
Non-Ir based catalysts for the electrocatalytic oxygen evolution reaction: progress and challenges
Lin
Lin
a,
Kai
Wei
a,
Xian
Wang
*a,
Wei
Ma
c,
Chunlei
Bian
a and
Junjie
Ge
 *ab
*ab
aSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: wangxian@ustc.edu.cn; gejunjie@ustc.edu.cn
bKey Laboratory of Low-Carbon Conversion Science & Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201204, China
cState Grid Anhui Electric Power Research Institute, Hefei, Anhui 230026, China
First published on 4th June 2024
Abstract
Oxygen evolution reaction (OER) electrocatalysis is the key to solve the problem of hydrogen production by hydrolyzing water and rechargeable metal–air battery. Therefore, the development of active and highly stable oxygen evolution catalyst materials has become a hot research topic. Ir-based catalysts for oxygen evolution are the most common electrocatalytic materials, but the high price and low yield of iridium greatly restrict its large-scale development. It is of great significance to develop efficient and cheap non-iridium-based catalysts for oxygen evolution. Herein, we first conclude the reaction mechanisms of the OER, and then classify the OER catalysts. In addition, some new research achievements in non-iridium catalysts in the past 5 years are also reviewed. On this basis, some improvement of OER catalysts and their future development were discussed.
 Lin Lin | Lin Lin received her B.E. degree from Anhui University in 2023. Currently, she is a postgraduate student at the University of Science and Technology of China under the supervision of Prof. Dr Junjie Ge. Her current research focuses on non-noble metal catalysts for the oxygen reduction reaction. |
 Xian Wang | Dr Xian Wang received his PhD in physical chemistry from the University of Science and Technology of China in 2021. Then, he worked at the Changchun Institute of Applied Chemistry as an assistant researcher. His research focuses on the anti-poisoning fuel cell anode catalysis. |
 Junjie Ge | Prof. Dr Junjie Ge received her PhD in physical chemistry from the Chinese Academy of Sciences in 2010. She worked at the University of South Carolina and University of Hawaii as a postdoc fellow for almost 5 years. She joined the Changchun Institute of Applied Chemistry in 2015 as a professor, where she was recruited in the Hundred Talents Program in CAS (2015). She joined the University of Science and Technology of China in 2022 as a professor. Her research interests include fuel cells, nanoscience, catalysis, and electrochemistry. |
1. Introduction
Due to the rapid growth of the global industry and the population explosion, the global demand for energy is increasing rapidly. Currently, the largest source of energy is fossil fuels, but they have a non-renewable nature and lead to the greenhouse effect due to the carbon dioxide gas produced by their utilization. Thus, the search for new clean renewable energy sources to replace fossil energy has become a focus of attention.1–3 Among many green energy sources, hydrogen is a promising clean energy carrier due to its zero-carbon content and the high gravimetric energy density,4,5 and water electrolysis is a promising new energy power generation technology. Compared with other harsh reactions involving high temperatures and pressures, the electrocatalytic reaction is mild and can be carried out under environmentally friendly conditions, and the energy conversion efficiency is extremely high, which makes it more suitable for industrial applications. Electrochemical water splitting can be carried out under acidic, alkaline and neutral conditions. Compared to the other two conditions, polymer electrolyte membrane water electrolysis (PEMWE) under acidic conditions has lower ohmic consumption, higher ionic conductivity, and fewer side reactions, allowing it to operate at current densities three to four times higher than that under alkaline conditions. The hydrogen production process of water electrolysis can be divided into two half electrodes: cathodic hydrogen evolution and anodic oxygen evolution.4 Compared with the 2e transfer in the cathode, the OER process has to go through four electron transfer steps, and the energy accumulation between the steps will lead to a larger overpotential and slower reaction rate.6,7 The development of efficient and stable electrocatalytic materials for oxygen evolution is a major problem at present.Ir-based catalysts, such as IrO2, due to their high durability, are usually considered as the most effective catalysts for the OER, especially under acidic conditions.8 However, as an important part of the electrolytic aquaculture hydrogen industry, Ir is in a state of shortage all over the world.9 Therefore, the development of other efficient and stable clay-rich catalysts is the key to large-scale application. The number of research studies on other non-Ir compounds with high efficiency and stability has increased, such as other noble metal-based (e.g. Ru, Rh, Pt, etc.), loaded catalysts with various metal nanoparticles embedded in different carbon supports, oxides, alloys, and hydroxides of non-precious metals (e.g. Co, Fe, Mn, etc.) and their complexes with carbon and spinel and chalcogenide systems with complex ternary structures.5,7,10–15
For further improving the performance of OER electrocatalysts, four factors need to be considered.16 The first is the composition of the electrocatalyst. Previous studies had shown that because of the synergistic and electronic interactions between metals, catalysts with multi-metal components exhibited better electrocatalytic activity.17 Secondly, more attention should be paid to the surface structure of electrode materials. Since the electrocatalytic performance depends on the surface of catalyst, it can be improved by constructing surface active sites on the catalyst.18 Moreover, it is important to carefully design and select the catalyst support. The dispersion of active components can be improved through the support, so as to improve the catalytic efficiency. Meanwhile, high porosity and larger specific surface area support promote uniform distribution of the catalyst.19 Finally, studies have shown that the active sites can be increased towards further improving metal utilization by reducing metal size.20 Single-atom catalysts (SACs) have received wide attention and have become a hot spot in electrocatalysis research because of their atomic-scale metal utilization (100% in theory), independent active sites, as well as special electronic structure. Moreover, SACs have high activity, selectivity and stability for many important reactions compared to conventional metal nanoparticles.21
Herein, we review the recent advances and breakthroughs in experimental results and theoretical mechanisms of various non-Ir-based OER catalysts in the last five years, focusing on SACs. First, we introduce the process and mechanism of the OER. And then, we classify the OER catalysts into no-single-atom and single-atom to introduce their research progress in recent years. We further summarize the methods used to improve catalyst performance. Finally, we provide an outlook on the development prospects in the field of the OER.
2. Oxygen evolution reaction (OER) mechanism
The OER does not involve the same chemical intermediates in acidic and neutral-basic solutions.22 In the mechanistic study of the OER, the thermodynamic potential of the oxygen precipitation reaction is constant at 1.23 V vs. RHE. Due to the reaction intermediate energy barrier, solution resistance, catalyst conductivity, etc., the actual OER voltage is much higher than the theoretical value, so an additional overvoltage is required to drive water electrolysis. Norskov et al. revealed the changes in the conversion energy barriers of intermediates during the OER by density functional theory (DFT) theoretical calculations, which profoundly revealed the changes in intermediates during the OER process, where the OER steps in acidic solutions and the intermediates involved are shown below.23| H2O + * → OHads + H+ + e− ΔG1 |
| OHads → Oads + H+ + e− ΔG2 |
| Oads + H2O → OOHads + H++ e− ΔG3 |
| OOHads → * + O2 + H+ + e− ΔG4 |
The OER process under alkaline conditions involves the following steps:
| OH−+ * → OHads + e− ΔG1 |
| OHads + OH− → Oads + H2O + e− ΔG2 |
| Oads + OH− → OOHads + e− ΔG3 |
| OOHads + OH− → * +O + H2O + e− ΔG4 |
From the reaction steps, it can be seen that in both acidic and neutral alkaline solutions, three chemical intermediates are involved, namely OH*, O* and OOH*. According to the previous study, there are two kinds of OER mechanisms: one is the evolution mechanism of the surface adsorption state (AEM), the other is lattice oxygen oxidation (LOM), based on the origin of the O atoms in the intermediates.24 In AEM, the O atom in the intermediate is derived from the oxygen atom of H2O in the electrolyte, while in LOM, the O atom is derived from the oxygen atom in the catalyst surface oxide. Moreover, the LOM does not involve the generation of the *OOH intermediate, and the formation of the O–O bond is by the direct coupling of the lattice oxygen on the metal in the adjacent double site (Fig. 1a and b).
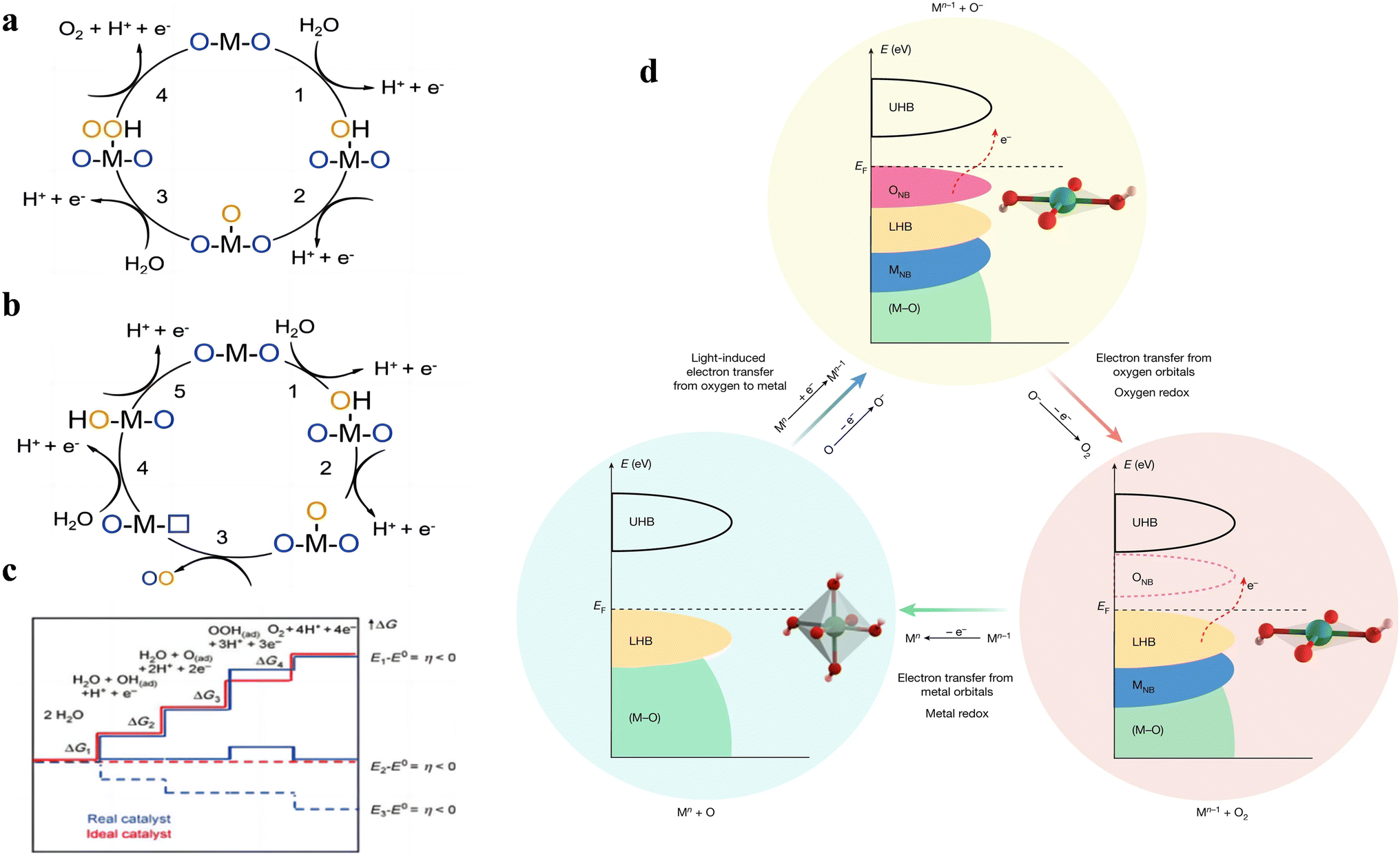 | ||
| Fig. 1 Proposed (a) AEM and (b) LOM for the OER. (c) The reaction course of the OER and the Gibbs free energy curve of the reaction products were given. Blue and red lines represent the actual chemical reaction pathway, while red lines represent the ideal path. Reproduced with permission.7 Copyright 2021, John Wiley & Sons. (d) Proposal for a light-induced electron transfer process with switchable metal and oxygen redox centres for the OER. Reproduced with permission.29 Copyright 2022, The Nature Publishing Group. | ||
The AEM mechanism plays an important role in OER theory, and there are corresponding intermediates in each basic stage. Among them, the step with largest free energy difference (G = max [ΔG1, ΔG2, ΔG3, ΔG4]) is used as the rate determining step (RDS)7 (Fig. 1c). It is worth noting that the energy potentials of each stage are related to each other, called the scaling relationship.25,26 According to the scaling relationship, the sum of the energy required for the OH* to O* process and the O* to OOH* process remains roughly at 3.2 eV. Thus, the overpotential is minimized if the O* energy level of the material can be optimally placed between OH* and OOH*. This scaling relationship can be described by a volcano curve between catalysts.25 The catalysts located on the left side of the volcano-shaped curve exhibit strong oxygen-binding adsorption, and the step of the OER in which the OOH* intermediate is generated is the RDS. The catalysts located on the right side of the volcano-shaped curve exhibit weak oxygen-binding adsorption, and the generation of O* intermediates is the RDS. At the top of the volcano curves, the energies of OH* to O* and the O* to OOH* steps are balanced, so a minimum overpotential is required. Therefore, the catalyst is most efficient when its adsorption site is closest to the apex of the volcano curve. The volcano curve can guide the design of electrocatalysts by adjusting their structure to alter the binding energy suitable at the catalytic site, so as to achieve the purpose of efficient OER performance.
LOM is the OER mechanism proposed in recent years, and compared to the AEM, the catalyst surface is no longer thermodynamically stable, but the catalyst surface is allowed to change with the oxygen release process. In the classical LOM process, the first and second steps are similar to the formation of O* in AEM, and the resulting O* reacts with oxygen atoms in the lattice to form oxygen defects in the crystal. Hydroxide moves outward from water, filling the gap. Because OOH* is not generated in the LOM cycle, it breaks through the restriction of the ratio of OH* to OOH*, and is expected to break through the limitation of the volcano–shaper curve of the AEM mechanism and obtain a smaller OER overpotential.27 The involvement of lattice oxygen in the reaction often occurs in the case where the metal d orbitals are highly hybridized with the 2p orbitals of oxygen. When the metal d-band center is lower than the 2p orbital center of O, some of the 2p electrons of O above the Fermi energy level will fall back into the metal d orbitals to maintain the stability of the crystal, resulting in the formation of O1− species, and the adjacent O1− will further form [O2]n– species, triggering the LOM mechanism. In 2017, J. T. Mefford et al. discovered the RDS of the LOM mechanism when investigating different metal–oxygen covalent 18O-labeled chalcogenides by on-line electrochemical mass spectrometry (OLEMS).28 They showed that these chalcogenides undergoing the LOM mechanism had pH-dependent OER kinetics, indicating the presence of uncoordinated proton-electron transfer. Therefore, the RDS of the LOM mechanism was judged to be a deprotonation step.
Inspired by the different RDSs of AEM and LOM, Wang et al. proposed a new light-triggered OER mechanism.29 It was shown that the designed NR-NiOOH could switch between octahedral (NiO6) and square planes (NiO4), thus modifying the electronic states near the Fermi energy level for the conversion between LOM and AEM mechanisms (Fig. 1d). The RDS can be bypassed using this electron transfer pathway. This work provides a new idea for OER research to break through the drawbacks of the conventional OER mechanism and further enhance the catalytic performance.
3. Non-single-atom catalysts for the oxygen evolution reaction
The catalysts can be classified into monometallic and polymetallic categories according to the number of metal species. The components in polymetallic catalysts are composed of two or more metals. The use of multiple metal combinations can optimize the oxygen intermediate adsorption energy or increase the conductivity of the catalyst to achieve high efficiency.30–32 For example, Ru, Co,33 Fe and Ni are widely used in development of multi-metal catalysts due to their strong coupling effect with each other. Peng et al.34 used a high-pressure and high temperature (HPHT) technique to dope Fe with Ni in CoS2, and various types of characterization studies demonstrated that the co-doping of Fe and Ni promotes the formation of the highly active species Co1−xFexOOH on the surface, and at the same time, the valence transition of Fe can inhibit the oxidation of Ni ions, which ensures the stability of the catalyst.Yan et al.35 doped CoOx clusters on rutile TiO2 carriers by the thermally induced phase segregation (TIPS) process and subsequent O2 plasma treatment. Due to the strong interactions between CoOx and TiO2, Co–Ti cooperative catalytic centres are formed with suitable adsorption energies for oxygen intermediates, which greatly enhanced the catalyst's OER activity because of the high concentration of protons in an acidic environment. RuO2 has difficulty in removing protons from oxygen intermediates in acidic environments. Wen et al. synthesized a binary oxide rutile Ru5W1Ox catalyst by using a sol–gel method,36 while the addition of W could introduce strong Brønsted acid sites to the RuO2 lattice. Combining the experiments and calculation, the author demonstrated that the constructed W–Obri–Ru bridging oxygen sites increased the catalyst surface proton mobility (Fig. 2a and b), attributed to a rapid bridging oxygen-assisted deprotonation process, which speeds up the rate of the OER. Compared to commercial RuO2, Ru5W1Ox exhibited a 20-fold higher intrinsic OER activity and great stability over 550 h.
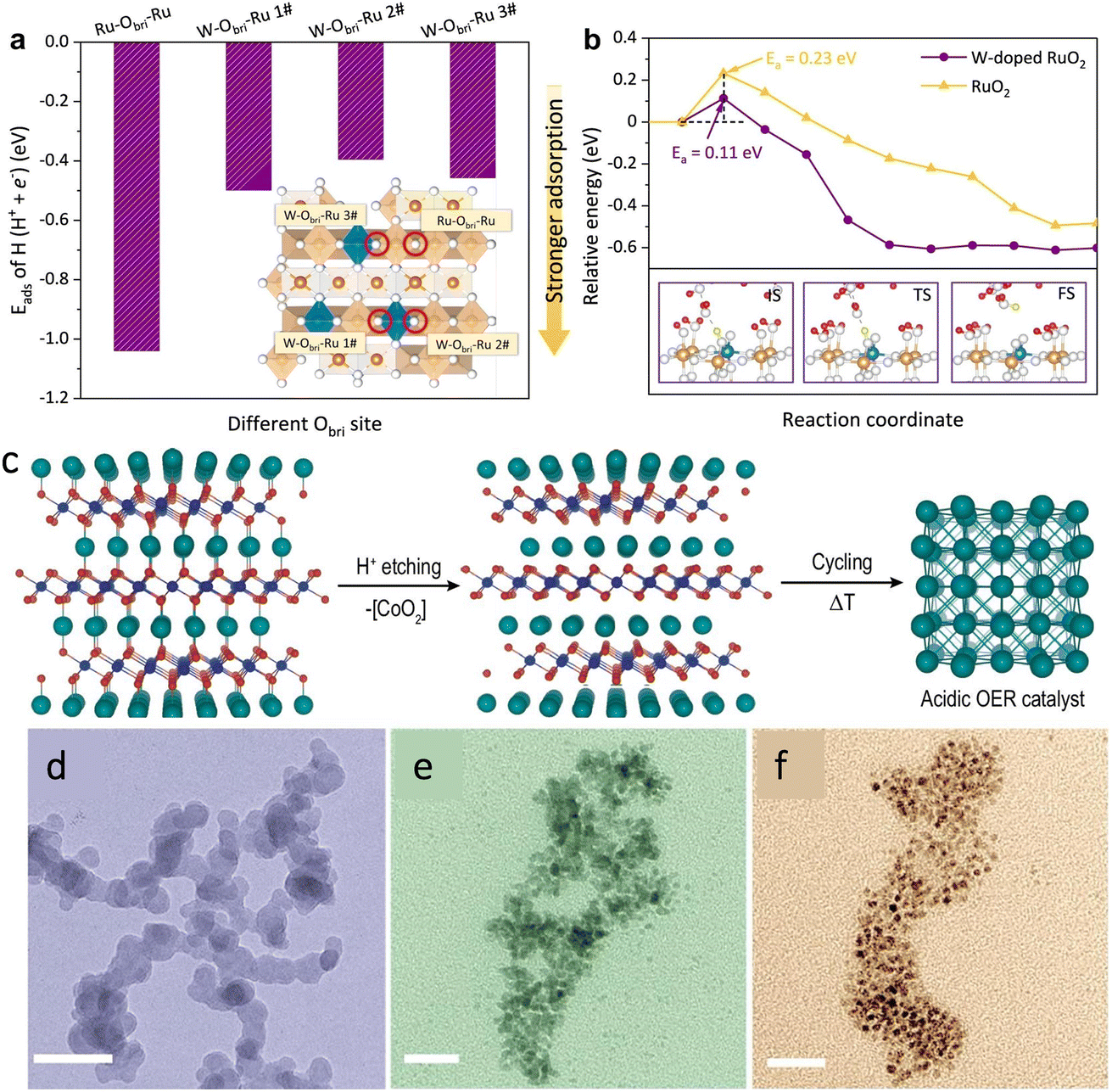 | ||
| Fig. 2 (a) The adsorption energy of the H atom on different Obri sites on the surface and (b) kinetic deprotonation barrier of OHbri on different solvent catalysts. Reproduced with permission.36 Copyright 2022, Springer Nature. (c) In 1MHClO4 solution, [CoO2] was separated from the PdCoO2 crystal by thermoelectric erosion technology to achieve high efficiency and high stability of palladium-based catalytic materials at a negative potential. (d–f) TEM of Pd nanoparticles at 25, 40 and 55 °C, respectively. Reproduced with permission.45 Copyright 2022, The Nature Publishing Group. | ||
Polymetallic systems have also been used to improve the stability of catalysts or to reduce the precious metal content that maintains catalytic activity.37,38 Nickel–iron layered double hydroxides (NiFe-LDHs) are one of the most active non-precious metal alkaline OER catalysts, but the stability of NiFe-LDHs is not satisfactory due to the extreme solubility of iron ions and the susceptibility of catalysts to lattice distortion during the OER process. Peng et al.39 introduced cationic vacancies into NiFe-LDHs to enhance the binding energy between iron and oxygen and alleviate the lattice distortion of NiFe-LDHs, which effectively suppressed the dissolution of the active sites during the OER process and greatly improved the stability of the catalysts. Li et al. reported an acidic OER catalyst with Mn doping in Co3O4.40 Along with the calculations and experiments, these results showed that Co2MnO4 not only maintained the same activity as Co3O4, but also prevented the dissolution of lattice oxygen showing great stability for over 1500 h at 200 mA cmgeo−2 due to the Mn–O bond formation. Owning to the tendency of Ru-based catalysts to undergo excessive oxidation and direct shedding of surface Ru metal, the stability of Ru-based OER catalysts in an acidic medium is not satisfactory.41 This is great importance to avoid excessive oxidation and metal shedding. Wu et al. reported a Ni-doping RuO2 OER catalyst.9 It was shown that Ni–RuO2 exhibited stable water electrolysis for >1000 h at 200 mA cm−2. According to the DFT calculations, the addition of Ni atoms not only increased the energy required for Ru shedding to improve the catalyst stability, but also promoted the active site density of the catalyst.
The loaded catalysts are the active component uniformly dispersed on a specially selected support. The support can provide an effective surface and suitable pore structure, so that the sintering and aggregation of active components can be greatly reduced. Furthermore, the mechanical strength, heat resistance and heat transfer properties of the catalyst can be enhanced. The reasonable structural design of the support and active site of the loaded catalyst can play an important role in enhancing the catalyst activity and stability.16,19,42 Previous studies have found that under steady-state high pressure, Pd can still maintain good stability at a weak pH, and its dissolution rate is only 0.03 ng cm−2 s−1, which is much lower than that of Ru and Ir, so it is a long-term OER catalyst with good application prospects. However, it is rarely used for water electrolysis because of the poor activity leading to the over-binding of oxygen-related intermediates.43,44 Through the regulation of oxygen binding energy, a OER catalyst with high activity and long-term stability is expected to be obtained. Peng et al. demonstrated a hierarchical Pd catalyst.45 By anchoring nano-Pd on a porous Pd backbone, the porous structure gave this layered Pd structure an ultra-high ECSA, which facilitated the catalytic reaction. Moreover, the size of nano-Pd is regulated by the control of the synthesis temperature (Fig. 2c–f). DFT calculations show that the reduction of the nano-Pd size will trigger lattice strain, which will reduce the binding energy of Pd to oxygen intermediates, decreasing the energy barrier of the PDS to improve the OER activity.
The support can not only serve as a scaffold to support the active component, but also play a role in enhancing the catalytic performance and stability of the catalyst through the spillover and strong interaction. Du et al. loaded RuO2 on CoOx as OER catalysts.10 CoOx, as a load, not only plays the role of a support, but also can replace RuO2 in the OER for preferential oxidation protecting the active sites. In addition, through theoretical simulation, we can form Ru/Co double sites with high activity on the surface of RuO2/CoOx, then regulate their binding energy with oxygen intermediate species, and then improve their OER performance.
4. Noble metal single-atom catalysts for the oxygen evolution reaction
Single atom catalysts (SACs) are formed by dispersing isolated unconnected atoms on a support. Due to their reduced size, SACs have several unique advantages over other nanoparticle catalysts.46,47 The monodisperse active sites in SACs can greatly enhance the utilization of metals, reducing the need for precious metals. This change also eliminates the gap in activity between noble and non-noble metals. The achievements in recent years in noble and non-noble metal SACs will now be discussed separately.4.1 Ru single-atom catalysts for the oxygen evolution reaction
Finding suitable supports is important to improve the stability of SACs. Due to the high complexity of the ligand sites and the supporting coordination environment of SACs, it has been a major challenge to precisely create active sites on the supports. Xiao et al.48 reported a method for electrochemical cation exchange in situ. The poor cycling stability and susceptibility to disproportionation of MnO2 was exploited by electrochemically cycling it to controllably construct Mn vacancies for Ru single-atom anchoring, and the number of cycles can be used to control the content of Ru. The process can realize precise regulation of metal ion defects in MnO2 and the Ru atom coordination environment. Combining the good OER catalytic activity of Ru and the strong synergistic effect of Ru and MnO2, the catalysts showed good OER performance and stability.Different modification methods have been studied to achieve the synthesis of catalysts with coordination environments and electronic configurations. Among these strategies, coordinated engineering is considered as an effective method for electronic structure adjustment. By precisely controlling the type and number of ligand atoms, the optimal absorption energy of the intermediate can be obtained. In addition to the popular coordination of O and N atoms, the search for other ligand atoms with different electronegativities has started in recent years. Chen,49 inspired by the unique electron-donatinf ability of halogen atoms, designed a RuCl2N2 coordination configuration catalyst anchored in the ZIF-67 template cavity. Since Ru atoms have a strong interaction with Cl, the 3d state peak of RuCl2N2/C splits above the Fermi level (Fig. 3a), so Ru is more favorable for electron transfer and promotes the catalytic reaction. DFT calculations showed that the free energy of RuCl2N2 was reduced from 1.74 V to 1.07 V compared to that of conventional RuN4 (Fig. 3b). The overpotential of Ru–Cl–N SACs is much lower than that of RuN4 SAC and IrO2. In addition, Ru can not only act as a catalytic site in the process of the catalytic reaction, but also change the coordination environment of the catalyst. Zheng et al.33 proposed that a Ru single atom is used to construct Ru–Co coordination Ru/LiCoO2 to realize the regulation of electronic properties of Ru/LiCoO2. First, he replaced Co with a single Ru atom in layered LiCoO2, and Ru was coordinated with 6 O atoms to make Ru–Co/LCO, which was then transformed into two-dimensional Ru–Co/ELCO by ultrasonic stripping. DFT calculation results show that the introduction of Ru atoms can induce the transfer of Co 3d and O 2p orbitals to the Fermi energy level (Fig. 3c) for enhancing Co–O covalency and facilitating the optimization of oxygen binding energy at the Co site, ultimately achieving a lower energy barrier for the OER. Electrochemical detection shows good OER activity of Ru–Co/ELCO (Fig. 3d). In addition, under the conditions of 50 and 100 mA cm−2, Ru–Co/ELCO exhibits good stability and no obvious aging phenomenon. This is primarily attributed to the strong structural stability, morphology and structure of Ru–Co/ELCO materials. Similarly, Mu et al.50 reported Ru doping of FeCo-LDH catalysts to improve the catalytic activity. Through DFT theoretical simulation and AC-STEM analysis, it is found that the local substitution of Ru in Fe–Co in FeCo-LDHs breaks the interface symmetry of FeCo-LDH. This symmetry breaking formed a strong coupled Schottky interface, which changed the electronic structure of the material, and a charge transfer region is constructed on the surface of heterogeneous atoms; thus the OER and overall water splitting electrochemical performance significantly improved. The RuxSACs@FeCo-LDH catalysts demonstrated remarkably low overpotentials for the OER, reaching 194 mV and 246 mV at 10 mA cm−2 and 1000 mA cm−2, respectively. Additionally, the catalysts exhibited high stability beyond 1000 hours at 1000 mA cm−2, surpassing commercial RuO2.
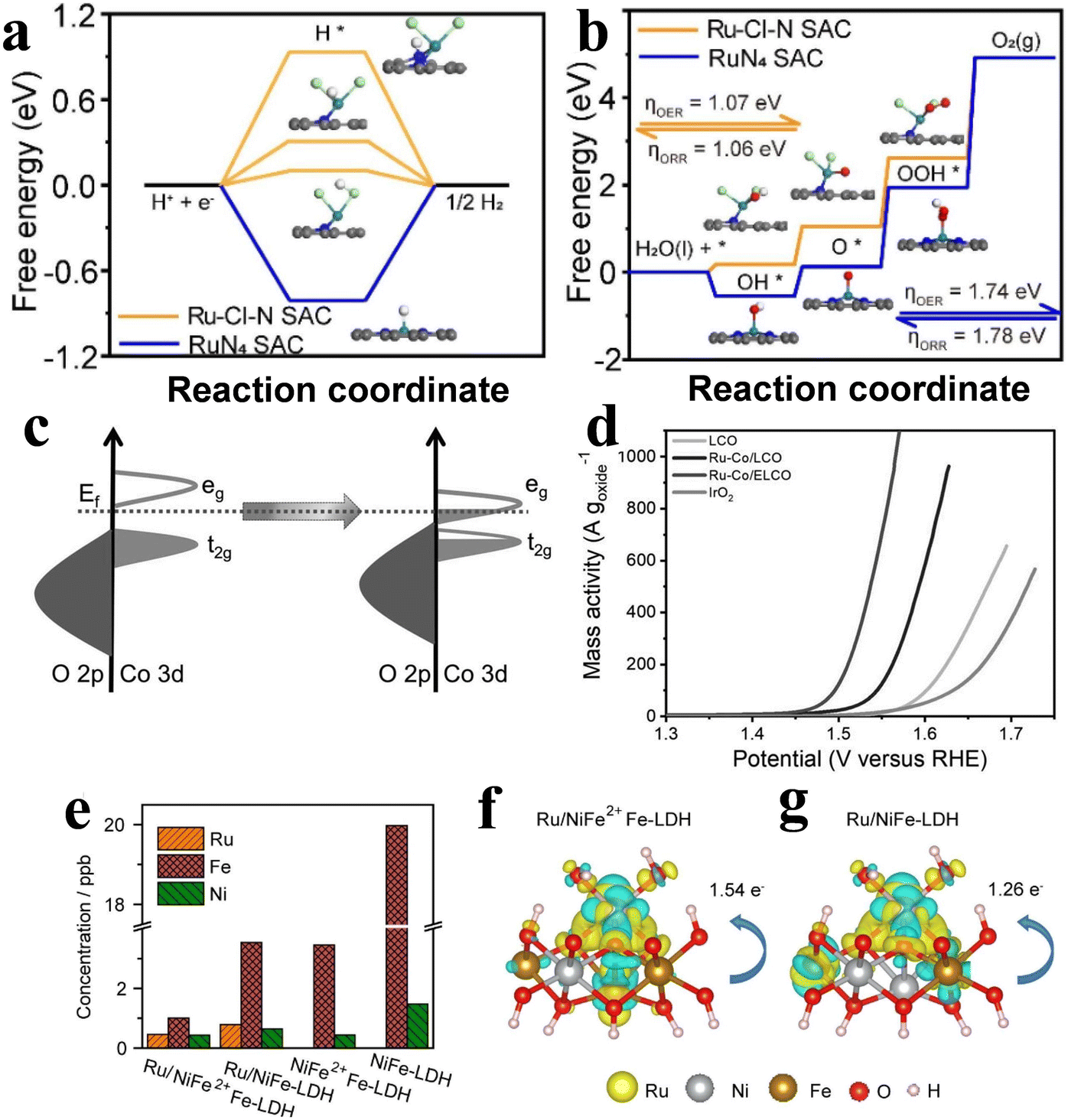 | ||
| Fig. 3 (a) Diagram of free energy for Ru–Cl–N SAC (orange) and RuN4 SAC (blue) in the HER. (b) Diagram of free energy for Ru–Cl–N SAC (orange) and RuN4 SAC (blue) in the OER and ORR. Reproduced with permission.49 Copyright 2022, Elsevier. (c) A qualitative primary energy spectrum is used to explain the electronic structure changes in Co and O. (d) LSV curves based on the mass of metal oxides. Reproduced with permission.33 Copyright 2022, John Wiley and Sons Ltd. (e) Difference in charge density in the comparison of the dissolution of Ru, Ni and Fe in electrolyte for (f) Ru/NiFe2+Fe-LDH and (g) Ru/NiFe-LDH. Yellow and blue represent the charge accumulation and depletion regions, respectively. Reproduced with permission. Reproduced with permission.51 Copyright 2022, Elsevier. | ||
When Ru catalysts are continuously operated at high oxidation potential for a long time, it is difficult for the Ru-based OER catalysts to maintain their stability because of their high oxidation state, and they can easily dissolve into the electrolyte. Great efforts have been made to improve catalyst stability by providing electron donors to lower the Ru valence state. It is well known that NiFe-LDH has strong OER activity in alkaline media, and Ru was often introduced into the surface of NiFe-LDH to increase its adsorption energy for oxygen and enhance its catalytic performance. However, during the electrooxidation process, Ru and Fe readily form high valence states and dissolve, which seriously affects the stability of the catalyst. Duan et al.51 anchored a single Ru atom on Fe2+ ion-doped NiFe layered double hydroxide, and XPS and XANES results showed that Ru/NiFe2+Fe-LDH has a relatively low Ru valence state. DFT results show that Ru and Fe2+ have strong electronic interaction, which can effectively reduce the valence state of Ru, improve the binding ability of the Fe–O bond and inhibit the precipitation of iron. In addition, the electron transfer between Ru and Fe increased, which significantly improved the catalytic performance and stability of Ru and NiFe-LDH carriers (Fig. 3e–g). The results of the alkaline medium test showed that its performance is better than that of most Ru and NiFe-based alkaline OER catalysts. In addition, Ru/NiFe2+Fe-LDH showed good stability, and its overpotential only increased by 16 mV after 100h at 100 mA cm−2.
4.2 Rh single-atom catalysts for the oxygen evolution reaction
The selection of a suitable reaction matrix is the key to improve the performance and stability of SACs. Carbon-based carriers are the most commonly used catalytic carrier at present, but their electrical conductivity is low, so they can only be used as a carrier for a catalytic reaction. Recently, researchers have shown that the OER performance can be effectively improved by introducing defect-rich metal oxides. By using a simple cation exchange strategy, Xu et al. successfully prepared Rh SAC-CuO NAs/CF.52 They showed that the addition of Rh could modulate the oxygen intermediate adsorption energy of CuO NAs/CF (Fig. 4a), and 6.8 wt% Rh SAC–CuO NAs/CF exhibited good OER catalytic performance with an overpotential of 197 mV at 10 mA cm−2. Similarly, Gu et al.53 anchored Rh single atoms on phosphorus-doped Co3O4 nanosheet arrays, and the addition of phosphorus enhanced the grain boundary conductivity of Co3O4 for further promoting the electron transport between Rh atoms, thus improving the electrocatalytic activity. EIS tests revealed that both the solution resistance and charge transfer resistance of P-doped Rh SAC-Co3O4 became smaller. LSV measurements showed that P-doped Rh SAC-Co3O4 NAs displayed much lower overpotential (Fig. 4b and c).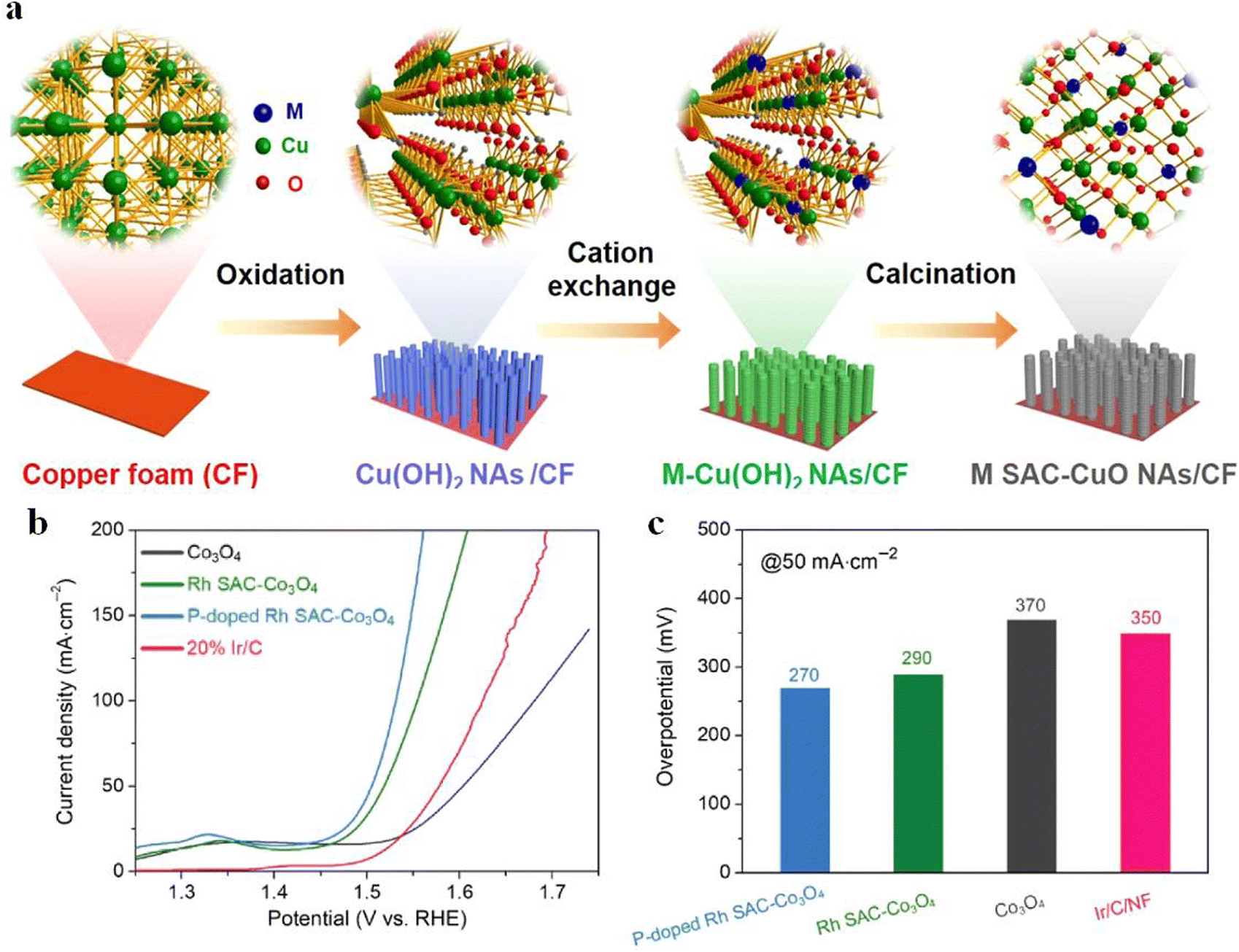 | ||
| Fig. 4 (a) Schematic representation and corresponding atomic structure of the products. Reproduced with permission.52 Copyright 2020 American Chemical Society (b) OER polarization curves and (c) overpotentials at 10 and 50 mA cm−2. Reproduced with permission.53 Copyright 2022, Springer Nature. | ||
4.3 Pt single-atom catalysts for the oxygen evolution reaction
Platinum (Pt) shows low OER efficiency at the anode due to its excessive binding energy to oxygen intermediates, and can be oxidized to soluble high-valent Ptx >4 species (e.g. PtO3) in an over-oxidizing environment with an anodic potential >1.4 V. This triggers the dissolution of the active Pt species, which leads to a drastic decrease in catalytic activity. Therefore, the research on this problem mainly focuses on how to increase the binding energy between Pt and oxygen and obtain better OER activity. The phosphomolybdic acid (PMA) cluster is a very good material for transition metal (TM) elements. By using DFT calculations, Talib et al.21 systematically investigated the electrocatalytic performance of a series PMA cluster-supported SACs for the OER(Fig. 5a). By calculating the ΔG values for each step of the OER, the overpotential values of the RDS of different TM-PMA SACs were derived, and the comparison revealed that the Pt1/PMA catalysts exhibited excellent OER activity, with the third step (O* to OOH*) as the RDS with a value of 0.49 V. The new two-dimensional materials composed of antimonene have shown high thermodynamic stability, electron mobility and appropriate band gap. Lu et al.54 embedded different TM atoms into a single Sb vacancy of TM@Sb to construct SACs. Calculating the Gibbs free energy differences of the adsorbed intermediates, using ΔGOOH* − ΔGO* as the descriptor of ηOER (Fig. 5b and c), the constructed volcano curves exhibited the electrocatalytic activity of different TM@Sb monolayers towards the OER (Fig. 5d). The results show that Pt@Sb monolayers have the smallest ηOER (0.48 V) showing its potential for the OER.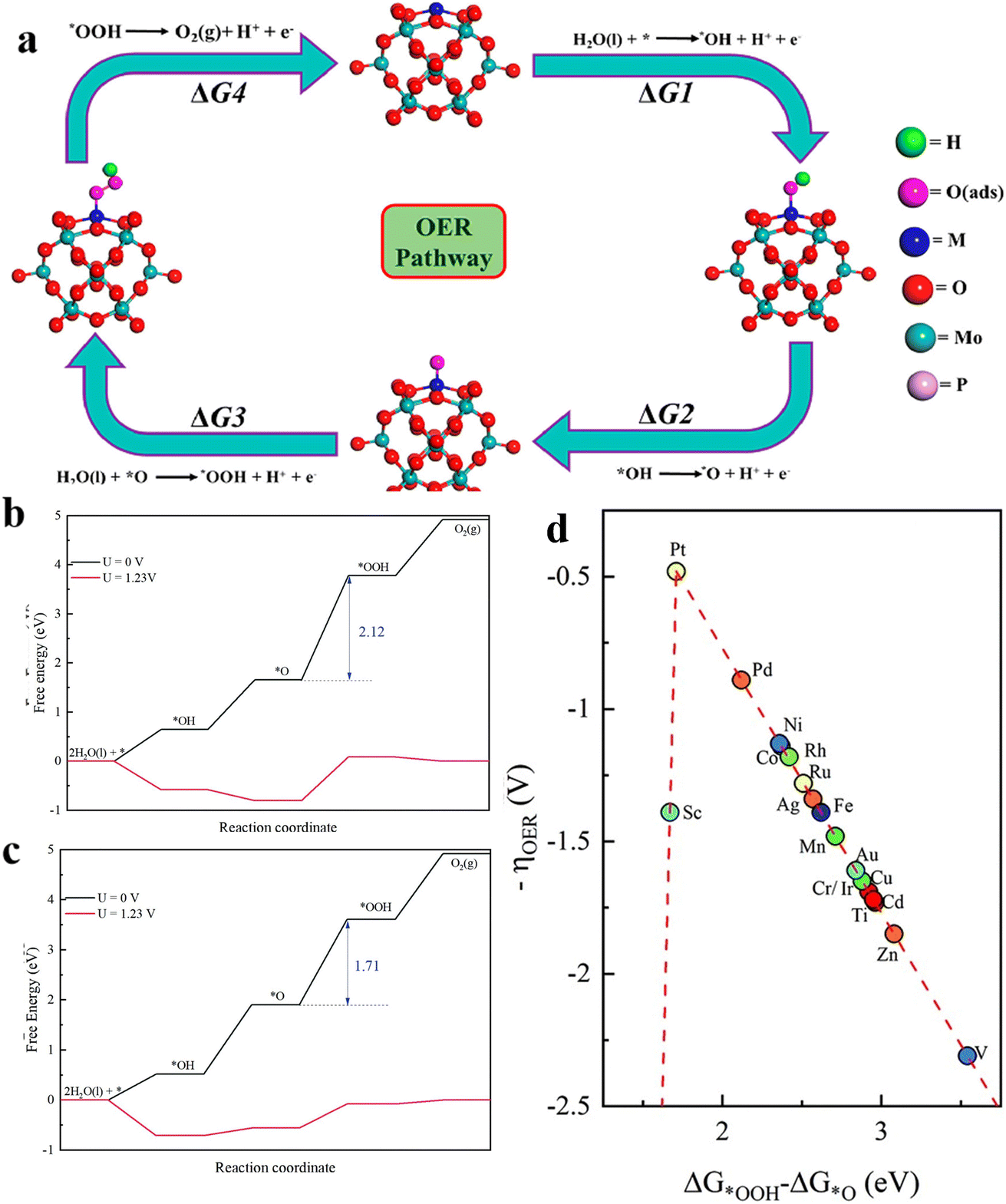 | ||
| Fig. 5 (a) Schematic of the 4e− OER pathway of the 4H site via the M1/PMA cluster, reproduced with permission.21 Copyright 2021, American Chemical Society. (b) Free energy diagrams of basic steps of the Pd@Sb monolayer and (c) Pd@Sb monolayer at different potentials. (d) The volcano curve between negative OER overpotential and ΔGOOH* − ΔGO* for TM@Sb monolayers. Reproduced with permission.54 Copyright 2021, Royal Society of Chemistry. | ||
MXenes are a new type of two-dimensional crystals composed of early transition metal carbides, nitrides and carbon-nitrides. They are well-suited as SACs due to their excellent electronic conductivity, high stability thermal conductivity, and large ion storage capacity. In addition, their surfaces also contain hydrophilic functional groups such as O and F, which makes them have both metal and hydrophobic conductivity and can significantly enhance both the catalytic activity and stability. In recent years, SACs prepared from Pt SAs and MXenes have shown great promise as OER catalysts, such as a Pt-doped Nb2CF2 catalyst designed by Kan et al. showing high OER performance,55 but the reason for the excellent performance of these Pt SAC-MXene catalysts is not well understood. Kan et al.56 conducted a comprehensive study of the OER performance of 26 representative Pt SAC-MXene catalysts using first-principles calculations to investigate their conformational relationships. Through the Bader charge analysis and the determination of different charge state concentrations, it was found that Pt SA obtained electrons from the Mxene substrate and transferred them to the intermediate products. Therefore, the more electrons Pt SA obtains, the faster the charge transfer and the better the catalyst OER performance. It is found that the number of charges on Pt SAs is related to the bond length between Pt and the submetal. Compared to the F-terminal MXenes, Pt SAs gain fewer electrons from the O-terminal MXenes, which is due to the fact that the 2p orbitals of O have one more set of lone pair electrons than F. O can accommodate more electrons than F, resulting in Pt gaining fewer electrons. In addition, the catalytic performance is also related to the Pt-5d state and work function of the SACs. These findings provide guidance for future Pt SAC-MXene catalysts. It is important to study the development of O-terminal Pt SAC-MXenes. Kan et al.57 proposed a strategy to leave Nb (or Cr) submetal in the outer layer and introduce submetal M* whose work function is lower than that of Nb (or Cr) in the middle layer to construct ordered MXenes. The addition of the second submetal can improve the charge density of Pt, and the intermediate product can gain more electrons during the reaction process, thus improving the efficiency of MXenes.
5. Non-noble metal single-atom catalysts for the oxygen evolution reaction
Although noble metals show excellent catalytic performance and stability in the OER, it is difficult to realize large-scale application due to their high cost and low abundance, which has forced researchers to search for non-noble metal-based OER catalysts. Since SACs reduce the size to the atomic level and provide a single active site with high activity and selectivity, they are expected to eliminate the activity difference between noble and non-noble metals. Recent studies have found that some non-noble metal SACs have been widely used in electrode materials because of their low cost, rich sources and strong corrosion resistance.58 In particular, because of their multivalent oxidation states (M2+/3+/4+ states have been shown to be the active sites of the OER), they are excellent candidates for the OER. The performance of the synthesized non-noble metal SACs may even be comparable to the performance of commercial noble metal catalysts through operations such as geometry improvement of non-noble metal catalysts, optimization of the active site coordination environment and enhancement of metal loading.5.1 Co single-atom catalysts for the oxygen evolution reaction
Sun et al.59 reported a sea urchin-like nanotube hierarchy (UNT Co SAs/N–C) cobalt single-atom OER electrocatalyst by using the “sacrificed-template” method (Fig. 6a). Owing to the unique metal node properties of MOFs, the metal components immobilized on the stable carbon matrix can achieve high quality single-atom dispersion during the high temperature pyrolysis treatment. Furthermore, according to the wavelet transformed EXAFS (WT-EXAFS), it was found that single Co atoms exist in the UNT SAs/N–C as CoN4 local structure, and the electron transfer of this structure was more easily realized, which made UNT Co SAs/N–C exhibit good OER catalytic properties with a Tafel slope of only 70 mV dec−1. Li et al.60 used atomic layer deposition (ALD) for synthesizing Co1Pt1/NCNS. The catalysis of Pt greatly enhanced the Co(Cp)2 dissociation on NCNS, allowing Co to be stabilized as a single atom on NCNS, and this synthetic strategy can also be extended to the preparation of Fe and Ni SACs.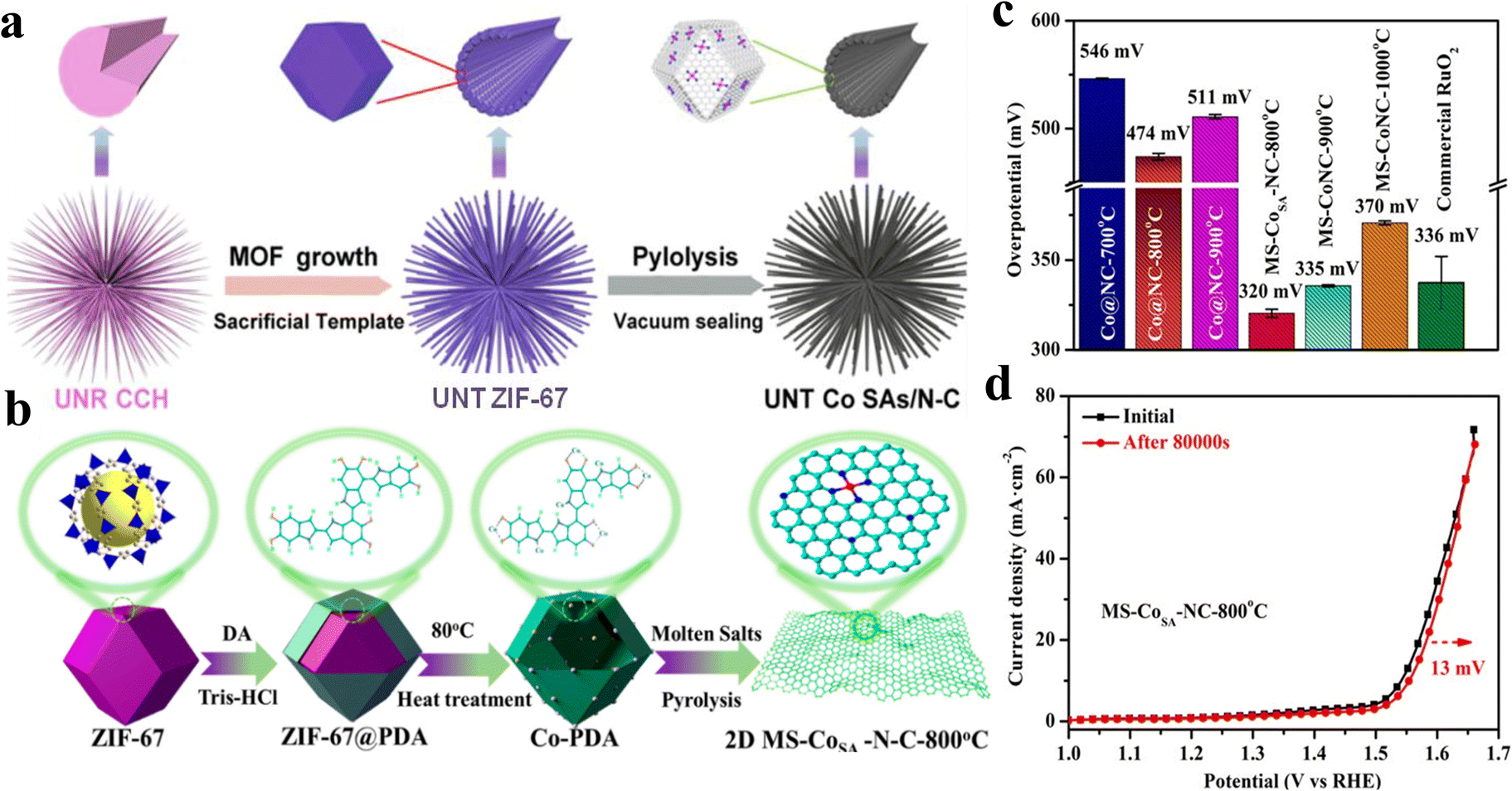 | ||
Fig. 6 (a) Schematic illustration of the synthesis method of UNT Co SAs/N–C catalysts. Reproduced with permission.59 Copyright 2019, Elsevier. (b) Principle of preparation of cobalt supported by metal nano-porous carbon by the molten salt method. (c) Comparison of the overpotential of MS-Co-N-C and Co@N–C, with a preparation temperature of 800, 900, and 1000 °C and commercial RuO2. (d) OER polarization curves before and after 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s were obtained for MS-CoSA-N-C-800 °C. Reproduced with permission.62 Copyright 2022, American Chemical Society. 000 s were obtained for MS-CoSA-N-C-800 °C. Reproduced with permission.62 Copyright 2022, American Chemical Society. | ||
The structure of the substrate has a great influence on the catalyst OER performance. Xie et al.61 synthesized a self-supporting nanosheet array called SS-Co-SAC. Because of its ordered arrangement, SS-Co-SAC has large surface area and high distribution of active sites, and close connection with the substrate and arrays is beneficial to promote electron transfer. Wang et al.62 employed various molten salt-assisted pyrolysis strategies for the hollow dodecahedral-like Co-PDA complexes by exfoliation to obtain ultrathin porous carbon-loaded Co single-atom catalysts (MS-CoSA-N-C-800 °C) (Fig. 6b). The ultrathin, porous carbon matrix produced by molten salt-assisted pyrolysis not only promotes the full exposure of metal atoms and removal of nanoparticles, but also achieves effective modulation of the metal-centered electronic structure by generating more defect sites. The modulated CoN4-GN sites promote the deprotonation of OH* in the OER process, which greatly enhances the OER performance. The overpotential is 320 mV at 10 mA cm−2 (Fig. 6c) and the Rct is 7.2 Ω, which is already smaller than that of commercial catalysts. The current density remained essentially unchanged after a 100000 s continuous stability test (Fig. 6d), indicating the superior durability of the MS-CoSA-N-C-800 °C catalysts.
The establishment of structure-effect relationships is important for OER catalyst study. Lv et al.63 developed an efficient screening process for SACs. According to this rule, the d-band center can describe the bonding ability of different catalysts to reactants. In addition, the authors further established the correlation between the required potentials (ΔGHO*, ΔGO* and ΔGHOO*) and d-band center for each basic step of the OER by investigating the d-band center of TM atoms. Adjusting the ratio of TM atoms can tune the d-band center of the catalyst to achieve precise control of the catalytic activity. Li et al.64 designed Co-SACs as a model system on a metal TaS2 monolayer with tunable loading and established the correlation between the active site spin state and the catalytic performance of the OER by varying the catalyst isolated CoHS, CoHS–CoTa and CoHS–CoHS site ratios to achieve changes in the active site spin density (Fig. 7a–c). The experiments and calculation results have shown that the neighboring CoTa can significantly increase the spin concentration of CoHS, thus improving its OER performance. In contrast, the neighboring CoHS will excessively increase the spin concentration of the CoHS center and make the O* bond between the O* and the CoHS center too large, thus affecting the subsequent catalytic process (Fig. 7d and e), affecting the catalytic performance of OER. This work indicates that the spin concentration of a single active site can regulate the OER performance, suggesting using spin concentration to characterize the OER performance, which is expected to have a certain guiding significance for further catalyst studies.
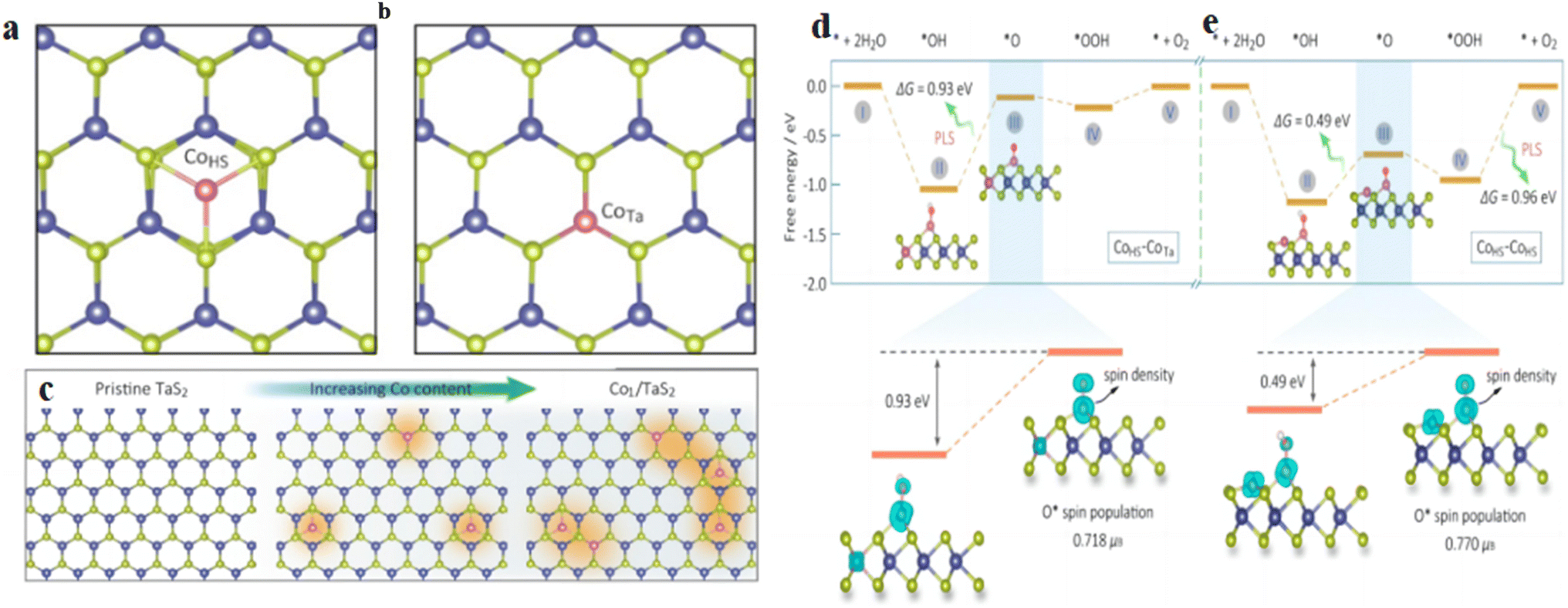 | ||
| Fig. 7 (a and b) Magnetic single-Co-atom catalysts (Co1/TaS2) with a controllable density of CoTa and CoHS sites. (c) Theoretical study for the OER of the spin-activity correlation on Co1/TaS2. Structural evolution of CoTa and CoHS sites in a TaS2 lattice with increasing Co loading. (d and e)Schematic of the free energy of the OER at the CoHS site with the CoTa and CoHS atoms in the vicinity. Reproduced with permission64 Copyright 2021, American Chemical Society. | ||
5.2 Fe single-atom catalysts for the oxygen evolution reaction
Du et al.65 devised a restricted recrystallization self-templating method to prepare nitrogen–carbon-doped hierarchical porous Fe SAC using precursors Fe(NO3)3, PVP and melamine(Fig. 8a), where PVP can restrict iron nitrate recrystallization into nanoparticles, and melamine can promote the further transformation of PVP into porous nitrogen-doped carbon with a large number of micropores and macropores. This method does not need additional templates for avoiding interference with the active site, and reduces the catalyst fabrication cost. The Fe SACs is rich in Fe–Nx sites and has a good mass/charge transport channel. The OER current density reaches 40 mA cm−2 at 1.8 V (Fig. 8b), which exceeds that of commercial RuO2. Zhong et al.66 invented a simple method allowing large-scale preparation of high-performance SACs using natural wood as the raw material. By pretreating with Lewis's acid FeCl3, the cellulose and hemicellulose in the wood underwent hydrolysis. The material not only has a multi-stage pore structure, but can also introduce dispersed atomic Fe–N sites into the multi-stage pores. The distribution of SACs in multi-stage pores is more uniform, thus improving the activity and durability of the OER.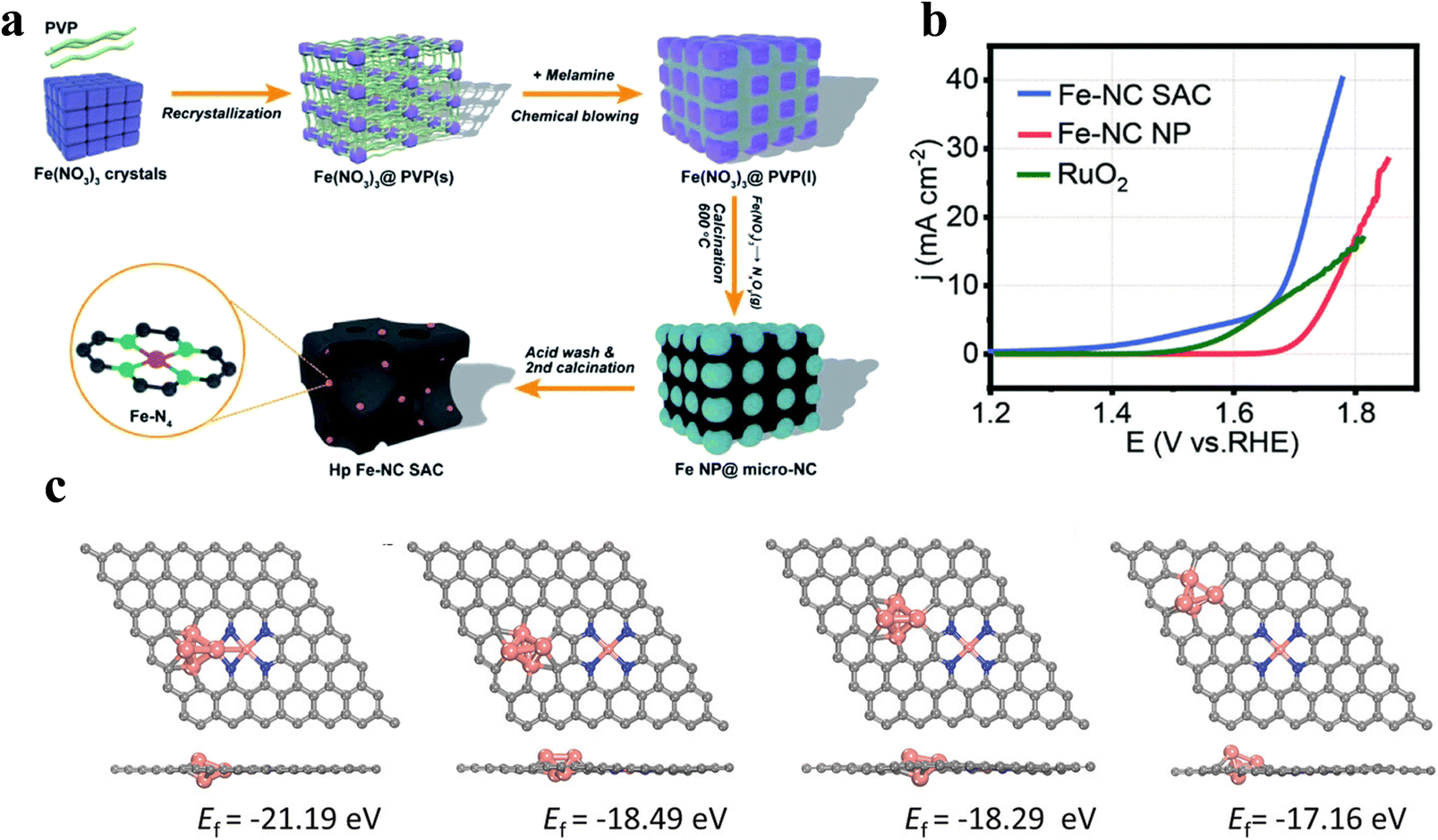 | ||
| Fig. 8 (a) Self-template synthesis process of the Fe-NC SAC. (b) The Fe-NC SAC, Fe-NC NP and Pt/C OER polarization curves of the OER at 1600 rpm; Reproduced with permission.65 Copyright 2020, Royal Society of Chemistry; (c) the formation energy of different configurations of Fe clusters and Fe–N4 sites. Reproduced with permission.67 Copyright 2022, Wiley-VCH Verla. | ||
The FeN4 coordination structure is a hot research topic for Fe single-atom catalysts because of its suitable binding strength of oxygen intermediates. It has been found that the catalytic performance of FeN4 sites is better when they are located at the edge than in the plane. However, the graphitization of carbon in edge site dominant samples is usually poor, which leads to poor electrochemical stability. Xiao et al.67 reported a new self-sacrificing template method to integrate FeN4 edge sites into highly graphitized layer graphene. DFT calculations showed that Fe clusters have a stabilizing effect on FeN4, and the FeN4 sites near the cluster are formed more easily (Fig. 8c). Therefore, the synthesis promoted the preferential deposition of FeN4 groups close to Fe clusters by using an extremely excessive amount of Fe precursors, and finally removed Fe clusters to form abundant FeN4 edge sites, which facilitated the mass/electron transfer and thus promoted the reaction kinetics.
5.3 Ni single-atom catalysts for the oxygen evolution reaction
Recent studies have found that the metal coordination environment in the SAC system and the electronegativity of the adjacent components have an important impact on its catalytic performance. Previous studies reported that Ni SACs were generally stabilized by metal or covalent bonds to single atoms. Furthermore, this leads to difficulties in the reaction of electrons with oxygen in the active site, resulting in their unsatisfactory OER catalytic activity.68,69 In recent years, scholars have found that the construction of SACs by using M–O complexes can effectively block the chemical bonds between metals, accelerate electron transfer, and enhance their oxygen evolution performance. However, the M–O bond is very weak, so it is very challenging to prepare an oxygen coordination catalyst with high stability. Xu et al.70 developed a bimetallic ion adsorption strategy. By using this strategy, the Ni–O–G SACs were successfully fabricated in two-dimensional ultrathin conductive graphene. FT- EXAFS tests revealed that Ni single atoms exist in the Ni–O coordination structure, which makes Ni exist in a highly oxidized state and facilitates the attraction and electron transport of hydroxide, and thus the free energy needed in the process of oxygen evolution is reduced accordingly. According to the electrochemical tests, the Ni–O–G SACs showed good OER catalytic ability. At the same time, Li et al.71 prepared Ni–O–G SACs by using NaCl as the template. NaCl provides a good template for constructing a three-dimensional hollow skeleton and two-dimensional layered materials. This project intends to use this technology to synthesize a series of 3D porous frameworks and 2D ultra-fine flake-like nanomaterials to achieve uniform dispersion of single Ni elements. Under the condition of 10 mA cm−2, the overpotential is 224 mV, and the Tafel slope is only 42 mV dec−1.Cations can regulate not only the coordination environment by constructing Ni–O bonds, but also can adjust the local electronic structure of the catalyst. Xu et al.72 prepared Ni and Fe bimetallic capsules on the surface of graphene by using ion adsorption and GO as the template (Fig. 9a). The author showed that the overpotential of Ni4Fe1–O–G SACs was as low as 247 mV with a TOF value of 1.35 compared to that of Ni–O–G SACs alone (329 mV) and Fe–O–G SACs (384 mV) at 10 mA cm−2. Similarly, Luo et al.73 designed atomically dispersed Fe and Ni anchored in nitrogen-doped carbon (FeNi–SAs@NC). It was found that compared with Ni–SAs@NC, the coordination environment and electronic structure of the surface changed obviously after the addition of Fe atoms, adjusting adsorption energy of the oxygen-containing intermediates and promoting the reaction kinetics (Fig. 9b–e).
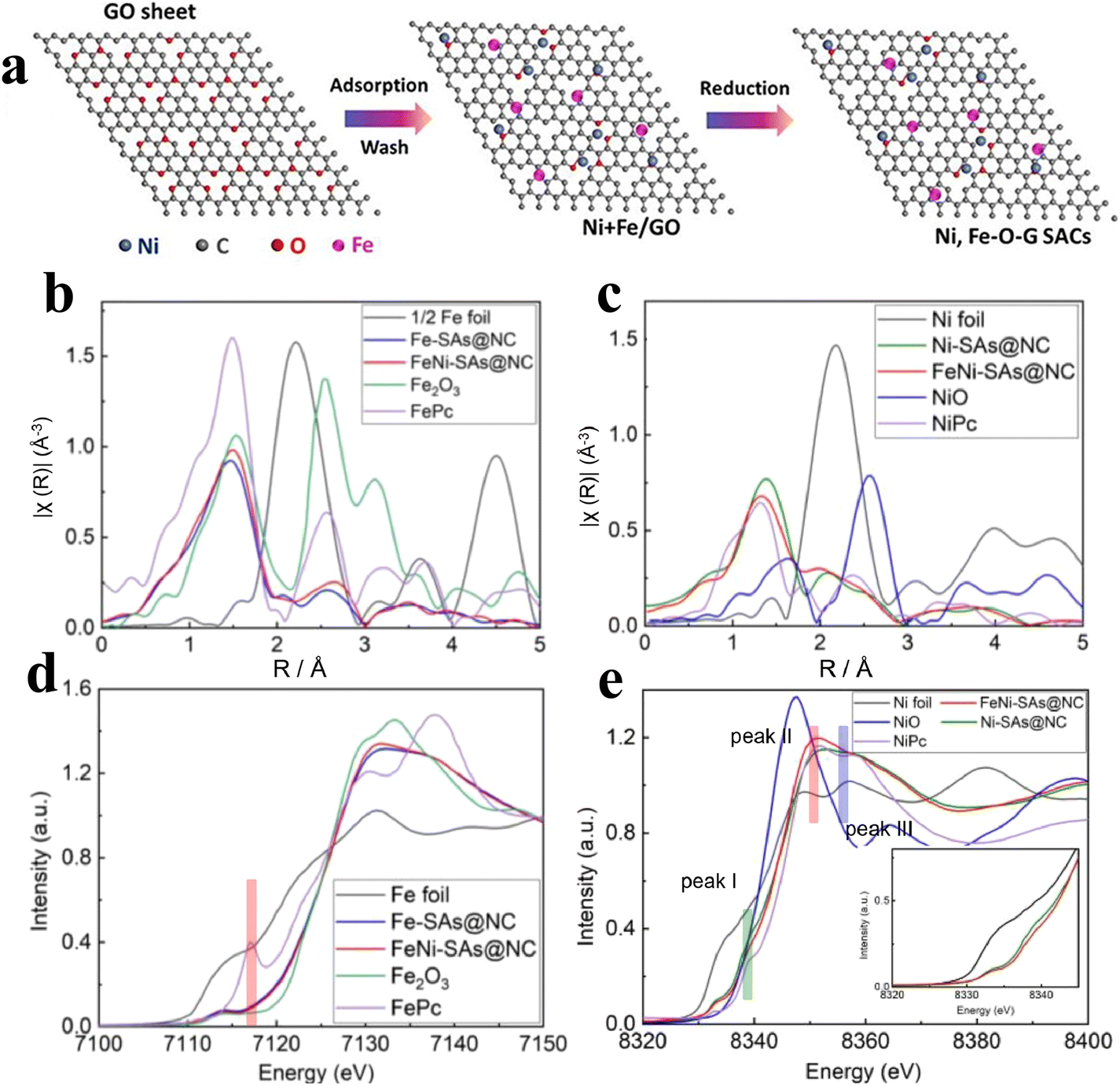 | ||
| Fig. 9 (a) Synthesis process of the Ni–O–G SACs. Reproduced with permission.72 Copyright 2019, Elsevier. (b) Fe K-edge FT-EXAFS spectra. (c) Ni K-edge FT-EXAFS spectra. (d and e) XANES at the Fe K-edge and Ni K-edge. Reproduced with permission.73 Copyright 2022, Elsevier. | ||
In order to improve the binding energy of catalysts to oxygen intermediates and the conductivity, non-noble metal SACs have been greatly promoted by improving the substrate structure, doping to change the ligand environment of active site and searching for conformational relationships. However, there is still a considerable gap compared to noble metal catalysts.
In order to clearly summarize the review, we compiled Table 1 to summarize the overpotential, Tafel slope and stability of the OER catalysts mentioned in the paper.
| Catalyst | Electrolyte | Overpotential/mV@10 mA cm−2 | Tafel slope/mV dec−1 | Degradation | Ref. |
|---|---|---|---|---|---|
| Fe,Ni–CoS2 | 1 M KOH | 242 | 35 | 8%@1.5 V after 500h | 34 |
| Co–TiO2 | 1 M KOH | 390 | 65 | 2.2 mV h−1 @10 mA cm−2 | 35 |
| Ru5W1Ox | 0.5 M H2SO4 | 227 | 42 | 0.014 mV h−1 @10 mA cm−2 | 36 |
| NivacFevac-LDH | 1 M KOH | 230 | 52 | 2.19% @1.6V after 1000CV | 39 |
| Co2MnO4 | 0.5 M H2SO4 | 290 | — | 1500 h at 200 mA cmgeo−2 (LT) | 40 |
| Ni–RuO2 | 0.1 M HClO4 | 214 | 42 | 0.098 mV h−1 @10 mA cm−2 | 9 |
| Pd-55 | 0.1 M HClO | 196 | 51 | 0.066 mV h−1 @10 mA cm−2 | 45 |
| RuO2/CoOx | 1.0 M PBS | 242 | 70 | 0.003 mV h−1 @10 mA cm−2 | 10 |
| Ru–MnO2 | 1.0 M KOH | 240 | 48 | Almost unchanged after 2000 CV@10 mA cm−2 | 48 |
| Ru–Cl–N SAC | 1.0 M KOH | 233 | 24 | — | 49 |
| Ru–Co/ELCO | 1.0 M KOH | 247 | 49 | Almost unchanged after 190 h@50 mA cm−2 | 33 |
| Rux SACs@FeCoLDH | 1.0 M KOH | 194 | 25 | 10 mV after 20000 CV@1 A cm−2 |
50 |
| Ru/NiFe2+Fe-LDH | 1.0 M KOH | 194 | 36 | 0.16 mV h−1 @100 mA cm−2 | 51 |
| Rh SAC-CuO NAs/CF | 1.0 M KOH | 197 | 71 | — | 52 |
| P-doped Rh SAC-Co3O4 | 1.0 M KOH | 268@50 mA cm−2 | 86 | 4.2% @1.5 V after 12h | 53 |
| UNT Co SAs/N–C | 1.0 M KOH | 380 | 70 | — | 59 |
| Ni1Pt1/NCNS SS-Co-SAC | 1.0 M KOH | 370 | 58 | Almost unchanged after 10 h@10 mA cm−2 | 60 |
| SS-Co-SAC | 1.0 M KOH | 348 | 94 | — | 61 |
| MS-CoSA-NC-800 °C | 1.0 M KOH | 320 | 94 | 0.46 mV h−1@10 mA cm−2 | 62 |
| Fe-NC SAC | 1.0 M KOH | 440 | 114 | 20 mV after 3000 CV@10 mA cm−2 | 65 |
| SAC-FeN-WPC | 1.0 M KOH | 400 | — | — | 66 |
| Fe/N-G-SAC | 1.0 M KOH | 370 | 73 | 6 mV after 2000 CV@10 mA cm−2 | 67 |
| Ni–O–G SACs | 1.0 M KOH | 328 | 84 | Almost unchanged after 50 h@10 mA cm−2 | 70 |
| Ni–O–G SACs | 1.0 M KOH | 224 | 42 | — | 71 |
| Ni4Fe1–O–G SACs | 1.0 M KOH | 247 | — | Little change after 48 h@20 mA cm−2 | 72 |
| FeNi–SAs@NC | 1.0 M KOH | 298 | 74 | No change after 1000 CV@10 mA cm−2 | 73 |
6. Outlook and conclusion
The development of new catalytic materials for the OER is the key to the realization of produce hydrogen. Different catalytic systems, such as polymetallic catalysts, loaded catalysts, metal oxides, and SACs, have their unique features and advantages as OER electrocatalysts, among which SACs were paid much attention by scholars in recent years due to their high metal utilization, isolated active sites, and unique electronic structures of active sites. At present, some promising results have been obtained in the study of noble/non-noble metal SACs in the OER. However, the practical applications also put higher requirements on the activity of these catalysts. For example:(1) For noble metal catalysts, how to reduce the use precious metals on the premise of ensuring efficient catalysis of precious metals?
(2) For non-noble metal catalysts, they are mainly focused on how to improve their catalytic activity. Increasing the loading of metal atoms, increasing the number of active sites and further optimizing the geometry of SACs to maximize the utilization of active centers lead to an improvement in performance.
(3) Not only are the high specific surface area, electrical conductivity, corrosion resistance, and abundant bonding sites of single atoms for stable loading required, it is also important to select some functional supports to increase the number and activity of active sites, such as a three-dimensional support doped with heteroatoms or defects. The presence of heteroatoms and defects in the support stabilizes the single atoms and improves the active site coordination environment, resulting in more efficient sites (e.g., M–Cx, M–Nx, etc.) and facilitating rapid electron transfer during the OER process.
(4) Although SMSI exists between single metal atoms and supports, how to achieve high stability of single metal atoms during use, especially under high potential or strong corrosive acidic solution conditions, is also an aspect to be developed and studied subsequently.
(5) At present, most of the development of SACs is still using the trial-and-error method for synthesis research, without any effective theories to guide scholars to conduct research in a particular direction. Therefore, it is very important to master and understand the conformational relationships of each type of SAC in order to establish descriptors that can accurately describe the conformational relationships to further guide the rational design of SACs.
(6) In terms of synthesis, the current catalysts used for the OER follow the same synthesis method used for the synthesis of SACs. However, how to synthesize SACs in a simpler way, precisely control the structure of catalysts, and achieve large-scale synthesis is still a huge challenge.
(7) Although non-Ir-based catalysts have made great progress through continuous research in recent years, they still have the disadvantages of slow reaction kinetics and excessive oxidation and dissolution under strong acidic and oxidizing conditions compared to Ir-based catalysts under acidic conditions, and how to stabilize the metal active sites under acidic conditions is also a difficulty that scholars have to address.
Based on the understanding of their action mechanism, guiding the research and development of monoatomic oxygen evolution electrocatalysts with high efficiency and wide application is the key to the development of high efficiency oxygen evolution electrocatalysts. Therefore, there is an urgent need for a deeper understanding of their catalytic mechanism to provide a theoretical basis for the research and development of efficient catalytic materials. We believe that through the understanding of the relevant mechanism, we hopefully optimize electrocatalytic materials for oxygen evolution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant: U22A20396 and 22209168), the Natural Science Foundation of Anhui Province (2208085UD04), the Liaoning Binhai Laboratory (Grant No. LBLF-2023-04), the Shandong Energy Institute (Grant: SEI U202307), and the Postdoctoral Fellowship Program of the China Postdoctoral Science Foundation (Grant: 2023M743375 and GZB20230698).References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- Y. Dou, T. Liao, Z. Ma, D. Tian, Q. Liu, F. Xiao, Z. Sun, J. Ho Kim and S. Xue Dou, Nano Energy, 2016, 30, 267–275 CrossRef CAS.
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83–110 CrossRef CAS.
- E. J. Park, C. G. Arges, H. Xu and Y. S. Kim, ACS Energy Lett., 2022, 7, 3447–3457 CrossRef CAS.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- K. Zhang and R. Zou, Small, 2021, 17, 2100129 CrossRef CAS PubMed.
- M. Kim, J. Park, M. Wang, Q. Wang, M. J. Kim, J. Y. Kim, H.-S. Cho, C.-H. Kim, Z. Feng, B.-H. Kim and S. W. Lee, Appl. Catal., B, 2022, 302, 120834 CrossRef CAS.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- K. Du, L. Zhang, J. Shan, J. Guo, J. Mao, C.-C. Yang, C.-H. Wang, Z. Hu and T. Ling, Nat. Commun., 2022, 13, 5448 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222–2232 RSC.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- Z.-Y. Yu, Y. Duan, Y. Kong, X.-L. Zhang, X.-Y. Feng, Y. Chen, H. Wang, X. Yu, T. Ma, X. Zheng, J. Zhu, M.-R. Gao and S.-H. Yu, J. Am. Chem. Soc., 2022, 144, 13163–13173 CrossRef CAS PubMed.
- J. Zhou, Y. Dong, Y. Ma and T. Zhang, ChemistrySelect, 2020, 5, 7311–7314 CrossRef CAS.
- Y. Tang, T. Zhang, X. Wu and S. Deng, Front. Chem., 2022, 10, 889470 CrossRef CAS PubMed.
- H. Li, C. Chen, D. Yan, Y. Wang, R. Chen, Y. Zou and S. Wang, J. Mater. Chem. A, 2019, 7, 23432–23450 RSC.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- Z. Wang, N. Heng, X. Wang, J. He and Y. Zhao, J. Catal., 2019, 374, 51–59 CrossRef CAS.
- M. Gliech, M. Klingenhof, M. Görlin and P. Strasser, Appl. Catal., A, 2018, 568, 11–15 CrossRef CAS.
- W. H. Lee, Y.-J. Ko, J.-Y. Kim, B. K. Min, Y. J. Hwang and H.-S. Oh, Chem. Commun., 2020, 56, 12687–12697 RSC.
- S. H. Talib, Z. Lu, X. Yu, K. Ahmad, B. Bashir, Z. Yang and J. Li, ACS Catal., 2021, 11, 8929–8941 CrossRef CAS.
- Y. Jia, Z. Xue, Y. Li and G. Li, Energy Environ. Mater., 2022, 5, 1084–1102 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Shan, Y. Zheng, B. Shi, K. Davey and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 2719–2730 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1085 CrossRef.
- Z. Xu, J. Rossmeisl and J. R. Kitchin, J. Phys. Chem. C, 2015, 119, 4827–4833 CrossRef CAS.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kötz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- X. Wang, S. Xi, P. Huang, Y. Du, H. Zhong, Q. Wang, A. Borgna, Y.-W. Zhang, Z. Wang, H. Wang, Z. G. Yu, W. S. V. Lee and J. Xue, Nature, 2022, 611, 702–708 CrossRef CAS PubMed.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- J. W. D. Ng, M. García-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- X. Zheng, J. Yang, Z. Xu, Q. Wang, J. Wu, E. Zhang, S. Dou, W. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202205946 CrossRef CAS PubMed.
- W. Peng, A. Deshmukh, N. Chen, Z. Lv, S. Zhao, J. Li, B. Yan, X. Gao, L. Shang, Y. Gong, L. Wu, M. Chen, T. Zhang and H. Gou, ACS Catal., 2022, 12, 3743–3751 CrossRef CAS.
- Y. Yan, C. Liu, H. Jian, X. Cheng, T. Hu, D. Wang, L. Shang, G. Chen, P. Schaaf, X. Wang, E. Kan and T. Zhang, Adv. Funct. Mater., 2021, 31, 2009610 CrossRef CAS.
- Y. Wen, C. Liu, R. Huang, H. Zhang, X. Li, F. P. García de Arquer, Z. Liu, Y. Li and B. Zhang, Nat. Commun., 2022, 13, 4871 CrossRef CAS PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102–121 CrossRef CAS PubMed.
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert, B. R. Cuenya, W. Schuhmann and M. Muhler, ChemSusChem, 2017, 10, 156–165 CrossRef CAS PubMed.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Angew. Chem., Int. Ed., 2021, 60, 24612–24619 CrossRef CAS PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- K. Klyukin, A. Zagalskaya and V. Alexandrov, J. Phys. Chem. C, 2019, 123, 22151–22157 CrossRef CAS.
- Y. Qin, M. Yang, C. Deng, W. Shen, R. He and M. Li, Nanoscale, 2021, 13, 5800–5808 RSC.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- F. D. Speck, A. Zagalskaya, V. Alexandrov and S. Cherevko, Angew. Chem., Int. Ed., 2021, 60, 13343–13349 CrossRef CAS.
- J. Peng, H. Sun, K. Ni, J. Wu, X. Sun, Y. Su, H. Cheng, Y. Liu, Y. Guo, W. Bi, Y. Zhu, C. Wu and Y. Xie, Natl. Sci. Rev., 2023, 10, nwac108 CrossRef CAS.
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- K. Xiao, R.-T. Lin, J.-X. Wei, N. Li, H. Li, T. Ma and Z.-Q. Liu, Nano Res., 2022, 15, 4980–4985 CrossRef CAS.
- J. Chen, J. Huang, R. Wang, W. Feng, H. Wang, T. Luo, Y. Hu, C. Yuan, L. Feng, L. Cao, K. Kajiyoshi, C. He, Y. Liu, Z. Li and Y. Feng, Chem. Eng. J., 2022, 441, 136078 CrossRef CAS.
- X. Mu, X. Gu, S. Dai, J. Chen, Y. Cui, Q. Chen, M. Yu, C. Chen, S. Liu and S. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC.
- X. Duan, P. Li, D. Zhou, S. Wang, H. Liu, Z. Wang, X. Zhang, G. Yang, Z. Zhang, G. Tan, Y. Li, L. Xu, W. Liu, Z. Xing, Y. Kuang and X. Sun, Chem. Eng. J., 2022, 446, 136962 CrossRef CAS.
- H. Xu, T. Liu, S. Bai, L. Li, Y. Zhu, J. Wang, S. Yang, Y. Li, Q. Shao and X. Huang, Nano Lett., 2020, 20, 5482–5489 CrossRef CAS.
- Y. Gu, X. Wang, A. Bao, L. Dong, X. Zhang, H. Pan, W. Cui and X. Qi, Nano Res., 2022, 15, 9511–9519 CrossRef CAS.
- S. Lu, H. L. Huynh, F. Lou, K. Guo and Z. Yu, Nanoscale, 2021, 13, 12885–12895 RSC.
- D. Kan, D. Wang, X. Zhang, R. Lian, J. Xu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 3097–3108 RSC.
- D. Kan, R. Lian, D. Wang, X. Zhang, J. Xu, X. Gao, Y. Yu, G. Chen and Y. Wei, J. Mater. Chem. A, 2020, 8, 17065–17077 RSC.
- D. Kan, D. Wang, Y. Cheng, R. Lian, B. Sun, K. Chen, W. Huo, Y. Wang, G. Chen and Y. Wei, ACS Appl. Mater. Interfaces, 2021, 13, 52508–52518 CrossRef CAS PubMed.
- Z.-P. Wu, X. F. Lu, S.-Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS.
- X. Sun, S. Sun, S. Gu, Z. Liang, J. Zhang, Y. Yang, Z. Deng, P. Wei, J. Peng, Y. Xu, C. Fang, Q. Li, J. Han, Z. Jiang and Y. Huang, Nano Energy, 2019, 61, 245–250 CrossRef CAS.
- J. Li, Y.-f. Jiang, Q. Wang, C.-Q. Xu, D. Wu, M. N. Banis, K. R. Adair, K. Doyle-Davis, D. M. Meira, Y. Z. Finfrock, W. Li, L. Zhang, T.-K. Sham, R. Li, N. Chen, M. Gu, J. Li and X. Sun, Nat. Commun., 2021, 12, 6806 CrossRef CAS PubMed.
- W. Xie, Y. Song, S. Li, J. Li, Y. Yang, W. Liu, M. Shao and M. Wei, Adv. Funct. Mater., 2019, 29, 1906477 CrossRef CAS.
- K. Wang, Z. Lu, J. Lei, Z. Liu, Y. Li and Y. Cao, ACS Nano, 2022, 16, 11944–11956 CrossRef CAS PubMed.
- X. Lv, W. Wei, H. Wang, B. Huang and Y. Dai, Appl. Catal., B, 2020, 264, 118521 CrossRef CAS.
- Z. Li, Z. Wang, S. Xi, X. Zhao, T. Sun, J. Li, W. Yu, H. Xu, T. S. Herng, X. Hai, P. Lyu, M. Zhao, S. J. Pennycook, J. Ding, H. Xiao and J. Lu, ACS Nano, 2021, 15, 7105–7113 CrossRef CAS PubMed.
- C. Du, Y. Gao, J. Wang and W. Chen, J. Mater. Chem. A, 2020, 8, 9981–9990 RSC.
- L. Zhong, C. Jiang, M. Zheng, X. Peng, T. Liu, S. Xi, X. Chi, Q. Zhang, L. Gu, S. Zhang, G. Shi, L. Zhang, K. Wu, Z. Chen, T. Li, M. Dahbi, J. Alami, K. Amine and J. Lu, ACS Energy Lett., 2021, 6, 3624–3633 CrossRef CAS.
- M. Xiao, Z. Xing, Z. Jin, C. Liu, J. Ge, J. Zhu, Y. Wang, X. Zhao and Z. Chen, Adv. Mater., 2020, 32, 2004900 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- Y. Xu, W. Zhang, Y. Li, P. Lu and Z.-S. Wu, J. Energy Chem., 2020, 43, 52–57 CrossRef.
- Y. Li, Z.-S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed.
- Y. Xu, W. Zhang, Y. Li, P. Lu, Y. Wang and Z.-S. Wu, Front. Mater., 2019, 6, 271 CrossRef.
- F. Luo, J. Zhu, S. Ma, M. Li, R. Xu, Q. Zhang, Z. Yang, K. Qu, W. Cai and Z. Chen, Energy Storage Mater., 2021, 35, 723–730 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |