
Lattice engineering of noble metal-based nanomaterials via metal–nonmetal interactions for catalytic applications
Long
Zheng†
,
Lei
Xu†
,
Ping
Gu†
and
Ye
Chen
 *
*
Department of Chemistry, The Chinese University of Hong Kong, Hong Kong, China. E-mail: yechen@cuhk.edu.hk
First published on 16th March 2024
Abstract
Noble metal-based nanomaterials possess outstanding catalytic properties in various chemical reactions. However, the increasing cost of noble metals severely hinders their large-scale applications. A cost-effective strategy is incorporating noble metals with light nonmetal elements (e.g., H, B, C, N, P and S) to form noble metal-based nanocompounds, which can not only reduce the noble metal content, but also promote their catalytic performances by tuning their crystal lattices and introducing additional active sites. In this review, we present a concise overview of the recent advancements in the preparation and application of various kinds of noble metal–light nonmetal binary nanocompounds. Besides introducing synthetic strategies, we focus on the effects of introducing light nonmetal elements on the lattice structures of noble metals and highlight notable progress in the lattice strain engineering of representative core–shell nanostructures derived from these nanocompounds. In the meantime, the catalytic applications of the light element-incorporated noble metal-based nanomaterials are discussed. Finally, we discuss current challenges and future perspectives in the development of noble metal–nonmetal based nanomaterials.
 Ye Chen | Dr Ye Chen is currently an assistant professor at the Department of Chemistry, The Chinese University of Hong Kong. She received her B. Eng. and Ph.D. degrees (Supervisor: Prof. Hua ZHANG) in materials science from Nanyang Technological University (Singapore) in 2015 and 2019, respectively. Her current research interest focuses on wet-chemical synthesis of novel low-dimensional nanomaterials and their applications in catalysis and clean energy. |
1. Introduction
Noble metal-based nanomaterials possess outstanding catalytic properties, including activity, selectivity, and durability, in a variety of catalytic reactions,1–4 which are important for developing sustainable energy storage and conversion technologies to mitigate global energy crisis and environmental pollution.5–7 For example, noble metal based electrocatalysts play key roles in proton exchange membrane fuel cells, which are promising clean energy conversion devices due to their high efficiency, high power density and low pollution emission.8,9 However, the low abundances and ever-increasing prices of noble metals still inhibit their commercial applications. Numerous strategies have been developed to fine-tune the noble metal-based nanostructures, including size,10 shape/facet,11 and composition control,12,13 to enhance their intrinsic catalytic properties and facilitate their sustainable use. Among these strategies, alloying noble metals with other elements to form nanoalloys or nanocompounds stands out as one of the most effective approaches to simultaneously reduce noble metal consumption, modify their electronic structure, and promote their catalytic properties.14,15Particularly, compared with the intensively studied metal–metal alloys, the incorporation of light nonmetal elements to form metal–nonmetal alloys or compounds has garnered increasing attention in the past decades because of several advantages.12,16 First, nonmetal elements have a wider range of electronegativities, which can lead to a variety of metal–nonmetal interactions that are covalent, metallic, or ionic. Second, a simultaneous change of the lattice structure, including strain and symmetry, of host noble metals could take place, causing local lattice distortion or even phase transformation. Depending on the insertion pathway, there are two common types of light nonmetal incorporation strategies in the noble metal.17 Lighter elements with smaller atomic radii, such as hydrogen (H), boron (B), carbon (C), and nitrogen (N), tend to occupy interstitial sites in the host metal, while for elements with larger atomic/ionic radii, such as phosphorus (P) and sulfur (S), they can also substitute the host atoms to constitute alloys or nanocompounds. Consequently, the binary nanoalloys or nanocompounds may exhibit distinct lattice strain or altered symmetry compared to the parent metal lattice (Fig. 1). Importantly, these modifications of the lattice structure of nanomaterials could affect their physicochemical properties and eventually impact their catalytic performances.18–20 Third, the abundant reserves of most light nonmetal elements make it cost-effective to develop noble metal–light nonmetal based nanomaterials.
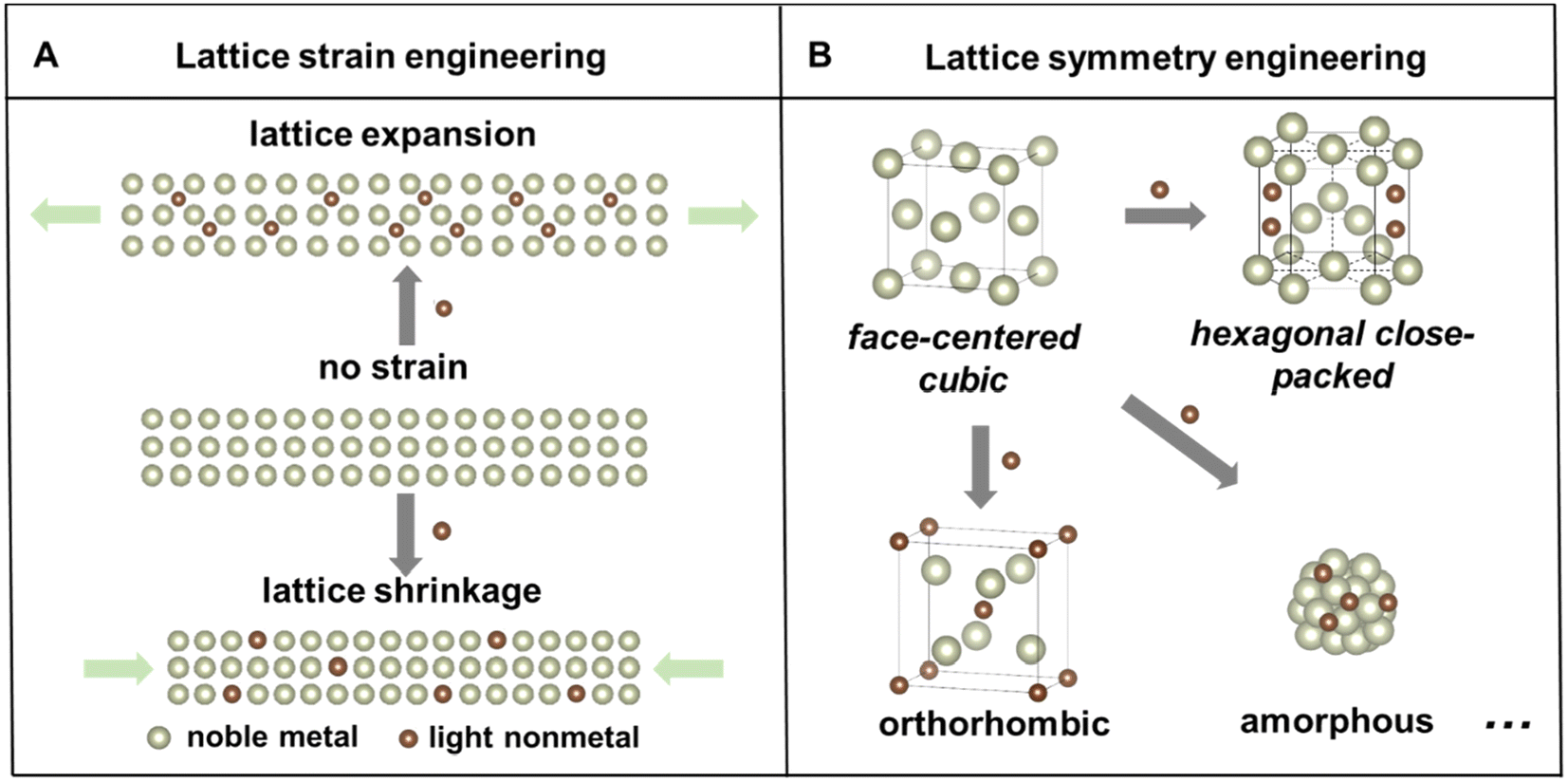 | ||
| Fig. 1 Schematic illustrations of lattice engineering of noble metal–light nonmetal binary nanocompounds via (A) lattice strain engineering and (B) lattice symmetry engineering from an fcc host metal. | ||
Strategies to achieve the incorporation of light nonmetal elements can be divided into two main categories, i.e. wet chemical methods and high-temperature solid-state methods. Compared with the harsh conditions in solid-state methods,21,22 wet chemical methods possess attractive advantages, including accurate control over the morphology and composition,23,24 more flexible synthetic routes for complex nano-architechtures,25,26 as well as good scalability. In the meantime, besides synthesis methods, the recent development of advanced characterization techniques, such as aberration-corrected transmission electron microscopy (TEM), neutron powder diffraction (NPD), extended X-ray absorption fine structure (EXAFS) spectroscopy, and X-ray absorption near-edge structure (XANES) spectroscopy, allows us to better understand the structure–property relationship and further optimize the nanostructures towards enhanced catalytic properties.
In this mini-review, we present an overview of the recent progress in the controlled synthesis, characterization, and catalytic performances of various noble metal-based nanomaterials, whose lattices were engineered by incorporating H, B, C, N, P, or S via wet chemical methods. We classify strategies to achieve the lattice engineering of noble metal-based nanomaterials into two categories: direct lattice engineering and indirect lattice engineering. First, in the direct lattice engineering, representative studies of lattice strain and symmetry engineering of various noble metal-based nanocompounds, including noble metal hydrides, borides, carbides, nitrides, phosphides and sulfides, and their catalytic performances are introduced. Then, typical examples of indirect lattice engineering via the formation of core–shell heterostructures are presented. Finally, some major challenges, opportunities and future directions in this emerging research field are discussed.
2.1. Direct lattice engineering of noble metal–light nonmetal nanocompounds
Based on the types of light nonmetal elements incorporated, in this section, we focus on lattice engineering of binary nanocompounds including noble metal hydrides, borides, carbides, nitrides, phosphides, and sulfides. Since the inserted light nonmetals may directly alter both lattice strain and symmetry of the host noble metals, we will separately discuss the strain and symmetry effects caused by the formation of these nanocompounds in two sections.Noble metal hydrides. Noble metal hydrides are key candidates in many catalytic reactions.14,16 H, as the smallest atom, can be easily incorporate into octahedral or tetrahedral interstitial sites of noble metal lattices, resulting in the formation of metal-H bonds and metal lattice expansion,32–35 The electrons from H atoms could directly contribute to a higher electron density of the host metal and therefore affect its physicochemical properties.36 In the meantime, the tensile strain caused by the expanded lattice can lead to the narrowing and center upshift the d-band of the host metal,37 which could be beneficial for improving the catalytic properties.38–41
To date, wet chemical preparation of noble metal hydrides only succeeded in the preparation of palladium hydride (PdHx)42,43 and rhodium hydride44 possibly due to their high affinity differences.45 The face-centered cubic (fcc) PdHx normally exists in two phases, depending on its H content (x). One is the α phase (0 < x < 0.02), in which the H atoms only slightly diffuse into Pd and cause lattice expansion. The other one is the β phase (0.6 < x < 1), in which the Pd lattice is inserted by more H atoms and further expanded. Sometimes, α and β phases can coexist in PdHx (0.02 ≤ x ≤ 0.6) nanocrystals.46,47 In general, the H interstitial sites are determined by the ratio of H/Pd in the Pd–H system, which can be derived from the lattice parameter of the PdHx nanocompounds.48–50 To distinguish the slight changes in lattice parameters of different PdHx, advanced characterization techniques, like aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-Ray diffraction (XRD) analysis, are often required.
The wet chemical preparation of PdHx nanocrystals often starts from pre-synthesized Pd nanocrystals, followed by H intercalation. The choice of a suitable H precursor is critical. Besides H2, H-containing chemicals that readily release H2 can also act as H sources, including sodium borohydride (NaBH4), hydrazine (N2H4), n-butylamine, and N,N-dimethylformamide (DMF). For example, the NaBH4-based thermal treatment method was used to synthesize PdHx nanocrystals by Phan et al.51 Specifically, pre-synthesized Pd nanocrystals and NaBH4 were dispersed in tetraethylene glycol solution, in which Pd further reacted with NaBH4 during heating to form PdHx. Compared with pure Pd nanocrystals, the PdHx samples prepared at different temperatures (from 90 °C to 190 °C) showed XRD peak shifts to lower 2θ angles (Fig. 2A). It worth mentioning that β-PdH0.7 could be obtained from Pd nanocrystals at reaction temperatures over 120 °C, while for bulk Pd, complete conversion of Pd to β-PdH0.7 occurred only at 190 °C.
