J. Jin, J. P. Theurillat, K. A. West, D. S. Leventhal, J. M. Lora, F. Sallusto and R. Geiger, Nature, 2021, 598, 662–666 CrossRef CAS PubMed.
- T. T. He, L. Wang, S. Q. Gou, L. Lu, G. H. Liu, K. Wang, Y. L. Yang, Q. J. Duan, W. B. Geng, P. Zhao, Z. Luo and K. Y. Cai, ACS Nano, 2023, 17, 9953–9971 CrossRef CAS PubMed.
- L. Liu, W. Xin, Q. Li, B. Huang, T. Yin, S. Hua, C. Yang, C. Chen, C. Han and Z. Hua, Adv. Sci., 2023, 10, e2301835 CrossRef PubMed.
- M. Li, S. Li, H. Zhou, X. Tang, Y. Wu, W. Jiang, Z. Tian, X. Zhou, X. Yang and Y. Wang, Nat. Commun., 2020, 11, 1126 CrossRef CAS PubMed.
- S. Z. Xie, P. Zhang, Z. L. Zhang, Y. Liu, M. H. Chen, S. Li and X. H. Li, Acta Biomater., 2021, 131, 172–184 CrossRef CAS PubMed.
- K. E. de Visser and J. A. Joyce, Cancer Cell, 2023, 41, 374–403 CrossRef CAS PubMed.
- S. M. Opal, P. J. Scannon, J. L. Vincent, M. White, S. F. Carroll, J. E. Palardy, N. A. Parejo, J. P. Pribble and J. H. Lemke, J. Infect. Dis., 1999, 180, 1584–1589 CrossRef CAS PubMed.
- R. Medzhitov, Nature, 2008, 454, 428–435 CrossRef CAS PubMed.
- S. Ding, Z. Liu, C. Huang, N. Zeng, W. Jiang and Q. Li, ACS Appl. Mater. Interfaces, 2021, 13, 10564–10573 CrossRef CAS PubMed.
- L. Q. Hua, Z. Q. Yang, W. R. Li, Q. S. Zhang, Z. L. Ren, C. Ye, X. Zheng, D. Li, Q. Long, H. M. Bai, W. J. Sun, X. Yang, P. Zheng, J. R. He, Y. J. Chen, W. W. Huang and Y. B. Ma, Adv. Mater., 2021, 33, 2103923 CrossRef CAS PubMed.
- D. N. Zhong, W. L. Li, Y. C. Qi, J. He and M. Zhou, Adv. Funct. Mater., 2020, 30, 1910395 CrossRef CAS.
- Z. Cao, S. Cheng, X. Wang, Y. Pang and J. Liu, Nat. Commun., 2019, 10, 3452 CrossRef PubMed.
- C. H. Luo, C. T. Huang, C. H. Su and C. S. Yeh, Nano Lett., 2016, 16, 3493–3499 CrossRef CAS PubMed.
- F. Chen, Z. Zang, Z. Chen, L. Cui, Z. Chang, A. Ma, T. Yin, R. Liang, Y. Han, Z. Wu, M. Zheng, C. Liu and L. Cai, Biomaterials, 2019, 214, 119226 CrossRef CAS PubMed.
- Q. W. Chen, X. H. Liu, J. X. Fan, S. Y. Peng, J. W. Wang, X. N. Wang, C. Zhang, C. J. Liu and X. Z. Zhang, Adv. Funct. Mater., 2020, 30, 1909806 CrossRef CAS.
- S. Suh, A. Jo, M. A. Traore, Y. Zhan, S. L. Coutermarsh-Ott, V. M. Ringel-Scaia, I. C. Allen, R. M. Davis and B. Behkam, Adv. Sci., 2019, 6, 1801309 CrossRef PubMed.
- S. Chowdhury, S. Castro, C. Coker, T. E. Hinchliffe, N. Arpaia and T. Danino, Nat. Med., 2019, 25, 1057–1063 CrossRef CAS PubMed.
- X. Wang, Z. Cao, M. Zhang, L. Meng, Z. Ming and J. Liu, Sci. Adv., 2020, 6, eabb1952 CrossRef CAS PubMed.
