
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards structurally versatile mesoionic N-heterocyclic olefin ligands and their coordination to palladium, gold, and boron hydride†
Tisa
Ževart
a,
Balazs
Pinter‡§
b,
Matic
Lozinšek
 c,
Damijana
Urankar
a,
Ross D.
Jansen-van Vuuren
a and
Janez
Košmrlj
*a
c,
Damijana
Urankar
a,
Ross D.
Jansen-van Vuuren
a and
Janez
Košmrlj
*a
aFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI 1000 Ljubljana, Slovenia. E-mail: Janez.Kosmrlj@fkkt.uni-lj.si
bThe University of Texas at El Paso, 500 West University Avenue, El Paso, TX 79968, USA
cJožef Stefan Institute, Jamova cesta 39, SI 1000 Ljubljana, Slovenia
First published on 28th March 2024
Abstract
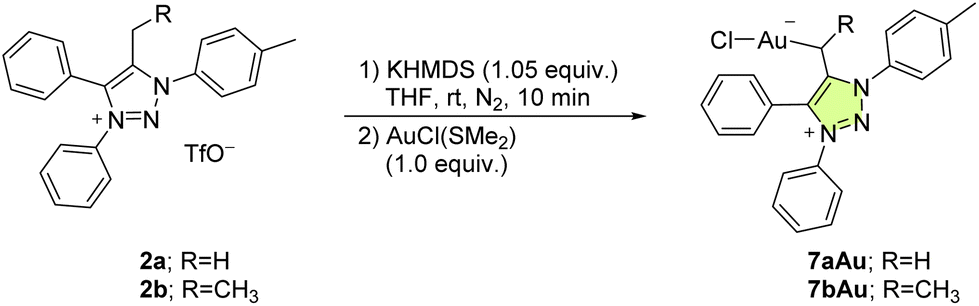
We have developed an efficient and versatile approach for the synthesis of a family of 1,2,3-triazole-based mesoionic N-heterocyclic olefin (mNHO) ligands and investigated their coordination to palladium, gold, and boron hydride experimentally and computationally. We reacted mNHOs obtained through deprotonation of the corresponding methylated and ethylated 1,3,4-triaryl-1,2,3-triazolium salts with [Pd(allyl)Cl]2 to give the corresponding [Pd(η3-allyl)Cl(mNHO)] coordination complexes. 13C NMR data revealed the strong σ-donor character of the mNHO ligands, consistent with the calculated bond orders and atom-condensed charges. Furthermore, we also synthesized [AuCl(mNHO)] and a BH3–mNHO adduct by reacting the triazolium salts with AuCl(SMe2) and BH3·THF, respectively. The BH3–mNHO adduct was tested in the reduction of select aldehydes and ketones to alcohols.
Introduction
N-Heterocyclic olefins were first prepared in 19931via deprotonation of methylated N,N′-dimethylimidazolium salts (NHOs, Fig. 1a) and have been identified as a promising new class of electron-rich and carbon-based ligands.2–10 Imidazole-based NHO coordination compounds with Mo,11 Rh,12–14 Au,10,13 W,11,15 Ir,5,16 Pd,8,14 and Ti17 have been reported. Their utility in catalysis is also documented.8,15,16,18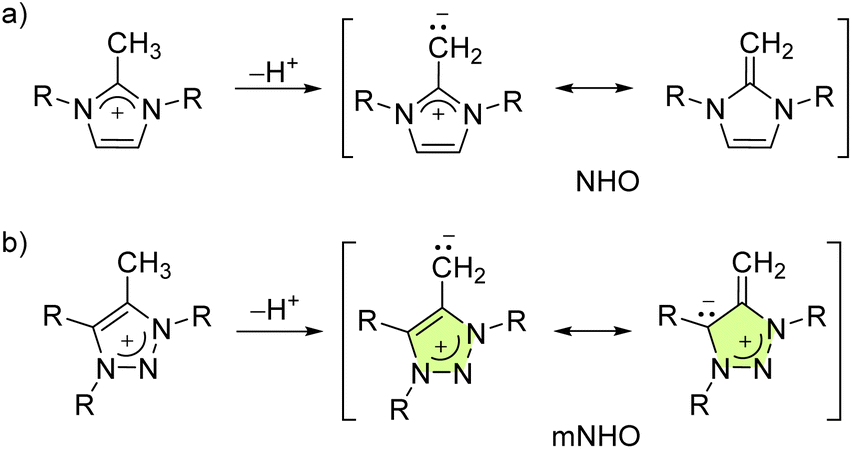 | ||
| Fig. 1 Formation and representative mesomeric structures of: (a) methylated N,N′-disubstituted imidazolium salts to form the corresponding imidazole-based N-heterocyclic olefins (NHOs), and (b) C5-methylated 1,3,4-trisubstituted-1,2,3-triazolium salts to form 1,2,3-triazole based mesoionic N-heterocyclic olefins (mNHOs). | ||
Subsequently, mesoionic N-heterocyclic olefins (mNHOs, Fig. 1b) were obtained in 2020 by the deprotonation of C5-methylated 1,3-diaryl-1,2,3-triazolium salts.19 Unlike NHOs, the ground state structures of mNHOs cannot be described by an uncharged mesomeric Lewis structure, giving them increased basicity and donor abilities (Fig. 1). These triazole-based mNHOs have been investigated as catalysts for hydroboration20 and N-methylation of primary amines.21 Reactions of C4-unsubstituted triazole-based mNHOs with Lewis acids (CO2, AlMe3, BH3·SMe2 and 9-borabicyclo[3.3.1]nonane),22,23 oxygen24 and aryl azides25 have also been described recently. mNHOs recently provided access to diazoolefins and the corresponding copper complexes.26–29
Five rhodium–carbonyl complexes with mNHO ligands have been reported, three of them being triazole-based [Fig. 2a(i)].3,19,30 To the best of our knowledge, these are the only known coordination compounds with mNHOs apart from the related but peculiar complexes with diazoolefin ligands [Fig. 2a(ii)].29 The studies indicated that mNHOs are more strongly basic compared to traditional NHOs and are much stronger σ-donors than both their NHOs and N-heterocyclic carbene (NHC) counterparts. However, unlike NHCs, they are unable to participate in π-backdonation.2,19