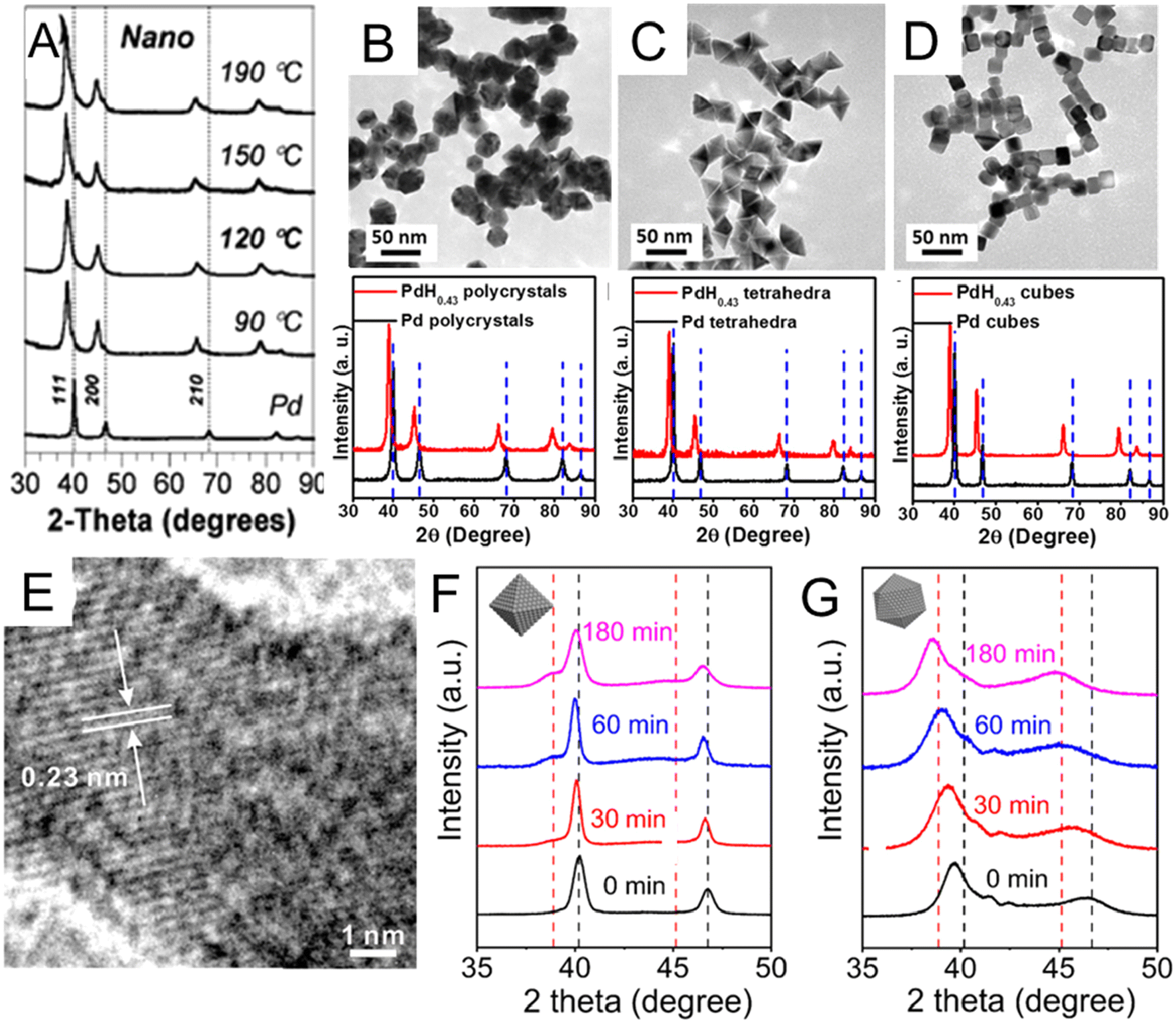 | ||
| Fig. 2 (A) X-ray diffraction (XRD) patterns of lattice-engineered PdHx nanocrystals. Vertical dashed lines indicate the peak positions of Pd. Reproduced with permission from ref 51 Copyright 2009, Royal Society of Chemistry. (B–D) Comparisons of transmission electron microscopy (TEM) images and XRD spectra of PdH0.43 (B) nanopolycrystals, (C) nanotetrahedra, and (D) nanocubes before and after conversion. Reproduced with permission from ref 23. Copyright 2015, American Chemical Society. (E) High resolution TEM (HRTEM) image of a typical PdH0.43 nanocrystals showing larger lattice distances than Pd. Reproduced with permission from ref 53. Copyright 2017, Elsevier. XRD patterns of (F) Pd octahedral nanocrystals and (G) Pd icosahedral nanocrystals after different treating times by N2H4. The black and red dash lines indicate the positions of the peaks associated with fcc-Pd and the β-phase PdH0.7, respectively. Reproduced with permission from ref 55. Copyright 2023, Wiley-VCH. | ||
Different from NaBH4, some organic molecules like DMF and n-butylamine release H2 by reacting with Pd surface to finally form PdHx. Via the organic molecule-based thermal method, shape-controlled synthesis of PdHx has been investigated. For instance, PdH0.43 nanopolycrystals, nanotetrahedra and nanocubes were synthesized by treating the corresponding Pd nanocrystals in DMF solution at 160 °C for 16 h.23 All XRD diffraction peaks of the three final PdH0.43 products shifted to lower angles with respect to those of original Pd nanocrystals, which proved the expansion of Pd lattice and successful incorporation of H atoms into Pd lattices (Fig. 2B–D). The PdH0.43 nanopolycrystals and PdH0.43 nanotetrahedra exhibited higher catalytic activities than their counterparts in the methanol oxidation reaction (MOR). Adopting the same two-step strategy, PdHx nanodendrites were synthesized by heating the pre-prepared Pd nanodendrites in DMF solution at 160 °C for 20 h.52 Compared with commercial Pd nanocrystals and Pd nanodendrites, PdHx nanodendrites showed enhanced electrocatalytic activities for both the formic acid oxidation reaction (FAOR) and hydrogen evolution reaction (HER). The authors applied theoretical analysis to show that PdHx may have weaker binding energies of intermediate catalytic species and toxic CO due to the ligand effect, which led to its higher catalytic activity. Besides shape controlled PdHx nanocrystals with a tunable H content of up to 0.43 have also been realized by the organic molecule-based thermal method. For example, a series of PdHx nanocatalysts with different H contents from 0.10 to 0.43 could be synthesized by heating the commercial Pd black in n-butylamine from 80 °C to 200 °C.53 The authors supposed that H2 was released by n-butylamine, a process catalyzed via Pd black. Then, H2 was adsorbed and dissociated on the Pd surface, enabling H diffusion into interstitial sites of the Pd lattice. The lattice distance (0.23 nm) of H-incorporated Pd (111)f nanoparticle were directly observed by high resolution TEM (HRTEM) to be larger than that of fcc Pd (0.22 nm) (Fig. 2E), proving the successful incorporation of H with an estimated content of 0.43.48,54 Compared with commercial Pd black and PdHx with other stoichiometries (0.10, 0.29, and 0.33), the PdH0.43 nanocatalysts showed lower overpotential, higher mass activity and better catalytic stability towards FAOR. The authors claimed that the excellent performance could be attributed to the weakened binding interactions between PdHx and reaction intermediates.
Another interesting aspect is to understand the phase transformation behavior of PdHx nanocrystals. It is commonly observed that the transformation of α-phase PdHx to β-phase PdHx, starts from the nucleation of the β-phase at the vertices of the nanocrystals, which then spreads to the whole nanocrystal.38 However, the process is considered difficult because the high energetic α/β phase interface is thermodynamically unfavorable for the formation of PdHx with intermediate α/β ratios, which often prevents the final formation of β-phase PdHx.38 Interestingly, a recent work reported a different transformation pathway. Zhou et al. found that the morphology of Pd nanocrystals could affect the transformation pathway of Pd to PdHx when using N2H4 as the H source.55 They observed that the Pd nano-icosahedron could transform into PdHx (0 < x ≤ 0.7) via a “single-phase pathway”, while Pd nano-octahedra with a similar size experienced a “two-phase pathway” to PdHx (0 < x ≤ 0.7) when treated by the same process. Specifically, the analyses of the XRD pattern showed that the Pd nano-octahedra were α phase at the initial stage. As the N2H4 treatment prolonged, a new (111)f peak belonging to the β-phase PdH0.7 appeared at around 38.8°, indicating that the α phase partially converted to the β phase. The two phases coexisted throughout the treatment, known as the “two-phase pathway” (Fig. 2F). As for Pd nano-icosahedra, after treatment with N2H4 for the same period of time, the Pd (111)f peak at about 39.84° from Pd nano-icosahedra left shifted continuously (without splitting into two peaks) until 38.46°, indicating that the PdHx icosahedral nanocrystals experienced a “single-phase pathway”. The H/Pd ratio increased with treatment time and reached 0.7. (Fig. 2G). Note that the icosahedral Pd nanoseeds may possess higher internal lattice strain than octahedral Pd, as indicated by the broader and left-shifted XRD peaks (Fig. 2F and G), which may still be preserved after incorporating H. As a possible result, the PdH0.7 nano-icosahedra demonstrated higher catalytic activity and better catalytic stability toward FAOR than the PdH0.7 nano-octahedra.
While PdHx is the most reported noble metal hydrides, recently, ambient-stable rhodium hydride (RhH) nanosheets with around 1 nm thickness have been reported by Fan et al.44 Different from the preparation of PdHx, a one-pot solvothermal method was applied to synthesize ultrathin RhH nanosheets via reducing the Rh precursor in the mixed solution of n-butylamine, formaldehyde, and benzyl-alcohol at 160 °C. Note that n-butylamine, formaldehyde, and benzyl-alcohol could all serve as the H source. The expanded lattice spacing of RhH (2.36 Å) than that of Rh (2.25 Å) suggested the successful incorporation of H. The existence of H was confirmed by solid-state 1H nuclear magnetic resonance spectra and the atomic ratio Rh/H was confirmed to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by the well-fitted XRD pattern with standard RhH. Compared with commercial Pt/C (30 mV) and Rh nanosheets (57 mV), the RhH nanosheets exhibited excellent HER performance with a lower overpotential (28 mV) and better stability. Theoretical calculations showed that the interstitial H atoms in RhH nanosheets can modulate the adsorption energy of H intermediates and promote H2 desorption to enhance HER activity.
:1 by the well-fitted XRD pattern with standard RhH. Compared with commercial Pt/C (30 mV) and Rh nanosheets (57 mV), the RhH nanosheets exhibited excellent HER performance with a lower overpotential (28 mV) and better stability. Theoretical calculations showed that the interstitial H atoms in RhH nanosheets can modulate the adsorption energy of H intermediates and promote H2 desorption to enhance HER activity.
Noble metal borides. B is located at the boundary between metals and nonmetals in the periodic table, which is well-known as a metalloid element with properties of both metals and nonmetals. Due to the unique electronic structure and small atomic radius of the B element, many different types of noble metal borides have been successfully synthesized via wet chemical methods by employing various B sources, including dimethylamine borane (DMAB), borane tetrahydrofuran (BH3-THF) and NaBH4.56,57
Organic B sources, including DMAB and BH3-THF, are stable and gentle B-releasing agents commonly used in organic solvent-based synthesis.58–60 In 2009, Wang et al. first developed a one-pot synthesis using DMAB to prepare B-doped Pd nanoparticles supported on carbon powder (PdBx/C) in alkaline aqueous solution.58 Importantly, in alkaline solutions, DMAB could react with OH− to generate BH3OH−, which further served as both a reductant and a B source to reduce Pd(II) ions and then release B to form PdBx.61 The obtained PdBx/C with 6.3 at% B content showed fcc Pd characteristic peaks with left shifts in the XRD pattern, indicating the expanded lattice distance in PdBx/C due to the incorporation of B atoms. PdBx/C showed enhanced activity and durability in catalyzing the formic acid electro-oxidation reaction than commercial Pd/C due to the electronic modification effect of B atoms. Then, the same group further expanded the application of the catalyst and applied the catalyst for formic acid decomposition reaction.59 Compared with commercial and homemade Pd/C, PdBx/C showed a much higher activity toward H2 production, possibly because the enlarged Pd lattice in PdBx (Fig. 3A) enhanced the adsorption of formate ions to reduce the activation energy of formic acid dehydrogenation. Due to the strong reduction ability of DMAB, the growth process is thermodynamically controlled, resulting in less morphology control of most reported PdBx nanomaterials.62–64 Therefore, other strategies, such as adding surfactants to control the reaction kinetics or adopting a two-step method to separate the nanocrystal growth and doping processes, have been developed to prepare PdBx nanocrystals with controlled morphologies, such as nanowires and mesoporous nanospheres.65,66 Recently, platinum (Pt) boride nanoparticles supported on carbon black (PtBx/C) have also been reported by using DMAB as the reductant and the B source.60 The content of B in PtBx/C detected by inductively coupled plasma atomic emission spectroscopy (ICP-AES) was around 6.6 at%, causing around 2% expansion of the Pt lattice in the XRD pattern. HRTEM also directly observed the slightly larger lattice distance of (111)f in Pt–B/C (0.231 nm) than that in Pt/C (0.227 nm). Notably, as the oxygen reduction reaction (ORR) catalyst in the single H2–O2 fuel cell, the maximum power density of PtBx/C was 1.24 times as high as that of Pt/C. Besides DMAB, BH3-THF is another organic B source.67,68 Since BH3-THF is liquid, it can serve as both a B source and solvent during the B doping process. For instance, Chan et al. reported PdBx/C with a high content of B (up to 20 at%) by heating Pd/C and BH3-THF in a THF solution at 100 °C for 1 h under a H2 atmosphere.67 During heating, BH3-THF would decompose to B2H6, catalyzed by Pd/C, and further release B atoms.14 After doping, the obtained PdBx/C maintained the fcc phase with 3.6% lattice expansion. EXAFS data showed that the insertion of B clearly led to an increase of the average Pd–Pd distance. Compared with Pd/C, PdBx/C displayed much enhanced selectivity (98%) for partial hydrogenation of 3-hexyn-1-ol to cis-hexen-1-ol without over-hydrogenation products. Theoretical calculations showed that B atoms could occupy the H adsorption sites on the Pd surface and reduce undesirable over-hydrogenation.
 | ||
| Fig. 3 (A) XRD patterns for PdB/C, commercial and homemade Pd/C. Reproduced with permission from ref 59. Copyright 2018, American Chemical Society. (B) Illustration of the synthesis procedure and formation mechanism of PdxB/C. (C) XRD patterns of B-Pd/C and Pd/C. Reproduced with permission from ref 69. Copyright 2017, American Chemical Society. HRTEM images of (D) Ir and (E) IrB nanosheets. Reproduced with permission from ref 74. Copyright 2023, Elsevier. (F) XRD pattern of PdC0.18 nanocubes. Reproduced with permission from ref 24 Copyright 2019, Royal Society of Chemistry. (G) XRD pattern of Ir@PAH metallene. Reproduced with permission from ref 86. Copyright 2022, Wiley-VCH. | ||
As an inorganic boronizing reagent, NaBH4 is a powerful B-source to synthesize a wide range of noble metal borides.69–71 For instance, Li et al. developed a one-pot method to prepare PdBx nanomaterials with different B contents in DMF solution, with NaBH4 serving as both a reductant and a B source (Fig. 3B).69 In DMF solution, NaBH4 can be dissociated into Na+ and BH4−, due to the relatively strong solvation of Na+ by DMF molecules.72 Then, relatively stable BH4− in the polar aprotic solvent such as DMF further was used as a reductant and dopant to synthesize PdBx nanomaterials. The content of B could be tuned by changing the reaction time and NaBH4 concentration. PdBx with the largest B content (20 at%) showed an apparent peak shift than pure Pd (Fig. 3C). Importantly, with the increase of the B content, the ORR activity of PdBx was gradually enhanced. Besides PdBx, NaBH4 could be used to synthesize other noble metal borides. For example, Li et al. prepared trace B-doped osmium (Os) (B–Os) aerogel in aqueous solution using NaBH4 as a reductant and a B source.70 XRD pattern and aberration-corrected HAADF STEM confirmed that the B–Os aerogels showed an enlarged lattice distance than pure hexagonal-close-packed (hcp) Os, attributed to the insertion of B atoms. The resultant B–Os aerogels demonstrated excellent HER catalytic activity in pH-universal electrolytes. The authors claimed that the strong electronic interaction between B and Os atoms could reduce the dissociation energy barrier of H2O and H absorption energy to promote HER activity. In addition, the B-doped rhodium (Rh) aerogel was also reported via one-pot synthesis by applying NaBH4 as the reductant and B source.73 In addition to the abovementioned one-pot methods, Duan et al. reported a two-step method to synthesize B-doped iridium (Ir) nanosheets (IrB NSs) supported on nickel (Ni) foam by using NaBH4 as the B source.74 Ir NSs on the Ni foam support were first prepared by reducing IrCl3. The Ir NSs/Ni form was then boronized in NaBH4-H2O solution in an ice bath for 5 h. After B doping, lattice spacing on the (111)f crystal plane of IrB NSs was 0.225 nm, larger than that of pure Ir (0.218 nm) (Fig. 3D and E). Moreover, the main diffraction peak, (111)f of IrB NSs slightly shifts to a lower angle region compared with that of Ir NSs, indicating the B doping induced expansion of Ir–Ir interplanar spacing. Compared with Ir NSs, IrB NSs showed enhanced performance for the HER and hydrazine oxidation reaction.
