 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the crystallisation of aspirin in a confined porous material using solid-state nuclear magnetic resonance†
Marie Juramy‡
 a,
Eric Bessona,
Stéphane Gastaldia,
Fabio Ziarellib,
Stéphane Vielac,
Giulia Mollicaa and
Pierre Thureau*a
a,
Eric Bessona,
Stéphane Gastaldia,
Fabio Ziarellib,
Stéphane Vielac,
Giulia Mollicaa and
Pierre Thureau*a
aAix-Marseille Univ., CNRS, ICR UMR 7273, 13397 Marseille, France. E-mail: pierre.thureau@univ-amu.fr
bAix-Marseille Univ., Centrale Mediterranée, CNRS, Fédération des Sciences Chimiques FR 1739, 13397 Marseille, France
cInstitut Universitaire de France, 75005 Paris, France
First published on 16th July 2024
Abstract
In this study, nuclear magnetic resonance (NMR) is used to investigate the crystallisation behaviour of aspirin within a mesoporous SBA-15 silica material. The potential of dynamic nuclear polarisation (DNP) experiments is also investigated using specifically designed porous materials that incorporate polarising agents within their walls. The formation of the metastable crystalline form II is observed when crystallisation occurs within the pores of the mesoporous structure. Conversely, bulk crystallisation yields the most stable form, namely form I, of aspirin. Remarkably, the metastable form II remains trapped within the pores of mesoporous SBA-15 silica material even 30 days after impregnation, underscoring its persistent stability within this confined environment.
Introduction
Despite the significance of crystallisation phenomena in various fields such as chemistry, materials science, and biology (e.g. biomineralization), gaining a comprehensive understanding of these processes can often be challenging.1–4 In fact, the intricate mechanistic pathway that leads from molecules dispersed in a solution to the formation of the initial crystalline assembly (referred to as the critical nucleation cluster) and subsequent growth towards the final crystalline phase is a complex phenomenon. Additionally, the process can be further complicated by the evolution of the system through different temporary phases, such as amorphous phases or metastable polymorphs, before achieving the desired crystallisation product. A thorough comprehension of the complete mechanistic pathway is crucial for advancing our knowledge of crystallisation processes and serves as a prerequisite for devising experimental strategies to effectively control the formation of specific polymorphs. Unfortunately, the observation of metastable crystalline forms can sometimes be difficult.For example, aspirin, alternatively also known as acetyl salicylic acid, is a long-standing medicinal drug that exists in various forms. The initial crystal structure of aspirin was determined in 1964 while the polymorphism of aspirin has long been speculated based on notable disparities in melting point and dissolution rates during experimental measurements.5,6
Subsequently, in 2004, Ouvrard and Price made a prediction regarding the presence of a second crystal structure of aspirin using crystal structure prediction techniques.7 Following this, experimental observation8 and successful crystal structure determination were achieved for form II of aspirin.9 Subsequently, Bond et al. confirmed the crystal structure by performing X-ray diffraction on higher purity crystals and showed that form I (i.e., the most stable form of aspirin) and form II had very similar structures.10 Hence, it is likely that forms I and II existed as inter-grown crystals.11 Later, in 2017, a third form of aspirin, known as form IV, was obtained from molten aspirin.12
In 2019, Peksa et al. explored the crystallisation of aspirin within the pores of porous glass composites characterized by different pore sizes. Raman spectroscopy and differential scanning calorimetry (DSC) experiments allowed them to identify that distinct crystalline forms of aspirin were present within the pores of the material.13 In fact, it is widely established that the process of crystallisation occurring within nanopores has the ability to generate and stabilize polymorphs that would otherwise be metastable under typical bulk crystallisation conditions.3,14–16
In this study, we used nuclear magnetic resonance (NMR) to study the crystallisation of aspirin within the pores of a mesoporous SBA-15 silica material. In fact, the precise measurement of solid-state NMR chemical shifts17–26 allowed for reliable identification of the different solid forms (e.g. polymorphs) present during crystallisation processes.27–29 However, it is worth noting that transient solid forms, which are typically present in minimal quantities, may not be detectable in NMR studies due to the inherently low sensitivity of the technique, thereby resulting in the potential inability to observe short-lived intermediate phases.
