
Water oxidation utilizing a ruthenium complex featuring a phenolic moiety inspired by the oxygen-evolving centre (OEC) of photosystem II†
Yugo
Kumagai
a,
Risa
Takabe
a,
Takashi
Nakazono
 b,
Mitsuo
Shoji
*c,
Hiroshi
Isobe
d,
Kizashi
Yamaguchi
ef,
Tomoyo
Misawa-Suzuki
g,
Hirotaka
Nagao
g and
Tohru
Wada
*a
b,
Mitsuo
Shoji
*c,
Hiroshi
Isobe
d,
Kizashi
Yamaguchi
ef,
Tomoyo
Misawa-Suzuki
g,
Hirotaka
Nagao
g and
Tohru
Wada
*a
aDepartment of Chemistry, College of Science, Rikkyo University, 3-34-1, Nishi-Ikebukuro, Toshima-ku, Tokyo, 171-8501, Japan. E-mail: twada@rikkyo.ac.jp
bResearch Center for Artificial Photosynthesis (ReCAP), Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: nakazono@omu.ac
cCenter for Computational Sciences, University of Tsukuba, Tennodai 1-1-1, Tsukuba 305-8577, Japan. E-mail: mshoji@ccs.tsukuba.ac.jp
dResearch Institute for Interdisciplinary Science, Okayama University, Okayama 700-8530, Okayama, Japan
eSANKEN, Osaka University, Ibaraki 567-0047, Osaka, Japan
fCenter for Quantum Information and Quantum Biology (QIQB), Osaka University, Toyonaka 560-0043, Osaka, Japan
gDepartment of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1, Kioi-cho, Chiyoda-ku, Tokyo, 102-8554, Japan
First published on 17th January 2024
Abstract
In this manuscript, we present the synthesis of a ruthenium complex [1]2+ bearing a phenolic moiety, of which design draws inspiration from the oxygen-evolving centre (OEC) of photosystem II. In comparison to its analogue, [Ru(OH2)(bpy)(trpy)]2+ ([2]2+, where bpy = 2,2′-bipyridine and trpy = 2,2′:6′,2′′-terpyridine), which lacks the phenolic moiety, [1]2+ exhibits a 50 mV reduction in onset-overpotential at pH 10, an augmented turnover number (TON = 1900) and frequency (TOF = 0.54 s−1) for electrochemical water oxidation at pH 9, clearly indicating the promoting influence of the phenolic moiety on catalytic activity. Electrochemical and electron paramagnetic resonance (EPR) experiments have elucidated the formation of a RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species with either a phenol cation radical or a phenoxyl radical under oxidative conditions. Theoretical investigations provided robust support for the water oxidation mechanism, predicated upon a nucleophilic attack by OH− onto RuIVO, concomitant with electron transfer to the phenol cation radical. This electron transfer effectively lowers the activation energy associated with the nucleophilic attack of OH− onto RuIVO.
O species with either a phenol cation radical or a phenoxyl radical under oxidative conditions. Theoretical investigations provided robust support for the water oxidation mechanism, predicated upon a nucleophilic attack by OH− onto RuIVO, concomitant with electron transfer to the phenol cation radical. This electron transfer effectively lowers the activation energy associated with the nucleophilic attack of OH− onto RuIVO.
Introduction
To harness solar energy effectively for our energy needs, it is imperative to achieve the conversion of solar energy into a chemically stored form that can be reliably accumulated and subsequently converted into electrical energy as needed.1–5 Solar-driven CO2 reduction using water as an electron source to produce industrially valuable compounds, such as CO, HCOOH, and CH3OH, holds significant promise as a solution to the problems of global warming and energy sustainability.6–11 This process not only mitigates atmospheric CO2 levels but also regenerates chemical energy.The four-electron water oxidation reaction serves as a pivotal step in the energy conversion system, both in natural photosynthesis and artificial counterparts.4,12–19 This reaction supplies electrons required for various reduction processes, including CO2 reduction, hydrogen evolution, and nitrogen reduction. Water oxidation necessitates a simultaneous four-electron transfer at a relatively high equilibrium electrode potential (1.23 V vs. NHE at pH 0) and involves a substantial activation energy. Consequently, the development of highly efficient water oxidation catalysts is recognized as a critical bottleneck in the field of artificial photosynthesis.20
In natural photosynthesis, the manganese cluster in photosystem II exhibits an extraordinary capacity to catalyse water oxidation, characterized by an exceptionally high quantum yield and reaction rate.21–24 Recently, the structure of photosystem II has been undergone meticulous scrutiny through X-ray diffraction techniques.25 Moreover, the mechanism underlying the generation of molecular oxygen through water oxidation has been explored with the aid of X-ray free electron lasers (XFEL).26,27 Within photosystem II, a tyrosine residue, Yz (D1-Y161), exists close to the manganese cluster through hydrogen bonds.25 The phenolic moiety of this tyrosine has been considered to serve as an intermediary in the transfer of electrons between the manganese cluster and the photo-excited P680.28–32 To put it differently, the phenolic moiety assumes the role of an electron acceptor for the manganese cluster. The involvement of tyrosine's phenolic group in the process of photosynthetic water oxidation has remained the subject of ongoing experimental and theoretical investigations.33,34 Yamaguchi and colleagues have undertaken a theoretical inquiry into the mechanism of water oxidation and the pivotal role played by tyrosine in the realm of photosynthesis.35
Presently, there are successive efforts in the research dedicated to the development of molecular catalysts for water oxidation, a fundamental pursuit in the field of artificial photosynthesis.14,16–18,36–42 Ever since Meyer's seminal work on the binuclear ruthenium complex, so called “the blue dimer,” many transition-metal complexes have been developed.43