Noble metal carbides. As one of the cheapest elements, C has attracted increasing attention for alloying metals to modify their physical, chemical, and mechanical properties.75–77 For example, inserting trace amounts of C into the iron lattice could make steel harder and stronger. However, only a few efforts in the past were made to develop noble metal carbides, especially by the wet chemical method, likely due to the often-harsh synthesis conditions to obtain noble metal carbides.78,79 For instance, Guo et al. reported a series of fcc PdCx nanocubes with tunable C/Pd ratios from 0.04 to 0.18 by heating the as-synthesized Pd nanocubes with α-D-glucose in oleylamine under 200 °C for different lengths of time.24 During heating, C atoms released by α-D-glucose would diffuse into interstitial sites of the Pd lattice. The resultant PdC0.18 had a larger lattice fringe of (200)f plane (0.2 nm) than that of pure Pd (0.19 nm) as suggested by the XRD patterns (Fig. 3F). In the alkyne semihydrogenation reaction, PdC0.18 nanocatalysts exhibited higher selectivity (>99%) towards alkenes compared with pure Pd and PdCx with other stoichiometries, due to the higher desorption rates of alkenes on the PdC0.18 surface evidenced by the Fourier transform infrared spectroscopy. Except for Pd, the insertion of C atoms in the gold (Au) lattice can also enhance the catalytic performance in the hydrogenation reaction.80 Sun et al. prepared C-doped Au (C–Au) nanoparticles supported on ordered mesoporous carbon by a hydrothermal method.80 The authors supposed that the added phenolic resins and triblock copolymer would decompose during heating to small molecules like CH4, and these monoatomic C precursors could accelerate the diffusion of the C adatom into the Au surface. The enlarged lattice distance of the Au (111)f plane was confirmed by aberration-corrected HAADF-STEM images, suggesting the interstitial occupation of C atoms. The C–Au nanocatalyst demonstrated a threefold increase in the turnover frequency (TOF) and superior chemoselectivity (>99%) for 3-vinylaniline in the hydrogenation of 3-nitrostyrene, surpassing the undoped Au/TiO2 catalyst. Theoretical calculations suggested that interstitial C atoms could reduce the dissociation energy barrier of H2 and promote the perpendicular adsorption of nitro groups on the C–Au surface, thereby enhancing the selectivity towards 3-vinylaniline.
Noble metal nitrides. Metal nitrides are non-stoichiometric and interstitial nanocompounds with metallic properties.16,81 N generally tends to incorporate into the interstitial sites in metal lattices with different stoichiometries and endow metals with unique properties like high structural stability under harsh conditions,82 superior electrical conductivities,83 and high melting point.84 However, the synthesis of metal nitrides, especially noble metal nitrides, usually requires extreme conditions like high pressure (above 58 kPa) and high temperature, due to the unreactive N source and high metal–N bond formation energy.12,85 Thus, there are a few reports of noble metal nitrides synthesized by the wet chemical method. In a recent work, Deng et al. reported ultrathin Ir@Poly (allylamine hydrochloride) (PAH) metallene nanosheets with a lateral dimension of about 100 nm by heating the IrCl3, PAH and HCOOH in deionized water at 95 °C for 4 h.86 Energy dispersive X-ray (EDX) elemental mapping proved that the N element from PAH mainly distributed in the exterior of the Ir@PAH metallene. In addition, the XRD pattern (Fig. 3G) showed that all diffraction peaks of Ir@PAH metallene nanosheets left shifted to a lower degree compared with those of fcc Ir. The lattice expansion in Ir@PAH metallene could be a combined result of N insertion and PAH-induced Ir surface hydroxylation. In EXAFS spectra, extra signals of Ir L3-edge in Ir@PAH metallene were observed, from which the Ir–N bonding could be derived with a bond length of around 2.13 Å. Compared with Ir nanosheets and commercial Pt/C, Ir@metallene exhibited better electrocatalytic activity and more robust stability for the HER in an acidic medium. The authors suggested that PAH molecules can optimize the electronic structure of Ir metallene due to strong N–Ir interaction and help Ir capture H ions to form proton-rich intermediates, which is favorable for the HER in an acidic environment.
Noble metal phosphides. Different from the above-mentioned light nonmetal elements with small atomic radii (including H, B, C, and N), P has a larger atomic radius and can either occupy the interstitial sites of the host metal lattice or partially substitute host metal atoms.12,14,87 The replacement/insertion of P into noble metals will affect their electronic structures and therefore physicochemical properties.14 This section mainly discusses the lattice strain effect on the host metals that originated from P incorporation and its impact on their catalytic performances in chemical reactions.14,32 For instance, Guo et al. developed three kinds of ultrathin Pd phosphide (PdPx) nanosheets exposed with different low-index facets ({100}f, {110}f and{111}f) by using different surfactants, employing sodium hypophosphite (NaH2PO4) as the P source (Fig. 4A).88Fig. 4B–D illustrate the typical TEM and XRD characterization of the PdPx nanosheet in the case of exposing{100}f facet. According to the XRD results (Fig. 4D), PdPx nanosheets with a {100}f basal plane exhibited typical fcc diffraction peaks and were left-shifted compared with the standard Pd peaks, indicating that tensile strain was induced after the introduction of P element. PdPx nanosheets with the {110}f and {111}f basal planes also exhibited similar lattice expansion (Fig. 4A). High-resolution X-ray photoelectron spectroscopy (XPS) showed that there exists charge transfer between Pd and P. Thanks to such reconstruction of electronic configuration by P doping, specific activity of PdPx nanosheets with {100}f exposed facets was as high as to 172.2 mA cm−2 towards the ethanol oxidation reaction (EOR), much higher than that of commercial Pd/C catalysts (30.8 mA cm−2). As an alternative to the inorganic P source, such as NaH2PO4, organic compounds may also provide P during the wet chemical synthesis. Guo et al. designed a phosphorization treatment to synthesize Pt phosphide (Pt2P) nanoparticles by treating Pt nanocubes with tri-n-octylphosphine (TOP) at 300 °C for 3 hours through a surface atomic diffusion process.89 The morphology changed from a cubic to spherical shape (Fig. 4E) after the introduction of P. HRTEM image of a single Pt2P nanoparticle along the [011]f zone axes demonstrated the interplanar distance of the (111) facet to be 0.27 nm (Fig. 4F), larger than that of metallic Pt (111). In addition, the XRD pattern indicated that the as-prepared Pt2P possessed the fcc phase with a noticeable peak shift to lower angles due to P insertion (Fig. 4G). In the ORR, the Pt2P catalyst showed 10.3 times enhanced mass activity compared to the commercial Pt/C catalyst. In addition to the lattice expansion of noble metals caused by P insertion, lattice shrinkage could also be induced by partially replacing noble metal atoms with P. For instance, Mao et al. synthesized Rh phosphide (RhPx) mesoporous nanoparticles by reducing potassium hexachlororhodate(III) (K2RhCl6) and sodium hypophosphite (NaH2PO2) via a wet chemical method. The XRD peak of RhPx is slightly right-shifted compared with that of Rh nanocrystals, indicating the shrinkage of the lattice in RhPx ascribed to the doping of P.90
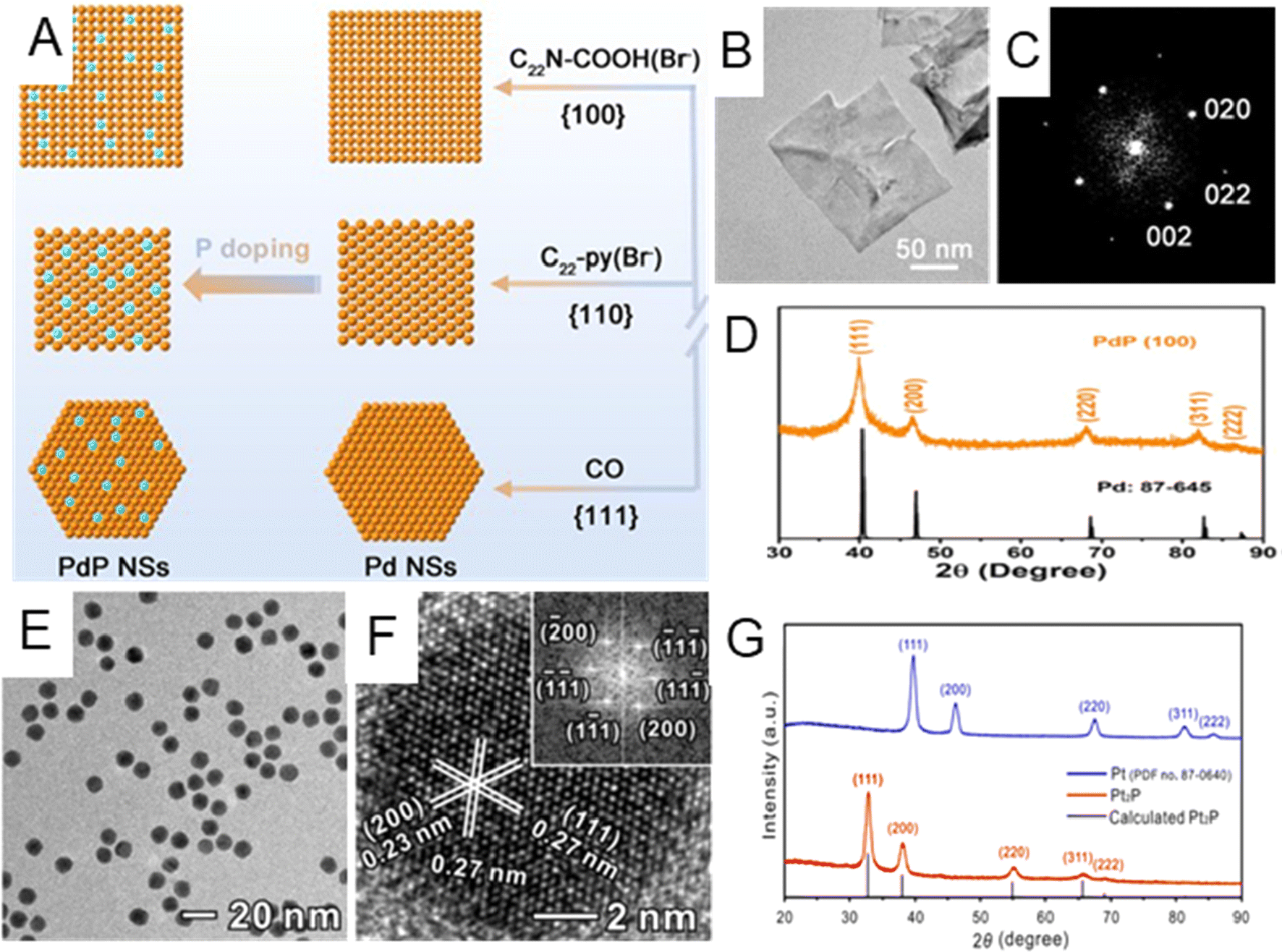 | ||
| Fig. 4 (A) Schematic illustration of the growth mechanism for three types of PdPx nanosheets with different exposed PdPx facets, namely {100}f, {110}f, and {111}f. (B) HRTEM image, (C) the corresponding Fourier diffractogram, and (D) XRD patterns of PdPx nanosheets with the {100}f facet. Reproduced with permission from ref 88. Copyright 2022, American Chemical Society. (E) Low-magnification TEM image of Pt2P nanocrystals. (F) HRTEM image of a single Pt2P nanoparticle. Inset: the corresponding fast Fourier transformation (FFT) pattern. (G) XRD patterns for Pt2P and Pt. Reproduced with permission from ref 89. Copyright 2019, American Chemical Society. | ||
Noble metal borides. In section 2.1.1, we summarized the recent works to prepare fcc PdBx nanomaterials by the wet chemical method. However, previous attempts could only achieve low B contents (<20 at%) and maintain the fcc Pd lattice.67,69 Density functional theory (DFT) calculations showed that on further increasing the B content in the PdBx compound, the hcp Pd2B phase could become thermodynamically stable.94 In 2017, Kobayashi et al. successfully prepared the first hcp Pd2B nanocrystals by inserting higher content of B atoms (∼35 at%) into as-synthesized fcc Pd nanocubes in BH3–THF solution below 80 °C for 6 days (Fig. 5A).68 The novel hcp structure of Pd2B was confirmed by the synchrotron powder XRD and aberration-corrected HAADF-STEM, where the Pd atoms showed characteristic hcp packing sequence of ABAB along the diagonal direction (Fig. 5B and C). In addition, electron-energy loss spectroscopy (EELS) demonstrated the homogeneous distribution of B atoms in the Pd lattice. Different from the synthesis method reported by Kobayashi et al.,68 Chen et al. used DMAB as the B source to synthesize hcp Pd2B nanosheets via heating DMAB and fcc Pd nanosheets in DMF solution at 120 °C with a shorter time (only 2 h).95 The authors further used DFT calculations to study the mechanism of B insertion into the Pd lattice and proposed that the insertion of B was accompanied by a layer-by-layer transition of the Pd lattice symmetry from fcc to hcp. Moreover, compared with fcc Pd, fcc PdBx and fcc/hcp PdBx, Pd2B with a pure hcp structure showed the best HER performance, due to the decreased formation energy of H2 caused by the formation of the hcp phase. In the above two cases, B atoms are randomly distributed in the Pd lattice. Lv et al. reported hcp Pd2B mesoporous nanocrystals with ordered sequences of Pd/B atoms (hcp-mesoPd2B-o) using DMAB as the B source.96 By gradually increasing the reaction temperature, lattice evolution from fcc mesoPd to fcc-mesoPd5B (100 °C), hcp-mesoPd2B-r (140 °C) with randomly distributed B, and finally hcp-mesoPd2B-o (160 °C) with atomically ordered B was achieved. The hcp-mesoPd2B-o showed faster full conversion (∼80s) to the reduction of p-nitrophenol than fcc-mesoPd5B (∼105s) and Pd black (∼235s).
