
In situ PET-RAFT polymerization to prepare guanidine-and-carbohydrate modified ZnO nanoparticles†
Junyan
Zhao
a,
Yu
Rao
a,
Hengyuan
Zhang
a,
Zhichen
Zhu
a,
Lihua
Yao
a,
Gaojian
Chen
 *ab and
Hong
Chen
*a
*ab and
Hong
Chen
*a
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, China. E-mail: chenh@suda.edu.cn
bCenter for Soft Condensed Matter Physics and Interdisciplinary Research & School of Physical Science and Technology, Soochow University, Suzhou, 215123, China. E-mail: gchen@suda.edu.cn
First published on 2nd May 2024
Abstract
ZnO–polymer core–shell nanoparticles were successfully prepared using a simple in situ open-to-air PET-RAFT method. The utilization of vinyltriethoxysilane (VTES) modified ZnO NPs as catalysts for polymerization, along with the grafting of polymers onto the ZnO NPs, offers significant antibacterial properties. The cationic monomer methacrylamide guanidine hydrochloride (MAGH) and the glycomonomer 2-methacrylamido glucopyranose (MAG) were grafted onto the ZnO NPs surface, further enhancing the antibacterial properties by promoting contact with bacteria and specific recognition of E. coli FimH proteins, leading to a significant improvement in the antibacterial ability compared with ZnO NPs. By combining the photocatalytic and antibacterial properties of ZnO NPs, the preparation of a core–shell material with good antibacterial properties was successfully achieved, providing a new strategy for the synthesis of antimicrobial materials.
Introduction
Since the discovery of antibiotics, they have been known as the terminator of bacteria due to their extremely high antibacterial effect.1 Unfortunately, the widespread use of antibiotics has led to the rapid spread of antimicrobial resistance genes between different strains, causing serious harm to human health.2,3 To address this challenge, in recent years, numerous antibacterial polymers have been developed. These polymers mimic the structure of antimicrobial peptides with broad-spectrum antibacterial properties, and do not easily cause drug resistance.4 Concurrently, nanomaterials with broad-spectrum antibacterial properties have drawn attention.Metal and metal oxide nanoparticles also have broad-spectrum antibacterial properties that can be applied in various applications due to their unique mechanical, magnetic, and photoelectric properties.5 Among them, ZnO NPs not only have high photochemical stability and photocatalytic activity,6 but also inhibit a variety of pathogens and fungi due to their antibacterial properties.7 Their antibacterial mechanisms are primarily categorized into three types: contact killing, ROS production, and release of zinc ions.8 Furthermore, the antibacterial activity of ZnO NPs can be modulated by modifying factors such as their size, shape, and concentration.9 However, given their small size, ZnO NPs tend to aggregate, which can be mitigated through surface modification10 or by compounding them with other materials11 to improve their antibacterial activity. A particularly effective method is surface modification with polymers,12–14 as it not only prevents aggregation of NPs but also introduces novel properties, making it an attractive option for various applications.
The use of light to drive polymerization offers advantages in providing spatial and temporal control over the reaction. Photoinduced electron/energy transfer–reversible addition–fragmentation chain transfer (PET-RAFT) polymerization has emerged as an excellent technique for developing well-defined polymers with advantages such as excellent spatial and temporal control, good selectivity and mild reaction conditions.15,16 In addition to antibacterial agents, ZnO NPs are also a photocatalyst17,18 and are used in PET-RAFT polymerization.19,20 Previously, we have successfully employed the photocatalytic properties of ZnO NPs to perform high throughput PET-RAFT polymerization in aqueous media.21
Among antibacterial polymers that are not prone to drug resistance, guanidine polymers are a good choice with certain advantages: the guanidine group can be ionized in a wider pH range, and it can bind to the cell membrane in the form of dihydro bonds, making the bacterial cell membrane firmly bound and the antibacterial ability stronger.22 Carbohydrates play a significant role in biological processes such as recognition and signaling,23–27 and they have a high affinity for proteins, making them suitable for applications that require target-specific binding.28–31 Different carbohydrates have been used to identify bacteria.32 For example, E. coli express FimH protein on their pili, allowing them to bind to glycopolymers on their surface.33 And synthesized glycopolymers immobilized on the surface of Fe NPs can capture and kill E. coli in water.34
Targeting antibacterial nanoparticles to specific bacteria or infected tissues presents an effective approach for treating infections, which can minimize side effects and improve antibacterial activity.35 In this paper, a simple, green and efficient method was designed to improve the antibacterial activity of ZnO NPs and endow them with specific antibacterial capabilities. The guanidine-cationic monomer MAGH36 and the carbohydrate monomer MAG37 were chosen to strengthen electrostatic interactions and facilitate specific recognition with E. coli, respectively. We defined the ZnO NPs grafted with the guanidine polymer as ZnO-G, and those grafted with the copolymer of guanidine and carbohydrate as ZnO-GC. Notably, the ZnO NPs themselves served as catalysts, enabling oxygen-tolerant in situ PET-RAFT polymerization on their silanized surface, to obtain a ZnO–polymer core–shell material with enhanced and selective antibacterial properties.