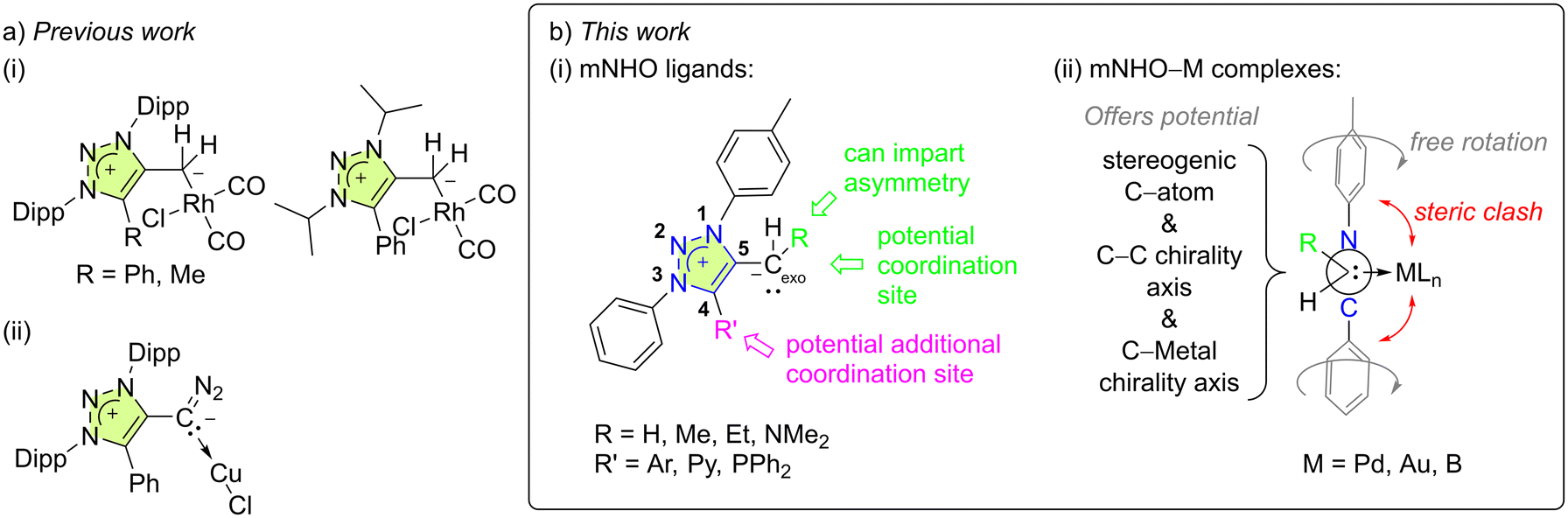 | ||
| Fig. 2 (a) (i) Previously reported complexes with triazole-based mNHOs Dipp = 2,6-diisopropylphenyl and (ii) triazole-based diazoolefin ligands. (b) This work, featuring: (i) mNHO ligand design, and (ii) mNHO–M coordination compounds that potentially exhibit non-biaryl C(sp2)–C(sp3) atropisomerism (providing R ≠ H) as represented by a Newman projection, viewed along the Cexo–C5 (also Cendo) vector. | ||
Thus, to further advance the field and reveal the hitherto unknown peculiarities of this ligand family, we have established novel efficient routes to structurally different 1,2,3-triazole-based mNHOs that can serve as versatile ligands to metals, and investigated their coordination properties towards palladium, gold, and boron hydride. Coordination of a prochiral mNHO (Fig. 2b (i), R ≠ H) to a square-planar palladium complex was anticipated to give a novel example of non-biaryl C(sp2)–C(sp3) atropisomer (Fig. 2b(ii)), and this was also investigated.
Results and discussion
mNHO ligands
We chose to investigate mNHOs with simple phenyl ring functionalities at the N1 and N3 positions, as shown in 1a (Fig. 3): the p-tolyl substituent at N1 was chosen to enable the compounds under investigation in the reaction mixtures to be easily followed by NMR spectroscopy via the distinct resonance of the methyl protons. Instead of employing triazene–alkyne cycloaddition, which is commonly used to prepare symmetric 1,3-diaryl-1,2,3-triazole based scaffolds,31–33 we chose to utilize copper-catalyzed click chemistry with subsequent N3 arylation as it allows for greater structural diversification.34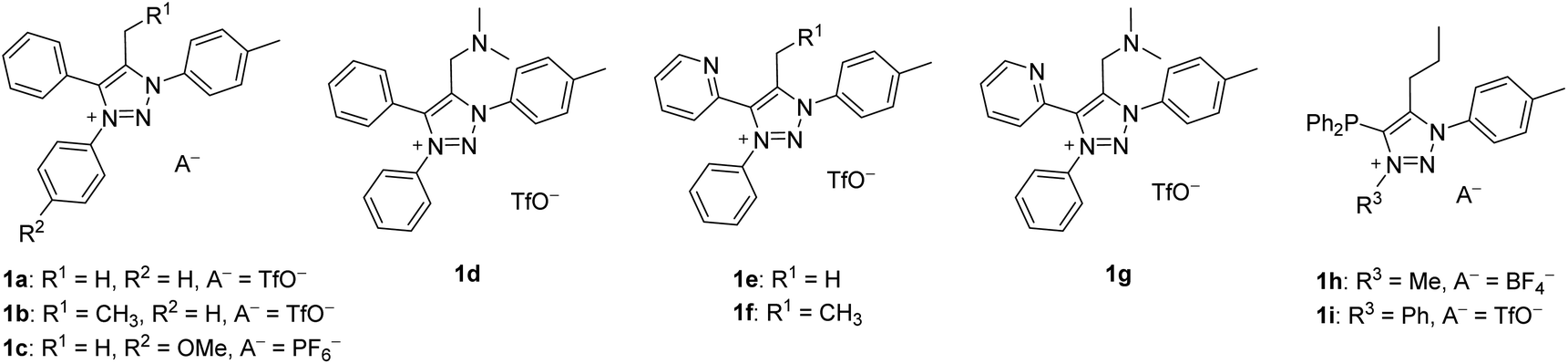 | ||
| Fig. 3 The chemical structures of compounds 1a–i initially planned for the synthesis. | ||
We designed two mNHO pro-ligands 1d and 1g, which potentially offer coordination through either the mNHO or the terminal nitrogen atom of the dimethylamine functionality. While compounds 1a–d can provide monodentate exocyclic carbon-ligation upon deprotonation, the general lack of stability of mNHO–metal compounds compelled us to consider pyridine- and phosphine-functionalized analogues 1e–g and 1h–i, which should enable C^N- and C^P-bidentate coordination, respectively, thus conferring additional stability to the complexes due to the chelating effect. Upon deprotonation and Cexo-ligation, the substrates 1a, c, and e with a methyl group at the triazole C5 would potentially form conformationally constrained structures with limited rotation around the Cexo–Cendo vector, leading to an interesting class of monodentate atropochiral ligands.35 In addition, in the same process, the prochiral substrates 1b, d, and f–i (Fig. 2b(i), R ≠ H) would acquire a new stereogenic centre.
The synthesis of C5-substituted triazolium salts 1a–g commenced from ‘click triazoles’ 2 which, upon treatment with iodonium salt, gave 1,3,4-triaryl substituted triazolium triflates 3a and b (Scheme 1a).34 Triazolium salts 3a and b were then alkylated at C5 to give 1a–b, and e–g. This approach for the target compounds enables the synthetic steps to be reversed, e.g., for the synthesis of 1c, starting with C5-methylated triazole 2a’36 could be N3-arylated as shown in Scheme 1b.