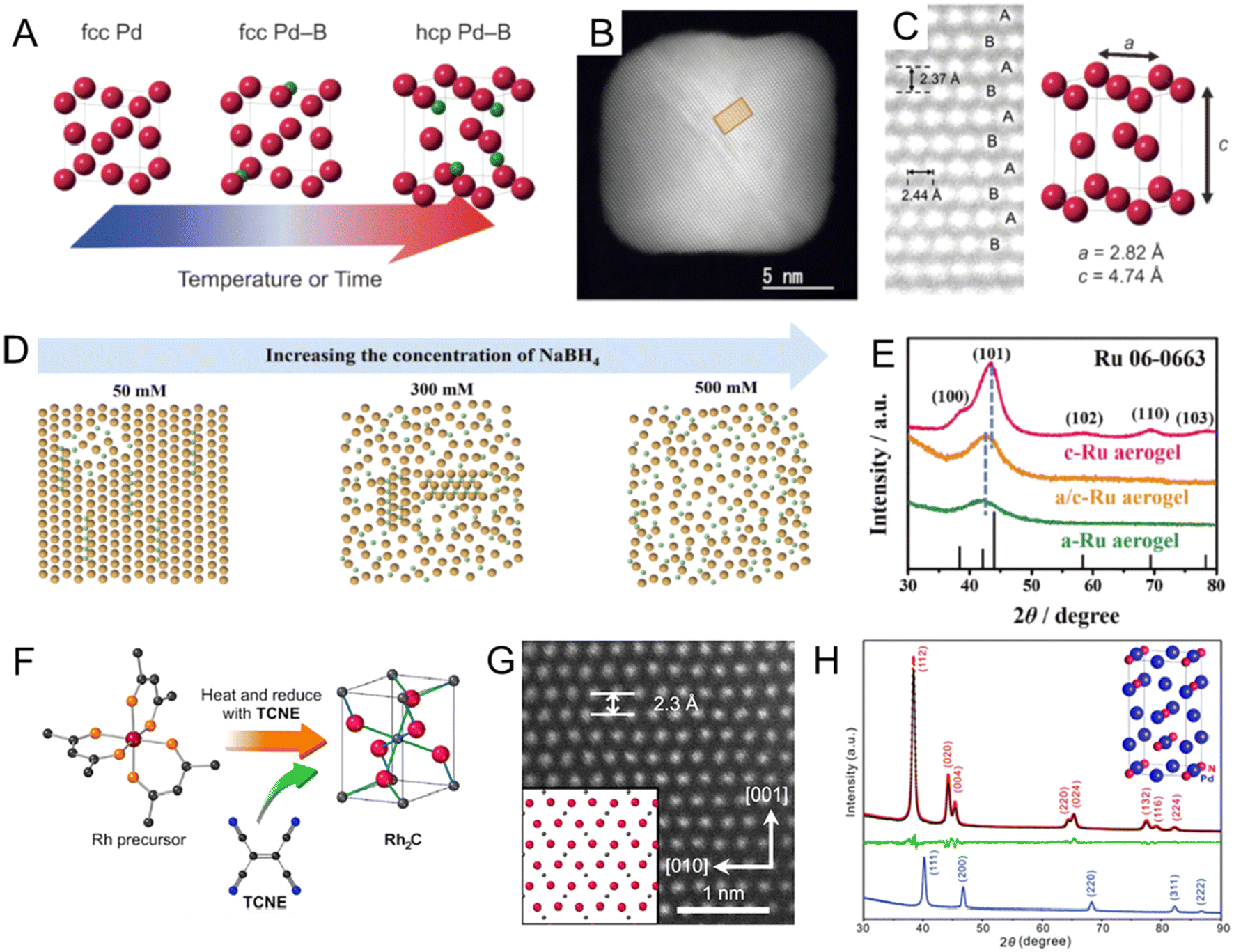 | ||
| Fig. 5 (A) Schematic illustration of structure rearrangement of Pd NCs induced by heavy B doping. (B) Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of hcp Pd2B nanocrystal and (C) magnified image of the rectangular region in (B). The bright dots correspond to Pd atoms. Reproduced with permission from ref 68. Copyright 2017, Wiley-VCH. (D) Schematic diagram of the atoms of the RuB aerogel when the concentration of the B source, was increased from 50 to 500 mM. (E) XRD patterns for a/c-RuB, a-RuB and c-RuB aerogels. Reproduced with permission from ref 97. Copyright 2023, Wiley-VCH. (F) Illustration of the synthesis of Rh2C nanocrystal. TCNE: tetracyanoethylene. (G) Aberration-corrected HAADF-STEM image of the Rh2C nanocrystal along the [100] zone axis. Reproduced with permission from ref 99. Copyright 2020, American Chemical Society. (H) XRD patterns of Pd nanocubes (blue line), Pd2N nanocrystals (black line), and Rietveld refinement of Pd2N nanocrystals. The red and green lines showed the calculated curve and the difference plot, respectively. Reproduced with permission from ref 106. Copyright 2021, Royal Society of Chemistry. | ||
Doping with B could also lead to the lattice distortion of the noble metal to eventually form amorphous structures.71,97 For example, amorphous RhB and amorphous/fcc heterophase RhB metallene were synthesized by mixing the crystalline fcc Rh metallene with NaBH4 dissolved in DMF solution in an ice bath.71 The crystallinity of RhB metallene could be tuned by the length of the boronization time, resulting in the formation of amorphous/fcc heterophase RhB (8 h) and amorphous RhB (16 h). No lattice fringe, diffusion ring, or diffraction peak could be observed in HRTEM, selected area diffraction (SAED) or XRD results, respectively, confirming the absence of crystalline structures in RhB. Due to the abundant phase boundaries in amorphous/fcc heterophase RhB metallene, it showed improved activity toward both the HER and H2O2 electrooxidation reaction as compared to crystalline fcc Rh metallene and amorphous RhB metallene. Recently, the same research group developed a general method to synthesize a series of amorphous/crystalline heterophase metal boride aerogels (RuB, PtB, PdB, and RhB) by reducing metal precursors with NaBH4.98 In particular, the crystallinity of the RuB aerogel could be controlled by the concentration of NaBH4 (Fig. 5D). When the concentration of NaBH4 was as high as 500 mM, an amorphous RuB aerogel could be obtained (Fig. 5E). The HER activity of the amorphous/crystalline heterophase RuB aerogel under alkaline conditions was superior to Pt/C, amorphous RuB, and crystalline RuB, owing to the most abundant catalytic sites in the amorphous/crystalline heterophase structure.
Noble metal carbides. C could form various carbides with non-noble metals and change their lattice symmetry, such as tungsten carbide (WC),75 iron carbide (Fe3C),76 and cobalt carbide (Co2C).77,98 However, noble metal carbides have not been observed in bulk size.99 As for noble metal carbides, although PdCx24 and AuCx80 nanocrystals have been synthesized as introduced in section 2.1.1, the lattice symmetry of Pd and Au in the noble metal carbides still maintained the fcc phase, probably due to the low C content. Recent studies showed that it is possible to change the lattice symmetry of noble metals in noble metal carbides when the C content increases, which showed enhanced catalytic performance than their parent metal counterparts.99,100 For example, Wakisaka et al. reported a novel rhodium carbide (Rh2C) nanocrystal with an orthorhombic phase by a liquid-phase synthesis (Fig. 5F).99 Naturally, the lattice symmetry of Rh is highly symmetric fcc. Interestingly, due to the existence of interstitial C in the Rh lattice, the Rh atoms in Rh2C had a hcp-like packing sequence and showed ABAB packing sequence along the [001] zone axis (Fig. 5G). The authors proposed that the organic oxidant, tetracyanoethylene, served as a C source and gradually decomposed to release C atoms during heating. fcc Rh nanocrystals were obtained without adding tetracyanoethylene. Synchrotron PXRD and NPD were performed to confirm the position of C atoms and the ratio of Rh to C. Both patterns fitted well with the hypothetical patterns of Rh2C with the orthorhombic phase. Moreover, the orthorhombic phase Rh2C nanocatalysts demonstrated higher HER performance than their fcc Rh counterpart in alkaline solution due to the unique packing sequence of Rh and the alloying effect of C. In another recent work, Cao et al. developed another strategy to synthesize orthorhombic phase RhCx nanomaterials via reducing the Rh precursor in a mixture of formaldehyde and oleylamine under 200 °C.100 The Rh lattice symmetry was transformed from fcc to an orthorhombic structure when half of the lattice octahedral sites were occupied by C atoms, which was confirmed by NPD. The existence of C atoms was further confirmed by low-frequency Raman spectral characterization. Interestingly, the same approach was further extended to insert C atoms into commercial fcc Rh black, leading to partial transformation into the orthorhombic RhCx phase. Compared with its fcc Rh counterpart, the RhCx nanocatalysts exhibited enhanced selectivity and activity toward the electrocatalytic EOR. The authors supposed that electronic and geometric effects induced by C insertion could decrease the energy barrier of the dehydrogenation step in the EOR and suppress side reactions, resulting in enhancement in both activity and selectivity.
Noble metal nitrides. Previous studies have shown that lattice symmetry engineering of metal nitrides could be achieved by the CVD method, in which the metal nitrides were prepared via the reaction between metal/metal precursors and ammonia gas (NH3),101–103 as well as using the magnetron sputtering method.104 In comparison, wet chemical synthesis was less reported. In 2011, Wu et al. successfully prepared the first transition metal nitride, i.e., cubic Cu3N nanocrystals by a wet chemical technique.105 However, there are limited reports on the synthesis of noble metal nitrides with novel crystal structures by the wet chemical method due to the inert nature of both the noble metal and the N–N triple bond. Recently, Guo et al. reported a facile hydrothermal method to synthesize a series of PdNx nanocubes with controllable stoichiometries (PdN0.08, PdN0.18, PdN0.26, PdN0.35, and Pd2N) by heating urea, PVP and pre-prepared Pd nanocubes in deionized water at 180 °C for 1 h.106 During heating, the decomposition of urea could release N atoms, which further diffused into the Pd lattice. Interestingly, on inserting more N atoms into the Pd lattice and reaching an N/Pd ratio of 0.5, the fcc Pd nanocubes transformed into tetragonal phase Pd2N nanocubes (Fig. 5H). In XPS, an extra peak of N 1s can be observed at 397.3 eV in the spectra for Pd2N nanocubes, indicating the presence of the Pd–N interaction. The enlarged lattice fringes of (100) (2.04 Å) for Pd2N measured b HRTEM also confirmed that N atoms diffused into the Pd lattice. Compared with undoped Pd/C and fcc PdNx with other stoichiometries, the tetragonal Pd2N exhibited much higher activity for the ORR under alkaline conditions. The authors believed that catalytic activity for the ORR in an alkaline medium were enhanced with an increasing N/Pd ratio in PdNx nanocatalysts. The introduction of N atoms into Pd lattices could downshift the d-band center of Pd and weaken the binding interaction between the Pd surface and the adsorbates. The excellent catalytic performance could also be attributed to the unique polycrystalline structure of tetragonal Pd2N. Cao et al. reported an AgN3-catalyzed hydroazidation reaction of terminal alkynes in which AgN3 was converted from Ag2CO3 during the reaction and simultaneously proved to be a robust catalyst.107 The authors supposed that Ag2CO3 was quickly and completely converted to AgN3 during the hydroazidation reaction of p-tolylacetylene. The fast conversion of Ag2CO3 to AgN3 took place within 5 min, as reflected by the pure orthorhombic AgN3 diffraction peaks in the XRD pattern. This work provided a novel and facile preparation strategy for the design and preparation of noble metal-based catalysts for organic reactions.
Noble metal phosphides. When alloying nonmetal elements with larger atomic radii, such as P, it is easier to change the original crystal symmetry of the host noble metal by forming metal phosphide compounds.16 Different lattice symmetries and varying P contents can modify physicochemical properties of noble metal phosphides.13 To date, many types of noble metal phosphides, including RhPx,108–111 RuPx,21,112,113 PdPx,114,115–117 and Au2P3,118 have been synthesized by the wet chemical method. For example, Wang et al. fabricated wrinkled Rh2P nanosheets by heating as-prepared fcc Rh nanosheets in tri-n-octylphosphine solution.108 The phosphating reaction occurred through the thermal decomposition of tri-n-octylphosphine. The wrinkled morphology of the Rh2P nanosheets and the elemental mapping of Rh and P was confirmed by TEM and EDX (Fig. 6A). The ratio of Rh to P was estimated to be ∼2
:1 (Fig. 6B). The XRD pattern of the sample matched well with that of cubic phase Rh2P (Fig. 6B). According to the XPS spectrum of Rh 2d, Rh in Rh2P showed a slightly positive valence state. The wrinkled Rh2P nanosheets exhibited better HER activity with an overpotential of 18.3 mV at a current density of 10 mA cm−2, much lower than commercial Pt/C (58.4 mV) and Rh nanosheets/C (68.9 mV) (Fig. 6C). In addition to the HER, noble metal phosphides have also been highly catalytically active toward the MOR. Li et al. developed a strategy to synthesize monodisperse Pt5P2 nanoparticles (Fig. 6D), which could serve as a MOR electrochemical catalyst.119 In the synthesis process of Pt5P2 nanoparticles, a mixed aqueous dispersion of carbon black, sodium hypophosphite, and dibasic sodium phosphate (P sources) was first prepared, followed by adding chloroplatinic acid hexahydrate (H2PtCl6·6H2O) as the Pt sources. The final product was obtained after heat treatment at 125 °C under reflux for 4 h. The diffraction peaks of pre-obtained Pt5P2 nanoparticles were indexed into the monoclinic phase of the Pt5P2 nanocrystal, and the fcc Pt characteristic peaks could not be observed in the XRD pattern, confirming the full phosphorization of Pt (Fig. 6E). In the MOR test, the mass activity of Pt5P2 (3.60 A mgPt−1) was found to be 11.0 and 9.1 times better than those of commercial Pt/C (0.33 A mgPt−1) and PtRu/C (0.39 A mgPt−1) (Fig. 6F), which indicated that the MOR kinetics may be accelerated on the Pt5P2 surface. Besides, the introduction of P atoms can also lead to lattice distortion of the original metal lattice to form amorphous materials, which are promising catalyst materials due to their abundant random directional bonds, surface unsaturated atoms, and abundant defects.120–122 One possible mechanism for the formation of amorphous noble metal phosphides is the simultaneous diffusion of the P element into the interstitial and substitutional positions of host metal lattice, destroying the previous long-range order of the crystal structure.14,123,124 Zhang et al. successfully prepared amorphous PdP (a-Pd) nanoparticles with enhanced EOR performance in comparison with crystalline Pd nanoparticles (c-Pd).125 The well-dispersed a-Pd nanoparticles (Fig. 6G) were synthesized by the reduction of the Pd salt in the mixture solution of TOP and oleylamine. The XRD pattern of a-PdP confirmed the amorphous structure, while clear diffraction peaks were observed in the XRD pattern of c-Pd nanoparticles (Fig. 6H). As for the EOR performance, a-PdP showed high reactivity of 4851 mA mg−1 at 0.75 V, which was higher than those of c-Pd and commercial Pt/C catalysts in Fig. 6I. The discovery of this process has important guiding significance for precious metal phosphorization, promoting nano-structural diversification.