One inherent advantage of using molecular catalysts is their capability to illuminate the intricate reaction mechanisms. The formation of the O–O bond, widely supposed as the rate-determining step within the catalytic cycle, can be roughly classified into two distinct categories: the nucleophilic attack of water onto a high-valent RuO entity (termed the water nucleophilic attack or WNA mechanism) and the coupling of two high-valent RuO species (referred to as the interaction of two metal-oxo species or I2M mechanism).38,40,44–48 In both cases, a pronounced electron deficiency in the oxo ligand is an essential prerequisite for O–O bond formation. The proton-coupled electron transfer (PCET) reaction plays a pivotal role in this process, wherein an oxidation of low-valent Ru(OH2) complex coupled with deprotonation gives rise to a RuIVO species.49–53 RuVO species generated from RuIVO is postulated to serve as the active species in O–O bond formation. The non-PCET pathways result in elevated oxidation potentials of RuIVO to RuVO species, consequently increasing the overall overpotential for the reaction. Ru(bda) complexes (bda = bipyridine-6,6′-di-carboxylato) reported by Sun as remarkable water oxidation catalysts implemented relatively low overpotential and a high reaction rate. In the catalytic mechanism involving Ru(bda), the RuVO species is generated through a PCET reaction of RuIV–OH.54–56 This mechanism is expected to underpin the remarkable catalytic activity of Ru(bda). Conversely, several catalysts have proposed catalytic mechanisms that incorporate the WNA process onto RuIVO. For instance, Fujita has presented a RuII(npm) complex (npm = 4-tert-butyl-2,6-di-1′,8′-(naphthyrid-2′-yl)pyridine) that favors the nucleophilic attack of OH− onto RuIVO coupled with electron transfer to the anode in neutral and basic media.57 Additionally, Meyer has proposed a mechanism involving the WNA process onto RuIVO for ruthenium complexes concreted with electron transfer from the catalyst's Ru centre to the RuIII(bpy)3 moiety (electron-atom-proton transfer:EAPT mechanism).58 It's overpotential (230 mV) for water oxidation is one of the lowest at pH 1 in water oxidation catalyst previously reported. In both cases, the WNA process is involved with electron transfer to an acceptor. Consequently, the presence of an electron acceptor proximate to the catalytic centre is expected to be indispensable for mechanisms encompassing a RuIVO species as the active reaction intermediate.
In this research, we have synthesized a ruthenium complex [1]2+ featuring a phenolic moiety, as a novel water oxidation catalyst (Chart 1). [Ru(OH2)(bpy)(trpy)]2+ (bpy = 2,2′-bipyridine and trpy = 2,2′:6′,2′′-terpyridine) as the foundational structure for [1]2+ is already recognized for its moderate water oxidation catalytic activity.59,60 As previously mentioned, within natural photosynthesis, the phenolic moiety associated with the tyrosine residue assumes a pivotal role as an electron acceptor, facilitating electron transfer to the manganese cluster. In a similar vein, the oxidized phenolic moiety of [1]2+ is anticipated to serve as an electron acceptor for the ruthenium reaction centre. In this paper, we present an evaluation of the catalytic activity of [1]2+ for electrochemical water oxidation, in direct comparison to [Ru(OH2)(bpy)(trpy)]2+ ([2]2+). Furthermore, we experimentally and theoretically researched on the water oxidation mechanism.
 | ||
| Chart 1 Structures of [1]2+ and [2]2+. | ||
Results and discussion
Synthesis and characterisation
A novel terpyridine ligand, 1-(2,2′:6′,2′′-terpylyd-4′-yl)-8-(2,6-di-tert-butyl-p-hydroxyphenyl)anthraquinone (thaq), has been successfully synthesized through a three-component Suzuki–Miyaura cross-coupling reaction of anthraquinone-1,8-dibronic acid with 4′-{(trifluoromethylsulfonyl)oxy}-2,2′:6′,2′′-terpyridine and 2,6-di-tert-butyl-4-bromophenol in the presence of Pd(PPh3)4 as the catalyst and K2CO3 as the base (Scheme S1†). This synthesis achieved a moderate yield of 59%, primarily due to the formation of byproducts, 1,8-bis(2,2′:6′,2′′-terpyd-4′-yl)anthraquinone and 1,8-bis(2,6-di-tert-butyl-p-hydroxyphenyl)anthraquinone. The synthesis of [RuCl(bpy)(thaq)](PF6) was achieved through the reaction of RuCl3(thaq), obtained by refluxing an ethanolic solution of RuCl3·3H2O and thaq, with bpy in the presence of NEt3 as a reductant. Subsequently, [RuCl(bpy)(thaq)](PF6) was treated with AgPF6 in an acetone/water mixed solvent, yielding the aqua complex [Ru(OH2)(bpy)(thaq)](PF6)2 ([1](PF6)2) with a 32% yield (Scheme S1†).X-ray analysis revealed the aqua ligand of [1]2+ establishes hydrogen bonds with water and acetone molecules, exhibiting bond lengths of 2.237 Å and 2.667 Å, respectively (Fig. 1). This interaction serves to stabilize the structure, with the aqua ligand oriented outward. The distance between Ru(1) and O(2) in the phenol moiety is measured at 7.051 Å, while the distance between the carbon atoms at positions 1 and 8 in the anthraquinone ring is 5.080 Å. Steric repulsion between the tBu group and the trpy causes this spacing. In fact, the closest carbon atoms of the tBu and trpy groups maintain a distance of 4.052 Å. Ru(1) adopts a hexa-coordinated coordination environment, which can be described as a slightly distorted octahedral structure. The bond length between Ru(1) and O(1) measures 2.125(2) Å, closely resembling 2.097(5) Å of [2](PF6)2.61
 | ||
| Fig. 1 Crystal structures of [1](PF6)2. The anisotropic displacement parameters are shown at 50% probability. Colors present atomic labels: gray (carbon), blue (nitrogen), red (oxygen), and purple (ruthenium). For clarity, we omitted two PF6 anions, all hydrogen atoms, and solvent molecules instead of a water and an acetone molecule bonding to the aqua ligand. Tables S1 and S2† list the crystal parameters, selected bond lengths, and angles. | ||
Analysis via1H-NMR of [1](PF6)2 in an acetone-d6/D2O mixture (in an 10/1 ratio) unequivocally revealed an independent conformation in solution (Fig. S3†). This observation suggests that the rotation of the Ru(trpy) moiety is impossible, a phenomenon attributed to the steric hindrance posed by the terpyridine and phenol moieties.