- Y. Li, S. Tang, Z. Cong, D. Lu, Q. Yang, Q. Chen, X. Zhang and S. Wu, Mater. Today Chem., 2022, 23, 100609 CrossRef CAS.
- M. H. Xiong, Y. Bao, X. J. Du, Z. B. Tan, Q. Jiang, H. X. Wang, Y. H. Zhu and J. Wang, ACS Nano, 2013, 7, 10636–10645 CrossRef CAS PubMed.
- H. Dai, Q. Fan and C. Wang, Exploration, 2022, 2, 20210157 CrossRef PubMed.
- L. F. Chen, R. Zhao, J. J. Shen, N. H. Liu, Z. X. Zheng, Y. Miao, J. F. Zhu, L. Zhang, Y. Y. Wang, H. P. Fang, J. Zhou, M. Y. Li, Y. Yang, Z. Liu and Q. Chen, Adv. Mater., 2023, 35, 2306281 CrossRef CAS PubMed.
- L. Wang, Z. P. Cao, M. M. Zhang, S. S. Lin and J. Y. Liu, Adv. Mater., 2022, 34, 2106669 CrossRef CAS PubMed.
- J. M. Pan, X. L. Li, B. F. Shao, F. N. Xu, X. H. Huang, X. Guo and S. B. Zhou, Adv. Mater., 2022, 34, 2106307 CrossRef CAS PubMed.
- Q. L. Hu, M. Wu, C. Fang, C. Y. Cheng, M. M. Zhao, W. H. Fang, P. K. Chu, Y. Ping and G. P. Tang, Nano Lett., 2015, 15, 2732–2739 CrossRef CAS PubMed.
- S. Uthaman, S. Pillarisetti, H. S. Hwang, A. P. Mathew, K. M. Huh, J. H. Rhee and I. K. Park, ACS Appl. Mater. Interfaces, 2021, 13, 4844–4852 CrossRef CAS PubMed.
- A. L. Siefert, M. J. Caplan and T. M. Fahmy, Biomaterials, 2016, 97, 85–96 CrossRef CAS PubMed.
- D. Z. Ni, S. Qing, H. Ding, H. Yue, D. Yu, S. Wang, N. N. Luo, Z. G. Su, W. Wei and G. H. Ma, Adv. Sci., 2017, 4, 1700083 CrossRef PubMed.
- L. Chen, H. Qin, R. F. Zhao, X. Zhao, L. R. Lin, Y. Che, Y. X. Lin, Y. Li, Y. T. Qin, Y. Y. Li, S. L. Liu, K. M. Cheng, H. Q. Chen, J. Shi, G. J. Anderson, Y. Wu, Y. L. Zhao and G. J. Nie, Sci. Transl. Med., 2021, 13, eabc2816 CrossRef CAS PubMed.
- R. B. Patel, M. Z. Ye, P. M. Carlson, A. Jaquish, L. Zangl, B. Ma, Y. Y. Wang, I. Arthur, R. S. Xie, R. J. Brown, X. Wang, R. Sriramaneni, K. Kim, S. Q. Gong and Z. S. Morris, Adv. Mater., 2019, 31, 1902626 CrossRef CAS PubMed.
- Q. Chen, G. J. Huang, W. T. Wu, J. W. Wang, J. W. Hu, J. M. Mao, P. K. Chu, H. Z. Bai and G. P. Tang, Adv. Mater., 2020, 32, 1908185 CrossRef CAS PubMed.
- F. G. Guo, J. K. Das, K. S. Kobayashi, Q. M. Qin, T. A. Ficht, R. C. Alaniz, J. X. Song and P. De Figueiredo, J. Immunother. Cancer, 2022, 10, 121 CrossRef.
- R. L. Vincent, C. R. Gurbatri, F. Li, A. Vardoshvili, C. Coker, J. Im, E. R. Ballister, M. Rouanne, T. Savage, K. de Los Santos-Alexis, A. Redenti, L. Brockmann, M. Komaranchath, N. Arpaia and T. Danino, Science, 2023, 382, 211–218 CrossRef CAS PubMed.
- Y. L. Yue, J. Q. Xu, Y. Li, K. M. Cheng, Q. Q. Feng, X. T. Ma, N. N. Ma, T. J. Zhang, X. W. Wang, X. Zhao and G. J. Nie, Nat. Biomed. Eng., 2022, 6, 898–909 CrossRef CAS PubMed.