 | ||
| Fig. 6 (A) HAADF-STEM image of wrinkled Rh2P nanosheets and energy dispersive X-ray (EDX) elemental mappings of Rh and P. (B) XRD pattern and EDX spectrum of Rh2P nanosheets. (C) The comparison of active current density of wrinkled Rh2P, Rh nanosheets and Pt/C for the hydrogen evolution reaction (HER) in 0.1M KOH solution. Reproduced with permission from ref 108. Copyright 2018, Wiley-VCH. (D) TEM image and (E) XRD pattern of the Pt5P2/C catalyst. (F) The comparison of mass activity of Pt5P2/C, commercial Pt/C, and PtRu/C catalysts for the methanol oxidation reaction (MOR) in 0.5 M H2SO4 solution containing 1.0 M CH3OH. Reproduced with permission from ref 119. Copyright 2020, Royal Society of Chemistry. (G) TEM image of amorphous PdP alloy nanoparticles (a-PdP0.1). Inset: the corresponding selected area electron diffraction (SAED) patterns showing no obvious diffraction spots. (H) XRD patterns of a-PdP0.1 and crystalline Pd nanoparticles (c-Pd). (I) Comparison of electrochemical performances of a-PdP0.1 and c-Pd in the ethanol oxidation reaction. Reproduced with permission from ref 125. Copyright 2022, Royal Society of Chemistry. | ||
Noble metal sulfides. Similar to P, incorporating S into noble metals can also significantly alter the crystal lattice of the host metal via the formation of noble metal sulfides. Theoretical studies have shown that electron transfer between d orbitals of precious metals and p orbitals of S can greatly adjust the electrocatalytic performance of noble metal sulfide catalysts.12,14,32,87 Motivated by such an advantage, numerous endeavors have been dedicated to the controlled synthesis of noble metal sulfide nanomaterials.126–133 Among the synthetic methods of noble metal sulfides, one-step synthesis is the most direct and efficient way. For instance, Du et al. synthesized monodisperse palladium sulfide (Pd4S) nanoparticles through a simple one-pot method.130 To be specific, the as-synthesized Pd4S was obtained by heating Pd(acac)2 and S powder in oleylamine at 320 °C via a one-pot sulfurization process. Monodisperse Pd4S nanoparticles had a uniform size distribution of about 10 nm (Fig. 7A), and the lattice spacing of Pd4S nanoparticles are 0.21 and 0.36 nm (Fig. 7B), which matched well with the (211) and (110) planes of the tetragonal Pd4S nanocrystal. In the alkaline solution, the as-obtained Pd4S/C catalyst exhibits good ORR performance, better than commercial Pt/C illustrated in Fig. 7C. In addition to the one-pot method to obtain noble metal sulfides, researchers also developed multi-step wet chemical methods, including template-assisted134,135 and cation displacement methods.135–137 Here, we only discuss a few representative reports. Park et al. have illustrated the synthesis of core@crown Cu1.81S@IrS2 hexagonal nanoplates via a template-assisted method.137 To prepare this core@crown structure, pre-obtained Cu1.8S nanoplates were dispersed in oleylamine, followed by adding iridium acetylacetonate and heat treatment at 240 °C for 0.5 h. As triclinic Cu1.81S and orthorhombic IrS2 have similar sulfur sublattices, epitaxial interfaces may be formed with IrS2 crowns at the Cu1.81S surfaces. In one systematic experimental study of the cation exchange method, Feng et al. developed a general synthesis strategy to prepare a library of noble metal chalcogenides using Cu chalcogenides as a sacrificial template for subsequent cation displacement.135 For example, two-dimensional Pd16S7 nanosheets were synthesized by cation exchange from CuxSy with the presence of ethylene glycol. The complete transformation of CuxSy into Pd16S7 was confirmed by XRD patterns and element mappings (Fig. 7D). Since both template synthesis and cation exchange can effectively control the shape and crystal facet, more efforts have been made to combine these methods for more diversified noble metal sulfide nanostructures. For example, Yoon et al. reported hollow rhodium sulphide (Rh2S3) nano-prisms as a HER catalyst with excellent catalytic activity and long-term durability.136 As shown in Fig. 7E, the hollow Rh2S3 nanoplate was obtained by etching Cu1.94S@Rh2S3 core–shell structures, which were first prepared by one-pot heating of copper(I) thiocyanate and Rh(acac)3 mixture. The whole process was believed to combine cation exchange, galvanic replacement, and the Kirkendall effect. The hexagonal faces of the orthorhombic Rh2S3 nano-prisms were assigned to {110} facets (Fig. 7F and G), and it was found that the six sides of hexagonal nanoprisms are {211} facets through HRTEM and FFT images. The Rh2S3 catalyst exhibited excellent stability after 10
000 cycles under strongly acidic conditions. This combination of multiple synthetic strategies greatly expanded the variety of complex sulfide nanostructures that could be designed and fabricated, providing novel electrochemical catalysts for advanced catalysis such as carbon dioxide electroreduction (CO2RR).
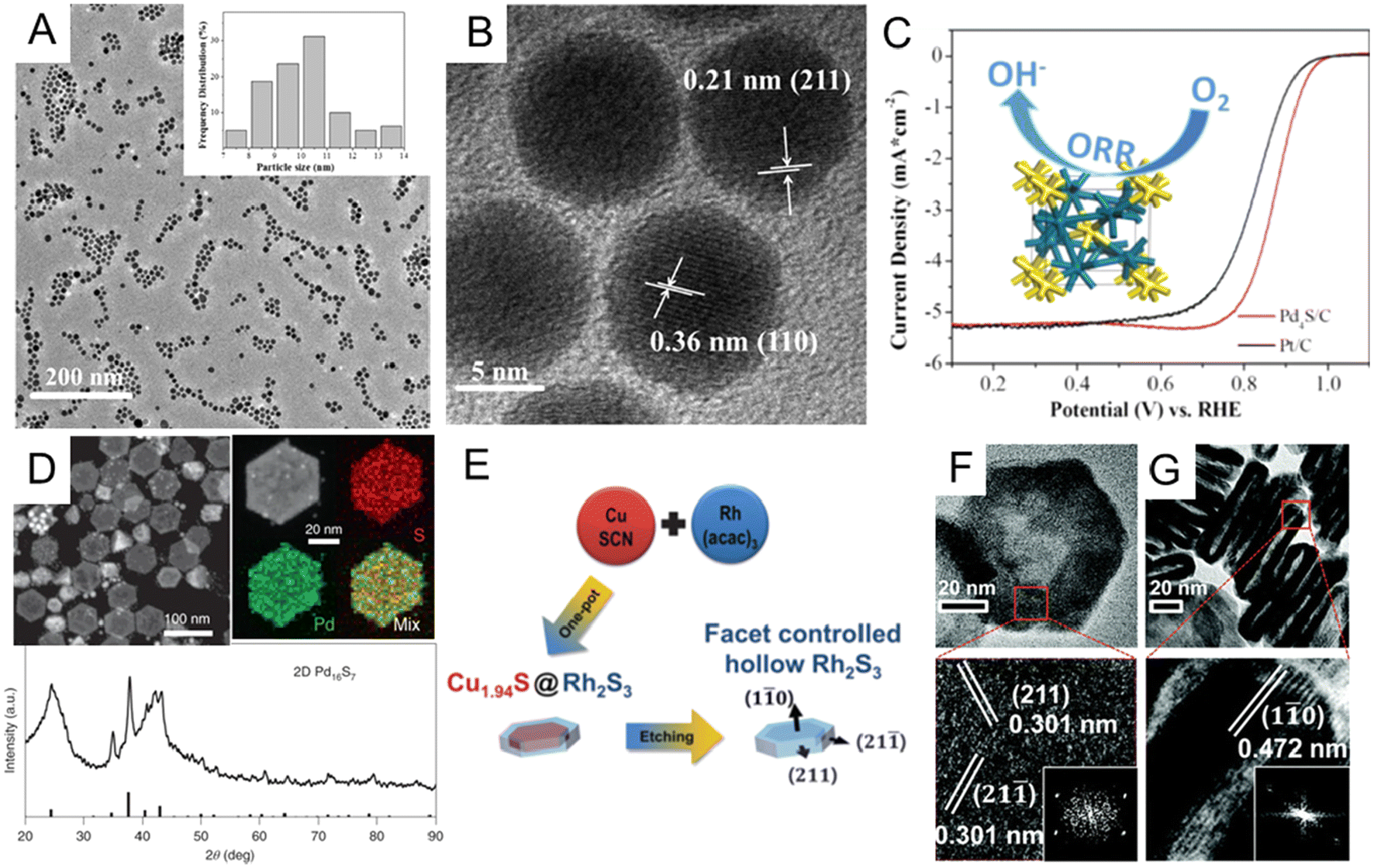 | ||
| Fig. 7 (A) Low-magnification TEM and (B) HRTEM images of Pd4S nanoparticles. (C) Comparison of oxygen reduction reaction (ORR) performances of the Pd4S/C and commercial Pt/C catalysts. Reproduced with permission from ref 130. Copyright 2018, American Chemical Society. (D) HAADF-STEM image, EDX element mappings, and X-ray diffraction pattern of Pd16S7 nanosheets prepared by cation exchange from the CuxSy template. Reproduced with permission from ref 135. Copyright 2022, Nature Publishing Groups. (E) Schematic illustration of the formation of hollow Rh2S3 catalysts. TEM and HRTEM images of the (F) top-view and (G) side-view of Rh2S3 hollow structures. Inset: corresponding FFT patterns. Reproduced with permission from ref 136. Copyright 2016, Royal Society of Chemistry. | ||
In addition, S doping into the noble metal could also cause heavy lattice distortion that may inevitably induce the reconstruction of the original electron configuration and generate a disordered lattice. For example, Xu et al. devised a way to prepare amorphous RuS2 (a-RuS2) nanoparticles dispersed on carbon spheres via a one-pot solvothermal method.138 To be specific, the a-RuS2 nanoparticles could be formed by the reduction of ruthenium(III) chloride trihydrate and L-cysteine (as the S source) in the aqueous dispersion of hollow carbon spheres at 160 °C for 9 h. The prepared a-RuS2 nanoparticles with a diameter of 2.6 nm were distributed uniformly on the carbon framework, which were beneficial for exposing more active sites and improving atomic utilization, enhancing the electrocatalytic HER. In addition to the direct one-pot method, amorphous noble metal sulfides can also be obtained by sulfurization progress in multiple steps. For instance, Xie et al. prepared amorphous S-doped Pd metallenes via a two-step solvothermal method.139 The Pd metallenes were first prepared and re-dispersed in oleylamine containing S powder under heat treatment at 210 °C for 1 h. After sulfurizing Pd metallene, the amorphous structure could be obtained, as no lattice fringes or diffraction peaks were observed in HRTEM images and XRD results, respectively. In the MOR, the amorphous S–Pd metallene catalyst showed better reaction activity compared with commercial Pd/C and Pt/C catalysts.