Acid-base equilibrium, redox behaviour, and water oxidation catalytic activity of [1]2+
Initially, we conducted an investigation into the acid–base equilibrium of [1]2+ in a H2O/MeOH (1/1) mixed solution, owing to the limited water solubility of [1](PF6)2. During this experiment, we documented pH values through the utilization of a pH meter in a H2O/MeOH (1/1) mixed solution that had been calibrated using a water buffer solution. When the pH was adjusted to 7.0, [1]2+ exhibited a conspicuous absorption band at 483 nm (ε = 1.03 × 104 M−1 cm−1, Fig. 2 and S5†). This band can be attributed to the metal-to-ligand charge transfer (MLCT) transition of RuII(bpy). The elevation of the pH level within the solution led to a redshift of the MLCT band to 515 nm, with isosbestic points clear at 450 and 495 nm (Fig. 2). This spectral alteration is entirely reversible, and upon reacidification of the solution, the spectrum reverts to its initial state at pH 7.0. The observed spectral alteration unequivocally is caused by the deprotonation of the aqua ligand, as there is an absence of consequential influence resulting from the deprotonation of the phenoic moiety upon the MLCT band of Ru(bpy). Moreover, the pKa value of [1]2+ closely approximates that of [2]2+ (pKa = 10). By virtue of constructing absorbance versus pH plots for a wavelength of 515 nm, we were able to discern the pKa value of [1]2+ is 10.3 (the inset of Fig. 2). | ||
| Fig. 2 UV-vis spectra of [1](PF6)2 in MeOH/water (1/1) at pH 6.0–12.0. Inset: plots of absorbance at 515 nm. | ||
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements of aqueous and organic solutions containing water of [1]2+ are rendered unfeasible due to the limited solubility of [1](PF6)2 in water. Consequently, we executed CV and DPV experiments employing a fluorine-doped tin oxide (FTO) coated glass electrode that had been modified with [1](PF6)2 with a cast method, as a working electrode in water (Fig. 3 and 4). CV analysis of [1]2+ at pH 10 unveiled a substantial catalytic current arising from water oxidation above Eon-set = 0.90 V vs. SCE, which enabled us to estimate the onset overpotential of [1]2+ at pH 10 to be 0.47 V (Fig. S6B†). In comparison with [2](BPhF4)2 (BPhF4 = tetrakis(tetrafluorophenyl)borate), [1]2+ exhibited a reduction in overpotential by 50 mV and manifested a five-fold amplification in catalytic current at 1.05 V. Indeed, a controlled-potential electrolysis of pH 9.0 water at 1.05 V employing FTO electrodes modified with either [1](PF6)2 or [2](BPhF4)2 resulted in the generation of molecular oxygen, yielding turnover numbers (TON) of 1900 for [1]2+ and 300 for [2]2+, accompanied by respective turnover frequencies (TOF) of 0.54 and 0.13 s−1 (Table 1 and Fig. S7†). The Faraday efficiency (FE) of [2]2+ was inferior to that of [1]2+. When utilizing [2]2+ as a catalyst, the volume of evolved oxygen displayed a minute magnitude of 0.7 μmol. A fraction of the generated oxygen may have leaked, permitting a slight ingress of Ar into the airtight sealed electrolysis cell. Even though the FE of [2]2+ being 100%, the TOF and TON of [2]2+ are conspicuously subpar compared to those of [1]2+.
 | ||
| Fig. 3 CVs using the FTO glass plate modified with [1](PF6)2 (red), [2](BPhF4)2 (blue) and the bare FTO electrode (blank test, black) as working electrodes in boronic acid buffer solution (100 mM) at pH 10. Counter electrode: Pt; reference electrode: SCE; scan rate: 50 mV s−1. Temperature: 293 K. | ||
 | ||
| Fig. 4 DPVs using FTO glass plate modified with [1](PF6)2 as a working electrode in boronic acid buffer solution (100 mM) at pH 2.0–7.0 (A) and 7.0–13.0 (B). Counter electrode: Pt; reference electrode: SCE; temperature: 293 K. | ||
| Catalyst | TON | TOF/s−1 | FE/% |
|---|---|---|---|
| [1](PF6)2 | 1900 | 0.54 | 83 |
| [2](BPhF4)2 | 300 | 0.13 | 45 |
Therefore, the experimental evidence corroborates that [1]2+ unequivocally surpasses [2]2+ in the realm of electrochemical water oxidation catalysis. Raman spectra of the FTO electrodes post-electrolysis for a duration of 30 minutes exhibited remarkable consistency to that of [1](PF6)2 before electrolysis (Fig. S8†), suggesting the stability of [1]2+ as a molecular catalyst, while obviating the formation of ruthenium oxide under these operative conditions. It is worthy of note that the presence of the phenolic moiety within the secondary coordination sphere of [1]2+ augments the catalytic efficacy of [2]2+ in the context of electrochemical water oxidation. The catalytic current and onset potential of water oxidation experienced a reduction and a positive shift, respectively, upon the acidification of the aqueous medium. The manifestation of catalytic current was scarcely discernible at pH levels below 6.0 (Fig. S6†). Therefore, [1]2+ expresses high catalytic activity for water oxidation in the alkaline region.
