
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGiant oligomeric porous cage-based molecules†
Alba
Cortés-Martínez
ab,
Cornelia
von Baeckmann
ab,
Laura
Hernández-López
ab,
Arnau
Carné-Sánchez
 *ab and
Daniel
Maspoch
*abc
*ab and
Daniel
Maspoch
*abc
aCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology, Campus UAB, Bellaterra, 08193 Barcelona, Spain. E-mail: arnau.carne@icn2.cat; daniel.maspoch@icn2.cat
bDepartament de Química, Facultat de Ciències, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Vallès, 08193 Barcelona, Spain
cICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 30th April 2024
Abstract
Most reported porous materials are either extended networks or monomeric discrete cavities; indeed, porous structures of intermediate size have scarcely been explored. Herein, we present the stepwise linkage of discrete porous metal–organic cages or polyhedra (MOPs) into oligomeric structures with a finite number of MOP units. The synthesis of these new oligomeric porous molecules entails the preparation of 1-connected (1-c) MOPs with only one available azide reactive site on their surface. The azide-terminated 1-c MOP is linked through copper(I)-catalysed azide–alkyne cycloaddition click chemistry with additional alkyne-terminated 1-c MOPs, 4-c clusters, or 24-c MOPs to yield three classes of giant oligomeric molecules: dimeric, tetrameric, or satellite-like, respectively. Importantly, all the giant molecules that we synthesised are soluble in water and permanently porous in the solid state.
Introduction
The overwhelming majority of known porous materials are either small (<5 nm) discrete cages (i.e. coordination and covalent cages),1–5 or infinite networks (i.e. metal- and covalent-organic frameworks).6–9 The chemical and dimensional (between 5 nm and 20 nm) space between these two classes has not been largely explored, due to the inherent synthetic and analytical challenges (Scheme 1).10–12 Specifically, using bottom-up approaches to assemble large porous discrete cages is not trivial.13 Another challenge lies in stopping the polymerisation reaction of extended networks at the oligomeric regime, which is not thermodynamically favoured, thus leading to polydisperse and metastable materials.14,15 Moreover, the down-sizing of crystalline porous networks below the 20 nm threshold often entails the accumulation of defects that are detrimental to their characterisation and function.16,17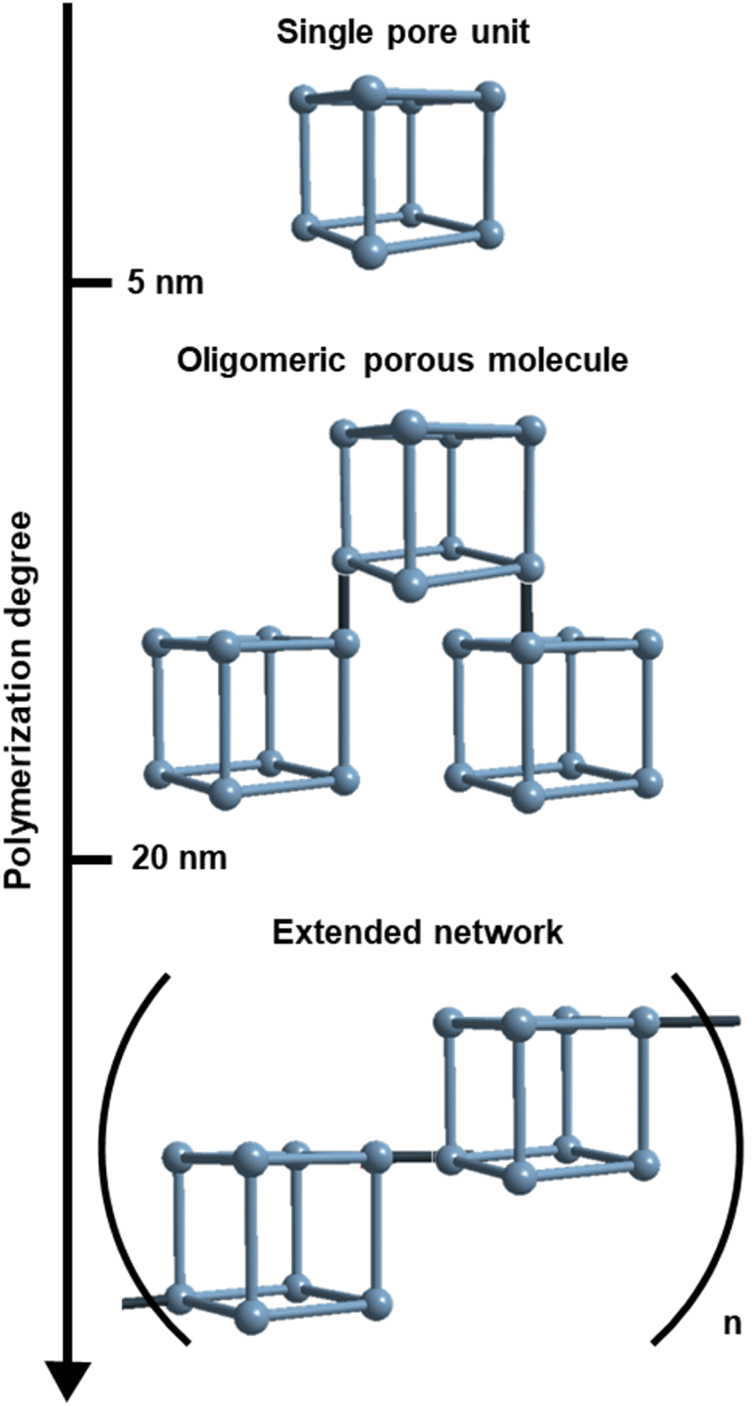 | ||
| Scheme 1 Illustrated scale of the regimes of reported porous materials. | ||
To overcome these challenges, one can take inspiration from the stepwise synthesis of giant organic18–22 and metal–organic23–25 molecules, in which each growing step proceeds through thermodynamic control. In the case of porous materials, this strategy entails the oligomerization of single pore units into giant multi-pore molecules. Pioneer studies have shown the viability of the pore oligomerization approach by either interlocking26–29 or linking a defined number of cages30–32 into multi-cage molecules. However, to the best of our knowledge, the permanent porosity of this giant molecules has not yet been demonstrated.