2.2. Indirect lattice engineering of noble metal–light nonmetal nanocompounds
Besides directly inserting foreign elements, we can also utilize a coherent metal–metal interface and engineer the lattice strain indirectly. For example, building core–shell structures has been proven as an effective strategy to tune the lattice strain of shell metals, which could be induced by the lattice mismatch between the core and the shell.20,140,141 Specifically, the exposed facets, surface morphology, and composition of the core metals can be utilized to modify the strain level in shell metals.142–144 Recently, by inserting or extracting light nonmetal elements into or from the core metal in a core–shell nanostructure, fine lattice strain engineering of different noble metals has been proven to be possible.145 For instance, a seed-mediated growth method was applied to synthesize PdHx@Ru metallenes via reducing RuCl3·xH2O in the formaldehyde solution with Pd metallenes as the seed.146 In the reaction process, while the H atoms were generated by the decomposition of formaldehyde and inserted into the lattice of Pd metallenes to form PdHx, Ru(III) was reduced to Ru(0) on the surface of PdHx to form atomically thin Ru layers (Fig. 8A). The average Pd–Pd/Ru bond length was estimated by FFT-extended EXAFS spectra of the Pd K-edge (Fig. 8B) to be 2.55 Å, which was 0.1 Å longer than that in Pd foil, indicating that lattice expansion in the Pd core came from inserting H atoms. The expanded lattice of PdHx relative to Pd resulted in a 4.5% tensile strained Ru skin. The formation of tensile strained Ru skin in PdHx@Ru was proven by the Ru–Ru/Pd bond length, which was 0.11 Å longer than that of Ru foil (Fig. 8C). The authors claimed that the tensile strained Ru skin enabled higher catalytic activity and better stability in the HER, which was evidenced by spectroscopic analyses and DFT calculations. In addition to the Ru skin, Pt skin in cubic and octahedral PdH0.43@Pt core–shell nanoparticles with exposed (200)f and (111)f facets, respectively, were synthesized via Pd-seeded epitaxial growth followed by the introduction of H into Pd to form PdH0.43.26 The enlarged lattice fringes of the Pt (200)f plane (2.01 Å, red rectangle in Fig. 8D and F) in cubic-PdH0.43@Pt NPs (c-PdH0.43@Pt NPs) measured from aberration-corrected STEM were larger than that of the (200)f plane (1.93 Å) in c-Pd@Pt NPs, and the octahedral-PdH0.43@Pt NPs (o-PdH0.43@Pt) had a larger Pt (111)f plane (2.45 Å, red rectangle in Fig. 8E and G) than that of the Pt (111)f plane (2.37 Å) in o-Pd@Pt NPs. These measurements proved that the Pt shells were stretched, likely caused by the lattice expansion of the Pd core during hydrogenation. In the XPS spectra of Pt 4f, the binding energies of c-PdH0.43@Pt NPs reduced by about 0.22 eV compared to that of c-Pd@Pt NPs (Fig. 8H) and the o-PdH0.43@Pt NPs exhibited lower binding energies (around 0.23 eV) compared to that of o-Pd@Pt NPs (Fig. 8I) both resulting from the successful insertion of H atoms into Pd shells. Compared with c-Pd@Pt and o-Pd@Pt, both c-PdH0.43@Pt and o-PdH0.43@Pt NPs exhibited higher catalytic activities in the MOR. Recently, few-layer Ir skins with different tensile strains were prepared using lattice engineered Pd-based octahedral nanocrystals with different H contents, namely o-Pd@Ir-1.2%, o-Pd@Ir-1.7% and o-Pd@Ir-2.1%.147 The XRD pattern showed that the peaks of o-Pd@Ir-1.2% matched well with those of the standard Pd pattern, while all the diffraction peaks of o-Pd@Ir-1.7% and o-Pd@Ir-2.1% left shifted to lower angles, confirming the presence of lattice expansion in the latter two samples. In the XPS spectrum of Ir 4f, the peak shifted to higher binding energy region with increasing tensile strain in Ir shells from 1.2% to 2.1%, indicating the gradually decreased electron density at the Ir shell with expanding lattices. The authors reported a volcano-shaped correlation between the strain of the Ir shell and its catalytic performances in the HER. The o-Pd/lr-1.7% NPs exhibited the highest mass activity and the lowest overpotential relative to other two catalysts. Based on theoretical analysis, the medium tensile strain on the Ir (111)f surface was believed to lower the adsorption strength of H intermediates. Interestingly, switching the sequence of epitaxial growth of the shell metal and doping of interstitial H in the core metal may yield different core–shell hydrides. Guo et al. reported the synthesis of Pd@Rh–H nanocubes by first epitaxial growth of Rh shells on Pd nanocubes and then H insertion, and in another experiment, PdH0.43@Rh nanocubes by first inserting H on Pd nanocubes followed by epitaxial growth of Rh.148 As shown in the scheme (Fig. 8J), to prepare Pd@Rh–H core–shell nanocubes, Pd seeds were first epitaxially coated with Rh to form Pd@Rh core–shell nanocubes, which were then treated by a H doping process, forming a Rh-doped PdH core and Pd-doped Rh shell along with lattice expansion. In another route to prepare PdH0.43@Rh core–shell nanocubes, epitaxial growth of Rh was achieved after transforming the Pd core into PdH0.43 nanocompounds (Fig. 8J). It is worth mentioning that the XPS peaks of Pd 3d in Pd@Rh–H showed lower binding energy than PdH0.43@Rh, suggesting that the intermixing of the less electronegative Rh with PdH may promote the electron transfer from Rh to Pd. The more effective charge transfer and formation of mixed alloy hydride on the surface of Pd@Rh–H made it a more active and stable MOR catalyst as compared to PdH0.43@Rh (Table 1).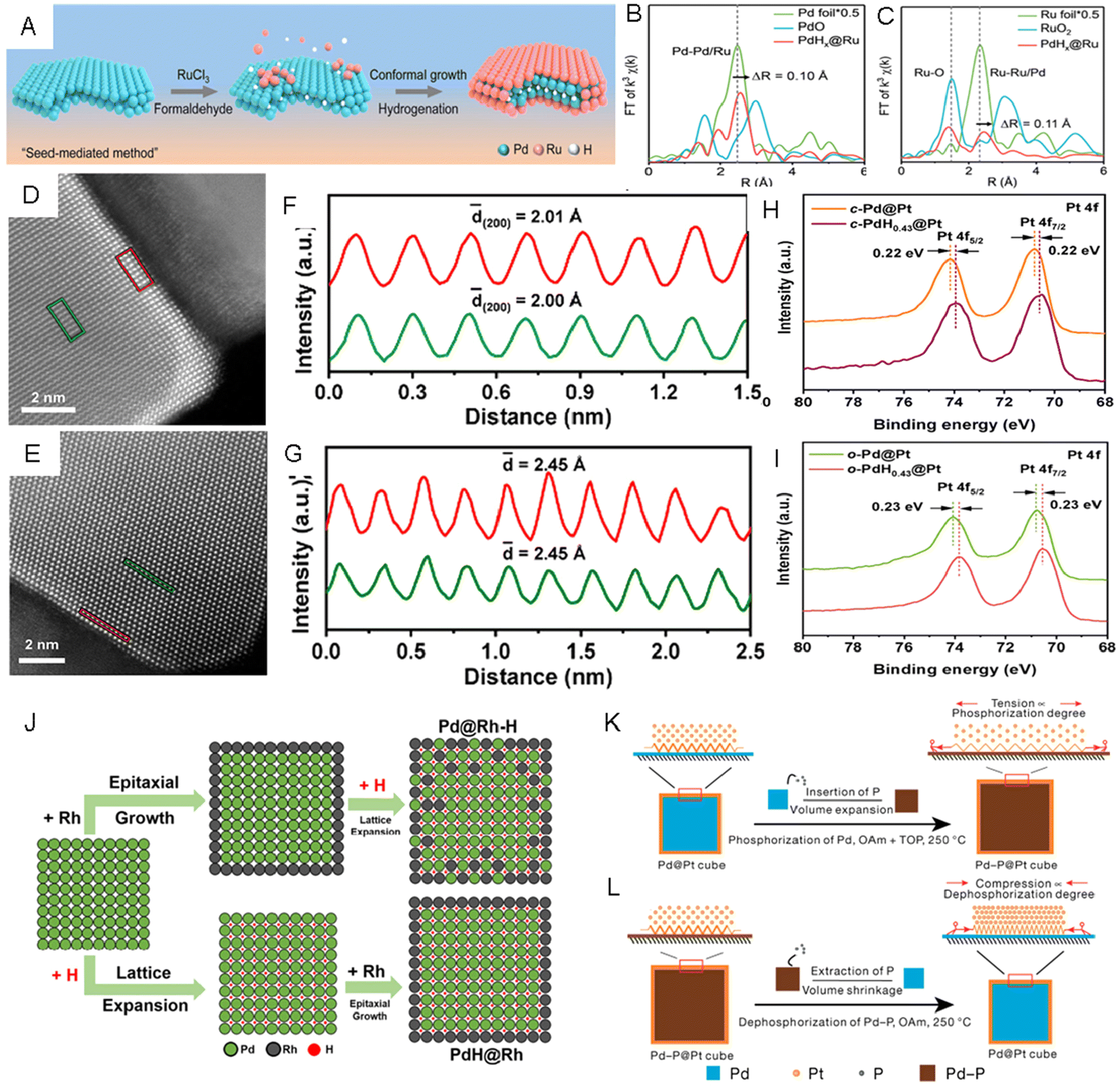 | ||
| Fig. 8 (A) Schematic illustration of the preparation of PdHx@Ru metallenes. (B and C) FFT-extended X-ray absorption fine structure (EXAFS) spectra of (B) Pd K-edge and (C) Ru K-edge in Ru foil, RuO2, and PdHx@Ru metallenes. Reproduced with permission from ref 146. Copyright 2023, American Chemical Society. (D) Aberration-corrected HAADF-STEM images taken from the (D) c-PdH0.43@Pt nanocubes and (E) o-PdH0.43@Pt nanooctahedra. (F and G) Integrated pixel intensities of the PdH0.43 core and Pt shell taken from the green and red rectangular regions in D and G, respectively. (H and I) X-ray photoelectron spectroscopy (XPS) spectra of Pt 4f in (H) c-Pd@Pt and c-PdH0.43@Pt nanocubes, as well as (I) in o-Pd@Pt and o-PdH0.43@Pt nanooctahedra. Reproduced with permission from ref 26. Copyright 2021, American Chemical Society. (J) Schematic illustration of the formation of Pd@Rh–H and PdH@Rh nanocubes with two different H insertion pathways. Reproduced with permission from ref 148. Copyright 2021, springer. (K and L) Schematic illustrations of (K) the tensile strain formation on the Pt shell by inserting P in Pd nanocubes and (L) compression strain formation on the Pt shell by extracting P from Pd-P nanocubes. Reproduced with permission from ref 25. Copyright 2021, Nature Publishing Groups. | ||
| Name of noble metal nanocompound | Wet chemical method | Types of lattice strain | Catalytic application | Ref |
|---|---|---|---|---|
| MOR: methanol oxidation reaction; FAOR: formic acid oxidation reaction; HER: hydrogen evolution reaction; ORR: oxygen reduction reaction; and EOR: ethanol oxidation reaction. | ||||
| PdHx nanocrystals | Solvothermal | Tensile strain | — | 51 |
| PdH0.43 nanopolycrystals | Solvothermal | Tensile strain | MOR | 23 |
| PdH0.43 nanotetrahedra; | Solvothermal | Tensile strain | MOR | 23 |
| PdH0.43 nanocubes | Hydrothermal + solvothermal | Tensile strain | MOR | 23 |
| PdHx nanodendrites | Solvothermal | Tensile strain | FAOR; HER | 52 |
| PdHx/C (x = 0.10, 0.29, 0.23, 0.43) | Solvothermal | Tensile strain | FAOR | 53 |
| PdHx nanooctahedra; | Hydrothermal + solvothermal | Tensile strain | FAOR | 55 |
| PdHx nanoicosahedra | Solvothermal | Tensile strain | FAOR | 55 |
| RhH nanosheet | Solvothermal | Tensile strain | HER | 44 |
| PdBx/C | Hydrothermal | Tensile strain | FAOR | 58 |
| PdBx/C | Hydrothermal | Tensile strain | Formic acid deposition reaction | 59 |
| PtBx/C | Solvothermal | Tensile strain | ORR | 60 |
| PdBx/C | Solvothermal | Tensile strain | Hydrogenation of 3-hexyn-1-ol | 67 |
| PdBx nanoparticles | Solvothermal | Tensile strain | ORR | 69 |
| OsBx aerogels | Hydrothermal | Tensile strain | HER | 70 |
| RhBx aerogels | Hydrothermal | Tensile strain | HER | 73 |
| IrBx nanosheets supported on Ni foam | Hydrothermal + solvothermal | Tensile strain | Hydrazine oxidation reaction; HER; | 74 |
| PdC0.18 nanocubes | Hydrothermal + solvothermal | Tensile strain | Alkyne semihydrogenation reaction | 24 |
| AuCx nanoparticles | Hydrothermal | Tensile strain | Hydrogenation of 3−nitrostyrene | 80 |
| Ir@PAH metallene | Hydrothermal | Tensile strain | HER | 86 |
| PdPx nanosheets | Hydrothermal | Tensile strain | EOR | 88 |
| Pt2P nanoparticles | Hydrothermal + solvothermal | Tensile strain | ORR | 89 |
| RhPx nanoparticles | Hydrothermal + solvothermal | Compressive strain | HER | 90 |
| PdHx@Ru core–shell metallenes | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | HER | 146 |
| PdH0.43@Pt core–shell nanocubes | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | MOR | 26 |
| PdH0.43@Pt core–shell nanooctahedra | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | MOR | 26 |
| PdHx@Ir core–shell nanoparticles | Solvothermal + epitaxial growth | Tensile strain | HER | 147 |
| Pd@Rh–H core–shell nanocubes | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | MOR | 148 |
| PdH0.43@Rh core–shell nanocubes | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | MOR | 148 |
| PdPx@Pt core–shell nanocubes | Hydrothermal + solvothermal + epitaxial growth | Tensile strain | HER; MOR | 25 |
| PdPx@Pt-derived Pd@Pt core–shell nanocubes | Hydrothermal + solvothermal + non-epitaxial growth | Compressive strain | HER; MOR | 25 |
Different from the above work, He et al. achieved both tensile strain and compression strain on Pt shells by inserting P into Pd@Pt core–shell nanocubes and extracting P from PdPx@Pt core–shell nanocubes, repectively.25 The authors claimed that P could diffuse into the Pd lattice of Pd@Pt core–shell nanocubes to form PdPx@Pt, while keeping Pt undoped. Therefore, the tensile strain in the Pt shells could be created due to the increased volume of the PdPx cores (Fig. 8K). With the increase of the phosphorization time, both the volume of the PdPx core and the tensile strain in Pt skin gradually increased with the increased P contents. On the other hand, compressive strain control was achieved by dephosphorizing PdPx@Pt nanocubes with different P contents via heat treatment. The Pd cores derived from PdPx would shrink after the dephosphorization process, causing compression strain in Pt (100)f shells (Fig. 8L). The lattice tensions and compressions in Pt (100)f were tailored from −5.1% to +5.9%. The authors supposed that both tensile and compressive strain could enhance the catalytic activity and provide excellent durability toward the MOR, but excessive strain could diminish the catalytic performance (Table 2).149
| Name of noble metal nanocompound | Wet chemical method | Type of lattice symmetry | Catalytic application | Ref |
|---|---|---|---|---|
| hcp: hexagonal-close-packed; fcc: face-centered cubic; MOR: methanol oxidation reaction; FAOR: formic acid oxidation reaction; HER: hydrogen evolution reaction; ORR: oxygen reduction reaction; and EOR: ethanol oxidation reaction. | ||||
| Pd2B nanocubes | Hydrothermal + solvothermal | hcp | — | 68 |
| Pd2B nanosheets | Solvothermal | hcp | HER | 95 |
| Pd2B mesoporous nanocrystals | Hydrothermal + solvothermal | hcp | P-Nitrophenol reduction reaction | 96 |
| RhB metallenes | Hydrothermal + solvothermal | Amorphous + amorphous/fcc heterophase | HER; hydrogen peroxide electrooxidation reaction | 71 |
| RuB aerogels | Hydrothermal | Amorphous/hcp heterophase | HER | 97 |
| Rh2C nanocrystals | Solvothermal | Orthorhombic | HER | 99 |
| RhCx | Solvothermal | Orthorhombic | EOR | 100 |
| Pd2N nanocubes | Hydrothermal | Tetragonal | ORR | 106 |
| AgN3 | Solvothermal | Orthorhombic | Hydroazidation reaction of p-tolylacetylene | 107 |
| Rh2P nanosheets | Solvothermal | Cubic | HER | 108 |
| Pt5P2 nanoparticles | Hydrothermal | Monoclinic | MOR | 119 |
| PdP nanoparticles | Solvothermal | Amorphous | EOR | 125 |
| Pd4S nanoparticles | Solvothermal | Tetragonal | ORR | 130 |
| IrS2 shells | Solvothermal + epitaxial growth | Orthorhombic | N/A | 137 |
| Rh2S3 nanoprisms | Solvothermal + acidic etching | Orthorhombic | HER | 136 |
| RuS2 nanoparticles | Solvothermal | Amorphous | HER | 138 |
| PdSx metallenes | Solvothermal | Amorphous | ORR | 139 |
3. Conclusion
In this mini-review, we present the recent development of lattice engineering in noble metal–light nonmetal binary nanocompounds together with some of their derived core–shell nanomaterials. Via wet chemical methods, lattice engineering of noble metal-based nanomaterials has been realized via either directly inserting light nonmetal elements into the metal lattice or indirectly tuning an epitaxial metal–metal compound interface. Their lattice variation in terms of strain and symmetry could be well characterized thanks to various advanced characterization technologies. Importantly, alloying noble metals with Earth-abundant light nonmetal elements has not only decreased the usage of noble metals in catalysts, but also improved their intrinsic catalytic performance by modifying their electronic states. With the great potential, there are still many challenges and opportunities in this direction.Firstly, although the wet chemical method has been successfully exploited to incorporate a light nonmetal with noble metal nanomaterials in some studies, there are still limitations of current approaches. (1) The diversity of wet chemically prepared noble metal–light nonmetal compounds is very limited compared to those prepared by other methods such as the gas–solid method and vapor transport method. There are especially limited methods to prepare C- and N-doped noble metal nanomaterials,150 probably because they are thermodynamically less favorable.151 Therefore, further development of feasible wet-chemical approaches to synthesize and stabilize a wider library of noble metal–light nonmetal compounds is desired. (2) Precisely controlled lattice engineering of the nanostructures, including morphology,152,153 composition,154 and crystal structure, remains a challenge. At most wet chemical synthesis temperatures, the light elements are often randomly distributed and unstable in noble metal lattices, which also highly depends on the type of precursors, solvents, temperature, and other synthetic parameters. Overall, the current understanding of the formation and stabilization mechanism of various noble metal compounds is preliminary and identifying the critical synthesis conditions remains important.
Secondly, although inserting light nonmetal elements into noble metal lattices provides an effective way to transform the lattice symmetry of host metals, it is still a great challenge to prepare noble metal nanomaterials with controlled unconventional phases. Recent studies show that phase engineering of nanomaterials plays an important role in tuning the physicochemical properties of noble metal nanomaterials.18,91,155 Inspired by this concept, indirect approaches by using noble metal nanocompounds with various crystal phases as the phase template to epitaxially grow shell metals with unconventional phases beome a promising solution. For example, orthorhombic phase Rh2C nanocompounds might be applied as the template to epitaxially grow noble metal shells or even non-noble metal shells with the unconventional orthorhombic phase by wet chemical epitaxial growth. In such way, a variety of heterostructured nanomaterials with unconventional phases could be precisely designed to further study their phase-dependent catalytic properties for enhanced performances.
Thirdly, the concept of inserting guest elements with smaller size to tune the lattice structures of host materials can also be extended to engineer the lattice of more kinds of nanomaterials, such as metal–organic frameworks, covalent organic frameworks, and high-entropy alloys. For example, small molecules could be inserted into the pores of metal–organic frameworks to cause lattice distortion and meanwhile modify their functions. In another recent study, N element could be inserted into Pt-based high-entropy alloy nanoparticles to cause tensile strain, resulting in improved ORR activity and durability.156 These examples show great potential for extending the lattice engineering concepts in this review to a broader range of nanomaterials.
Lastly, although superior catalytic performances have been reported in the HER, ORR, and MOR, deeper insights into the relationship between the electrochemical performance and lattice structure of noble metal compounds are still required. It is therefore important to study in-depth factors that affect the performance of the catalytic system by combining advanced characterization strategies with theoretical calculations, aiming for rational design and reliable explanation. Additionally, there are a number of burgeoning electrochemical reactions that may unlock more potential of noble metal compounds for various applications, such as the nitrogen reduction reaction (NRR), nitrate reduction reaction (NO3RR) and CO2RR. For example, Pd-based nanocompounds are intensively studied for fuel cell applications,116 but there are few research reports on their applications in other reactions such as the CO2RR, in which metallic Pd-based nanocatalysts have been proven to be promising.157 Besides electrocatalytic applications, more efforts could be made to explore other important categories of catalytic reactions, such as thermal catalysis and photocatalysis, to expand the potential applications of lattice-engineered noble metal compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.C. thanks the support from Start-up Fund (Project No. 4930977) and the Direct Grant for Research (Project No. 4053444) from the Chinese University of Hong Kong.References
- M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. V. Merinov and Z. Lin, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- Q. Guan, C. Zhu, Y. Lin, E. I. Vovk, X. Zhou, Y. Yang, H. Yu, L. Cao, H. Wang, X. Zhang, X. Liu, M. Zhang, S. Wei, W.-X. Li and J. Lu, Nat. Catal., 2021, 4, 840–849 CrossRef CAS.
- M. Zhao, Z. Chen, Y. Shi, Z. D. Hood, Z. Lyu, M. Xie, M. Chi and Y. Xia, J. Am. Chem. Soc., 2021, 143, 6293–6302 CrossRef CAS PubMed.
- X. Ren, X. Dong, L. Liu, J. Hao, H. Zhu, A. Liu and G. Wu, SusMat, 2023, 3, 442–470 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522–11569 CrossRef CAS PubMed.
- J. Xie, Y. Jiang, S. Li, P. Xu, Q. Zheng, X. Fan, H. Peng and Z. Tang, Acta Phys.-Chim. Sin, 2023, 39, 2306037 Search PubMed.
- S. Guo and E. Wang, Nano Today, 2011, 6, 240–264 CrossRef CAS.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Nano Energy, 2016, 29, 198–219 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- H. Chen, B. Zhang, X. Liang and X. Zou, Chin. J. Catal., 2022, 43, 611–635 CrossRef CAS.
- J. Liu, C. Lee, Y. Hu, Z. Liang, R. Ji, X. Y. D. Soo, Q. Zhu and Q. Yan, SmartMat, 2023, 4, e1210 CrossRef CAS.
- C. Zhang, W. Liu, C. Chen, P. Ni, B. Wang, Y. Jiang and Y. Lu, Nanoscale, 2022, 14, 2915–2942 RSC.
- H.-l. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- R. Guo, K. Zhang, S. Ji, Y. Zheng and M. Jin, Chin. Chem. Lett., 2021, 32, 2679–2692 CrossRef CAS.
- T. Chen, C. Foo and S. C. E. Tsang, Chem. Sci., 2021, 12, 517–532 RSC.
- Y. Chen, Z. Lai, X. Zhang, Z. Fan, Q. He, C. Tan and H. Zhang, Nat. Rev. Chem., 2020, 4, 243–256 CrossRef CAS PubMed.
- H. Li, X. Zhou, W. Zhai, S. Lu, J. Liang, Z. He, H. Long, T. Xiong, H. Sun, Q. He, Z. Fan and H. Zhang, Adv. Energy Mater., 2020, 10, 2002019 CrossRef CAS.
- X. Yang, Y. Wang, X. Tong and N. Yang, Adv. Energy Mater., 2022, 12, 2102261 CrossRef CAS.
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed.
- X. Ai, X. Zou, H. Chen, Y. Su, X. Feng, Q. Li, Y. Liu, Y. Zhang and X. Zou, Angew. Chem., Int. Ed., 2020, 59, 3961–3965 CrossRef CAS PubMed.
- Z. Zhao, X. Huang, M. Li, G. Wang, C. Lee, E. Zhu, X. Duan and Y. Huang, J. Am. Chem. Soc., 2015, 137, 15672–15675 CrossRef CAS PubMed.
- R. Guo, Q. Chen, X. Li, Y. Liu, C. Wang, W. Bi, C. Zhao, Y. Guo and M. Jin, J. Mater. Chem. A, 2019, 7, 4714–4720 RSC.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P.-F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS PubMed.
- V. J. Parks and A. J. Durelli, Exp. Mech., 1967, 7, 279–280 CrossRef.
- H. Wang, S. Xu, C. Tsai, Y. Li, C. Liu, J. Zhao, Y. Liu, H. Yuan, F. Abild-Pedersen and F. B. Prinz, Science, 2016, 354, 1031–1036 CrossRef CAS PubMed.
- L. Nguyen, H.-P. Komsa, E. Khestanova, R. J. Kashtiban, J. J. Peters, S. Lawlor, A. M. Sanchez, J. Sloan, R. V. Gorbachev and I. V. Grigorieva, ACS Nano, 2017, 11, 2894–2904 CrossRef CAS PubMed.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- Z. Xi, X. Cheng, Z. Gao, M. Wang, T. Cai, M. Muzzio, E. Davidson, O. Chen, Y. Jung and S. Sun, Nano Lett., 2019, 20, 272–277 CrossRef PubMed.
- Z. Li, X. Lu, J. Teng, Y. Zhou and W. Zhuang, Nanoscale, 2021, 13, 11314–11324 RSC.
- J. Norton and J. Sowa, Chem. Rev., 2016, 116, 8315–8317 CrossRef PubMed.
- S. Kato, S. K. Matam, P. Kerger, L. Bernard, C. Battaglia, D. Vogel, M. Rohwerder and A. Züttel, Angew. Chem., Int. Ed., 2016, 55, 6028–6032 CrossRef CAS PubMed.
- T. Zhang, H. Miyaoka, H. Miyaoka, T. Ichikawa and Y. Kojima, ACS Appl. Energy Mater., 2018, 1, 232–242 CrossRef CAS.
- J. Fan, X. Cui, S. Yu, L. Gu, Q. Zhang, F. Meng, Z. Peng, L. Ma, J.-Y. Ma, K. Qi, Q. Bao and W. Zheng, ACS Nano, 2019, 13, 12987–12995 CrossRef CAS PubMed.
- Y. Xiong, Y. Ma, L. Zou, S. Han, H. Chen, S. Wang, M. Gu, Y. Shen, L. Zhang, Z. Xia, J. Li and H. Yang, J. Catal., 2020, 382, 247–255 CrossRef CAS.
- Y. Shi, R. Schimmenti, S. Zhu, K. Venkatraman, R. Chen, M. Chi, M. Shao, M. Mavrikakis and Y. Xia, J. Am. Chem. Soc., 2022, 144, 2556–2568 CrossRef CAS PubMed.
- M. K. Kabiraz, J. Kim, W.-J. Lee, B. Ruqia, H. C. Kim, S.-U. Lee, J.-R. Kim, S.-M. Paek, J. W. Hong and S.-I. Choi, Chem. Mater., 2019, 31, 5663–5673 CrossRef CAS.
- X. Guo, Z. Hu, J. Lv, J. Qu and S. Hu, Dalton Trans., 2021, 50, 10359–10364 RSC.
- J. Wu, X. Cui, J. Fan, J. Zhao, Q. Zhang, G. Jia, Q. Wu, D. Zhang, C. Hou, S. Xu, D. Jiao, L. Gu, D. J. Singh and W. Zheng, ACS Energy Lett., 2021, 6, 1912–1919 CrossRef CAS.
- C. Zhan, H. Li, X. Li, Y. Jiang and Z. Xie, Sci. China Mater., 2020, 63, 375–382 CrossRef CAS.
- Y. Zhu, C. Gao, S. Bai, S. Chen, R. Long, L. Song, Z. Li and Y. Xiong, Nano Res., 2017, 10, 3396–3406 CrossRef CAS.
- J. Fan, Y. Zhang, W. Liu, M. Li, Y. Cai, Q. Ji, Z. Zhao, G. Hou, A. Li, W. Zhou, L. Yu and D. Deng, Matter, 2023, 6, 3877–3888 CrossRef CAS.
- G. Wang, J. Liu, Y. Sui, M. Wang, L. Qiao, F. Du and B. Zou, J. Mater. Chem. A, 2019, 7, 14876–14881 RSC.
- R. Lässer and K. H. Klatt, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 748–758 CrossRef.
- F. D. Manchester, A. San-Martin and J. M. Pitre, J. Phase Equilib., 1994, 15, 62–83 CrossRef CAS.
- J. E. Schirber and B. Morosin, Phys. Rev. B: Solid State, 1975, 12, 117–118 CrossRef CAS.
- J. E. Worsham, M. K. Wilkinson and C. G. Shull, J. Phys. Chem. Solids, 1957, 3, 303–310 CrossRef CAS.
- J. A. Eastman, L. J. Thompson and B. J. Kestel, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 84–92 CrossRef CAS PubMed.
- T.-H. Phan and R. E. Schaak, Chem. Commun., 2009, 3026–3028, 10.1039/B902024A.
- H.-Y. Sun, Y. Ding, Y.-Q. Yue, Q. Xue, F.-M. Li, J.-X. Jiang, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 13149–13157 CrossRef CAS PubMed.
- J. Zhang, M. Chen, H. Li, Y. Li, J. Ye, Z. Cao, M. Fang, Q. Kuang, J. Zheng and Z. Xie, Nano Energy, 2018, 44, 127–134 CrossRef CAS.
- Y. Wang, S. N. Sun and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1–4 CrossRef CAS PubMed.
- S. Zhou, M. Figueras-Valls, Y. Shi, Y. Ding, M. Mavrikakis and Y. Xia, Angew. Chem., Int. Ed., 2023, 62, e2023069 Search PubMed.
- H. Li, X. Qin, X.-G. Zhang, K. Jiang and W.-B. Cai, ACS Catal., 2022, 12, 12750–12764 CAS.
- M. Fan, X. Liang, Q. Li, L. Cui, X. He and X. Zou, Chin. Chem. Lett., 2023, 34, 107275 CAS.
- J.-Y. Wang, Y.-Y. Kang, H. Yang and W.-B. Cai, J. Phys. Chem. C, 2009, 113, 8366–8372 CAS.
- B. Jiang, X.-G. Zhang, K. Jiang, D.-Y. Wu and W.-B. Cai, J. Am. Chem. Soc., 2018, 140, 2880–2889 CAS.
- Z. Mao, C. Ding, X. Liu, Q. Zhang, X. Qin, H. Li, F. Yang, Q. Li, X.-G. Zhang, J. Zhang and W.-B. Cai, ACS Catal., 2022, 12, 8848–8856 CAS.
- S. S. Djokić, in Modern aspects of electrochemistry, Springer, 2002, pp. 51–133 Search PubMed.
- M. Wang, X. Qin, K. Jiang, Y. Dong, M. Shao and W.-B. Cai, J. Phys. Chem. C, 2017, 121, 3416–3423 CAS.
- K. Jiang, J. Chang, H. Wang, S. Brimaud, W. Xing, R. J. Behm and W.-B. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 7133–7138 CAS.
- S. Hong, S. Chung, J. Park, J. P. Hwang, C. H. Lee, S. Uhm, S. Bong and J. Lee, ACS Catal., 2021, 11, 4722–4729 CAS.
- H. Lv, L. Sun, D. Xu, J. Henzie, Y. Yamauchi and B. Liu, J. Mater. Chem. A, 2019, 7, 24877–24883 CAS.
- Y. Wang, H. Lv, L. Sun, X. Guo, D. Xu and B. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 17599–17607 CAS.
- C. W. A. Chan, A. H. Mahadi, M. M.-J. Li, E. C. Corbos, C. Tang, G. Jones, W. C. H. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. E. Tsang, Nat. Commun., 2014, 5, 5787 CAS.
- K. Kobayashi, H. Kobayashi, M. Maesato, M. Hayashi, T. Yamamoto, S. Yoshioka, S. Matsumura, T. Sugiyama, S. Kawaguchi, Y. Kubota, H. Nakanishi and H. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 6578–6582 CAS.
- J. Li, J. Chen, Q. Wang, W.-B. Cai and S. Chen, Chem. Mater., 2017, 29, 10060–10067 CAS.
- Y. Li, C.-K. Peng, H. Hu, S.-Y. Chen, J.-H. Choi, Y.-G. Lin and J.-M. Lee, Nat. Commun., 2022, 13, 1143 CAS.
- K. Deng, W. Wang, Q. Mao, H. Yu, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Small, 2022, 18, 2203020 CAS.
- F. A. Carey and R. J. Sundberg, Advanced organic chemistry: part A: structure and mechanisms, Springer Science & Business Media, 2007.
- K. Deng, T. Ren, Y. Xu, S. Liu, Z. Dai, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2020, 8, 5595–5600 CAS.
- Z. Duan, T. Ren, Y. Xu, Z. Wang, H. Yu, K. Deng, L. Wang and H. Wang, Int. J. Hydrogen Energy, 2023, 48, 37045–37052 CAS.
- L. H. Bennett, J. R. Cuthill, A. J. McAlister, N. E. Erickson and R. E. Watson, Science, 1974, 184, 563–565 CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CAS.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CAS.
- J. Cored, A. García-Ortiz, S. Iborra, M. J. Climent, L. Liu, C.-H. Chuang, T.-S. Chan, C. Escudero, P. Concepción and A. Corma, J. Am. Chem. Soc., 2019, 141, 19304–19311 CAS.
- J. Kim, H. J. Kim, B. Ruqia, M. J. Kim, Y.-J. Jang, T. H. Jo, H. Baik, H.-S. Oh, H.-S. Chung, K. Baek, S. Noh, M. Jung, K.-j. Kim, H.-K. Lim, Y.-S. Youn and S.-I. Choi, Adv. Mater., 2021, 33, 2105248 CAS.
- Y. Sun, Y. Cao, L. Wang, X. Mu, Q. Zhao, R. Si, X. Zhu, S. Chen, B. Zhang, D. Chen and Y. Wan, Nat. Commun., 2020, 11, 4600 CAS.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J.-M. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 CAS.
- S. Tan, B. M. Tackett, Q. He, J. H. Lee, J. G. Chen and S. S. Wong, Nano Res., 2020, 13, 1434–1443 CAS.
- B. Avasarala, T. Murray, W. Li and P. Haldar, J. Mater. Chem., 2009, 19, 1803–1805 CAS.
- P. D. D. Singh, Z. V. P. Murthy and S. K. Kailasa, Coord. Chem. Rev., 2023, 481, 215046 CAS.
- J. C. Crowhurst, A. F. Goncharov, B. Sadigh, J. M. Zaug, D. Aberg, Y. Meng and V. B. Prakapenka, J. Mater. Res., 2008, 23, 1–5 Search PubMed.
- K. Deng, T. Zhou, Q. Mao, S. Wang, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Adv. Mater., 2022, 34, 2110680 CAS.
- T. Chen, C. Foo and S. C. E. Tsang, Chem. Sci., 2021, 12, 517–532 Search PubMed.
- K. Guo, Y. Meng, R. Guo, H. Guo, S. Qiu, D. Fan, D. Xu and Y. Li, ACS Appl. Energy Mater., 2022, 5, 10758–10766 CAS.
- R. Guo, W. Bi, K. Zhang, Y. Liu, C. Wang, Y. Zheng and M. Jin, Chem. Mater., 2019, 31, 8205–8211 CAS.
- Q. Mao, S. Jiao, K. Ren, S. Wang, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 426, 131227 Search PubMed.
- Q. Yun, Y. Ge, Z. Shi, J. Liu, X. Wang, A. Zhang, B. Huang, Y. Yao, Q. Luo, L. Zhai, J. Ge, Y. Peng, C. Gong, M. Zhao, Y. Qin, C. Ma, G. Wang, Q. Wa, X. Zhou, Z. Li, S. Li, W. Zhai, H. Yang, Y. Ren, Y. Wang, L. Li, X. Ruan, Y. Wu, B. Chen, Q. Lu, Z. Lai, Q. He, X. Huang, Y. Chen and H. Zhang, Chem. Rev., 2023, 123, 13489–13692 CAS.
- Y. Zhai, P. Han, Q. Yun, Y. Ge, X. Zhang, Y. Chen and H. Zhang, eScience, 2022, 2, 467–485 Search PubMed.
- J. Hong, J.-H. Bae, H. Jo, H.-Y. Park, S. Lee, S. J. Hong, H. Chun, M. K. Cho, J. Kim, J. Kim, Y. Son, H. Jin, J.-Y. Suh, S.-C. Kim, H.-K. Roh, K. H. Lee, H.-S. Kim, K. Y. Chung, C. W. Yoon, K. Lee, S. H. Kim, J.-P. Ahn, H. Baik, G. H. Kim, B. Han, S. Jin, T. Hyeon, J. Park, C. Y. Son, Y. Yang, Y.-S. Lee, S. J. Yoo and D. W. Chun, Nature, 2022, 603, 631–636 Search PubMed.
- G.-F. Wei, L.-R. Zhang and Z.-P. Liu, Phys. Chem. Chem. Phys., 2018, 20, 27752–27757 CAS.
- L. Chen, L.-R. Zhang, L.-Y. Yao, Y.-H. Fang, L. He, G.-F. Wei and Z.-P. Liu, Energy Environ. Sci., 2019, 12, 3099–3105 CAS.
- H. Lv, D. Xu, C. Kong, Z. Liang, H. Zheng, Z. Huang and B. Liu, ACS Cent. Sci., 2020, 6, 2347–2353 CAS.
- K. Deng, W. Wang, Z. Lian, H. Yu, Z. Wang, Y. Xu, H. Wang and L. Wang, Small, 2023, 19, 2304181 CAS.
- F. Yu, T. Lin, Y. An, K. Gong, X. Wang, Y. Sun and L. Zhong, Chem. Commun., 2022, 58, 9712–9727 CAS.
- T. Wakisaka, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, H. Akiba, O. Yamamuro, K. Ikeda, T. Otomo, N. Palina, Y. Chen, L. S. R. Kumara, C. Song, O. Sakata, W. Xie, M. Koyama, Y. Kubota, S. Kawaguchi, R. L. Arevalo, S. M. Aspera, E. F. Arguelles, H. Nakanishi and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 1247–1253 CAS.
- Z. Cao, H. Li, Q. Fan, Z. Liu, Z. Chen, Y. Sun, J. Ye, M. Cao, C. Shen, Y. Jiang, M. Chi, J. Cheng, H. Chen, Z. Xie and Y. Xia, Nano Energy, 2023, 113, 108597 CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CAS.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CAS.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 CAS.
- G. M. Veith, A. R. Lupini, L. Baggetto, J. F. Browning, J. K. Keum, A. Villa, L. Prati, A. B. Papandrew, G. A. Goenaga, D. R. Mullins, S. E. Bullock and N. J. Dudney, Chem. Mater., 2013, 25, 4936–4945 CAS.
- H. Wu and W. Chen, J. Am. Chem. Soc., 2011, 133, 15236–15239 Search PubMed.
- R. Guo, K. Zhang, Y. Liu, Y. He, C. Wu and M. Jin, J. Mater. Chem. A, 2021, 9, 6196–6204 Search PubMed.
- S. Cao, Q. Ji, H. Li, M. Pang, H. Yuan, J. Zhang and X. Bi, J. Am. Chem. Soc., 2020, 142, 7083–7091 Search PubMed.
- K. Wang, B. Huang, F. Lin, F. Lv, M. Luo, P. Zhou, Q. Liu, W. Zhang, C. Yang and Y. Tang, Adv. Energy Mater., 2018, 8, 1801891 Search PubMed.
- S. Yang, X. Yang, Q. Wang, X. Cui, H. Zou, X. Tong and N. Yang, Chem. Eng. J., 2022, 449, 137790 Search PubMed.
- F. Yang, Y. Zhao, Y. Du, Y. Chen, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2018, 8, 1703489 Search PubMed.
- H. Duan, D. Li, Y. Tang, Y. He, S. Ji, R. Wang, H. Lv, P. P. Lopes, A. P. Paulikas and H. Li, J. Am. Chem. Soc., 2017, 139, 5494–5502 CAS.
- J. Yu, Y. Guo, S. She, S. Miao, M. Ni, W. Zhou, M. Liu and Z. Shao, Adv. Mater., 2018, 30, 1800047 Search PubMed.
- R. Wang, D. Li, T. Li, W. Sun and W. Hu, Colloids Surf., A, 2023, 668, 131452 CAS.
- K. C. Poon, D. C. Tan, T. D. Vo, B. Khezri, H. Su, R. D. Webster and H. Sato, J. Am. Chem. Soc., 2014, 136, 5217–5220 CAS.
- F. Luo, Q. Zhang, X. Yu, S. Xiao, Y. Ling, H. Hu, L. Guo, Z. Yang, L. Huang and W. Cai, Angew. Chem., Int. Ed., 2018, 57, 14862–14867 CAS.
- H. Xie, Q. Geng, X. Zhu, Y. Luo, L. Chang, X. Niu, X. Shi, A. M. Asiri, S. Gao and Z. Wang, J. Mater. Chem. A, 2019, 7, 24760–24764 CAS.
- H. Lv, Y. Teng, Y. Wang, D. Xu and B. Liu, Chem. Commun., 2020, 56, 15667–15670 CAS.
- A. E. Henkes, Y. Vasquez and R. E. Schaak, J. Am. Chem. Soc., 2007, 129, 1896–1897 CAS.
- M. Li, Y. Fang, G. Zhang, P. Cui, Z. Yang and J. He, J. Mater. Chem. A, 2020, 8, 10433–10438 Search PubMed.
- G. Wu, X. Zheng, P. Cui, H. Jiang, X. Wang, Y. Qu, W. Chen, Y. Lin, H. Li and X. Han, Nat. Commun., 2019, 10, 4855 Search PubMed.
- G. Wu, X. Han, J. Cai, P. Yin, P. Cui, X. Zheng, H. Li, C. Chen, G. Wang and X. Hong, Nat. Commun., 2022, 13, 4200 Search PubMed.
- N. Yang, H. Cheng, X. Liu, Q. Yun, Y. Chen, B. Li, B. Chen, Z. Zhang, X. Chen and Q. Lu, Adv. Mater., 2018, 30, 1803234 Search PubMed.
- Q. Yao, Z. Yu, L. Li and X. Huang, Chem. Rev., 2023, 123, 9676–9717 CAS.
- M. Wang, L. Li, M. Wang and X. Huang, NPG Asia Mater., 2022, 14, 78 CAS.
- Y. Zhang, J. Fang, L. Zhang, D. Wei, W. Zhu and Z. Zhuang, Chem. Commun., 2022, 58, 4488–4491 Search PubMed.
- P. Li, W. Li, Y. Huang, J. Li, Q. Huang, S. Zhao and S. Tian, Nanoscale, 2022, 14, 6258–6267 CAS.
- Y. Xu, K. Ren, T. Ren, M. Wang, S. Yu, Z. Wang, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2020, 8, 19873–19878 Search PubMed.
- R. G. Chaudhuri and S. Paria, J. Colloid Interface Sci., 2012, 369, 117–122 Search PubMed.
- Y. Feng, Q. Yao, J. Li, N. Goswami, J. Xie and J. Yang, Nano Res., 2016, 9, 942–950 Search PubMed.
- C. Du, P. Li, F. Yang, G. Cheng, S. Chen and W. Luo, ACS Appl. Mater. Interfaces, 2018, 10, 753–761 CAS.
- R. Nandan and K. Nanda, Nanoscale, 2017, 9, 12628–12636 CAS.
- A. Li, J. Lin, Z. Huang, X. Wang and L. Guo, iScience, 2018, 10, 1–10 CAS.
- H. Gao, H. Zhou, Y. Hao, G. Zhou, H. Zhou, F. Gao, J. Xiao, P. Tang and G. Hao, J. Semicond., 2023, 44, 1–8 Search PubMed.
- Z. Fan, X. Zhang, J. Yang, X.-J. Wu, Z. Liu, W. Huang and H. Zhang, J. Am. Chem. Soc., 2015, 137, 10910–10913 CAS.
- Y. Feng, Y. Ji, Y. Zhang, Q. Shao, Y. Xu, Y. Li and X. Huang, Nat. Synth., 2022, 1, 626–634 Search PubMed.
- D. Yoon, B. Seo, J. Lee, K. S. Nam, B. Kim, S. Park, H. Baik, S. H. Joo and K. Lee, Energy Environ. Sci., 2016, 9, 850–856 CAS.
- J. Park, J. Park, J. Lee, A. Oh, H. Baik and K. Lee, ACS Nano, 2018, 12, 7996–8005 CAS.
- Y. Xu, C. Du, Q. Shen, J. Huang, X. Zhang and J. Chen, Chem. Eng. J., 2021, 417, 129318 CAS.
- M. Xie, B. Zhang, Z. Jin, P. Li and G. Yu, ACS Nano, 2022, 16, 13715–13727 CAS.
- X. Zhang and G. Lu, J. Phys. Chem. Lett., 2014, 5, 292–297 CAS.
- P. Moseley and W. A. Curtin, Nano Lett., 2015, 15, 4089–4095 CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CAS.
- G. Meng, W. Sun, A. A. Mon, X. Wu, L. Xia, A. Han, Y. Wang, Z. Zhuang, J. Liu, D. Wang and Y. Li, Adv. Mater., 2019, 31, 1903616 Search PubMed.
- F. Shi, J. Peng, F. Li, N. Qian, H. Shan, P. Tao, C. Song, W. Shang, T. Deng, H. Zhang and J. Wu, Adv. Mater., 2021, 33, 2101511 CAS.
- Y. Liu, Z. Chen, C. Liu, J. Zhang, W. Hu and Y. Deng, J. Mater. Sci. Technol., 2022, 98, 205–211 CAS.
- J. Fan, Z. Feng, Y. Mu, X. Ge, D. Wang, L. Zhang, X. Zhao, W. Zhang, D. J. Singh, J. Ma, L. Zheng, W. Zheng and X. Cui, J. Am. Chem. Soc., 2023, 145, 5710–5717 CAS.
- H. Guo, L. Li, Y. Chen, W. Zhang, C. Shang, X. Cao, M. Li, Q. Zhang, H. Tan, Y. Nie, L. Gu and S. Guo, Adv. Mater., 2023, 35, 2302285 CAS.
- X. Guo, Z. Hu, J. Lv, H. Li, Q. Zhang, L. Gu, W. Zhou, J. Zhang and S. Hu, Nano Res., 2022, 15, 1288–1294 CAS.
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870–874 CAS.
- L. Sun and B. Liu, Adv. Mater., 2023, 35, 2207305 CAS.
- Z. Li, X. Ai, H. Chen, X. Liang, X. Li, D. Wang and X. Zou, Chem. Commun., 2021, 57, 5075–5078 CAS.
- H. Lv, L. Sun, D. Xu, W. Li, B. Huang and B. Liu, CCS Chem., 2022, 4, 2854–2863 CAS.
- Y. Wang, L. Sun, H. Lv, C. Zheng and B. Liu, CCS Chem., 2023, 5, 1896–1907 CAS.
- Y. Wang, H. Lv, L. Sun and B. Liu, ACS Nano, 2021, 15, 18661–18670 CAS.
- M. Zhao and Y. Xia, Nat. Rev. Mater., 2020, 5, 440–459 CAS.
- X. Zhao, H. Cheng, X. Chen, Q. Zhang, C. Li, J. Xie, N. Marinkovic, L. Ma, J. C. Zheng and K. Sasaki, J. Am. Chem. Soc., 2024, 146, 3010–3022 CAS.
- A. Mustafa, Y. Shuai, B. G. Lougou, Z. Wang, S. Razzaq, J. Zhao and J. Shan, Chem. Eng. Sci., 2021, 245, 116869 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |