
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borylation and rearrangement reactions of azasilaanthracenes to afford B,N-doped nanographenes†
Elena
Zender
a,
Danillo
Valverde
 b,
Robert
Neubaur
a,
Sebastian
Karger
a,
Alexander
Virovets
a,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Yoann
Olivier
*b and
Matthias
Wagner
*a
b,
Robert
Neubaur
a,
Sebastian
Karger
a,
Alexander
Virovets
a,
Michael
Bolte
a,
Hans-Wolfram
Lerner
a,
Yoann
Olivier
*b and
Matthias
Wagner
*a
aInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt (Main), Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bLaboratory for Computational Modeling of Functional Materials, Namur Institute of Structured Matter, University of Namur, Rue de Bruxelles, 61, 5000 Namur, Belgium. E-mail: yoann.olivier@unamur.be
First published on 8th May 2024
Abstract
An air-stable B3,N3-containing dibenzobisanthene (8) was prepared in 29% yield by heating a 1,3,5-tri(azasilaanthryl)benzene (5) with BBr3 (180 °C). Under these conditions, the reaction does not stop after threefold SiMe2/BBr exchange but proceeds further via two rearrangement and two intramolecular C–H borylation steps. Some mechanistic details were unveiled by using smaller model systems and applying lower reaction temperatures. According to X-ray crystallography, compound 8 has a helically distorted scaffold. Due to its multiple resonance structure, it shows a narrow-band blue-green emission (λem = 493 nm; ΦPL = 84%; FWHM = 0.20 eV; THF); samples measured in PMMA gave prompt and delayed fluorescence lifetimes of 10.7 ns and 136 μs, respectively. The optical properties of 8 and of structurally related species were also investigated by quantum-chemical means: most of these compounds exhibit a small energy gap ΔEST between the lowest excited singlet (S1) and triplet (T1) states and a non-negligible spin–orbit coupling (SOC) between S1 and T1/T2, demonstrating their potential as thermally activated delayed fluorescence (TADF) emitters.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) containing π-electron-accepting B atoms and π-electron-donating N atoms in mutual 1,4-positions find increasing applications as OLED materials.1 The main reason is that such heteroatom doping patterns can lead to a decoupling of the PAH's frontier orbitals, which, in turn, results in a small energy gap (ΔEST) between the lowest excited singlet and triplet states, facilitating triplet-to-singlet upconversion. As a consequence, corresponding Bn,Nm-PAHs are ideal candidates for use as MR-TADF emitters (MR-TADF: multiple resonance thermally activated delayed fluorescence). In theory, MR-TADF compounds can utilize all electrogenerated excitons in an OLED for luminescence, whereas conventional fluorescence emitters are only driven by the 25% of singlet excitons.2–8 In terms of synthesis, many representatives of MR-TADF Bn,Nm-PAHs are notable for their convenient accessibility from suitable aryl amines through electrophilic aromatic borylation with BX3 (X = Br, I).3,9–12 We recently contributed to this field the B3,N2-PAH C (Scheme 1), which is one of the few examples that contain more B than N atoms and thus have a formally electron-deficient π system.13 Remarkably, in this case the established threefold electrophilic C–H borylation failed for the introduction of the central B atom. Therefore, we had to use the halogenated starting materials A (X = Cl) or B (X = I) and form the first of the three B–C bonds (marked in red) via a lithiation/borylation approach.2,13–17 Herein, we show that no C is formed in the reaction of parent 1 (X = H) with BBr3, because a skeletal rearrangement to the ladder-type azaborine12Br takes place instead (Scheme 2). We provide insight into some mechanistic details of this rearrangement and expand the scope of the reaction from the 1,3-disubstituted precursor 1 to its 1,3,5-trisubstituted congener 5 (Scheme 3).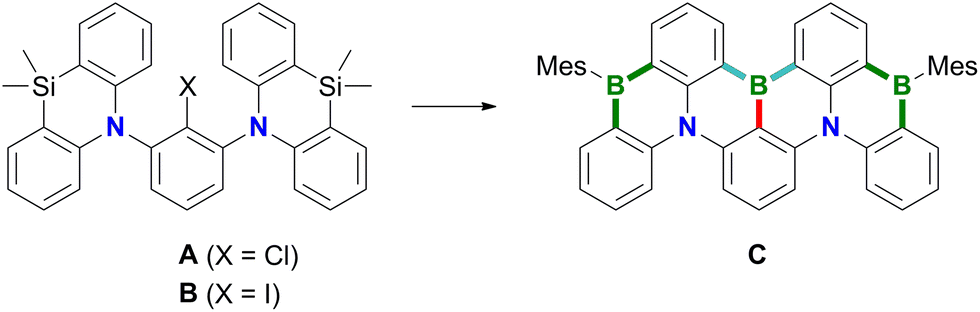 | ||
| Scheme 1 The B3,N2-PAH C was synthesized from A or Bvia a sequence of lithiation/borylation (red bond), electrophilic aromatic borylation (blue bonds), and Si/B-exchange reactions (green bonds). | ||
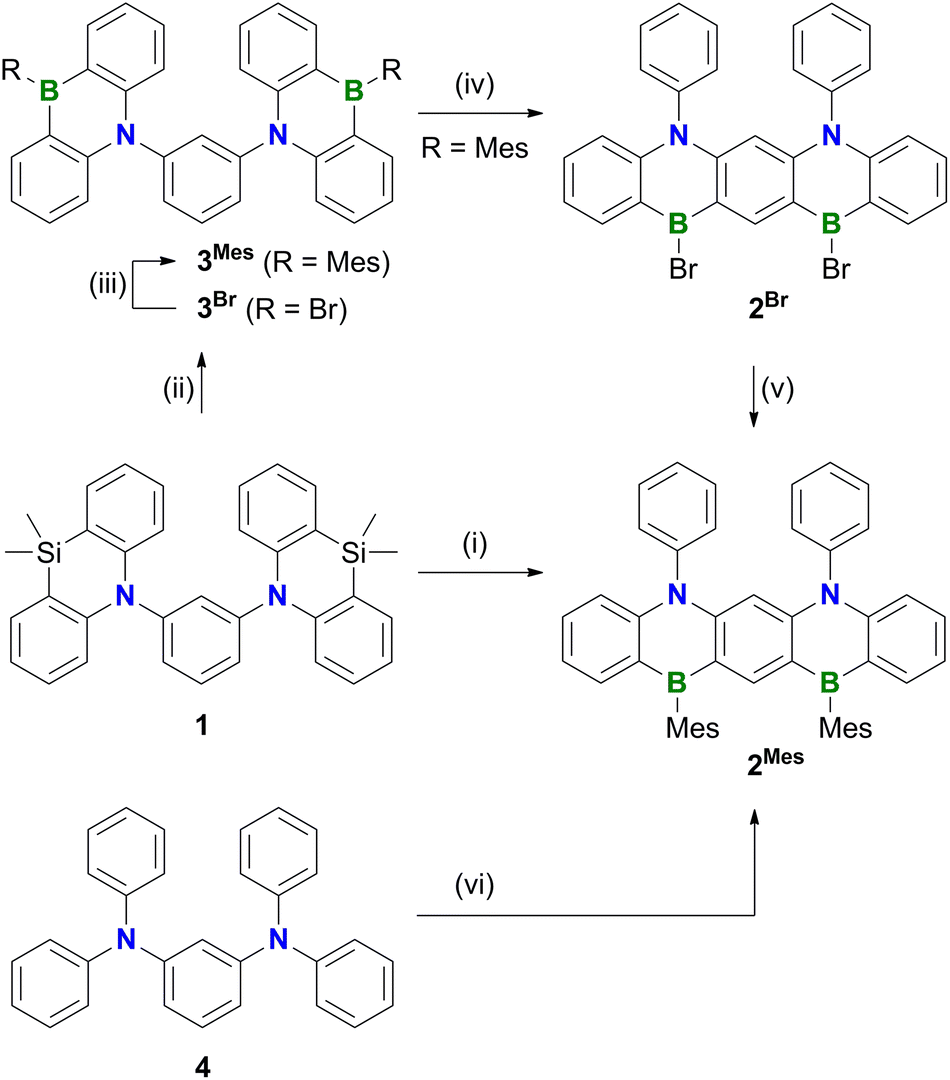 | ||
| Scheme 2 Synthetic pathways to 2Mes. Conditions: (i) 1. BBr3 (12 equiv.), C6H6, 180 °C, 116 h, sealed ampoule; 2. MesMgBr (3 equiv.), toluene, −78 °C to room temperature, 39% over two steps; (ii) BBr3 (5 equiv.), CH2Cl2, room temperature, 1 d, closed J. Young vessel, product was used after solvent exchange without further purification; (iii) MesMgBr (3 equiv. relative to 1), toluene, −78 °C to room temperature, 42% over two steps; (iv) BBr3 (12 equiv.), C6H6, 180 °C, 92 h, sealed ampoule, product was used after solvent exchange without further purification; (v) MesMgBr (3 equiv. relative to 3Mes), toluene, −78 °C to room temperature, 44% over two steps; (vi) 1. BBr3 (12 equiv.), o-dichlorobenzene, 180 °C, 112 h, sealed ampoule; 2. MesMgBr (3 equiv.), toluene, −78 °C to room temperature, 58% over two steps. | ||
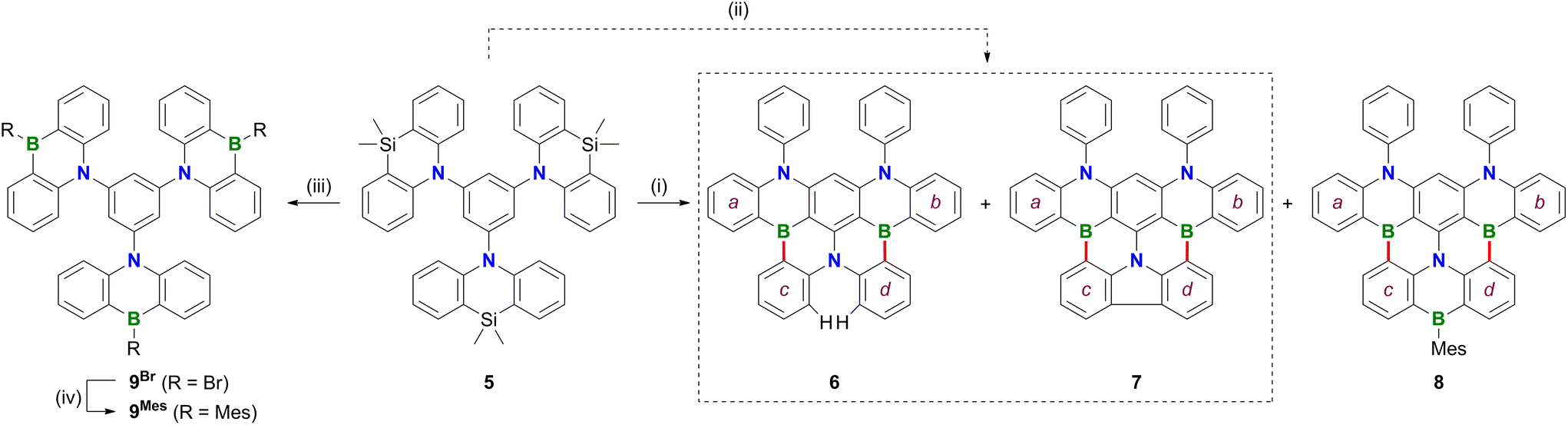 | ||
| Scheme 3 Borylation and rearrangement reactions of 5. Conditions: (i) 1. BBr3 (28 equiv.), C6H6, 180 °C, 7 d, sealed ampoule; 2. MesMgBr (6 equiv.), toluene, −78 °C to room temperature, the product mixture contained 6–8, but only 8 could be isolated in 29% yield over two steps; (ii) 1. BBr3 (30 equiv.), C6H6, 180 °C, 11 d, sealed ampoule; 2. iPrOH, CH2Cl2, 6% (6), 9% (7); (iii) BBr3 (neat), room temperature, 1 d, closed J. Young vessel, product was used after solvent exchange without further purification; (iv) MesMgBr (5 equiv. relative to 5), toluene, −78 °C to room temperature, 56% over two steps. | ||
Results and discussion
Treatment of 1 with excess BBr3 in C6H6 (12 equiv., 180 °C, 116 h) results in the formation of a characteristically red precipitate. Mesitylation of this primary product with MesMgBr affords 2Mes in 39% overall yield (Mes: mesityl; Scheme 2). Compound 2Mes was characterized by NMR spectroscopy, X-ray crystallography (Fig. S61†), and optoelectronic measurements (see below). Closely related B2,N2-pentacenes containing N–CH2Ph or N–Me and aryl–nBu moieties are known, but have been prepared from tetrabrominated m-phenylene diamines.18–20 To gain insight into the mechanism underlying the formation of 2Mes, 1 was stirred with BBr3 (5 equiv.) in CH2Cl2 at room temperature; subsequent mesitylation gave 3Mes in 42% yield. This suggests that its B–Br precursor 3Br is likely present to a significant extent also during the synthesis of 2Mes before the red B2,N2-pentacene 2Br precipitates. Consistent with the postulated role of 3Br as a key intermediate, 2Br is also accessible from 3Mes and BBr3via