To elucidate the redox properties inherent to [1]2+, we conducted DPV examinations (Fig. 4). DPV of [1]2+ at pH 2.0 unveiled an oxidation wave at Epa = 0.84 V. The alkalization of the solution caused the division of this oxidation wave into two waves. The more negative potential wave underwent a pronounced negative shift, while the more positive potential wave remained unaltered as the solution's pH increased. A substantial catalytic current, arising from the process of water oxidation, became conspicuous beyond a pH of 8.0, and its potential underwent a negative shift with the progressive elevation of pH. Ultimately, at pH 10, the second oxidation wave dissipated due to the overlap with the catalytic current. A Pourbaix diagram for [1]2+, as derived from Fig. 4, has been represented in Fig. 5. The gradients of the first oxidation potential within the pH range from 2.0 to 9.8 and from 9.8 to 13.0 were determined to be −53 mV/pH and −26 mV/pH, respectively. The singular point at pH 9.8 is almost consistent of the pKa of [2]2+ determined by pH titration (Fig. 2). The gradient values denote that the initial oxidation events of [1]2+ are attributed to the two-electron oxidation of RuII(OH2), occurring concurrently with doubly deprotonation, resulting in RuIVO within the pH range of 2.0 to 9.8. Furthermore, the two-electron oxidation of RuII(OH), simultaneous with single deprotonation, gives rise to RuIVO beyond pH 10. In the context of homogenous aqueous surroundings, stepwise proton-coupled electron transfer (PCET) processes involving [2]2+ yield RuIII(OH) and RuIVO species.62
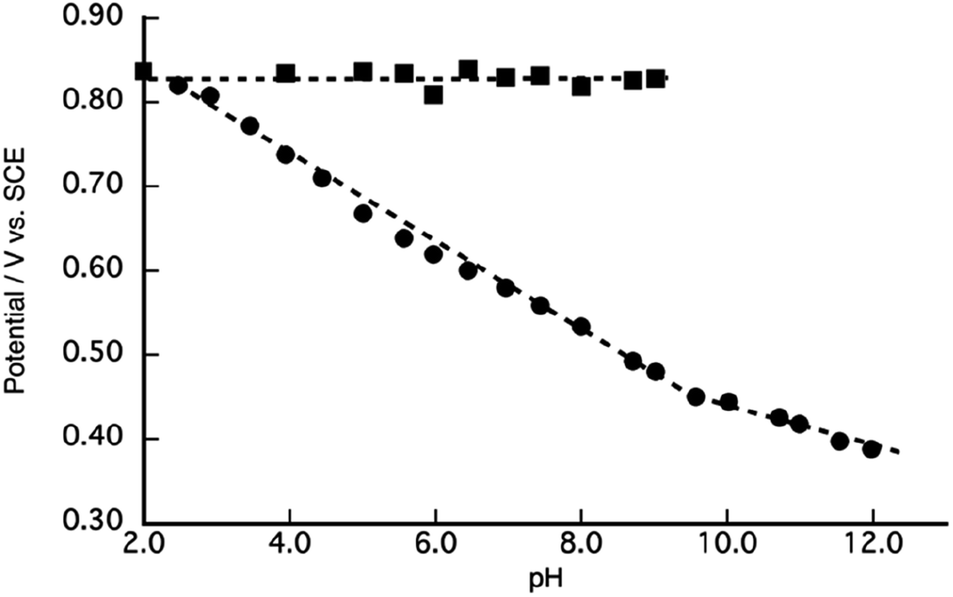 | ||
| Fig. 5 A Pourbaix diagram of [1](PF6)2. | ||
The second oxidation wave at 0.82 V demonstrates a notable pH-independence. The redox potential associated with RuVO/RuIVO in [2]2+ has been previously reported to fall within the range of Epa = 1.73 V vs. NHE, which corresponds to 1.49 V vs. SCE.47 The modest potential of 0.82 V of the second oxidation wave, therefore, stands as inadequate for the attribution to RuVO/RuIVO. Consequently, this second oxidation wave can be assigned to the oxidation of the phenolic moiety. The pH-invariance exhibited by the phenol oxidation potential suggests that it does not entail a PCET reaction. Notably, the reported pKa values for 2,6-di- and 2,4,6-tri-tert-butylphenol stand at 17.3 and 17.8,63 respectively, thereby implying a substantial pKa for the 2,6-di-tert-butylphenol moiety, which, in turn, inhibits PCET reactions within the pH range of 2.0 to 9.5. Although PCET response associated with the phenolic moiety remains unclear beyond pH 9.8, primarily due to the overlapping of the oxidation wave with catalytic currents, the redox characteristics of [1]2+ can be summarized in Scheme 1. It is reasonable to conjecture that the redox process of the phenolic moiety is likely to underpin the enhanced catalytic efficiency of [1]2+ in comparison to [2]2+.