Herein we report the stepwise synthesis of permanently porous oligomeric molecules by the concatenation of a defined, finite number of metal–organic cages or polyhedra (MOPs). The resultant giant oligomeric porous molecules merge the properties that arise from linking pore-units (i.e. extrinsic porosity and inter-cavity cooperativity)33,34 to the those typically observed in molecules, such as defined molecular weight, stoichiometric reactivity, and solubility in liquids, including water.21–23,35
In our synthetic route to oligomeric porous molecules, the building blocks are robust Rh(II)-based MOPs (Rh-MOPs).36 Rh-MOPs can have up to 24 covalent reactive sites on their external surfaces, stemming from the 5-position of the 1,3-benzendicarboxylate (BDC) derivative used in their synthesis.37 Consequently, the high connectivity of Rh-MOPs complicates the control of their polymerisation into well-defined oligomeric structures rather than extended networks.38–41 To address this challenge, we aimed to create Rh-MOPs with only one reactive site on their surface. By employing protecting groups, we selectively masked the reactivity of 23 of the 24 reactive sites to yield 1-connected (1-c) Rh-MOPs.42 Next, using orthogonal chemistry, we assembled these 1-c MOPs with other 1-c MOPs or with 4-c clusters or 24-c Rh-MOPs to yield giant oligomeric molecules of three types: dimeric, tetrameric, or satellite-like (Fig. 1).
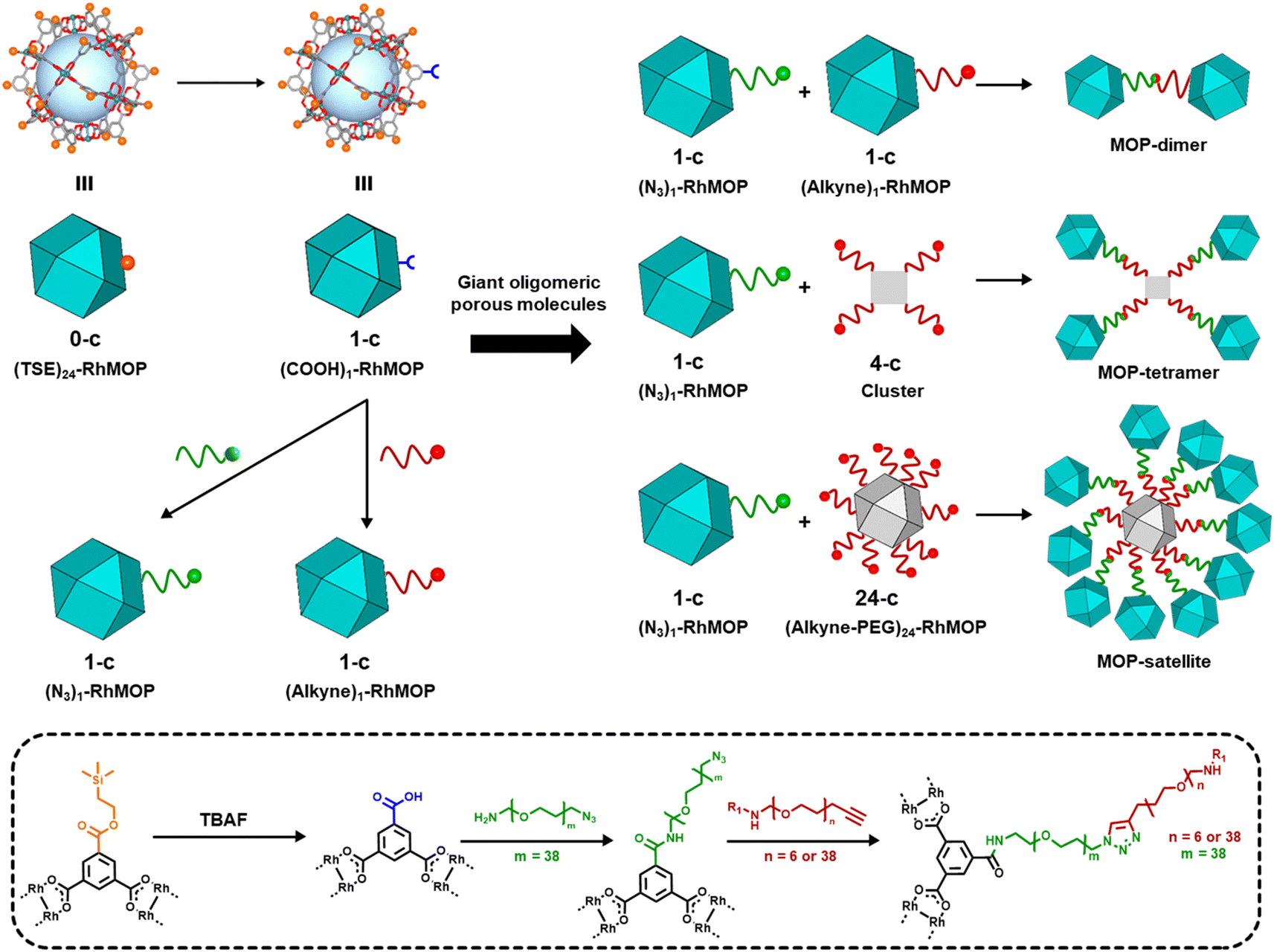 | ||
| Fig. 1 (Top) Schematic of the synthesis of 1-c MOPs terminated with azide or alkyne groups, and their subsequent linkage to other MOPs or clusters through click chemistry to form oligomeric porous dimeric, tetrameric or satellite-like molecules. (Bottom) Representative reaction schematic for the synthesis of giant, oligomeric, MOP-based molecules. | ||
Results and discussion
Synthesis of 1-connected Rh-MOPs
We began the synthesis of 1-c Rh-MOP from a Rh-MOP in which all of its 24 peripheral carboxylic acid groups were protected with 2-(trimethylsilyl)ethyl (TSE) groups. One of the 24 TSE groups was selectively cleaved by treating the fully protected Rh-MOP with 1.5 mol eq. (per MOP) of the deprotecting agent tetrabutylammonium fluoride (TBAF). This reaction afforded a new Rh-MOP having only one available surface carboxylic group with the formula (COOTSE-BDC)23(BTC)1Rh24 (where BTC = 1,3,5-benzenetricarboxylate; and COOTSE-BDC24 = 5-((2-(trimethylsilyl)ethoxy)carbonyl)-1,3-benzendicarboxylate). The integrity and formula of the 1-c Rh-MOP (hereafter, (COOH)1-RhMOP) was confirmed by Matrix-Assisted Laser Desorption/Ionisation-Time-Of-Flight (MALDI-TOF) mass spectroscopy, which showed a peak centered at 9770 m/z that corresponds to the expected molecular formula of [(COOTSE-BDC)23(BTC)1Rh24 + H+]+·2H2O (expected mass of 9776 g mol−1) (Fig. 2a and S4†). Analysis of the 1H-NMR spectrum of the acid digested (COOH)1-RhMOP confirmed the expected ratio between the aromatic and the aliphatic protons of the protected ligand, thus confirming the removal of one protecting group (Fig. S3†). Furthermore, Diffusion-Ordered Spectroscopy (DOSY) NMR analysis of the (COOH)1-RhMOP in CDCl3 revealed the same diffusion coefficient of 1.9 × 10−10 m2 s−1 for both aliphatic and aromatic signals, which further corroborated that the product had retained 23 of its original 24 TSE groups (Fig. S2†).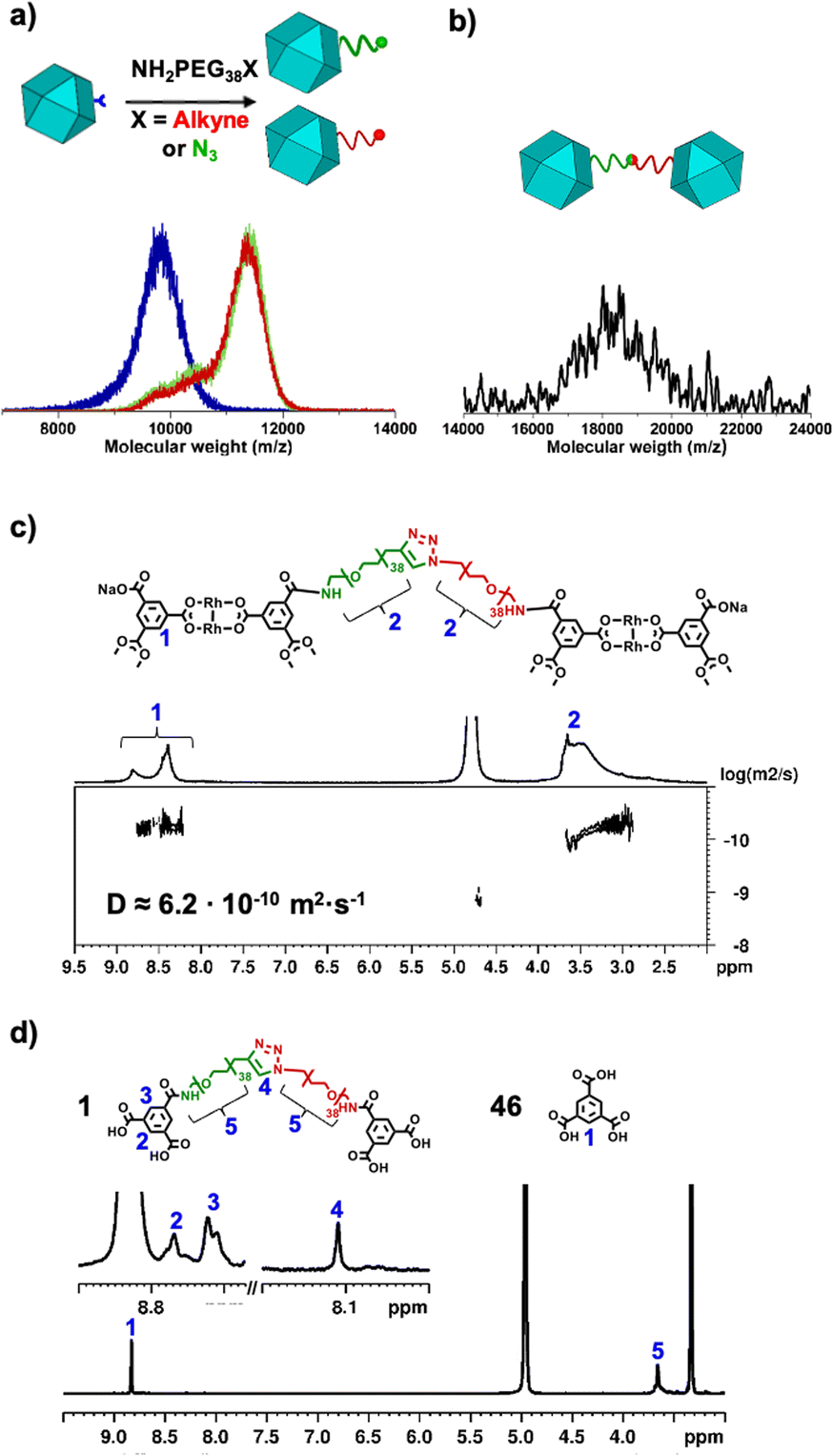 | ||
Fig. 2 (a) Comparison of the MALDI-TOF spectra of (COOH)1-RhMOP (blue), (N3)1-RhMOP (green) and (alkyne)1-RhMOP (red) which evidences an increase in mass due to the attachment of a chain of NH2PEG38X to the MOP (where X = N3 or alkyne). (b) MALDI-TOF spectrum of the MOP-dimer, showing the single broad peak centered at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 538 m/z. (c) DOSY-NMR spectrum of the MOP-dimer. (d) 1H-NMR spectrum of the acid-digested MOP-dimer. 538 m/z. (c) DOSY-NMR spectrum of the MOP-dimer. (d) 1H-NMR spectrum of the acid-digested MOP-dimer. | ||
Next, to confer (COOH)1-RhMOP with the orthogonal reactivity required to oligomerise it with additional MOPs, we functionalised its surface with a single polyethylene glycol (PEG) chain terminated with either alkyne or azide group. Thus, (COOH)1-RhMOP was reacted with a PEG chain terminated at one end with a primary amine (for coupling to the surface carboxylic acid), and at the other end, with either an alkyne or azide moiety (for the oligomerisation). The coupling reactions between (COOH)1-RhMOP and either NH2-PEG38-N3 or NH2-PEG38-alkyne proceeded homogenously in N,N-dimethylformamide (DMF), using 1-hydroxybenzotriazole (HOBt), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and N,N-diisopropylethylamine (DIPEA) as coupling agents. They yielded two distinct MOPs, each with only one PEG chain attached at its surface: an azide-terminated one, having the formula (COOTSE-BDC)23(N3-PEG38-BDC)Rh24 (hereafter, (N3)1-RhMOP); and an alkyne-terminated one, having the formula (COOTSE-BDC)23(alkyne-PEG38-BDC)Rh24 (hereafter, (alkyne)1-RhMOP). The coupling of a single functionalised PEG chain on the surface of (COOH)1-RhMOP was first demonstrated by MALDI-TOF mass spectroscopy, which showed the expected mass for each product: 11540 m/z for (N3)1-RhMOP and 11521 m/z for (alkyne)1-RhMOP (Fig. 2a, S10 and S16† respectively). Both values agree with the respective calculated molecular weights for the corresponding expected molecular formulae of the 1-c Rh-MOPs: 11544 ± 480 g mol−1 for [(COOTSE-BDC)23(N3-PEG38-BDC)Rh24 + H+]+ and 11521 ± 370 g mol−1 for [(COOTSE-BDC)23(N3-PEG38-BDC)Rh24 + H+]+. The formation of a single amide bond on the surface of the MOP was further supported by the 1H-NMR analysis of the corresponding acid-digested samples: the spectra revealed the quantitative transformation of the initial BTC linker into a PEG functionalised BDC linker via amide-bond formation (Fig. S9 and S15†). The ratio between the PEG-functionalised BDC and COOTSE-BDC was found to be the expected value of 1:23 in both (N3)1-RhMOP and (alkyne)1-RhMOP (Fig. S7 and S13†). The DOSY analysis of (N3)1-RhMOP and (alkyne)1-RhMOP revealed a single diffusion-coefficient for the MOP core and the PEG chain in both products: 1.8 × 10−10 m2 s−1 and 1.7 × 10−10 m2 s−1 respectively (Fig. S8 and S14†). Interestingly, the remaining 23 protected carboxylic acid groups on the surface of (N3)1-RhMOP and (alkyne)1-RhMOP could be removed by treating both 1-c RhMOPs with excess of TBAF. Under these conditions, both deprotected (N3)1-RhMOP and (alkyne)1-RhMOP were functionalized with 23 carboxylic acid groups (Fig. S19–S30†).
Finally, to corroborate the synthesis of pure (COOH)1-RhMOP and derived mono-PEGylated compounds, we performed a control experiment consisting of reacting (COOH)1-RhMOP (obtained by treating a TSE-protected Rh-MOP with 1.5 mol eq. of TBAF) with an excess amount of NH2-PEG38-N3 (5 mol eq. per Rh-MOP). The product obtained from this reaction was analysed through MALDI-TOF mass spectrometry, showing only the peak corresponding to (N3)1-RhMOP that contains one attached PEG chain on the MOP surface (Fig. S31†). Conversely, when NH2-PEG38-N3 was reacted with Rh-MOPs containing a higher percentage of free carboxylic acid groups on their surface, a distribution of multi-PEGylated Rh-MOPs was obtained as a product. Specifically, PEGylated Rh-MOPs with 2 and 3 PEG chains on their surface were obtained when 5 mol eq. of NH2-PEG38-N3 was reacted with TSE-protected Rh-MOPs treated with 3 and 5 mol eq. of TBAF, respectively (Fig. S32†). These experiments confirm the successful synthesis of pure (COOH)1-RhMOP as the presence of Rh-MOPs with a higher number of available carboxylic acids on their surface would yield a distribution of PEGylated Rh-MOPs in the presence of excess of NH2-PEG38-N3.
Synthesis of the oligomeric dimeric MOP-based molecule
Next, we synthesised a dimeric MOP-based molecule (hereafter, MOP-dimer), by coupling (N3)1-RhMOP to (alkyne)1-RhMOP through a copper(I)-catalysed, azide–alkyne cycloaddition (CuAAC) click reaction.43 To this end, both MOPs were reacted under homogenous conditions in a mixture of CH2Cl2/DMF (1:1), using copper sulphate and sodium acetate as catalysts, to afford the corresponding crude products as a green solid. To facilitate the purification of the MOP-dimer, all surface TSE protecting groups were cleaved, which yielded a dimeric MOP in which each MOP unit had 23 available carboxylic acid groups. Next, the MOP-dimer was purified through successive washing with 0.3 M HCl and basic MeOH, in which the dimer is insoluble, but the catalyst and unreacted precursors are soluble. The obtained purified product exhibited pH-dependent aqueous solubility, which we ascribed to the presence of up to 46 available carboxylic groups. Once deprotonated, the carboxylate groups imparted negative charge to the resulting MOP-dimer, as confirmed by Z-potential measurements performed in basic water, which revealed a value of – 48.5 ± 6.9 mV for the MOP-dimer (Fig. S41†). The successful dimerization of two different MOPs was first evidenced by MALDI-TOF. The spectrum exhibited a single broad peak centred at 18538 m/z, in good agreement with the expected mass (18539 ± 850 g mol−1) for the MOP-dimer having a molecular formula of [(COOH-BDC)46(BDC-PEG38-1H-1,2,3-triazol-4-yl-PEG38-BDC)1Rh48-H+]−·DMF (Fig. 2b and S38†). DOSY NMR spectroscopy in basic D2O revealed a decrease in the diffusion coefficient from the deprotected 1-c MOP precursors (6.6 × 10−10 m2 s−1 and 6.9 × 10−10 m2 s−1) to the MOP-dimer (6.2 × 10−10 m2 s−1) (Fig. 2c, S20, S26 and S34†).44 Analogously, Dynamic Light Scattering (DLS) measurements revealed that the molecule size in solution had increased from that of the deprotected (N3)1-RhMOP (2.6 ± 0.2 nm) or the deprotected (alkyne)1-RhMOP (2.3 ± 0.3 nm), to that of the MOP-dimer (6.3 ± 1.1 nm) (Fig. 3a, S24, S30 and S40†).
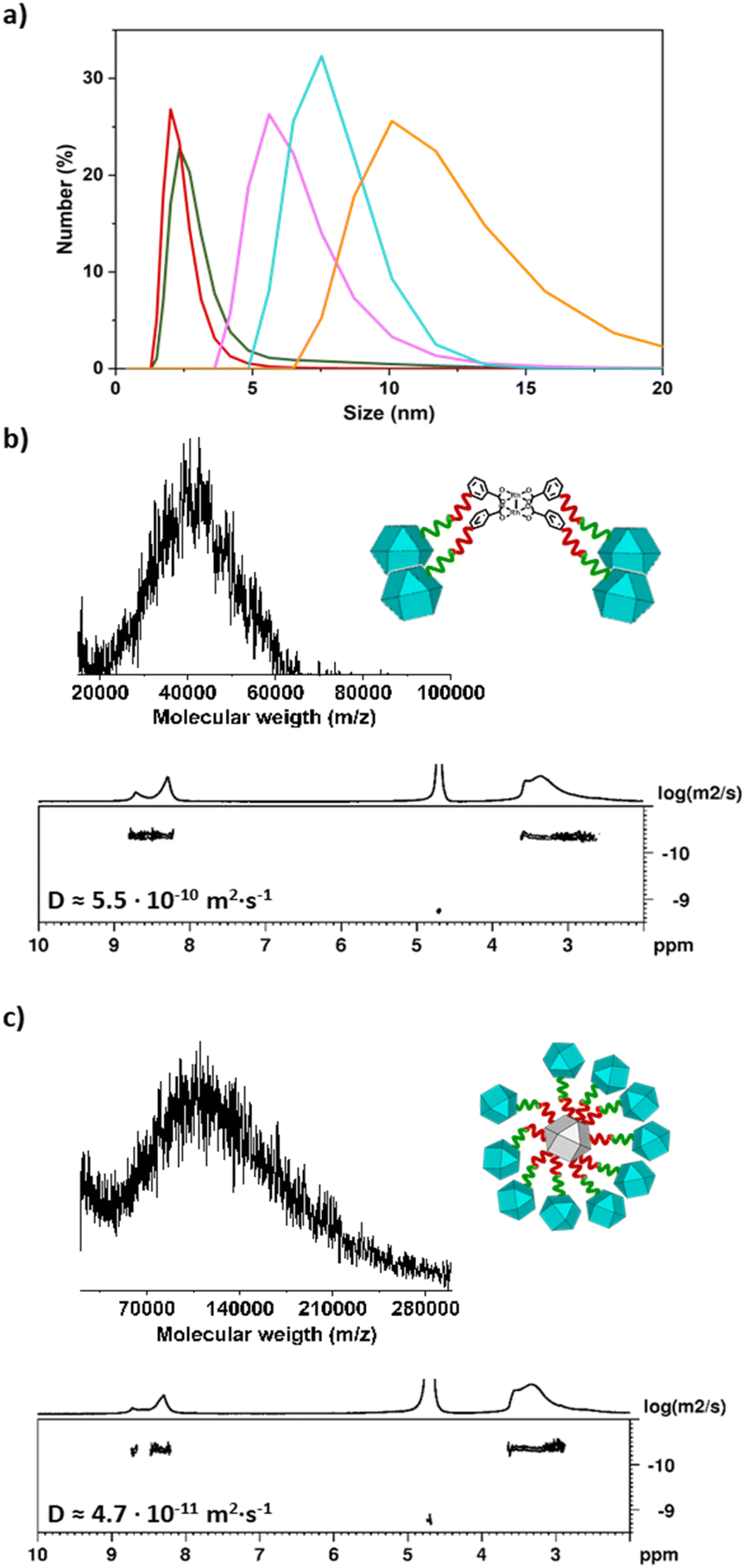 | ||
| Fig. 3 (a) DLS spectra illustrating the differences in particle size (hydrodynamic-diameter) among the deprotected (N3)1-RhMOP (green), the deprotected (alkyne)1-RhMOP (red), the MOP-dimer (violet), the MOP-tetramer (cyan) and the MOP-satellite (orange). (b and c) MALDI-TOF spectra (top) and DOSY-NMR spectra (bottom) of MOP-tetramer (b) and MOP-satellite (c). | ||
To confirm that the two MOPs in MOP-dimer were linked through a covalent bond (i.e. the triazole ring formed upon the CuAAC reaction) and not simply entangled through supramolecular PEG-MOP interactions, we submitted it to acid digestion and then, analysed the resultant ligands through 1H-NMR (Fig. 2d). The spectrum of the isolated PEG linker clearly showed a peak at 8.11 ppm, which can be ascribed to the proton of the expected triazole ring (Fig. S35–S37†). Further analysis of the relative integration of the triazole ring and the aromatic core belonging to the Rh-MOP confirmed that every 1-c Rh-MOP was linked through a triazole ring. Together, these data confirmed that the two MOPs had indeed been assembled into one new dimer, thus corroborating our original idea that this could be accomplished by strategically controlling the number of reactive sites on the surface of the MOP precursors, and then subjecting them to orthogonal click chemistry.
Increasing the connectivity in the synthesis of oligomeric MOP-based molecules: a MOP-tetramer and a MOP-satellite
Having confirmed the viability of the MOP oligomerisation, we next targeted oligomeric structures having a higher number of MOP units. We began by synthesising a tetrameric structure in which four 1-c MOPs are linked to a single 4-c node that comprises a dirhodium paddlewheel unit (Fig. 1). The starting point was the Rh2(bdc)4 cluster, which has four available carboxylic acid groups,45 which were used to attach, via amide coupling, four alkyne-terminated NH2-PEG6-alkyne chains (Fig. S42–S45†). Next, (N3)1-RhMOP (10 mol eq.) was reacted with the synthesised alkyne-functionalised 4-c cluster in a mixture of CH2Cl2/DMF (1:1), using copper sulfate and sodium ascorbate as CuAAC catalysts. The reaction proceeded homogenously and, after 48 hours, it was quenched by extracting the crude reaction with 0.3 M HCl and water to remove the catalysts. The remaining organic solvent was removed in vacuo, and the resultant crude product was treated with TBAF to deprotect all the carboxylic acid groups present in the mixture. To purify the carboxylic acid-functionalised tetramer (hereafter, MOP-tetramer) from any unreacted 4-c cluster or (N3)1-RhMOP, the crude product was dissolved in basic water, and then filtered using a centrifugal filter with a molecular weight cut-off of 30 kDa. The MOP-tetramer was retained, whereas the (smaller) 4-c cluster and (N3)1-RhMOP passed through the filter. The purified MOP-tetramer was characterised by MALDI-TOF, which confirmed the successful attachment of four MOPs to the 4-c cluster, as evidenced by a broad peak centred at 39027 m/z, in agreement with the expected mass of a MOP-tetramer having a molecular formula of [[(COOH-BDC)23(BDC-PEG38-1H-1,2,3-triazol-4-yl-PEG6-BDC)1Rh24]4Rh2 + H+]+ (39047 ± 1480 g mol−1) (Fig. 3b and S48†). Moreover, the 1H-NMR spectrum of MOP-tetramer in basic D2O confirmed the expected ratio of aliphatic PEG protons to aromatic protons in the 1-c MOP and the 4-c cluster, which had the same diffusion coefficient of 5.5 × 10−10 m2 s−1 (Fig. 3b, S46 and S47†). Importantly, the higher oligomeric degree of MOP-tetramer compared to MOP-dimer was corroborated analytically by its lower diffusion coefficient (5.5 × 10−10 m2 s−1vs. 6.2 × 10−10 m2 s−1) and by its greater particle-size (7.2 ± 0.6 nm vs. 6.3 ± 1.1 nm), as revealed by DLS (Fig. 3a and S50†).
Finally, to further extend the oligomeric degree of this family of MOP-based giant molecules, we sought to prepare a highly connected node. We reasoned that such a node could be synthesised from a cuboctahedral Rh-MOP, which can contain up to 24 reactive sites on its surface. Thus, a 24-c node was synthesised using a Rh-MOP with all its carboxylic acid groups available (hereafter, COOH24-RhMOP). This entailed attaching 24 NH2-PEG6-alkyne chains, via amide-coupling chemistry, onto the surface of the COOH24-RhMOP to afford a 24-c node (hereafter, (alkyne)24-RhMOP). MALDI-TOF and 1H-NMR spectroscopy confirmed the quantitative functionalisation of COOH24-RhMOP into (alkyne)24Rh-MOP (Fig. S52–S55†). Next, 24-c (alkyne)24Rh-MOP was reacted with an excess of 1-c (N3)1-RhMOP (240 mol eq. per (alkyne)24-RhMOP) in a CuAAC click reaction to yield a satellite-like, MOP-based, giant molecule. The reaction proceeded homogenously in a mixture of CH2Cl2/DMF (1:1), using the same CuAAC catalysts as above with the addition of tris-(hydroxypropyltriazolylmethyl)amine (THPTA), for up to 14 days. Note that the chelating agent THPTA was used to further accelerate the reaction.46,47 The solvent was removed in vacuo to afford the crude product, which was then treated with TBAF to yield a deprotected, satellite-like molecule (hereafter, MOP-satellite) in which all the peripheral MOPs contained 23 surface carboxylic-acid groups available for further reactions. MOP-satellite is soluble in basic water and could be separated from unreacted (N3)1-RhMOPs using a centrifugal filter with a molecular weight cut-off of 50 kDa. The isolated, purified MOP-satellite was first analysed through mass spectrometry, which revealed a peak centred at 107346 m/z, which we ascribed to a satellite structure having 10 peripheral MOPs and a molecular formula of [[((COOH-BDC)23(BDC-PEG38-1H-1,2,3-triazol-4-yl-PEG6-BDC)1Rh24)10(alkyne-PEG6-BDC)14Rh24] + H+]+ (expected molecular weight: 107678 ± 3700 g mol−1; Fig. 3c and S59†). This molecular composition was further supported by the 1H-NMR spectrum of the MOP-satellite in basic D2O, in which the ratio of aromatic protons to aliphatic PEG protons was 0.39; in agreement with the expected value for a satellite with 10 peripheral MOPs (0.41) (Fig. S57†). The aromatic and aliphatic signals displayed the same diffusion coefficient of 4.7 × 10−11 m2 s−1 (Fig. 3c and S58†), which is the smallest value among those of the synthesised oligomeric MOP-based molecules, consistent with the MOP-satellite having the highest oligomeric degree and the largest molecule size (calculated: 8.6 nm; DLS value: 9.6 ± 0.8 nm (Fig. 3a and S61†). Finally, we also analysed the Na(I) to Rh(II) ratio of the fully deprotonated MOP-satellite salt through inductively coupled plasma mass spectrometry (ICP-MS). The experimentally observed ratio of Na:Rh molar ratio was 1.19, which agrees with the expected value (1.15) (Table S1†).
Study of the adsorption capabilities
Having prepared a family of giant oligomeric MOP-based molecules of increasing oligomeric degree, we next endeavoured to explore the functionality of the intrinsic voids stemming from their respective MOP cavities. We had envisioned that their oligomerisation would give rise to the first set of intrinsically porous oligomeric molecules. To demonstrate the permanent porosity of the three oligomeric MOP-based molecules, we subjected them to CO2-adsorption/desorption experiments at 195 K (Fig. S63–S68†). The measurements confirmed that each giant molecule had retained the microporosity of its parent MOP, as evidenced by the corresponding isotherms, all of which exhibit a type-1 shape (Fig. 4). These isotherms were characterized by a sharp increase in the low-pressure range, followed by a plateau in the middle-to-high pressure range. This shape of the isotherm is consistent with the type-1 adsorption characteristic of microporous materials. The total CO2-uptakes at 1 bar were: 54.4 mol mol−1 MOP unit (MOP-dimer), 30.8 mol mol−1 MOP unit (MOP-tetramer), and 35.1 mol mol−1 MOP unit (MOP-satellite). We ascribed the higher uptake of MOP-dimer to its lack of dangling free PEG chains, which can block porosity; indeed, such chains are found in MOP-satellite. It can also be attributed to its absence of non-porous structuring units (i.e. cluster); for example, these units are found in the MOP-tetramer. Interestingly, CO2-uptake was markedly higher for all the giant oligomeric molecules than for the deprotected 1-c (N3)1-RhMOP (6.1 mol mol−1 MOP unit), the deprotected 1-c (alkyne)1-RhMOP (8.3 mol mol−1 MOP unit) and the 24-c (alkyne)24-RhMOP (3.6 mol mol−1 MOP unit) precursors. We ascribed the greater porosity of the oligomeric molecules relative to their precursors to fact that the PEG chains are less mobile when they act as linkers in the former, than when they are dangling from the surface of the latter.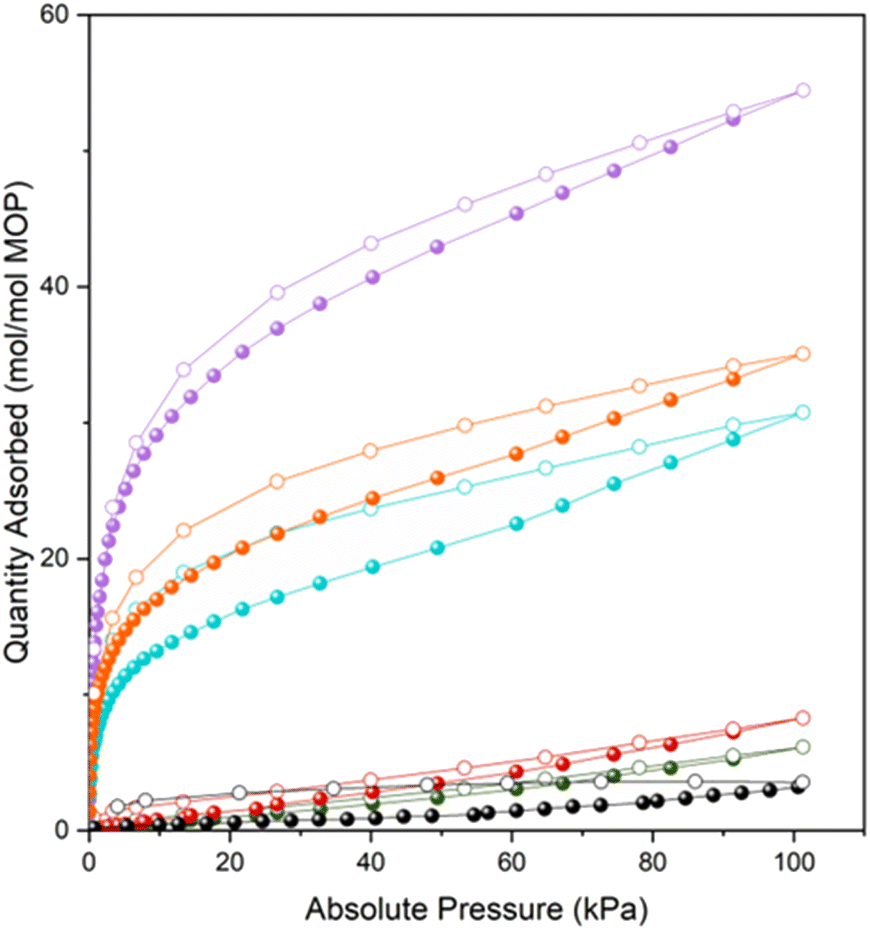 | ||
| Fig. 4 Isotherms of CO2-adsorption at 195 K for the deprotected (N3)1-RhMOP (green), the deprotected (alkyne)1-RhMOP (red), the (alkyne)24-RhMOP (black), the MOP-dimer (purple), the MOP-tetramer (blue), and the MOP-satellite (orange). | ||
Conclusions
In summary, we have synthesised, characterised, and functionally validated a new class of giant oligomeric porous cage-based molecules. Namely, we developed a new method for the stepwise assembly of individual MOP cavities into oligomeric molecules, based on two factors that enable oligomerisation, rather than polymerisation. Firstly, the use of 1-c MOPs as building blocks enables termination of the linkage reaction at the oligomeric regime. Secondly, the use of an orthogonal reaction (in our case, CuAAC click chemistry) to link these blocks with other 1-c MOPs or with 4-c nodes or 24-c MOPs to yield molecules of increasing oligomeric degrees, precluding self-condensation between the precursors. Furthermore, gas-sorption experiments revealed that the giant oligomeric molecules retain the intrinsic porosity of the cavities of their parent MOP. We are confident that our results should inform the future design of new porous materials that will occupy the chemical and dimensional space between purely monomeric cavities and extended networks.Author contributions
A. C. M.: conceptualization, methodology, investigation, and writing-original draft. C. v. B.: methodology and investigation. L. H. L.: validation and investigation. A. C. S: funding acquisition, conceptualization, supervision and writing-review and editing. D. M.: funding acquisition, conceptualization, supervision and writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the Europa Excelencia grant (EUR2021-121997) and the Catalan AGAUR (project 2017 SGR 238). It was also funded by the CERCA Programme/Generalitat de Catalunya. ICN2 is supported by the Severo Ochoa Centres of Excellence programme, Grant CEX2021-001214-S, funded by MCIN/AEI/10.13039.501100011033. A. C. S. is indebted to the Ramón y Cajal Program (RYC2020-029749-I Fellowship). C. v. B. thanks the Austrian Science Fund (FWF), Erwin Schrödinger fellowship for supporting the project J 4637.Notes and references
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528 RSC.
- D. Zhang, T. K. Ronson, Y. Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934 RSC.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS PubMed.
- G. Maurin, C. Serre, A. Cooper and G. Férey, Chem. Soc. Rev., 2017, 46, 3104 RSC.
- A. V. Virovets, E. Peresypkina and M. Scheer, Chem. Rev., 2021, 121, 14485 CrossRef CAS PubMed.
- J. V. Rival, P. Mymoona, K. M. Lakshmi, Nonappa, T. Pradeep and E. S. Shibu, Small, 2021, 17, 1 CrossRef.
- H. Wang, Y. Li, N. Li, A. Filosa and X. Li, Nat. Rev. Mater., 2021, 6, 145 CrossRef CAS.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563 CrossRef CAS PubMed.
- X. G. Wang, Q. Cheng, Y. Yu and X. Z. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7836 CrossRef CAS.
- F. Caddeo, R. Vogt, D. Weil, W. Sigle, M. E. Toimil-Molares and A. W. Maijenburg, ACS Appl. Mater. Interfaces, 2019, 11, 25378 CrossRef CAS PubMed.
- X. Cai, Z. Xie, D. Li, M. Kassymova, S.-Q. Zang and H.-L. Jiang, Coord. Chem. Rev., 2020, 417, 213366 CrossRef CAS.
- J. Andreo, R. Ettlinger, O. Zaremba, Q. Peña, U. Lächelt, R. F. de Luis, R. Freund, S. Canossa, E. Ploetz, W. Zhu, C. S. Diercks, H. Gröger and S. Wuttke, J. Am. Chem. Soc., 2022, 144, 7531 CrossRef CAS PubMed.
- D. T. S. Rijkers, G. W. Van Esse, R. Merkx, A. J. Brouwer, H. J. F. Jacobs, R. J. Pieters and R. M. J. Liskamp, Chem. Commun., 2005, 13, 4581 RSC.
- C. N. Urban, C. A. Bell, M. R. Whittaker and M. J. Monteiro, Macromolecules, 2008, 41, 1057 CrossRef.
- Z. Liu, S. Wang, G. Li, Z. Yang, Z. Gan and X. H. Dong, Macromolecules, 2022, 55, 5954 CrossRef CAS.
- Y. Li, R. Biswas, W. P. Kopcha, T. Dubroca, L. Abella, Y. Sun, R. A. Crichton, C. Rathnam, L. Yang, Y. Yeh, K. Kundu, A. Rodríguez-Fortea, J. M. Poblet, K. Lee, S. Hill and J. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202211704 CrossRef CAS PubMed.
- A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J. S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J. F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50 CrossRef PubMed.
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, J. Am. Chem. Soc., 2015, 137, 5662 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, B. Song, Y. Zhang, X. Jiang, M. Wang, R. Tumbleson, C. Liu, P. Wang, X.-Q. Hao, T. Rojas, A. T. Ngo, J. L. Sessler, G. R. Newkome, S. W. Hla and X. Li, Nat. Chem., 2020, 12, 468 CrossRef CAS PubMed.
- G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2013, 52, 9932 CrossRef CAS PubMed.
- T. Hasell, X. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750 CrossRef CAS PubMed.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem.–Eur. J., 2016, 22, 14104 CrossRef CAS.
- R. Zhu, I. Regeni, J. J. Holstein, B. Dittrich, M. Simon, S. Prévost, M. Gradzielski and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 13652 CrossRef CAS PubMed.
- B. P. Benke, T. Kirschbaum, J. Graf, J. H. Gross and M. Mastalerz, Nat. Chem., 2023, 15, 413 CrossRef CAS PubMed.
- C. Yu, P. Yang, X. Zhu and Y. Wang, Sci. China: Chem., 2022, 65, 858 CrossRef CAS.
- C. Yu, Y. Yang and Y. Wang, New J. Chem., 2021, 45, 22049 RSC.
- R. L. Greenaway, V. Santolini, F. T. Szczypiński, M. J. Bennison, M. A. Little, A. Marsh, K. E. Jelfs and A. I. Cooper, Chem.–Eur. J., 2020, 26, 3718 CrossRef CAS PubMed.
- A. Khobotov-Bakishev, L. Hernández-López, C. von Baeckmann, J. Albalad, A. Carné-Sánchez and D. Maspoch, Adv. Sci., 2022, 9, e2104753 CrossRef PubMed.
- E. Sánchez-González, M. Y. Tsang, J. Troyano, G. A. Craig and S. Furukawa, Chem. Soc. Rev., 2022, 51, 4876–4889 RSC.
- A. Fernandez, J. Ferrando-Soria, E. M. Pineda, F. Tuna, I. J. Vitorica-Yrezabal, C. Knappke, J. Ujma, C. A. Muryn, G. A. Timco, P. E. Barran, A. Ardavan and R. E. P. Winpenny, Nat. Commun., 2016, 7, 1 Search PubMed.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843 CrossRef CAS PubMed.
- J. Albalad, L. Hernández-López, A. Carné-Sánchez and D. Maspoch, Chem. Commun., 2022, 58, 2443 RSC.
- T. Grancha, A. Carné-Sánchez, F. Zarekarizi, L. Hernández-López, J. Albalad, A. Khobotov, V. Guillerm, A. Morsali, J. Juanhuix, F. Gándara, I. Imaz and D. Maspoch, Angew. Chem., 2021, 133, 5793 CrossRef.
- J. Troyano, S. Horike and S. Furukawa, J. Am. Chem. Soc., 2022, 144, 19475 CrossRef CAS PubMed.
- A. Khobotov-Bakishev, C. von Baeckmann, B. Ortín-Rubio, L. Hernández-López, A. Cortés-Martínez, J. Martínez-Esaín, F. Gándara, J. Juanhuix, A. E. Platero-Prats, J. Faraudo, A. Carné-Sánchez and D. Maspoch, J. Am. Chem. Soc., 2022, 144, 15745 CrossRef CAS PubMed.
- A. J. Gosselin, G. E. Decker, A. M. Antonio, G. R. Lorzing, G. P. A. Yap and E. D. Bloch, J. Am. Chem. Soc., 2020, 142, 9594 CrossRef CAS PubMed.
- J. Albalad, A. Carné-Sánchez, T. Grancha, L. Hernández-López and D. Maspoch, Chem. Commun., 2019, 55, 12785 RSC.
- L. Hernández-López, C. von Baeckmann, J. Martínez-Esaín, A. Cortés-Martínez, J. Faraudo, C. Caules, T. Parella, D. Maspoch and A. Carné-Sánchez, Chem.–Eur. J., 2023, 29, e202301945 CrossRef PubMed.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586 RSC.
- C. von Baeckmann, S. Ruiz-Relaño, I. Imaz, M. Handke, J. Juanhuix, F. Gándara, A. Carné-Sanchez and D. Maspoch, Chem. Commun., 2023, 59, 3423 RSC.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879 CrossRef CAS PubMed.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01974a |
| This journal is © The Royal Society of Chemistry 2024 |