Experimental
Materials
Zinc oxide (ZnO, 50 ± 10 nm) and zinc standard solution (1000 μg mL−1 Zn in 1% HNO3) were purchased from Shanghai Macklin Biochemical. Ethanol (≥AR), sodium hydroxide (NaOH, ≥AR, 97%) and sodium chloride (NaCl, ≥AR, 99.5%) were purchased from Shanghai Aladdin Biochemical Technology. Vinyltriethoxysilane (VTES) was purchased from Energy Chemical. 4-Cyano-4-(phenylcarbonothioylthio)-pentanoic acid (CPADB) was purchased from Sigma-Aldrich. Yeast extract and tryptone were purchased from Oxoid. Nutrient broth and agar powder were purchased from Solarbio. 2-Deoxy-2-(methacrylamido)-glucopyranose21 (MAG) and methacrylamide guanidine hydrochloride4 (MAGH) were prepared as reported previously. The bacterial strains used were E. coli MG1655 (ATCC-700926) and S. aureus (ATCC-6538) from Biofeng. They were stored in a −80 °C ultralow-temperature refrigerator. A xenon lamp was purchased from Beijing Zhongjiao Jinyuan Company (model: CEL-HXF300, wavelength range: 320–2500 nm, light intensity = 4.7 mW cm−2@420 nm).Preparation of ZnO-VTES
Zinc oxide nanoparticles (1 g) were first sonicated with 20 mL ethanol for 5 min and then transferred to a beaker for surface modification. The amount of ultrapure water (1.76 g) required for the hydrolysis of the silane coupling agent was calculated and added to the beaker, and then 121 μL of vinyltriethoxysilane was added dropwise to react for 1 h. To catalyze the condensation reaction, sodium hydroxide was added to raise the pH value to 10 followed by reaction for another 2 h. Then, the nanoparticles were centrifuged, washed, and dried.ZnO-mediated in situ PET-RAFT polymerization
Typically, to synthesize ZnO-G, ZnO-VTES (50 mg), MAGH (115 mg, 0.7 mM), and CPADB (1 mg, 3.5 μM) were slowly added in 2 mL of ultrapure water into the sample vials. The molar ratio of the monomers was set as [MAGH]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[MAG]0:[CPADB]0 = 200:0:1.
:[MAG]0:[CPADB]0 = 200:0:1.
Similarly, to synthesize ZnO-GC, ZnO-VTES (50 mg), MAGH (58 mg, 0.35 mM), MAG (87 mg, 0.35 mmol), and CPADB (1 mg, 3.5 μM) were slowly added in 2 mL of ultrapure water into the sample vials. The molar ratio of the monomers was set as [MAGH]0:[MAG]0:[CPADB]0 = 100:100:1.
The sample vials were exposed to air with the reaction solution under simulated sunlight (light intensity = 47 mW cm−2) at 25 °C in a dry bath thermostat for 6 h. Then, we turned off the light to stop the polymerization and the nanoparticles were centrifuged, washed, and dried.
In addition, towards researching the effect of different polymerization chain lengths, we conducted the photo-polymerization of ZnO-G with different illumination times, 20, 40, 60, 180, and 360 min, and obtained ZnO-G at different photo-polymerization times.
Characterization
A Varian Mercury-400 spectrometer (Agilent Technologies) was used to acquire 1H NMR spectra of the monomers and polymers. Gel permeation chromatography (E2695 Waters) was used to measure molecular weights and molecular weight distributions. A TGA550 thermogravimetric analyzer (Waters) was used to determine the quantity of polymers grafted onto the particle surfaces. Total internal reflection Fourier transform infrared spectra of samples were acquired on a Nicolet 6700FT-IR spectrometer (Thermo Fisher Scientific). Water contact angles (WCAs) were measured on a contact angle measuring instrument (SL-200C, KINO Industry) with ultrapure water. Zeta potentials were tested on a Nano-ZS90 model Zetasizer instrument (Malvern) in ultrapure water. X-ray photoelectron spectroscopy (XPS, EXCALAB 250 XI, Thermo Scientific) was used to determine the chemical composition of the modified ZnO. A scanning electron microscope (SEM, S-4700, Hitachi) equipped with an energy dispersive spectroscopy (SEM-EDS) elemental composition analyzer was used to characterize the morphology of samples and the bacteria after interaction with the modified ZnO. The morphology of samples was also characterized using transmission electron microscopy (TEM, HT-7700, voltage: 120 kV, Hitachi). Inductively coupled plasma atomic emission spectrometry (ICP-OES, ICAP-7200, Thermo Fisher Scientific) was used to quantitatively analyze the Zn content in 1 mg ZnO-G with photo-polymerization times of 0, 20, 40, 60, 180, and 360 min and the ratio of PMAGH to ZnO in ZnO-G was calculated and plotted.Antibacterial assay
The antibacterial properties of the samples were evaluated by the flat colony counting method. E. coli MG1655 frozen at −80 °C were streaked on LB plates and incubated for 12 h at 37 °C to achieve activation. Single colonies of activated bacteria were added to 2 mL of LB liquid medium and cultured for 8–12 h in a shaker at 37 °C and 190 rpm. S. aureus ATCC-6538 was cultured in broth medium for 18–24 h. Based on the OD values at 600 nm (E. coli MG1655) and 650 nm (S. aureus ATCC-6538), the densities of the bacterial suspensions were reduced to 107 CFU mL−1 by dilution with sterilized PBS.To research the antibacterial properties of ZnO-G with different polymeric chain lengths, three ZnO-G solutions and ZnO solution were prepared in PBS at a concentration of 1 mg mL−1. 100, 200 and 500 μL aliquots of the ZnO, ZnO-G solutions were diluted with PBS to a final volume of 900 μL, and 100 μL of bacterial suspension (107 CFU mL−1) was added to each diluted solution, resulting in final concentration of 100, 200, and 500 μg mL−1. The mixtures were incubated at 37 °C for 3 h. After 50-fold dilution, 50 μL of the suspensions were added onto LB plates and distributed evenly. The nutrient agar plates were incubated in a 37 °C incubator for 8–12 h. Samples without ZnO and ZnO-G were used as controls. Furthermore, we researched the specific antibacterial performance of ZnO-GC against E. coli MG1655. The antibacterial experiment was the same as above.
The number of single colonies was counted. The antibacterial efficiency was estimated from eqn (1)
| Antibacterial effiency = (A − B)/A × 100% | (1) |
For observing the bacterial morphology, silicon wafers were fixed in a mixture with 2.5% glutaraldehyde solution for 2 h and washed with PBS. They were dehydrated with ethanol solutions in a concentration gradient of 30%, 50%, 70%, 90%, and 100% for 10 min at each step. The morphology of the bacterial membranes was observed by SEM.
A mixture of E. coli (107 CFU mL−1) and S. aureus (106 CFU mL−1) was prepared based on the optical density (OD) to visually reflect the specific antibacterial ability of ZnO-GC. The mixtures consisted of 100 μL of ZnO-G or ZnO-GC at a concentration of 1 mg mL−1, 800 μL of PBS and 100 μL of the mixed bacterial solution. The mixtures were incubated at 37 °C for 3 h. After 50-fold dilution, 50 μL of the suspensions were added onto LB plates and distributed evenly. The nutrient agar plates were incubated in a 37 °C incubator for 8–12 h. Samples without ZnO-G and ZnO-GC were used as controls.
The number of single colonies was counted, and the total number of colonies was obtained from eqn (2):
| Total number of colonies = A × B | (2) |
Statistical analysis
To ensure validity, each experiment was repeated three times and statistically analyzed. The statistical differences were evaluated using one-way ANOVA tests. A value of P ≤ 0.05 was considered statistically significant (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).Results and discussion
Double-bond functional groups were effectively incorporated onto the surface of ZnO NPs through the aldol condensation reaction between the silane coupling agent vinyltriethoxysilane (VTES) and the hydroxyl groups present on the ZnO surface, resulting in the formation of ZnO-VTES. Subsequently, a grafting-through method was employed, without the need for oxygen removal through degassing, utilizing ZnO-VTES mediated PET-RAFT to graft polymers containing guanidine and carbohydrate moieties onto the ZnO surface, as illustrated in Scheme 1.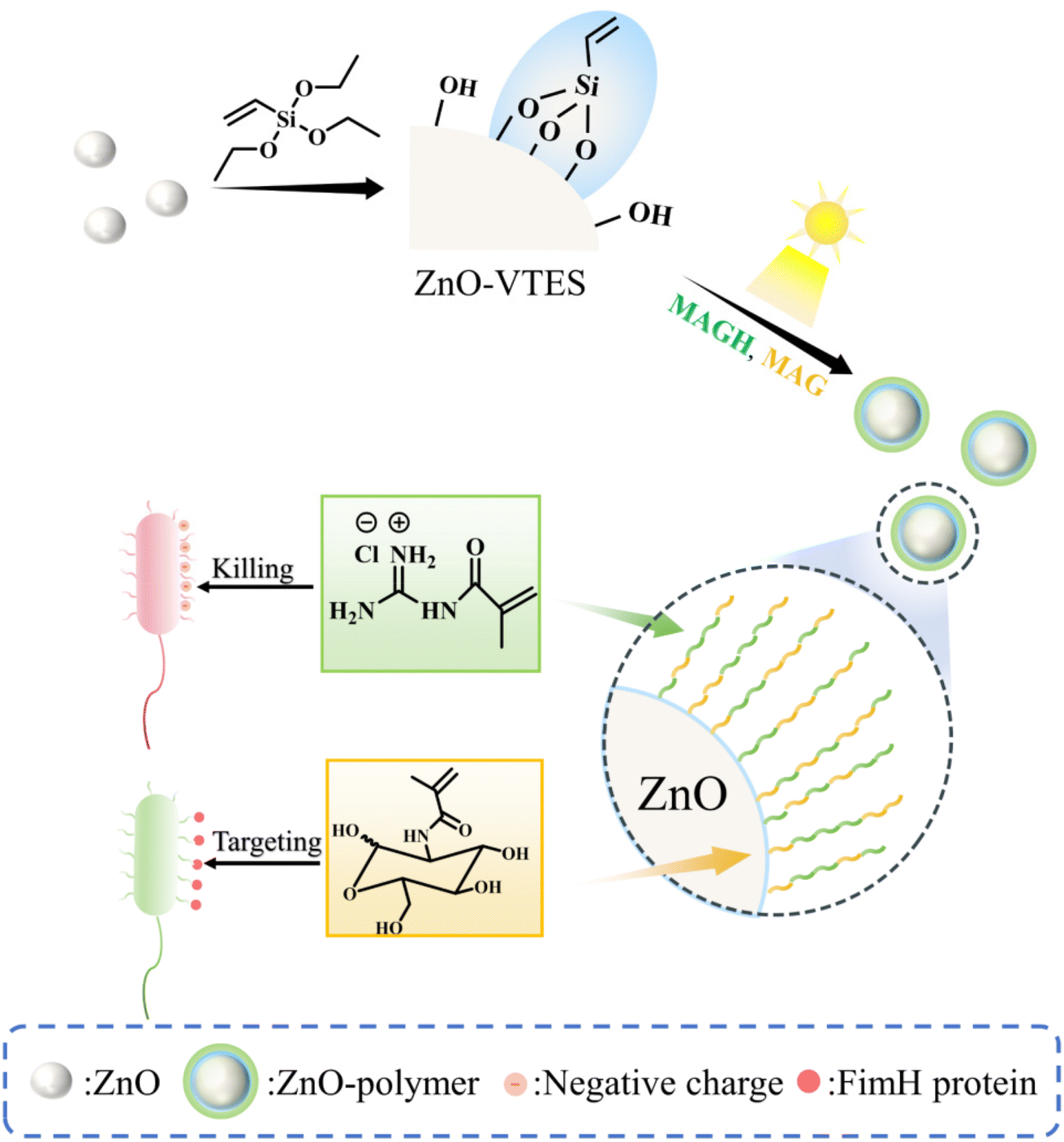 | ||
| Scheme 1 Scheme of ZnO-mediated in situ PET-RAFT to synthesize NPs with enhanced and targeted antibacterial ability. | ||
The confirmation of the synthesis of ZnO-VTES was achieved through the utilization of infrared spectroscopy (IR) and water contact angle (WCA) measurements. A comparison of the FT-IR spectra of ZnO and ZnO-VTES revealed distinct peaks at 1588 cm−1 and 1050 cm−1, which corresponded to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and Si–O bond, respectively, indicating the presence of vinyltriethoxysilane in the latter (Fig. 1A). The WCA measurements of ZnO and ZnO-VTES are displayed in Fig. 1B. The surface of ZnO exhibited superhydrophilicity, as evidenced by an almost zero water contact angle. In contrast, ZnO-VTES displayed a WCA of 8.6 ± 0.2°, which can be attributed to the abundance of hydrophilic hydroxyl groups on the surface of the ZnO NPs and the incorporation of hydrophobic silane coupling agents.38
C bond and Si–O bond, respectively, indicating the presence of vinyltriethoxysilane in the latter (Fig. 1A). The WCA measurements of ZnO and ZnO-VTES are displayed in Fig. 1B. The surface of ZnO exhibited superhydrophilicity, as evidenced by an almost zero water contact angle. In contrast, ZnO-VTES displayed a WCA of 8.6 ± 0.2°, which can be attributed to the abundance of hydrophilic hydroxyl groups on the surface of the ZnO NPs and the incorporation of hydrophobic silane coupling agents.38
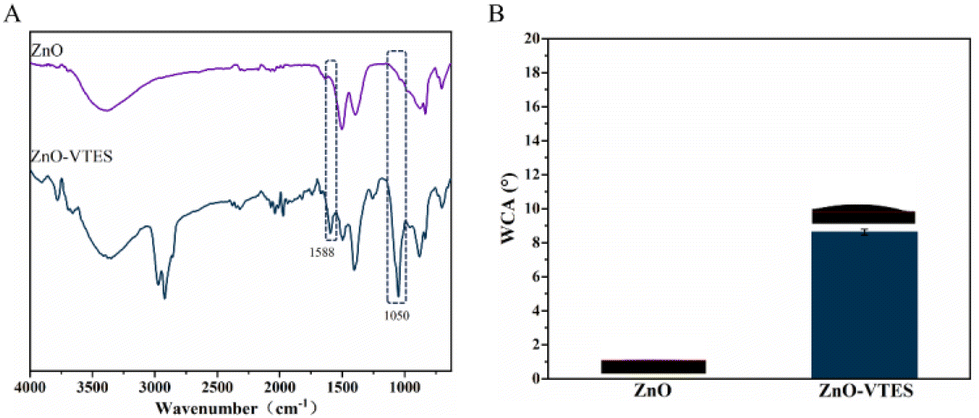 | ||
| Fig. 1 (A) FT-IR spectra of ZnO and ZnO-VTES. (B) WCAs of ZnO and ZnO-VTES. | ||