To address these challenges, employing low-temperature solid-state NMR and dynamic nuclear polarisation (DNP) experiments can prove to be a valuable strategy.30,31 In principle, using low temperatures can trap transient forms, while DNP experiments can overcome the inherent challenges associated with the low sensitivity of NMR, especially when monitoring transient forms.32,33
In DNP experiments, microwave radiation is applied to the sample, facilitating the transfer of electron spin polarisation from unpaired electrons to the nuclear spins, thereby enhancing the strength of the NMR signal.34–42 However, the addition of paramagnetic polarising agents43–51 to the typically diamagnetic sample may alter the properties of the system being investigated, posing a potential concern when applying DNP experiments to the study of crystallisation processes. Usually, the polarising agents are dissolved in specific solvents, such as water/glycerol or tetrachloroethane, to promote glass formation during freezing, which helps to maximise the efficiency of DNP by ensuring a homogeneous dispersion of the radical across the sample. Nevertheless, using glass forming agents in crystallisation experiments can potentially impact the outcome of the crystallisation process.52,53
In the case of crystallisation processes occurring in confined conditions, these issues can be overcome by integrating the polarising agent within the walls of nanostructured porous materials.54–56 This ensures that (i) the polarising agent and the molecule undergoing crystallisation in the solution exist in separate phases, and (ii) the polarising agents are uniformly distributed throughout the entire sample. Notably, DNP polarising agents, such as TEMPO nitroxide radicals, were incorporated into the pore walls of silica materials.54,55,57,58 Interestingly, these materials proved useful for monitoring crystallisation processes using DNP experiments.59
In this study, we investigated the crystallisation of aspirin within the pores of a mesoporous SBA-15 silica material, which possesses pores with a diameter of approximately 7–8 nm. Our experiments used two kinds of mesoporous SBA-15 silica materials, i.e. with and without polarising agents incorporated into their walls, enabling us to explore the potential of DNP experiments in the case of aspirin. Our results indicate that, under confinement, crystallisation of form II is favoured compared to bulk conditions.
Results and discussion
Fig. 1 illustrates our strategy for monitoring the crystallisation of aspirin within mesoporous SBA-15 silica material. The mesoporous material is impregnated with an undersaturated solution of aspirin dissolved in ethanol (0.8 M) at ambient temperature and stored in an open vial within a desiccator at a fixed humidity of 43%, allowing crystallisation to proceed through solvent evaporation. After 2 days, the material is extracted from the vial and transferred to a solid-state NMR rotor. The rotor is then inserted into the DNP solid-state NMR spectrometer at a temperature of 98 K, rapidly quenching the crystallisation process. Subsequently, no further evolution of the crystallisation process occurs, and the resulting sample represents the crystallisation system frozen at the moment of quenching. A variety of NMR experiments can then be conducted on this so-called “frozen” crystallisation system.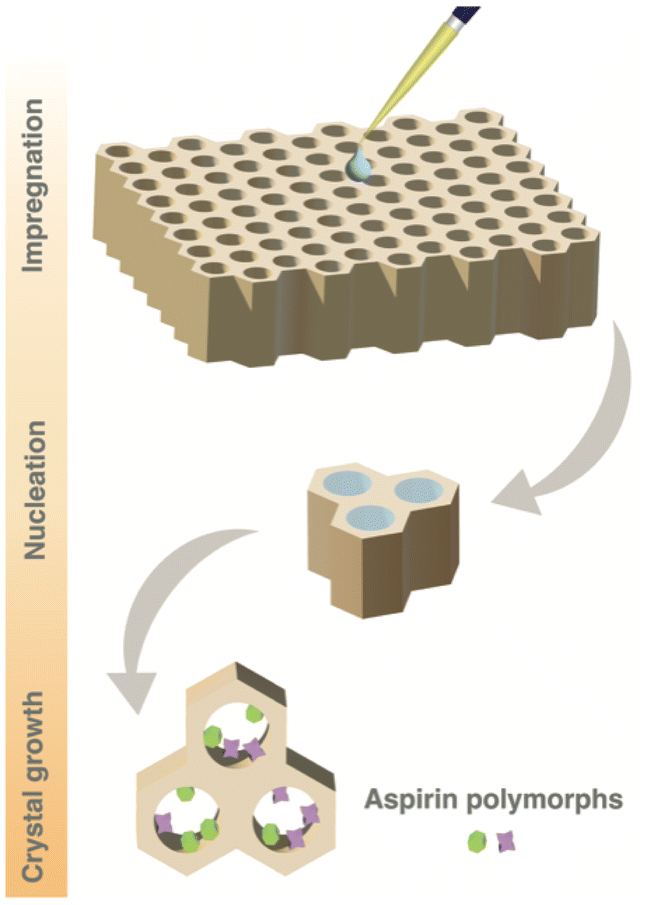 | ||
| Fig. 1 Schematic representation of the crystallisation of aspirin within the pores of mesoporous SBA-15 silica material (with or without wall-embedded TEMPO radicals). The mesoporous silica material is impregnated with a solution of aspirin in ethanol (0.8 M). Evaporation of solvent (ethanol) leads to crystallisation of aspirin within the pores of the material. | ||
Concurrently, an identical solution of 0.8 M aspirin dissolved in ethanol was allowed to crystallise under bulk conditions to compare the favoured crystalline forms within confined conditions with those obtained through bulk crystallisation. After 2 days of crystallisation, 13C CPMAS NMR experiments were performed on both the mesoporous silica impregnated with the aspirin/ethanol solution and the crystals obtained from bulk crystallisation. Furthermore, a 13C CPMAS NMR experiment was performed on the commercial aspirin used to prepare the solution.
Fig. 2 displays the 13C CPMAS NMR spectra recorded at a temperature of 98 K for these three samples. Specifically, Fig. 2c corresponds to the spectrum recorded for the commercial aspirin. The observed 13C chemical shifts matched with the expected 13C chemical shifts of form I. Fig. 2b shows the 13C CPMAS NMR spectrum recorded on the crystals obtained from the bulk crystallisation; the spectrum is identical to the 13C CPMAS NMR spectrum obtained for the commercial form. Therefore, the bulk crystallisation of aspirin in ethanol also led to the formation of form I.
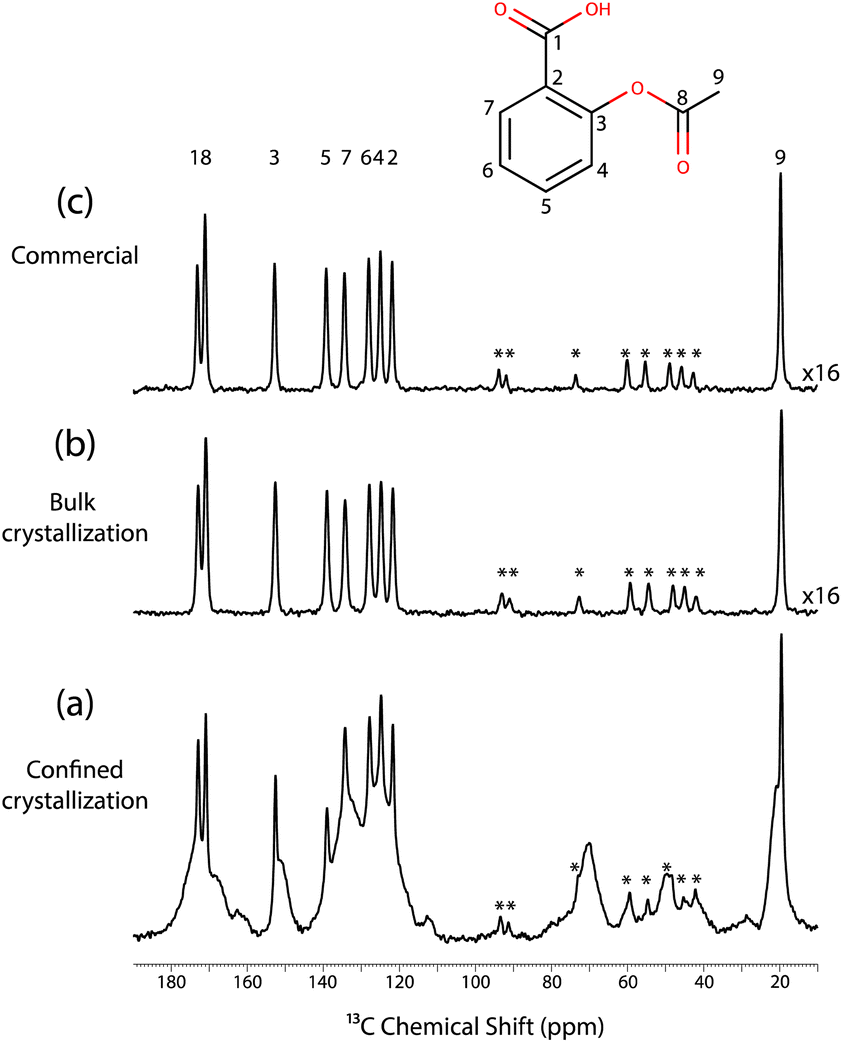 | ||
| Fig. 2 (a) 13C CPMAS NMR spectrum recorded for a mesoporous SBA-15 silica material without wall-embedded TEMPO radicals and impregnated at room temperature with a solution of aspirin in ethanol (0.8 M). The spectrum was recorded 2 days after impregnation. (b) 13C CPMAS NMR spectrum recorded for a powder sample of aspirin subsequently obtained from a solution of aspirin in ethanol. (c) 13C CPMAS NMR spectrum of commercial aspirin. All spectra were recorded without microwave irradiation and at a temperature of 98 K. Signals labelled with “*” correspond to spinning side bands. | ||
Finally, Fig. 2a shows the spectrum recorded on the impregnated mesoporous SBA-15 silica material, where 3 distinct phases can be observed. The sharp signals matched with the signals of both the commercial and the bulk crystals, and hence correspond to the 13C NMR signals of form I. Moreover, the broad signals are probably due to the amorphous aspirin/ethanol frozen glass phase. Interestingly, a third signal is observed in the region corresponding to the methyl group, as shown in Fig. 3.
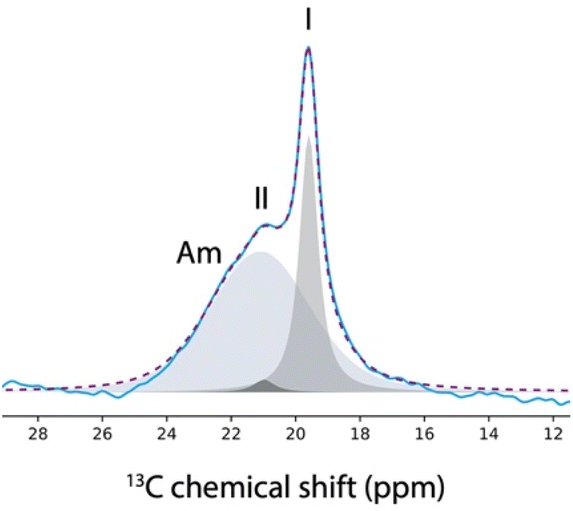 | ||
| Fig. 3 13C NMR resonances corresponding to the methyl group of the aspirin spectrum (blue curve) recorded at 98 K on a mesoporous SBA-15 silica material 2 days after impregnation with a solution of aspirin in ethanol (0.8 M). The Gaussian peaks used for the deconvolution are shown as filled grey shapes and their sum is shown as a dotted purple line. The 3 Gaussian peaks correspond to the individual 13C NMR resonances of the amorphous phase (Am), and the crystalline phases of form I and II of aspirin. | ||
Notably, this third methyl 13C NMR signal appears as a low intensity shoulder on the main peak and has a chemical shift of 20.9 ppm, which may correspond to form II of aspirin.60,61
In fact, it has been shown that the 13C CPMAS spectra of form I and form II exhibited variations primarily in the methyl 13C peak.8,60,61 Specifically, at room temperature, the 13C chemical shift of the methyl group in form I was found to be 20.0 ppm, compared to 20.7 ppm in form II. At a temperature of 105 K, the chemical shift slightly shifted with the methyl group of form I being at 19.8 ppm and form II at 20.9 ppm.61 Importantly, it is not possible to reproduce the 13C NMR spectrum shown in Fig. 3 when only two signals are used in the deconvolution, as shown in the ESI.†
As a result, the confined crystallisation of aspirin in a mesoporous SBA-15 silica material seems to have led to the detection of 3 different phases, aspirin molecules in solution, form I and form II.
It is well known that NMR allows amorphous phases to be distinguished from crystalline phases on the basis of signal line widths, with amorphous phases giving rise to significantly broader lines. In fact, the signal from amorphous phases of aspirin/ethanol frozen solution can be suppressed through a T1ρ relaxation filter experiment. This method relies on differences in the relaxation times of aspirin in crystalline and amorphous phases.59,62 The T1ρ-filtered 13C CPMAS NMR spectrum is shown in Fig. 4 where the 13C NMR resonances arising from the amorphous phase have been completely suppressed, whereas the 13C NMR resonances corresponding to the crystalline forms I and II of aspirin are still observed. Overall, the results presented in Fig. 2–4 clearly demonstrate that form II of aspirin is formed when crystallisation occurs in a confined environment.
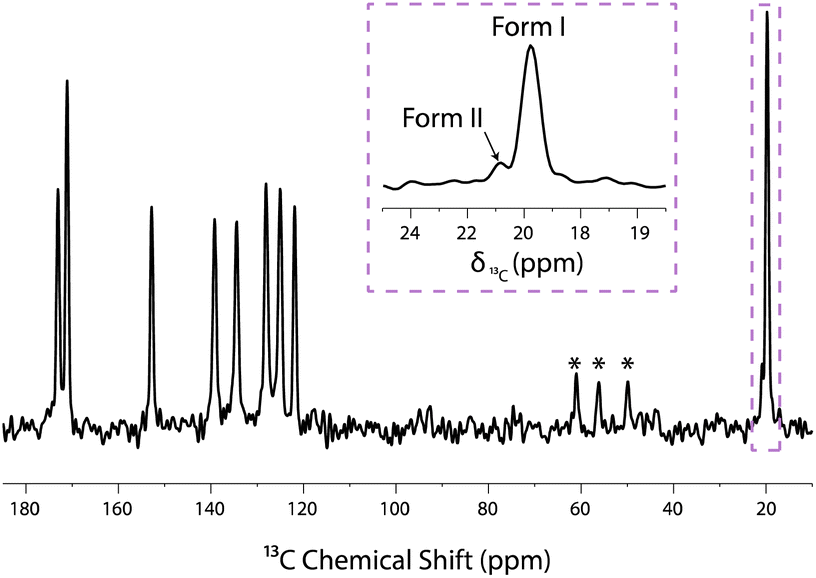 | ||
| Fig. 4 T1ρ-filtered 13C CPMAS NMR spectrum recorded on a mesoporous SBA-15 silica material impregnated with a 0.8 M solution of aspirin in ethanol. This experiment was recorded 2 days after material impregnation, without microwave irradiation and at a temperature of 98 K. Signals labelled with “*” correspond to spinning side bands. The 13C NMR chemical shift of the methyl group signal of forms I and II are at 19.8 ppm and 20.9 ppm, respectively. | ||
It should be noted that all the spectra shown in Fig. 2–4 were recorded using a mesoporous SBA-15 silica material without polarising agents incorporated into its walls. As a result, no DNP experiments were conducted in this case.
In order to assess whether DNP experiments could improve the sensitivity of NMR experiments conducted on aspirin, similar experiments were performed utilising a mesoporous SBA-15 silica material with polarising agents incorporated into its walls. In principle, under microwave irradiation, the large polarisation of the electronic spins of the TEMPO radicals located within the walls of the material should be transferred to the different phases of aspirin. Unfortunately, no DNP enhancement was observed in our case.
These results are consistent with those reported by Hanrahan et al., where a relatively modest DNP enhancement of 7.5 was measured for an aspirin powder impregnated with a 16 mM solution of TEKPol in 1,3-dibromobutane.61 The TEKPol radical typically provides much higher DNP signal enhancements than TEMPO, which may explain why we did not observe any DNP signal enhancement in our experiments. Another potential explanation for the lack of DNP enhancement in our experiment could arise from the polarisation transfer that occurs in the mesoporous SBA-15 silica material, which is still poorly understood. It may be speculated that the low density of 1H spins in our inorganic materials may not correspond to the optimal polarisation transfer through 1H spin diffusion from the electronic spins located within the walls of the material towards the pores.63 Moreover, this polarisation transfer is further reduced in our experiments by the fast rotation of the methyl groups in aspirin molecules, acting as a potential relaxation sink, thus leading to poor DNP polarisation transfer.64–66 In fact, at a temperature of 98 K, a 1H T1 relaxation time of ≈3 s was measured in the case of aspirin form I. As a result, the enhanced polarisation quickly dissipates through 1H relaxation before it can spread throughout the sample.66–68
Conclusions
Although DNP experiments performed with tailored porous materials proved ineffective in the case of aspirin, these results nonetheless demonstrate that form II of aspirin can be obtained when the crystallisation occurs within the pores of a mesoporous SBA-15 silica material. This result confirmed that unstable crystalline forms can be stabilised in a confined environment. Importantly, evaporation of ethanol from the crystallisation system does not reach completion in our experiments. For a sample left in a sealed desiccator at room temperature and fixed humidity (43%) for 30 days after impregnation, the 13C CPMAS NMR spectrum recorded at 98 K still contains a broad signal due to the amorphous aspirin/ethanol frozen solution, together with sharp signals corresponding to forms I and II of aspirin. Interestingly, even 30 days after the start of crystallisation, the metastable form II of aspirin remains trapped and stabilized within the mesoporous silica material at room temperature. We also note that, although CPMAS experiments are intrinsically nonquantitative, the significant difference between the signal intensities of both forms allowed us to conclude that form II was present in a smaller amount than form I, but no attempt was made at this point to precisely quantify their respective amounts (using, for instance, multi-CP experiments).69Importantly, the experimental strategy used here is confirmed to be a powerful method to reveal the existence and possibly to investigate the structure of transient crystalline forms. These forms typically evolve too rapidly in bulk solution to be detected using typical NMR measurement conditions. Moreover, this approach is adaptable for exploring various crystallising substances and solvents, offering a versatile means to gain fresh insights into a wide array of crystallisation systems. In future studies, it would be interesting to monitor whether the DNP NMR sensitivity could be improved by using alternative materials, such as porous polymers with nitroxide radicals grafted in the bulk matrix.70 In fact, it can be anticipated that the higher 1H density in this kind of porous material may yield more efficient polarisation transfer from the polarising agents towards the target, potentially enhancing the sensitivity of DNP solid-state NMR experiments for studying crystallisation in confined media.
Experimental
Mesoporous silica
The SBA-15 mesoporous silica materials were synthesized by the method of Besson et al. (particle size < 10 μm).54 The mesopores have cylindrical shape (diameter 7–8 nm) and are connected through micropores.The materials with wall-embedded TEMPO radicals were obtained through heating of the corresponding bis-alkoxyamine.54,56 The radical concentration (0.52 ± 0.03 mmol g−1) and the uniform distribution of radicals were established by electron paramagnetic resonance.71
DNP enhancements (εDNP) measured on a 0.2 M aqueous solution of U–13C/15N proline were used to evaluate the radical concentration and homogeneous distribution in the SBA material. Values of εDNP measured for the material used in this study are comparable to those reported previously for SBA materials with similar radical concentrations.54
Sample preparation
Aspirin was purchased from Sigma-Aldrich and used without further purification. Absolute ethanol was purchased from Carlo Erba.An undersaturated solution of aspirin (0.8 M) in ethanol was prepared in a glass vial (2 ml), at room temperature. The solution was then stirred for 10 min, until aspirin molecules were completely dissolved. The solution was passed through a 0.45 μl poly (tetrafluoroethylene) syringe filter in order to remove any insoluble particles left in the solution.
A volume of 40 μl of the aspirin solution was used to impregnate 20 mg of mesoporous silica material. The amount of aspirin solution used for impregnation was approximately equal to the total pore volume of the mesoporous material, based on the total mass and specific surface area of the material (determined by the Brunauer–Emmett–Teller (BET) method), while the pore size distribution was calculated from desorption isotherms using the Barrett–Joyner–Halenda (BJH) method. This procedure maximises the loading of aspirin within the pores while minimizing the residual amount of aspirin on the exterior surfaces of the mesoporous material.
The sample was stored in a glass vial (2 ml) left open in a desiccator at a humidity level of 43% and a temperature of 22 °C. Humidity was controlled by a supersaturated aqueous solution of potassium carbonate. A few days after impregnation of the silica (i.e., 2 days), the sample was packed in a 3.2 mm sapphire rotor and quenched in the MAS DNP spectrometer at 100 K in order to identify the crystalline form of aspirin favoured under confined conditions. The remaining 110 μl of the solution of aspirin was stored in a glass vial (2 ml) left open in a desiccator at a humidity level of 43% and a temperature of 22 °C. A few days later, when the crystals are formed (i.e., 2 days), the crystals were packed in a 3.2 mm sapphire rotor and quenched in the MAS DNP spectrometer at 100 K in order to identify the crystalline form of aspirin favoured under bulk conditions in ethanol.
NMR experiments
DNP solid-state NMR experiments were carried out on a Bruker 9.4 T wide-bore magnet (1H and 13C Larmor frequencies were 400 MHz and 100 MHz, respectively) operated using an AVANCE-III HD spectrometer and equipped with a Bruker 3.2 mm low-temperature double-resonance DNP 1H/{29Si–13C} CPMAS probe with MAS frequency of 8 kHz. The spectrometer was equipped with a microwave source, a gyrotron, allowing the microwave irradiation of the sample. The field sweep coil of the NMR magnet was set so that the microwave irradiation corresponds to the maximum DNP enhancement of AMUPol (i.e., 263.334 GHz). At the output of the probe waveguide, the estimated power of the microwave beam was 4 W.The 1H–13C CPMAS experiments NMR spectra were recorded with a MAS frequency of 8 kHz (i.e., standard MAS frequency for MAS DNP experiments performed with 3.2 mm probe). The 1H 90 °C pulse duration was 2.5 μs. 1H decoupling was performed using SPINAL64 and with a nutation frequency of 100 kHz. The contact time was set to 2 ms and the contact pulse on 1H was linearly ramped from 60 to 90 kHz. The 1H T1 relaxation time was measured using a saturation–recovery 1H–13C CP pulse sequence. The 1H–13C CPMAS spectra were recorded with a relaxation delay of 9 s and 7168 repetitions.
The T1ρ-filtered 1H–13C CPMAS experiment was performed using the same parameters as the 1H–13C CPMAS experiment and with a 1H spinlock r. f. field amplitude of 40 kHz and a duration τSL = 20 ms.
Data availability
ESI† contains the signal deconvolution of the data shown in Fig. 3 using only 2 Gaussian peaks. The raw data presented in the manuscript is available at https://doi.org/10.5281/zenodo.12688845.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from ERC under the European Union Horizon 2020 research and innovation programme (Grant Agreement No. 758498). The authors acknowledge the Agence Nationale de la Recherche for funding (ANR-18-CE29-007).References
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002 Search PubMed.
- B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. R. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241–274 CrossRef PubMed.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed.
- S. M. Woodley, G. M. Day and R. Catlow, Philos. Trans. R. Soc., A, 2020, 378, 20190600 CrossRef CAS PubMed.
- A. G. Mitchell and D. J. Saville, J. Pharm. Pharmacol., 1967, 19, 729–734 CrossRef CAS PubMed.
- R. Tawashi, Science, 1968, 160, 76 CrossRef CAS PubMed.
- C. Ouvrard and S. L. Price, Cryst. Growth Des., 2004, 4, 1119–1127 CrossRef CAS.
- E. J. Munson, presented in part at Polymorphism in Crystals, ACS ProSpectives Conference Series, Tampa, FL, 2004 Search PubMed.
- P. Vishweshwar, J. A. McMahon, M. Oliveira, M. L. Peterson and M. J. Zaworotko, J. Am. Chem. Soc., 2005, 127, 16802–16803 CrossRef CAS PubMed.
- A. D. Bond, R. Boese and G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 615–617 CrossRef CAS PubMed.
- E. J. Chan, T. R. Welberry, A. P. Heerdegen and D. J. Goossens, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 696–707 CrossRef CAS PubMed.
- A. G. Shtukenberg, C. T. Hu, Q. Zhu, M. U. Schmidt, W. Xu, M. Tan and B. Kahr, Cryst. Growth Des., 2017, 17, 3562–3566 CrossRef CAS.
- P. Peksa, J. Trzmiel, M. Ptak, M. Kostrzewa, R. Szatanik, A. Barascu, D. Enke and A. Sieradzki, J. Mater. Sci., 2019, 54, 404–413 CrossRef CAS.
- B. D. Hamilton, J.-M. Ha, M. A. Hillmyer and M. D. Ward, Acc. Chem. Res., 2012, 45, 414–423 CrossRef CAS PubMed.
- F. C. Meldrum and C. O’Shaughnessy, Adv. Mater., 2020, 32, 2001068 CrossRef CAS PubMed.
- N. Fellah, I. J. C. Dela Cruz, B. G. Alamani, A. G. Shtukenberg, A. V. Pandit, M. D. Ward and A. S. Myerson, Cryst. Growth Des., 2024, 24, 3527–3558 CrossRef CAS.
- L. E. Díaz, L. Frydman, A. C. Olivieri and B. Frydman, Anal. Lett., 1987, 20, 1657–1666 CrossRef.
- R. K. Harris, Analyst, 2006, 131, 351–373 RSC.
- J. M. Griffin, D. R. Martin and S. P. Brown, Angew. Chem., Int. Ed., 2007, 46, 8036–8038 CrossRef CAS PubMed.
- M. Geppi, G. Mollica, S. Borsacchi and C. A. Veracini, Appl. Spectrosc. Rev., 2008, 43, 202–302 CrossRef CAS.
- P. Cerreia Vioglio, G. Mollica, M. Juramy, C. E. Hughes, P. A. Williams, F. Ziarelli, S. Viel, P. Thureau and K. D. M. Harris, Angew. Chem., Int. Ed., 2018, 57, 6619–6623 CrossRef CAS PubMed.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- R. Mathew, K. A. Uchman, L. Gkoura, C. J. Pickard and M. Baias, Magn. Reson. Chem., 2020, 58, 1018–1025 CrossRef CAS PubMed.
- T. Whewell, V. R. Seymour, K. Griffiths, N. R. Halcovitch, A. V. Desai, R. E. Morris, A. R. Armstrong and J. M. Griffin, Magn. Reson. Chem., 2022, 60, 489–503 CrossRef CAS PubMed.
- M. Cordova and L. Emsley, J. Am. Chem. Soc., 2023, 145, 16109–16117 CrossRef CAS PubMed.
- M. Rahman, H. R. W. Dannatt, C. D. Blundell, L. P. Hughes, H. Blade, J. Carson, B. P. Tatman, S. T. Johnston and S. P. Brown, J. Phys. Chem. A, 2024, 128, 1793–1816 CrossRef CAS PubMed.
- C. E. Hughes, P. A. Williams, T. R. Peskett and K. D. M. Harris, J. Phys. Chem. Lett., 2012, 3, 3176–3181 CrossRef CAS PubMed.
- C. E. Hughes, P. A. Williams, V. L. Keast, V. G. Charalampopoulos, G. R. Edwards-Gau and K. D. M. Harris, Faraday Discuss., 2015, 179, 115–140 RSC.
- K. D. M. Harris, C. E. Hughes, P. A. Williams and G. R. Edwards-Gau, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2017, 73, 137–148 CrossRef CAS PubMed.
- J. Jeon, W.-M. Yau and R. Tycko, J. Am. Chem. Soc., 2020, 142, 21220–21232 CrossRef CAS PubMed.
- T. Schmidt, J. Jeon, W.-M. Yau, C. D. Schwieters, R. Tycko and G. M. Clore, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122308119 CrossRef CAS PubMed.
- P. Vioglio Cerreia, P. Thureau, M. Juramy, F. Ziarelli, S. Viel, P. A. Williams, C. E. Hughes, K. D. M. Harris and G. Mollica, J. Phys. Chem. Lett., 2019, 10, 1505–1510 CrossRef PubMed.
- M. Juramy, P. C. Vioglio, F. Ziarelli, S. Viel, P. Thureau and G. Mollica, Solid State Nucl. Magn. Reson., 2022, 122, 101836 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- A. J. Rossini, J. Phys. Chem. Lett., 2018, 9, 5150–5159 CrossRef CAS PubMed.
- L. Zhao, A. C. Pinon, L. Emsley and A. J. Rossini, Magn. Reson. Chem., 2018, 56, 583–609 CrossRef CAS PubMed.
- A. G. M. Rankin, J. Trébosc, F. Pourpoint, J.-P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2019, 101, 116–143 CrossRef CAS PubMed.
- R. W. Hooper, B. A. Klein and V. K. Michaelis, Chem. Mater., 2020, 32, 4425–4430 CrossRef CAS.
- T. Biedenbänder, V. Aladin, S. Saeidpour and B. Corzilius, Chem. Rev., 2022, 122, 9738–9794 CrossRef PubMed.
- W. Y. Chow, G. De Paëpe and S. Hediger, Chem. Rev., 2022, 122, 9795–9847 CrossRef CAS PubMed.
- I. B. Moroz and M. Leskes, Annu. Rev. Mater. Res., 2022, 52, 25–55 CrossRef CAS.
- C. Song, K.-N. Hu, C.-G. Joo, T. M. Swager and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 11385–11390 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- G. Mathies, M. A. Caporini, V. K. Michaelis, Y. Liu, K.-N. Hu, D. Mance, J. L. Zweier, M. Rosay, M. Baldus and R. G. Griffin, Angew. Chem., Int. Ed., 2015, 54, 11770–11774 CrossRef CAS PubMed.
- D. Wisser, G. Karthikeyan, A. Lund, G. Casano, H. Karoui, M. Yulikov, G. Menzildjian, A. C. Pinon, A. Purea, F. Engelke, S. R. Chaudhari, D. Kubicki, A. J. Rossini, I. B. Moroz, D. Gajan, C. Copéret, G. Jeschke, M. Lelli, L. Emsley, A. Lesage and O. Ouari, J. Am. Chem. Soc., 2018, 140, 13340–13349 CrossRef CAS PubMed.
- A. Lund, G. Casano, G. Menzildjian, M. Kaushik, G. Stevanato, M. Yulikov, R. Jabbour, D. Wisser, M. Renom-Carrasco, C. Thieuleux, F. Bernada, H. Karoui, D. Siri, M. Rosay, I. V. Sergeyev, D. Gajan, M. Lelli, L. Emsley, O. Ouari and A. Lesage, Chem. Sci., 2020, 11, 2810–2818 RSC.
- R. Yao, D. Beriashvili, W. Zhang, S. Li, A. Safeer, A. Gurinov, A. Rockenbauer, Y. Yang, Y. Song, M. Baldus and Y. Liu, Chem. Sci., 2022, 13, 14157–14164 RSC.
- R. Harrabi, T. Halbritter, F. Aussenac, O. Dakhlaoui, J. van Tol, K. K. Damodaran, D. Lee, S. Paul, S. Hediger, F. Mentink-Vigier, S. T. Sigurdsson and G. De Paëpe, Angew. Chem., Int. Ed., 2022, 61, e202114103 CrossRef CAS PubMed.
- T. Halbritter, R. Harrabi, S. Paul, J. van Tol, D. Lee, S. Hediger, S. T. Sigurdsson, F. Mentink-Vigier and G. De Paëpe, Chem. Sci., 2023, 14, 3852–3864 RSC.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed.
- E. Meirzadeh, S. Dishon, I. Weissbuch, D. Ehre, M. Lahav and I. Lubomirsky, Angew. Chem., Int. Ed., 2018, 57, 4965–4969 CrossRef CAS PubMed.
- E. Besson, F. Ziarelli, E. Bloch, G. Gerbaud, S. Queyroy, S. Viel and S. Gastaldi, Chem. Commun., 2016, 52, 5531–5533 RSC.
- D. L. Silverio, H. A. van Kalkeren, T.-C. Ong, M. Baudin, M. Yulikov, L. Veyre, P. Berruyer, S. Chaudhari, D. Gajan, D. Baudouin, M. Cavaillès, B. Vuichoud, A. Bornet, G. Jeschke, G. Bodenhausen, A. Lesage, L. Emsley, S. Jannin, C. Thieuleux and C. Copéret, Helv. Chim. Acta, 2017, 100, e1700101 CrossRef.
- P. Nabokoff, G. Brulay, C. Dol, G. Gerbaud, B. Guigliarelli, E. Etienne, E. Bloch, F. Ziarelli, E. Besson and S. Gastaldi, J. Phys. Chem. C, 2023, 127, 9699–9706 CrossRef CAS.
- M. Cavaillès, A. Bornet, X. Jaurand, B. Vuichoud, D. Baudouin, M. Baudin, L. Veyre, G. Bodenhausen, J.-N. Dumez, S. Jannin, C. Copéret and C. Thieuleux, Angew. Chem., Int. Ed., 2018, 57, 7453–7457 CrossRef PubMed.
- E. Besson, A. Vebr, F. Ziarelli, E. Bloch, G. Gerbaud, S. Queyroy, P. Thureau, S. Viel and S. Gastaldi, Phys. Chem. Chem. Phys., 2022, 24, 25279–25286 RSC.
- M. Juramy, R. Chèvre, P. Cerreia Vioglio, F. Ziarelli, E. Besson, S. Gastaldi, S. Viel, P. Thureau, K. D. M. Harris and G. Mollica, J. Am. Chem. Soc., 2021, 143, 6095–6103 CrossRef CAS PubMed.
- D. Sperger, PhD Dissertation, University of Kansas, 2010 Search PubMed.
- M. P. Hanrahan, A. Venkatesh, S. L. Carnahan, J. L. Calahan, J. W. Lubach, E. J. Munson and A. J. Rossini, Phys. Chem. Chem. Phys., 2017, 19, 28153–28162 RSC.
- J. R. Yarava, S. R. Chaudhari, A. J. Rossini, A. Lesage and L. Emsley, J. Magn. Reson., 2017, 277, 149–153 CrossRef CAS PubMed.
- O. Lafon, A. S. L. Thankamony, T. Kobayashi, D. Carnevale, V. Vitzthum, I. I. Slowing, K. Kandel, H. Vezin, J.-P. Amoureux, G. Bodenhausen and M. Pruski, J. Phys. Chem. C, 2013, 117, 1375–1382 CrossRef CAS.
- G. Mollica, D. Le, F. Ziarelli, G. Casano, O. Ouari, T. N. T. Phan, F. Aussenac, P. Thureau, D. Gigmes, P. Tordo and S. Viel, ACS Macro Lett., 2014, 3, 922–925 CrossRef CAS PubMed.
- A. C. Pinon, A. J. Rossini, C. M. Widdifield, D. Gajan and L. Emsley, Mol. Pharm., 2015, 12, 4146–4153 CrossRef CAS PubMed.
- A. Zagdoun, A. J. Rossini, M. P. Conley, W. R. Grüning, M. Schwarzwälder, M. Lelli, W. T. Franks, H. Oschkinat, C. Copéret, L. Emsley and A. Lesage, Angew. Chem., Int. Ed., 2013, 52, 1222–1225 CrossRef CAS PubMed.
- A. Abragam, Principles of Nuclear Magnetism, Oxford University Press, Oxford, 1961 Search PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- R. L. Johnson and K. Schmidt-Rohr, J. Magn. Reson., 2014, 239, 44–49 CrossRef CAS PubMed.
- T. El Daraï, S. F. Cousin, Q. Stern, M. Ceillier, J. Kempf, D. Eshchenko, R. Melzi, M. Schnell, L. Gremillard, A. Bornet, J. Milani, B. Vuichoud, O. Cala, D. Montarnal and S. Jannin, Nat. Commun., 2021, 12, 4695 CrossRef PubMed.
- P. Nabokoff, S. Gastaldi and E. Besson, Microporous Mesoporous Mater., 2021, 311, 110674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00123k |
| ‡ Present address: Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, U.K. |
| This journal is © The Royal Society of Chemistry 2024 |