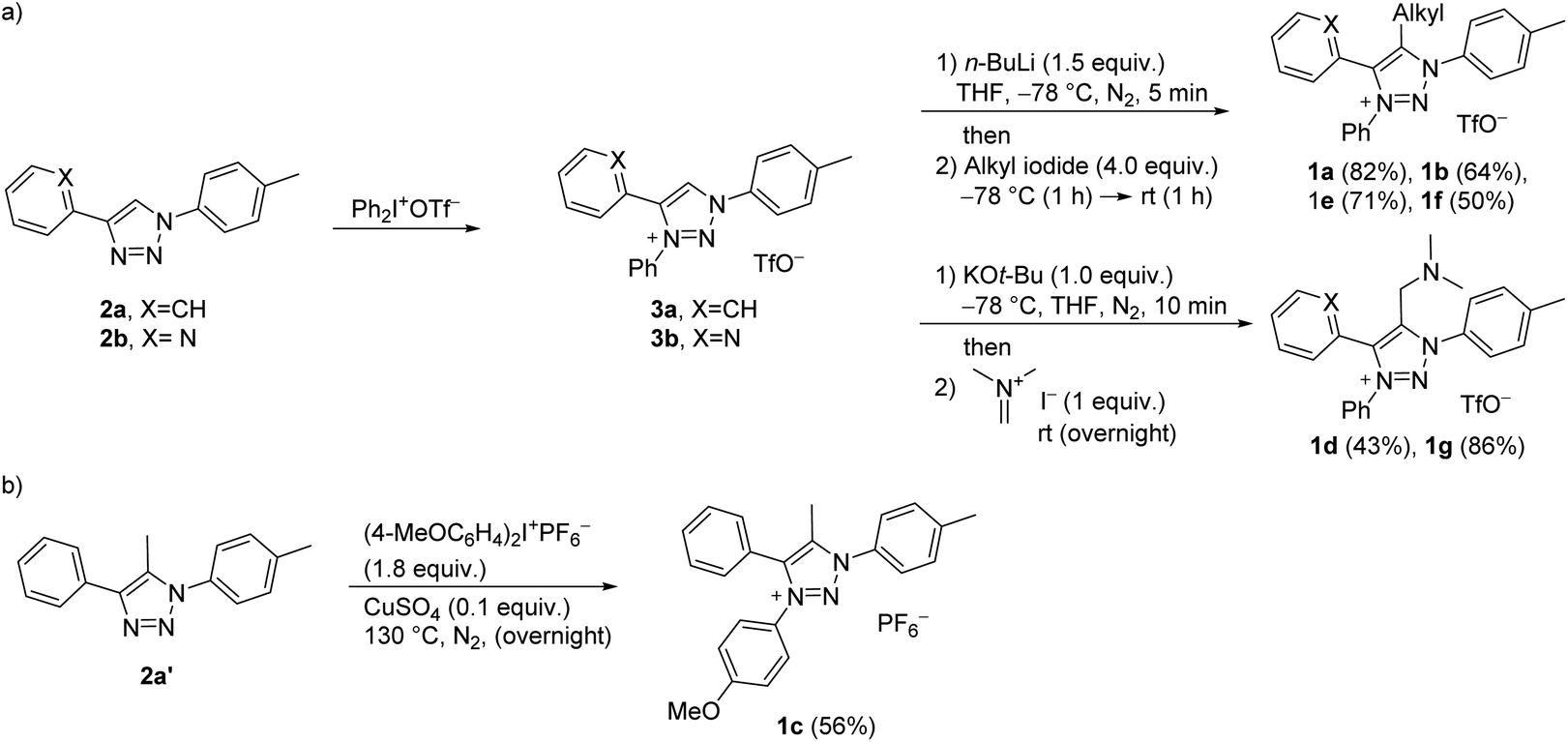 | ||
| Scheme 1 Two alternative approaches for 1,3,4,5-tetrasubstituted triazolium salts 1 from click triazoles 2: (a) N3-arylation of triazoles 2a,b with diphenyliodonium triflate gives triazolium salts 3a,b, which upon treatment with a base (n-BuLi or KOt-Bu) and electrophile (alkyl iodide or N,N-dimethylmethyleneammonium iodide) affords target compounds 1a–b, and e–g. (b) N3-arylation of 1,4,5-trisubstituted triazole 2a′ with bis(4-methoxyphenyl)iodonium hexafluorophosphate to give the target 1c. Percentage yields of isolated pure products are reported. | ||
For the synthesis of diphenylphosphine functionalized triazolium salt 1h, pent-1-yne (4) was converted to triazole 5 according to the modified literature procedure,37 which was then N3 methylated with Merwein salt (Scheme 2a). For the arylation of 5 with iodonium salt, the phosphorus atom was BH3-protected to form 6 (Scheme 2b), as it was presumed that the relatively harsh reaction conditions would otherwise lead to undesired P- rather than N-arylation. It turned out, however, that BH3 protection in 6 was not tolerated by the arylation protocol; in situ phosphine deprotection occurred prior to arylation and we isolated the phosphonium salt 1j instead of the desired product 1i (Scheme 2b).
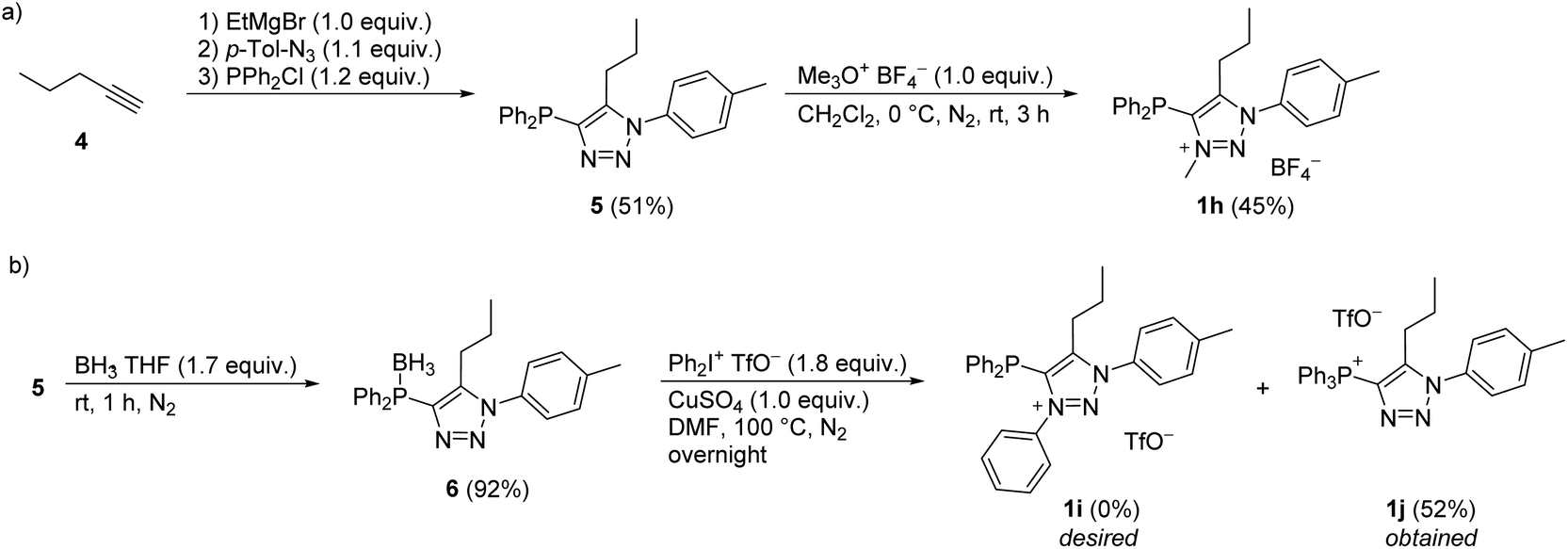 | ||
| Scheme 2 Synthesis of compounds 1h and attempted preparation of 1i, forming 1j instead. Percentage yields of isolated pure products are reported. | ||
To obtain the corresponding mNHO derivatives, deprotonation of this set of triazolium salts 1 was attempted using an equimolar amount of potassium bis(trimethylsilyl)amide (KHMDS, Scheme 3) in dry THF at room temperature. For 1a, this resulted in an immediate colour change, with the reaction mixture turning purple, indicating the deprotonation and formation of mNHO 7a [Scheme 3(a)]. This colour change was consistent with what was previously reported19 indicating the resonance stabilisation of the negatively polarised Cexo− moiety with the cationic triazole unit. With the other triazolium salts (1b–1g), the reaction mixtures instantly turned purple (7b and 7c), dark blue (7d, 7e, and 7f) or green (7g) [the structures of corresponding mNHOs are shown in Scheme 3(b)]. According to 1H NMR analyses of the reaction mixtures, the deprotonation of triazolium salts 1a–1g with KHMDS to 7a–7g was quantitative.
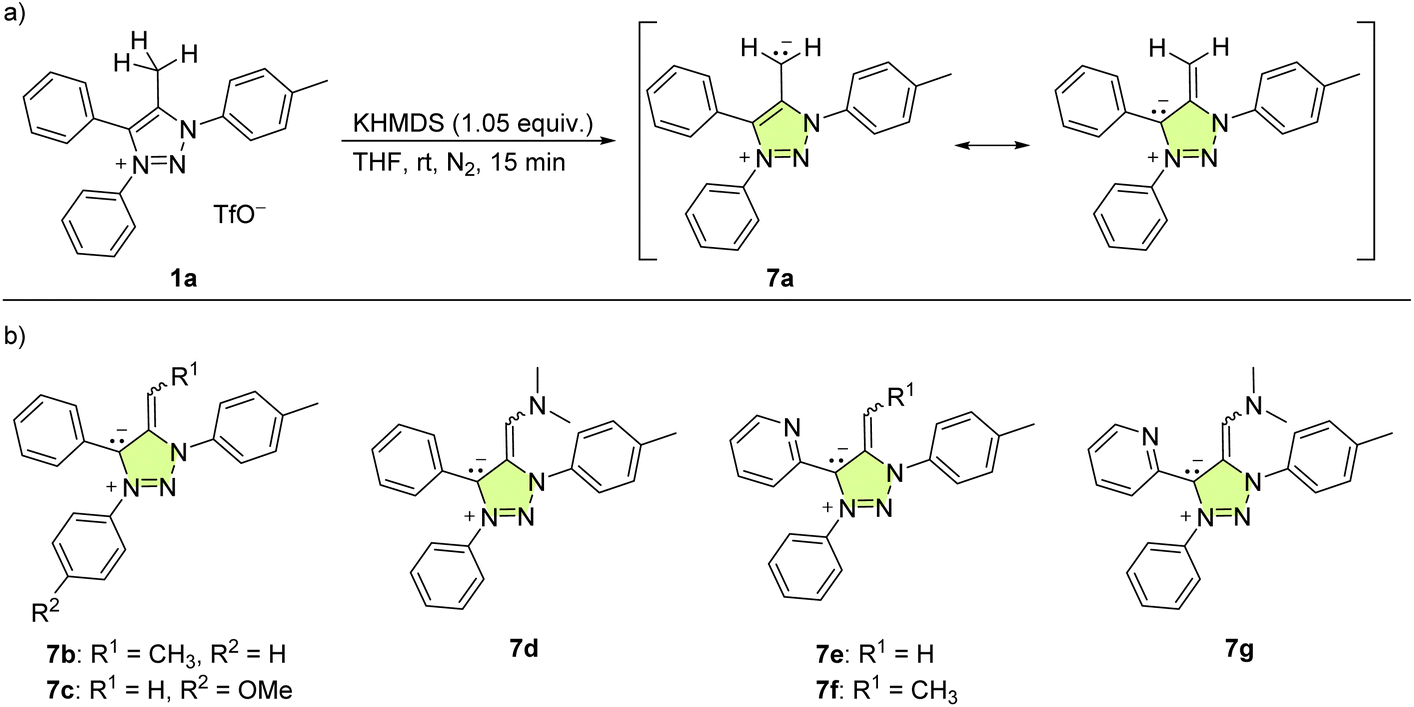 | ||
| Scheme 3 (a) Deprotonation of 1a to mNHO 7a; (b) the structures of the other mNHOs (7b–7g) that were formed. | ||
Deprotonation of 1a to 7a with KHMDS in THF-d8 was monitored by 1H NMR spectroscopy. The spectra indicated the appearance of two doublets at δ = 3.18 and 3.25 ppm corresponding to non-equivalent geminal CexoH2 protons. The chemical shifts were in the range observed for olefinic protons and in agreement with those reported previously for CexoH2 protons in related compounds.19 In the case of 7b, obtained by in situ KHMDS deprotonation of 1b, the 1H NMR spectra showed the appearance of two overlapping quartets at δ = 3.87 and 3.84 ppm (J = 7.4 and 7.3 Hz) in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7 ratio that could be assigned to the methine CexoH protons of the two geometric isomers E and Z at the exocyclic double bond (Fig. 4). The resonances for the methyl groups appeared as two overlapping doublets at δ = 1.26 and 1.24 ppm (J = 7.4 and 7.3 Hz). A similar observation of the geometric isomers was made for 7d, 7f and 7g (obtained from 1d, 1f and 1g, respectively). On the other hand, the aromatic regions of the 1H NMR spectra were in all cases overcrowded with overlapping resonances; those for the methyl groups of the p-tolyls unambiguously showed the presence of the two isomers. All mNHOs 7 were found to be sensitive to moisture, instantly reforming the triazolium salts 1 upon exposure to the atmosphere.
:0.7 ratio that could be assigned to the methine CexoH protons of the two geometric isomers E and Z at the exocyclic double bond (Fig. 4). The resonances for the methyl groups appeared as two overlapping doublets at δ = 1.26 and 1.24 ppm (J = 7.4 and 7.3 Hz). A similar observation of the geometric isomers was made for 7d, 7f and 7g (obtained from 1d, 1f and 1g, respectively). On the other hand, the aromatic regions of the 1H NMR spectra were in all cases overcrowded with overlapping resonances; those for the methyl groups of the p-tolyls unambiguously showed the presence of the two isomers. All mNHOs 7 were found to be sensitive to moisture, instantly reforming the triazolium salts 1 upon exposure to the atmosphere.
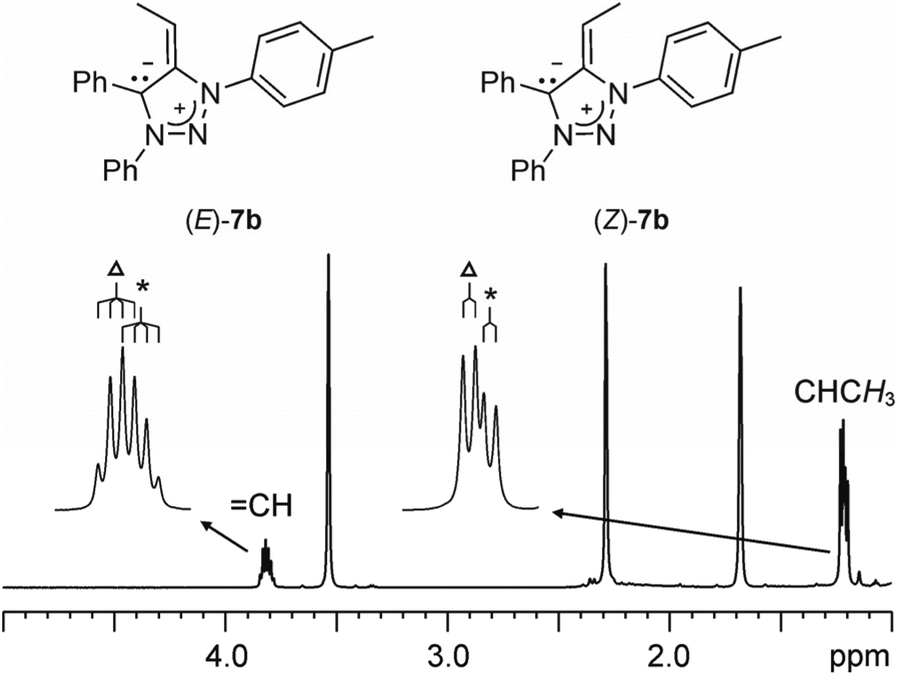 | ||
| Fig. 4 The selected region of 1H NMR (THF-d8) spectrum of in situ generated 7b. Δ and * denote the major and minor isomers, respectively. | ||
The treatment of 1j with KHMDS resulted in the formation of triphenyl(1,2,3-triazol-4-yl)phosphonium ylide (8j) based on the NMR analysis (Scheme 4a, and Fig. S1†). Interestingly, related N-unsubstituted 4-aryl-5-triphenylphosphonium-l,2,3-triazole ylides were reported in 1973,38 and since then have not been investigated further.
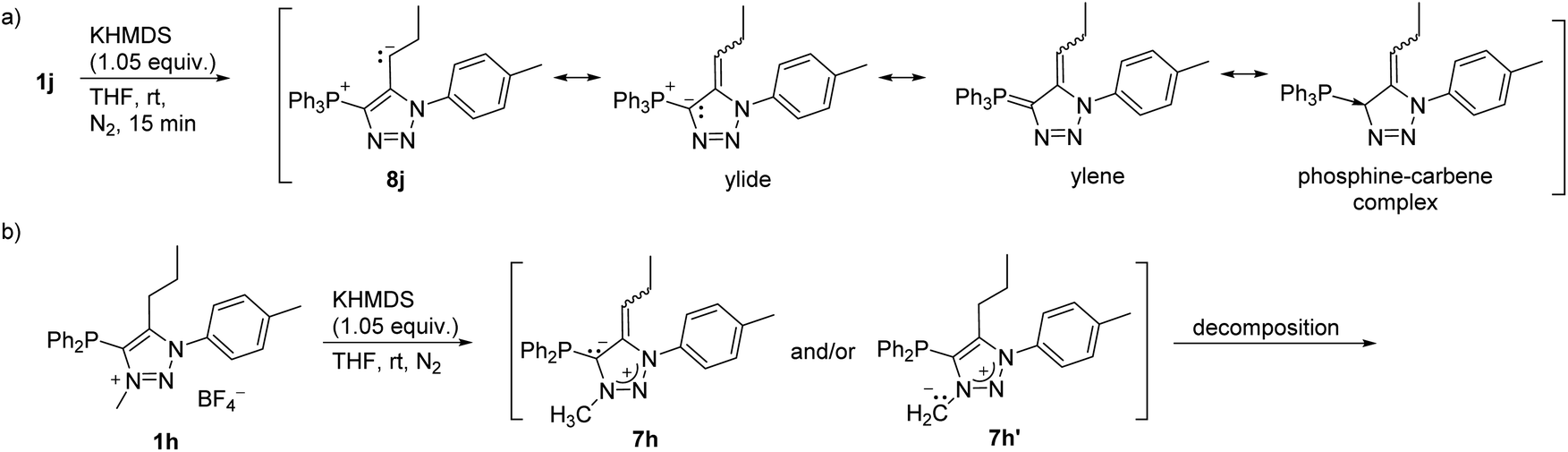 | ||
| Scheme 4 (a) Deprotonation of 1j to 8j, showing the canonical structures of 8j; (b) deprotonation of 1h. | ||
The treatment of 1h with KHMDS was accompanied by an immediate colour change to dark red, indicating deprotonation (Scheme 4b); however, the NMR spectra of the reaction mixture acquired shortly after the addition of a base revealed decomposition to a complex mixture of unidentified products that likely proceeds through 7h and/or 7h′.
mNHO–palladium coordination compounds
We selected 7a as a model compound to probe coordination abilities toward palladium. Treatment of in situ formed 7a with [Pd(allyl)Cl]2 in dry THF at room temperature instantly afforded 7aPd (Table 1). The crude product was dissolved in diethyl ether and the solution layered with pentane at −20 °C to form a yellow precipitate, which was isolated by filtration. This yellow solid was stable for several days under a nitrogen atmosphere at room temperature but began to decompose after prolonged storage. Various attempts to prepare crystals of 7aPd suitable for X-ray analysis (including using different solvents, solvent mixtures, temperatures, and crystallisation techniques) were unsuccessful. Thus, 7aPd was characterized by a combination of mass spectrometry (Fig. S2) and NMR spectroscopy (see the ESI†) and its structure was corroborated by density functional theory (DFT) calculations (see below).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | NMR solvent | δ C1(allyl) | δ C2(allyl) | δ C3(allyl) | δ CexoH | δ Cexo | δ C4 | δ C5 | δ N1 |
| 13C NMR chemical shifts were extracted from the 1H–13C gs-HSQC and 1H–13C gs-HMBC spectra.a Two resonances for the two stereoisomers are given. The first and second figures refer to the upfield and downfield resonances, respectively. | |||||||||
| 1a | CDCl3 | 2.51 | 11.0 | 140.9 | 140.1 | 250 | |||
| 7a | THF-d8 | 3.18, 3.25 | 46.8 | 120.1 | 146.1 | 192 | |||
| 7aPd | THF | 63.6 | 109.9 | 53.7 | 2.01, 2.33 | −1.6 | 130.2 | 161.6 | 238 |
| 7bPd | THF | 62.4, 63.4 | 109.8, 111.4 | 56.0, 56.1 | 2.98, 3.30a | 10.7, 10.9a | 130.7, 130.7a | 162.2, 162.3a | |
High-resolution (HRMS) electrospray ionization mass spectrometry in a positive ion mode (ESI+) revealed a [M − Cl]+ ion at m/z 472.0997 (calcd for C25H24N3Pd+: 472.1005) and was interpreted as the result of in-source collision-induced dissociation of 7aPd (Fig. S2a†). The peak that corresponds to [(1a–TfO)]+m/z 326.1647 (calcd for C22H20N3+: 326.1652) was the most intense in the spectrum, likely the result of hydrolysis of 7aPd during the sample preparation.