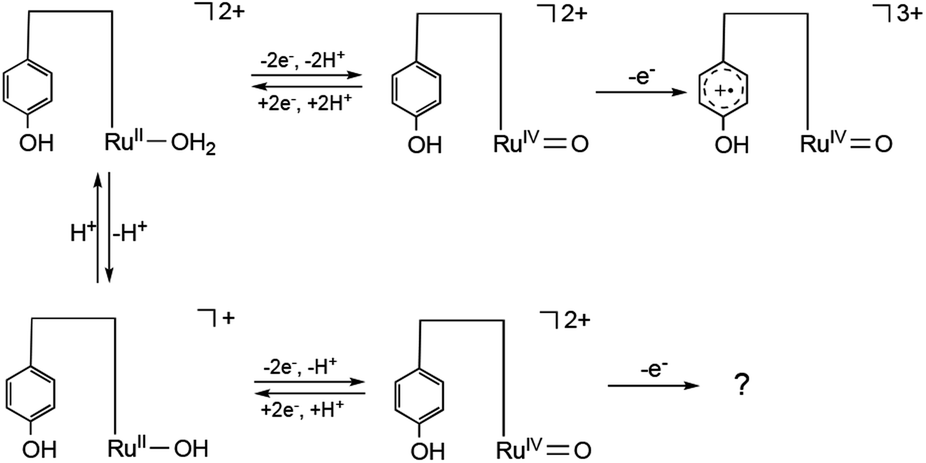 | ||
| Scheme 1 Redox behavior of [1]2+ at pH < 9.8 (upper) and pH > 9.8 (lower). | ||
Assignment of the oxidized form of [1]2+
To scrutinize the redox behaviour, we executed a chemical oxidation of [1]2+, employing tris(2,4-dibromophenyl)aminium hexafluoroantimonate, commonly referred to as “Magic Green” (MG), renowned for its robust oxidizing properties. Initially, we monitored the oxidation reaction of [1]2+ with MG through UV-visible spectroscopy (Fig. 6). Gradual additions of MG to the CH2Cl2 solution of [1]2+ (100 μM) under Ar at room temperature led to a notable reduction in absorbance, accompanied by a slight shift of the MLCT band of RuII(bpy) from 485 nm to 490 nm. Up to 2.0 equivalents, MG-adding induced a progressive attenuation in the intensity of the MLCT band, concomitant with the augmentation of intensity around 370 nm. Masaoka's work theoretically elucidated that RuIVO species, derived from [2]2+, is associated with an absorption maximum at 362 and 381 nm.64 The observed decrement in absorbance, coupled with the redshift of the absorption peak, is indicative of the deprotonation of RuII(OH2) to RuII(OH), facilitated by the tris(2,4-dibromophenyl)amine present in the MG solution. Indeed, the deprotonated form of [1]2+ manifests a MLCT band at 515 nm (Fig. 2). These spectroscopic changes indicate the occurrence of a two-electron oxidation process of RuII(OH2) in conjunction with deprotonation, giving rise to the formation of RuIVO in an organic medium similarly to in an aqueous medium. Furthermore, with the incremental addition of MG, a new absorption band emerged at 634 nm, similar to that of the 2,3,6-tri-tert-butylphenoxyl radical,65 supporting the assignment that the second oxidation wave observed in the CVs originates from the oxidation of the phenolic moiety of [1]2+. Subsequent introduction of 3,5-di-tert-butylcatechol as a reductant served to regenerate the MLCT band of RuII(bpy) at 507 nm (Fig. S10†). Subsequent acidification of the solution with HClO4 led to the near-complete restoration of the original spectral profile of [1]2+, thus substantiating the stability of [1]2+ under oxidative conditions (Fig. S11†).
 | ||
| Fig. 6 UV-vis spectra of the CH2Cl2 solution of [1](PF6)2 in the absence (blue) and presence of MG (2.0 eq.: purple, 3.0 eq.: red) at room temperature. | ||
Subsequently, we conducted Electron Paramagnetic Resonance (EPR) analyses on the frozen CH2Cl2 solution containing [1]2+ both in the absence and presence of MG at 77 K. [1]2+ exhibited an absence of any EPR signal because of its diamagnetic property as a consequence of its 4d6 electronic configuration with a low-spin state. Upon the addition of 3.0 equivalents of MG, an EPR signal was discerned, exhibiting relatively sharp isotropic features, at giso = 2.0039 (Fig. 7), which is distinct from that of MG (giso = 2.0099) observed under identical experimental conditions (Fig. S12†). Although the inherent paramagnetism of the hexacoordinated RuIVO species (S = 1), X-band EPR of RuIVO generally exhibit no EPR signal because of a large spin coupling exhibited in a zero magnetic field.65–76 Mukai reported that a powder sample of 2,6-di-tert-butyl-4-phenyl phenoxyl radical displayed a sharp EPR signal at g = 2.0047.77 In solution, both the 2,6-di-tert-butyl-4-phenyl phenoxyl radical and the 2,4,6-di-tert-butyl phenoxyl radical demonstrated EPR signals at g = 2.0044 and 2.0046, respectively, each revealing hyperfine coupling with the 3,5-H atoms, characterized by coupling constants Ha = 1.73 and 1.71 G, respectively.78 Despite our exhaustive exploration of various measurement conditions, any triplet signal was not observed, primarily due to the solvent effect on EPR signal.79 However, it is a well-established fact that RuIII (4d5, S = 1/2) and RuV (d3, S = 1/2) complexes typically manifest anisotropic EPR signals.67,68,80,81 Notably, this EPR signal was unequivocally abolished upon the introduction of 3,5-di-tert-butylcatechol into the solution. Therefore, this EPR spectrum provides compelling evidence for the formation of phenoxyl radical species.
 | ||
| Fig. 7 X-band EPR spectra of [1](PF6)2 in glassy CH2Cl2 at 77 K (liquid N2) in the absence (black) and presence of 3.0 equiv. (red) of MG. Microwave frequency, 9.029006 GHz; microwave power, 0.9980 mW; modulation width, 100 mT; modulation amplitude, 200 mT. | ||
A proposed water oxidation mechanism
Based on these experimental findings, we propose a water oxidation mechanism catalysed by [1]2+ in alkaline water (Scheme 2). The PCET reaction involving [1]2+ results in the formation of [RuIVO, phenol]2+, which subsequently undergoes further oxidation to give rise to either [RuIVO, phenoxyl radical]2+ or [RuIVO, phenol cation radical]3+. Results of DPV analyses (Fig. 4) and the Pourbaix diagram (Fig. 5) distinctly indicate a one-electron oxidation of the phenolic moiety, devoid of any deprotonation, under a pH of 9. However, it remains equivocal whether the phenoxyl radical or phenol cation radical emerges because the redox wave of phenolic moiety of [1]2+ was not observed due to overlapping with catalytic current. In an aqueous medium at a higher pH, a nucleophilic attack by OH− on RuIVO coupled with an electron transfer from the Ru centre to the phenoxyl radical or phenol cation radical forms the O–O bond, characterizing the mechanism as WNA mechanism. The electron transfer from Ru to the oxidized state of the phenolic moiety is considered to enhance the nucleophilic attack of OH− on RuIV, a species traditionally perceived as inert. Furthermore, the subsequent PCET reaction of [RuIII(OOH), phenol]2+ is anticipated to result in the generation of O2 and the regeneration of [1]2+.
 | ||
| Scheme 2 A proposed mechanism of water oxidation catalysed by [1]2+. | ||
Theoretical density functional theory (DFT) study
We theoretically investigated to identify the active RuO and substrate species enough to proceed the O–O bond formation. For the oxidation of the RuO side, four possibilities exist: RuIVO, RuVO, [RuIVO, phenol cation radical] and [RuIVO, phenoxyl radical]. Regarding the substrate, hydroxide anion (OH−) or water (H2O) are the candidates. We constructed all the eight models and we have evaluated their relative energies in the transition states and peroxide products. The molecular structure of the representative Ru complex with a phenol cation radical was shown in Fig. S13.† For the RuIVO species (labelled a and e), both the OH− and water substrates are not enough to form peroxide species because the products are higher in energy than the reactants by more than 10 kcal mol−1. For the RuVO species (labelled b and f), both the OH− and water substrates are enough to form peroxide species because the products are lower in energy than the reactants and the transition states is not higher than 25 kcal mol−1. This shows that the RuVO species is very reactive, however, the oxidation potential is high and it requires substantial oxidation to be formed. In fact, the oxidation potential of [RuIVO, phenol cation radical] is lower in energy than the RuVO species by 12.9 kcal mol−1. Even though this species still processes the RuIVO centre, OH− substrate can react into peroxide species with a low formation energy (ΔE(c) = −1.9 kcal mol−1) and with a low activation energy of ΔE≠(c) = 22.3 kcal mol−1. We evaluated that H2O substrate is not enough to accomplish the reaction for [RuIVO, phenol cation radical] because of a high activation barrier (ΔE≠(g) = 49.3 kcal mol−1), where this model was labelled g. The RuIVO species with phenoxyl radicals (labelled d and h) do not produce stable peroxide species (ΔE(d) = 22.7 kcal mol−1 and ΔE(h) = 22.9 kcal mol−1). This means that deprotonated phenol (phenoxyl) radical is not enough to oxidize to form the Ru(III)OOH product and to lower the activation barrier during the O–O bond formation by inducing the one electron transfer. The high reactivity of the phenol cation radical reminds us the short hydrogen bond observed between Yz and His190 in PSII, where deprotonated H atom upon Yz oxidation is transferred back from His190 to the original Yz upon the Yz reduction.34 The present theoretical calculations showed that O–O bond formation can take place in RuIVO state just after the phenol cation radical formation as in the situation of the model c. Optimized molecular structures of all these models a–h were depicted in Fig. S14.† Their relative energies along the O–O bond formation step were summarized in Table S3.†
Mechanism of the O–O bond formation
As we have identified the RuIVO species with the phenol cation radical in c is in a low oxidation state but still reactive, the detailed reaction mechanism of the O–O bond formation is further analyzed theoretically. Relative energy profile along the O–O bond formation in c was shown in Fig. S15.† Atomic distances participating in the reaction were plotted in Fig. S16.† By combining the energy and distance changes, early reaction before reaching the transition state, before NEB step 5, is OH− closing to RuIVO center via proton transfer step. This reaction (OH− + H2O → H2O + OH−) is clearly described by the alteration of O–H distances (R2 and R3) between the hydrogen bond and O–H covalent bond. At the transition state (NEB step 5 in Fig. S16†), the O atoms of OH− and RuO come close to R1 = 2.21 Å. This indicates that O–O bond formation is surely the rate-limiting step of the reaction.
The O–O bond formation is also characterized from the electronic structures. The spin densities in the reactant and product states are shown in Fig. 8. In the reactant state, positive spin densities concentrate to the RuIVO moieties and negative spin densities spread over the phenol aromatic ring. This spin density distribution shows the two alpha spins in π* orbital of RuIVO and one beta spin over the phenol radical. The beta spin on the phenol radical disappears after the transition state and spin density localizes only at the RuO in the product state (Fig. S17†). This indicates that RuIII is formed and the phenol moiety becomes a non-radial state by one electron (alpha spin) transfer from RuOOH moiety to the phenol moiety along the O–O bond formation. This O–O bond formation mechanism is well described by using the orbital correlation diagram as illustrated in Fig. 9.
 | ||
| Fig. 8 Plots of spin densities before (A) and after (B) O–O bond formation of c in [RuIVO, phenol cation radical]. The blue surfaces represent the positive spin densities while the red surfaces show the negative spin densities (isovalue = 0.01). | ||
 | ||
| Fig. 9 O–O bond formation mechanism of 1. (A) Schematic molecular orbital diagram and (B) drawing in their formal charges. The O–O bond formation reaction is accomplished by (i) the σ bond formation between RuIVO and HO− and (ii) single electron transfer from the substrate (OH−) to the phenol radical (L˙). | ||
In fact, the oxidation potential of [RuIVO, phenol cation radical]3+ is lower in energy than the RuVO species by 12.9 kcal mol−1. Even though this species still processes the RuIVO centre, OH− substrate can react into peroxide species with a low formation energy (ΔE(c) = −1.9 kcal mol−1) and with a low activation energy of ΔE≠(c) = 22.3 kcal mol−1. We evaluated that H2O substrate is not enough to accomplish the reaction for [RuIVO, phenol cation radical]3+ because of a high activation barrier (ΔE≠(g) = 49.3 kcal mol−1), where this model was labelled g.