- Z. X. Chen, T. Y. Yong, Z. H. Wei, X. Q. Zhang, X. Li, J. Q. Qin, J. Y. Li, J. Hu, X. L. Yang and L. Gan, Adv. Sci., 2024, 11, 2305081 CrossRef CAS PubMed.
- B. C. Wei, J. M. Pan, R. T. Yuan, B. F. Shao, Y. Wang, X. Guo and S. B. Zhou, Nano Lett., 2021, 21, 5905 CrossRef CAS PubMed.
- W. F. Chen, Y. N. Zhu, J. H. Chen, X. D. Jing, Y. M. Xiong, L. Zou, J. H. Li, Q. H. Liu, Y. P. Li, Y. Huang and J. H. He, Adv. Funct. Mater., 2023, 33, 2307001 CrossRef CAS.
- S. Leschner, K. Westphal, N. Dietrich, N. Viegas, J. Jablonska, M. Lyszkiewicz, S. Lienenklaus, W. Falk, N. Gekara, H. Loessner and S. Weiss, PLoS One, 2009, 4, e6692 CrossRef PubMed.
- L. N. Liu, X. Liu, W. J. Xin, L. L. Zhou, B. L. Huang, C. Han, Z. T. Cao and Z. C. Hua, Signal Transduction Targeted Ther., 2023, 8, 134 CrossRef CAS PubMed.
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- M. Vétizou, J. M. Pitt, R. Daillère, P. Lepage, N. Waldschmitt, C. Flament, S. Rusakiewicz, B. Routy, M. P. Roberti, C. P. M. Duong, V. Poirier-Colame, A. Roux, S. Becharef, S. Formenti, E. Golden, S. Cording, G. Eberl, A. Schlitzer, F. Ginhoux, S. Mani, T. Yamazaki, N. Jacquelot, D. P. Enot, M. Bérard, J. Nigou, P. Opolon, A. Eggermont, P. L. Woerther, E. Chachaty, N. Chaput, C. Robert, C. Mateus, G. Kroemer, D. Raoult, I. G. Boneca, F. Carbonnel, M. Chamaillard and L. Zitvogel, Science, 2015, 350, 1079–1084 CrossRef PubMed.
- V. Gopalakrishnan, C. N. Spencer, L. Nezi, A. Reuben, M. C. Andrews, T. V. Karpinets, P. A. Prieto, D. Vicente, K. Hoffman, S. C. Wei, A. P. Cogdill, L. Zhao, C. W. Hudgens, D. S. Hutchinson, T. Manzo, M. P. de Macedo, T. Cotechini, T. Kumar, W. S. Chen, S. M. Reddy, R. S. Sloane, J. Galloway-Pena, H. Jiang, P. L. Chen, E. J. Shpall, K. Rezvani, A. M. Alousi, R. F. Chemaly, S. Shelburne, L. M. Vence, P. C. Okhuysen, V. B. Jensen, A. G. Swennes, F. McAllister, E. M. R. Sanchez, Y. Zhang, E. Le Chatelier, L. Zitvogel, N. Pons, J. L. Austin-Breneman, L. E. Haydu, E. M. Burton, J. M. Gardner, E. Sirmans, J. Hu, A. J. Lazar, T. Tsujikawa, A. Diab, H. Tawbi, I. C. Glitza, W. J. Hwu, S. P. Patel, S. E. Woodman, R. N. Amaria, M. A. Davies, J. E. Gershenwald, P. Hwu, J. E. Lee, J. Zhang, L. M. Coussens, Z. A. Cooper, P. A. Futreal, C. R. Daniel, N. J. Ajami, J. F. Petrosino, M. T. Tetzlaff, P. Sharma, J. P. Allison, R. R. Jenq and J. A. Wargo, Science, 2018, 359, 97–103 CrossRef CAS PubMed.
