
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the adsorption tendency of film-forming additives to enable fast-charging hard carbon anodes with regulated Li plating†
Yongteng
Dong
,
Yuanmao
Chen
,
Xinyang
Yue
* and
Zheng
Liang
 *
*
Frontiers Science Center for Transformative Molecules, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: xinyangyue@sjtu.edu.cn; liangzheng06@sjtu.edu.cn
First published on 6th March 2024
Abstract
Regulating lithium (Li) plating with high reversibility on hard carbon (HC) anodes is a practical approach to breaking through the bottleneck of the fast-charging lithium-ion batteries (LIBs). However, the solid electrolyte interphase (SEI) working in the complex interfacial Li intercalation/deposition processes is unstable, leading to unsafe Li plating with rapid capacity fading. Herein, we gauge the adsorption tendency of film-forming additives (ethylene carbonate (EC) and fluoroethylene carbonate (FEC)) on a carbon matrix containing defect features, thus achieving a robust SEI for HC to facilitate uniform and reproducible Li plating. The results demonstrate that the tilted conformation of FEC induced by the asymmetric electrostatic interaction leads to weaker adsorption between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of FEC and the defect site compared to that of EC. Therefore, the FEC encourages the SEI with more uniform and excellent space continuity irrespective of the distribution of the defect on the HC. Combined with local high-concentration electrolyte systems, the FEC-assisted SEI is excellent in regulating the Li plating morphology and maintaining self-structural stability over cycling. Under Li plating occupation greater than 16%, an average Coulombic efficiency (CE) of 99.8% for the HC anode over 250 cycles is retained by FEC, while that for the EC system is merely 97.9%. These findings are helpful in guiding the selection of film-forming additives for different anodes in achieving reversible Li plating for fast-charging LIBs.
O of FEC and the defect site compared to that of EC. Therefore, the FEC encourages the SEI with more uniform and excellent space continuity irrespective of the distribution of the defect on the HC. Combined with local high-concentration electrolyte systems, the FEC-assisted SEI is excellent in regulating the Li plating morphology and maintaining self-structural stability over cycling. Under Li plating occupation greater than 16%, an average Coulombic efficiency (CE) of 99.8% for the HC anode over 250 cycles is retained by FEC, while that for the EC system is merely 97.9%. These findings are helpful in guiding the selection of film-forming additives for different anodes in achieving reversible Li plating for fast-charging LIBs.
Broader contextAddressing the time-consuming charging process has become the top priority in battery industries. However, the graphite anode accompanied by unsafe lithium plating has been considered the major obstacle for fast-charging lithium-ion batteries. Many efforts attempt to eliminate Li plating through electrode or electrolyte modifications but are limited by the dynamic migration of the rate-limiting step that determines the fast-charging capability. In this work, by understanding the adsorption tendency of film-forming additives on hard carbon, the FEC-assisted SEI is excellent not only in regulating the Li plating morphology but also in maintaining self-structural stability over cycling. The average Li plating reversibility of the hard carbon anode can retain 99.8% over 250 cycles. As a result, the pouch cell can deliver a higher SOC (82.2%) and operate stably over 1700 cycles under 6C-rate charging. This work offers a guideline for the selection of film-forming additives in achieving reversible Li plating for fast-charging batteries. |
Introduction
The booming market of high energy density lithium-ion batteries (LIBs) in transportation, portable electronics, and energy storage systems has been witnessed in recent years, driven largely by the urgency to combat energy shortage and carbon emission.1,2 The consumer acceptance and market penetration of electric vehicles (EVs) remain low, mainly attributed to the time-consuming charging of EV batteries. For instance, it usually takes 2–6 hours for EV batteries to be fully charged, while it merely takes several minutes to refuel a gasoline vehicle.3,4 Fast charging has thus become one of the critical initiatives in the battery or EV industries in the long run. The major barrier to fast-charging LIBs lies in the restriction of the graphite anode susceptible to lithium (Li) plating.5 Due to the low reaction kinetics of graphite, the polarization inside LIBs grows and gradually reaches the critical value where the anode potential is lower than 0 V (vs. Li/Li+) under fast charging,6,7 and then Li+ ions are prone to depositing onto the anode surface rather than intercalating into the graphite. Uncontrolled Li plating causes a series of fast-charging concerns, including fast capacity decay, battery volume deformation, thermal runaway, and dendrite-induced safety hazards.8–18Many efforts have attempted to overcome Li plating behavior, involving a metal-based anode,19–21 expansion of graphite layer spacing,22–24 surface and defect engineering,25–27 and the design of a weak solvation electrolyte.28–31 These methods are somewhat useful for a single-step kinetic improvement under specific conditions but are limited by the dynamic migration of the rate-limiting step that varies instantaneously with cycling conditions (state of charge (SOC), rates, temperature, etc.).32 Therefore, whether Li plating can be eliminated while maintaining a high SOC for anodes during fast charging is still unknown. Releasing and then appropriately utilizing the Li plating capacity on the anode can be considered a “win–win” strategy, in which Li plating is no longer unsafe, and the SOC of the anode after fast charging can approach 100%. Liang's group has demonstrated that Li plating reversibility on the graphite anode under high-rate charging, where the Li plating capacity accounts for 40% of the overall areal intercalation capacity of the graphite anode, can reach up to 99.5% in local high-concentration electrolyte (LHCE) systems.32 Xu et al. further constructed a robust and LiF/Li3N-rich SEI on the graphite anode for regulating the Li plating layer.33 Accordingly, the compatibility of the SEI with both Li metal plating and Li-ion intercalation chemistry at the anode interfaces is a prerequisite to facilitate uniform and reproducible Li plating under fast charging.
Unlike graphite, hard carbon (HC) featuring abundant defect sites, nanopores, and short-range graphitized domains can enrich Li active sites, shorten Li-ion diffusion, and reduce local current density, allowing better rate performance.34,35 Therefore, achieving high Li plating reversibility on the HC anode should be more meaningful for fast-charging LIBs. Although Li plating regulation on HC materials can be referred to as the SEI strategy of the graphite, the structural difference of SEI derived on graphite and the HC anode using the same electrolyte remains uncertain. This is because the defects on the HC surface have a higher density than that of graphite and are also distributed randomly.34 The defect regions with high electrochemical activity adsorb the electrolyte components and hasten their reduction to form an SEI island, so the stability and space continuities of the SEI on the HC heavily depend on film-forming additives. However, the adsorption/decomposition tendency of film-forming additives in LHCE systems on HC surfaces and their impact on SEI structures remains elusive. Additionally, the evolution of the SEI before and after Li plating on HC has not yet been thoroughly explored.
In this work, we demonstrated that film-forming additives with a low adsorption tendency to defect sites can facilitate a uniform SEI on the HC anode to regulate the Li plating morphology under fast-charging conditions. Two classic additives, ethylene carbonate (EC) and fluoroethylene carbonate (FEC), are compared and investigated on the carbon surface with defect structures in the LHCE system. The adsorption behavior of EC and FEC in the presence of abundant anions is analyzed through density functional theory (DFT) calculations. The results reveal that interfacial FEC molecules with a tilted configuration have lower adsorption energy than EC on a single defect site. Thus, a uniform and LiF-rich SEI can be formed on the HC by FEC regardless of the effect of surficial defects, while the EC-induced SEI is relatively uneven and easily broken during Li-plating cycles. When Li plating accounts for more than 16% of the total areal intercalation capacity of the anode, the average Li plating reversibility of the HC anode in FEC-containing LHCE retains 99.8% over 250 cycles. Whereas to EC, the reversibility of their uncontrolled Li plating is merely 97.9%, resulting in the accumulation of dead Li and a rapid Coulombic efficiency (CE) decay. The self-made LiFePO4‖HC pouch cells with a low N/P ratio are 6C-cycled in the FEC-containing LHCE, exhibiting long-term stability over 1700 cycles. This study provides an alternative approach to address the Li plating issue and sheds light on the effect of additives in LHCE systems on the HC anode for fast-charging LIBs.
Results and discussion
Analysis of Li plating reversibility
Local high-concentration electrolyte (LHCE) systems have been confirmed to regulate Li plating on the graphite anode by its unique solvation structure and anion-derived SEI.36–39 Incorporating film-forming additives into electrolytes can establish a qualitative solid electrolyte interphase (SEI) with excellent space continuity, compactness, high ionic conductivity, and stability.11 Since the electrolyte compatible with Li-metal/Li-ion operation is required to regulate the Li plating morphology, a typical LHCE system containing 1.4 M lithium bis(fluorosulfonyl)imide (LiFSI) salt, 25 vol% dimethyl carbonate (DMC) solvent, and 40 vol% 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (HFE) diluent, was prepared as the baseline LHCE. Two typical film-forming additives, 1.0 vol% ethylene carbonate (EC) and fluoroethylene carbonate (FEC), were separately added into baseline LHCE to yield additive-containing LHCEs (denoted as LHCE-EC and LHCE-FEC).A constant lithiation cycling (CLC) test was carried out to evaluate the Coulombic efficiency (CE) of the hard carbon (HC) anode for the three studied LHCEs in half cells.33,40 A CLC test involves a complete lithiation process with a constant capacity and a de-lithiation process with a cutoff voltage of 2.0 V (vs. Li/Li+). An areal capacity of 0.9 mA h cm−2 (1C = 0.9 mA h cm−2) intercalation capacity based on a mass loading of 2.5 mg cm−2 and initial Coulombic efficiency (ICE) was selected as the constant lithiation capacity (Fig. S1, ESI†). As shown in Fig. 1a, cells containing LHCE and LHCE-EC exhibit low average CEs of 96.1% and 97.9% with a sudden drop after nearly 100 cycles at 1C. By comparison, the cell using LHCE-FEC shows stable cycling and maintains a 99.8% average CE over 250 cycles without abrupt decline if the capacity decay of sole Li intercalation/de-intercalation is deducted (Fig. S2, ESI†). This demonstrates that introducing FEC to LHCE can extend the cycling performance of the HC with Li plating. Similar trends were also observed in LHCE-FEC cells at 2 and 3C with average CEs of 99.3% (250 cycles) and 98.9% (200 cycles) which far exceeds the performances of LHCE and LHCE-EC cells. As examined by CLC rate tests, a high CE of >97% can still be maintained for LHCE-FEC cells at 10C (Fig. S3, ESI†). Such high performance, however, is not available for either LHCE or LHCE-EC cells. The stepwise increasing overpotential and premature onset of Li plating follow the order of LHCE > LHCE-EC > LHCE-FEC. Furthermore, the initial voltage curve of constant lithiation reveals that Li plating occurs at the earliest stage in LHCE cells at different C-rates (Fig. 1b). A prolonged Li-ion intercalation process was found for LHCE cells in the presence of EC, showing improved storage performance. Notably, the proportion of Li-ion interaction capacity in LHCE-FEC cells is the highest, accounting for 0.75, 0.59, and 0.53 mA h cm−2 at 1–3C, respectively.
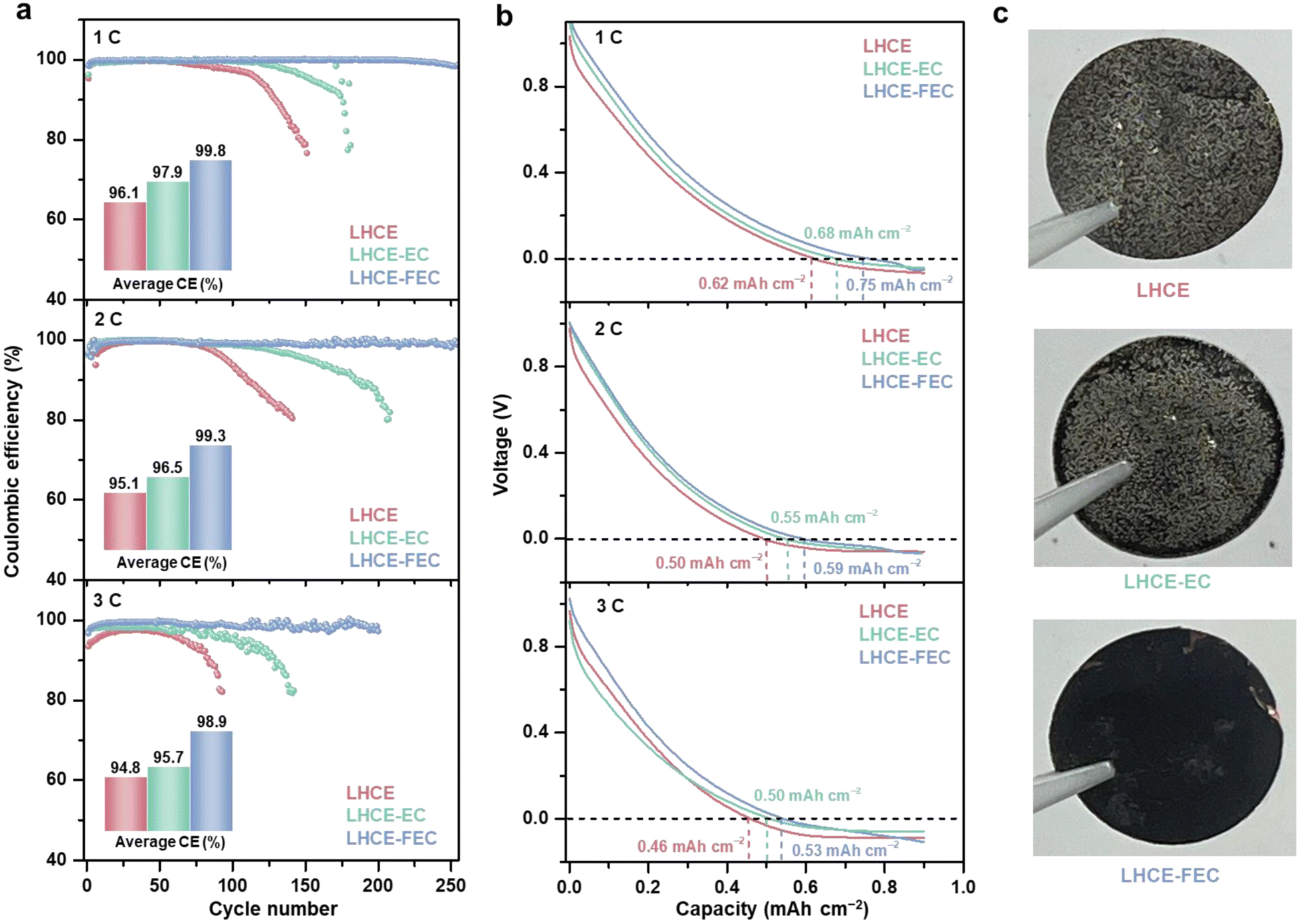 | ||
| Fig. 1 (a) CE of half-cells cycled with LHCE, LHCE-EC and LHCE-FEC at 1–3C, respectively, in the CLC test. (b) Initial voltage curves of the HC anode in the above-noted electrolytes during the CLC process. (c) Digital photos of HC anodes in the studied electrolytes after 100 cycles at 1C. | ||
Since Li plating contributes the lowest proportion of the total lithiation capacity, the lifespan of LHCE-FEC cells can be significantly extended. After disassembling cells, it was seen through digital photos that anodes cycled in LHCE and LHCE-EC are covered with a dense layer of Li metal, indicating that HC anodes are subjected to the damage of uneven Li plating with low reversibility (Fig. 1c). Conversely, much less Li deposit macroscopically appears on the anode cycled in LHCE-FEC, indicating uniform Li plating under FEC additive regulation. This result was further proved by the Li deposition/stripping performance of the Li metal‖bare Cu cells. In Fig. S4 (ESI†), compared to LHCE or LHCE-EC, LHCE-FEC renders the cell with a higher CE (98.5%) and lower overpotential, demonstrating the crucial role of the FEC additive in reducing the energy barrier of Li nucleation and growth.
In baseline LHCE, HC anodes may be prone to suffering from severe anion co-intercalation and graphene exfoliation due to insufficient protection of the SEI after repeated lithiation and de-lithiation.41 It was previously reported that EC-containing LHCE can form a LiF-rich SEI to regulate the Li plating morphology on the graphite anode surface.32,37 However, massive Li deposits were found on the HC anode surface in LHCE-EC cells under CLC tests, which was not the case in LHCE-FEC cells. Besides, the CE of the LHCE-EC cells does not exhibit satisfactory performance and is instead inferior to that of LHCE-FEC cells, as examined by CLC tests. Furthermore, Li+ transference number does not show an obvious difference among the three LHCEs after potentiostatic polarization tests of Li‖Li symmetrical cells (Fig. S5 and Table S1, ESI†). This is probably due to the similar solvation structure for all studied LHCEs as described by molecular dynamics (MD) simulations and radial distribution function (RDF) (Fig. S6, ESI†). Although FEC undergoes preferential reduction over EC,11 the inferior performance is least likely to be caused by the slightly higher LiF content from the reduction of FEC during SEI formation. The decomposition of abundant FSI− anions also contributes to the LiF generation in all studied LHCEs. This brought a concern about whether the addition of EC or FEC to baseline LHCE could influence the SEI structure and Li plating behavior on the HC surface.
Adsorption tendency of additives in LHCE
Several works have demonstrated that the surface defect regions of carbon substrates have a significant impact on the SEI composition and morphology.42–45 Effective adsorption occurring on the defective sites would meliorate the surface reactivity of the carbon substrate when electrolytes are further decomposed/reduced.46,47 For this reason, a fundamental understanding of SEI formation and its evolution on the HC surface in LHCE systems needs to be provided on the atomic scale. Adsorption structures of EC and FEC molecules in the presence of an FSI− anion and a Li+ cation on the graphene plane with a single vacancy (SV, assumed as a defect site) were determined using density functional theory (DFT) calculations. The adsorption tendency of EC and FEC molecules near the SV region was investigated based on various adsorption configurations on HC slabs (calculation details are given in the ESI†). Fig. 2a shows both top and side views of a single FSI on defect-free and SV graphene surfaces. When lithiation starts at the anode side, the FSI tends to neutralize the charge on the graphene surface with its negatively charged atoms. On a defect-free graphene surface, four O atoms in a single FSI will draw close to the surface as much as possible. However, on the SV graphene surface, two O atoms in proximity to each other will preferentially adsorb to the SV region. The adsorption energy determined from DFT calculations is −2.60 eV for FSI on defect-free surfaces, compared to −2.48 eV on SV graphene surfaces. A more negative adsorption implies a more stable configuration.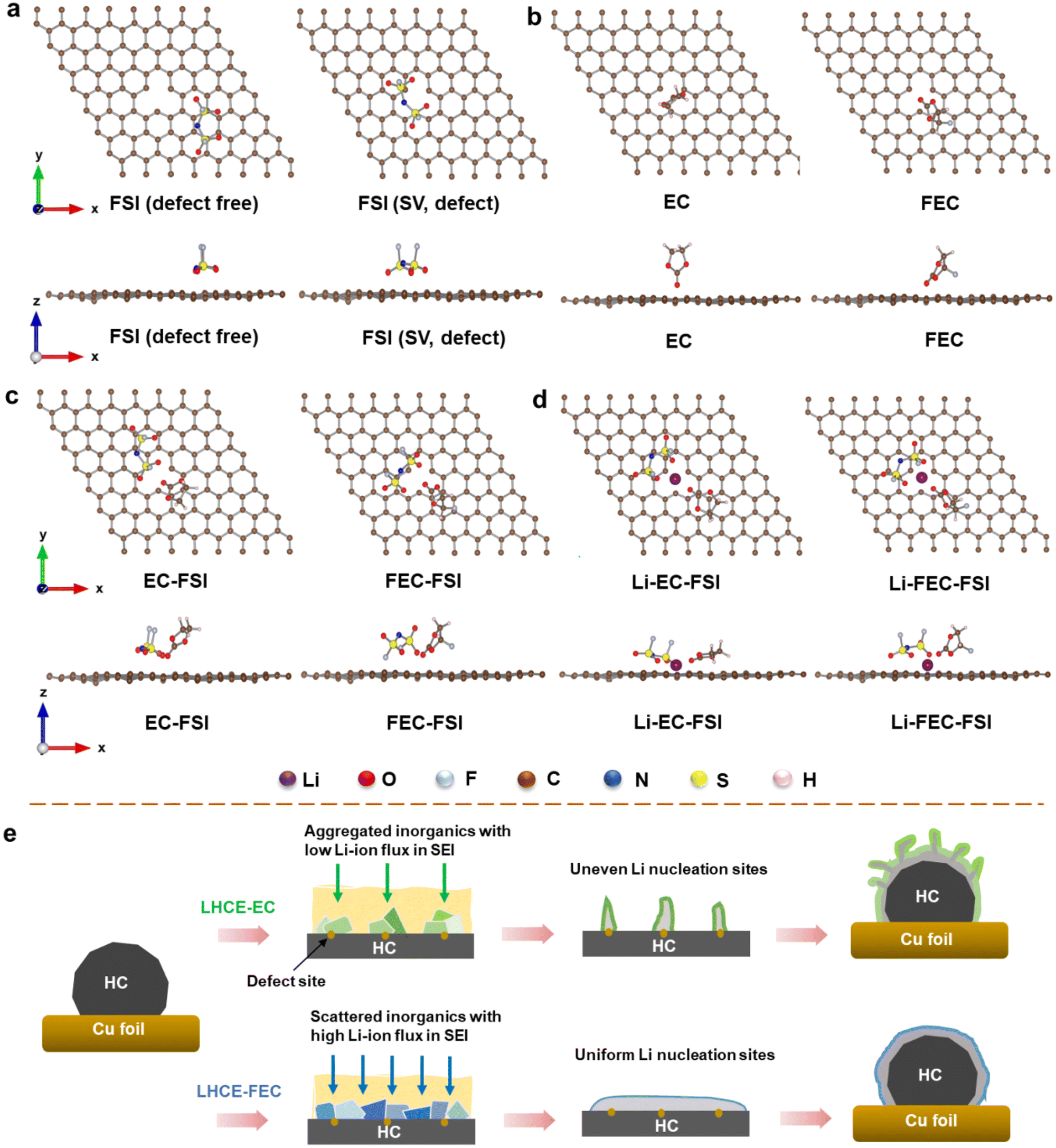 | ||
| Fig. 2 Top and side views of the optimized adsorption structures of (a) a single FSI on defect-free and SV graphene surfaces, (b) pure EC and FEC on the SV graphene surface, (c) EC-FSI and FEC-FSI on the SV graphene surface, and (d) Li-EC-FSI and Li-FEC-FSI on the SV graphene surface. (e) Schematic illustration of the effects of EC and FEC on SEI structures and Li plating morphology on the HC surface in LHCE systems. | ||
Furthermore, the adsorption configurations of pure EC and FEC molecules on SV were also modeled by optimized structures (Fig. 2b). The EC molecule in a reduction boundary exhibits a vertical conformation near the SV region with an O atom in the carbonyl CO group having interactions with the SV surface. In contrast, the FEC molecule demonstrates a tilted conformation with three atoms (two O atoms and one F atom) in contact with the radical groups near SV. In addition, the O atoms in carbonyl groups in EC are closer to the SV region than those in FEC due to their stronger interactions with the SV surface. This is further verified from the adsorption energy with −1.01 eV for EC and −0.92 eV for FEC on the SV surface.
Since the solvent-Li-FSI-additive ion-pairs or clusters tend to migrate as an entirety in LHCE systems, they have a higher chance to overcome the electrostatic repelling force of negatively charged anode and participate in SEI formation.41 With the existence of an FSI near SV, the adsorption structure of EC and FEC will change to some extent. The carbonyl O atoms of EC and FEC will adsorb to the SV region together with the FSI anion. In particular, EC shifts from a vertical to a tilted conformation near the SV region, whereas FEC still maintains its previous conformation (Fig. 2c). The established adsorption structure of FSI in the EC-FSI configuration shows that two O atoms from each OSO group along with two F atoms adsorb to the SV region, in comparison with one O atom from each OSO group of FSI adsorbing to the SV region in FEC-FSI. The adsorption energies of EC-FSI and FEC-FSI were calculated to be −3.25 eV and −3.08 eV, respectively.
The adsorption configuration of additive-FSI systems on the SV surface was also studied in the presence of a Li atom. In Fig. 2d, three O atoms from OSO bonds of an FSI anion in Li-EC-FSI are prone to aggregation with the Li atom and collectively approach the SV region. Meanwhile, EC transitions from tilted to almost horizontal conformations with their carbonyl O atom drawing closer to the Li atom. The adsorption energy of Li-EC-FSI was calculated to be −4.32 eV. As for FSI in the Li-FEC-FSI adsorption configuration, there are also three O atoms from OSO groups approaching the SV region but in a more scattered form around the Li atom. Unlike EC with different adsorption configurations, FEC remains in its tilted state induced by asymmetric electrostatic interaction at all times, and two O atoms and one F atom are less close to the Li atom near the SV region as well (−3.97 eV). In short, EC-involved adsorption is generally more exothermic and stable than FEC-involved adsorption in various configurations near the SV region. The adsorption energies for configurations are summarized in Table S2 (ESI†).
From DFT calculations, it has been proved that the rich defect sites of the HC anode would influence the adsorption tendency of additives in LHCE systems. The tilted conformation of FEC induced by the asymmetric electrostatic interaction leads to weaker adsorption between the CO of FEC and the defect site than that of EC. Taken in this sense, the reduction of EC is more likely to produce an aggregated structure if there are multiple EC molecules existing near the SV site, while the reduction products of FEC are dispersed more uniformly at the beginning of cell operation. This process plays a critical role in SEI formation. As illustrated in Fig. 2e, after the reduction of EC, despite the formed SEI being rich in inorganic components, they possibly aggregate and form local clumps in the inner layer of the SEI on the HC anode. This inhibits ion diffusion stemming from low Li-ion flux in the SEI under fast charging conditions, giving rise to uneven nucleation sites and serious Li dendrite growth. By comparison, the decomposition of FEC on the HC surface is uniform and shows a low response to defects, thus promoting the uniform growth of the anion-derived SEI. Consequently, the Li deposition beneath the SEI induced by LHCE-FEC can be more controllable and denser due to the homogeneous LiF-rich SEI featuring high Li-ion flux and stability.
Structural characterization of the SEI
The thickness of the SEI was measured on the nanoscale by using transmission electron microscopy (TEM). The initial SEI formed on the HC anode using a LHCE, before Li plating, is the thickest (≈31 nm) among all studied LHCEs, which may be assigned to the absence of a film-forming agent (Fig. 3a–c). After introducing EC and FEC, the thickness of the SEIs was reduced by 3–4 nm and 14–16 nm for the LHCE-EC and LHCE-FEC, respectively. The interfacial chemistry on the HC surface was detected by surface X-ray photoelectron spectroscopy (XPS). Compared with the interface in the LHCE, the intensity of inorganic LiF, Li2O, and Li2CO3 from F 1s and O 1s spectra evidently enhances at the interface in LHCE-EC and LHCE-FEC (Fig. 3d–f). The surface XPS spectra of F 1s show LiF (685.3 eV) peaks that are attributed to the synergistic decomposition of FSI, EC, and FEC.32,41 The higher content of CO bond in LHCE-EC and LHCE-FEC is indicative of more organic lithium ethylene decarbonate (LEDC, represented by C−O, CO, and C−C bonds), as noted by C 1s spectra, serving as an electron insulator to block the further reduction of electrolytes. This is primarily caused by the reduction of EC and FEC.
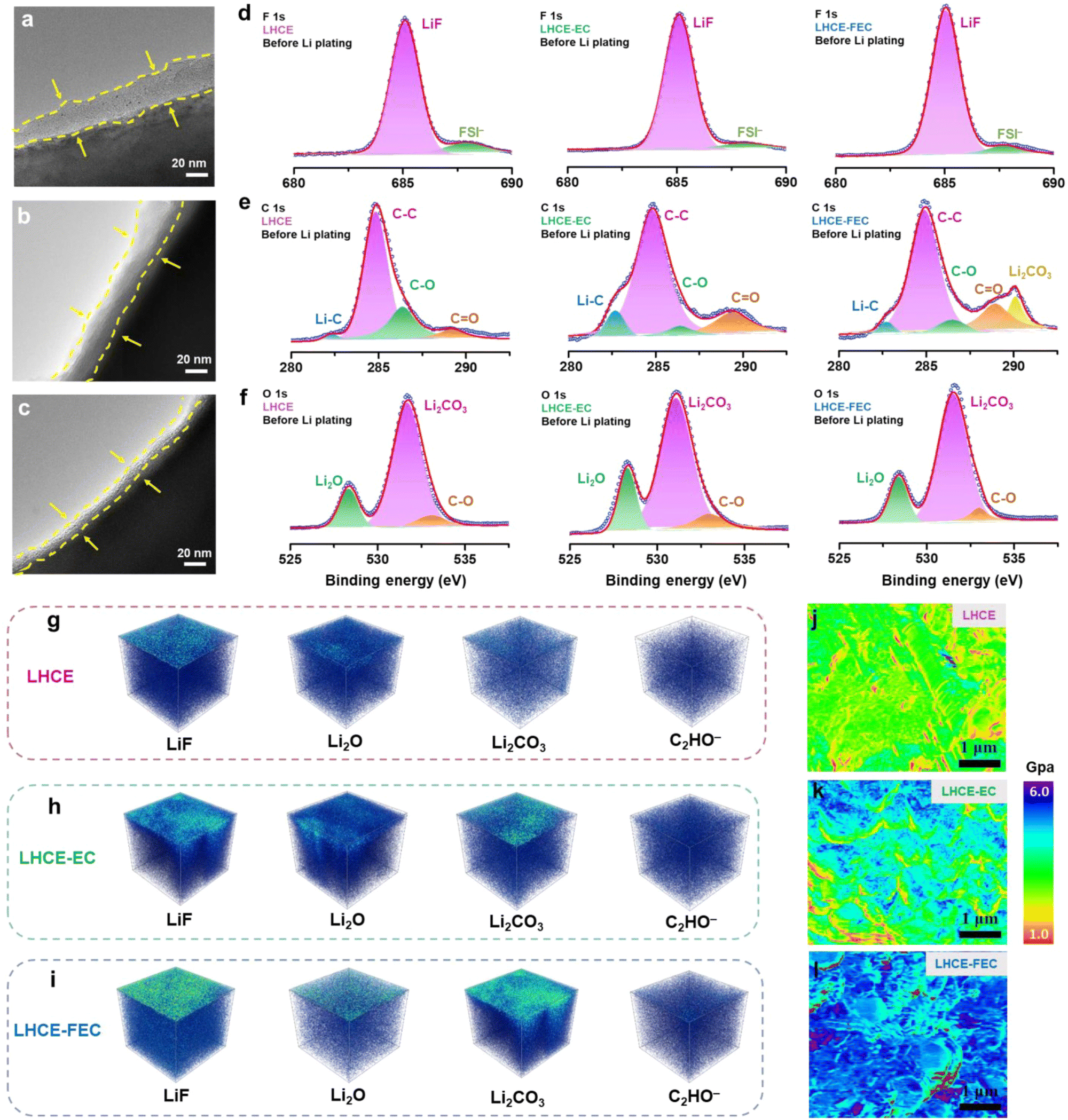 | ||
| Fig. 3 TEM images of the SEI before Li plating on the HC anode using (a) LHCE, (b) LHCE-EC, and (c) LHCE-FEC. The surface XPS spectra of (d) F 1s, (e) C 1s, and (f) O 1s on HC anodes using LHCE, LHCE-EC, and LHCE-FEC before Li plating at 0 nm depth, respectively. TOF-SIMS 3D reconstruction models of SEI components formed in (g) LHCE, (h) LHCE-EC, and (i) LHCE-FEC. Young's modulus of the SEI formed in (j) LHCE, (k) LHCE-EC, and (l) LHCE-FEC. | ||
The density and spatial uniformity of the SEI components were analyzed through the 3D depth profiling of time-of-flight secondary ion mass spectrometry (TOF-SIMS). Signals of inorganic species (LiF, Li2O, and Li2CO3) distribute uniformly throughout the LHCE-FEC derived SEI, which is in sharp contrast to the heterogeneous and cluster-like inorganics in EC-induced and additive-free SEIs (Fig. 3g–i). The higher concentrations of LiF and Li2CO3 are also in accordance with the results of XPS spectra. The above results suggest that chemical uniformity and spatial distribution of SEI species were strongly influenced by the introduction of EC and FEC. Combining a thinner inorganic-rich and chemically homogeneous SEI, high/uniform Li-ion flux and diffusion channels can be formed on the surface of the HC anode.
The mechanical properties of the SEI formed in the studied LHCEs were examined through Atomic Force Microscopy (AFM). Owing to the lack of an additive agent, the SEI formed in the LHCE is of the lowest modulus and the highest mechanical fragility (Fig. 3j–l). It is brittle and prone to crack by the volume change during lithiation.32,48 The mechanical stability of the SEI formed in LHCE-EC remains at a moderate level while suffering from uneven stress distribution. Remarkably, the FEC-induced SEI maintains the highest modulus that can better withstand the volume change and stress fluctuation caused by dendrite formation.
Li plating morphology and behavior
In addition to SEI evolution, elemental Li distribution before Li plating and after complete lithiation (Li plating)/de-lithiation on the HC anode surface was analyzed via TOF-SIMS mapping. In Fig. 4a–c, Li− mapping indicates an aggregated pattern of Li distribution on the HC surface in LHCE before Li plating. After complete lithiation and de-lithiation, numerous areas of Li enrichment can be found on the anode. The inhomogeneous Li plating aggravates dendritic Li growth and dead Li formation.49 After the introduction of EC, the distribution of Li becomes less irregular on the anode (Fig. 4d–f), but some local Li enrichments can still be observed after complete de-lithiation. For LHCE-FEC, uniform Li distribution patterns were observed on the HC anode (Fig. 4g–h). The less accumulation of elemental Li confirms that the evenly scattered inorganics induced from FEC provide uniform Li-ion diffusion channels on the HC anode before Li plating. The overall weaker intensity of the Li signal after complete lithiation/de-lithiation explains the thinner and uniform deposition layer along the HC surface. Such a Li plating behavior would decrease the danger of fast-charging and improve the reversibility of the Li plating capacity, which explains the high average CE of the HC||Li half cells with LHCE-FEC during CLC tests. | ||
| Fig. 4 Li− TOF-SIMS mapping of HC anodes (a) before Li plating, (b) after complete lithiation, and (c) after complete delithiation using LHCE. Li− TOF-SIMS mapping of HC anodes (d) before Li plating, (e) after complete lithiation, and (f) after complete delithiation using LHCE-EC. Li− TOF-SIMS mapping of HC anodes (g) before Li plating, (h) after complete lithiation, and (i) after complete delithiation using LHCE-FEC after 50 cycles of the CLC test at 1C. (j)–(l) COMSOL models of the evolution of Li plating behavior on HC particles using LHCE, LHCE-EC, and LHCE-FEC, respectively. | ||
To visualize the evolution of Li plating behavior on the HC surface, the stepwise lithiation process for the studied LHCEs was modelled using COMSOL Multiphysics software. In Fig. 4j–l, the low Li-ion flux of the interphase derived in the LHCE or LHCE-EC exacerbates the “tip effect”, resulting in the severe growth of Li dendrites. In contrast, high Li-ion flux interphase induced from the FEC enables uniform Li deposition with low surface areas. As the dendritic structures are indicative of Li plating morphology, ex-situ scanning electron microscopy (SEM) was applied to monitor the Li plating morphology at the microscale. Random dendritic Li and filamentous structures become the predominant morphology on the HC anode using an LHCE and LHCE-EC and start to grow laterally across HC particles at the interface when cells reach a fixed capacity (Fig. S7a–j, ESI†). By comparison, Li plating on the HC anode using LHCE-FEC tends to form a dense and rounded morphology. No dead Li was found after complete de-lithiation (Fig. S7k–o, ESI†).
Because of the continuous Li plating, the internal resistance of cells would gradually increase with higher lithiation capacity of the anode as it cycles to different states of charge (SOCs). Electrochemical impedance spectroscopy (EIS) measurements reveal that the higher resistance of the SOC in the LHCE and LHCE-EC cells was primarily caused by the declining active surface area of the anode from Li plating (Fig. S8, ESI†). The lower resistance of LHCE-FEC cells stands for the expanded surface area, resulting from the growth of dense Li bumps around HC particles under deep lithiation. The homogeneous Li plating on the HC surface ensures a high/uniform Li-ion flux of the LHCE-FEC-derived SEI with sufficient reaction areas and ion diffusion activity in the HC bulk phase.
Effects of Li plating on SEI
After Li plating, the thickness of the SEIs was examined and compared with their initial states. As noted in Fig. 5a–c, the SEIs formed in the LHCE and LHCE-EC, after recasting, are much thicker and rougher than their initial structures with evident fragments. However, the restructured SEI in LHCE-FEC remains smooth and almost intact in comparison to its original structure with a thickness of around 16–17 nm. In-depth XPS spectra show that the intensity of these organic/inorganic SEI species decreases in the LHCE and LHCE-EC after Li plating on the HC anode (Fig. 5d–f). This is ascribed to the SEI recasting by side reactions of continuous Li dendrites exposure to electrolytes. Conversely, no significant signal drop of species in LHCE-FEC-derived SEI before and after Li plating indicates the function of a stable SEI to effectively suppress Li dendrite growth and the volume change.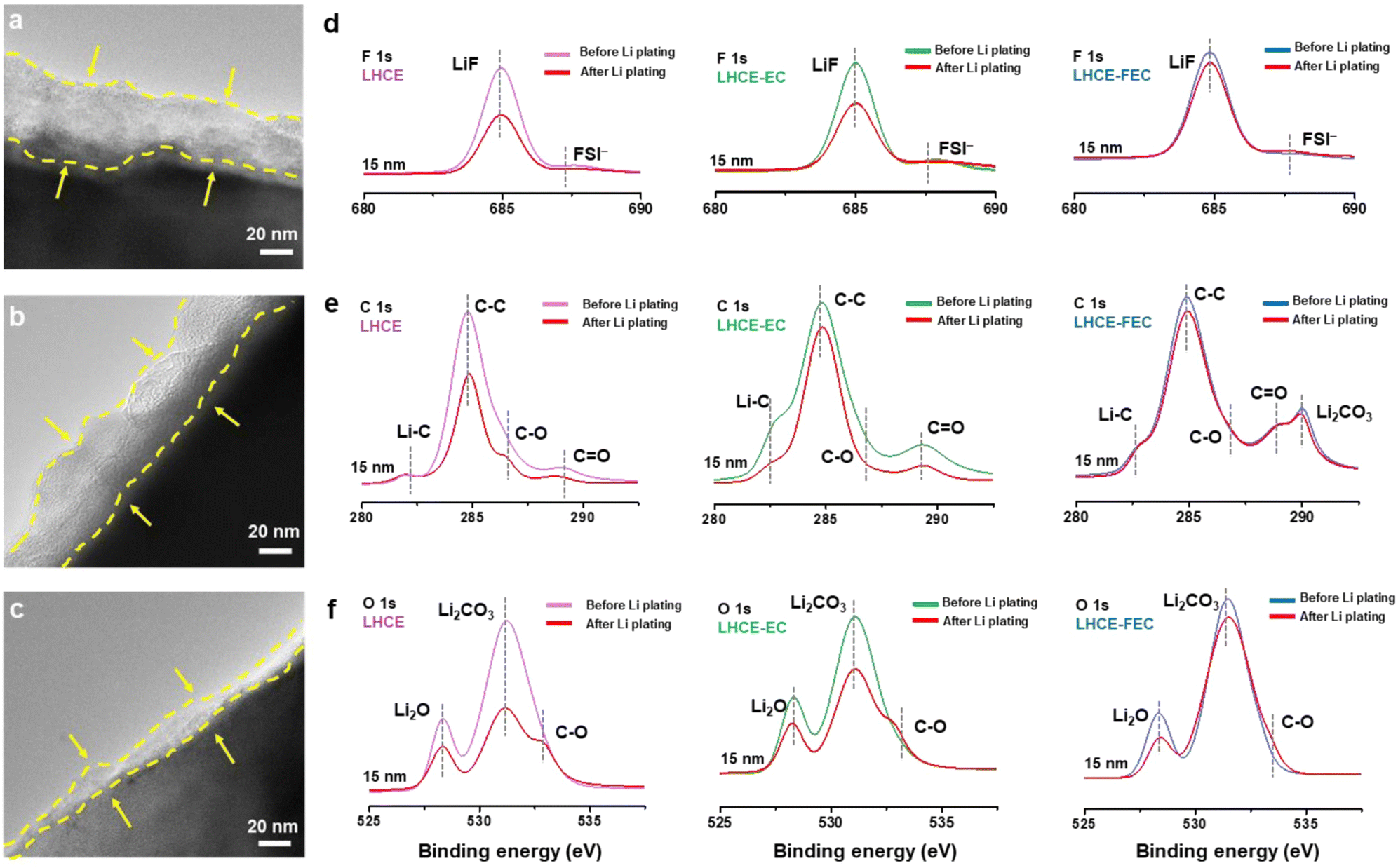 | ||
| Fig. 5 TEM images of the SEI after Li plating on the HC anode using (a) LHCE, (b) LHCE-EC, and (c) LHCE-FEC. In-depth XPS spectra of (d) F 1s, (e) C 1s, and (f) O 1s of the SEIs formed in LHCE, LHCE-EC, and LHCE-FEC before and after Li plating on the HC anode, respectively. | ||
Li+ ions normally have to overcome the energy barrier of the interphase under lithiation. The SEI containing a large number of fragments would increase the energy barrier of Li-ion diffusion. A thinner and dense SEI can improve its stability and shorten the path for Li-ion diffusion. For this reason, the internal resistance of SEI regeneration and anode kinetics before and after Li plating were subsequently determined by temperature-dependent impedance tests (Fig. S9, ESI†). The values of activation energy (Ea) were calculated based on the Arrhenius law.50 By fitting the EIS spectra in the Arrhenius plot, both charge-transfer resistance (Rct) and SEI resistance (Rf) can be quantified for cells using the three LHCEs (Fig. S10, ESI†), in which the SEI formed in LHCE-FEC exerts the lowest diffusion (46.1/34.3 kJ mol−1 for Rct,before/after) and de-solvation resistance (42.8/27.5 kJ mol−1 for Rf,before/after) before and after Li plating, allowing the fast kinetics of Li+ ions (Fig. S11, ESI†). As would be expected, the diffusion polarization and random dendrites can be greatly reduced and suppressed with the advantages of high/uniform Li-ion flux in the FEC-induced SEI.
Full cell proof-of-concept
The practicability of the studied electrolytes under fast charging conditions was evaluated by full-cell tests using HC anode and LiFePO4 cathode (LFP‖HC). The discharging rate was set to be 1C constantly (excluding C-rate <1). To verify the reversible Li plating, the ratio of negative areal capacity to positive areal capacity (N/P ratio) was set as 0.8 for intentional Li plating on the HC anode. The ICE of cells using LHCE, LHCE-EC and LHCE-FEC were tested to be 68.5%, 64.1%, and 67.8%, respectively (Fig. S12, ESI†). Charging full cells with a low N/P ratio would lead to excessive cathodic Li inventory plated on the top of the anode.32,51 Thus, three-electrode cells were assembled with Li metal chips as a reference electrode to probe Li plating behavior in full cells (Fig. S13, ESI†). At 1–3C (1C = 0.9 mA h cm−2), Li plating capacity accounts for 0.31, 0.38, and 0.41 mA h cm−2 in LHCE cells, respectively (Fig. S14, ESI†). With the existence of EC additive, the proportion of Li plating capacity drops a bit corresponding to 0.25, 0.29, and 0.36 mA h cm−2 in LHCE-EC cells, respectively. Among the three electrolytes, Li plating capacity retains the lowest values for LHCE-FEC cells of merely 0.17, 0.2, and 0.22 mA h cm−2, respectively. In addition, the internal resistance of LHCE-FEC cells remains the lowest (Fig. S15, ESI†). Fig. 6a and b present the cell cycling performance and capacity retention for various electrolytes. LHCE and LHCE-EC cells have undergone dramatic capacity decay at the early stage of cycling with capacity retention values of 81.46% and 87.74% after 500 cycles at 1C, respectively. The LHCE-FEC cells could maintain a reversible capacity of 0.68 mA h cm−2 with a high retention of 99.21% over 500 cycles. Voltage profiles of LHCE-FEC cells at the 5th and the 400th cycles further prove the stable cycling and small polarization (Fig. 6c). Cells using LHCE-FEC also have a better rate performance, showing high SOC and stable CE at 0.1–5C (Fig. 6d and Fig. S16, ESI†). The corresponding SEM images after 1C-rate cycling confirm the nearly dendrite-free HC surface using LHCE-FEC, whereas the uncontrolled Li dendrite growth can be noticed on HC anodes using LHCE and LHCE-EC (Fig. S17, ESI†). Similar trends of cycling performance have also been observed at 2 and 3C (Fig. S18, ESI†). When increasing the mass load of the HC anode to 4 mg cm−2, LHCE-FEC cells still deliver a much higher reversible capacity and retention than LHCE and LHCE-EC cells at 1–3C (Fig. S19, ESI†).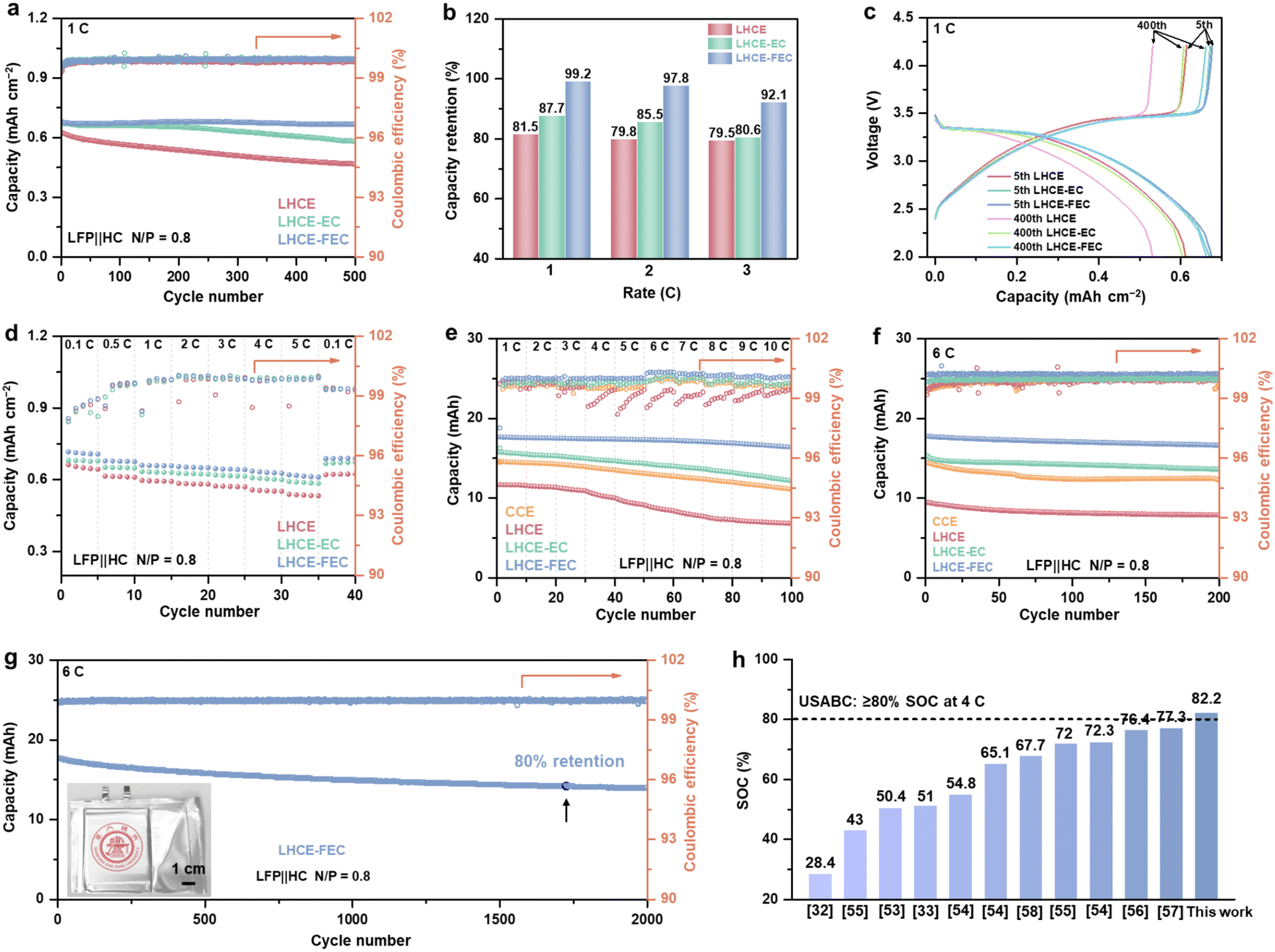 | ||
| Fig. 6 (a) Cycling performance of the LFP‖HC full cells in LHCE, LHCE-EC, and LHCE-FEC at 1C. (b) Capacity retention of full-cells at 1–3C after 500 cycles. (c) Voltage curves of the full cells in LHCE, LHCE-EC, and LHCE-FEC at the 5th and the 400th cycles. (d) Rate performance of full cells at 0.1–5C. (e) Rate and (f) cycling performance of pouch cells in LHCE, LHCE-EC, and LHCE-FEC at 1–10C and 6C, respectively. (g) Long-term cycling stability of the pouch cell using LHCE-FEC at 6C. (h) Comparison of the fast-charging capability of LHCE-FEC pouch cells with reported carbonaceous anodes and electrolytes at 6C. 1C = 0.9 mA h cm−2 (coin-type full cell). 1C = 20 mA h (pouch cell). Voltage range: 2.0–4.2 V. | ||
To validate the availability of the HC anode under extreme fast-charging operations using the studied LHCEs, the self-made 20-mA h LFP‖HC pouch cells with the same N/P ratio (0.8) were assembled using two single-layer electrodes in the dimensions of 4.1 × 5.1 cm (Fig. S20, ESI†). Detailed parameters of pouch cells are presented in Table S3 (ESI†). A constant–current constant–voltage (CCCV) charging mode was selected for pouch cell tests and limited by a cut-off current density of 0.05 C (1C = 20 mA h). The SOC of pouch cells was obtained based on the ratio of the capacity of the CC stage to the cell discharging capacity at 0.2C (Fig. S21, ESI†). Fig. 6e shows that LHCE-FEC cells can maintain a high-rate stability of up to 10C and can be charged from 90.1% to 71.2% SOC at 1–10C before the CV process. In the case of the LHCE-EC cell, the capacity deterioration occurs at a rate of 4C as well as rapid capacity fading when the charging C-rate is further increased (only from 78.5% to 50.2% SOC at 1–10C). Pouch cells with the LHCE exhibit the worst rate performance, which is inferior to those of conventional concentration electrolytes (CCE, 1 M LiPF6 in EC/DMC 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume). The high proportion of CC stage by voltage curves suggests that the lower polarization of LHCE-FEC cells is primarily attributed to the uniform Li plating and fast diffusion kinetics on the reacting interphase (Fig. S22, ESI†). Under extreme cycling at 6C, LHCE-FEC cells could also deliver a much higher SOC (82.2%) at the CC stage and cycling stability than LHCE and LHCE-EC cells with a capacity retention of 93.7% after 200 cycles (Fig. 6f). The high SOC of 82.2% exceeds the United States Advanced Battery Consortium (USABC) metrics for high-performance batteries (SOC ≥ 80% under 4C-rate).52 In addition, the long-term cycling stability (over 1700 cycles) is maintained when capacity retention reaches 80%, and the voltage evolution of the LHCE-FEC cell from the 1st to the 2000th cycles imply a lower cell polarization under 6-C cycling (Fig. 6g and Fig. S23, ESI†). The SOC corresponding to the CC stage for pouch cells was also compared with those carbonaceous anodes and electrolytes reported in the literature under practical 6-C cycling (Fig. 6h and Table S4, ESI†), exhibiting the greater CC charge acceptance of pouch cells using LHCE-FEC.53–58 Additionally, the 200-mA h laminated double-side pouch cells were prepared to examine its fast-charging performance (Fig. S24, ESI†). The SOC of 80.6% from the CC stage still can be achieved with capacity retention of 92.1% over 200 cycles under 6-C cycling (Fig. S25, ESI†).
:1 by volume). The high proportion of CC stage by voltage curves suggests that the lower polarization of LHCE-FEC cells is primarily attributed to the uniform Li plating and fast diffusion kinetics on the reacting interphase (Fig. S22, ESI†). Under extreme cycling at 6C, LHCE-FEC cells could also deliver a much higher SOC (82.2%) at the CC stage and cycling stability than LHCE and LHCE-EC cells with a capacity retention of 93.7% after 200 cycles (Fig. 6f). The high SOC of 82.2% exceeds the United States Advanced Battery Consortium (USABC) metrics for high-performance batteries (SOC ≥ 80% under 4C-rate).52 In addition, the long-term cycling stability (over 1700 cycles) is maintained when capacity retention reaches 80%, and the voltage evolution of the LHCE-FEC cell from the 1st to the 2000th cycles imply a lower cell polarization under 6-C cycling (Fig. 6g and Fig. S23, ESI†). The SOC corresponding to the CC stage for pouch cells was also compared with those carbonaceous anodes and electrolytes reported in the literature under practical 6-C cycling (Fig. 6h and Table S4, ESI†), exhibiting the greater CC charge acceptance of pouch cells using LHCE-FEC.53–58 Additionally, the 200-mA h laminated double-side pouch cells were prepared to examine its fast-charging performance (Fig. S24, ESI†). The SOC of 80.6% from the CC stage still can be achieved with capacity retention of 92.1% over 200 cycles under 6-C cycling (Fig. S25, ESI†).
Until now, the unsafe Li plating encountered in anode has long been considered as the major hurdle for fast-charging LIBs. A single-step kinetic modification can only eliminate Li plating at a specific condition while losing hold of the other. Appropriately utilizing reversible Li plating capacity on the HC anode has shown to be a promising approach for fast-charging LIBs. By constructing an SEI with high Li-ion flux at the anode interface using LHCE-FEC, the Li plating morphology was actively regulated on the anode surface. The safety risk caused by dendrite formation was minimized to a great extent, and meanwhile, the high SOC could be maintained for anodes. On the other hand, using the HC anode allows it to completely exert its high-rate superiority while mitigating its deficiency, namely low ICE, rendering HC a potential candidate for a fast-charging anode. With the studied SEI of high compatibility at the anode/electrolyte interface, the Li plating reversibility of the LHCE system and the high-rate capability of the HC for fast-charging LIBs can be integrated to their best advantage.
Conclusions
In this work, the adsorption tendency of EC and FEC and their functions for the SEI structure on the HC surface were elucidated in a typical LHCE system. DFT calculations unveil that the FEC molecule can maintain the tilted conformation that results in weaker adsorption between the CO of FEC and the defect site in the presence of abundant FSI− anions and Li+ ions. The inorganic products of FEC are distributed uniformly within the LHCE-FEC derived SEI, compared to the unevenly aggregated inorganic clumps of the SEI formed in LHCE-EC, irrespective of the defect distribution on HC. Accordingly, the FEC-assisted SEI not only provides high Li-ion flux of the interphase and uniform Li nucleation sites but also maintains its self-structural stability over cycles. Cells using LHCE-FEC could maintain an excellent average CE of 99.8% over 250 cycles. Owing to the robust SEI, the uniform Li plating morphology with high reversibility was achieved during repeated Li plating/stripping on the HC surface. The self-exacerbated dendrite/dead Li formation can be well restricted. Meanwhile, diffusion polarization was also greatly reduced before and after Li plating, allowing the fast kinetics of Li+ ions. As a consequence, the self-made LFP‖HC pouch cell with a low N/P ratio exerts a high SOC (82.2%) and maintains a cycling stability of 6C for over 1700 cycles until reaching 80% retention, which further confirms the applicability of the reversible Li plating strategy under extreme fast charging conditions. This work has demonstrated a new approach to address Li plating issues by understanding the adsorption tendency of EC and FEC on the carbon matrix. More selections for film-forming agents to achieve the reversible Li plating for fast-charging LIBs need to be further explored.
Author contributions
Y. D.: conceptualization, investigation, methodology, data analysis, resources, and writing – original draft. Y. C.: investigation, visualization, resources, and methodology. X. Y.: conceptualization, methodology, and supervision. Z. L.: conceptualization, supervision, and project administration. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) under Grant No. 22379093, the Fundamental Research Funds for the Central Universities (22X010201631, 23X010301599), and CATL Future Energy Research Institute (22H010102023).References
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. Xie and Y. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS PubMed.
- P. Nambisan, P. Saha and M. Khanra, J. Energy Storage, 2021, 41, 102918 CrossRef.
- A. Burnham, E. J. Dufek, T. Stephens, J. Francfort, C. Michelbacher, R. B. Carlson, J. Zhang, R. Vijayagopal, F. Dias, M. Mohanpurkar, D. Scoffield, K. Hardy, M. Shirk, R. Hovsapian, S. Ahmed, I. Bloom, A. N. Jansen, M. Keyser, C. Kreuzer, A. Markel, A. Meintz, A. Pesaran and T. R. Tanim, J. Power Sources, 2017, 367, 237 CrossRef CAS.
- W. Cai, Y. Yao, G. Zhu, C. Yan, L. Jiang, C. He, J. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806 RSC.
- E. Logan and J. Dahn, Trends Chem., 2020, 2, 354 CrossRef CAS.
- M. Colclasure, A. R. Dunlop, S. E. Trask, B. J. Polzin, A. N. Jansen and K. Smith, J. Electrochem. Soc., 2019, 166, A1412 CrossRef.
- R. Tian, S. H. Park, P. J. King, G. Cunningham, J. Coelho, V. Nicolosi and J. N. Coleman, Nat. Commun., 2019, 10, 1933 CrossRef PubMed.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926 RSC.
- A. S. Mussa, A. Liivat, F. Marzano, M. Klett, B. Philippe, C. Tengstedt, G. Lindbergh, K. Edström, R. W. Lindström and P. Svens, J. Power Sources, 2019, 422, 175 CrossRef CAS.
- Y. Liu, H. Shi and Z. Wu, Energy Environ. Sci., 2023, 16, 4834 RSC.
- G. Sikha, B. N. Popov and R. E. White, J. Electrochem. Soc., 2004, 151, A1104 CrossRef CAS.
- N. Ogihara, Y. Itou, T. Sasaki and Y. Takeuchi, J. Phys. Chem. C, 2015, 119, 4612 CrossRef CAS.
- T. Waldmann, M. Wilka, M. Kasper, M. Fleischhammer and M. Wohlfahrt-Mehrens, J. Power Sources, 2014, 262, 129 CrossRef CAS.
- S. J. Zhang, Y. M. Zhang, Z. Y. Zhang, H. L. Wang, Y. Cao, B. S. Zhang, X. Y. Liu, C. Mao, X. P. Han, H. C. Gong, Z. X. Yang and J. Sun, Adv. Energy Mater., 2022, 12, 2103888 CrossRef CAS.
- A. Tomaszewska, Z. Y. Chu, X. N. Feng, S. O'Kane, X. H. Liu, J. Y. Chen, C. Z. Ji, E. Endler, R. H. Li, L. S. Liu, Y. L. Li, S. Q. Zheng, S. Vetterlein, M. Gao, J. Y. Du, M. Parkes, M. G. Ouyang, M. Marinescu, G. Offer and B. Wu, eTransportation, 2019, 1, 100011 CrossRef.
- X. B. Han, L. G. Lu, Y. J. Zheng, X. N. Feng, Z. Li, J. Q. Li and M. G. Ouyang, eTransportation, 2019, 1, 100005 CrossRef.
- C. Bommier, W. Chang, Y. F. Lu, J. Yeung, G. Davies, R. Mohr, M. Williams and D. Steingart, Cell Rep. Phys. Sci., 2020, 1, 100035 CrossRef.
- A. Vasileiadis, N. J. J. de Klerk, R. B. Smith, S. Ganapathy, P. Harks, M. Z. Bazant and M. Wagemaker, Adv. Funct. Mater., 2018, 28, 1 CrossRef.
- X. D. Zheng, C. C. Dong, B. Huang and M. Lu, Ionics, 2013, 19, 385 CrossRef CAS.
- L. C. Qiu, Q. C. Wang, X. Y. Yue, Q. Q. Qiu, X. L. Li, D. Chen, X. J. Wu and Y. N. Zhou, Electrochem. Commun., 2020, 112, 106684 CrossRef CAS.
- Y. X. Lin, Z. H. Huang, X. L. Yu, W. C. Shen, Y. P. Zheng and F. Y. Kang, Electrochim. Acta, 2014, 116, 170 CrossRef CAS.
- D. K. Son, J. Kim, M. R. Raj and G. Lee, Carbon, 2021, 175, 187 CrossRef CAS.
- Y. J. Oh, J. H. Park, J. S. Park, S. S. Kim, S. J. Hong, Y. W. Na, J. H. Kim, S. Nam and S. J. Yang, Energy Storage Mater., 2022, 44, 416 CrossRef.
- D. S. Kim, Y. E. Kim and H. Kim, J. Power Sources, 2019, 422, 18 CrossRef CAS.
- M. Shi, Z. G. Tai, N. Li, K. Y. Zou, Y. Z. Chen, J. J. Sun and Y. N. Liu, Sustain, Energy Fuels, 2019, 3, 3116 CAS.
- M. M. Wang, J. R. Wang, J. C. Xiao, N. Q. Ren, B. C. Pan, C. S. Chen and C. H. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 16279 CrossRef CAS PubMed.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160 CrossRef CAS.
- M. Okoshi, Y. Yamada, S. Komaba, A. Yamada and H. Nakai, J. Electrochem. Soc., 2017, 164, A54 CrossRef CAS.
- L. F. Zhang, L. L. Chai, L. Zhang, M. Shen, X. L. Zhang, V. S. Battaglia, T. Stephenson and H. H. Zheng, Electrochim. Acta, 2014, 127, 39 CrossRef CAS.
- Z. J. Li, J. Liu, Y. A. Qin and T. Gao, ACS Appl. Mater. Interfaces, 2022, 14, 33004 CrossRef CAS PubMed.
- X. Y. Yue, J. Zhang, Y. T. Dong, Y. M. Chen, Z. Q. Shi, X. J. Xu, X. L. Li and Z. Liang, Angew. Chem., Int. Ed., 2023, 62, e202302285 CrossRef CAS PubMed.
- X. J. Xu, X. Y. Yue, Y. M. Chen and Z. Liang, Angew. Chem., Int. Ed., 2023, 62, e202306963 CrossRef CAS PubMed.
- L. J. Xie, C. Tang, Z. H. Bi, M. X. Song, Y. F. Fan, C. Yan, X. M. Li, F. Y. Su, Q. Zhang and C. M. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef CAS.
- H. X. Gong, Y. L. Chen, S. C. Chen, C. Y. Xu, Y. F. Yang, Y. S. Ye, Z. J. Huang, R. Ning, Y. Cui and Z. N. Bao, ACS Energy Lett., 2022, 7, 4417 CrossRef CAS.
- L. L. Jiang, C. Yan, Y. X. Yao, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3402 CrossRef CAS PubMed.
- W. L. Cai, C. Yan, Y. X. Yao, L. Xu, X. R. Chen, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13007 CrossRef CAS PubMed.
- H. Jia, Y. B. Xu, S. D. Burton, P. Y. Gao, X. H. Zhang, B. E. Matthews, M. H. Engelhard, L. R. Zhong, M. E. Bowden, B. W. Xiao, K. S. Han, C. M. Wang and W. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 54893 CrossRef CAS PubMed.
- X. Cao, Y. B. Xu, L. C. Zhang, M. H. Engelhard, L. R. Zhong, X. D. Ren, H. P. Jia, B. Liu, C. J. Niu, B. E. Matthews, H. P. Wu, B. W. Arey, C. M. Wang, J. G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 2529 CrossRef CAS.
- J. Sieg, J. Bandlow, T. Mitsch, D. Dragicevic, T. Materna, B. Spier, H. Witzenhausen, M. Ecker and D. U. Sauer, J. Power Sources, 2019, 427, 260 CrossRef CAS.
- H. Jia, J. M. Kim, P. Y. Gao, Y. B. Xu, M. H. Engelhard, B. E. Matthews, C. M. Wang and W. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218005 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky, A. Ulus and V. Yufit, Electrochim. Acta, 2004, 50, 391 CrossRef CAS.
- L. J. Zhou, Z. F. Hou and L. M. Wu, J. Phys. Chem. C, 2012, 116, 21780 CrossRef CAS.
- X. F. Fan, W. T. Zheng and J. L. Kuo, ACS Appl. Mater. Interfaces, 2012, 4, 2432 CrossRef CAS PubMed.
- Y. Okamoto, J. Phys. Chem. C, 2016, 120, 14009 CrossRef CAS.
- H. Y. Zhu, J. A. Russell, Z. T. Fang, P. Barnes, L. Li, C. M. Efaw, A. Muenzer, J. May, K. Hamal, I. F. Cheng, P. H. Davis, E. J. Dufek and H. Xiong, Small, 2021, 17, 2105292 CrossRef CAS PubMed.
- L. Mandeltort and J. T. Yates, J. Phys. Chem. C, 2012, 116, 24962 CrossRef CAS.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, u9245 CrossRef PubMed.
- D. C. Lin, J. Zhao, J. Sun, H. B. Yao, Y. Y. Liu, K. Yan and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4613 CrossRef CAS PubMed.
- P. W. Atkins, Physical Chemistry, Oxford University Press, New York, 1990 Search PubMed.
- C. Martin, M. Genovese, A. J. Louli, R. Weber and J. R. Dahn, Joule, 2020, 4, 1296 CrossRef CAS.
- A. Tomaszewska, Z. Chu, X. Feng, S. O’Kane, X. Liu, J. Chen, C. Ji, E. Endler, R. Li and L. Liu, eTransportation, 2019, 1, 100011 CrossRef.
- Y. X. Yao, X. Chen, N. Yao, J. H. Gao, G. Xu, J. F. Ding, C. L. Song, W. L. Cai, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202214828 CrossRef CAS PubMed.
- K. H. Chen, V. Goel, M. J. Namkoong, M. Wied, S. Müller, V. Wood, J. Sakamoto, K. Thornton and N. P. Dasgupta, Adv. Energy Mater., 2021, 11, 2003336 CrossRef CAS.
- S. Tu, B. Zhang, Y. Zhang, Z. Chen, X. Wang, R. Zhan, Y. Ou, W. Wang, X. Liu, X. Duan, L. Wang and Y. Sun, Nat. Energy, 2023, 8, 1365 CrossRef CAS.
- K. H. Chen, M. J. Namkoong, V. Goel, C. Yang, S. Kazemiabnavi, S. M. Mortuza, E. Kazyak, J. Mazumder, K. Thornton, J. Sakamoto and N. P. Dasgupta, J. Power Sources, 2020, 471, 228475 CrossRef CAS.
- T. R. Tanim, Z. Yang, A. M. Colclasure, P. R. Chinnam, P. Gasper, Y. Lin, L. Yu, P. J. Weddle, J. Wen, E. J. Dufek, I. Bloom, K. Smith, C. C. Dickerson, M. C. Evans, Y. Tsai, A. R. Dunlop, S. E. Trask, B. J. Polzin and A. N. Jansen, Energy Storage Mater., 2021, 41, 656 CrossRef.
- N. Gao, S. Kim, P. Chinnam, E. J. Dufek, A. M. Colclasure, A. Jansen, S. B. Son, I. Bloom, A. Dunlop, S. Trask and K. L. Gering, Energy Storage Mater., 2022, 44, 296 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization, calculation details, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d4ee00119b |
| This journal is © The Royal Society of Chemistry 2024 |