
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination of Al(C6F5)3vs. B(C6F5)3 on group 6 end-on dinitrogen complexes: chemical and structural divergences†
Léon
Escomel
 a,
Frederico F.
Martins
b,
Laure
Vendier
a,
Anaïs
Coffinet
a,
Nicolas
Queyriaux
a,
Vera
Krewald
*b and
Antoine
Simonneau
*a
a,
Frederico F.
Martins
b,
Laure
Vendier
a,
Anaïs
Coffinet
a,
Nicolas
Queyriaux
a,
Vera
Krewald
*b and
Antoine
Simonneau
*a
aLCC-CNRS, Université de Toulouse, CNRS, UPS 205 Route de Narbonne, BP44099, F-31077 Toulouse Cedex 4, France. E-mail: antoine.simonneau@lcc-toulouse.fr
bDepartment of Chemistry, Quantum Chemistry, TU Darmstadt, Peter-Grünberg-Str. 4, 6, 4287 Darmstadt, Germany
First published on 17th June 2024
Abstract
The coordination of the Lewis superacid tris(pentafluorophenyl)alane (AlCF) to phosphine-supported, group 6 bis(dinitrogen) complexes [ML2(N2)2] is explored, with M = Cr, Mo or W and L = dppe (1,2-bis(diphenylphosphino)ethane), depe (1,2-bis(diethylphosphino)ethane), dmpe (1,2-bis(dimethylphosphino)ethane) or 2 × PMe2Ph. Akin to tris(pentafluorophenyl)borane (BCF), AlCF can form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts by coordination to one distal nitrogen of general formula trans-[ML2(N2){(μ-η1:η1-N2)Al(C6F5)3}]. The boron and aluminium adducts are structurally similar, showing a comparable level of N2 push–pull activation. A notable exception is a bent (BCF adducts) vs. linear (AlCF adducts) M–N–N–LA motif (LA = Lewis acid), explained computationally as the result of steric repulsion. A striking difference arose when the formation of two-fold adducts was conducted. While in the case of BCF the 2:1 Lewis pairs could be observed in equilibrium with the 1:1 adduct and free borane but resisted isolation, AlCF forms robust 2:1 adducts trans-[ML2{(μ-η1:η1-N2)Al(C6F5)3}2] that isomerise into a more stable cis configuration. These compounds could be isolated and structurally characterized, and represent the first examples of trinuclear heterometallic complexes formed by Lewis acid–base interaction exhibiting p and d elements. Calculations also demonstrate that from the bare complex to the two-fold aluminium adduct, substantial decrease of the HOMO–LUMO gap is observed, and, unlike the trans adducts (1:1 and 1:2) for which the HOMO was computed to be a pure d orbital, the one of the cis-trinuclear compounds mixes a d orbital with a π* one of each N2 ligands. This may translate into a more favourable electrophilic attack on the N2 ligands instead of the metal centre, while a stabilized N2-centered LUMO should ease electron transfer, suggesting Lewis acids could be co-activators for electro-catalysed N2 reduction. Experimental UV-vis spectra for the tungsten family of compounds were compared with TD-DFT calculations (CAM-B3LYP/def2-TZVP), allowing to assign the low extinction bands found in the visible spectrum to unusual low-lying MLCT involving N2-centered orbitals. As significant red-shifts are observed upon LA coordination, this could have important implications for the development of visible light-driven nitrogen fixation.
:1 adducts by coordination to one distal nitrogen of general formula trans-[ML2(N2){(μ-η1:η1-N2)Al(C6F5)3}]. The boron and aluminium adducts are structurally similar, showing a comparable level of N2 push–pull activation. A notable exception is a bent (BCF adducts) vs. linear (AlCF adducts) M–N–N–LA motif (LA = Lewis acid), explained computationally as the result of steric repulsion. A striking difference arose when the formation of two-fold adducts was conducted. While in the case of BCF the 2:1 Lewis pairs could be observed in equilibrium with the 1:1 adduct and free borane but resisted isolation, AlCF forms robust 2:1 adducts trans-[ML2{(μ-η1:η1-N2)Al(C6F5)3}2] that isomerise into a more stable cis configuration. These compounds could be isolated and structurally characterized, and represent the first examples of trinuclear heterometallic complexes formed by Lewis acid–base interaction exhibiting p and d elements. Calculations also demonstrate that from the bare complex to the two-fold aluminium adduct, substantial decrease of the HOMO–LUMO gap is observed, and, unlike the trans adducts (1:1 and 1:2) for which the HOMO was computed to be a pure d orbital, the one of the cis-trinuclear compounds mixes a d orbital with a π* one of each N2 ligands. This may translate into a more favourable electrophilic attack on the N2 ligands instead of the metal centre, while a stabilized N2-centered LUMO should ease electron transfer, suggesting Lewis acids could be co-activators for electro-catalysed N2 reduction. Experimental UV-vis spectra for the tungsten family of compounds were compared with TD-DFT calculations (CAM-B3LYP/def2-TZVP), allowing to assign the low extinction bands found in the visible spectrum to unusual low-lying MLCT involving N2-centered orbitals. As significant red-shifts are observed upon LA coordination, this could have important implications for the development of visible light-driven nitrogen fixation.
Introduction
Since the discovery of the first transition metal (TM) dinitrogen complex in 1965,1 the quest for an efficient and mild process for dinitrogen transformation embodies an ultimate goal for chemists. Although much progress has been made in the last two decades in the field of artificial nitrogen fixation, the number of catalytic systems for N2 conversion under homogeneous conditions remains limited.2,3 Therefore, new molecular design strategies must be explored to overcome the current scientific barriers and to gain access to optimised N2 conversion.Donor–acceptor activation is a strategy that has not been largely implemented in N2 chemistry involving molecular complexes. This parallels neither its success for other small molecules activation, e.g., CO24–6 or H2,7–9 both well exemplified through frustrated Lewis pair (FLP) chemistry10–17 and metal–ligand cooperativity,18,19 nor the fact that this concept finds echo in the two main processes responsible for nitrogen fixation. With regard to the nitrogenase enzymes, the “push–pull hypothesis” surmises that the acidic residues found in the active site build H-bonds with the distal N of N2 bound to the FeMo-cofactor,20–24 thus increasing polarization of the diatomic molecule and facilitating its protonation.25,26 Besides, promotion of the Haber–Bosch catalysts with electropositive elements lowers the barrier for N2 dissociation due to electrostatic effects, which can be seen as another manifestation of N2 donor–acceptor activation.27–30
At the molecular level, it can be achieved by the Lewis acid–base pairing of terminal N2 complexes with Lewis acids,31–33 which results in increased N2 polarisation due to enhanced back-bonding from the donor metal. This was pioneered by the Chatt group34–36 with neutral main-group Lewis acids, and was later further exemplified by the Fryzuk,37 Tuczek,38 Szymczak25 and Simonneau39,40 groups. Main group41,42 and transition metal cations43 have also been shown to participate in such type of donor–acceptor systems. The “donor” partner is generally an early-to-mid transition metal with a low formal oxidation state, although a model of purely main-group N2 donor–acceptor activation system was proposed by the Stephan group.44 By providing an access to a highly polarised N2 unit, opportunities for the discovery of new reactivity patterns for dinitrogen complexes can arise, for instance N2 protonation,25 silylation or borylation.39 In this context, the team of Szymczak and ours have focused on the coordination of the strong boron Lewis acid tris(pentafluorophenyl)borane, B(C6F5)3 (BCF) with a series of group 6 and 8 (M = Mo, W, and Fe) phosphine N2-complexes and have studied with DFT the implications for the N2 ligand.25,39,43 We have recently shown in a computational study that binding LAs to transition-metal N2-complexes may shift their molecular orbital ordering.45 Thus, by levelling basicity and redox potentials, Lewis acid coordination to the N2 ligand may be an interesting way to mitigate overpotentials in homogeneous ammonia synthesis (electro)catalysed with metal complexes. Recently, we turned our interest towards Lewis Super Acids (LSA),46 driven by the curiosity of gauging the push–pull effect when the acceptor is an extremely electron-deficient species. We have shown that a two-channel activation by the means of a strongly electrophilic bis(borane) C6F4{B(C6F5)2}2 (B2CF) imparts significant activation to the diatomic molecule, up to the diazene-diide (N22−) state.40 In the continuation of this work, we decided to study the coordination of the aluminium analogue of BCF, tris(pentafluorophenyl)alane – Al(C6F5)3 (AlCF)47–56 – to group 6 dinitrogen complexes, in order to assess how the resulting Lewis pairs differ or not in terms of structure and reactivity with respect to BCF.
AlCF is structurally close to BCF as they both feature three C6F5 ligands in their coordination sphere and a central trivalent group 13 element, differing by their metal radii and electronegativity.57 This apparently anecdotic distinction turns out to change quite significantly their chemical properties. As a matter of fact, while BCF is notably stable in a trigonal planar geometry and do not interact with weak and even moderate donors (such as non-polar and aromatic molecules and even oxygen-based compounds),58,59AlCF is highly reactive (thermal and shock sensitive) in this configuration and is only stable in a tetrahedral environment where the 4th position is occupied by a weak donor49,51,53,60 or, in its unsolvated dimeric form, through double Al–F interactions between Al and the ortho-F atom of one C6F5 ring.53 This singular aspect to form adducts with very weak donors (vide infra) suggests indeed higher electrophilic properties of AlCFvs.BCF, and it is now widely accepted that AlCF has a much stronger Lewis acid character than BCF.56 From computational studies and compiled experimental data, AlCF is considered as an LSA, having a Fluoride Ion Affinity (FIA)61 – acknowledged to be a way to estimate Lewis acidity – of 530 kJ mol−1. In ascending order, the latter has an FIA higher than B2CF (523 kJ mol−1), SbF5 (490 kJ mol−1) (the reference of the LSA scale), and much higher than BCF (450 kJ mol−1).53,56,62–65
In this work, we describe a new family of AlCF adducts with Chatt-Hidai type group 6 dinitrogen complexes, by the means of spectroscopy (NMR, IR, UV-vis), single crystal X-ray diffraction (sc-XRD), and DFT calculations. Notable chemical and structural discrepancies are observed by comparison to BCF (see Fig. 1), which are duly highlighted throughout the article. Remarkably, the switch from boron to aluminium allowed us to isolate bis(μ-η1:η1-N2–AlCF) adducts that remained elusive in the case of BCF. These are the first examples of neutral two-fold adducts of a main group Lewis acid with a bis(dinitrogen) complex.
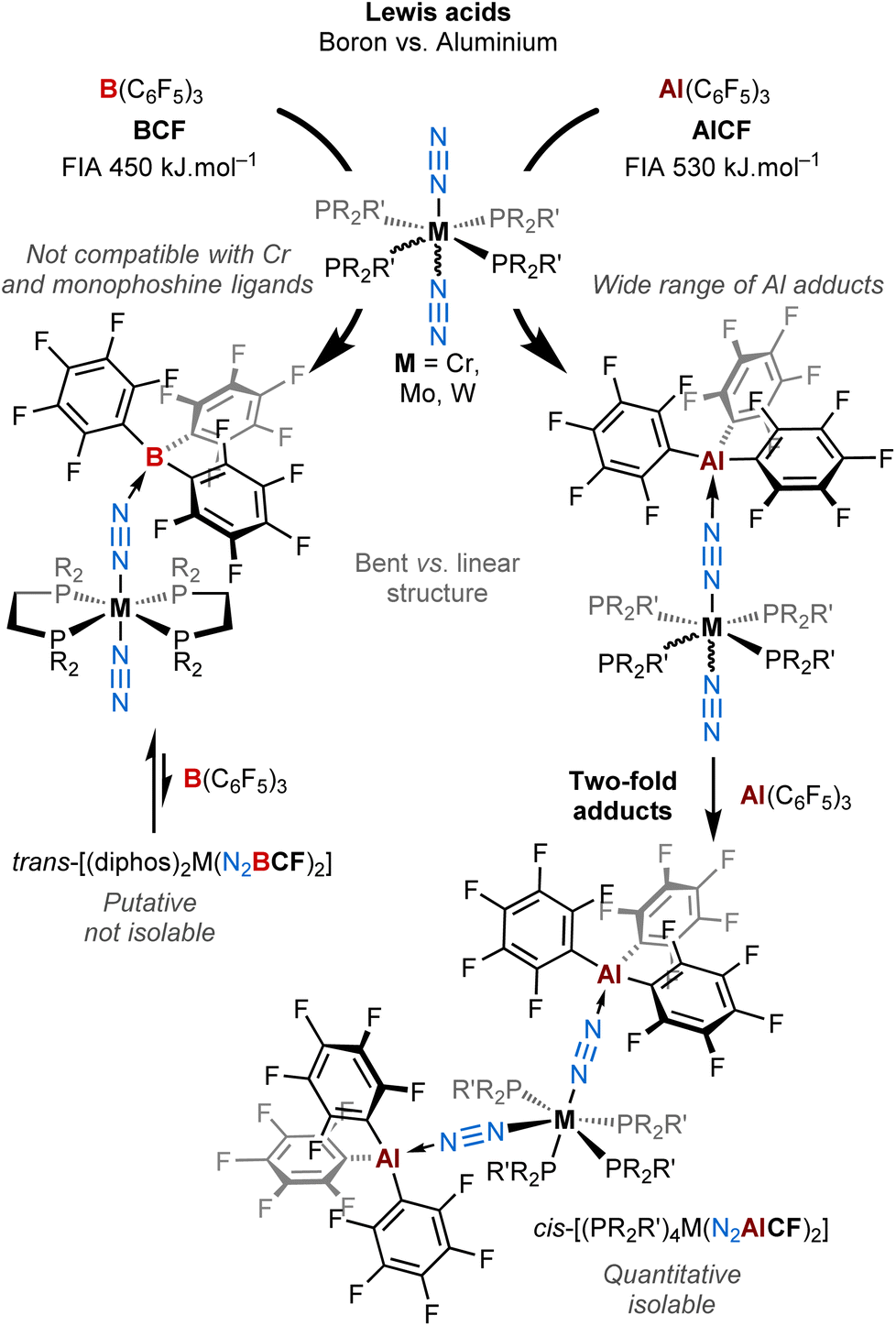 | ||
| Fig. 1 Coordination of AlCFversusBCF on Group 6 end-on dinitrogen complexes leading to a new family of mono and double Al dinitrogen adducts. | ||
Results and discussion
Syntheses of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 adducts supported with bis(phosphines)
:1 adducts supported with bis(phosphines)
Stoichiometric treatment (1:1) of tris(pentafluorophenyl)alane toluene adduct51 with a series of dinitrogen complexes trans-[ML2(N2)2] in toluene (M = W, Mo, and Cr; L = 1,2-bis(diethylphosphino)ethane [depe] or 1,2-bis(diphenylphosphino)ethane [dppe] or 1,2-bis(dimethylphosphino)ethane [dmpe])43,66–75 under a dinitrogen atmosphere produced new 1:1 adducts [ML2(N2)(μ-N2)Al(C6F5)3] 1Al, 2Al, 5Al, 6Al, and 7Al of trans configuration in moderate to excellent yields (Scheme 1 and Table 1). Note that better results in terms of analytical purity, yields, and reproducibility have been obtained with the depe and dmpe series (see ESI†). Adducts 1Al, 2Al, 5Al, 6Al and 7Al were characterised in solution and in the solid-state by multi-nuclei NMR and IR spectroscopies as well as single-crystal XRD analysis. Similarities are found between the depe-supported complexes 1Al, 2Al and their boron analogues 1B, 2B. Indeed, NMR signatures of these species are very close especially when considering their 31P NMR resonance (see Table 3). Coordination of the LA (BCF or AlCF) at the distal nitrogen of the N2 fragment induces a nearly equal bathochromic shift of the μ-N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N IR band and hypsochromic shift of the terminal NN stretching mode (see Table 3).