- B. Routy, E. Le Chatelier, L. Derosa, C. P. M. Duong, M. T. Alou, R. Daillère, A. Fluckiger, M. Messaoudene, C. Rauber, M. P. Roberti, M. Fidelle, C. Flament, V. Poirier-Colame, P. Opolon, C. Klein, K. Iribarren, L. Mondragón, N. Jacquelot, B. Qu, G. Ferrere, C. Clémenson, L. Mezquita, J. R. Masip, C. Naltet, S. Brosseau, C. Kaderbhai, C. Richard, H. Rizvi, F. Levenez, N. Galleron, B. Quinquis, N. Pons, B. Ryffel, V. Minard-Colin, P. Gonin, J. C. Soria, E. Deutsch, Y. Loriot, F. Ghiringhelli, G. Zalcman, F. Goldwasser, B. Escudier, M. D. Hellmann, A. Eggermont, D. Raoult, L. Albiges, G. Kroemer and L. Zitvogel, Science, 2018, 359, 91–97 CrossRef CAS PubMed.
- M. J. Lin, J. Svensson-Arvelund, G. S. Lubitz, A. Marabelle, I. Melero, B. D. Brown and J. D. Brody, Nat. Cancer, 2022, 3, 911–926 CrossRef CAS PubMed.
- M. Cronin, R. M. Stanton, K. P. Francis and M. Tangney, Cancer Gene Ther., 2012, 19, 731–740 CrossRef CAS PubMed.
- A. Hyrenius-Wittsten and K. T. Roybal, Trends Cancer, 2019, 5, 583–592 CrossRef CAS PubMed.
- W. A. Lim and C. H. June, Cell, 2017, 168, 724–740 CrossRef CAS PubMed.
- S. A. Richman, S. Nunez-Cruz, B. Moghimi, L. Z. Li, Z. T. Gershenson, Z. Mourelatos, D. M. Barrett, S. A. Grupp and M. C. Milone, Cancer Immunol. Res., 2018, 6, 36–46 CrossRef CAS PubMed.
- K. R. Parker, D. Migliorini, E. Perkey, K. E. Yost, A. Bhaduri, P. Bagga, M. Haris, N. E. Wilson, F. Liu, K. Gabunia, J. Scholler, T. J. Montine, V. G. Bhoj, R. Reddy, S. Mohan, I. Maillard, A. R. Kriegstein, C. H. June, H. Y. Chang, A. D. Posey and A. T. Satpathy, Cell, 2020, 183, 126–142 CrossRef CAS PubMed.
- R. G. Majzner and C. L. Mackall, Cancer Discovery, 2018, 8, 1219–1226 CrossRef CAS PubMed.
- D. M. O'Rourke, M. P. Nasrallah, A. Desai, J. J. Melenhorst, K. Mansfield, J. J. D. Morrissette, M. Martinez-Lage, S. Brem, E. Maloney, A. Shen, R. Isaacs, S. Mohan, G. Plesa, S. F. Lacey, J. M. Navenot, Z. H. Zheng, B. L. Levine, H. Okada, C. H. June, J. L. Brogdon and M. V. Maus, Sci. Transl. Med., 2017, 9, eaaa0984 CrossRef PubMed.
- K. M. Cappell and J. N. Kochenderfer, Nat. Rev. Clin. Oncol., 2023, 20, 359–371 CrossRef CAS PubMed.
- A. Jakovija and T. Chtanova, Front. Immunol., 2023, 14, 1060258 CrossRef CAS PubMed.
- Z. Mi, Q. Yao, Y. Qi, J. H. Zheng, J. H. Liu, Z. G. Liu, H. P. Tan, X. Q. Ma, W. H. Zhou and P. F. Rong, Acta Pharm. Sin. B, 2023, 13, 819–833 CrossRef CAS PubMed.
- J. Chen, Y. Qiao, G. Chen, C. Chang, H. Dong, B. Tang, X. Cheng, X. Liu and Z. Hua, Acta Pharm. Sin. B, 2021, 11, 3165–3177 CrossRef CAS PubMed.
- D. S. Leventhal, A. Sokolovska, N. Li, C. Plescia, S. A. Kolodziej, C. W. Gallant, R. Christmas, J. R. Gao, M. J. James, A. Abin-Fuentes, M. Momin, C. Bergeron, A. Fisher, P. F. Miller, K. A. West and J. M. Lora, Nat. Commun., 2020, 11, 2739 CrossRef CAS PubMed.
- Y. Qiu, M. X. Su, L. Y. Liu, Y. Q. Tang, Y. Pan and J. B. Sun, Drug Des., Dev. Ther., 2021, 15, 2269–2287 CrossRef PubMed.
- V. Nicaise, M. Roux and C. Zipfel, Plant Physiol., 2009, 150, 1638–1647 CrossRef CAS PubMed.
- H. L. Yin, N. Pu, Q. D. Chen, J. C. Zhang, G. C. Zhao, X. F. Xu, D. S. Wang, T. T. Kuang, D. Y. Jin, W. H. Lou and W. C. Wu, Cell Death Dis., 2021, 12, 1033 CrossRef CAS PubMed.
- D. S. Leventhal, A. Sokolovska, N. Li, C. Plescia, S. A. Kolodziej, C. W. Gallant, R. Christmas, J. R. Gao, M. J. James, A. Abin-Fuentes, M. Momin, C. Bergeron, A. Fisher, P. F. Miller, K. A. West and J. M. Lora, Nat. Commun., 2020, 11, 2739 CrossRef CAS PubMed.
- B. Wei, J. Pan, R. Yuan, B. Shao, Y. Wang, X. Guo and S. Zhou, Nano Lett., 2021, 21, 4231–4240 CrossRef CAS PubMed.
- H. Zhang, Y. Wang, L. Zhu, Z. Qi, K. Cao, J. Chang and L. Hou, J. Controlled Release, 2023, 360, 660–671 CrossRef CAS PubMed.
- M. Du, T. Wang, R. J. Feng, P. H. Zeng and Z. Y. Chen, Front. Bioeng. Biotechnol., 2023, 11, 1144963 CrossRef PubMed.
- K. Liu, L. Wang, J. Peng, Y. Lyu, Y. Li, D. Duan, W. Zhang, G. Wei, T. Li, Y. Niu and Y. Zhao, Adv. Mater., 2024, 36, e2310735 CrossRef PubMed.
- Y. Zhou, Q. Li, Y. Wu, W. Zhang, L. Ding, C. Ji, P. Li, T. Chen, L. Feng, B. Z. Tang and X. Huang, Adv. Mater., 2024, 36, e2313953 CrossRef PubMed.
- Q. N. Ye, Y. Wang, S. Shen, C. F. Xu and J. Wang, Adv. Healthcare Mater., 2021, 10, 2002139 CrossRef CAS PubMed.
- J. Martinez-Quintanilla, I. Seah, M. Chua and K. Shah, J. Clin. Invest., 2019, 129, 1407–1418 CrossRef PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. W. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. S. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- R. Ngoune, A. Peters, D. von Elverfeldt, K. Winkler and G. Pütz, J. Controlled Release, 2016, 238, 58–70 CrossRef CAS PubMed.
- R. Liu, Z. P. Cao, L. Wang, X. Y. Wang, S. S. Lin, F. Wu, Y. Pang and J. Y. Liu, Nano Today, 2022, 45, 101537 CrossRef CAS.
- Q. J. Chen, T. Sun and C. Jiang, Nano-Micro Lett., 2021, 13, 1–47 CrossRef PubMed.
- J. J. Li, Q. Xia, H. Y. Guo, Z. Z. Fu, Y. Liu, S. S. Lin and J. Y. Liu, Angew. Chem., Int. Ed., 2022, 134, e202202409 CrossRef.
- N. Sethi and Y. B. Kang, Nat. Rev. Cancer, 2011, 11, 735–748 CrossRef CAS PubMed.
- C. Munguti, M. C. Mutebi, M. Ng'ang'a and R. Wasike, J. Clin. Oncol., 2020, 38, 12508 Search PubMed.
- L. Bracci, G. Schiavoni, A. Sistigu and F. Belardelli, Cell Death Differ., 2014, 21, 15–25 CrossRef CAS PubMed.
- L. Chen, W. Q. Hong, W. Y. Ren, T. Xu, Z. Y. Qian and Z. Y. He, Signal Transduction Targeted Ther., 2021, 6, 225 CrossRef PubMed.
- D. S. Leventhal, A. Sokolovska, N. Li, C. Plescia, S. A. Kolodziej, C. W. Gallant, R. Christmas, J. R. Gao, M. J. James, A. Abin-Fuentes, M. Momin, C. Bergeron, A. Fisher, P. F. Miller, K. A. West and J. M. Lora, Nat. Commun., 2020, 11, 2739 CrossRef CAS PubMed.