Conclusions
This research substantiated that the ruthenium complex [1]2+, featuring an appended phenolic moiety, notably evinces superior catalytic efficacy concerning electrochemical water oxidation compared to the phenolic-devoid analogue [2]2+. Electrochemical analyses and EPR investigations have unveiled the formation of either phenol cation radicals or phenoxyl radical species under the oxidative conditions. DFT calculations have furnished a compelling explication that the electron transfer from Ru to the phenol cation radical concomitant with the nucleophilic attack by OH− on RuIVO renders the O–O bond formation exothermic with low activation energy, whereas the nucleophilic attack by OH− on RuIVO is endothermic in nature. The O–O bond formation mechanism of 1 is clearly different from the two major categories: WNA and I2M mechanisms, and it is rather classified as a third mechanism, called EAPT mechanism proposed by Meyer58 or nonadiabatic one-electron transfer assisted O–O bond formation mechanism.35 This study represents an inaugural illustration wherein the redox reaction involving the phenolic moiety in the second coordination sphere augments the water oxidation catalytic activity of transition-metal complexes. The discerning elucidation of these observations imparts novel perspectives on the involvement of tyrosine residues in the contexts of both synthetic and natural photosynthetic processes.
Author contributions
Yugo Kumagai and Risa Takabe: all practical experiments: syntheses and characterizations of the complexes, electrochemical and chemical oxidation experiments. Takashi Nakazono: supervision of all practical experiments. Mitsuo Shoji: all the DFT calculations and original draft preparation, and supervision. Hiroshi Isobe and Kizashi Yamaguchi: supervision of computational experiments. Tomoyo Misawa-Suzuki and Hirotaka Nagao: EPR experiments. Tohru Wada: original draft preparation and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by JSPS KAKENHI (Grant 20H05116) in Scientific Research on Innovative Areas ‘Innovations for Light-Energy Conversion (I4LEC), Kato foundation for Promotion of Science (KJ-3203) (to TW), JSPS KAKENHI (20H05453, 20H05088 and 22H04916) (to MS). Computational resources used in this study were supported by Multidisciplinary Cooperative Research Program in CCS, University of Tsukuba. The authors would like to applicate for Dr Akiko Sekine of Tokyo Institute of Technology support X-ray analysis.Notes and references
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W.-H. Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K.-S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper, R. Van De Krol and F. Houle, J. Phys. D: Appl. Phys., 2022, 55, 323003 CrossRef.
- H. Dau, E. Fujita and L. Sun, ChemSusChem, 2017, 10, 4228–4235 CrossRef CAS PubMed.
- B. Battistella, L. Iffland-Muhlhaus, M. Schutze, B. Cula, U. Kuhlmann, H. Dau, P. Hildebrandt, T. Lohmiller, S. Mebs, U. P. Apfel and K. Ray, Angew Chem. Int. Ed. Engl., 2023, 62, e202214074 CrossRef CAS PubMed.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS PubMed.
- R. H. Crabtree, Energy Production and Storage, John Wiley & Sons Ltd, West Sussex, 2010 Search PubMed.
- T. K. Michaelos, D. Y. Shopov, S. B. Sinha, L. S. Sharninghausen, K. J. Fisher, H. M. C. Lant, R. H. Crabtree and G. W. Brudvig, Acc. Chem. Res., 2017, 50, 952–959 CrossRef CAS.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2016, 7, 70–88 CrossRef.
- D. G. Nocera, Inorg. Chem., 2009, 48, 10001–10017 CrossRef CAS PubMed.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503–2520 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS.
- R. Matheu, P. Garrido-Barros, M. Gil-Sepulcre, M. Z. Ertem, X. Sala, C. Gimbert-Suriñach and A. Llobet, Nat. Rev. Chem, 2019, 3, 331–341 CrossRef CAS.
- P. Garrido-Barros, C. Gimbert-Suriñach, R. Matheu, X. Sala and A. Llobet, Chem. Soc. Rev., 2017, 46, 6088–6098 RSC.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS.
- A. Llobet, Molecular Water Oxidation Catalysis, John Wiley & Sons Ltd, West Sussex, 2014 Search PubMed.
- R. Bofill, J. Garcia-Anton, L. Escriche, X. Sala and A. Llobet, Compr. Inorg. Chem. II, 2013, 8, 505–523 CAS.
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CrossRef CAS.
- M. M. Najafpour, I. Zaharieva, Z. Zand, S. Maedeh Hosseini, M. Kouzmanova, M. Holynska, I. Tranca, A. W. Larkum, J.-R. Shen and S. I. Allakhverdiev, Coord. Chem. Rev., 2020, 409, 213183 CrossRef CAS.
- M. M. Najafpour, G. Renger, M. Holynska, A. N. Moghaddam, E. M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J. R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef CAS PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- J. Barber, Philos. Trans. R. Soc., B, 2008, 363, 2665–2674 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L.-J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J.-R. Shen, Science, 2019, 366, 334–338 CrossRef CAS.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L. J. Yu, T. Sakamoto, T. Motomura, J. H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J. R. Shen, Nature, 2017, 543, 131–135 CrossRef CAS PubMed.
- G. Renger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1164–1176 CrossRef CAS PubMed.
- B. A. Barry, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 46–54 CrossRef CAS PubMed.
- B. A. Barry, J. Photochem. Photobiol., B, 2011, 104, 60–71 CrossRef CAS PubMed.
- H. Ishikita and E. W. Knapp, Biophys. J., 2006, 90, 3886–3896 CrossRef CAS PubMed.
- L. P. Candeias, S. Turconi and J. H. A. Nugent, Biochim. Biophys. Acta, Bioenerg., 1998, 1363, 1–5 CrossRef CAS PubMed.
- H. Matsuoka, J. R. Shen, A. Kawamori, K. Nishiyama, Y. Ohba and S. Yamauchi, J. Am. Chem. Soc., 2011, 133, 4655–4660 CrossRef CAS PubMed.
- M. Ibrahim, T. Fransson, R. Chatterjee, M. H. Cheah, R. Hussein, L. Lassalle, K. D. Sutherlin, I. D. Young, F. D. Fuller, S. Gul, I. S. Kim, P. S. Simon, C. de Lichtenberg, P. Chernev, I. Bogacz, C. C. Pham, A. M. Orville, N. Saichek, T. Northen, A. Batyuk, S. Carbajo, R. Alonso-Mori, K. Tono, S. Owada, A. Bhowmick, R. Bolotovsky, D. Mendez, N. W. Moriarty, J. M. Holton, H. Dobbek, A. S. Brewster, P. D. Adams, N. K. Sauter, U. Bergmann, A. Zouni, J. Messinger, J. Kern, V. K. Yachandra and J. Yano, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12624–12635 CrossRef CAS.