an initial demesitylation21 step (Fig. S1†). Overall, our experimental observations agree with quantum-chemical calculations (SMD(C6H6)/ωB97XD/def2-TZVP//SMD(C6H6)/ωB97XD/def2-SVP), which predict that 3Br is less stable by 4.3 kcal mol−1 than its ladder-type isomer 2Br (see the ESI† for more details). We finally emphasize that if the focus is not on the reactivity of 1 but on the preparation of 2Mes, it is advisable to subject the readily available diamine 4 to C–H borylation with BBr3, as this protocol gives a yield of 58%.In an attempt to prepare larger Bn,Nm-doped PAHs, the protocol of the reaction 1 → 2Mes was subsequently adapted to tri(azasilaanthryl)benzene 5 (Scheme 3). Again, the borylation step in C6H6 led to the precipitation of a red solid, which was isolated and mesitylated. Besides 2Mes, which stems from unwanted C–N-bond cleavage,22 the resulting product mixture included 6, 7, and 8 (the latter was isolated in 29% yield). To suppress the formation of 8 and 2Mes and to obtain pure 6 (6%) and 7 (9%), we found it advantageous to perform an iPrOH quench instead of the mesitylation step. All three compounds 6–8 contain a B2,N2-pentacene fragment analogous to 2Mes, which presumably originates again from a rearrangement of two azasilaanthryl substituents. The third Ar2N moiety is incorporated into the main π system through twofold intramolecular borylation (red bonds in Scheme 3). In the case of 8, the third SiMe2/BBr exchange was also successful, and the resulting bromoborane persisted until the kinetically stabilizing Mes substituent was introduced. To rationalize the formation of 6 and 7, the following points need to be considered: (i) azasilaanthracenes in general and 5 in particular can be handled under ambient atmosphere without decomposition. It is therefore unlikely that 6 and 7 were generated by protodesilylation and oxidative SiMe2 extrusion, respectively. (ii) Similar to the 1 → 3Mes conversion, treatment of 5 with BBr3 at room temperature, followed by mesitylation, gave 9Mes in 56% yield (Scheme 3). (iii) A 1H NMR spectrum recorded on the above-mentioned red precipitate after removal of all volatiles showed no signals assignable to residual SiMe groups (CD2Cl2; Fig. S7†). (iv) Moreover, the spectrum did not contain the characteristic proton resonances of 6 and 7 (Fig. S7†), indicating that 6 and 7 are only produced during mesitylation/iPrOH quenching. This process is presumably promoted by the formation of tetracoordinated boron intermediates,23 which undergo either protodeborylation or reductive elimination at the B(III) center.
In contrast to 2Mes, which is synthesized most efficiently through C–H borylation of the diaminobenzene 4, 6 and 8 are not accessible starting from the corresponding triaminobenzene 1,3,5-(Ph2N)3C6H3 and BBr3 at temperatures below 200 °C. Instead, a literature-known B,N3-PAH is generated, which is formally derived from 6 by replacing one C3B fragment with three C–H moieties (see the ESI† for more details).24
The NMR spectra of 6–8 show the following characteristic features: (i) a doublet at δ(1H) = 8.31 (2H), assignable to the two extra protons of 6, (ii) a sharp 13C resonance at 125.4 ppm for the quaternary atoms constituting the central C–C bond of the carbazole moiety in 7, and (iii) broadened MesBC2 signals and Mes resonances in the case of 8.
The proposed molecular structures of 6 (Fig. S63†), 7 (Fig. S64†), and 8 (Fig. 1) were confirmed by X-ray crystallography (see the ESI† for the solid-state structures of 1, 2Br, 5, and 9Mes).25 The pentacene fragments of 6–8 show only small twists with dihedral angles between the two outer 1,2-phenylene rings in the range Ar(a)//Ar(b) = 4.10(5)–13.98(4)° (see Scheme 3 for the ring labeling). Compound 6 features three [4]helicene substructures (2 × N2,B- and 1 × B2,N-doped) that are linked through the central benzene ring. A large dihedral angle Ar(c)//Ar(d) of 48.97(7)° is caused by steric repulsion of the two extra H atoms in the cove26 region. Replacement of these H atoms with a C–C bond in 7 renders Ar(c) and Ar(d) essentially coplanar. Concomitantly, the substantial dihedral angles Ar(a)//Ar(c) and Ar(b)//Ar(d) of avg. 29.41° in 6 are reduced to values of 3.43(5)–17.99(4)° in 7. Once Ar(c) and Ar(d) are not connected by a direct C–C bond but by a BMes bridge, the reduced contraction and increased conformational flexibility in the newly formed BNC4 ring restores the helicity in 8 with angles Ar(a)//Ar(c) and Ar(b)//Ar(d) of avg. 29.01° and Ar(c)//Ar(d) of 23.57(15)°. In the crystal lattice, the molecules of 8 are pairwise connected by π–π-stacking interactions with an off-set geometry (Fig. 1c). The intradimer distance between the B(3) atom and the C6H4 plane C(51)-to-C(56) is 3.403(4) Å (see pink dashed line in Fig. 1c). The (8)2 dimers are arranged into infinite one-dimensional arrays with shortest interdimer contacts of 3.610(3) Å (Fig. S67†).
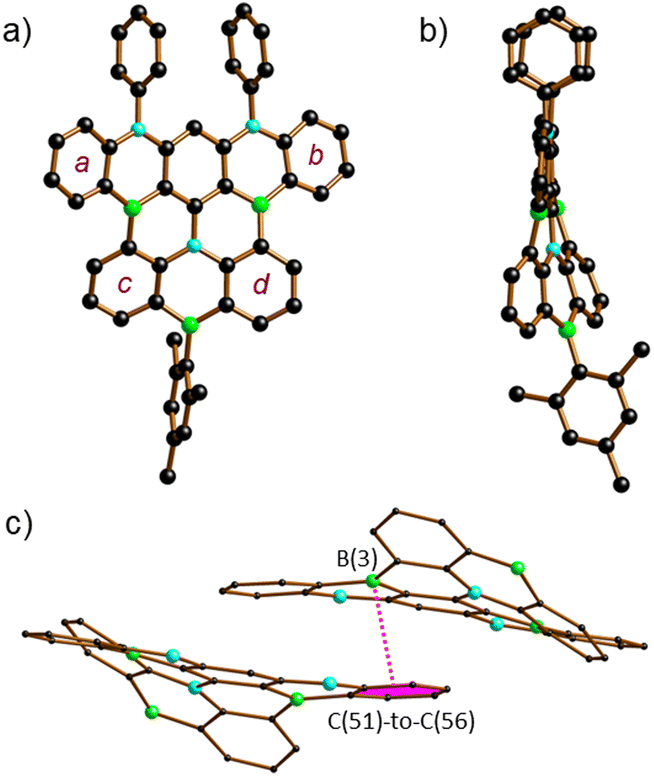 | ||
| Fig. 1 Molecular structure of 8 in the solid state; (a) front view, (b) side view, and (c) section of the crystal lattice showing the pairwise π–π-stacking. H atoms are omitted for clarity; in (c), the Mes and Ph substituents are also omitted. B: green, C: black, N: cyan. | ||
Of the molecules 6–8, only 8 represents a new structural motif, while derivatives of 6 and 7 with alkyl and/or aryl substituents have already been reported.22,27,28 However, the series of maximally comparable compounds 6–8 now makes it possible to study the effects of intramolecular linking (by a C–C bond or a BMes bridge) on the optoelectronic properties of the core structure 6 without the result being influenced by subtle effects of peripheral groups.
Cyclic voltammograms of 2Mes and 6–8 showed a reversible first (least cathodic) reduction wave in all four cases (THF, [nBu4N][PF6], vs. FcH/FcH+; Fig. S53–S57†). The B2,N2-PAH 2Mes is the worst electron acceptor with E1/2 = −2.60 V (Table 1). The B2,N3-PAH 6 is easier to reduce by 40 mV. Here, the higher degree of delocalization seems to outcompete the presence of a formally π-electron-rich third N atom. The increased planarity of 7 due to the presence of a direct C–C bond further improves the electron-accepting ability by 210 mV. Finally, the presence of a third B(sp2) center in the B3,N3-PAH 8 facilitates reduction to the largest extent (E1/2 = −2.19 V). The general trend found for the experimentally determined redox potentials is consistent with the trend in the calculated LUMO-energy levels (Table S7†).
E
1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [V] a [V] |
λ
absb [nm] |
λ em [nm] |
Φ
PLc [%] |
FWHMd [eV] |
|
|---|---|---|---|---|---|
| a THF, [nBu4N][PF6], vs. FcH/FcH+. b Only the most bathochromically shifted absorption band is given. c Quantum yields were determined by using a calibrated integrating sphere. d Full-width at half-maximum. | |||||
| 6 | −2.56 | 424 | 447 | 18 | 0.22 |
| 7 | −2.35 | 456 | 479 | 84 | 0.19 |
| 8 | −2.19 | 461 | 493 | 84 | 0.20 |
| 9Mes | −2.72 | 388 | 398 | 96 | 0.23 |
| 2Mes | −2.60 | 419 | 440 | 66 | 0.18 |
Absorption and emission spectra were recorded in THF. Compound 6 has its longest-wavelength absorption maximum and its emission maximum at λabs = 424 nm and λem = 447 nm (Fig. 2 top, Table 1). The presence of the C–C bond in 7 results in pronounced bathochromic shifts of Δλabs = 0.20 eV and Δλem = 0.18 eV. In comparison, the effect of an introduced BMes bridge (8vs. 6) is even larger with band shifts of Δλabs = 0.23 eV and Δλem = 0.26 eV. While 6 shows a moderate photoluminescence quantum yield of ΦPL = 18%, 7 and 8 have large ΦPL values of 84%. We attribute the lower ΦPL of 6 to two possible reasons: (i) the larger singlet–triplet (S1–T1) spin–orbit coupling (SOC) of 6 compared to 7 and 8 (see below) could lead to a faster intersystem crossing process and potentially generate a large population of the non-emissive triplet state. (ii) The ready inversion of the B2,N-[4]helicene subunit of 6 should open efficient non-radiative decay channels.
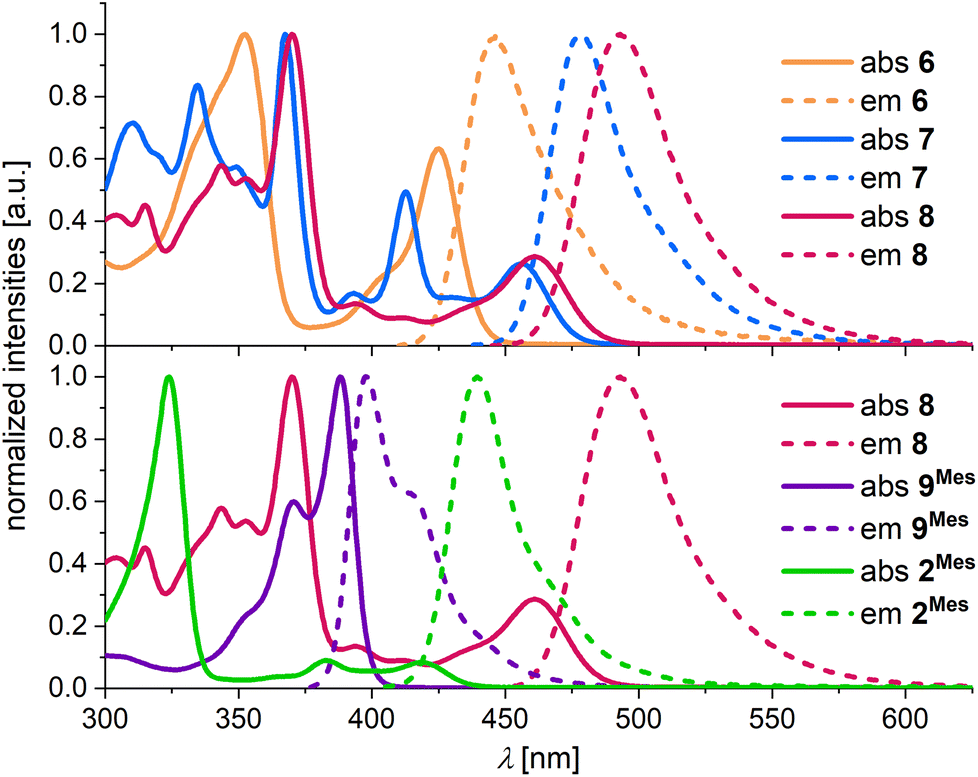 | ||
| Fig. 2 Normalized UV/vis absorption and emission spectra of 6 (orange), 7 (blue), 8 (red), 9Mes (purple), and 2Mes (green), measured in THF. | ||
A comparison of the approximately isomeric B3,N3-PAHs 9Mes and 8 reveals that the rearrangement-induced expansion of the π-electron system causes a huge bathochromic shift of the absorption and emission bands by 0.51 and 0.61 eV, respectively (Fig. 2 bottom, Table 1). The B2,N2-PAH 2Mes adopts an intermediate position with λabs = 419 nm and λem = 440 nm.
Importantly, the full-width at half-maximum values (FWHM; Table 1) measured for the emission bands of all synthesized compounds meet the standards expected for MR-TADF emitters.29 Furthermore, the transient emission-decay curves of the most extended π-electron systems 6–8 show both prompt (range: 4.2–14.8 ns) and delayed components (range: 35–136 μs; poly(methyl methacrylate) (PMMA) matrix, 0.1 wt% of the respective emitter, 300 K; Table S1†), suggesting TADF behavior.
Further insight into key excited-state properties of 6–8 and 2Mes was gained by quantum-chemical calculations using the spin-component scaling second-order algebraic-diagrammatic construction (SCS-ADC(2)) method together with the def2-SVP basis set. For all four compounds, we find that the lowest excited singlet state (S1) is mostly described by a HOMO → LUMO transition (>70% weight) with considerable oscillator strength (f > 0.1; Table S8 and Fig. S69†). The trend in computed S1 energies follows the one experimentally observed in the electronic spectra, with a decrease in energy along the sequence 2Mes → 6 → 7 → 8. The lowest excited triplet state (T1) is also predominantly described by a HOMO → LUMO transition for all compounds except for molecule 6, where T1 is mainly described by a HOMO → LUMO+1 transition (85% weight). In addition, all molecules exhibit short-range charge-transfer (SRCT) excited states that are characterized by an alternating pattern of increasing and decreasing electron density on adjacent atoms, which is a typical feature of multiple resonance TADF compounds. The CT character of an excited state can be quantified by analyzing the CT distance (dCT) between the positive and negative electron density variation (Δρ) and the charge transferred upon excitation (qCT). The computed dCT for 6 has the lowest value of ≈0.80 Å for both the S1 and the T1 state; dCT increases to 2.35 Å (1.86 Å) for S1 (T1) in 7 and 2.45 Å (2.13 Å) for S1 (T1) in 8, likely due to increased π conjugation (Table S9†). These results point toward a larger CT character for 7 and 8 as compared to 6. For 2Mes, dCT is intermediate [S1 (T1): 1.85 Å (1.63 Å)]. Excited states with large qCT and/or large dCT are expected to be more strongly influenced by solvent effects. In line with this expectation and the computed dCT values, a comparison of the calculated gas-phase emission spectra and our experimentally obtained spectra (THF, relative permittivity ε = 7.58 at T = 298.15 K)30 shows the smallest solvent-induced red-shift for 6, followed by 2Mes, 7, and 8 (Fig. S70† and Table 1). The vertical energy gap between the S1 and T1 states (ΔEST) is predicted to be small for compounds 6 (0.06 eV), 7 (0.15 eV), and 8 (0.12 eV), but slightly larger for 2Mes (0.27 eV; Table S8†). The adiabatic ΔEST values do not change much compared to the vertical ones for compounds 7 (0.15 eV), 8 (0.12 eV), and 2Mes (0.34 eV; Table S10†). A more pronounced change from 0.06 to 0.20 eV is computed for 6 (Table S10†) as a consequence of the different nature of its S1 and T1 states, which leads to an asymmetric relaxation toward the minima of their respective potential energy surfaces. Our computed adiabatic ΔEST values are in line with experimentally determined data for derivatives of 6 (0.18 eV) and 7 (0.11 eV).15,22,27 To further elaborate on a possible (reverse) intersystem-crossing [(r)ISC] mechanism, we emphasize that the computed spin–orbit coupling (SOC) between the S1 and T1 states of 6 is as large as 0.662 cm−1, and thus about one order of magnitude higher than the SOCs of 7, 8, and 2Mes (Table S11†). As the (r)ISC rate constant is proportional to the SOC,31 the lower ΦPL of 6 can be rationalized by a high ISC rate that successfully competes with the rate of radiative decay. We hypothesize that, despite the large SOC, the rISC rate might be limited by the high ΔEST, thereby hampering triplet upconversion. Interestingly, the second excited triplet state T2 is very close in energy to the S1 state in 6 (Δ = 0.01 eV), 7 (Δ = −0.01 eV), and 2Mes (Δ = 0.01 eV), whereas this energy gap is larger in 8 (Δ = 0.16 eV). Furthermore, the SOC between the S1 and T2 states is comparable to the SOC between S1 and T1 for 2Mes and 7, but larger (smaller) for 8 (6). The reason lies in the fact that S1 and T2 of 8 (6) have different (similar) electron-density distribution patterns, as can be seen in the difference-density plots between S1–T1 and S1–T2 (Fig. 3). In view of the SOC values and the energy difference of T2 relative to S1, we propose that T2 could play a major role for the emission behavior of compounds 7 and 8, but less so for that of 6. In the case of compound 2Mes, both the S1–T1 and T1–T2 energy gaps (≈0.3 eV) are quite large, thereby limiting reverse intersystem crossing and supporting the observed absence of delayed fluorescence for this compound.
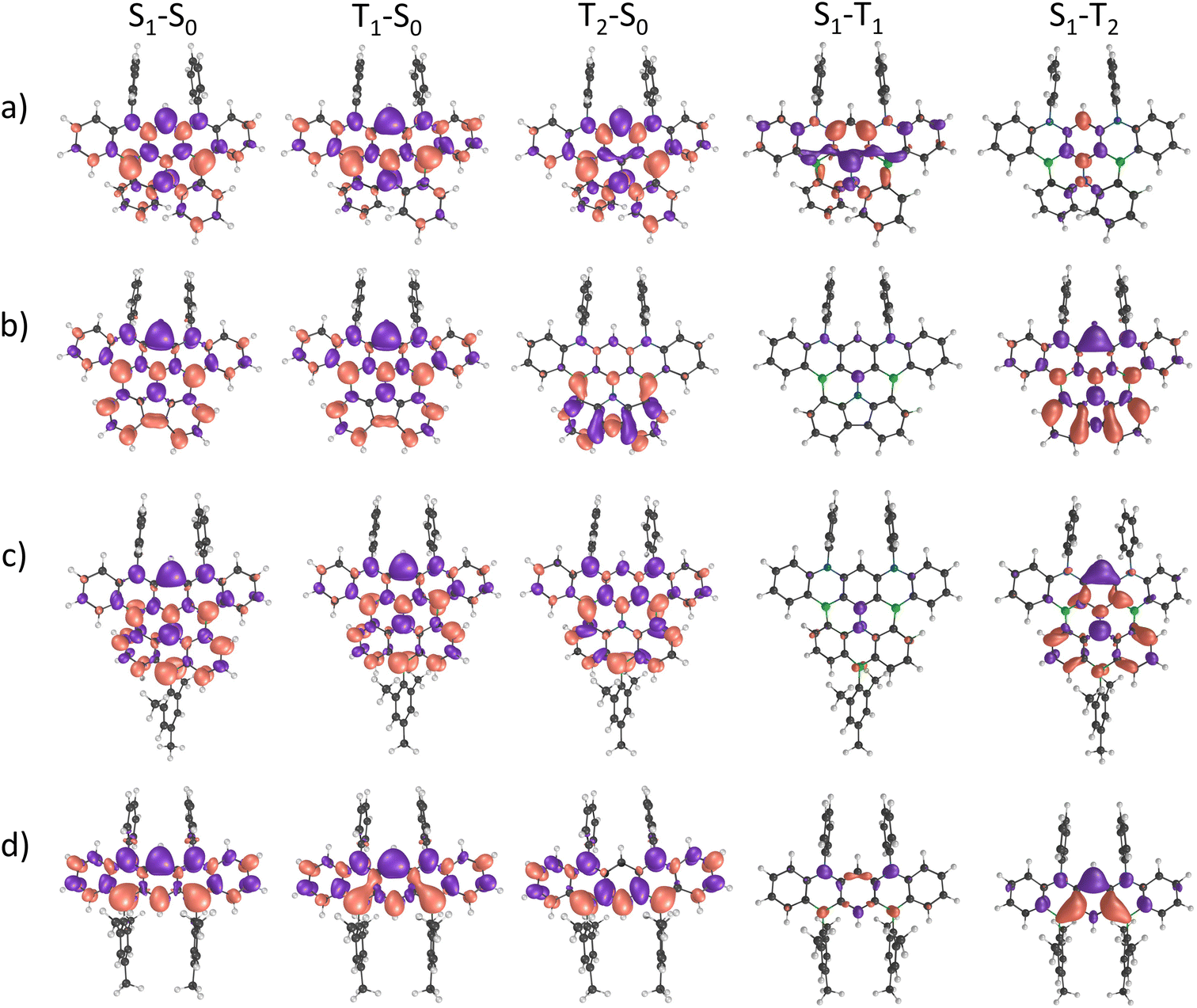 | ||
| Fig. 3 Difference-density plots between the excited states (S1, T1, and T2) and the ground state (S0) for (a) 6, (b) 7, (c) 8, and (d) 2Mes calculated at the SCS-ADC(2)/def2-SVP level. An isovalue of 0.001 e au−3 was used to plot the densities. | ||
The vibronically resolved absorption and emission spectra of 6–8 and 2Mes were simulated within the Franck–Condon approximation using the normal modes computed at the time-dependent density functional (TD-DFT) level (see the ESI† for further details). As can be seen in Fig. S70a,† the experimentally observed trend in the energy of the most bathochromic absorption peak is well reproduced by the S1 vertical excitation energy computed from the optimized ground-state structure (Table S8†). The FWHM values obtained from the simulation of the emission spectra (0.24 eV (31 nm) for 6, 0.20 eV (26 nm) for 7, 0.21 eV (28 nm) for 8, and 0.19 eV (23 nm) for 2Mes; Fig. S70b†) are in excellent agreement with the experiments (Table 1). Moreover, the FWHMs are rather small, in line with the small computed reorganization energies of around 0.1 eV, highlighting the rigidity of all compounds (see Fig. S71†). Apart from molecule 7 whose emission spectrum is broadened by both low-frequency and high-frequency modes, the emission spectra of the other molecules are mainly broadened by low-frequency modes (see Huang–Rhys factor plot in terms of the frequency in Fig. S72†).
Conclusions
1,3- and 1,3,5-azasilaanthryl-substituted benzenes (1 and 5) contain only small, locally constrained π-conjugated systems, due to their sterically enforced propeller-type conformations. We have now demonstrated that a one-pot sequence of Si/B exchange, skeletal rearrangement, and electrophilic borylation reactions leads from 1 or 5 to Bn,Nm-doped nanographenes with extended π-electron clouds. The series of closely related products (6–8) enabled us to elucidate the profound effects of linking peripheral aryl fragments in 6 by either a direct C–C bond (7) or a borylene bridge (8). Apart from substantial structural changes (6 is a trifold [4]helicene whereas 7 is essentially planar), especially the optoelectronic properties are affected: upon going from 6 to 8, electrochemical reduction becomes more facile by 0.37 V; the most bathochromic absorption band and the emission band shift from 424 nm to 461 nm and from 447 nm to 493 nm, respectively. The distribution of the π-electron-accepting B atoms and the π-electron-donating N atoms in the individual polycyclic aromatic hydrocarbons fulfils the formal prerequisite for multiple resonance thermally activated delayed fluorescence (MR-TADF). Indeed, the transient emission-decay curves of 6–8 show a delayed component and quantum-chemical calculations reveal short-range charge-transfer excited states with alternating patterns of increasing and decreasing electron density on adjacent atoms. The energy gaps between the S1 and T1 states (ΔEST) are predicted to be small (≤0.2 eV). Also, the computed spin–orbit coupling between the S1 and T1 states of 6–8 is appreciably high. In summary, we herein disclose a straightforward route to potentially useful, energy-efficient OLED emitters.Author contributions
E. Z. carried out all compound syntheses, characterizations of new compounds, and quantum-chemical calculations of 2Br and 3Br. All other computational studies were performed by D. V. and Y. O.; R. N. assisted with the synthesis of 2Mes, 2Br, and 3Mes. S. K. assisted with the synthesis of 1, 5, and 9Mes. M. B. performed the X-ray crystal structure analysis of 2Br. A. V. performed the X-ray crystal structure analyses of all other compounds. H.-W. L., Y. O., and M. W. supervised the project. The manuscript was written by E. Z., D. V., Y. O., and M. W. and edited by all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Parts of this research (project I-20220822) were carried out at PETRA III at DESY (Hamburg), a member of the Helmholtz Association (HGF). We would like to thank Dr Martin Tolkiehn and Dr Eugenia Peresypkina for their assistance regarding the use of the beamline P24 at DESY. We also wish to thank Prof. Dr Thomas Jüstel and Dr David Enseling from the University of Applied Sciences Münster, Department of Chemical Engineering for the photoluminescence decay and emission measurements in PMMA. D. V. and Y. O. acknowledge funding from Fonds de la Recherche Scientifique-FNRS under grant no. F.4534.21. This research used resources of the “Plateforme Technologique de Calcul Intensif (PTCI)” (https://www.ptci.unamur.be) located at the University of Namur, Belgium, which is supported by the FNRS-FRFC, the Walloon Region, and the University of Namur (conventions no. 2.5020.11, GEQ U.G006.15, 1610468, RW/GEQ2016 et U.G011.22). The PTCI is member of the “Consortium des Équipements de Calcul Intensif (CÉCI)” (https://www.ceci-hpc.be).References
- I. Shin, H. N. Lim and W. P. Hong, Synthesis, 2022, 54, 570–588 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- Y. Sano, T. Shintani, M. Hayakawa, S. Oda, M. Kondo, T. Matsushita and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 11504–11511 CrossRef CAS PubMed.
- R. K. Konidena and K. R. Naveen, Adv. Photonics Res., 2022, 3, 2200201 CrossRef CAS.
- S. M. Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef.
- F. Huang, X.-C. Fan, Y.-C. Cheng, H. Wu, X. Xiong, J. Yu, K. Wang and X.-H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202306413 CrossRef CAS PubMed.
- T. Hua, J. Miao, H. Xia, Z. Huang, X. Cao, N. Li and C. Yang, Adv. Funct. Mater., 2022, 32, 2201032 CrossRef CAS.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef CAS PubMed.
- X. Lv, J. Miao, M. Liu, Q. Peng, C. Zhong, Y. Hu, X. Cao, H. Wu, Y. Yang, C. Zhou, J. Ma, Y. Zou and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202201588 CrossRef CAS PubMed.
- H. J. Cheon, Y.-S. Shin, N.-H. Park, J.-H. Lee and Y.-H. Kim, Small, 2022, 18, 2107574 CrossRef CAS PubMed.
- X.-C. Fan, F. Huang, H. Wu, H. Wang, Y.-C. Cheng, J. Yu, K. Wang and X.-H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202305580 CrossRef CAS PubMed.
- E. Zender, S. Karger, R. Neubaur, A. Virovets, H.-W. Lerner and M. Wagner, Org. Lett., 2024, 26, 939–944 CrossRef CAS PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- Y. Liu, X. Xiao, Z. Huang, D. Yang, D. Ma, J. Liu, B. Lei, Z. Bin and J. You, Angew. Chem., Int. Ed., 2022, 61, e202210210 CrossRef CAS PubMed.
- P. Jiang, L. Zhan, X. Cao, X. Lv, S. Gong, Z. Chen, C. Zhou, Z. Huang, F. Ni, Y. Zou and C. Yang, Adv. Opt. Mater., 2021, 9, 2100825 CrossRef CAS.
- C. M. van Beek, A. M. Swarbrook, C. E. Creissen, C. S. Hawes, T. A. Gazis and P. D. Matthews, Chem. – Eur. J., 2024, 30, e202301944 CrossRef CAS PubMed.
- T. Agou, J. Kobayashi and T. Kawashima, Org. Lett., 2006, 8, 2241–2244 CrossRef CAS PubMed.
- For corresponding B3,N3-heptacenes, see: (a) S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed; (b) K. Stavrou, S. M. Suresh, D. Hall, A. Danos, N. A. Kukhta, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, A. Monkman and E. Zysman-Colman, Adv. Opt. Mater., 2022, 10, 2200688 CrossRef CAS.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed.
- C–N-bond cleavage reactions in comparable systems have been reported: K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed.
- Precedence for such reactivity exists in the form of a 9,10-dihydro-9,10-diboraanthracene → 9-borafluorene conversion: S. Kirschner, S.-S. Bao, M. K. Fengel, M. Bolte, H.-W. Lerner and M. Wagner, Org. Biomol. Chem., 2019, 17, 5060–5065 RSC.
- Y. Wang, Y. Duan, R. Guo, S. Ye, K. Di, W. Zhang, S. Zhuang and L. Wang, Org. Electron., 2021, 97, 106275 CrossRef CAS.
- Compound 6 crystallizes with 0.25 toluene solvate molecules. The crystal lattice contains two molecules 6A and 6B in the asymmetric unit; since the key metrical parameters of 6A and 6B are sufficiently similar considering the experimental error margins (cf. the overlay in Fig. S63b†), it is justified to discuss only the data of 6A in the main text. Compound 7 crystallizes as a C6H6 solvate with two crystallographically unique molecules, 7A and 7B, in general positions. Some key metrical parameters of 7A and 7B differ significantly, so both molecules are considered in the discussion whenever necessary. Compound 8 crystallizes with two CHCl3 solvate molecules.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CrossRef CAS.
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882–2886 CrossRef CAS PubMed.
- T. Hatakeyama, A. Mizutani and T. Koike, WO2017188111A1, 2017.
- During borylation experiments on 3Mes with BBr3 in C6D6, we observed essentially quantitative H/D exchange at the positions ortho and para to the N atoms, while exchange at the meta positions was far less pronounced (see the ESI† for more details). This can be considered as an experimental mapping of the charge-density distribution in the HOMO of 3Mes/2Br.
- J. F. Coetzee and T.-H. Chang, Pure Appl. Chem., 1985, 57, 633–638 CrossRef CAS.
- Y. Olivier, B. Yurash, L. Muccioli, G. D'Avino, O. Mikhnenko, J. C. Sancho-García, C. Adachi, T.-Q. Nguyen and D. Beljonne, Phys. Rev. Mater., 2017, 1, 075602 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, analytical, computational, and crystallographic data. CCDC 2338843–2338851. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01166j |
| This journal is © The Royal Society of Chemistry 2024 |