- R. B. Patel, M. Z. Ye, P. M. Carlson, A. Jaquish, L. Zangl, B. Ma, Y. Y. Wang, I. Arthur, R. S. Xie, R. J. Brown, X. Wang, R. Sriramaneni, K. Kim, S. Q. Gong and Z. S. Morris, Adv. Mater., 2019, 31, 1902626 CrossRef CAS PubMed.
- J. W. Jiang, Y. J. Huang, Z. S. Zeng and C. S. Zhao, ACS Nano, 2023, 17, 843–884 CrossRef CAS PubMed.
- Z. Y. Liao, W. T. Zhang, H. Q. Zheng, Y. F. Wang, J. C. Yu, H. J. Li and Z. Gu, J. Controlled Release, 2022, 344, 272–288 CrossRef PubMed.
- P. Xu, C. Clark, T. Ryder, C. Sparks, J. P. Zhou, M. Wang, R. Russell and C. Scott, Biotechnol. Prog., 2017, 33, 478–489 CrossRef CAS PubMed.
- C. Flowers, B. Chastek, L. Becker, D. Mahmoud and E. Dulac, Haematologica, 2014, 99, 784–785 Search PubMed.
- J. X. Fan, M. Y. Peng, H. Wang, H. R. Zheng, Z. L. Liu, C. X. Li, X. N. Wang, X. H. Liu, S. X. Cheng and X. Z. Zhang, Adv. Mater., 2019, 31, 1808278 CrossRef PubMed.
- T. Danino, A. Prindle, G. A. Kwong, M. Skalak, H. Li, K. Allen, J. Hasty and S. N. Bhatia, Sci. Transl. Med., 2015, 7, 289ra284 Search PubMed.
- V. Staedtke, R. Y. Bai, K. Kim, M. Darvas, M. L. Davila, G. J. Riggins, P. B. Rothman, N. Papadopoulos, K. W. Kinzler, B. Vogelstein and S. B. Zhou, Nature, 2018, 564, 273–277 CrossRef CAS PubMed.
- W. J. Luo, C. G. Li, Y. Q. Zhang, M. Y. Du, H. M. Kou, C. Lu, H. Mei and Y. Hu, BMC Cancer, 2022, 22, 98 CrossRef CAS PubMed.
- X. H. Huang, J. M. Pan, F. N. Xu, B. F. Shao, Y. Wang, X. Guo and S. B. Zhou, Adv. Sci., 2021, 8, 2003572 CrossRef CAS PubMed.
- M. X. Hao, S. Y. Hou, W. S. Li, K. M. Li, L. J. Xue, Q. F. Hu, L. L. Zhu, Y. Chen, H. B. Sun, C. Y. Ju and C. Zhang, Sci. Transl. Med., 2020, 12, eaaz6667 CrossRef CAS PubMed.
- L. Guo, H. Chen, J. Ding, P. Rong, M. Sun and W. Zhou, Exploration, 2023, 3, 20230017 CrossRef CAS PubMed.
- S. Lin, F. Wu, Y. Zhang, H. Chen, H. Guo, Y. Chen and J. Liu, Chem. Soc. Rev., 2023, 52, 6617–6643 RSC.
- Z. P. Cao and J. Y. Liu, Acc. Mater. Res., 2024, 23, 1162 Search PubMed.
- D. Hu, H. Xu, W. Zhang, X. Xu, B. Xiao, X. Shi, Z. Zhou, N. K. H. Slater, Y. Shen and J. Tang, Biomaterials, 2021, 277, 121130 CrossRef CAS PubMed.
- H. Pan, M. Zheng, A. Ma, L. Liu and L. Cai, Adv. Mater., 2021, 33, e2100241 CrossRef PubMed.
- D. Lin, X. Feng, B. Mai, X. Li, F. Wang, J. Liu, X. Liu, K. Zhang and X. Wang, Biomaterials, 2021, 277, 121124 CrossRef CAS PubMed.
- X. Guan, J. Chen, Y. Hu, L. Lin, P. Sun, H. Tian and X. Chen, Biomaterials, 2018, 171, 198–206 CrossRef CAS PubMed.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2024 |