
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation and antimicrobial activity of supramolecular cobalt-peptide conjugates†
Liudmila
Janzen
,
Reece G.
Miller
 and
Nils
Metzler-Nolte
*
and
Nils
Metzler-Nolte
*
Faculty of Chemistry and Biochemistry, Inorganic Chemistry I – Bioinorganic Chemistry, Ruhr University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: nils.metzler-nolte@rub.de; Web: https://www.chemie.rub.de/ac1
First published on 14th June 2024
Abstract
Herein, we describe the synthesis and characterisation of four new supramolecular cobalt conjugates of antimicrobial peptides functionalised with terpyridine ligands (L). Peptides were chosen based on the well-established arginine-tryptophan (RW)3 motif, with terpyridine-derivatized lysine (Lys(tpy)) added to the sequence, or replacing tryptophan residues. Self-assembly of the antimicrobial peptides with Co(BF4)2·6H2O formed exclusively CoL2 dimers (for peptides with one tpy ligand each) and Co2L4 metallo-macrocycles (for peptides with two tpy ligands for each peptide), which could be ‘locked’ by oxidation of Co(+II) to Co(+III) with ammonium ceric nitrate. The Co-peptide complexes were characterised by mass spectrometry and in solution by NMR spectroscopy, including 2D diffusion ordered NMR spectroscopy (DOSY) which confirmed the proposed stoichiometries. The antimicrobial activity of the novel peptides and their metallo-supramolecular assemblies was investigated by determination of their minimal inhibitory concentration (MIC) against a panel of Gram-positive and Gram-negative bacteria. Complexation with cobalt increases the activity of the peptides in almost every case. Most of the new metal–peptide conjugates showed good activity against Gram-positive bacteria, including a multi-resistant S. aureus strain and the opportunistic pathogenic yeast C. albicans (down to 7 μmol l−1 for the most active Co2L4 derivate), a value that is increased five-fold compared to the lysine-derivatized peptide ligand alone. Interestingly, conjugates of the CoL2 type also showed decent activity against Gram-negative bacteria including the WHO-flagged problematic A. baumannii strain (down to 18 μmol l−1 for the most active derivative).
Introduction
Metal ions play a key role in numerous biological processes. Due to their interaction with macromolecules such as enzymes, pro-hormones, membranes and presecretory granules, a variety of biological functions including catalytic, structural, and regulatory activities are realized. Certain metals may also exhibit toxicity towards bacteria and yeast, which has led to their application as antimicrobial agents since ancient times.1 More recently, mechanistic studies have helped elucidate the toxic effects of metals on microbial cells, leading to an increased interest in the application of metal-based complexes as antimicrobial agents.2–9 It has been demonstrated that certain metals damage antibiotic resistance biofilms,10 inhibit metabolic pathways in a selective manner,11 and can kill multidrug-resistant bacteria.12 Beyond small molecular compounds, large self-assembled metallosupramolecular helicates with promising antimicrobial activity have attracted greater interest due to their DNA-binding properties.13,14 Interestingly, further studies showed that changing the metal centre in the helicate structure influences the bactericidal activity against certain bacterial strains.15 Despite some promise in these early applications, similar systems were not explored further.With fast emergence in antibiotic resistance, combined with the decline in novel antibiotic therapies, the need to discover and develop new natural as well as synthetic antibiotic agents has increased significantly. One attractive option is the combination of antimicrobial peptides (AMPs) and metal ions. AMPs are a promising antibiotic class with some structural diversity and broad activity spectrum.16 Formally, they can also be distinguished in natural (typically with longer peptide sequences and post-synthetic modifications) and synthetic AMPs (synAMPs), the latter typically having shorter sequences, and possibly non-natural modifications.17,18 One of these modifications can be the covalent modification of peptides with metal complexes to enhance their antimicrobial activity, or alter their spectrum of activity.2,19,20 The presence of metals could lead to conformational changes in the AMPs followed by change of their properties and biologically activity. In natural systems, metallopeptides such as bacitracin (isolated from Bacillus subtilis), coordinated with several divalent transition metal ions (e.g. Zn, Mn, Co and Cu), and bleomycin (isolated from Streptomyces verticillatus), coordinated with Cu(II) or Fe(II), showed enhanced biological activity in inhibiting bacterial growth than in their pure form.21–25 Herein, we report the coordination-driven supramolecular conjugates of cobalt and short cationic peptide derivatives of a known antimicrobial H-(RW)3-NH2 peptide (Fig. 1). The (RW)3 motif was first reported and its structure–activity relationship (SAR) investigated by Svendsen and coworkers.26 Numerous modifications of this peptide were investigated, including N-terminal and side chain modification with organometallic complexes and fatty acids, often leading to spectacular activity e.g. against cystic fibrosis.27 Modifications of the (RW)3 peptide with unnatural transition metal complexes like ruthenocene were also crucial to finally elucidate the mode of action of this highly attractive class of synAMPs. This membrane-targeting peptide causes delocalization of peripheral membrane proteins, ultimately limiting cellular energy and undermining cell-wall integrity.28
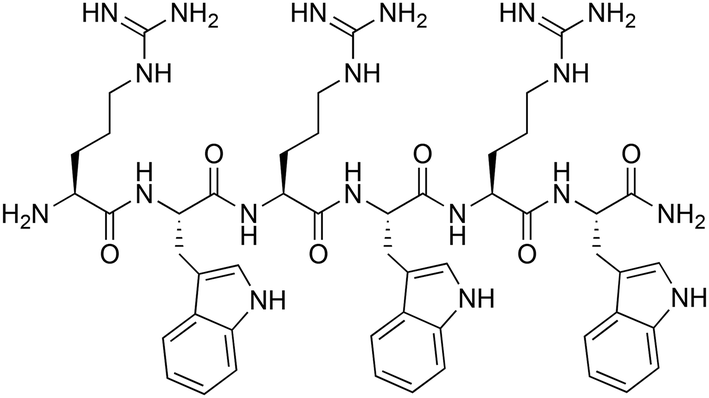 | ||
| Fig. 1 Structure of the parent H-(RW)3-NH2 peptide.6 | ||
The essential transition metal cobalt (Co) exhibits a unique redox chemistry and is an integral part of the vitamin B12 group that is essential for healthy nervous system function, production of red blood cells, and metabolism of folic and fatty acids.29 Compared to some other transition metals like Pt and Au, the biological activity of cobalt compounds has not been studied in details. There are only a few cobalt-based compounds which exhibit activity against several bacteria and viruses.30–39 Nagababu et al. investigated a series of Co(III) mixed ethylenediamine complexes, which exhibited antibacterial activity against nonpathogenic bacteria. Out of those, the Co complexes with bipyridine, 1,10-phenanthroline, dimethylimidazole, and pyrazole exhibited the highest antibacterial activity.
Further investigation by these authors of the fungicidal properties of these complexes revealed also high antifungal activity.35 Of relevance for the present work is Co in oxidation states +II (d7-Co), which is kinetically labile with rapid ligand exchange, and its +III oxidation state (d6-Co) that supports complexes with similar geometry, but is substitutionally inert.
Characterisation and structural investigation in the field of supramolecular chemistry is always a major challenge, especially in self-organizing systems with potentially multiple stoichiometries. Increasingly, diffusion-ordered NMR spectroscopy (DOSY) is gaining popularity, being used to glean information on the size and shape, as well to probe intermolecular complex formation. For example, diffusion measurement has been successfully applied to characterize the structure of metallosupramolecular helicates and rotaxane, and to study their behaviour in solution.40,41 Therefore, for the characterization of herein synthesized supramolecular systems the diffusion-ordered NMR spectroscopy (DOSY) is the method of choice. DOSY provides a reasonable approximation for the determination of the possible complex stoichiometry by using the Stokes–Einstein equation (eqn (1)).42–44 In this work, we report the synthesis of antimicrobial peptides featuring terpyridine (tpy) ligands for metal complexation and the subsequent selective self-assembly of these peptides into ‘oxidatively locked’ metallosupramolecular architectures using the redox-dependent lability of Co(tpy)2 complex formation. After characterisation of the complexes, including their exact composition and stoichiometry, the influence of stoichiometry and metal presence or absence on antimicrobial activity will be investigated by determining the minimal inhibitory concentration (MIC).
Results and discussion
Design strategy
In order to obtain different (RW)3 peptide derivatives with metal-chelating abilities, a new terpyridine-containing amino acid was synthesized. To this end, a carboxylic acid derivative of the metal binding 2,2′:6′,2′′-terpyridine (tpy) ligand was coupled to the side-chain amino group of the hydrophilic basic amino acid lysine (Lys), following a modified literature procedure.45,46 Then, this Lys(tpy) amino acid either substituted the tryptophan (W) of the (RW)3 peptide or was added to the sequence (Fig. 2). Either one or two Lys(tpy) derivatives were incorporated into the sequences to explore structure–activity relationships (SAR). All peptides and peptide-terpyridine derivatives for metal conjugation were prepared by solid phase synthesis techniques.47 The tpy ligand forms stable bis-complexes with pseudo octahedral coordination at the metal centre with metal ion in low oxidation states.48 The σ-donor and π-acceptor character of the terpyridine ligands as well as the chelate effect contribute to the complex stability.49,50 Additionally, both bivalent cobalt and pyridine belong to the borderline group according to Pearson's hard and soft Lewis acids and bases (HSAB) theory.51 So, the affinity of the metal to the nitrogen atoms of the tpy ligand outcompetes other potential donor atoms in the peptide structure, which leads to a high probability of the desired complex formation. However, because ligands around the kinetically labile Co(II) ion can still rearrange, the preferred Co(tpy)2 product was subsequently “locked” by oxidation of the metal ion to the inert Co(III) oxidation state.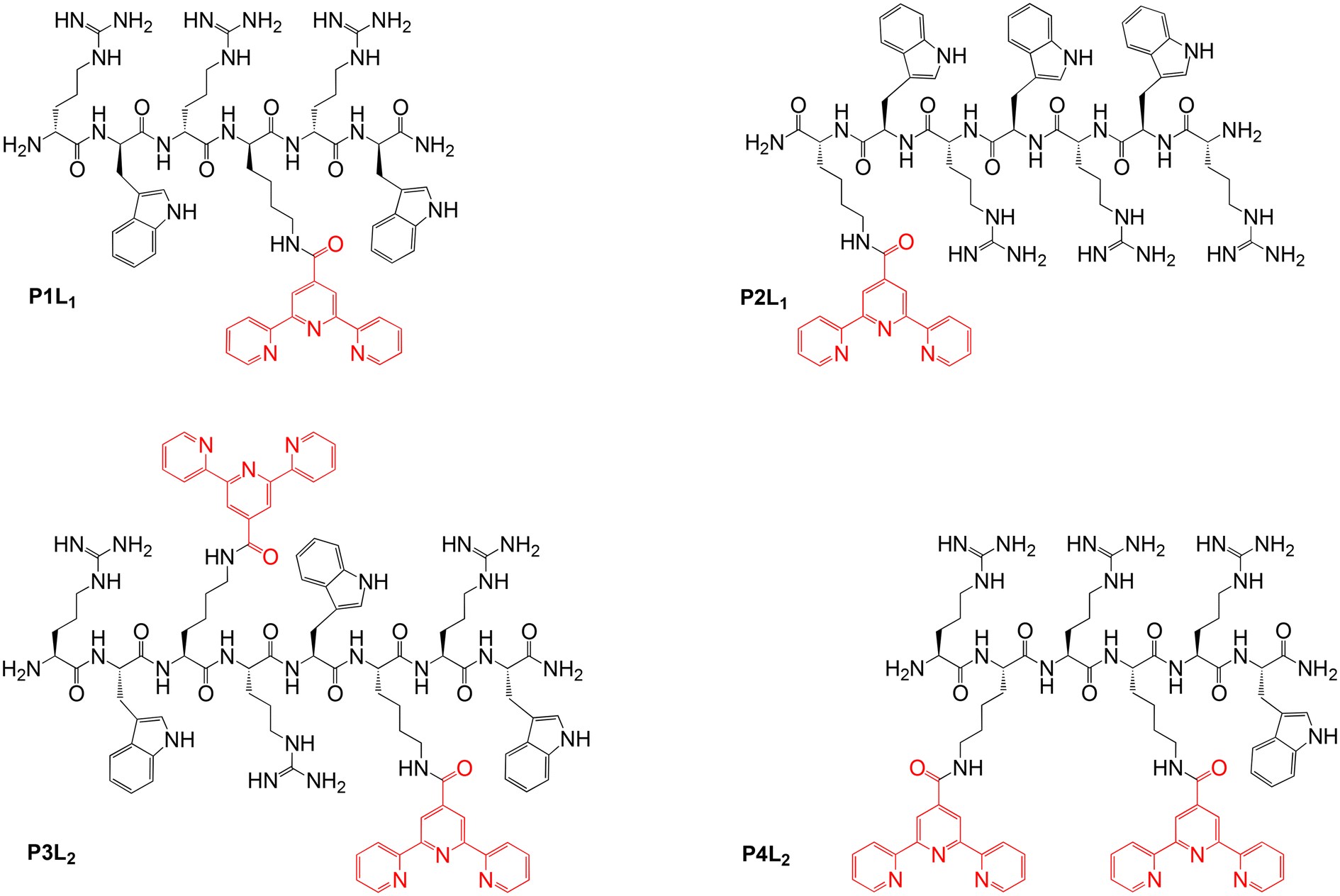 | ||
| Fig. 2 Overview of the isolated peptides (P1–P4) coupled with tpy ligands (Ln, n = 1,2 Lys(tpy) residues). | ||
Synthesis
The peptide synthesis was performed manually at room temperature using an fluorenylmethyloxycarbonyl (Fmoc) protection strategy on Rink amide resin as solid support. The coupling reaction of amino acids was carried out by using the coupling reagent 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) and the additional base diisopropylethylamine (DIPEA) in excess (see ESI† for details).46 The further attachment of the ligands on the peptides was also carried out on the solid support. For that purpose, Lys with the tert-butyloxycarbonyl (Boc) protecting group on the side chain was chosen, because of deprotection condition with low concentrated trifluoroacetic acid (TFA) solution (30%). By contrast, the peptide cleavage from the Rink amide resin and deprotection of arginine and tryptophan side chains require a higher concentration of TFA (95%).52 Thus, coupling of terpyridine carboxylic acid to the lysine amino side chain could be carried out selectively, minimizing the formation of undesired by-products. The tpy ligand was attached to the Lys side chain on the solid phase using a coupling reagent mixture of diisopropylcarbodiimide (DIC) and hydroxybenzotriazole (HOBt). Isolation and purification of the tpy-derivatized peptides after cleavage from the Rink amide resin was performed by semi-preparative reverse phase HPLC followed by lyophilisation. Analytical data of the peptides are summarized in Table 1, with structures shown in Fig. 2.| Peptide | Calcd Mass [M] (g mol−1) | MS MALDI-TOF [M + H]+ | Retention time HPLC (min) | Diffusion coefficient in DMSO-d6 (m2 sec−1) | Yield after purification via HPLC (%) |
|---|---|---|---|---|---|
| P1L1 | 1244.66 | 1245.75 | 7.2 | 8.81 × 10−11 | 4 |
| P2L1 | 1430.74 | 1431.7 | 7.4 | 8.5 × 10−11 | 4 |
| P3L2 | 1817.91 | 1818.17 | 5.6 | 7.78 × 10−11 | 3 |
| P4L2 | 1445.75 | 1446.91 | 5.4 | 7.86 × 10−11 | 7 |
The cobalt peptides conjugates are made by one pot reaction of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 or 1:1:1 ratio of prepared peptides, CoII(BF4)2·6H2O and later oxidizing agent NH4[CeIV(NO3)6], in DMSO-d6 (Table 2). Formation of the complexes was accompanied by a colour change. After addition of the purple solution of high-spin Co(II) complex to the colourless peptide solution, product formation was accompanied by the immediate change to dark red orange colour. After chemical oxidation to the more stable Co(III)-peptide complexes by Cer(IV) salt, a second colour change to pale orange was observed (for more details, including electronic absorption spectra, see ESI†). Reactions were carried out in deuterated DMSO to enable NMR monitoring of the reaction progress, however products were later isolated by precipitation (see ESI† for exact procedure). The schematic formation of cobalt-peptide conjugates is presented in Scheme 1.
:1:1 or 1:1:1 ratio of prepared peptides, CoII(BF4)2·6H2O and later oxidizing agent NH4[CeIV(NO3)6], in DMSO-d6 (Table 2). Formation of the complexes was accompanied by a colour change. After addition of the purple solution of high-spin Co(II) complex to the colourless peptide solution, product formation was accompanied by the immediate change to dark red orange colour. After chemical oxidation to the more stable Co(III)-peptide complexes by Cer(IV) salt, a second colour change to pale orange was observed (for more details, including electronic absorption spectra, see ESI†). Reactions were carried out in deuterated DMSO to enable NMR monitoring of the reaction progress, however products were later isolated by precipitation (see ESI† for exact procedure). The schematic formation of cobalt-peptide conjugates is presented in Scheme 1.
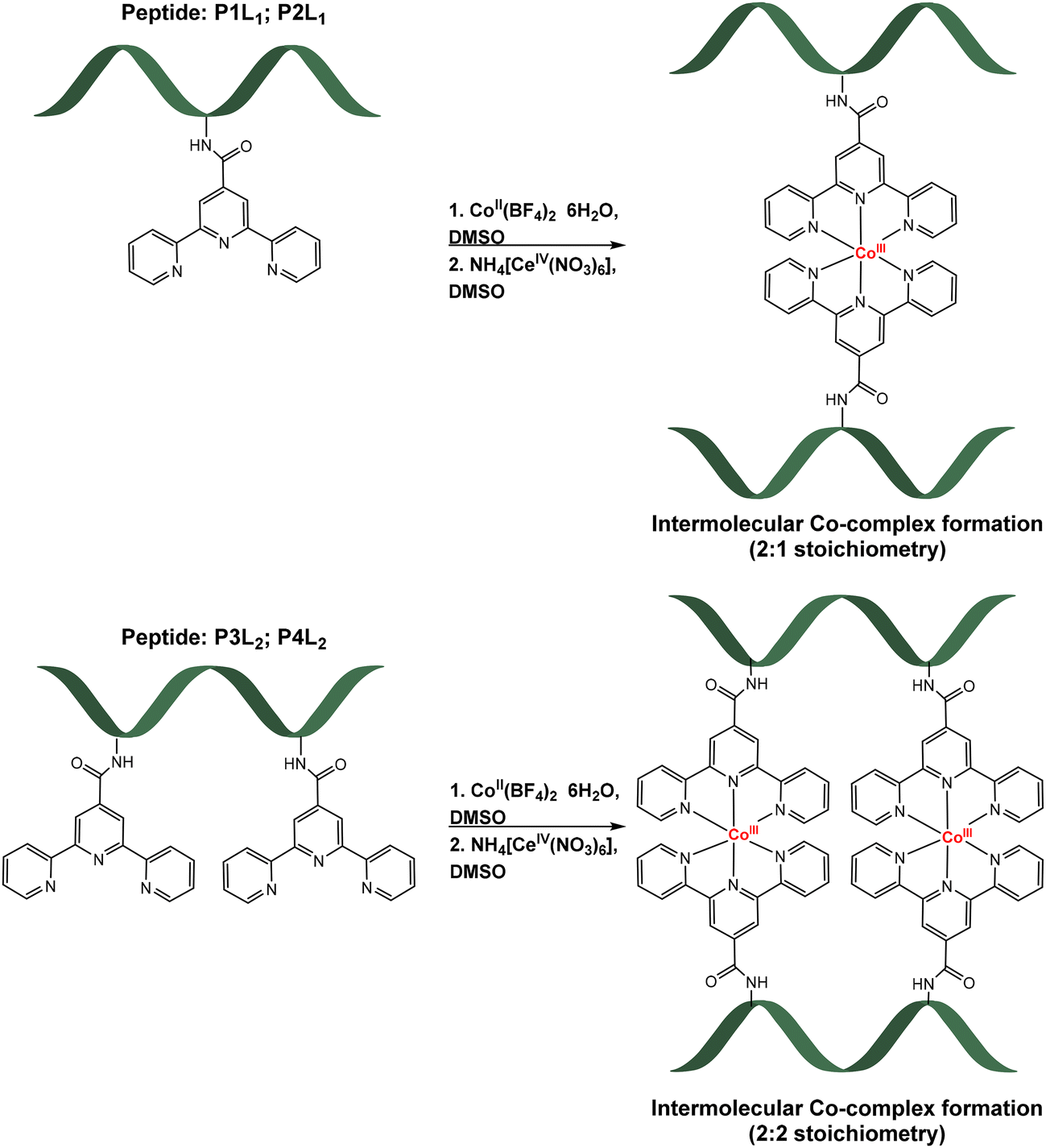 | ||
| Scheme 1 Formation of Co-peptide conjugates with one tpy ligand (top, [CoIII(P1L1)2]3+; [CoIII(P2L1)2]3+) and two tpy ligands (bottom, [(CoIII(P3L2))2]6+; [(CoIII(P4L2))2]6+). | ||
| Co-peptide conjugates | Calcd mass [M] (g mol−1) | MS MALDI-TOF | Diffusion coefficient in DMSO-d6 (m2 sec−1) | Yield (%) |
|---|---|---|---|---|
| [CoIII(P1L1)2]3+ | 2548.25 | 1303.84 [CoIII(P1L1)]+; 1245.88 [P1L1 + H]+ | 6.42 × 10−11 | 90 |
| [CoIII(P2L1)2]3+ | 2920.14 | 1489.37 [CoIII(P2L1)]+; 1431.8 [P2L1 + H]+ | 6.14 × 10−11 | 93 |
| [(CoIII(P3L2))2]6+ | 3753.68 | 1935.07 [Co2III(P3L2)]+; 1876.15 [CoIII(P3L2) + H]+; 1818.15 [P3L2 + H]+ | 5.42 × 10−11 | 90 |
| [(CoIII(P4L2))2]6+ | 3009.36 | 1563.7 [Co2III(P4L2)]+; 1505.05 [CoIII(P4L2) + H]+; 1446.97 [P4L2 + H]+ | 5.56 × 10−11 | 92 |
In order to verify the correct binding of the cobalt to the nitrogen of terpyridine ligands in the peptide structure, product formation was monitored by higher frequency (400 MHz) 1H-NMR. The first spectrum was made after the peptide-ligand synthesis, then after the CoII-complex formation, and then again after oxidation by Cer(IV). The characteristic terpyridine signals in the 1H-NMR spectrum of P1L1 were identified by comparison with the 1H-NMR spectrum of 2,2′:6′,2′′-terpyridine-4′-carboxylic acid (L) (Fig. 3).
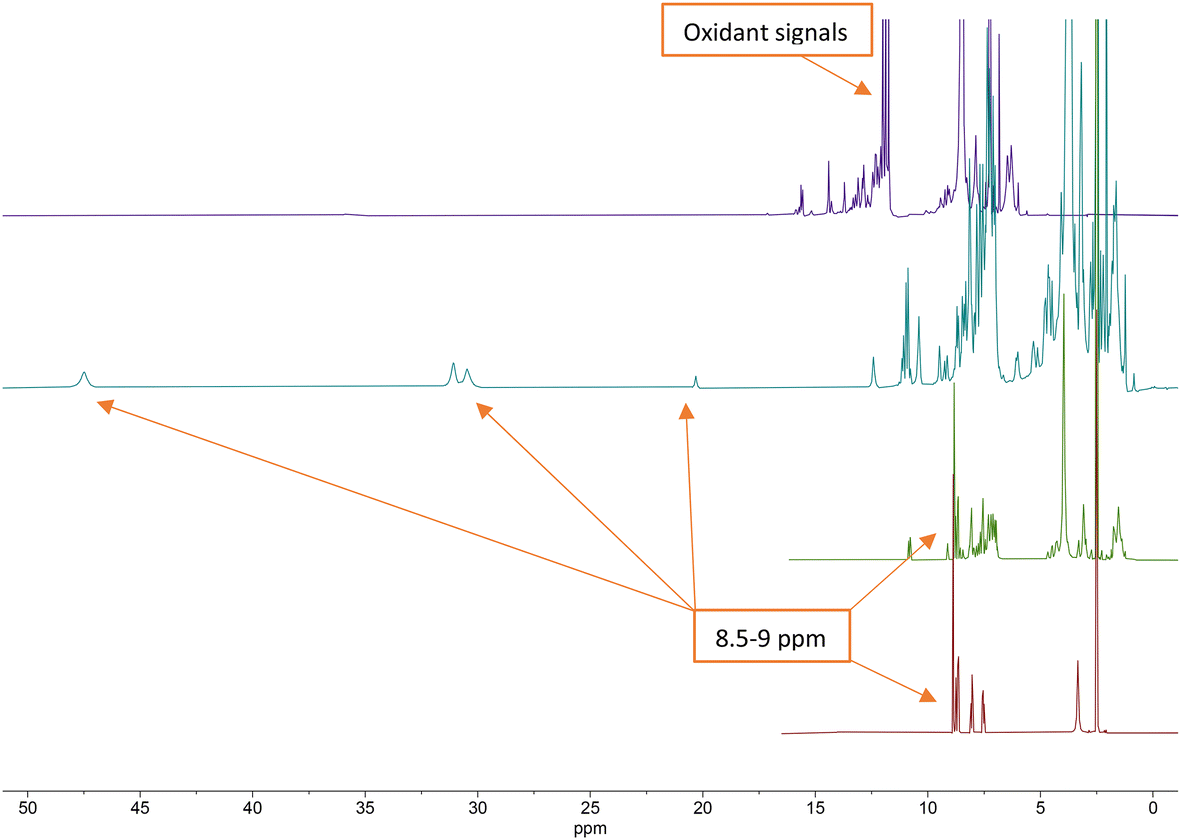 | ||
| Fig. 3 Stacked 1H NMR (400 MHz) spectra of terpyridine ligand (bottom, red), P1L1 (middle, green), [CoII(P1L1)2]2+ complex (middle, teal) and [CoIII(P1L1)2]3+ complex (top, purple). | ||
From the 1H NMR spectra in Fig. 3, it can be seen that the aromatic protons of the tpy ligand (8.5 ppm to 9 ppm) were broadened and shifted to low field in the spectrum of the Co(II)-complex, consistent with coordination to a paramagnetic metal centre.23 After oxidation to Co(III), the paramagnetic effect vanished, which led to the absence of signals in the low field and increased the quality of the spectrum (Fig. 3, top, purple). However, the tpy signals remained shifted in the spectrum of the Co(III)-peptide complex. To ensure the correct coordination of the peptide to cobalt, a higher frequency 1H NMR (700 MHz) was recorded (Fig. 4). In the stacked spectra, the characteristic terpyridine ligand signals at 7.5 ppm, 8.0 ppm, and 8.5 ppm to 9.0 ppm (Fig. 4, red spectrum) appeared in the 1H NMR of P4L2 with a slight shift to the higher field (Fig. 4, teal spectrum). After complexation and oxidation to Co(III), the terpyridine signals could be approximately assigned in the 1H NMR spectrum (Fig. 4, top, purple spectrum) if compare with the 1H NMR of tpy-Co(III) complex (Fig. 4, middle green spectrum). However, the NH-proton of the indole remained at 10.79 ppm in both spectra, in P4L2 and Co(III)-P4L2; therefore, it can be assumed that the tpy ligand was coordinated to the cobalt ion.
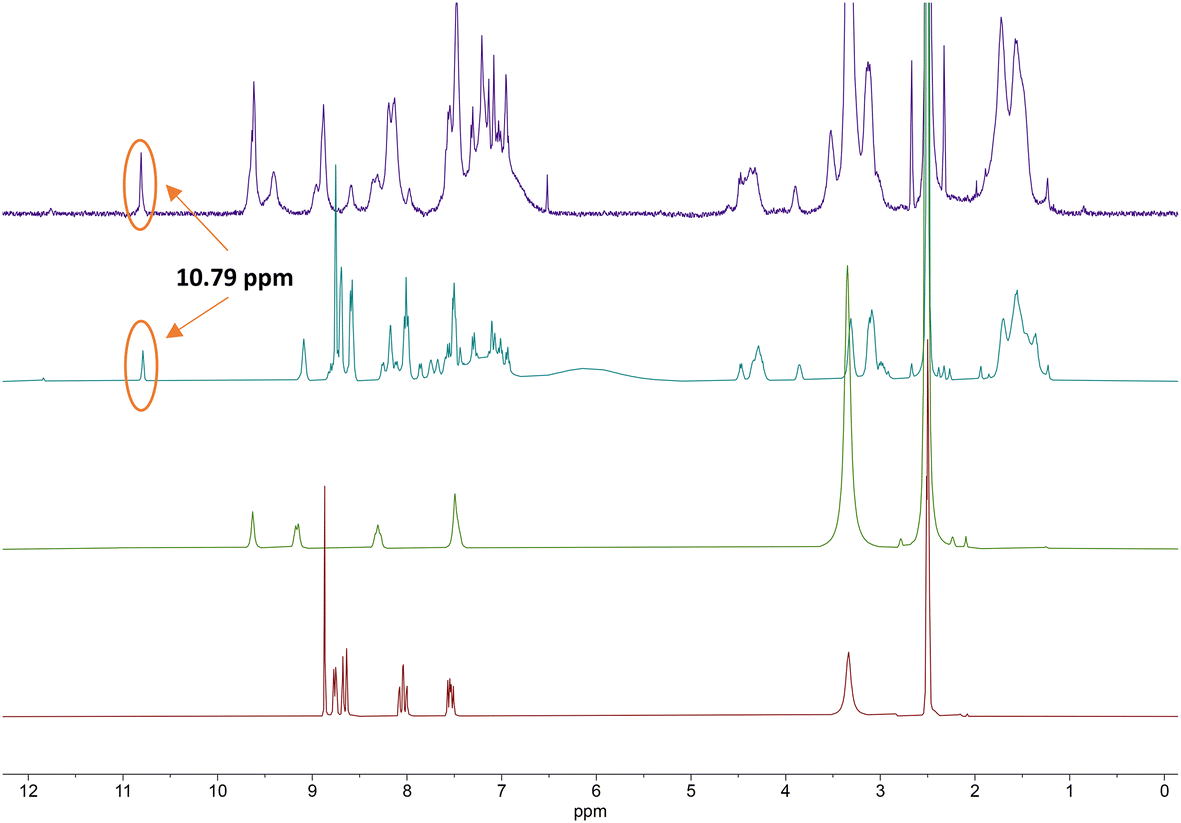 | ||
| Fig. 4 Stacked 1H NMR (700 MHz) spectra of terpyridine ligand (tpy-COOH, bottom, red), [CoIII(tpy-COOH)2]3+ (middle, green), P4L2 (middle, teal) and [(CoIII(P4L2))2]6+ complex (top, purple). | ||
To purify the final product from ceric ammonium nitrate, precipitation with ethyl acetate/diethyl ether (1:1) and lyophilization was carried out. The 1H-NMR spectrum confirmed the absence of the oxidant, since ceric ammonium nitrate has characteristic signals in the range between 6.9 ppm and 7.2 ppm (for more details see ESI,† including assignment of 1H NMR signals).
Characterization of the cobalt-peptide conjugates
The previous investigation of the formation of the cobalt peptide conjugates using 1H NMR (Fig. 3 and 4) has demonstrated that cobalt is coordinated to the terpyridine ligands. To investigate and possibly confirm the predicted 2:1 stoichiometry of the formed supramolecular complexes with peptides P1L1 and P2L1, and the 2:2 stoichiometry of complex formation for peptides with two tpy ligands (P3L2 and P4L2), mass spectrometry measurements were performed using MALDI TOF (see suppl. information for mass spectra of all complexes). For the cobalt conjugates with two tpy entities the masses of characteristic fragments were detected, such as 1935.07 for [CoIII2(P3L2)]+ and 1563.7 for [CoIII2(P4L2)]+, respectively (Fig. 5). The presence of peaks originating from fragments with two metal centres and one peptide entity rules out intramolecular complex formation (i.e. two tpy ligands from the same peptide binding one Co center) and rather suggests intermolecular complex formation as indicated in Scheme 1 (bottom). However, these mass spectrometry results could only be taken as a first indication and further studies are required to confirm the preferred stoichiometry of the complexes formed (see Scheme 2, and discussion below).
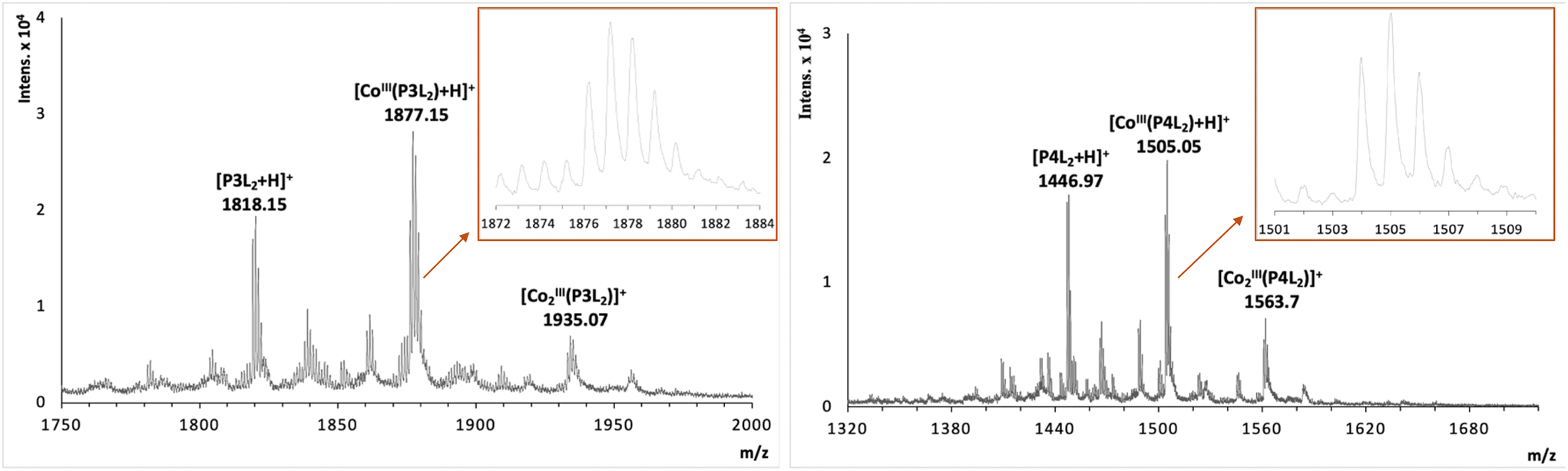 | ||
| Fig. 5 MALDI TOF spectrum of cobalt-peptide conjugate [(CoIII(P3L2))2]6+ (left) and [(CoIII(P4L2))2]6+ (right). Note that all peaks in the mass spetra are singly charged (as proven by the peak distance of isotopomers in the inserts); this is a consequence of the ionization technique rather than a molecular property. | ||
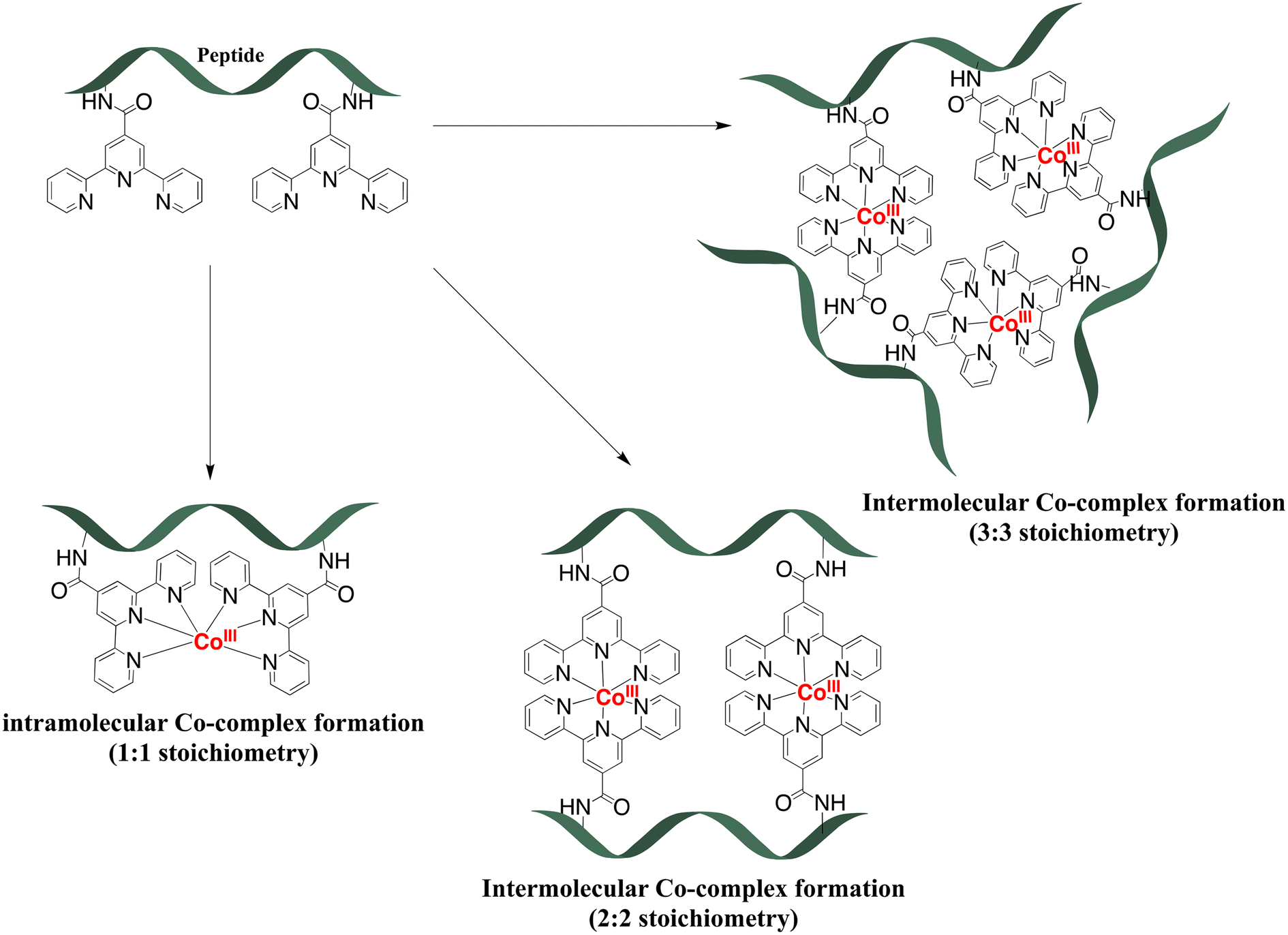 | ||
| Scheme 2 Possible stoichiometry of complexes during the coordination of peptides with two tpy ligands (P3L2 and P4L2) to the cobalt ion. | ||
The detected masses for the cobalt complexes with peptides P1L1 and P2L1 correspond to fragments with one peptide unit and one cobalt centre (see ESI†), 1303.84 for [CoIII(P1L1)]+ and 1489.37 for [CoIII(P2L1)]+, respectively. Unfortunately, characterization of the Co conjugates by HPLC was impossible. After HPLC purification of the cobalt conjugates, only the peptides could be isolated, indicating the instability of the complexes under HPLC conditions. To further verify the purity and confirm the predicted stoichiometry of the formed complexes, the diffusion-ordered NMR spectroscopy (DOSY NMR) was applied (Fig. 6 and 7, for more spectra see ESI†).
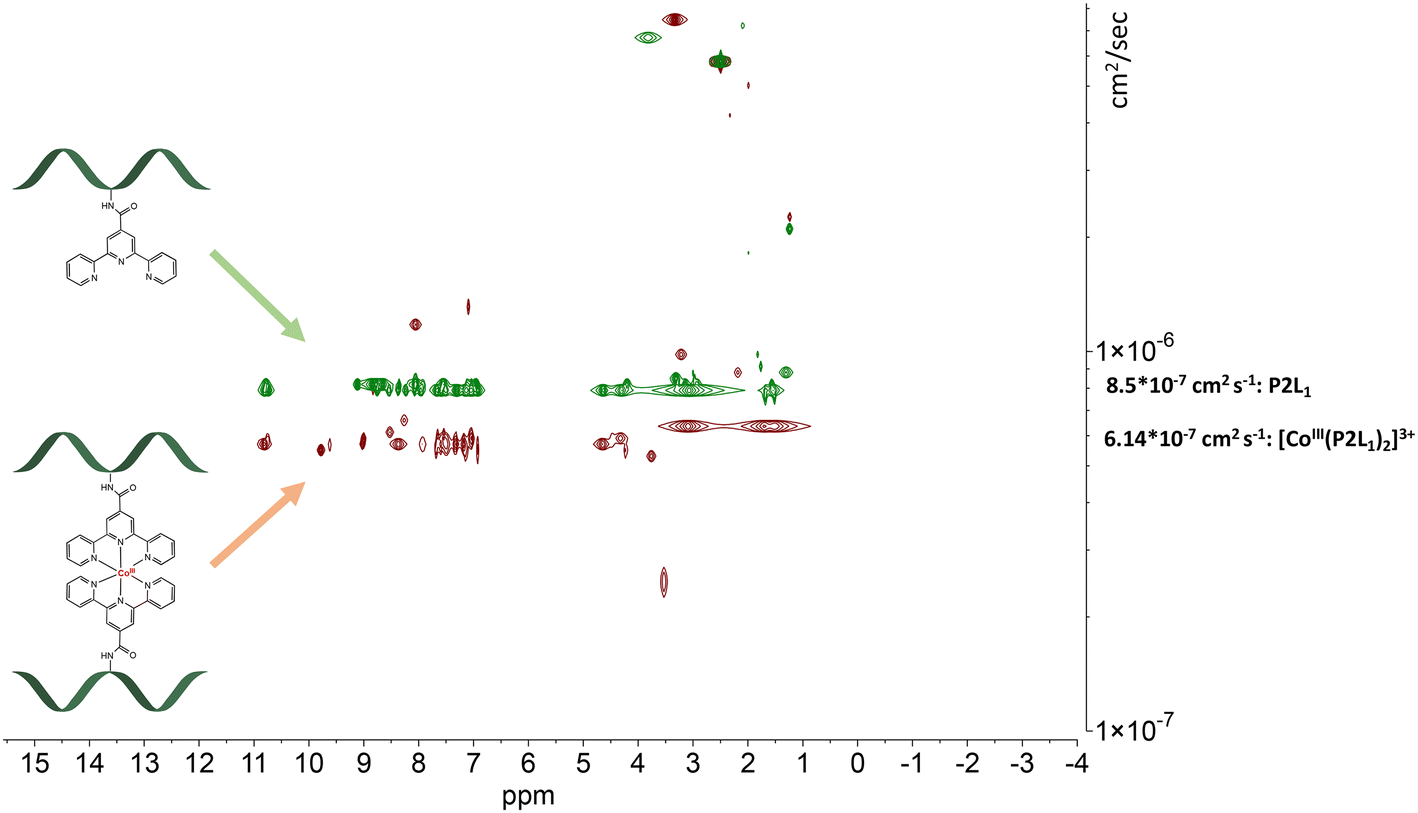 | ||
| Fig. 6 Stacked DOSY NMR spectrum of P2L1 (green) and [CoIII(P2L1)]3+(red) directly after the oxidation reaction in DMSO-d6 (2.50 ppm), water peak is at 3.33 ppm. | ||
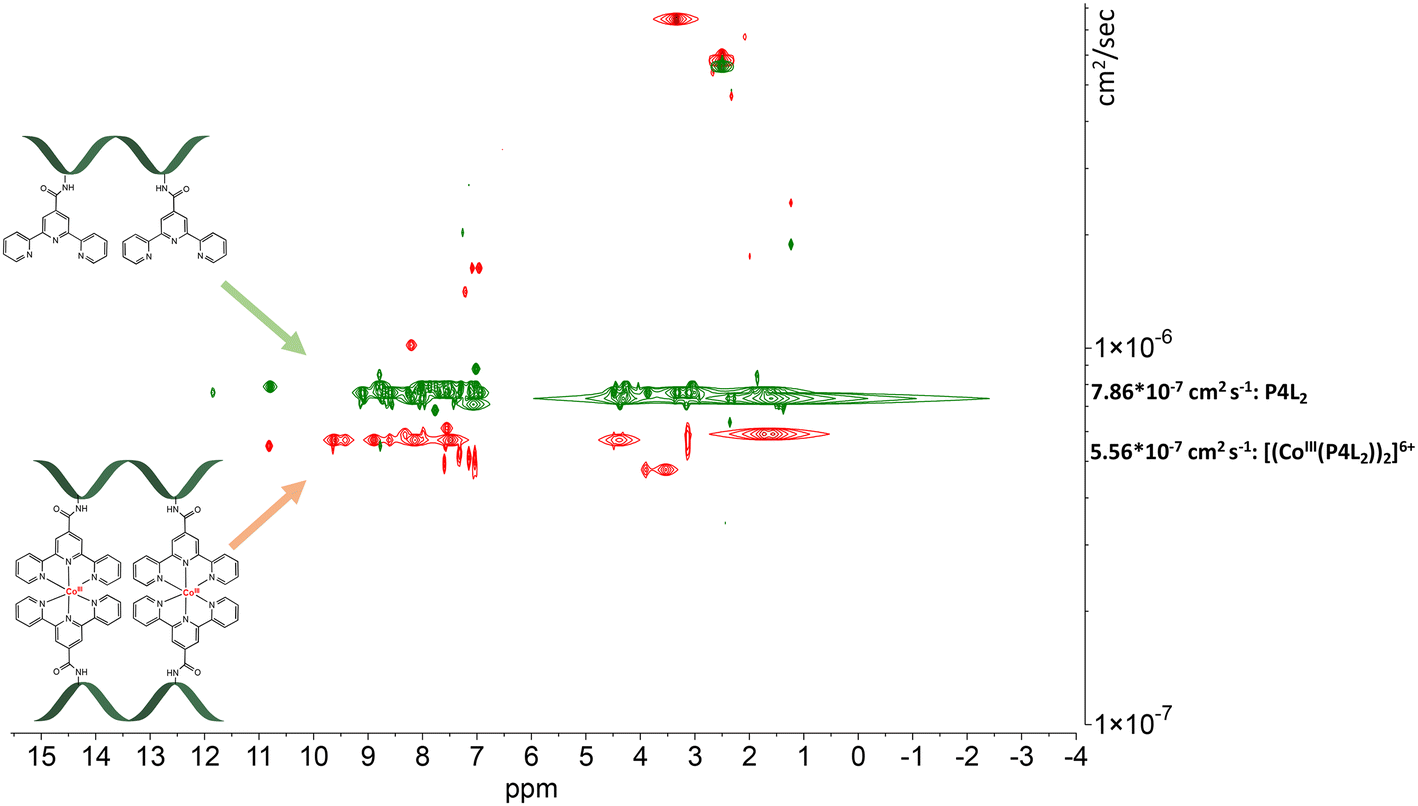 | ||
| Fig. 7 Stacked DOSY NMR spectrum of P4L2 (green) and [(CoIII(P4L2))2]6+ (red) directly after the oxidation reaction in DMSO-d6 (2.50 ppm), water peak is at 3.33 ppm. | ||
The DOSY spectral results demonstrate that the complexation reaction with cobalt led to the clean formation of only one product. Since each species has a different diffusion coefficient in the solvent, they move at different rates and therefore appear on different horizontal levels of a 2D DOSY NMR spectrum. This can be observed in the stacked DOSY NMR spectra of the metal-free peptides P2L1 and P4L2 (Fig. 6 and 7, green), which has a greater diffusion coefficient than its cobalt conjugates (Fig. 6 and 7, red) because of the faster movement in the solvent. Since no respective signals of metal-free peptides in the 2D DOSY NMR spectrum of cobalt complexes were observed, it can be assumed that the reaction proceeds to completion.
The ligand-to-metal ratio (stoichiometry) by the complex formation can vary due to different factors (e.g. reduction of entropy, sterically influence, affinity of the metal centre to other donor atoms within the peptide structure), these lead to differences in the size of the molecules, which in turn leads to differences in the rate of diffusion in the solution. In this case, the observed diffusion coefficients from the DOSY NMR experiments were used to calculate the hydrodynamic size of the prepared compounds with the aim of determining the stoichiometry of the complexes (Schemes 1 and 2). Since the observed diffusion coefficients of the complexes are lower than the diffusion coefficients of the peptides, it can be assumed that the formed cobalt conjugates are larger and move slower in the solution than the peptides. Diffusion coefficients (D) were reported in DMSO-d6 for each individual signal and were averaged to obtain molecular diffusion coefficients for the metal-free peptides and the cobalt conjugates, respectively. Then, the Stokes radius as well as a spherical volume ( ) of the molecules using the Stokes–Einstein equation (eqn (1)) were calculated (Table 3).
) of the molecules using the Stokes–Einstein equation (eqn (1)) were calculated (Table 3).
 | (1) |
| Peptides and their cobalt conjugates | Calcd mass (g mol−1) | D (average) (m2 sec−1) | Stokes radius (Angstrom, Å) | Volume (Å3) |
|
|---|---|---|---|---|---|
| P1L1 | 1244.66 | 8.81 | 12.2 | 7590 | 2.58 |
| [CoIII(P1L1)2]3+ | 2548.25 | 6.42 | 16.7 | 19629 |
|
| P2L1 | 1430.77 | 8.5 | 12.6 | 8469 | 2.66 |
| [CoIII(P2L1)2]3+ | 2920.14 | 6.14 | 17.5 | 22516 |
|
| P3L2 | 1817.91 | 7.78 | 13.8 | 11050 |
2.95 |
| [(CoIII(P3L2))2]6+ | 3753.68 | 5.42 | 19.8 | 32634 |
|
| P4L2 | 1445.75 | 7.86 | 13.7 | 10683 |
2.83 |
| [(CoIII(P4L2))2]6+ | 3009.36 | 5.56 | 19.34 | 30317 |
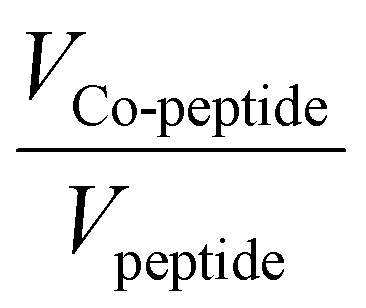
In eqn (1), kB is the Boltzmann constant, η is the viscosity of the solvent in which diffusion occurs at temperature T (for DMSO η = 1.996 Pa s at 293 K), and rs is the Stokes radius of the diffusing species. The following calculation of the volume ratios of cobalt-free peptides and Co-peptide conjugates allows the prediction of the complex stoichiometry.
As can be seen in Table 3, the calculated values of the volume ratios of peptides with one tpy entity to their cobalt complexes are greater than two, which means that two peptides coordinate to one cobalt centre and therefore the stoichiometry of the complex is 2:1 (Scheme 1). Ideally, this volume ratio would be approximately two, if it is assumed that the structure of the molecules is a hard sphere moving in a continuum fluid without any external contributions by solvation or other interactions. However, a deviation from the ideal value can be expected. A study by Allouche et al. demonstrated that the calculated hydrodynamic radius by the modification of the Stokes–Einstein equation for non-spherical species has an approximately similar deviation from the hydrodynamic size (rs) calculated by the general eqn (1) for different non-spherical molecules.53 Therefore, it was confirmed that the Stokes–Einstein approximation is also valid for non-spherical molecules and the derived hydrodynamic size can also be applied for further investigation of molecular systems. The observed deviation of the volume ratio from the ideal ratio can be explained not only by the non-spherical structure of the prepared compounds, but also by the high charge, which is distributed over the whole system, due to the presence of the basic amino acid arginine and the trivalent metal centre. Each positive charge requires an associated anion, which is TFA− or BF4− in the case of the compounds prepared herein. These anions do not diffuse completely independently and therefore contribute to the measured volume. Additionally, a higher charge on the whole molecules leads to the stronger solvation, which in turn leads to the reduction of the diffusion rate. A slight contribution of peptide size to the diffusion coefficient of the peptides and their cobalt conjugates is even detected between very similar compounds of one stoichiometry, e.g. of the CoL2 type. Peptide P2L1 has seven amino acids in its structure and P1L1 only six, and consequently diffusion of P1L1 (and its Co complex) is slightly faster than P2L1 (compare entries 1 vs. 3, and 2 vs. 4 in Table 3). For peptides with two tpy entities, the size effect is more remarkable, as can be seen by the fact that both P1L1 and P4L2 are six amino acid peptides, but P4L2 has two lysine residues and a significantly larger Stokes volume (Table 3).