N IR band and hypsochromic shift of the terminal NN stretching mode (see Table 3).
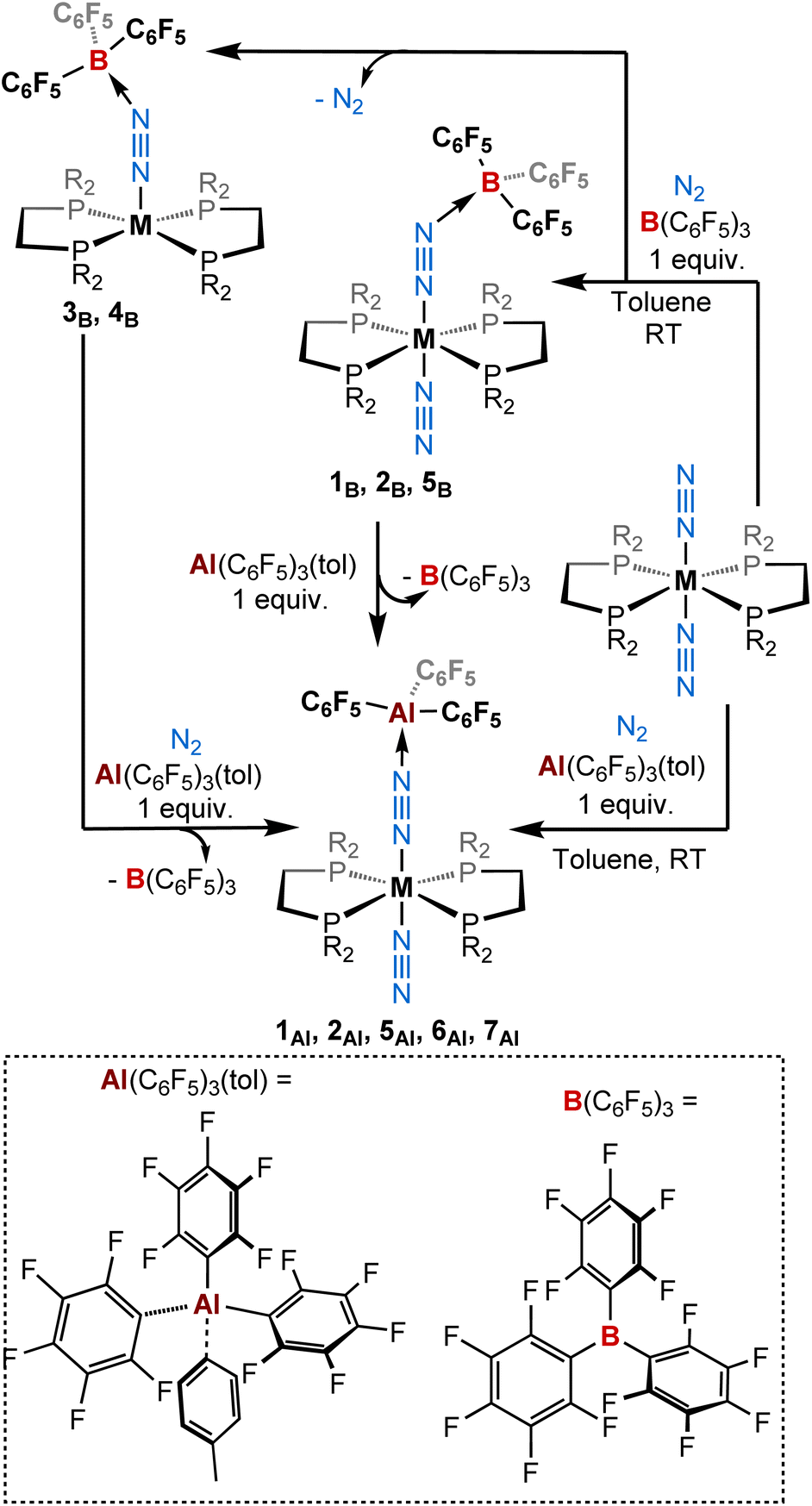 | ||
| Scheme 1 Reactivity of ML2(N2)2 (M = W, Mo, Cr; L = depe, dppe, dmpe) complexes with (top) B(C6F5)3 and (bottom) Al(C6F5)3(tol) (1 equiv.) under a dinitrogen atmosphere. | ||
| Compound | LAa | M | R | Config. | N2 motifs | Yield (%) |
|---|---|---|---|---|---|---|
| a LA = Lewis acid. b NMR yield. | ||||||
| 1AL | AlCF | W | Et | trans | μ-N2, η-N2 | 89 |
| 1B 43 | BCF | W | Et | trans | μ-N2, η-N2 | 62 |
| 2AL | AlCF | Mo | Et | trans | μ-N2, η-N2 | 100 |
| 2B 43 | BCF | Mo | Et | trans | μ-N2, η-N2 | 53 |
| 3B | BCF | W | Ph | trans | μ-N2 | {31}b |
| 4B | BCF | Mo | Ph | trans | μ-N2 | {95}b |
| 5AL | AlCF | W | Ph | trans | μ-N2, η-N2 | 51 |
| 5B | BCF | W | Ph | trans | μ-N2, η-N2 | {69}b |
| 6AL | AlCF | Mo | Ph | trans | μ-N2, η-N2 | 81 |
| 7AL | AlCF | Cr | Me | trans | μ-N2, η-N2 | 79 |
Suitable single-crystals of 1Al and 2Al for XRD studies have been grown from a cold and saturated solution of toluene/n-pentane. The solid-state structures of 1Al and 2Al (see Fig. 3, left, for 1Al and ESI† for 2Al) depict a similar octahedral geometry around the group 6 metal to that of 1B and 2B. Expectedly, coordination of AlCF to the distal N atom in complexes 1Al and 2Al imparts a tetrahedral geometry around the Al center (angles averaged at 109.5° for 1Al and 108.0° for 2Al). The N1–N2 bond lengths are similar between the aluminium and boron analogues (see Table 3). The TM-N1 distance is slightly shortened in the case of aluminium adducts (W–N1 = 1.855 Å for 1Alvs. 1.909 Å for 1B and Mo–N1 = 1.869 Å for 2Alvs. 1.894 Å for 2B).
Overall, these experimental data point to a diminished bond order for the N2 unit as a result of enhanced back-donation with a similar “push–pull” activation level of μ-N2 in species 1Al, 2Al and 1B, 2B. However, we noticed structural divergences between 1Al, 2Al and their boron analogues 1B, 2B. According to the Cambridge Structural Database (CSD), the Al1–N2 bond lengths – 1.817 Å for 1Al and 1.842 Å for 2Al – are found to be the shortest ones compared to the expected Al–N distances range for similar reported N–Al(C6F5)3 motifs (from 1.853 Å76,77 to 2.167 Å78) and N2–AlR3 (R = alkyl) fragments (from 1.929 Å79 to 2.089 Å80). On the other hand, the B–N2 length for the boron congeners (1.549 Å for 1B and 1.562 Å for 2B) are found in the expected B–N distances range for similar reported N–B(C6F5)3 moieties (from 1.492 Å81 to 1.807 Å82) but are slightly below the B–N distances range for comparable bridging diazo borane (μ-N2)–B(C6F5)3 and azido borane (μ-N3)–B(C6F5)3 fragments (from 1.575 Å25 to 1.678 Å38). These results thus advocate for the presence of robust interactions between the bridging N2 and the LA centre, more prominent in the case of aluminium.
Experimentally, we demonstrated the stronger affinity of μ-N2 motif for AlCFvs.BCF by treating adduct 1B with one equivalent of Al(C6F5)3(tol) that leads to the formation of 1Al and free BCF with an NMR yield higher than 90% (see Scheme 1 and ESI†). Note that over time this equilibrium does not evolve showing that the formation of 1Al from 1B is thermodynamically favourable. This set of clues led us to analyse the N1–N2–LA angle. In the case of the aluminium adducts, a nearly straight N1–N2–Al angle is found – 168.4° and 167.8° for 1Al and 2Al, respectively. These data conflict with similar reported N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Al angles of bridging diazenido trialkylaluminum (μ-NN)–AlR3 and azido trialkylaluminum (μ-NNN)–AlR3 species featuring values ranging from 105.5°80 to 158.9°.84 These results also contrast with the bent N1–N2–B angle found for the boron analogues – 148.4° for 1B and 150.9° for 2B. While [M(depe)2(N2)2] and [M(dppe)2(N2)2] cleanly reacted with BCF to form quantitatively 1:1 adducts, we observed significant divergent behaviours when we engaged trans-[Cr(dmpe)2(N2)2] with BCF. Indeed, this leads to a partial and unselective reaction towards a complex mixture of species (starting materials in equilibrium with other species, see ESI†) that we were not able to isolate from each other in the solid-state. Among them, we can assume that the 1:1 adduct is partly formed. On the opposite, the stoichiometric reaction of Al(C6F5)3(tol) with [Cr(dmpe)2(N2)2] cleanly produced a new 1:1 adduct 7Al in good yields (Scheme 1, bottom). Spectroscopic and crystallographic parameters of 7Al are nearly identical to those of tungsten and molybdenum analogues 1Al and 2Al (see Fig. 3 and Table 3) showing a similar N2 activation degree (close N–N distances and NN IR stretches) and AlCF coordination (close Al–N distances and Al–N–N angles in particular).
N–Al angles of bridging diazenido trialkylaluminum (μ-NN)–AlR3 and azido trialkylaluminum (μ-NNN)–AlR3 species featuring values ranging from 105.5°80 to 158.9°.84 These results also contrast with the bent N1–N2–B angle found for the boron analogues – 148.4° for 1B and 150.9° for 2B. While [M(depe)2(N2)2] and [M(dppe)2(N2)2] cleanly reacted with BCF to form quantitatively 1:1 adducts, we observed significant divergent behaviours when we engaged trans-[Cr(dmpe)2(N2)2] with BCF. Indeed, this leads to a partial and unselective reaction towards a complex mixture of species (starting materials in equilibrium with other species, see ESI†) that we were not able to isolate from each other in the solid-state. Among them, we can assume that the 1:1 adduct is partly formed. On the opposite, the stoichiometric reaction of Al(C6F5)3(tol) with [Cr(dmpe)2(N2)2] cleanly produced a new 1:1 adduct 7Al in good yields (Scheme 1, bottom). Spectroscopic and crystallographic parameters of 7Al are nearly identical to those of tungsten and molybdenum analogues 1Al and 2Al (see Fig. 3 and Table 3) showing a similar N2 activation degree (close N–N distances and NN IR stretches) and AlCF coordination (close Al–N distances and Al–N–N angles in particular).
DFT investigation on the N–N–LA angle
To shed light into these results, DFT calculations at the BP86/def2-TZVP level of theory were employed, including implicit solvation and dispersion corrections, starting from the crystal structures of the BCF and AlCF adducts. One explicit solvent molecule was added to the molecular models due to the aforementioned greater stability of AlCF in a tetrahedral environment; this is required for the analysis of the thermodynamics behind the LA binding.The potential energy surface (PES) minima found upon geometry relaxation match well with experiment: M–N1 and N2–LA bonds were only ca. 0.050 Å longer than the experimental ones and other deviations were even smaller. The N1–N2–LA angles obtained for the computed structures of AlCF and BCF adducts were 170° and 148°, respectively. A detailed comparison of the computational and experimental structural and spectroscopic features is included in the ESI (Tables S5 and S6).† To further elucidate the PES regarding the binding angle of the LAs, constrained geometry optimisations with varying N–N–LA angles (148° to 172°) were carried out for both LA adducts (Fig. 2). The NN–B angle is in fact extremely flexible: in the case of the least sterically impeded adduct, we observe the energy minimum at ca. 150° (in agreement with experimental data) and a small dent close to 165°, separated by less than 0.5 kcal mol−1. The latter is not a local minimum as it is due to a ca. 10° rotation of one ethyl phosphine substituent. The increase in energy along the bending motion is, overall, meagre, with an energetic cost of less than 1 kcal mol−1. For the AlCF 1:1 adduct, in contrast, a continuous and steeper increase in energy is observed as the NN–LA angle is decreased.