- M. Shoji, H. Isobe, Y. Shigeta, T. Nakajima and K. Yamaguchi, Chem. Phys. Lett., 2018, 698, 138–146 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- W. C. Hsu and Y. H. Wang, ChemSusChem, 2021, 15, e202102378 CrossRef.
- J. M. Kamdar and D. B. Grotjahn, Molecules, 2019, 24, 494 CrossRef PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, Dalton Trans., 2019, 48, 779–798 RSC.
- M. D. Kaerkaes, O. Verho, E. V. Johnston and B. Aakermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef CAS.
- T. Okada and M. Kaneko, Molecular Catalysts for Energy Conversion, Springer Science and Buisiness Media Inc., New York, 2009 Search PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–35 CrossRef CAS.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS.
- T. A. Betley, Q. Wu, T. Van Voorhis and D. G. Nocera, Inorg. Chem., 2008, 47, 1849–1861 CrossRef CAS PubMed.
- T. Kikuchi and K. Tanaka, Eur. J. Inorg. Chem., 2014, 2014, 607–618 CrossRef CAS.
- D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218–227 RSC.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed.
- L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Saveant, Organic Electrochemistry, CRC Press, Boca Raton, 5th edn, 2016 Search PubMed.
- M. Okamura, M. Yoshida, R. Kuga, K. Sakai, M. Kondo and S. Masaoka, Dalton Trans., 2012, 41, 13081–13089 RSC.
- C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704–7717 RSC.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- T. Fan, L. Duan, P. Huang, H. Chen, Q. Daniel, M. S. G. Ahlquist and L. Sun, ACS Catal., 2017, 7, 2956–2966 CrossRef CAS.
- J. Yang, L. Wang, S. Zhan, H. Zou, H. Chen, M. S. G. Ahlquist, L. Duan and L. Sun, Nat. Commun., 2021, 12, 373 CrossRef CAS PubMed.
- L. Wang, L. Duan, Y. Wang, M. S. G. Ahlquist and L. Sun, Chem. Commun., 2014, 50, 12947–12950 RSC.
- Y. M. Badiei, D. E. Polyansky, J. T. Muckerman, D. J. Szalda, R. Haberdar, R. Zong, R. P. Thummel and E. Fujita, Inorg. Chem., 2013, 52, 8845–8850 CrossRef CAS PubMed.
- M. R. Norris, J. J. Concepcion, Z. Fang, J. L. Templeton and T. J. Meyer, Angew Chem. Int. Ed. Engl., 2013, 52, 13580–13583 CrossRef CAS PubMed.
- M. Yoshida, S. Masaoka, J. Abe and K. Sakai, Chem.–Asian J., 2010, 5, 2369–2378 CrossRef CAS PubMed.
- S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS.
- X. J. Yang, F. Drepper, B. Wu, W. H. Sun, W. Haehnel and C. Janiak, Dalton Trans., 2005, 256–267 RSC.
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 1845–1851 CrossRef CAS.
- F. G. Bordwell and J. Cheng, J. Am. Chem. Soc., 1991, 113, 1736–1743 CrossRef CAS.
- A. Kimoto, K. Yamauchi, M. Yoshida, S. Masaoka and K. Sakai, Chem. Commun., 2012, 48, 239–241 RSC.
- A. Yadav and P. Mathur, Inorg. Chim. Acta, 2015, 435, 206–214 CrossRef CAS.
- W. K. Seok and T. J. Meyer, J. Am. Chem. Soc., 2002, 110, 7358–7367 CrossRef.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 2002, 20, 436–444 CrossRef.
- C. M. Che, T. F. Lai and K. Y. Wong, Inorg. Chem., 1987, 26, 2289–2299 CrossRef CAS.
- C. M. Che, W. T. Tang, W. T. Wong and T. F. Lai, J. Am. Chem. Soc., 1998, 111, 9048–9056 CrossRef.
- W.-C. Cheng, W.-Y. Yu, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1994,(1), 57–62 RSC.
- C.-M. Che, C. Ho and T.-C. Lau, J. Chem. Soc., Dalton Trans., 1991, 1901–1907 RSC.
- J. M. Power, K. Evertz, L. Henling, R. Marsh, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1990, 29, 5058–5065 CrossRef CAS.
- C.-M. Che, K.-Y. Wong and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1985, 546–548 RSC.
- B. A. Moyer and T. J. Meyer, Inorg. Chem., 1981, 20, 436–444 CrossRef CAS.
- T. Ishizuka, S. Ohzu and T. Kojima, Synlett, 2014, 25, 1667–1679 CrossRef.
- T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692–11700 CrossRef CAS PubMed.
- K. Mukai, K. Ueda, K. Ishizu and S. Yamauchi, Bull. Chem. Soc. Jpn., 1984, 57, 1151–1152 CrossRef CAS.
- G. Brigati, M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 4828–4832 CrossRef CAS PubMed.
- K. Mukai, H. Nishiguchi, K. Ishizu, Y. Deguchi and H. Takaki, Bull. Chem. Soc. Jpn., 1967, 40, 2731–2739 CrossRef CAS.
- C. M. Che, V. W. W. Yam and T. C. W. Mak, J. Am. Chem. Soc., 1990, 112, 2284–2291 CrossRef CAS.
- S. Miyazaki, T. Kojima, T. Sakamoto, T. Matsumoto, K. Ohkubo and S. Fukuzumi, Inorg. Chem., 2008, 47, 333–343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterizations, electrochemical experiments, chemical oxidation of [1](PF6)2, computational details. CCDC 2312432. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3se01610b |
| This journal is © The Royal Society of Chemistry 2024 |