For the determination of the expected 2:2 stoichiometry of the conjugates with two tpy entities (P3L2 and P4L2), the same calculation as previous was made (Scheme 2 and Table 3). The observed volume ratio is only slightly greater than the volume ratio of the peptides with one tpy entity and 2:1 stoichiometry (compare [CoIII(P1L1)2]3+ and [(CoIII(P4L2))2]6+ for peptides with identical number of amino acids). The reason is very likely the second Co(III) centre, which increases the overall charge of the structure. Furthermore, more tpy entities in the structure of peptides and their cobalt conjugates, as well as more amino acids in case of P3L2 also contribute to a higher molecular weight, but likely also to a greater deviation from the ideal spherical shape.
Potentially, a number of other structures are conceivable especially for P3L2 and P4L2 peptides with more than one tpy ligand on the peptide backbone, some of which are shown in Scheme 2. First, if formation of an intramolecular Co-complex with 1:1 stoichiometry would have occurred (which might be considered entropically favourable), then it would be expected to have a much greater diffusion coefficient due to the smaller size of the molecules. Additionally, during MALDI TOF measurement the mass of the fragment containing only one cobalt and one peptide should be detected, as in the case of cobalt conjugates of P1L1 and P2L1 (Table 2). Neither DOSY nor mass spectra gave a hint to this 1:1 stoichiometry, indicating that intramolecular complex formation does not occur. Possibly, it might be sterically hindered due to the close proximity of the terpyridine entities on the peptide. By contrast, the 3:3 stoichiometry of the complex (Scheme 2) would result in a supramolecular complex with a higher molecular weight. Such a molecule would have a much lower diffusion coefficient and higher volume ratio (in comparison to the two 2:2 complexes) – which again was not observed in our DOSY experiments. Additionally, such complexes with a higher ligand-to-cobalt ratio are entropically unfavorable.54 Therefore, from the existing MS and 2D DOSY NMR data we conclude that P3L2 and P4L2 both form exclusively intermolecular Co complexes with 2:2 stoichiometry.
Influence of metal centre and complex stoichiometry on antimicrobial activity of the peptides
Determination of the minimum inhibitory concentration (MIC) is usually a starting point for more extensive preclinical evaluation of tested antibacterial substances. The MIC value represents the lowest concentration of antimicrobial agents required for visible hindrance of bacterial growth in vitro. The determination of the MIC values was performed as described by Albada et al.,55,56 see experimental section for a detailed description of procedures and Table 4 for results. The MIC data were used in this work to investigate the changes in antimicrobial activity of peptides upon coordination to the cobalt ions. For the calculation of the MIC values in μM, the molecular weights of the peptides together with one TFA counter ion for each basic amino acid residue were used. The molecular weight of the cobalt-peptide conjugates was calculated together with BF4 counter ions.| Peptides and their cobalt conjugates | Minimal inhibitory concentration (μmol l−1) | ||||||
|---|---|---|---|---|---|---|---|
| Gram − | Gram + | Yeast | |||||
| E. coli DSM 30083 | A. baumannii DSM 30007 | P. aeruginosa DSM 50071 | B. subtilis 168 DSM402 | S. aureus DSM 20231 | S. aureus ATCC 43300 | C. albicans DSM1386 | |
| (RW)3 | 21 | 85 | >100 | 3 | 11 | 6 | — |
| P1L1 | 75.2 | >75.2 | >75.2 | 9.4 | >75.2 | >75.2 | 18.8 |
| [CoIII(P1L1)2]3+ | 18.3 | 18.3 | >146.1 | 9.1 | 36.5 | 18.3 | 4.6 |
| P2L1 | 67.8 | >67.8 | 67.8 | 33.9 | 67.8 | 67.8 | 16.9 |
| [CoIII(P2L1)2]3+ | 16.5 | 33 | 66 | 8.2 | 33 | 16.3 | 4.1 |
| P3L1 | >56.3 | >56.3 | >56.3 | 7 | >56.3 | >56.3 | 14.1 |
| [(CoIII(P3L2))2]6+ | >51.5 | >51.5 | >51.5 | 25.7 | 51.5 | 51.5 | 51.5 |
| P4L2 | 134.5 | 67.3 | 67.3 | 33.6 | 33.6 | 33.6 | 4.2 |
| [(CoIII(P4L2))2]6+ | >60.6 | >60.6 | >60.6 | 7.6 | 7.6 | 7.6 | 7.6 |
| [CoIII-(tpy-COOH)2]3+ | >292.9 | >292.9 | >292.9 | >292.9 | >292.9 | >292.9 | >292.9 |
By comparing the observed biological results, it can be concluded that not only double peptide entities in the structure of conjugates but also the complexation with cobalt increases the activity of peptides almost in every case. By and large, peptides and their Co complexes are more active against Gram-positive bacteria than against Gram-negative ones. Importantly however, peptides P1L1 and P2L1 demonstrate higher activity against Gram-negative pathogens upon complexation with cobalt. The obtained values are even smaller than the MIC of the parent (RW)3 peptide against E. coli, A baumannii, and P. aeruginosa. Within the range of peptide-ligand structures tested herein, the greatest activity against Gram-negative pathogens is found for [CoIII(P2L1)2]3+. For instance, introduction of cobalt through the formation of an octahedral complex with two tpy ligands attached on the lysine residue of P1L1 and P2L1 increases the activity against E. coli twofold (considering that the cobalt complexes contain two bactericidal peptide units). In contrast, the peptides P3L2 and P4L2 with two tpy ligands each demonstrate low activity against Gram-negative pathogens, and complexation to cobalt does not make a significant difference. In the case of Gram-positive pathogens (two S. aureus strains, including the multi-resistant ATCC 43300 strain), the greatest activity (7.6 μM) is observed for [(CoIII(P4L2))2]6+. Again, this is approximately twofold greater than the activity of the cobalt-free peptide P4L2 (considering that [(CoIII(P4L2))2]6+ has two peptides in its structure). This activity matches that of the parent (RW)3 peptide against the same S. aureus strains, with 11 μM and 6 μM, respectively.
As one would expect, exchanging Trp for Lys(tpy) influences the activity of the metal-free peptides, but not in a uniform way. Exchanging one or more Trp residues strongly decreases activity of the peptide against Gram-positive bacteria. Surprisingly however, P1L1 and P2L1 with only one Lys(tpy) moiety instead of Trp in their structures, are less active against S. aureus strains than P4L2 with two Trp exchanged against Lys(tpy) moieties. On the other hand, (RW)3 has low activity against any Gram-negative strain, and swapping Trp for Lys(tpy) makes very little difference overall. It is also interesting to note that the compounds exhibit a peculiar bactericidal behaviour against the Gram-positive bacterium B. subtilis. For example, the presence of cobalt in the P1L1 structure has only a slight influence on the activity, while adding cobalt to peptides P4L2 and P2L1 significantly increases the activity. In the case of P3L2, the bactericidal activity even decreases threefold through the insertion of cobalt. It remains for future experiments to determine whether these remarkably non-uniform tendencies are determined by the hydrophobic character, modified membrane interactions due to less Trp residues, or simply a consequence of differential bacterial uptake in the different species.
Remarkably, minimum inhibitory concentrations against the opportunistic pathogenic yeast C. albicans range between 4.13 μM and 51.5 μM, thereby indicating a potent antimicrobial activity of peptides and their cobalt conjugates. The greatest activity against C. albicans was observed for [CoIII(P2L1)2]3+ (4.1 μM), which overall exhibits the best broad-band activity across all tested pathogens. Again, the insertion of cobalt also reduces the activity of P3L2, as in the case of Gram-positive B. subtilis, making it less active than the metal-free peptide. Finally, the [CoIII(tpy-COOH)2]3+ complex is not active against any of the tested bacteria and yeast (MIC values > 293 μM), thereby ruling out an activity of the Co(tpy)2 complex, or the metal ion itself after decomplexation.
Conclusion
In this work, the synthesis of four new antimicrobial peptides featuring terpyridine ligands as metal binding sites was achieved. Addition of [CoII(H2O)](BF4)2 resulted in self-assembly to selectively form L2M and L4M2 bioconjugates, depending on the number of tpy ligands on the peptide. The intended complex formation and stoichiometry could be confirmed by MALDI-TOF mass spectrometry. Further characterisation by DOSY NMR experiment showed the purity of the conjugates and indicated the intermolecular complex formation based on the obtained diffusion coefficients. Preliminary biological tests (determination of minimum inhibitory concentration) of the cobalt-peptide conjugates showed that the complexation increased almost in every case the antimicrobial activity of peptides. Interestingly, the presence of cobalt ion in the structure of P1L1 and P2L1 increased activity against E. coli, S. aureus ATCC 43300, yeast C. albicans and B. subtilis 168 DSM402 in case of P1L1 twofold (considering that in the cobalt complexes are two bactericidal peptide units). The greatest activity against C. albicans was observed for [CoIII(P2L1)2]3+ (4.1 μM).Future research will focus on further biological tests to understand the bactericidal role of cobalt ion in the structure of peptides. Since the mode of action of (RW)3 peptide is well studied, the proteomic profiling of synthesized conjugates can provide the information about the influence of cobalt ion on mechanism of action of peptides.
Author contributions
L. J.: investigation, visualization, writing – original draft; R. G. M.: supervision, writing – review and editing; N. M.-N.: conceptualization, supervision, funding acquisition, writing – review and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to Pascal Dietze and Prof. Dr Julia Bandow for the Measurement of minimal inhibitory concentration (MIC) of isolated peptides and their cobalt conjugates. R. G. M. is grateful to the Alexander von Humboldt Foundation for a postdoctoral fellowship.References
- J. W. Alexander, Surg. Infect., 2009, 10, 289–292 CrossRef PubMed.
- B. Albada and N. Metzler-Nolte, Acc. Chem. Res., 2017, 50, 2510–2518 CrossRef CAS PubMed.
- J. Liang, D. Sun, Y. Yang, M. Li, H. Li and L. Chen, Eur. J. Med. Chem., 2021, 224, 113696 CrossRef CAS PubMed.
- A. Evans and K. A. Kavanagh, J. Med. Microbiol., 2021, 70, 001363 CrossRef CAS PubMed.
- P. Biegański, Ł. Szczupak, M. Arruebo and K. Kowalski, RSC Chem. Biol., 2021, 2, 368–386 RSC.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- Y. Lin, H. Betts, S. Keller, K. Cariou and G. Gasser, Chem. Soc. Rev., 2021, 50, 10346–10402 RSC.
- A. Pandey and E. Boros, Chem. – Eur. J., 2021, 27, 7340–7350 CrossRef CAS PubMed.
- M. Claudel, J. V. Schwarte and K. M. Fromm, Chemistry, 2020, 2, 849–899 CrossRef CAS.
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877–888 CrossRef CAS PubMed.
- L. Macomber and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8344–8349 CrossRef CAS PubMed.
- J. B. Wright, K. Lam and R. E. Burrell, Am. J. Infect. Control, 1998, 26, 572–577 CrossRef CAS PubMed.
- L. J. Childs, J. Malina, B. E. Rolfsnes, M. Pascu, M. J. Prieto, M. J. Broome, P. M. Rodger, E. Sletten, V. Moreno, A. Rodger and M. J. Hannon, Chem. – Eur. J., 2006, 12, 4919–4927 CrossRef CAS PubMed.
- A. D. Richards, A. Rodger, M. J. Hannon and A. Bolhuis, Int. J. Antimicrob. Agents, 2009, 33, 469–472 CrossRef CAS PubMed.
- S. V. Kumar, W. K. C. Lo, H. J. L. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1–6 CrossRef CAS.
- K. V. R. Reddy, R. D. Yedery and C. Aranha, Int. J. Antimicrob. Agents, 2004, 24, 536–547 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- E. F. Haney, S. C. Mansour and R. E. W. Hancock, in Antimicrobial Peptides, ed. P. R. Hansen, Springer New York, New York, NY, 2017, vol. 1548, pp. 3–22 Search PubMed.
- B. Albada and N. Metzler-Nolte, Chem. Rev., 2016, 116, 11797–11839 CrossRef CAS PubMed.
- H. B. Albada, A.-I. Chiriac, M. Wenzel, M. Penkova, J. E. Bandow, H.-G. Sahl and N. Metzler-Nolte, Beilstein J. Org. Chem., 2012, 8, 1753–1764 CrossRef CAS PubMed.
- E. G. Seebauer, E. P. Duliba, D. A. Scogin, R. B. Gennis and R. L. Belford, J. Am. Chem. Soc., 1983, 105, 4926–4929 CrossRef CAS.
- S. Rao and G. Venkateswerlu, Curr. Microbiol., 1989, 19, 253–258 CrossRef CAS.
- L.-J. Ming and J. D. Epperson, J. Inorg. Biochem., 2002, 91, 46–58 CrossRef CAS PubMed.
- J. T. Garbutt, A. L. Morehouse and A. M. Hanson, J. Agric. Food Chem., 1961, 9, 285–289 CrossRef CAS.
- Y. Sugiura, Biochem. Biophys. Res. Commun., 1979, 90, 375–383 CrossRef CAS PubMed.
- M. B. Strøm, B. E. Haug, M. L. Skar, W. Stensen, T. Stiberg and J. S. Svendsen, J. Med. Chem., 2003, 46, 1567–1570 CrossRef PubMed.
- R. A. Murphy, M. Coates, S. Thrane, A. Sabnis, J. Harrison, S. Schelenz, A. M. Edwards, T. Vorup-Jensen and J. C. Davies, Microbiol. Spectrum, 2022, 10, e00813–e00822 Search PubMed.
- M. Wenzel, A. I. Chiriac, A. Otto, D. Zweytick, C. May, C. Schumacher, R. Gust, H. B. Albada, M. Penkova, U. Kramer, R. Erdmann, N. Metzler-Nolte, S. K. Straus, E. Bremer, D. Becher, H. Brotz-Oesterhelt, H.-G. Sahl and J. E. Bandow, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1409–E1418 CrossRef CAS PubMed.
- R. Banerjee and S. W. Ragsdale, Annu. Rev. Biochem., 2003, 72, 209–247 CrossRef CAS PubMed.
- A. Abebe, Y. Bayeh, M. Belay, T. Gebretsadik, M. Thomas and W. Linert, Future J. Pharm. Sci., 2020, 6, 13 CrossRef.
- M. N. Akhtar, W. T. A. Harrison, M. Shahid, I.-U. Khan, E. Iqbal and J. Iqbal, Transition Met. Chem., 2016, 41, 325–330 CrossRef CAS.
- E. L. Chang, C. Simmers and D. A. Knight, Pharmaceuticals, 2010, 3, 1711–1728 CrossRef CAS PubMed.
- Z. H. Chohan, A. Rauf, S. Noreen, A. Scozzafava and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2002, 17, 101–106 CrossRef CAS PubMed.
- J. T. P. Matshwele, S. Odisitse, O. Mazimba, T. B. Demissie, M. O. Koobotse, D. T. Mapolelo, K. Bati, L. G. Julius, D. O. Nkwe, M. Jongman and F. M. Nareetsile, Inorg. Chim. Acta, 2024, 563, 121911 CrossRef CAS.
- P. Nagababu, J. N. L. Latha, P. Pallavi, S. Harish and S. Satyanarayana, Can. J. Microbiol., 2006, 52, 1247–1254 CrossRef CAS PubMed.
- M. Patel, M. Chhasatia and B. Bhatt, Med. Chem. Res., 2011, 20, 220–230 CrossRef CAS.
- N. Şahin, E. Üstün, İ. Özdemir, S. Günal, N. Özdemir, H. Bülbül, N. Gürbüz, İ. Özdemir and D. Sémeril, Inorg. Chem. Commun., 2023, 157, 111396 CrossRef.
- A. Rambabu, N. Ganji and S. Daravath, J. Fluoresc., 2020, 30, 1397–1410 CrossRef CAS PubMed.
- S. A. Khan, S. A. A. Nami, S. A. Bhat, A. Kareem and N. Nishat, Microb. Pathog., 2017, 110, 414–425 CrossRef CAS PubMed.
- L. Allouche, A. Marquis and J. Lehn, Chem. – Eur. J., 2006, 12, 7520–7525 CrossRef CAS PubMed.
- C. A. J. Hooper, L. Cardo, J. S. Craig, L. Melidis, A. Garai, R. T. Egan, V. Sadovnikova, F. Burkert, L. Male, N. J. Hodges, D. F. Browning, R. Rosas, F. Liu, F. V. Rocha, M. A. Lima, S. Liu, D. Bardelang and M. J. Hannon, J. Am. Chem. Soc., 2020, 142, 20651–20660 CrossRef CAS PubMed.
- J. T. Edward, J. Chem. Educ., 1970, 47, 261 CrossRef CAS.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520–554 CrossRef CAS PubMed.
- J. Husson, J. Dehaudt and L. Guyard, Nat. Protoc., 2013, 9, 21–26 CrossRef PubMed.
- J. T. Chantson, M. Vittoria Verga Falzacappa, S. Crovella and N. Metzler-Nolte, ChemMedChem, 2006, 1, 1268–1274 CrossRef CAS PubMed.
- G. Dirscherl and B. König, Eur. J. Org. Chem., 2008, 597–634 CrossRef CAS.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- N. Zhang, J. Yang, R.-X. Hu and M.-B. Zhang, Z. Anorg. Allg. Chem., 2013, 639, 197–202 CrossRef CAS.
- H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373–399 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- L. Allouche, A. Marquis and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 7520–7525 CrossRef CAS PubMed.
- P. W. Atkins and J. De Paula, Physikalische Chemie. Hauptband, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 5. Auflage, 2013 Search PubMed.
- H. B. Albada, A.-I. Chiriac, M. Wenzel, M. Penkova, J. E. Bandow, H.-G. Sahl and N. Metzler-Nolte, Beilstein J. Org. Chem., 2012, 8, 1753–1764 CrossRef CAS PubMed.
- H. B. Albada, P. Prochnow, S. Bobersky, S. Langklotz, J. E. Bandow and N. Metzler-Nolte, ACS Comb. Sci., 2013, 15, 585–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00907j |
| This journal is © The Royal Society of Chemistry 2024 |