We have determined the quantity of polymers grafted onto the particle surface using TGA (Fig. 2A). As the polymerization time increased, the overall weight loss in polymer-modified ZnO NPs rose from 8.9 to 26.7%. We also monitored the conversion of monomers in solution using 1H NMR. As depicted in Fig. S5,† the alkene hydrogens of MAGH and MAG decreased gradually in the reaction, which confirms the successful polymerization of the two monomers. The polymerization rate of MAG was slightly slower than that of MAGH, consistent with previously reported literature indicating that sugar monomers typically polymerize at a slower rate due to bulk hindrance.39,40 The overall conversion of monomers determined using 1H NMR was comparable to that obtained using TGA, indicating that the majority of the polymer formed was grafted onto the NPs (Fig. 2B). This observation suggests that most of the free radicals were generated near the ZnO NPs, leading to the grafting of polymers onto the ZnO NP surfaces. It is also worth noting that the rate of polymer grafting decelerated and plateaued after 60 minutes, as a result of the polymer coating on the ZnO NPs affecting the photocatalytic activity and hindering the polymerization reaction, a characteristic phenomenon observed in grafting-through methodologies.41Fig. 2C displays representative scanning electron microscopy (SEM) images of ZnO-G at various polymerization durations. As the polymerization time increased, there was an augmentation in the particle size of the ZnO-G core–shell particles. This observation suggested an increase in the amount of grafted polymer on the ZnO surface, confirming the success of the grafting-through polymerization.
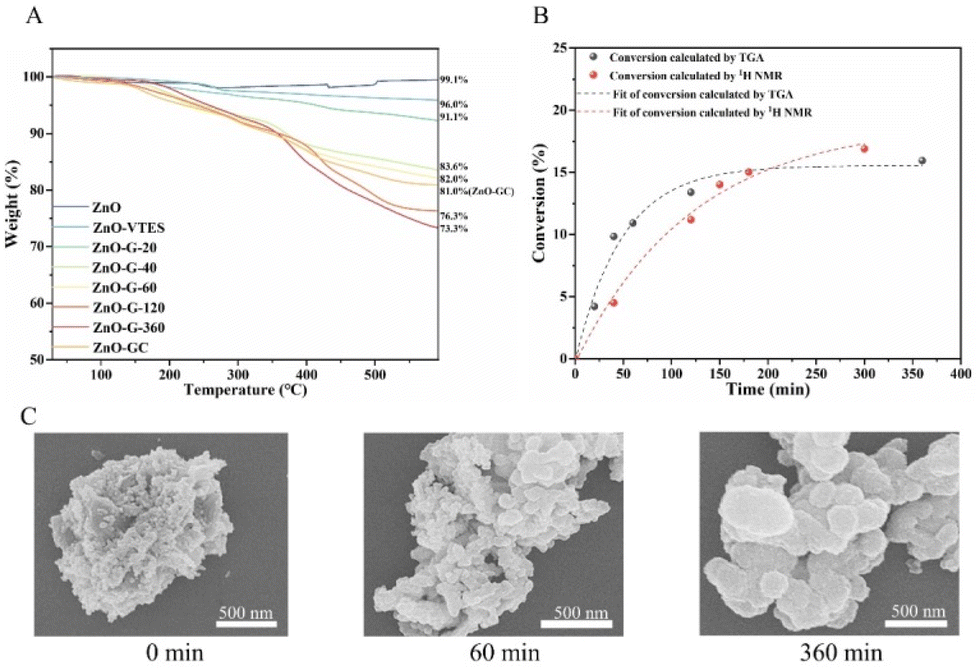 | ||
| Fig. 2 (A) TGA of ZnO, ZnO-VTES, ZnO-GC and ZnO-G with different polymerization times. (B) Conversion vs. time for the polymerization of MAGH and MAG determined by TGA and 1H NMR. (C) SEM images of ZnO-G with polymerization times of 0, 60 and 360 min. | ||
In order to achieve specific antibacterial properties for the ZnO–polymer, we employed the copolymerization of guanidine monomer MAGH and carbohydrate monomer MAG to synthesize ZnO-GC (Fig. 3A). The ZnO-GC sample exhibited characteristic FT-IR peaks at 2935 cm−1 and 1658 cm−1, corresponding to the sugar ring on PMAG and CO on the ester bond, respectively. These peaks indicate the successful grafting polymerization of MAGH and MAG onto ZnO (Fig. 3B). The zeta potentials of ZnO-G and ZnO-GC were measured to be +29.5 mV and +20.3 mV, respectively (Fig. S8†). The higher zeta potential of ZnO-G can be attributed to the presence of the positively charged guanidine group on PMAGH. On the other hand, ZnO-GC exhibited a lower zeta potential compared to ZnO-G due to the replacement of half of the positively charged PMAGH with uncharged PMAG.
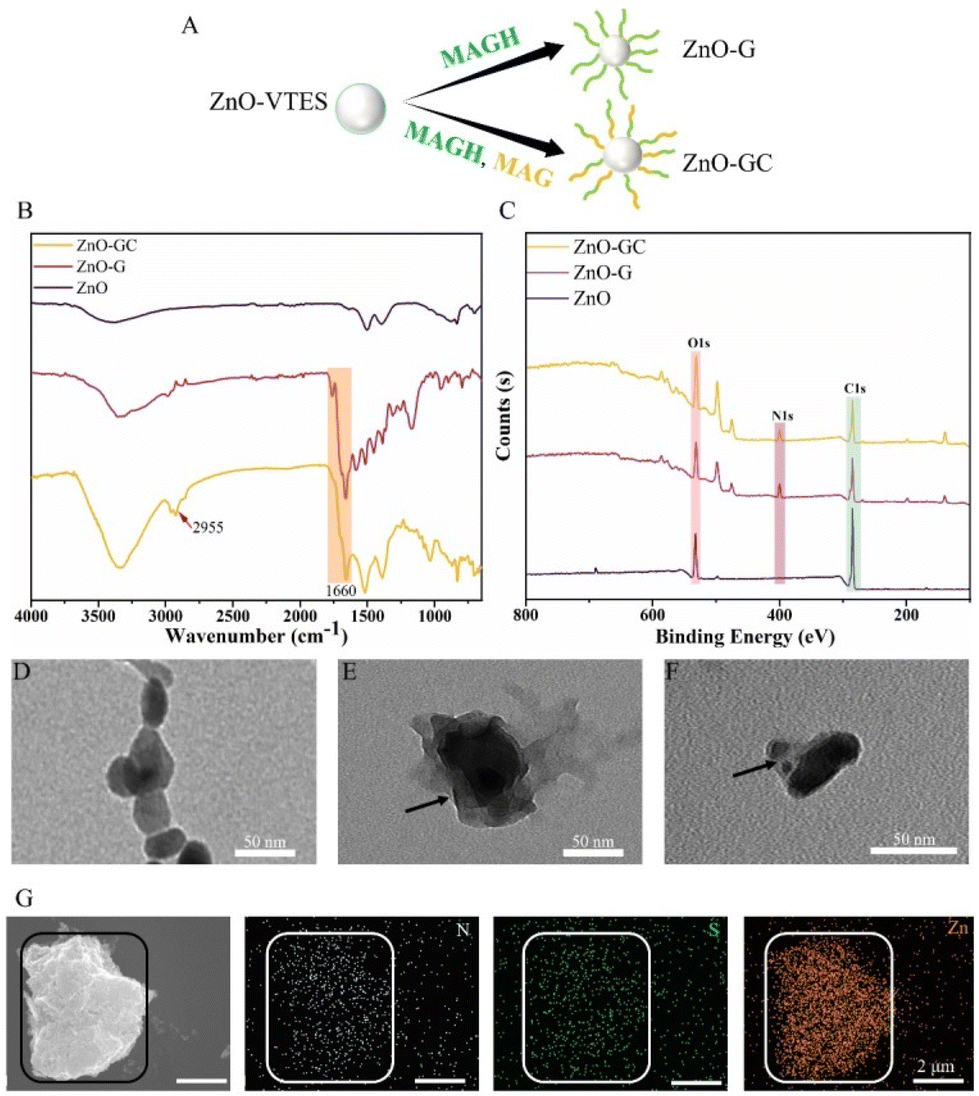 | ||
| Fig. 3 (A) Scheme showing ZnO-G and ZnO-GC. (B) FT-IR spectra of ZnO, ZnO-G and ZnO-GC. (C) C, N, O contents of ZnO, ZnO-G and ZnO-GC determined by XPS. TEM images of (D) ZnO, (E) ZnO-G and (F) ZnO-GC. SEM and SEM-EDS images of (G) ZnO-GC. | ||
The X-ray photoelectron spectroscopy (XPS) technique was employed to analyze the ZnO–polymer core–shell particles (Fig. 3C and Fig. S9†). The N and O contents in ZnO-G were 11.36% and 21.08%, respectively, whereas the N and O contents in ZnO-GC were 7.5% and 28.21%, respectively. The disparity in the N and O contents between ZnO-G and ZnO-GC can be attributed to the introduction of carbohydrates, which possess a higher proportion of O, and the reduced concentration of guanidine groups, which possess a higher proportion of N, in ZnO-GC.
Additionally, the morphologies of the ZnO NPs were compared using transmission electron microscopy (TEM). Notably, both ZnO-G and ZnO-GC exhibited a distinct contrast, with the lighter polymer portion enveloping the darker ZnO core, in comparison to the initial ZnO nanoparticles (Fig. 3D–F). Additionally, the SEM-EDS analysis of the distribution of N, S and Zn in ZnO-GC closely corresponded to their respective morphologies (Fig. 3G). This suggests the presence of MAGH/MAG and CPABD on the surface of the ZnO NPs, further confirming the successful synthesis of ZnO–polymer core–shell nanoparticles. Thereby providing further evidence for the successful synthesis of ZnO–polymer core–shell nanoparticles.
Antibacterial assay
With the introduction of guanidine and carbohydrate polymers, the antibacterial efficacy of ZnO was enhanced (Fig. 4A–C). ZnO-G exhibited a concentration-dependent effect, displaying a stronger bactericidal efficacy with 96% antibacterial efficacy at a concentration of 500 μg mL−1. This can be attributed to the increased bacterial interaction with ZnO-G due to the presence of guanidine cations. Additionally, the guanidine groups formed stronger bidentate hydrogen bonds with bacterial cell membranes, leading to cell wall rupture and subsequent bacterial death. Specifically, when the carbohydrate MAG was introduced to form ZnO-GC, the antibacterial efficiency against E. coli was further improved. At a concentration of 100 μg mL−1, it exhibited a strong antibacterial effect, approaching 100%. This finding suggests that the glycopolymers and cationic guanidine polymers on ZnO NPs synergistically contribute to the capture and killing of bacteria.43
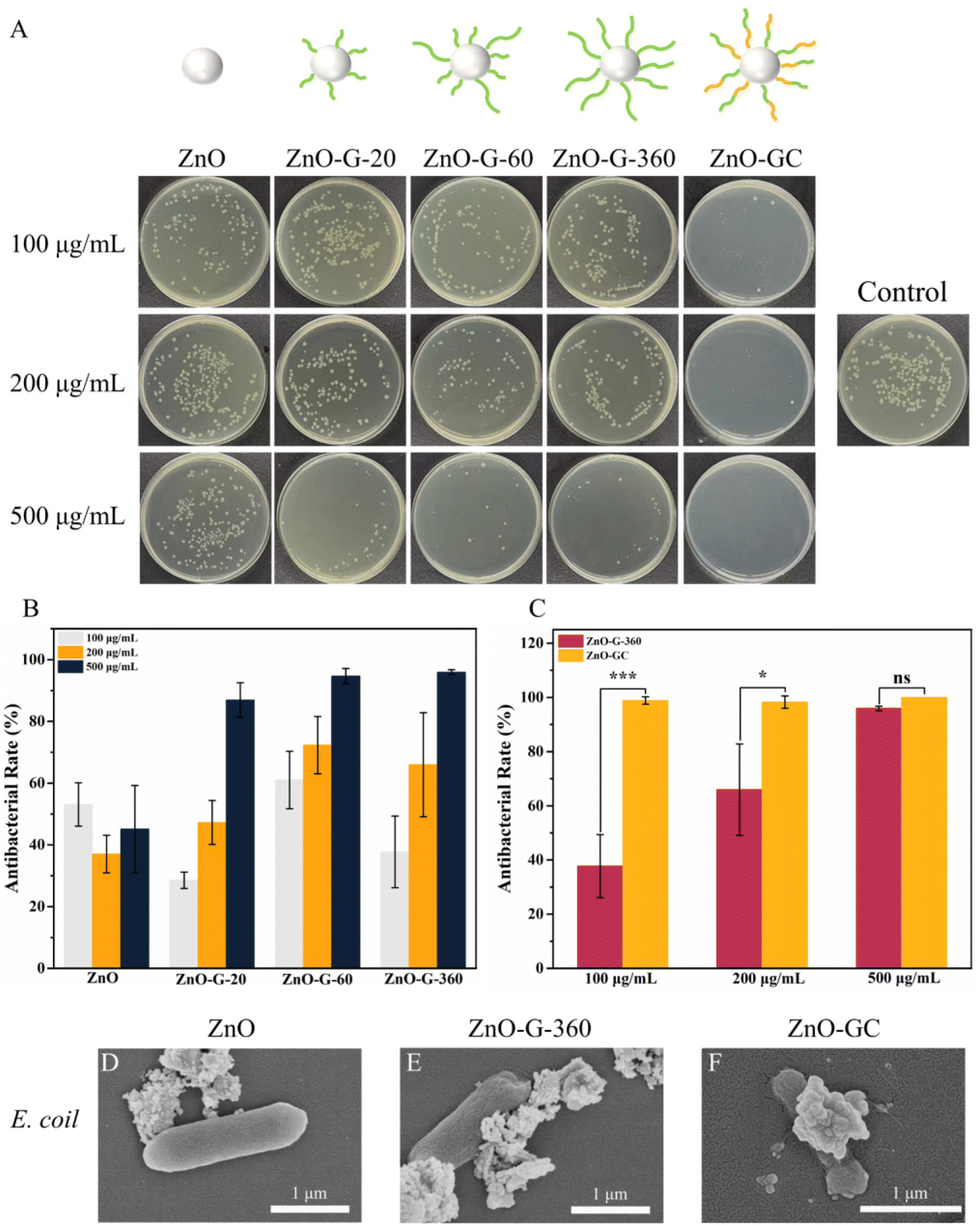 | ||
| Fig. 4 (A) Antibacterial effects of ZnO, ZnO-G and ZnO-GC at concentrations 100, 200 and 500 μg mL−1 against E. coli. (B) Influence of polymerization time on the antibacterial effects of ZnO-G at different concentrations against E. coli. (C) Influence of introducing glycopolymer on antibacterial effects of ZnO-G-360 and ZnO-GC against E. coli. Data are reported as mean ± SD (n = 3). SEM images of E. coli treated with (D, E and F) ZnO, ZnO-G and ZnO-GC at 100 μg mL−1. | ||
To elucidate the antibacterial mechanism and understand the impact of ZnO-G and ZnO-GC core–shell NPs on bacterial morphology, the interaction between E. coli and ZnO, ZnO-G-360 and ZnO-GC at a concentration of 100 μg mL−1 was investigated by SEM. ZnO NPs exhibited a limited association with the bacteria, whereas ZnO-G-360 showed significantly enhanced interaction. Notably, ZnO-GC not only exhibited pronounced contact with the bacteria's surface, but there was also evident bacterial collapse, indicating substantial damage inflicted upon the bacteria (Fig. 4D–F). Therefore, the introduction of guanidine and carbohydrate polymers can effectively increase the antibacterial performance against E. coli.
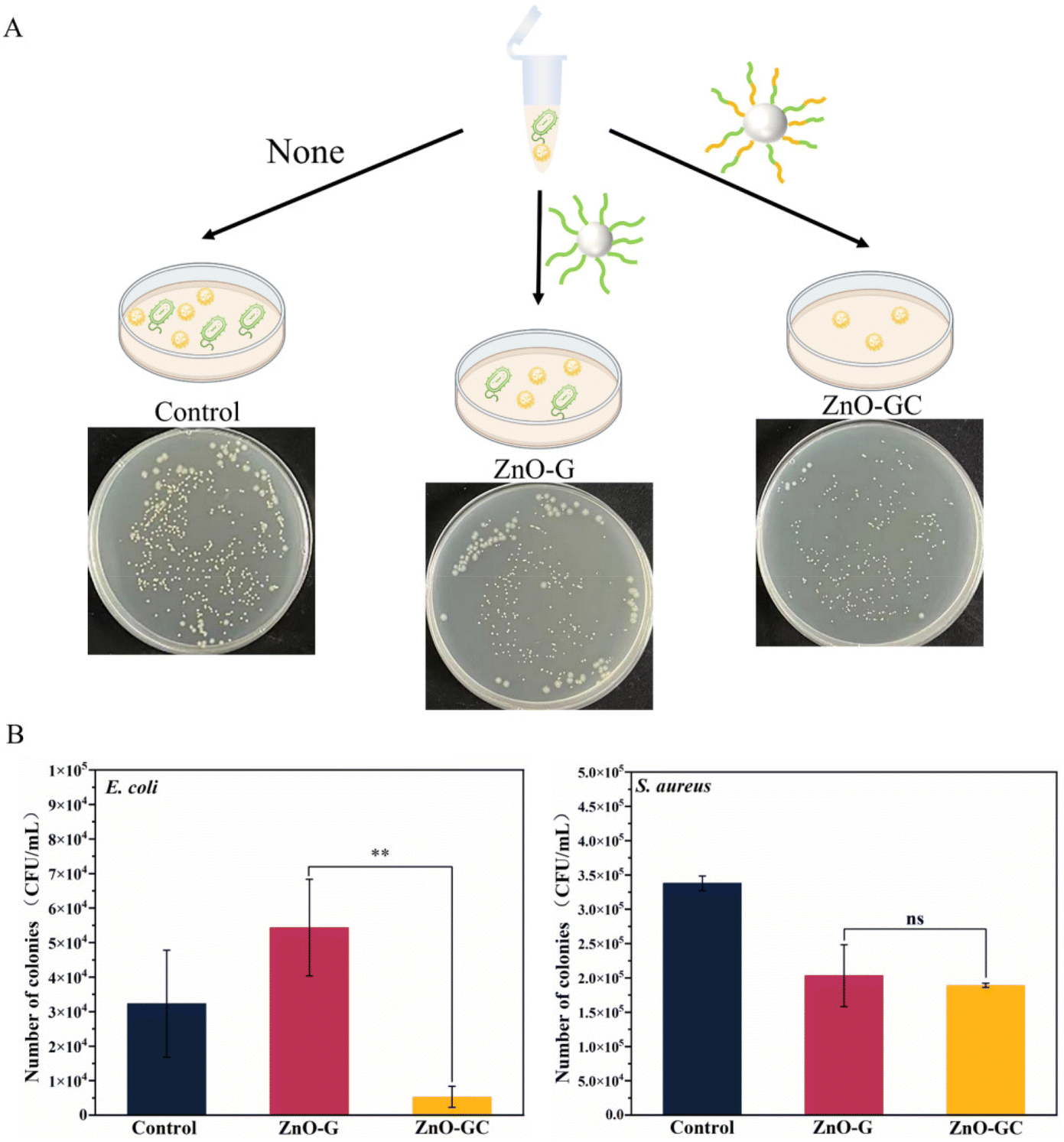 | ||
| Fig. 5 (A) Scheme showing ZnO-G and ZnO-GC treatment of mixed bacterial solutions of E. coli and S. aureus. (B) Antibacterial effects of ZnO-G and ZnO-GC at 100 μg mL−1 on mixed bacterial solutions and statistics on the number of colonies. Data are reported as mean ± SD (n = 3). | ||
Conclusions
In this study, we successfully synthesized novel ZnO–polymer core–shell nanoparticles utilizing a green and highly effective in situ photo-polymerization technique. Remarkably, the ZnO nanoparticles acted as catalysts, facilitating oxygen-tolerant in situ PET-RAFT polymerization on their silanized surface. The incorporation of double bonds on the ZnO surface ensures a grafting-through mechanism for the growth of guanidine and carbohydrate polymers on its surface. The incorporation of guanidine and carbohydrates significantly augmented the interaction between ZnO NPs and bacteria, resulting in improved antibacterial efficacy. Additionally, this modification conferred specific antibacterial properties against E. coli. The grafting of polymers greatly enhanced the antibacterial capabilities of ZnO. In order to achieve a bactericidal effect on over 95% of E. coli, a concentration of 10 mg mL−1 of ZnO is required, whereas ZnO-G necessitates a concentration of 500 μg mL−1, and ZnO-GC only requires 100 μg mL−1. Moreover, aside from the enhanced antibacterial efficacy, the findings revealed that in a mixed bacterial solution, ZnO-GC exhibited a preferential bactericidal effect on E. coli in the presence of S. aureus. In summary, the successful preparation of a core–shell material with potent antibacterial properties has been accomplished. The incorporation of guanidine has enhanced the broad-spectrum antibacterial efficacy without any selectivity, while the addition of carbohydrates has conferred improved and distinctive antibacterial capabilities towards specific bacterial species. This innovative approach has presented a promising strategy for the synthesis of novel antibacterial materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (No. 22171203 and 21935008), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Key Laboratory of Polymeric Materials Design and Synthesis for Biomedical Function, Soochow University. The authors also thank the website of app.Biorender.com for the assistance in creating the illustration figures.References
- J. Jutkina, N. P. Marathe, C. F. Flach and D. G. J. Larsson, Sci. Total Environ., 2018, 616–617, 172–178 CrossRef CAS PubMed.
- A. H. van Hoek, D. Mevius, B. Guerra, P. Mullany, A. P. Roberts and H. J. Aarts, Front. Microbiol., 2011, 2, 203 Search PubMed.
- D. N. Wilson, Nat. Rev. Microbiol., 2014, 12, 35–48 CrossRef CAS PubMed.
- Y. Rao, J. Wang, H. Wang, H. Wang, R. Gu, J. Shen, Q. Hao, J. L. Brash and H. Chen, Biomacromolecules, 2022, 23, 2170–2183 CrossRef CAS PubMed.
- G. M. Whitesides, Small, 2005, 1, 172–179 CrossRef CAS PubMed.
- Y. Li, C. Liao and S. C. Tjong, Int. J. Mol. Sci., 2020, 21, 8836 CrossRef CAS PubMed.
- A. Hui, R. Yan, W. Wang, Q. Wang, Y. Zhou and A. Wang, Carbohydr. Polym., 2020, 247, 116685 CrossRef CAS PubMed.
- H. R. Kim, D. Lee, G. H. Lee, S. K. Kim, S. J. Choi, E. T. Hwang, A. Maharjan, B. S. Kim, D. Kim and J. H. Joo, Part. Part. Syst. Charact., 2019, 36, 1900141 CrossRef CAS.
- B. L. da Silva, B. L. Caetano, B. G. Chiari-Andreo, R. Pietro and L. A. Chiavacci, Colloids Surf., B, 2019, 177, 440–447 CrossRef PubMed.
- X. Zhu, J. Wang, L. Cai, Y. Wu, M. Ji, H. Jiang and J. Chen, J. Hazard. Mater., 2022, 430, 128436 CrossRef CAS PubMed.
- J. Liu, J. Shao, Y. Wang, J. Li, H. Liu, A. Wang, A. Hui and S. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16264–16273 CrossRef CAS.
- C. Bressy, V. G. Ngo, F. Ziarelli and A. Margaillan, Langmuir, 2012, 28, 3290–3297 CrossRef CAS PubMed.
- A. Hui, R. Yan, W. Wang, Q. Wang, Y. Zhou and A. Wang, Carbohydr. Polym., 2020, 247, 116685 CrossRef CAS PubMed.
- Y. J. Jo, E. Y. Choi, N. W. Choi and C. K. Kim, Ind. Eng. Chem. Res., 2016, 55, 7801–7809 CrossRef CAS.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, Chem. Commun., 2018, 54, 6591–6606 RSC.
- M. Cvek, A. M. Jazani, J. Sobieski, T. Jamatia and K. Matyjaszewski, Macromolecules, 2023, 56, 5101–5110 CrossRef CAS PubMed.
- S. Dadashi-Silab, A. M. Asiri, S. B. Khan, K. A. Alamry and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1500–1507 CrossRef CAS.
- X. Li, L. Zhang, Z. Wang, S. Wu and J. Ma, Carbohydr. Polym., 2021, 259, 117752 CrossRef CAS PubMed.
- E. Liang, M.-s. Liu, B. He and G.-X. Wang, Adv. Polym. Technol., 2018, 37, 2879–2884 CrossRef CAS.
- Y. Zhou, C. Gu, L. Zheng, F. Shan and G. Chen, Polym. Chem., 2022, 13, 989–996 RSC.
- G. Zhang, M. Zhou, Z. Xu, C. Jiang, C. Shen and Q. Meng, J. Colloid Interface Sci., 2019, 540, 295–305 CrossRef CAS PubMed.
- M. Kufleitner, L. M. Haiber and V. Wittmann, Chem. Soc. Rev., 2023, 52, 510–535 RSC.
- J. I. Quintana, U. Atxabal, L. Unione, A. Ardá and J. Jiménez-Barbero, Chem. Soc. Rev., 2023, 52, 1591–1613 RSC.
- M. Nagao, H. Matsumoto and Y. Miura, Chem. – Asian J., 2023, 18, e202300643 CrossRef CAS PubMed.
- A. Monaco, B. D. Barbieri, G. Yilmaz, R. J. Shattock and C. R. Becer, Polym. Chem., 2023, 14, 2750–2761 RSC.
- M. H. Stenzel, Macromolecules, 2022, 55, 4867–4890 CrossRef CAS.
- C. von der Ehe, C. Weber, M. Gottschaldt and U. S. Schubert, Prog. Polym. Sci., 2016, 57, 64–102 CrossRef CAS.
- M. Nagao, T. Masuda, M. Takai and Y. Miura, J. Mater. Chem. B, 2024, 1782–1787, 10.1039/D3TB02663A.
- J. Sun, J. Guo, L. Zhang, L. Gong, Y. Sun, X. Deng and W. Gao, J. Controlled Release, 2023, 356, 175–184 CrossRef CAS PubMed.
- L. Su, Y. Feng, K. Wei, X. Xu, R. Liu and G. Chen, Chem. Rev., 2021, 121, 10950–11029 CrossRef CAS PubMed.
- N. Sharon, FEBS Lett., 1987, 217, 145–157 CrossRef CAS PubMed.
- T. J. Cuthbert, B. Hisey, T. D. Harrison, J. F. Trant, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2018, 57, 12707–12710 CrossRef CAS PubMed.
- Z. Miao, D. Li, Z. Zheng and Q. Zhang, Polym. Chem., 2019, 10, 4059–4066 RSC.
- M. J. Hajipour, K. M. Fromm, A. A. Ashkarran, D. J. d. Aberasturi, I. R. d. Larramendi, T. Rojo, V. Serpooshan, W. J. Parak and M. Mahmoudi, Trends Biotechnol., 2012, 30, 499–511 CrossRef CAS PubMed.
- H. Zhang, Y. Liu, T. Luo, Q. Zhao, K. Cui, J. Huang, T. Jiang and Z. Ma, Polym. Chem., 2018, 9, 3922–3930 RSC.
- W. Lu, W. Ma, J. Lu, X. Li, Y. Zhao and G. Chen, Macromol. Rapid Commun., 2014, 35, 827–833 CrossRef CAS PubMed.
- N. Khurana, P. Arora, A. S. Pente, K. C. Pancholi, V. Kumar, C. P. Kaushik and S. Rattan, Inorg. Chem. Commun., 2021, 124, 108347 CrossRef CAS.
- L. Yin, M. C. Dalsin, A. Sizovs, T. M. Reineke and M. A. Hillmyer, Macromolecules, 2012, 45, 4322–4332 CrossRef CAS.
- J. R. Lamb, K. P. Qin and J. A. Johnson, Polym. Chem., 2019, 10, 1585–1590 RSC.
- C. Lin, T. Lü, D. Qi, Z. Cao, Y. Sun and Y. Wang, Ind. Eng. Chem. Res., 2018, 57, 15280–15290 CAS.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS PubMed.
- Y. Zheng, Y. Luo, K. Feng, W. Zhang and G. Chen, ACS Macro Lett., 2019, 8, 326–330 CrossRef CAS PubMed.
- N. Sharon, FEBS Lett., 2001, 217, 145–157 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00223g |
| This journal is © The Royal Society of Chemistry 2024 |