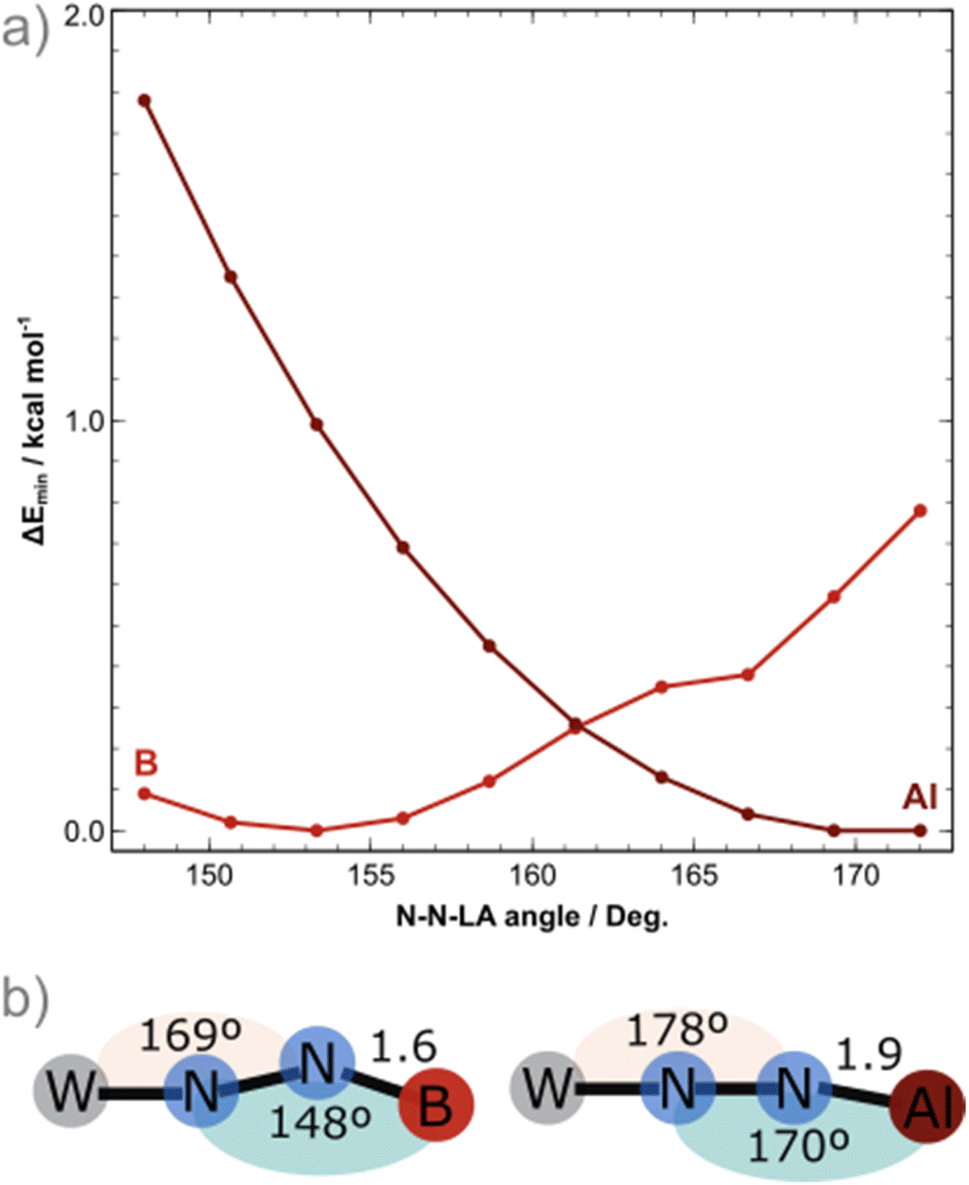 | ||
| Fig. 2 (a) Potential energy surface along the N–N–Lewis acid angle coordinate for the AlCF (crimson) and BCF (red) 1:1 adducts, energies reported relative to PES minimum; and (b) relevant angles of the optimized structures. | ||
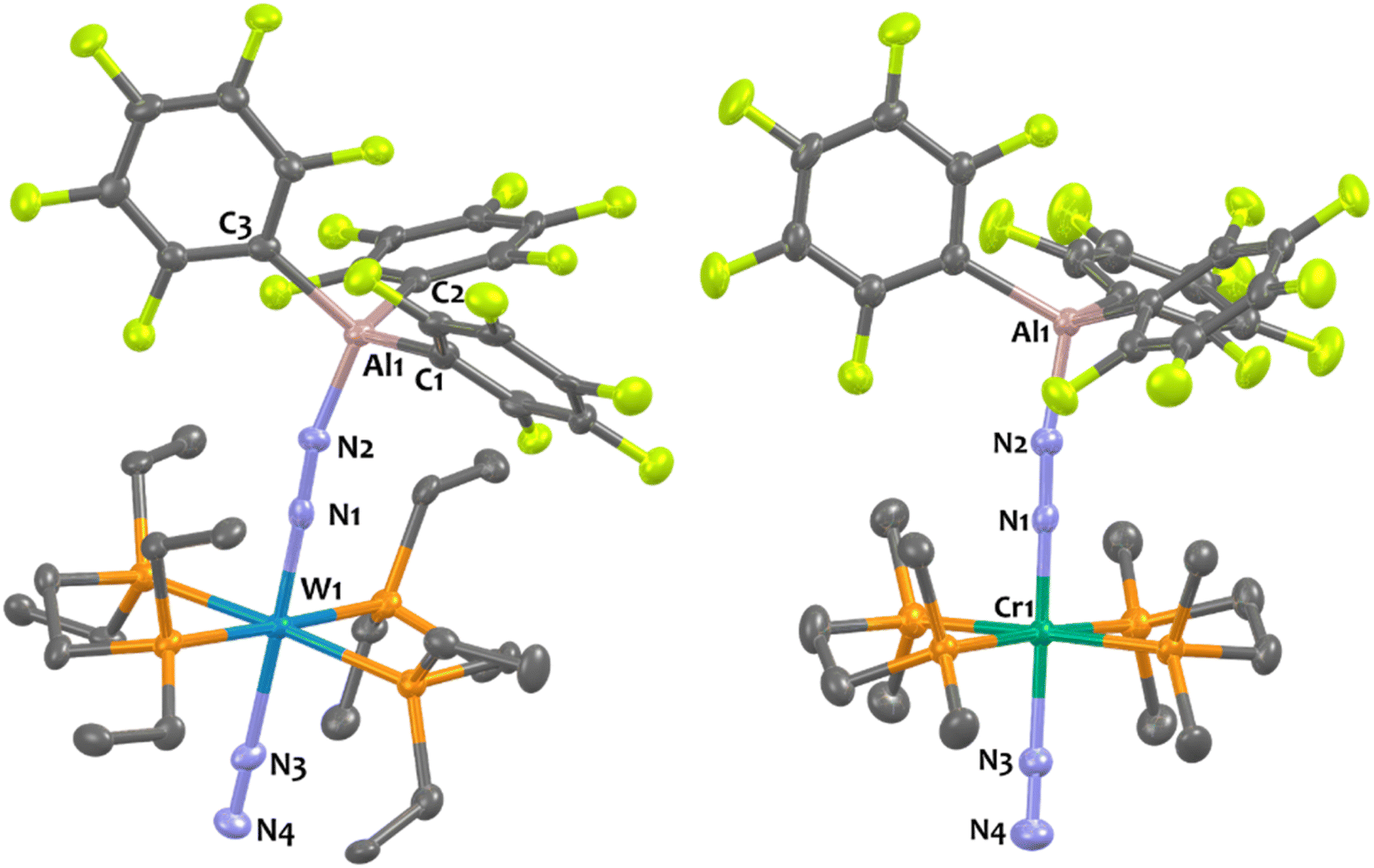 | ||
| Fig. 3 Solid-state structures of 1Al and 7Al. Ellipsoids are represented with 30% probability. Hydrogen atoms have been omitted for clarity. Two independent molecules were found in the asymmetric unit (Z′ = 2) of 1Al but one of them has been omitted for clarity. Selected bond distances (Å) and angles (°) have been averaged between both independent molecules for 1Al: Al1–N2 1.816(7), W1–N1 1.855(2), W1–N3 2.113(2), N1–N2 1.203(6), N3–N4 1.114(1), W1–N1–N2 178.6(6), W–N3–N4 177.2(6), N1–W1–N3 177.3(6), N1–N2–Al1 168.3(6). For 7Al: Al1–N2 1.8473, Cr1–N1 1.7507, Cr1–N3 1.9766, N1–N2 1.177(2), N3–N4 1.100(3), N3–Cr1–N1 177.47(7), N2–N1–Cr1 178.55(2), N4–N3–Cr1 178.02(18), N1–N2–Al1 170.26(2). | ||
There is a marginal stabilisation of the frontier occupied orbitals in both adducts as the angle is bent from 148° to 172° (Fig. S93†), suggesting that the differing angles obtained in the crystal structures are not rooted in electronic structure stabilisation effects but are instead mainly due to steric hindrances. Note that for the NN–LA angle to bend (blue in Fig. 1b), a simultaneous bending of the M–N1N2 angle (peach, in Fig. 1b) by 9° occurs to better accommodate the LA around the phosphine ligand arms. This is true for both LAs.
Influence of the atmosphere: N2vs. Ar
It is important to mention that for adducts in the depe and dmpe series we observed the same reactivity whether working under dinitrogen or argon. Nevertheless, we noticed significant divergences for the dppe series. Indeed, when using B(C6F5)3, our group had previously observed the elimination of one dinitrogen molecule during the reaction leading to the formation of [M(dppe)2(μ-N2)B(C6F5)3] adducts where the apical site (left vacant by N2 dissociation) is occupied by an agostic interaction with an ortho hydrogen of one of the phenyl groups in the solid-state.39 This process occurred under argon (Scheme 2, middle). Under dinitrogen, we noticed the same reactivity for trans-[Mo(dppe)2(N2)2] (i.e. loss of one of the N2 ligands) but the stoichiometric treatment of trans-[W(dppe)2(N2)2] with BCF leads, after one night, to a mixture of [W(dppe)2(μ-N2)B(C6F5)3] 3B and trans-[W(dppe)2(N2)(μ-N2)B(C6F5)3] 5B in a 31:69 3B:5B ratio, respectively. Of note, species 5B was observed in solution but we did not succeed to isolate it (see Scheme 1, top left, and ESI†).
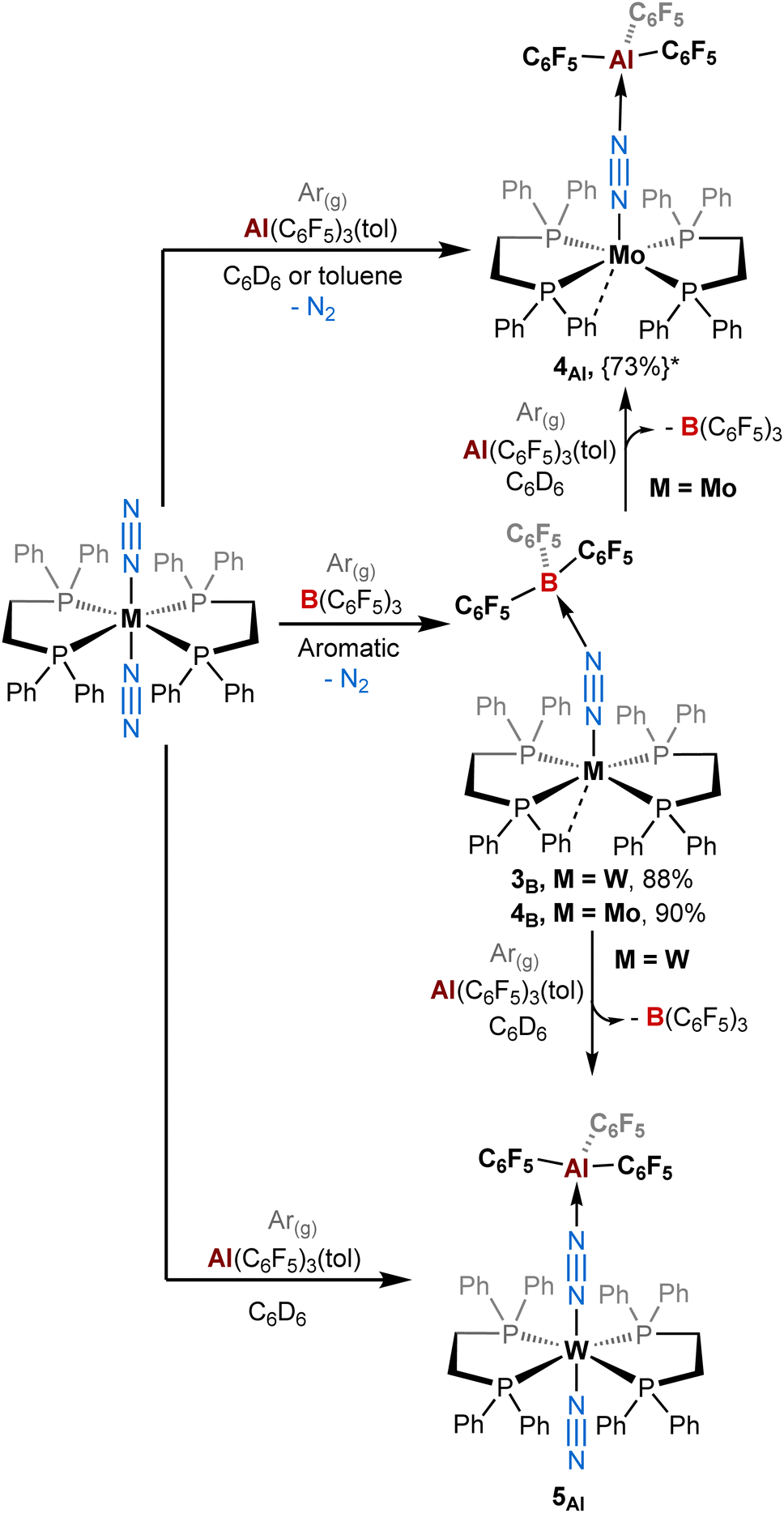 | ||
| Scheme 2 Reactivity of ML2(N2)2 (M = W, Mo; L = dppe) complexes with B(C6F5)3 and Al(C6F5)3(tol) under an argon atmosphere. {The yield in brackets followed by a star*} represents the NMR yield. The other complexes (L = depe or dmpe) reacted similarly as under a dinitrogen atmosphere (see Scheme 1). | ||
By contrast, the reaction of Al(C6F5)3 with trans-[M(dppe)2(N2)2] (M = Mo and W) does not promote the elimination of N2 when working under a dinitrogen atmosphere. This leads instead to a similar reactivity to that of the depe series i.e. the formation of products 5Al and 6Al where the terminal N2 stays bonded to the metal centre (Scheme 1). Under an inert atmosphere of argon, the stoichiometric treatment of AlCF with trans-[Mo(dppe)2(N2)2] leads predominantly (73% NMR yield, see ESI†) to the formation of the aluminium analogue of 4B, [Mo(dppe)2(μ-N2)AlCF] 4Al, in which the second dinitrogen ligand is lost during the reaction (Scheme 2, top). Identity of adduct 4Al is successfully established by XRD studies. It should be noted, however, that the quality of XRD data was not good enough to discuss the metrical parameters in great detail but confirmed the atom connectivity and loss of one N2 ligand (see ESI†).
Surprisingly, changing from Mo to W drastically impacts this chemistry since the 1:1 reaction of trans-[W(dppe)2(N2)2] with AlCF under argon does not trigger N2 dissociation and instead promotes the quantitative formation of 5Al as under a dinitrogen atmosphere (see Scheme 2, bottom, and ESI†). This highlights the sensitivity of these species towards the retention of their second N2 ligand, depending whether the reaction medium is N2-saturated or not. We assumed that formation of adducts 3–4 involves first the formation of 5–6 as intermediates (coordination of the LA at the distal N), which can then lose their terminal N2 ligand depending on the LA and the atmosphere. In this case, this second step is more feasible (in ascending order) for 4B > 3B > 4Al > 3Al. This translates into Mo- and/or B-containing species having a greater tendency to labilise the trans-N2 ligand. Spectroscopic and crystallographic data of 3–4vs.5–6 revealed distinct features (Table 3). The IR μ-NN stretching mode is shifted to lower wavenumbers for adducts 3–4vs.5–6 and the bridging N–N distances are elongated in adducts 3–4vs.5–6. Therefore, the elimination of the terminal dinitrogen molecule induces a stronger polarisation of the M–NN–LA fragment (3B, 4Bvs.5Al and 6Al). Notably, comparable crystallographic data between 5Al, 6Al and 1Al, 2Al are found when it comes to the LA–N2 distance and N1–N2–LA angle i.e. a short Al–N separation and a nearly linear Al–NN array, again contrasting with the boron adducts 3B and 4B featuring bent B–NN angles (see Table 3).
Also, similarly to the depe series, the stronger affinity of N2 metal complexes for AlCFvs.BCF in the dppe series was verified experimentally by treating the BCF adducts 3B–4B with one equivalent of AlCF producing instantly (whether working under argon or dinitrogen) the aluminium adducts 4Al, 5Al, and 6Al and free BCF (see Scheme 1, left, Scheme 2, and ESI†). These reactions demonstrate the stronger affinity of AlCFvs.BCF for the dinitrogen ligand.
Syntheses of 2:1 adducts
Since we employed bis(dinitrogen) complexes as Lewis base partner, we were curious to know whether the reaction of [ML2(N2)2] with two equivalents of the Lewis acid (AlCF or BCF) could provide 2:1 adducts. When we added two equivalents of B(C6F5)3 to [M(depe)2(N2)2] (M = Mo, W) we noticed an immediate colour change from orange-brown to deep purple-blue. While in the case of molybdenum, NMR analyses suggested some degradation occurring upon addition of the second equivalent of BCF, the spectra recorded when the W species was employed suggests the formation of a new putative complex 8B (see Scheme 3, top, and Table 2). This is evidenced by a gain in symmetry as indicated by undifferentiated alkyl protons in the 1H NMR spectrum, as opposed to 1B (see ESI†). However, we also cannot exclude that such 1H NMR spectrum results from signal coalescence of 1B due to a concentration phenomenon as already observed in the case of 1Al (Fig. S1–S3 and S40†). This could explain why the 31P NMR spectrum showed no change with respect to the mono adduct 1B (δ = 34.7 ppm). Surprisingly, only two large signals are observed in 19F NMR, contrasting with the well-resolved multiplets characterizing ortho, meta and para fluorine resonances in 1B. This may suggest either a fluxional behaviour of 8B or that a fast 1B + BCF ⇌ 8B equilibrium takes place at room temperature. Measuring 1H and 19F NMR at low temperature (down to −60 °C) resulted in de-coalescence of the signals. In particular, broad resonances, which chemical shifts match those of 1B and free BCF, are found in the 19F NMR spectrum, pointing to an equilibrated mixture. Shoulders on the peaks of the para and meta fluorine of 1B might be assigned to the two-fold adduct 8B (Scheme 1, top-right, and Fig. S40†). Unfortunately, our attempts to isolate such a two-fold adduct were unsuccessful: crystals of 1B were systematically collected from the purple solutions.
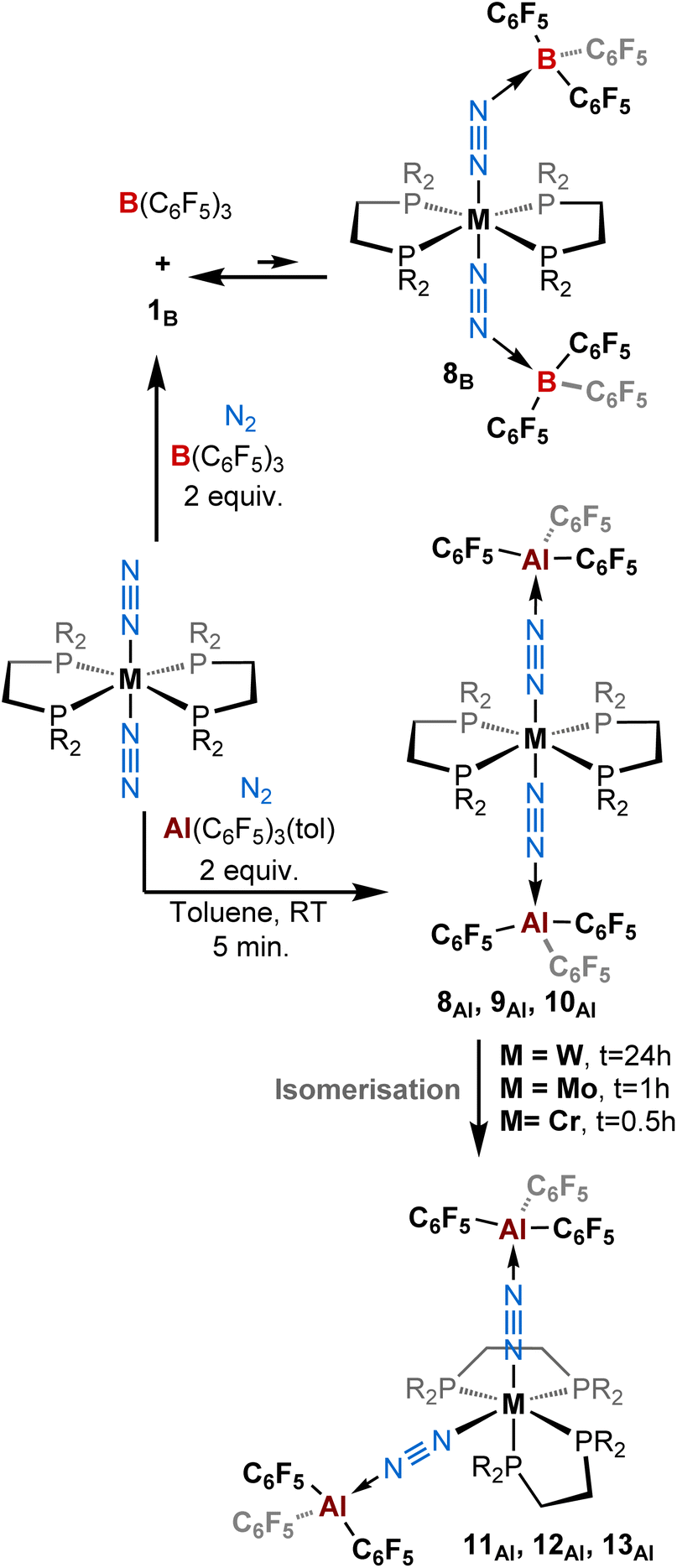 | ||
| Scheme 3 Reactivity of [ML2(N2)2] (M = W, Mo, Cr; L = depe, dppe, dmpe) complexes with (top) two equivalents of B(C6F5)3 and (bottom) two equivalents of Al(C6F5)3(tol) under a dinitrogen atmosphere. | ||
| Compound | LAa | M | R | Config. | N2 motifs | Yield (%) |
|---|---|---|---|---|---|---|
| a LA = Lewis acid. b n.i. = not isolated. | ||||||
| 8Al | AlCF | W | Et | trans | 2× μ-N2 | 96 |
| 8B | BCF | W | Et | trans | 2× μ-N2 | n.i.b |
| 9Al | AlCF | Mo | Et | trans | 2× μ-N2 | n.i.b |
| 10Al | AlCF | Cr | Me | trans | 2× μ-N2 | n.i.b |
| 11Al | AlCF | W | Et | cis | 2× μ-N2 | 77 |
| 12Al | AlCF | Mo | Et | cis | 2× μ-N2 | 94 |
| 13Al | AlCF | Cr | Me | cis | 2× μ-N2 | 77 |
Gratifyingly, treatment of trans-[M(depe)2(N2)2] (M = Mo, W) and trans-[M(dmpe)2(N2)2] (M = Cr) with two equivalents of AlCF·toluene instantly triggered a quantitative reaction characterised by a colour change from reddish (ML2(N2)2 starting materials) to blue azure/greenish within seconds. We attributed this colour change to the formation of a 2:1 adduct with a trans-configuration – species 8Al (M = W), 9Al (M = Mo), and 10Al (M = Cr) (Scheme 3, Table 2, and Fig. 4, top). With time, we noticed an additional colour change from blue/green to brown/orange corresponding to the formation of another 2:1 adduct this time with a cis-configuration, namely products 11Al (M = W), 12Al (M = Mo), and 13Al (M = Cr) (Scheme 3, bottom, and Fig. 4, bottom).
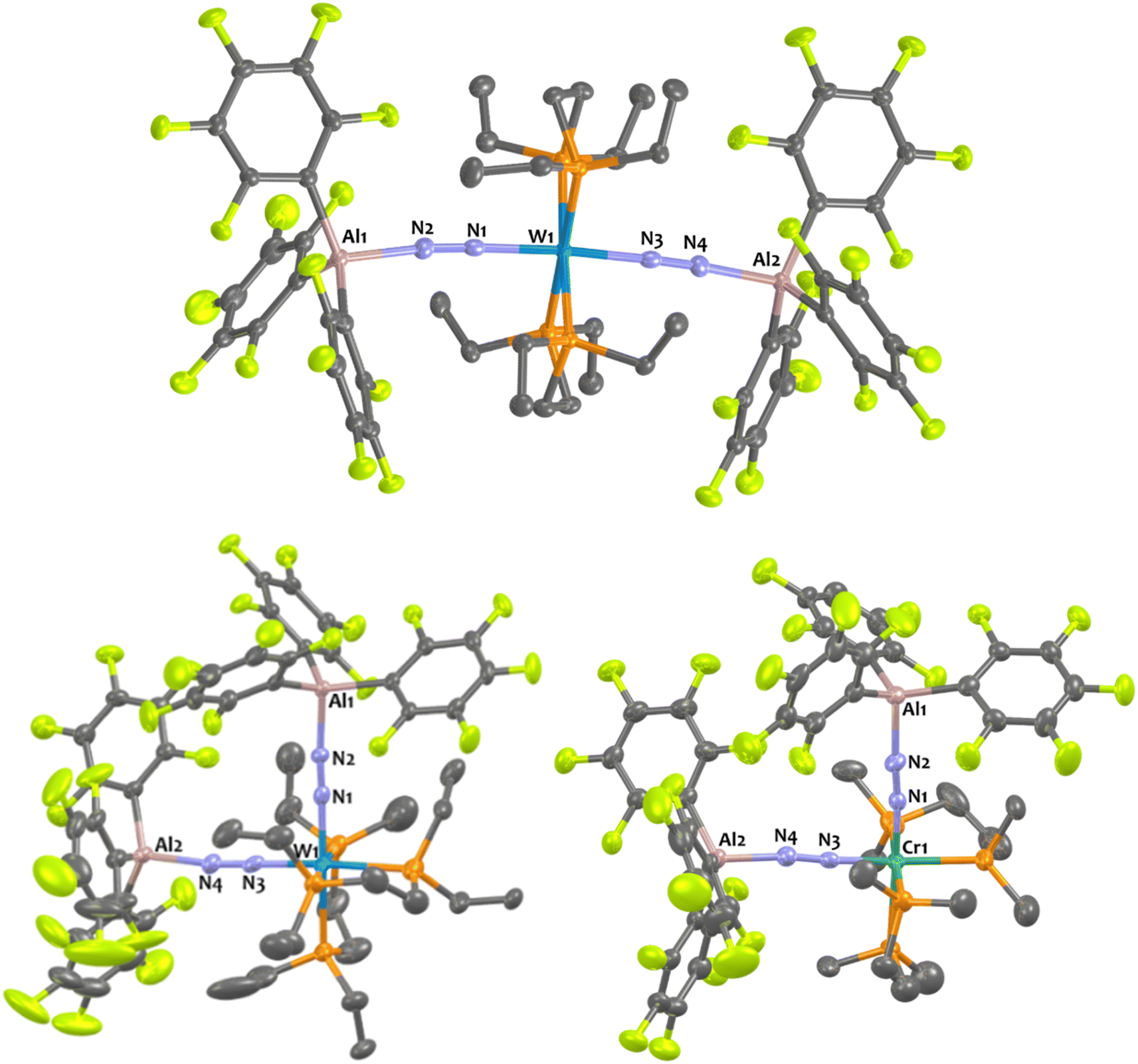 | ||
| Fig. 4 Solid-state structures of 8Al, 11Al, and 13Al. Ellipsoids are represented with 30% probability. Hydrogen atoms have been omitted for clarity. Two independent molecules were found in the asymmetric unit (Z′ = 2) of 11Al, and 13Al but one of them has been omitted for clarity. Selected bond distances (Å) and angles (°) have been averaged between both independent molecules for 11Al and 13Al. For 8Al: Al1–N2 1.927(2), Al2–N4 1.919(2), W1–N1 1.964(2), W1–N3 1.956(3), N1–N2 1.113(3), N3–N4 1.114(3), N3–W1–N1 174.13(7), N2–N1–W1 176.88(2), N4–N3–W1 177.09(2), N1–N2–Al1 168.64(2), N3–N4–Al2 175.22(2). For 11Al: Al1–N2 1.901(4), Al2–N4 1.894(0), W1–N1 1.919(9), W1–N3 1.901(0), N1–N2 1.144(6), N3–N4 1.156(6), W1–N1–N2 175.5(4), W1–N3–N4 175.8(4), N1–W1–N3 89.20(7), Al1–N2–N1 175.7(9), Al2–N4–N3 168.7(4). For 13AL Al1–N2 1.884(6), Al2–N4 1.899(0), Cr1–N1 1.784(2), Cr1–N3 1.778(9), N1–N2 1.156(1), N3–N4 1.157(1), Cr1–N1–N2 175.6(9), Cr2–N3–N4 176.0(9), N1–Cr1–N3 89.19(2), Al1–N1–N2 170.8(4), Al2–N4–N3 172.0(0). | ||
In the case of tungsten, intermediate 8Al is stable enough (for one or two hours at room temperature) so that we succeeded to isolate it and analyse it by IR, XRD, and NMR. Then, 8Al is progressively (within one day) converted into product 11Al. However, intermediates 9Al (M = Mo) and 10Al (M = Cr) evolved within minutes towards products 12Al and 13Al, precluding their isolation (see ESI† for further details). The trans geometry of intermediate 8Al is first evidenced by its 1H NMR spectrum that exhibits 3 centrosymmetric signals (δ = 1.44, 1.13, and 0.68 ppm) and by its 31P NMR spectrum that displays a shielded pseudo-triplet (1JW–P = 141 Hz) at δ = 31.0 ppm (vs. 34.6 ppm for 1Al). This configuration is confirmed by its structure in the solid-state (Fig. 4, top). Here, the Al1–N2–N1–W1–N3–N4–Al2 atoms are almost perfectly aligned. Also, the coordination of a second AlCF moiety imparts a significant shortening of the N–N bonds (1.11 Å vs. 1.20 Å in 1Al) and elongation of the W–N1 (1.96 Å vs. 1.86 Å in 1Al) and Al–N (1.92 Å vs. 1.82 Å in 1Al) bonds (see Table 3) showing a decreased activation of the bridging dinitrogen fragments. These features are verified by IR where the ATR spectrum of 8Al displays a single bridging N2 stretch at higher wavenumber to that of 1Al (1808 vs. 1778 cm−1). Based on these data, we propose a formal bridging Al–NN–M depiction. The cis arrangement of products 11Al and 12Al is first demonstrated by NMR spectroscopy as their 1H NMR spectra display asymmetrical depe resonances (see Fig. S44 and S54†) and their 31P NMR spectra feature two triplets (2JP–P = 6 Hz and 14 Hz for 11Al and 12Al), each integrating for 2P (see Table 3 and ESI†). We could not analyse 13Al (M = Cr, L = dmpe) by NMR spectroscopy as this species was not soluble in chemically compatible deuterated solvents (even ortho-dichlorobenzene). IR-ATR spectra of 11Al, 12Al, and 13Al display two intense NN bands (see Table 3) assigned to symmetric and asymmetric N2 stretches. Eventually, solid-state structures of 11Al, 12Al, and 13Al (see Fig. 4-bottom and ESI†) confirmed the cis arrangement, with almost orthogonal N1–M–N3 angles. Of note, an elongation of the N–N bond is observed for 11Al when compared to the trans adduct 8Al (1.152 Å vs. 1.113 Å, respectively).
| Adduct | δ 31P NMRa | ν 1 (μ-N2) | ν 2 (N2) | N1–N2 | N3–N4 | N2–LAb | N4–LAb | M–N1 | M–N3 | N1–M–N3 | N1–N2–LAb | N3–N4–LAb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded in C6D6. b LA = Lewis acid. | ||||||||||||
| 1Al | 34.6 | 1778 | 2088 | 1.204 | 1.114 | 1.817 | — | 1.855 | 2.113 | 177.4 | 168.4 | — |
| 1B | 34.7 | 1767 | 2076 | 1.181 | 1.082 | 1.549 | — | 1.909 | 2.015 | 175.7 | 148.4 | — |
| 2Al | 52.6 | 1790 | 2137 | 1.168 | 1.103 | 1.842 | — | 1.869 | 2.128 | 177.7 | 167.8 | — |
| 2B | 53.0 | 1789 | 2120 | 1.175 | 1.093 | 1.562 | — | 1.894 | 2.129 | 176.3 | 150.9 | — |
| 3B | 69.2 | 1717 | — | 1.212 | — | 1.571 | — | 1.841 | — | — | 140.3 | — |
| 4Al | 70.9 | — | — | — | — | — | — | — | — | — | — | — |
| 4B | 73.1 | 1744 | — | 1.197 | — | 1.568 | — | 1.841 | — | — | 141.5 | — |
| 5Al | 45.4 | 1773 | 2121 | 1.181 | 1.090 | 1.865 | — | 1.885 | 2.108 | 177.2 | 169.3 | — |
| 6Al | 63.1 | 1786 | 2161 | 1.174 | 1.094 | 1.876 | — | 1.894 | 2.139 | 173.8 | 176.6 | — |
| 7Al | 62.3 | 1802 | 2122 | 1.177 | 1.100 | 1.847 | — | 1.751 | 1.977 | 177.5 | 170.3 | — |
| 8Al | 31.0 | 1808 | — | 1.113 | 1.114 | 1.927 | 1.919 | 1.964 | 1.956 | 174.1 | 168.6 | 175.2 |
| 11Al | 28.6, 16.6 | 1903, 1802 | — | 1.145 | 1.157 | 1.901 | 1.894 | 1.920 | 1.901 | 89.2 | 175.8 | 168.7 |
| 12Al | 44.4, 29.2 | 1927, 1821 | — | 1.148 | 1.138 | 1.892 | 1.907 | 1.910 | 1.921 | 88.6 | 173.9 | 170.7 |
| 13Al | — | 1948, 1833 | — | 1.156 | 1.157 | 1.885 | 1.899 | 1.784 | 1.779 | 89.2 | 170.8 | 172.0 |
| 15Al | −18.8, −22.1, −25.5 | 1776 | 2037 | — | — | — | — | — | — | — | — | — |
| 16Al | −24.3, −26.5 | 1901, 1804 | — | 1.149 | 1.150 | 1.915 | 1.891 | 1.917 | 1.906 | 91.3 | 168.5 | 179.1 |
DFT investigation on the 2:1 adduct formation
DFT calculations show that sequential binding of two equivalents of BCF or AlCF is thermodynamically favourable (Fig. 5), although binding of the second LA is associated with a relatively lower stabilisation of the adduct as may be expected from the trans effect. The individual contributions to the Gibbs energies can be found in the ESI, Tables S7 and S8.†
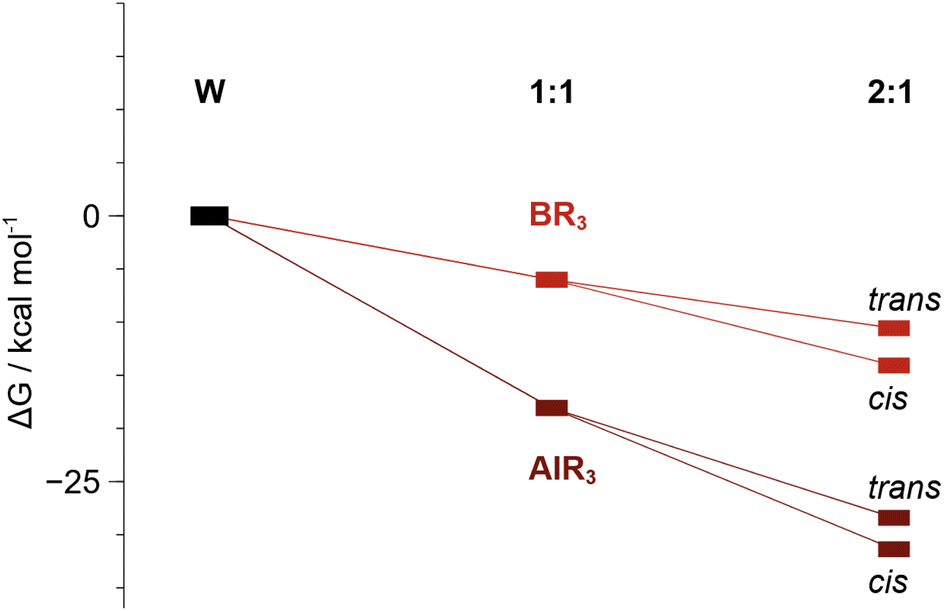 | ||
| Fig. 5 Relative Gibbs energies (kcal mol−1) of 1:1 (1B and 1Al) and 2:1 adduct formation. The cis isomer is more stable than the trans by ca. 3 kcal mol−1. | ||
The AlCF adducts are lower in relative energy than the BCF analogues. Conversion from trans to cis adducts was observed and indeed the cis isomeric form is shown to be more stable by 3.0 kcal mol−1 over the trans 2:1 adduct. We analysed the MO diagrams of the AlCF adduct series to rationalise the degree of dinitrogen activation observed (Fig. 6). The complete frontier MO diagram as well as the depiction of the orbitals for the bare tungsten depe complex can be found in the ESI (Fig. S92).†
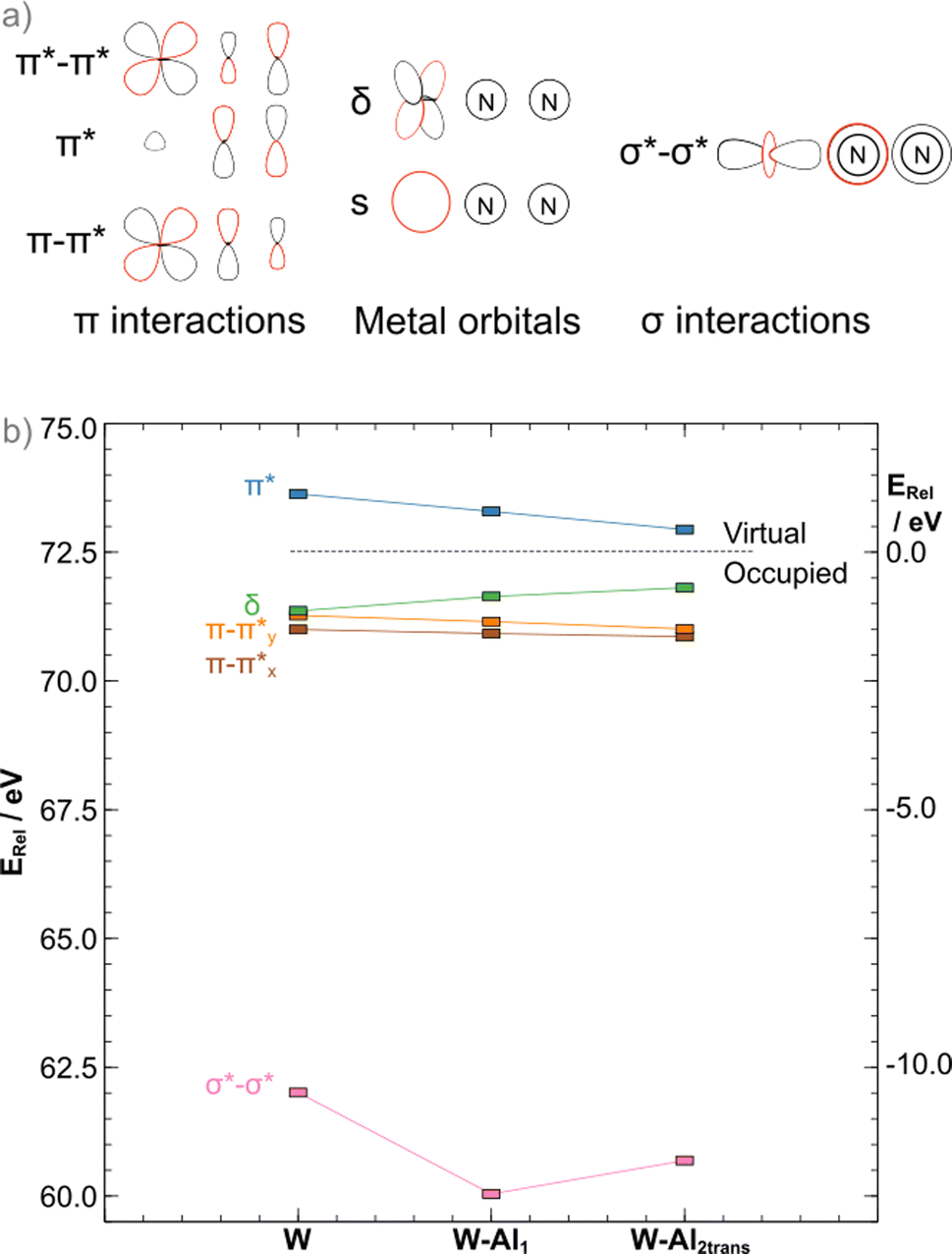 | ||
| Fig. 6 (a) 2D depiction of the orbitals involved in the “push–pull” activation and metal s orbital used as reference and (b) MO diagram for the Al series of adducts (blue: LUMO/LUMO+1 average; green: HOMO; orange: HOMO−1, brown: HOMO−2, pink: σ antibonding interaction within the bridge). Orbital energies are plotted relative to the tungsten s orbital and thus all are positive (left vertical axis). Energies relative to proximally the midpoint of the HOMO/LUMO gap shown on the right vertical axis. Nomenclature of the orbitals considers symmetry. | ||
The binding of LAs to the terminal atom of the nitrogen ligand has been shown to stabilise π* interactions in the N–N bridge, resulting in bond25 weakening. In the case of the depe complexes, the same is observed when the formation of the 1:1 adduct occurs as a ca. 0.10 eV stabilisation from the bare complex to the AlCF 1:1 adduct (νN–N = 1778 cm−1) is observed. However, in apparent contradiction to experimental results (νN–N = 1808 cm−1 measured for the 2:1 trans adduct), a further ca. 0.08 eV stabilisation is noted upon binding of the second LA. As we have shown in previous work,45 an analysis of the π interactions is insufficient to explain dinitrogen activation in such complexes. A concomitant destabilisation (0.65 eV) of the σ*–σ* orbital is observed that greatly exceeds the π stabilisation, explaining the increase in N–N stretching frequency (from the computed 1864 cm−1 in W–Al1 to 1880 cm−1 in W–Al2trans). The more stable cis adduct showed a slightly decreased bond strength (νN–N = 1802 cm−1). The MO diagram for the cis 2:1 adduct shows a further 0.21 eV destabilisation of the σ*–σ* orbital, which should result in a stronger dinitrogen bond. It is not the case here, however, as the different coordination geometry allows for mixing of the metal d orbital that would form the δ MO in the trans adducts with the π orbitals of the dinitrogen bridge (Fig. 7). Therefore, the nature of the HOMO – a metal-centred orbital in the trans isomer – is significantly modified, forming an additional π–π* interaction in the cis isomer. This yields a total population of 6 electrons in N–N antibonding frontier orbitals instead of 4, leading to a greater overall activation of the N–N bond. The higher extent of the overlap between the metal and ligand orbitals is also likely responsible for the greater stability of this form.
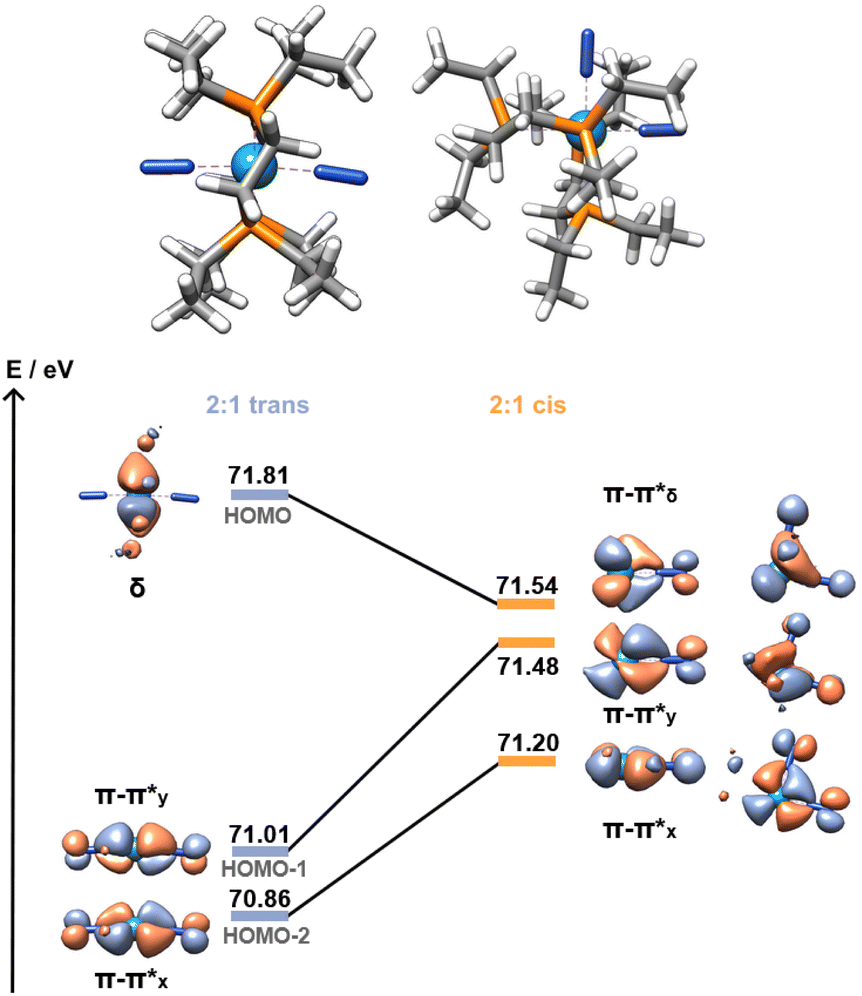 | ||
| Fig. 7 Cis (11Al) vs. trans (8Al) frontier orbitals (HOMO to HOMO−2). AlCF omitted for clarity. Orbitals of the cis isomer are shown in side and top views. | ||
Case of a monophosphine-supported W–N2 complex
To get more insights about the divergent chemical behaviours of AlCFvs.BCF towards bis-dinitrogen complexes, we also investigated their reactivity with cis-[ML′4(N2)2] species (M = Mo or W, L′ = dimethylphenylphosphine). Stoichiometric treatment of BCF with cis-[WL′4(N2)2] leads to the partial abstraction of one PMe2Ph ligand to form a BCF-phosphine adduct – species 14 – (Scheme 4-top left) with a complex mixture of species (see ESI†) that we were not able to identify (except some remaining starting dinitrogen complex). From this experiment we concluded that adjunction of the Lewis acid mainly triggered decomposition. Furthermore, this highlights the ease for BCF to dissociate a monophosphine ligand suggesting its stronger affinity for PMe2Ph vs. N2. On the opposite, using similar conditions to that of the [M(depe)2(N2)2] series, the reaction of AlCF with cis-[ML′4(N2)2] (M = W) produces new LA-dinitrogen adducts – products 15Al and 16Al (Scheme 4, bottom). First clues about the identity of the mono adduct 15Al is evidenced by NMR spectroscopy. Indeed, its 31P NMR spectrum displays three signals at chemical shifts of −18.8, −22.1, and −25.5 ppm integrating respectively for 1, 2 and 1 phosphorus nuclei. This NMR signature suggests that 15Al is cis-[W(PMe2Ph)4(N2){μ-N2–Al(C6F5)3}] having one terminal dinitrogen motif and one bridging dinitrogen fragment. These above aspects are confirmed by IR spectroscopy where coordination of one AlCF molecule at one distal nitrogen induces an averaged bathochromic shift of −171 cm−1 of the μ-NN IR band −1776 vs. 1947 cm−1 in cis-[W(PMe2Ph)4(N2)2]— and an averaged hypsochromic shift of +89 cm−1 of the terminal NN stretching mode −2037 vs. 1947 cm−1 in cis-[W(PMe2Ph)4(N2)2]. Unfortunately, despite the good purity of 15Al verified by elemental and spectroscopic analysis, our attempts to get single crystals were unsuccessful.
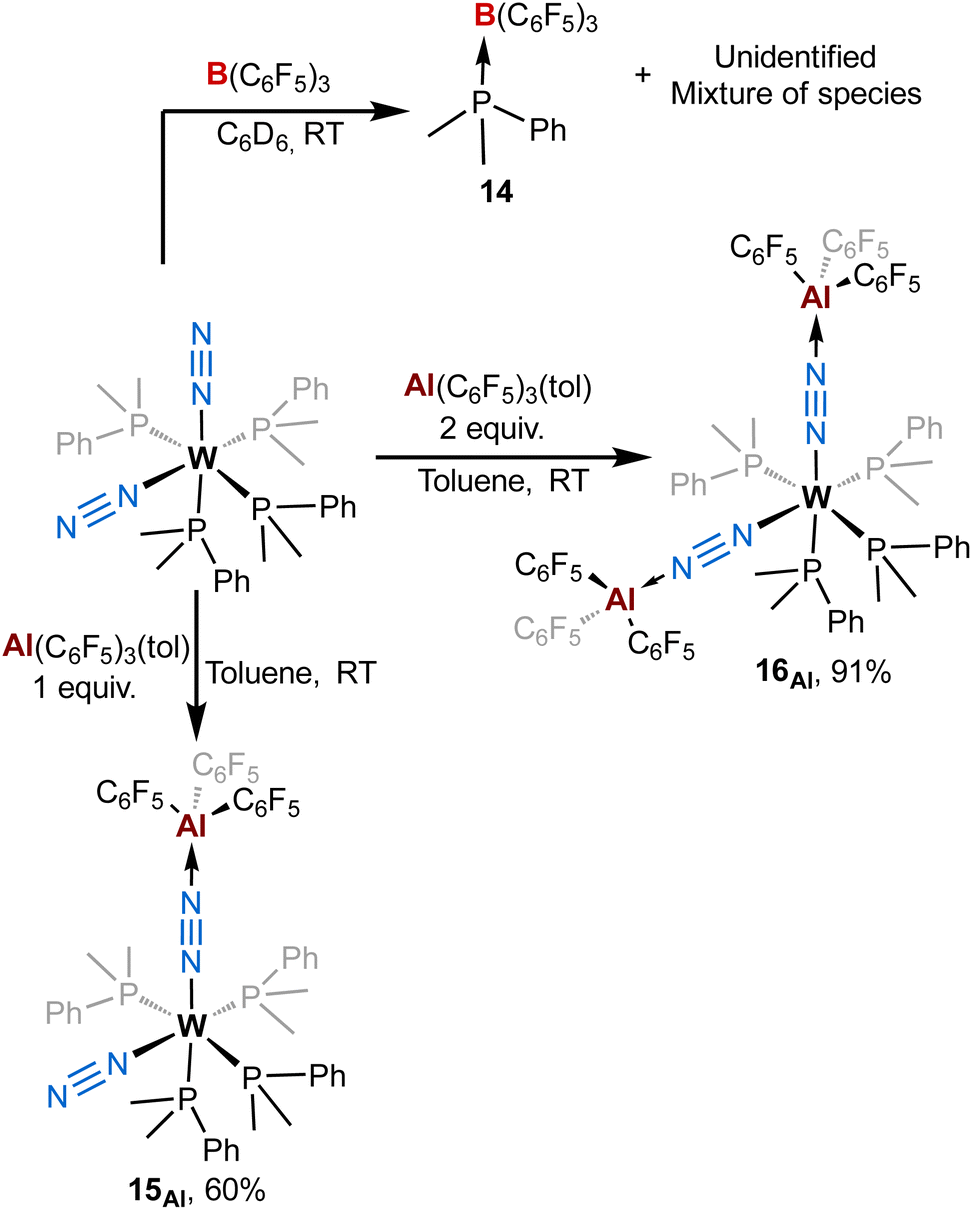 | ||
| Scheme 4 Reactivity of [M(PMe2Ph)4(N2)2] (M = W or Mo) complexes with (top) B(C6F5)3 and (bottom) Al(C6F5)3(tol). | ||
Addition of two equivalents of Al(C6F5)3(tol) on cis-[W(PMe2Ph)4(N2)2] produced a new two-fold adduct 16Al —cis-[W(PMe2Ph)4{μ-N2–Al(C6F5)3}2]— that was fully characterised in solution and in the solid-state. Spectroscopic and crystallographic data of 16Al are very close to those of its congeners 11Al (M = W), 12Al (M = Mo), and 13Al (M = Cr), showing a cis geometry for the AlCF-(μ-N2) fragments (see Table 3). Indeed, aside from their 31P NMR chemical shifts, the IR and XRD data of 16Alvs.11Al are almost identical (see Table 3 and Fig. 8).
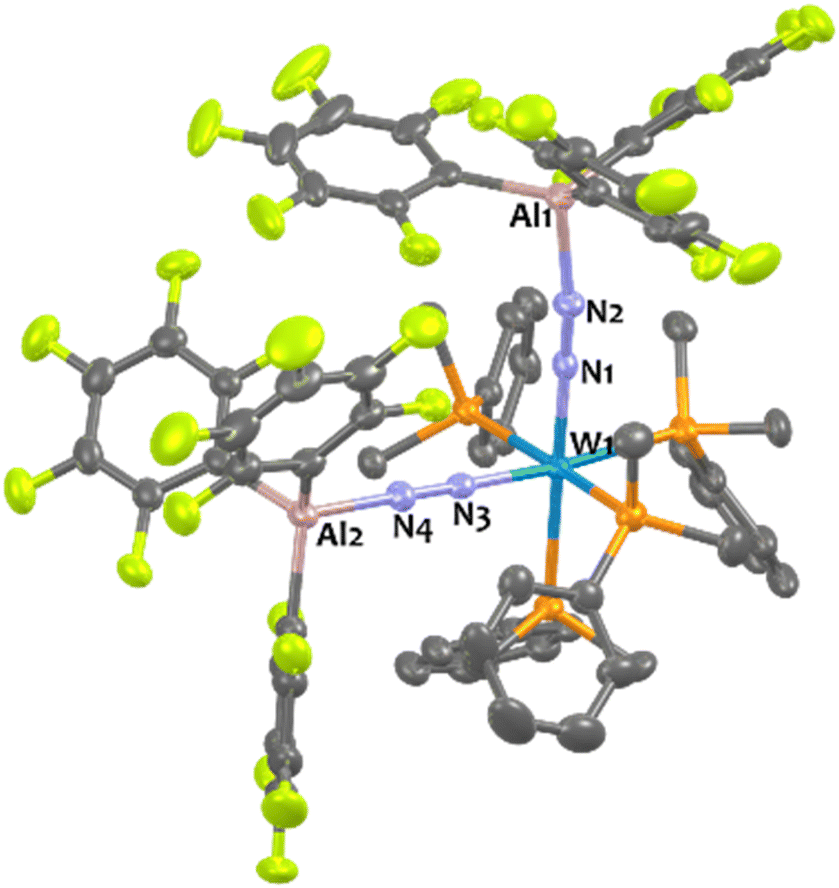 | ||
| Fig. 8 Solid-state structure of 16Al. Ellipsoids are represented with 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°): Al1–N2 1.915(3), Al2–N4 1.891(3), W1–N1 1.917(3), W1–N3 1.906(3), N1–N2 1.149(4), N3–N4 1.150(4), W1–N1–N2 179.2(3), W1–N3–N4 177.5(3), N1–W1–N3 91.34(1), Al1–N2–N1 168.5(3), Al2–N4–N3 179.1(3). | ||
Electronic spectroscopy of the depe-supported W complexes
The recorded UV-vis absorption spectra of trans-[M(depe)2(N2)2] (M = W and Mo) at 298 K display two types of bands, an intense transition (320–330 nm, ε ≈ 105 M−1 cm−1) assigned to metal-to-ligand charge transfer (MLCT) involving a ligand phosphorus atom, and a less intense transition (440–500 nm, ε ≈ 103 M−1 cm−1) assigned to a ligand field (LF) d–d transition.85The MLCT transition of the aluminium and boron adducts does not shift substantially (Δλ < 3 nm) compared to that of the W starting complex (Fig. 9). However, their intensities are about two times lower compared to the W starting complex. We thus assign these energetically similar UV signatures to the chemical environment around the W–P that does not change substantially upon coordination of the LA (unlike the dinitrogen ligand where the coordination of AlCF or BCF takes place). Fig. 10 (left side) displays the visible spectra of each sample at a concentration of 10−3 M. The trans-W(depe)2(N2)2 starting complex displayed two LF (d–d transitions) bands at λ1 = 467 nm and λ2 = 509 nm in agreement with literature data.85 For the Al mono adduct (W–Al, crimson line) we observed a significant blue shift of the first band —λ1 = 411 nm— and a small red shift of the second band —λ2 = 513 nm. For the boron mono-adduct (red line), we observed a slight blue shift for the first and second bands (λ1 = 437 nm, λ1 = 506 nm). The Al double adducts of trans configuration display two new bands (crimson dotted line), one at a wavenumber of 499 nm and the other at a high wavenumber of 633 nm (this complex has a green-cyan colour). For the cis-double adduct, the spectrum displays a single maximum in the visible region at 497 nm. Note that for all the Al and B adducts, we noticed absorption in the [550–700 nm] spectral window (unlike the W starting complex where there is no absorption at all in this area).
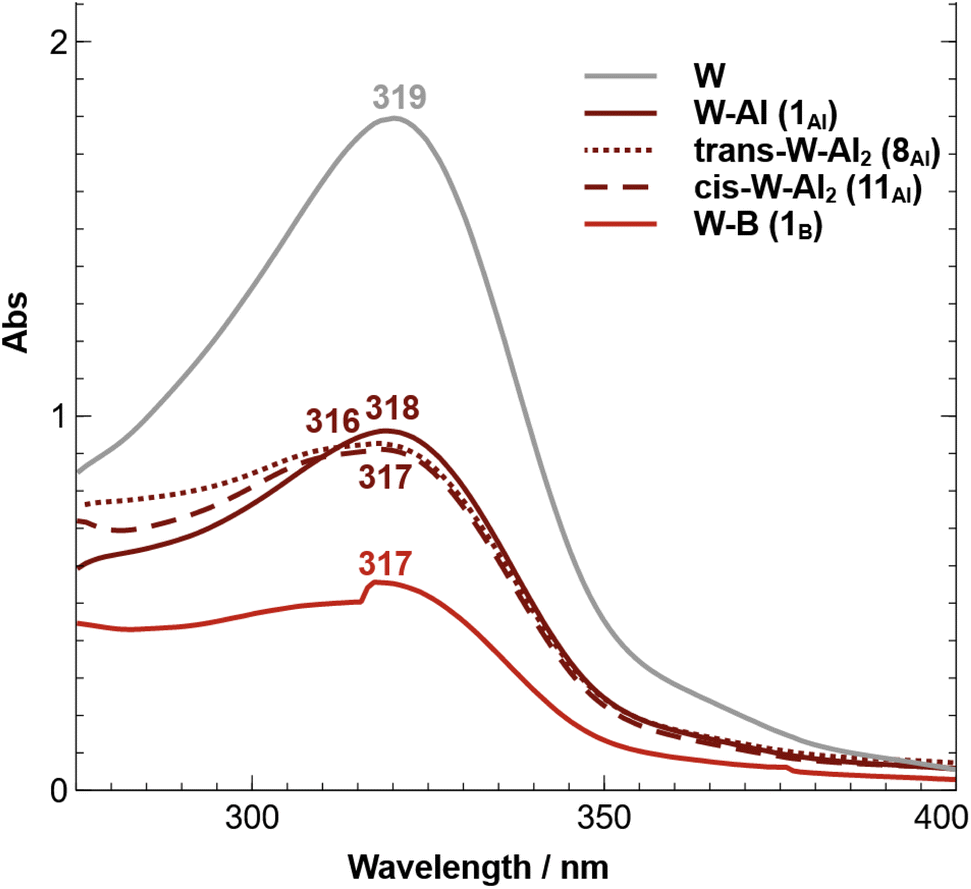 | ||
| Fig. 9 Absorption spectra of trans-[W(depe)2(N2)2] (W, grey line), trans-[W(depe)2(N2)(μ-N2–Al(C6F5)3)] (W–Al, crimson line), trans-[W(depe)2{(μ-N2–Al(C6F5)3}2] (trans-W–Al2, dotted crimson line), cis-[W(depe)2{(μ-N2–Al(C6F5)3}2] (cis-W–Al2, dashed crimson line), and trans-[W(depe)2(N2)(μ-N2–B(C6F5)3] (W–B, red line). The concentration of each sample is about 30 μM. | ||
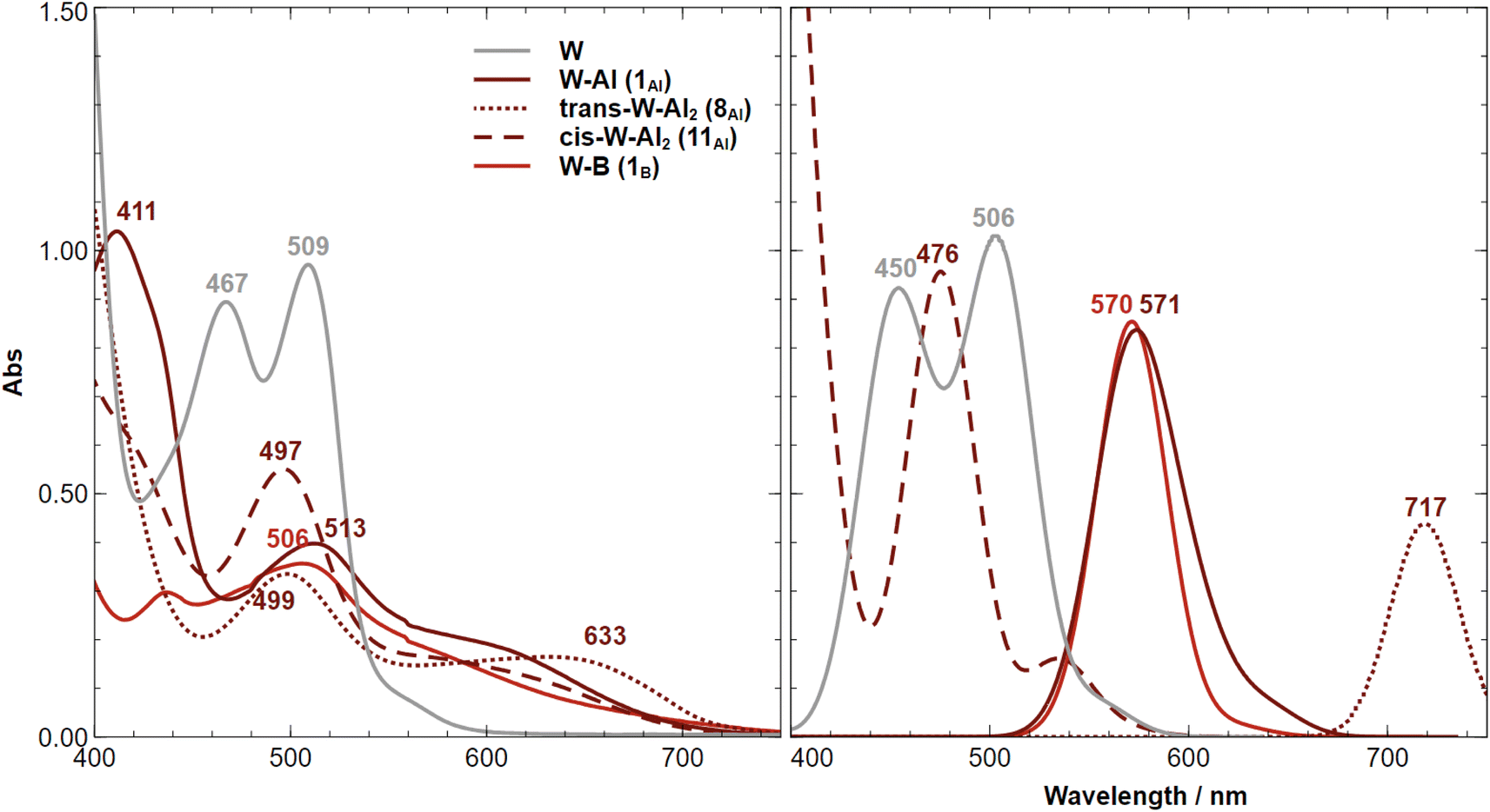 | ||
| Fig. 10 Experimental (left) and computational (CAM-B3LYP, right) absorption spectra of trans-[W(depe)2(N2)2] (W, grey line), trans-[W(depe)2(N2)(μ-N2–Al(C6F5)3)] (W–Al, crimson line), trans-[W(depe)2{(μ-N2–Al(C6F5)3}2] (trans-W–Al2, dotted crimson line), cis-[W(depe)2{(μ-N2–Al(C6F5)3}2] (cis-W–Al2, dashed crimson line), and trans-[W(depe)2(N2)(μ-N2–B(C6F5)3] (W–B, red line). The concentration of each sample is about 1000 μM. A Lorentzian line broadening with FWHM of 8 was applied to the computed peaks. | ||
Computing the electronic excitation spectra of transition-metal complexes with a high degree of quantitative accuracy is far from trivial.86 Nevertheless TD-DFT calculations are key to provide understanding into the nature of the transitions responsible for the UV-vis bands. The peaks obtained via TD-DFT calculations (CAM-B3LYP/def2-TZVP) are in qualitative agreement with the recorded spectra, albeit being generally red shifted by ca. 30–60 nm, except for the bare tungsten complex in which an almost exact match is obtained. The mono-substituted adducts have almost overlapping spectra in both experiment and computations. Two peaks are observed in the experimental UV spectrum of 11Al (trans-W–Al2, Fig. 10-left) while the computed one displays just one. A second, considerably red-shifted peak is visible in the computed spectrum at wavelengths greater than 750 nm (Fig. S97†). An analysis of the Natural Transition Orbitals (NTOs) shows, however, that these do not correspond to d–d transitions, but instead to low-lying MLCT transitions from the metal to both nitrogen ligands (Fig. 11). Such low-lying charge transfer transitions had already been identified in a Ru(II) complex87 that has, like the compounds studied here, a ligand-based LUMO orbital. Remaining relevant NTOs as well as the calculated peaks and associated difference densities can be found in Fig. S94–S96.†
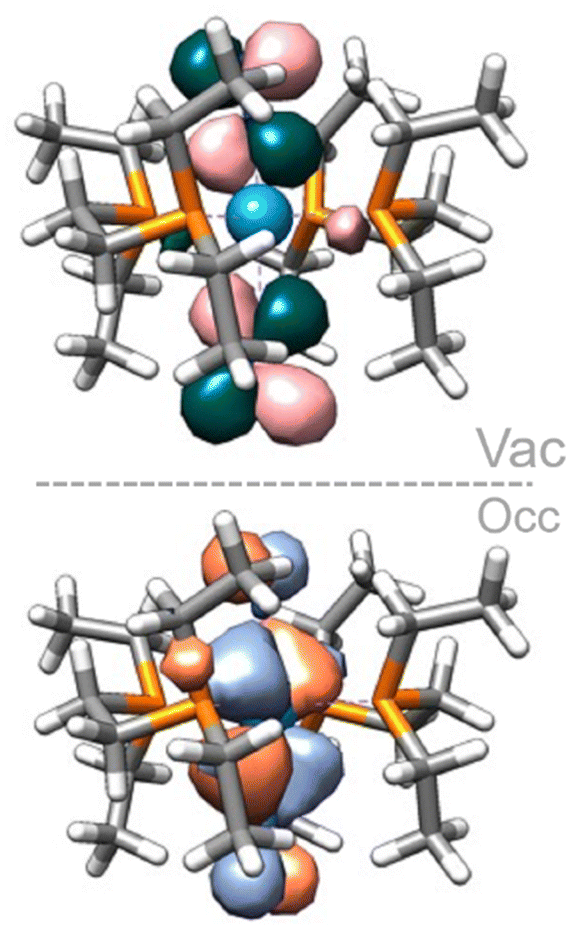 | ||
| Fig. 11 NTOs (occupied – bottom, vacant – top) of the 450 nm band of the bare tungsten complex. | ||
Conclusions
This work was motivated by previous results from our groups having thoroughly investigated, experimentally on the one hand, the coordination of tris(pentafluorophenyl)borane, BCF, to formally zerovalent group 6 bis(dinitrogen) complexes supported with phosphine ligands, and computationally on the other hand, the influence of LA binding to a dinitrogen ligand. This combined experimental/theoretical study explores similar chemistry employing tris(pentafluorophenyl)alane, AlCF. The shift to a structurally comparable but more Lewis acidic species led to the isolation of related 1:1 adducts of an extensive family of dinitrogen complexes, including a chromium-based and monophosphine-based ones that could not be selectively formed when BCF was employed. A notable difference on the structural point of view is the linear N–N–Al vs. bent N–N–B motif that is explained by steric repulsion between the C6F5 groups with the ethyl substituents of the phosphines built up as a result of longer Al–C bonds.
Unlike BCF, AlCF makes robust two-fold μ-N2 adducts with the bis(dinitrogen) complexes. They form with an initial trans arrangement that evolves in solution to a more stable cis one with a rate depending on the metal (Cr > Mo > W). To the best of our knowledge, these compounds are the first examples of trinuclear heterometallic complexes formed by Lewis acid–base interaction exhibiting p and d elements. Among the handful of N2-bridged trinuclear heterobimetallic species88–98 of general formula M1(μ-N2)M2(μ-N2)M1 (M1 = Cr,89 Mo,95–97 Re,92 Fe,93 Co;90,91,93,94,98 M2 = Na,95 Mg,90,91,93–95,97,98 Ti,89 Zr,92,96 V,96 Fe96), many are based on a low diversity of metal/metal couples, typically on magnesium/early transition metals pairs, as a result of a formally anionic dinitrogen complex formed by reduction with an alkaline or alkaline-earth metal. For the synthesis of d-block-only congeners, a general strategy consists in halide substitution by an electron-rich N2 ligand, a transformation that accompanies with formal oxidation of the N2-ligated metal centre concomitant with reduction of N2. Here, the novelty of our bis(μ-η1:η1-N2–AlCF) specimens resides in the use of a p-block metal that interacts with neutral group 6 N2 complexes through Lewis acid–base pair formation, through straightforward syntheses (no redox state change, no by-products, and no workup). Note that this synthetic approach parallels a recent work published by Mazzanti and coworkers where they reported the coordination of f-elements (lanthanides and uranium) to an end-on dinitrogen iron complex leading to the formation of N2 bridged heterobimetallic adducts.99 Last but not least, the close proximity of the two activated dinitrogen motifs in these adducts (imparted by their cis-configuration) may pave the way towards new type of N2 reactivity. DFT calculations show that the diminished level of N2 activation in these systems, evidenced experimentally by comparison of IR and XRD data to those of the 1:1 adducts, can be interpreted by a destabilisation of a σ-symmetric, W–N antibonding component of the W–N–N bonding. While the “bare” N2 complexes, their 1:1 and trans-2:1 Lewis acid adducts have a HOMO of pure d character, in the cis-2:1 adducts this orbital overlaps with a π* orbital of each N2 ligands. This could result, in terms of reactivity, into a selective reactivity of the N2 ligands towards electrophiles vs. the metal centre. From the bare [W(depe)2(N2)2] complex to the two-fold aluminium adduct, substantial decrease of the HOMO–LUMO gap is noticed. In particular, the stabilized N2-centered LUMO should more easily accept electrons, suggesting Lewis acids could be co-activators for (electro) catalysed N2 reduction.
Electronic spectroscopy was examined for the depe-supported W–N2 complex and its adducts both experimentally and computationally. This investigation suggests that the nature of the observed absorptions in the visible spectrum is an unusual low-lying MLCT involving N2-centered orbitals that significantly red-shifts upon LA coordination. This could have important implication for visible light-driven nitrogen fixation, and we are currently exploring the reactivity of LA-adducts of N2 complexes towards this end.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† All the computational data have been uploaded (https://www.iochem-bd.org/handle/10/229000) onto the ioChem-BD platform (https://www.iochem-bd.org/) to facilitate data exchange and dissemination, according to the FAIR principles of OpenData sharing.Author contributions
Conceptualization: A. S.; formal analysis: L. E., L. V., A. C., N. Q.; funding acquisition: A. S., V. K.; investigation L. E., A. C., N. Q., F. M.; methodology: L. E., A. C., N. Q., F. M., V. K., A. S.; project administration: V. K., A. S.; supervision V. K., A. S.; validation: L. E., F. M.; visualization: L. E., F. M.; writing – original draft: L. E., F. M.; writing – review & editing: V. K., A. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. E. and A. S. thanks the French Agence Nationale de la Recherche for funding (grant ANR-21-CE07-0003). N. Q. and A. S. are indebted to the European Research Council for funding (ERC Starting Grant no 757501). A. C. is grateful to the French Ministry of National and Superior Education and Research (MENESR) for a PhD fellowship. F. F. M. gratefully acknowledges a Career Bridging Grant from TU Darmstadt. We are grateful to the CNRS (Centre National de la Recherche Scientifique) and the Technische Universität Darmstadt for providing access to facilities, specifically the Lichtenberg II high performance computer at TU Darmstadt on which all calculations for this work were performed.Notes and references
- A. D. Allen and C. V. Senoff, Nitrogenopentammineruthenium(II) complexes, Chem. Commun., 1965, 621 RSC.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- Y. Roux, C. Duboc and M. Gennari, Molecular Catalysts for N2 Reduction: State of the Art, Mechanism, and Challenges, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
- M. Pérez-Jiménez, H. Corona, F. de la Cruz-Martínez and J. Campos, Donor–Acceptor Activation of Carbon Dioxide, Chem.–Eur. J., 2023, 29, e202301428 CrossRef PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis pair chemistry of carbon, nitrogen and sulfur oxides, Chem. Sci., 2014, 5, 2625–2641 RSC.
- A. E. Ashley and D. O'Hare, FLP-Mediated Activations and Reductions of CO2 and CO, Top. Curr. Chem., 2013, 334, 191–217 CrossRef PubMed.
- R. M. Bullock and G. M. Chambers, Frustration across the periodic table: heterolytic cleavage of dihydrogen by metal complexes, Philos. Trans. R. Soc., A, 2017, 375, 20170002 CrossRef PubMed.
- D. W. Stephan, Diverse Uses of the Reaction of Frustrated Lewis Pair (FLP) with Hydrogen, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- M. A. Stevens and A. L. Colebatch, Cooperative approaches in catalytic hydrogenation and dehydrogenation, Chem. Soc. Rev., 2022, 51, 1881–1898 RSC.
- J. C. Slootweg and A. R. Jupp, Frustrated Lewis Pairs, Springer Nature, 2020 Search PubMed.
- F.-G. Fontaine and D. W. Stephan, On the concept of frustrated Lewis pairs, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- D. W. Stephan, The broadening reach of frustrated Lewis pair chemistry, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan, Frustrated Lewis Pairs: From Concept to Catalysis, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis Pair Chemistry: Development and Perspectives, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- Frustrated Lewis Pairs II: Expanding the Scope, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, 2013 Search PubMed.
- Frustrated Lewis Pairs I: Uncovering and Understanding, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, 2013 Search PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, Implementation of Cooperative Designs in Polarized Transition Metal Systems—Significance for Bond Activation and Catalysis, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- E. R. M. Habraken, A. R. Jupp, M. B. Brands, M. Nieger, A. W. Ehlers and J. C. Slootweg, Parallels between Metal-Ligand Cooperativity and Frustrated Lewis Pairs, Eur. J. Inorg. Chem., 2019, 2019, 2436–2442 CrossRef CAS PubMed.
- P. C. Dos Santos, R. Y. Igarashi, H.-I. Lee, B. M. Hoffman, L. C. Seefeldt and D. R. Dean, Substrate Interactions with the Nitrogenase Active Site, Acc. Chem. Res., 2005, 38, 208–214 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed.
- S. M. Keable, J. Vertemara, O. A. Zadvornyy, B. J. Eilers, K. Danyal, A. J. Rasmussen, L. De Gioia, G. Zampella, L. C. Seefeldt and J. W. Peters, Structural characterization of the nitrogenase molybdenum-iron protein with the substrate acetylene trapped near the active site, J. Inorg. Biochem., 2018, 180, 129–134 CrossRef CAS PubMed.
- D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, A bound reaction intermediate sheds light on the mechanism of nitrogenase, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed.
- W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed.
- J. B. Geri, J. P. Shanahan and N. K. Szymczak, Testing the Push–Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS PubMed.
- J. P. Shanahan and N. K. Szymczak, Hydrogen Bonding to a Dinitrogen Complex at Room Temperature: Impacts on N2 Activation, J. Am. Chem. Soc., 2019, 141, 8550–8556 CrossRef CAS PubMed.
- J. J. Mortensen, B. Hammer and J. K. Nørskov, Alkali Promotion of N2 Dissociation over Ru(0001), Phys. Rev. Lett., 1998, 80, 4333–4336 CrossRef CAS.
- S. Dahl, A. Logadottir, C. J. H. Jacobsen and J. K. Nørskov, Electronic factors in catalysis: the volcano curve and the effect of promotion in catalytic ammonia synthesis, Appl. Catal., A, 2001, 222, 19–29 CrossRef CAS.
- G. P. Connor and P. L. Holland, Coordination chemistry insights into the role of alkali metal promoters in dinitrogen reduction, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed.
- Q. Wang, J. Guo and P. Chen, The impact of alkali and alkaline earth metals on green ammonia synthesis, Chem, 2021, 7, 3203–3220 CAS.
- A. Coffinet, A. Simonneau and D. Specklin, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, Wiley, 2nd edn, 2020, pp. 1–25 Search PubMed.
- A. J. Ruddy, D. M. C. Ould, P. D. Newman and R. L. Melen, Push and pull: the potential role of boron in N2 activation, Dalton Trans., 2018, 47, 10377–10381 RSC.
- A. Simonneau and M. Etienne, Enhanced Activation of Coordinated Dinitrogen with p-Block Lewis Acids, Chem. - Eur. J., 2018, 24, 12458–12463 CrossRef CAS PubMed.
- J. Chatt, J. R. Dilworth, R. L. Richards and J. R. Sanders, Chemical Evidence concerning the Function of Molybdenum in Nitrogenase, Nature, 1969, 224, 1201–1202 CrossRef CAS PubMed.
- J. Chatt, R. H. Crabtree and R. L. Richards, Dinitrogen- and carbonyl-complexes as bases towards trimethylaluminium, J. Chem. Soc. Chem. Commun., 1972, 534 RSC.
- J. Chatt, R. H. Crabtree, E. A. Jeffery and R. L. Richards, The basic strengths of some dinitrogen complexes of molybdenum(0), tungsten(0), rhenium(I), and osmium(II), J. Chem. Soc., Dalton Trans., 1973, 1167–1172 RSC.
- F. Studt, B. A. MacKay, S. A. Johnson, B. O. Patrick, M. D. Fryzuk and F. Tuczek, Lewis adducts of the side-on end-on dinitrogen-bridged complex [{(NPN)Ta}2(μ-H)2(μ-η1:η2-N2)] with AlMe3, GaMe3, and B(C6F5)3: Synthesis, structure, and spectroscopic properties, Chem.–Eur. J., 2005, 11, 604–618 CrossRef CAS PubMed.
- H. Broda, S. Hinrichsen, J. Krahmer, C. Nather and F. Tuczek, Molybdenum dinitrogen complexes supported by a silicon-centred tripod ligand and dppm or dmpm: tuning the activation of N2, Dalton Trans., 2014, 43, 2007–2012 RSC.
- A. Simonneau, R. Turrel, L. Vendier and M. Etienne, Group 6 Transition-Metal/Boron Frustrated Lewis Pair Templates Activate N2 and Allow its Facile Borylation and Silylation, Angew. Chem., Int. Ed., 2017, 56, 12268–12272 CrossRef CAS PubMed.
- D. Specklin, M.-C. Boegli, A. Coffinet, L. Escomel, L. Vendier, M. Grellier and A. Simonneau, An orbitally adapted push–pull template for N2 activation and reduction to diazene-diide, Chem. Sci., 2023, 14, 14262–14270 RSC.
- A. D. Piascik, P. J. Hill, A. D. Crawford, L. R. Doyle, J. C. Green and A. E. Ashley, Cationic silyldiazenido complexes of the Fe(diphosphine)2(N2) platform: structural and electronic models for an elusive first intermediate in N2 fixation, Chem. Commun., 2017, 53, 7657–7660 RSC.
- L. G. Pap, A. Couldridge, N. Arulsamy and E. Hulley, Electrostatic polarization of nonpolar substrates: a study of interactions between simple cations and Mo-bound N2, Dalton Trans., 2019, 48, 11004–11017 RSC.
- D. Specklin, A. Coffinet, L. Vendier, I. del Rosal, C. Dinoi and A. Simonneau, Synthesis, Characterization, and Comparative Theoretical Investigation of Dinitrogen-Bridged Group 6-Gold Heterobimetallic Complexes, Inorg. Chem., 2021, 60, 5545–5562 CrossRef CAS PubMed.
- C. Tang, Q. Liang, A. R. Jupp, T. C. Johnstone, R. C. Neu, D. Song, S. Grimme and D. W. Stephan, 1,1-Hydroboration and a Borane Adduct of Diphenyldiazomethane: A Potential Prelude to FLP-N2 Chemistry, Angew. Chem., Int. Ed., 2017, 56, 16588–16592 CrossRef CAS PubMed.
- F. F. Martins and V. Krewald, Cooperative Dinitrogen Activation: Identifying the Push–Pull Effects of Transition Metals and Lewis Acids in Molecular Orbital Diagrams, Eur. J. Inorg. Chem., 2023, 26, e202300268 CrossRef CAS.
- L. Greb, Lewis Superacids: Classifications, Candidates, and Applications, Chem.–Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- J. L. W. Pohlmann and F. E. Brinckmann, Preparation and Characterization of Group III A Derivatives, Z. Naturforsch. B, 1965, 20, 5–11 CrossRef.
- T. Belgardt, J. Storre, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Tris(pentafluorophenyl)alane: A Novel Aluminum Organyl, Inorg. Chem., 1995, 34, 3821–3822 CrossRef CAS.
- G. S. Hair, A. H. Cowley, R. A. Jones, B. G. McBurnett and A. Voigt, Arene Complexes of Al(C6F5)3. Relationship to a Déjà Vu Silylium Ion, J. Am. Chem. Soc., 1999, 121, 4922–4923 CrossRef CAS.
- J. Klosin, G. R. Roof, E. Y.-X. Chen and K. A. Abboud, Ligand Exchange and Alkyl Abstraction Involving (Perfluoroaryl)boranes and -alanes with Aluminum and Gallium Alkyls, Organometallics, 2000, 19, 4684–4686 CrossRef CAS.
- S. Feng, G. R. Roof and E. Y.-X. Chen, Tantalum(V)-Based Metallocene, Half-Metallocene, and Non-Metallocene Complexes as Ethylene−1-Octene Copolymerization and Methyl Methacrylate Polymerization Catalysts, Organometallics, 2002, 21, 832–839 CrossRef CAS.
- N. G. Stahl, M. R. Salata and T. J. Marks, B(C6F5)3- vs Al(C6F5)3-Derived Metallocenium Ion Pairs. Structural, Thermochemical, and Structural Dynamic Divergences, J. Am. Chem. Soc., 2005, 127, 10898–10909 CrossRef CAS PubMed.
- J. Chen and E. Y.-X. Chen, Unsolvated Al(C6F5)3: structural features and electronic interaction with ferrocene, Dalton Trans., 2016, 45, 6105–6110 RSC.
- D. M. C. Ould, J. L. Carden, R. Page and R. L. Melen, Synthesis and Reactivity of Fluorinated Triaryl Aluminum Complexes, Inorg. Chem., 2020, 59, 14891–14898 CrossRef CAS PubMed.
- I. V. Kazakov, A. S. Lisovenko, N. A. Shcherbina, I. V. Kornyakov, N. Y. Gugin, Y. V. Kondrat'ev, A. M. Chernysheva, A. S. Zavgorodnii and A. Y. Timoshkin, Structural and Energetic Features of Group 13 Element Trispentafluorophenyl Complexes with Diethyl Ether, Eur. J. Inorg. Chem., 2020, 2020, 4442–4449 CrossRef CAS.
- A. Y. Timoshkin, The Field of Main Group Lewis Acids and Lewis Superacids: Important Basics and Recent Developments, Chem.–Eur. J., 2024, 30, e202302457 CrossRef CAS PubMed.
- L. Pauling, Atomic Radii and Interatomic Distances in Metals, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- W. E. Piers and T. Chivers, Pentafluorophenylboranes: from obscurity to applications, Chem. Soc. Rev., 1997, 26, 345 RSC.
- J. R. Lawson and R. L. Melen, Tris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- J. Chen and E. Y. -X. Chen, Elusive Silane–Alane Complex [Si H⋯Al]: Isolation, Characterization, and Multifaceted Frustrated Lewis Pair Type Catalysis, Angew. Chem., Int. Ed., 2015, 54, 6842–6846 CrossRef CAS PubMed.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, On a quantitative scale for Lewis acidity and recent progress in polynitrogen chemistry, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS.
- J. C. Haartz and D. H. McDaniel, Fluoride ion affinity of some Lewis acids, J. Am. Chem. Soc., 1973, 95, 8562–8565 CrossRef CAS.
- E. Y.-X. Chen, W. J. Kruper, G. Roof and D. R. Wilson, Double Activation of Constrained Geometry and ansa-Metallocene Group 4 Metal Dialkyls: Synthesis, Structure, and Olefin Polymerization Study of Mono- and Dicationic Aluminate Complexes, J. Am. Chem. Soc., 2001, 123, 745–746 CrossRef CAS PubMed.
- A. Y. Timoshkin and G. Frenking, Gas-Phase Lewis Acidity of Perfluoroaryl Derivatives of Group 13 Elements, Organometallics, 2008, 27, 371–380 CrossRef CAS.
- J. F. Kögel, D. A. Sorokin, A. Khvorost, M. Scott, K. Harms, D. Himmel, I. Krossing and J. Sundermeyer, The Lewis superacid Al[N(C6F5)2]3 and its higher homolog Ga[N(C6F5)2]3 – structural features, theoretical investigation and reactions of a metal amide with higher fluoride ion affinity than SbF5, Chem. Sci., 2017, 9, 245–253 RSC.
- T. A. George and M. E. Noble, A direct one-step preparation of bis(dinitrogen) complexes of molybdenum(0) from molybdenum(V) chloride, Inorg. Chem., 1978, 17, 1678–1679 CrossRef CAS.
- J. R. Dilworth, R. L. Richards, G. J.-J. Chen and J. W. Mcdonald, in Inorganic Syntheses, John Wiley & Sons, Ltd, 1990, pp. 33–43 Search PubMed.
- R. Poli and H. D. Mui, True nature of trihalotris(tetrahydrofuran)molybdenum(III), MoX3(THF)3 (X = Cl, Br, I). A paramagnetic proton NMR study, J. Am. Chem. Soc., 1990, 112, 2446–2448 CrossRef CAS.
- F. Stoffelbach, D. Saurenz and R. Poli, Improved Preparations of Molybdenum Coordination Compounds from Tetrachlorobis(diethyl ether)molybdenum(IV), Eur. J. Inorg. Chem., 2001, 2699–2703 CrossRef CAS.
- A. C. Filippou, G. Schnakenburg, A. I. Philippopoulos and N. Weidemann, Ge2 trapped by triple bonds between two metal centers: the germylidyne complexes trans,trans-[Cl(depe)2M≡Ge—Ge≡M(depe)2Cl] (M=Mo, W) and bonding analyses of the M≡Ge—Ge≡M chain, Angew. Chem., Int. Ed., 2005, 44, 5979–5985 CrossRef CAS PubMed.
- E. M. Zolnhofer, A. A. Opalade, T. A. Jackson, F. W. Heinemann, K. Meyer, J. Krzystek, A. Ozarowski and J. Telser, Electronic Structure and Magnetic Properties of a Low-Spin CrII Complex: trans-[CrCl2(dmpe)2] (dmpe = 1,2-Bis(dimethylphosphino)ethane), Inorg. Chem., 2021, 60, 17865–17877 CrossRef CAS PubMed.
- G. S. Girolami, G. Wilkinson, A. M. R. Galas, M. Thornton-Pett and M. B. Hursthouse, Synthesis and properties of the divalent 1,2-bis(dimethylphosphino)ethane (dmpe) complexes MCl2(dmpe)2 and MMe2(dmpe)2 (M = Ti, V, Cr, Mn, or Fe). X-Ray crystal structures of MCl2(dmpe)2 (M = Ti, V, or Cr), MnBr2(dmpe)2, TiMe1.3Cl0.7(dmpe)2, and CrMe2(dmpe)2, J. Chem. Soc., Dalton Trans., 1985, 1339 RSC.
- A. J. Kendall, S. I. Johnson, R. M. Bullock and M. T. Mock, Catalytic Silylation of N2 and Synthesis of NH3 and N2H4 by Net Hydrogen Atom Transfer Reactions Using a Chromium P4 Macrocycle, J. Am. Chem. Soc., 2018, 140, 2528–2536 CrossRef CAS PubMed.
- J. E. Salt, G. S. Girolami, G. Wilkinson, M. Motevalli, M. Thornton-Pett and M. B. Hursthouse, Synthesis and characterisation of 1,2-bis(dimethylphosphino)ethane (dmpe) complexes of chromium-(0) and -(IV): X-ray crystal structures of trans-Cr(N2)2(dmpe)2, cis-Cr(CO)2(dmpe)2, Cr(C2Ph2)2(dmpe), and CrH4(dmpe)2, J. Chem. Soc., Dalton Trans., 1985, 685 RSC.
- L. A. Berben and S. A. Kozimor, Dinitrogen and Acetylide Complexes of Low-Valent Chromium, Inorg. Chem., 2008, 47, 4639–4647 CrossRef CAS PubMed.
- J. He, Y. Zhang, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Chain Propagation and Termination Mechanisms for Polymerization of Conjugated Polar Alkenes by [Al]-Based Frustrated Lewis Pairs, Macromolecules, 2014, 47, 7765–7774 CrossRef CAS.
- L. L. Cao, J. Zhou, Z. Qu and D. W. Stephan, Single Electron Transfer to Diazomethane–Borane Adducts Prompts C−H Bond Activations, Angew. Chem., Int. Ed., 2019, 58, 18487–18491 CrossRef CAS PubMed.
- T. Asada, Y. Hoshimoto and S. Ogoshi, Rotation-Triggered Transmetalation on a Heterobimetallic Cu/Al N-Phosphine-Oxide-Substituted Imidazolylidene Complex, J. Am. Chem. Soc., 2020, 142, 9772–9784 CrossRef CAS PubMed.
- Z. Yu, M. J. Heeg and C. H. Winter, A bridging ethyl complex of aluminium, Chem. Commun., 2001, 353–354 RSC.
- W. Uhl and F. Hannemann, A methylene bridged dialuminium compound as a chelating Lewis acid—complexation of azide and acetate anions by R2Al–CH2–AlR2 [R=CH(SiMe3)2], J. Organomet. Chem., 1999, 579, 18–23 CrossRef CAS.
- J. Liu, T.-L. Lam, M.-K. Sit, Q. Wan, C. Yang, G. Cheng and C.-M. Che, Pure blue phosphorescent platinum(ii) emitters supported by NHC-based pincer type ligands with unitary emission quantum yields, J. Mater. Chem. C, 2022, 10, 10271–10283 RSC.
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Frustrated Lewis Pair Behavior of Intermolecular Amine/B(C6F5)3 Pairs, Organometallics, 2012, 31, 2367–2378 CrossRef CAS.
- K. Bläsing, J. Bresien, R. Labbow, D. Michalik, A. Schulz, M. Thomas and A. Villinger, Borane Adducts of Hydrazoic Acid and Organic Azides: Intermediates for the Formation of Aminoboranes, Angew. Chem., Int. Ed., 2019, 58, 6540–6544 CrossRef PubMed.
- W. Uhl, F. Hannemann, W. Saak and R. Wartchow, Diazomethane Derivatives Bearing Dialkylaluminium or DialkylgalliumSubstituents – The Isomeric Diazomethane and Nitrile Imine Structures Realized by the Different Coordination Behavior of Aluminium and Gallium, Eur. J. Inorg. Chem., 1999, 1999, 771–776 CrossRef.
- B. L. Nordwig, D. J. Ohlsen, K. D. Beyer, A. S. Wruck and J. G. Brummer, The Effect of the Diphosphine Basicity on the Excited-State Properties of trans-(N2)2W(R2PCH2CH2PR2)2 : Identification of Near-Degenerate, Luminescent 3MLCT and 3LF Terms, Inorg. Chem., 2006, 45, 858–867 CrossRef CAS PubMed.
- F. Maschietto, M. Campetella, J. Sanz García, C. Adamo and I. Ciofini, Chasing unphysical TD-DFT excited states in transition metal complexes with a simple diagnostic tool, J. Chem. Phys., 2021, 154, 204102 CrossRef CAS PubMed.
- Y. Sun, S. N. Collins, L. E. Joyce and C. Turro, Unusual Photophysical Properties of a Ruthenium(II) Complex Related to [Ru(bpy)2 (dppz)]2+, Inorg. Chem., 2010, 49, 4257–4262 CrossRef CAS PubMed.
- T. Knoell, J. Polanco, S. N. MacMillan, J. A. Bertke, C. Foroutan-Nejad, K. M. Lancaster and A. Gus Bakhoda, Alkaline earth metal-assisted dinitrogen activation at nickel, Dalton Trans., 2024, 53, 4689–4697 RSC.
- X. Wang, Y. Wang, Y. Wu, G.-X. Wang, J. Wei and Z. Xi, Syntheses and Characterizations of Hetero-Bimetallic Chromium-Dinitrogen Transition-Metal Complexes, Inorg. Chem., 2023, 62, 18641–18648 CrossRef CAS PubMed.
- B. A. Suslick and T. D. Tilley, Mechanistic Interrogation of Alkyne Hydroarylations Catalyzed by Highly Reduced, Single-Component Cobalt Complexes, J. Am. Chem. Soc., 2020, 142, 11203–11218 CrossRef CAS PubMed.
- S. L. Apps, P. W. Miller and N. J. Long, Cobalt(-i) triphos dinitrogen complexes: activation and silyl-functionalisation of N2, Chem. Commun., 2019, 55, 6579–6582 RSC.
- T. D. Lohrey, R. G. Bergman and J. Arnold, Controlling dinitrogen functionalization at rhenium through alkali metal ion pairing, Dalton Trans., 2019, 48, 17936–17944 RSC.
- T. R. Dugan, K. C. MacLeod, W. W. Brennessel and P. L. Holland, Cobalt–Magnesium and Iron–Magnesium Complexes with Weakened Dinitrogen Bridges, Eur. J. Inorg. Chem., 2013, 2013, 3891–3897 CrossRef CAS PubMed.
- T. A. Betley and J. C. Peters, Dinitrogen Chemistry from Trigonally Coordinated Iron and Cobalt Platforms, J. Am. Chem. Soc., 2003, 125, 10782–10783 CrossRef CAS PubMed.
- G. E. Greco and R. R. Schrock, Synthesis, Structure, and Electrochemical Studies of Molybdenum and Tungsten Dinitrogen, Diazenido, and Hydrazido Complexes That Contain Aryl-Substituted Triamidoamine Ligands, Inorg. Chem., 2001, 40, 3861–3878 CrossRef CAS PubMed.
- M. B. O'Donoghue, W. M. Davis, R. R. Schrock and W. M. Reiff, Heterobimetallic Dinitrogen Complexes That Contain the {[N3N]Mo−NN}− Ligand, Inorg. Chem., 1999, 38, 243–252 CrossRef.
- M. B. O'Donoghue, W. M. Davis and R. R. Schrock, Derivatization of Dinitrogen by Molybdenum in Triamidoamine Complexes, Inorg. Chem., 1998, 37, 5149–5158 CrossRef.
- H. F. Klein, H. Koenig, S. Koppert, K. Ellrich and J. Riede, Cobalt diazenides of Main Group 1-3 metals: X-ray structure of a Grignard compound containing (dinitrogen)(trimethylphosphine)cobaltate anions, Organometallics, 1987, 6, 1341–1345 CrossRef CAS.
- N. Jori, J. J. Moreno, R. A. K. Shivaraam, T. Rajeshkumar, R. Scopelliti, L. Maron, J. Campos and M. Mazzanti, Iron promoted end-on dinitrogen-bridging in heterobimetallic complexes of uranium and lanthanides, Chem. Sci., 2024, 15, 6842–6852 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General methods, experimental procedures, NMR spectra, crystallographic and computational details and CIF files. CCDC 2346952–2346961. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02713b |
| This journal is © The Royal Society of Chemistry 2024 |