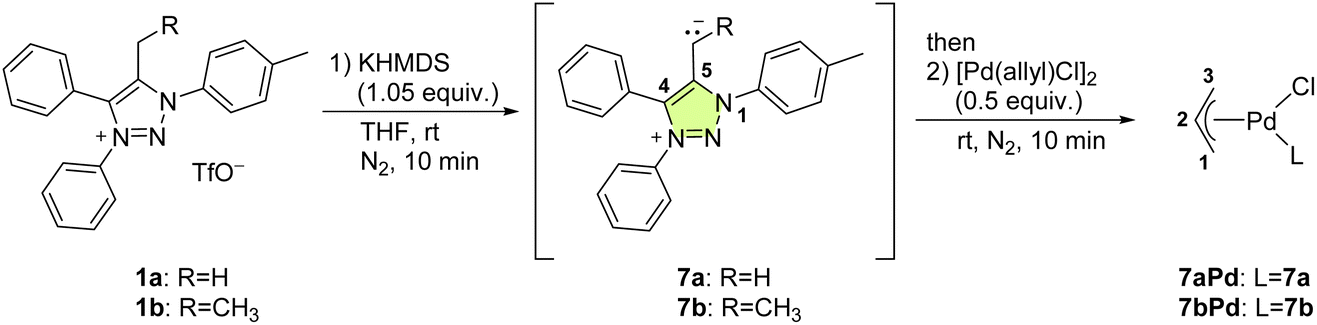
The structure of 7aPd consists of a neutral square planar complex of Pd(II) in which mNHO monodentately coordinates to Pd through the exocyclic carbon (Cexo) atom. The allyl ligand binds to palladium in an η3-fashion with its two terminal atoms occupying cis-positions and the coordination sphere is completed by a chloride (Fig. 5a), also shown in Table 1.
 | ||
| Fig. 5 (a) Graphical representation of planes and possible stereogenic axes in 7aPd. (b) Axial chirality of a square-planar complex with 2-methylpyridine as an exemplary ligand from the literature.39 | ||
A square-planar complex may be chiral under certain conditions even if it does not consist of chiral ligand(s).39 The chirality in a generic d8cis-M(MePy)AB2 platform, shown in Fig. 5b with 2-methylpyridine (a monodentate planar ring with only Cs local symmetry) as one of the ligands, originates from the rotation around the M–N bond that generates the opposite enantiomer; this pair of enantiomers can be described in terms of axial chirality.40,41 In addition to pyridine and some other heterocyclic ligands, square-planar metal complexes incorporating carbene ligands with the heterocyclic ring perpendicular to the coordination plane showing axial chirality are well documented.42
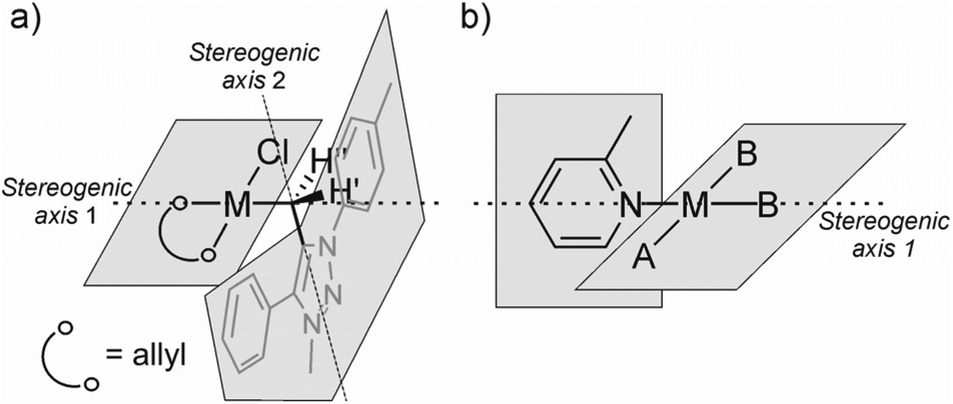
In line with the experimentally deduced structure of 7aPd, DFT calculations demonstrate such an arrangement and configuration to be a stable, ideal d8 square-planar molecular structure.43 The computed equilibrium structure, shown in Fig. 6, in addition, reveals the in-plane orientation of the Cexo–Cendo bond of the mNHO ligand with respect to the plane of the complex. The emerging two conformers, namely Cexo–Cendo pointing towards or away from Cl−, differ by 6 kcal mol−1 in Gibbs free energy, the former isomer being the more stable. Relaxed scan calculations show that the two isomers are inter-connected through a shallow potential energy surface with a rotational maximum at 8 kcal mol−1 to the more stable isomer, indicating free rotation along the Pd–Cexo bond at room temperature.
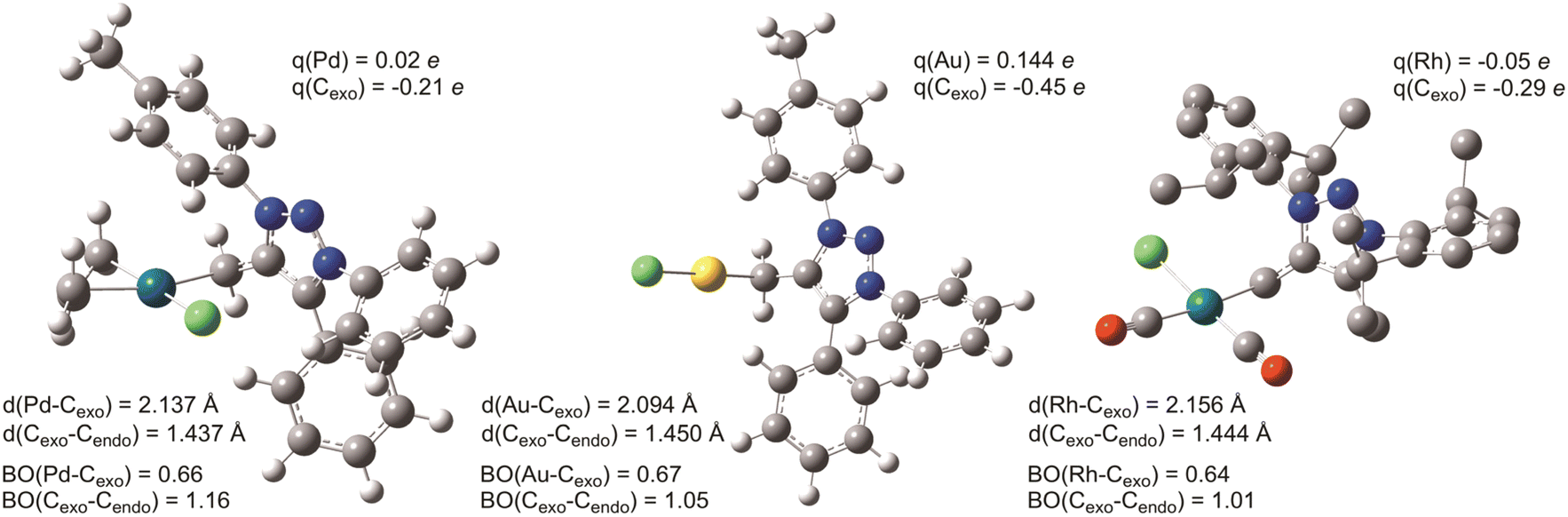 | ||
| Fig. 6 Equilibrium structures of 7aPd (left), 7aAu (middle) and [RhCl(CO)2mNHO] (right) obtained at the PBE0/def2-SV(p) level of theory, given together with characteristic structural metrics (metal–Cexo and Cexo–Cendo bond distances), corresponding bond orders and atom condensed charges. | ||
In contrast, the rotation along the Cexo–Cendo bond is sterically hindered: as can be deduced intuitively from the structure shown in Fig. 5a and 6, rotation along this bond leads to the clash of the phenyl and tolyl groups of mNHO with the Pd–Cl functionality. Relaxed surface scans imply sharply increasing potential energy surfaces as these groups approach the chloride ligand, triggering the distortion of the structure and causing linearization of the Pd–Cexo–Cendo angle to avoid close contact with these groups. This leads to unstable wavefunctions and high electronic energies. The hindered rotation creates a stereogenic axis characteristic of the molecular platform, as shown in Fig. 5a, which has implications also for the NMR spectra as discussed below.
A limited rotation around Stereogenic axis 2 leads to the formation of a pair of enantiomers P and M (Fig. 7a) which, in a non-chiral environment (like in this study), should lead to the observation of one species in the NMR spectra. However, the constitutionally equivalent atoms H′ and H′′ of the exocyclic methylene group are not symmetry related and should resonate at different chemical shifts in the 1H NMR spectrum. Indeed, the 1H spectrum of compound 7aPd shows two doublets at δ = 2.33 ppm (J = 9.6 Hz) and δ = 2.01 ppm (J = 9.6 Hz) belonging to the diastereotopic H′ and H′′ (Table 1, and Fig. S3), while only one set of carbon resonances was observed in the 1H–13C gs-HSQC and 1H–13C gs-HMBC spectra (Fig. S48–S52†).
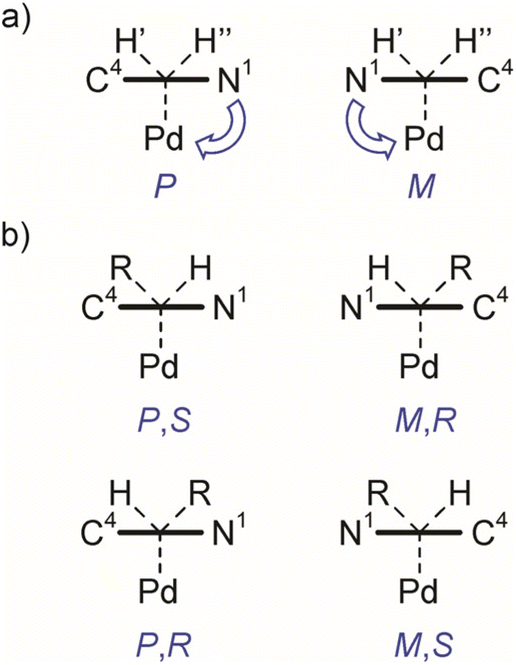 | ||
| Fig. 7 (a) Viewed along the Cendo–Cexo vector (Stereogenic axis 2), the triazole ring lies in front of the tetrahedral carbon atom giving stereoisomers P and M. (b) The combined conformationally stable Cendo–Cexo stereogenic axis (Stereogenic axis 2) and C(sp3) stereogenic centre (Cexo) give rise to two diastereomeric pairs of enantiomers (P,S and M,R; and P,R and M,S) of 7bPd (R = Me, descriptors R,S refer to the absolute configuration at Cexo). | ||
Now, if a proton of the Cexo methylene group in 7aPd is substituted by another group, say, a methyl group, this introduces a C(sp3) stereocentre into the molecule. Provided that fast rotation is present around Stereogenic axis 1, this results in four stereoisomers, i.e., two diastereomeric pairs of enantiomers (P,S and M,R; and P,R and M,S, Fig. 7b) and, thus, two observable species in the NMR spectra. This was confirmed by the experimental data for 7bPd, the preparation of which is shown in Table 1. Analyses of the 1H NMR and 13C NMR chemical shifts revealed the presence of two species of 7bPd in the THF solution as evident from the two sets of resonances for the allyl ligand and two sets for the mNHO ligand (Fig. S4†). Two sets of 13C resonances could also be extracted from the 1H–13C gs-HSQC and 1H–13C gs-HMBC spectra (Fig. S53–S55†). Remarkably, this molecular scaffold with a conformationally stable C(sp2)–C(sp3) stereogenic axis is a novel example of this rare stereogenic element.44
For 7aPd, the 13C resonance of Cexo was found at δ = −1.6 ppm (Table 1), which is different from those of the previously reported triazole based mNHO-Rh complexes (δ = 7.0–8.3 ppm (ref 19) and 7.9 ppm (ref. 30)) yet similar to that of the imidazole-based-NHO-Pd (δ = −7.0 − (+0.99) ppm).14 Increasing the substitution of Cexo when going from 7aPd to 7bPd caused a downfield shift of about +11 ppm for the methyl group; in diastereomeric 7bPd, Cexo resonated at δ = 10.7 and 10.9 ppm, which is close to the value expected in alkane series (+9 ppm).45
In addition, the η3-allyl ligand can adopt an exo or endo configuration; the designations refer to isomers in which the vectors Cexo–Cendo and C2ally–H point in the same or in opposite directions, respectively. Sharp proton peaks of the allyl ligand in both 7aPd and 7bPd, in conjunction with solution NMR investigations reported in the literature,46 suggest fast isomerisation47,48 between the exo and endo isomers of (η3-allyl)Pd and, thus, this dynamic phenomenon has no influence on the above-discussed diastereomerism.
Although 13C NMR chemical shifts result from a complex interplay between shielding and deshielding effects,45 themselves stemming from the intramolecular electron flows stimulated by the applied external magnetic field, it has been suggested (on the basis of both experimental49 and theoretical investigations50) that there is an empirical correlation between the 13C NMR shifts and the relative charge (and thus reactivity) at the η3-allyl termini in the (η3-allyl)palladium complexes. Namely, when the ligands go from pure donors to those with some acceptor character, the 13C shift at the η3-allyl termini trans to the ligand shifts downfield. For example, the η3-allyl termini trans to purely donor TMEDA (tetramethylethylenediamine) and chloride were found to resonate at ca. δ = 60–62 ppm whereas, for example, for the corresponding triphenylphosphine ligand with some π-acceptor character, the 13C shift can be found at ca. 79 ppm.49 Besides guiding the deduction of a tentative structure, the 13C NMR shifts of C3allyl at δ = 53.7 ppm and δ = 56.0 ppm for 7aPd and 7bPd, respectively (Table 1), indicate the strong σ donor character of the mNHO ligands.19,30 The calculated bond orders and atom-condensed charges, listed in Fig. 6, corroborate this finding; a bond order of 0.66 indeed indicates a strong Pd–Cexo interaction and a Lowdin charge analysis revealed almost identical atomic charges on the allyl terminal carbons trans to mNHO (0.00e) and trans to chloride (−0.03e).
Another interesting feature of 7aPd revealed by calculations is a Cexo–Cendo bond order of 1.16 that implies a minor but non-negligible π interaction between the triazole ring and the exo carbon, i.e. the reactivity of Cexo's lone-pair and localized negative charge is quenched not only by the interaction with the metal but also by some delocalization to the aromatic heterocycle, which is consistent with the resonance structures of mNHO in Fig. 1. The corresponding π interaction is portrayed clearly by HOMO and HOMO−6 of 7aPd (Fig. S5†).
The allyl ligand in 7aPd and 7bPd appears to stabilize the mNHO–metal bond, probably by the electron-withdrawing effect originating from its π-accepting ability. This may explain why the treatment of 7a and b with non-allyl metal precursors such as [PdCl2(NCPh)2] did not give stable products. Nevertheless, even keeping 7aPd or 7bPd dissolved overnight in dry THF under an inert atmosphere was detrimental and led to the formation of a complex mixture of unidentified products as revealed by NMR analysis.
mNHO–gold coordination compounds
Gold is known to be particularly carbophilic and a strong Lewis acid13,51–53 and, concomitantly, it has been successfully coordinated to NHOs.10,13 Accordingly, in a similar fashion to the synthesis of the Pd complexes, 7a was prepared in situ from 1a and reacted with chloro(dimethylsulfide)gold(I), AuCl(SMe2), to give a product that was identified as 7aAu (Table 2) according to NMR spectroscopy and ESI+ HRMS spectrometry. In the 1H NMR spectrum (THF solution), the methylene protons of 7aAu appeared as a singlet at δ = 2.15 ppm. In the carbon NMR spectrum, Cexo was found to resonate at δ = 15.2 ppm, which is considerably downfield as compared to the equivalent Cexo in 7aPd (see above). The positive-ion ESI-HRMS revealed the [7aAu–Cl]+ ion at m/z 522.1236 (calcd for C22H19AuN3+ [7aAu–Cl]+ 522.1239) and was interpreted as the result of in source collision-induced dissociation of 7aAu (Fig. S6†). In addition, the [7aAu–Cl + CH3CN]+ ion at m/z at 563.1491 (calcd for C24H22AuN4+: 563.1505) was found to be the most intense peak. The acetonitrile in this cluster came from the solvent used for the sample preparation. As in the case of 7aPd, the peak that corresponded to the triazolium cation [(1a–TfO)]+ was also found in the ESI+ HRMS spectrum of 7aAu. As shown in Fig. 6, 7aAu adopted a linear structure, corresponding to a d10 metal centre, with a rather strong (BO = 0.67) and short (2.09 Å) Au–mNHO bond. Another gold–mNHO derivative, complex 7bAu (Table 2), was prepared in the same way as 7aAu. As with the palladium derivatives, the gold complexes 7aAu and 7bAu were stable in the solid state for several days under an inert atmosphere but decomposed overnight under an inert atmosphere in a THF solution.|
|
|||||
|---|---|---|---|---|---|
| Cmpd. | NMR solvent | δ CexoH | δ Cexo | δ C4 | δ C5 |
| 13C NMR chemical shifts were extracted from the 1H–13C gs-HSQC and 1H–13C gs-HMBC spectra.a Two resonances for the two stereoisomers are given. The first and second figures refer to the upfield and downfield resonances, respectively. For the NMR data for 1a, see Table 1. | |||||
| 7aAu | THF | 2.15 | 15.2 | 132 | 153 |
| 7bAu | THF | 2.83, 2.89a | 15.6, 26.2a | 133.0, 133.2a | 154.7, 155.4a |
Phosphine-functionalized triazolium salt coordinated to palladium
The coordination properties of the phosphine-functionalized analogue 1h, which may provide C^P-bidentate coordination, were also investigated. Since the attempts to prepare mNHO 7h were unsuccessful (Scheme 4b), we decided to test a stepwise approach, and compound 1h was first reacted with PdCl2 in dichloromethane to give a pale yellow bis-phosphine–Pd complex 9hPd (Scheme 5), the structure of which was confirmed by NMR and X-ray diffraction analysis (for crystallographic data, see ESI Fig. S7, S8 and Tables S1, S2†). Treatment of the isolated 9hPd with KHMDS in the next step to produce a C^P-bidentate coordination was unsuccessful and resulted in the decomposition of 9hPd and the formation of black palladium. Similar to 1h (vide supra), based on NMR spectra, it appeared that the triazole N3-Me part of the ligand in 9hPd did not survive treatment with a base.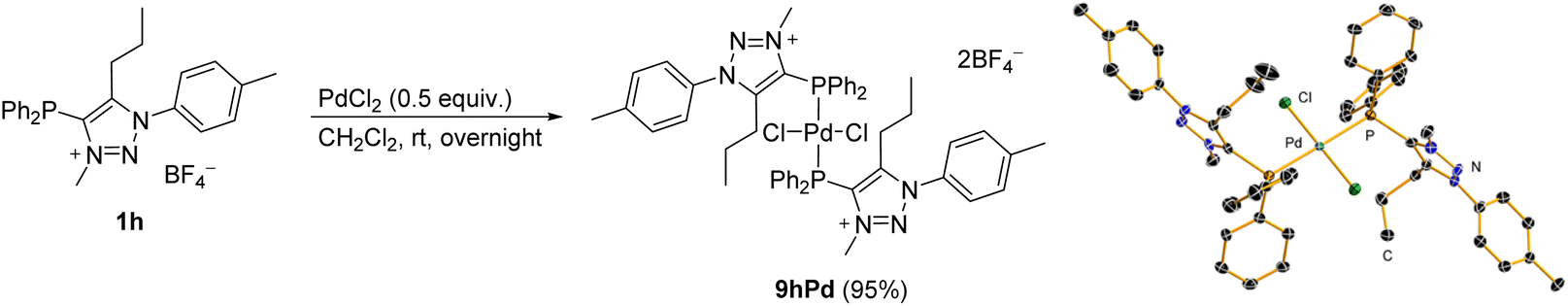 | ||
| Scheme 5 Synthesis of the phosphine-functionalized Pd complex 9hPd (for crystallographic data; see the ESI†). | ||
mNHO–boron hydride adduct
In comparison with Pd and Au, mNHO Lewis adducts with trihydridoborane (BH3) turned out to be exceedingly stable. Compound 10aBH3 was easily prepared starting from 1a through in situ formation of mNHO 7a with subsequent addition of BH3·THF (Scheme 6) and isolated as a light-yellow air-stable solid. Crystals suitable for X-ray diffraction analysis were obtained by layering a dichloromethane solution of the product with hexane at −20 °C (Fig. 8). For crystallographic data, see ESI Fig. S9, S10 and Tables S1 and S3.†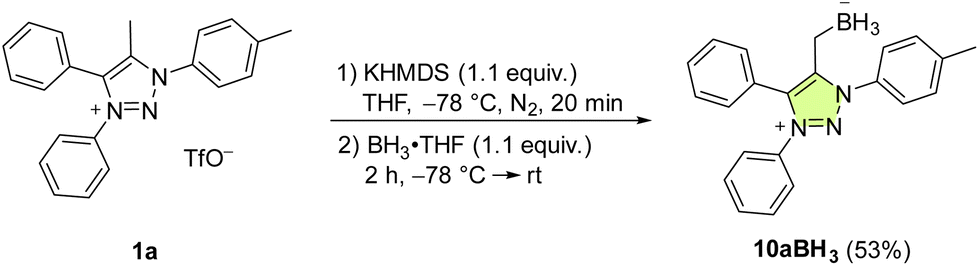 | ||
| Scheme 6 The synthesis of mNHO–boron hydride adduct 10aBH3. | ||
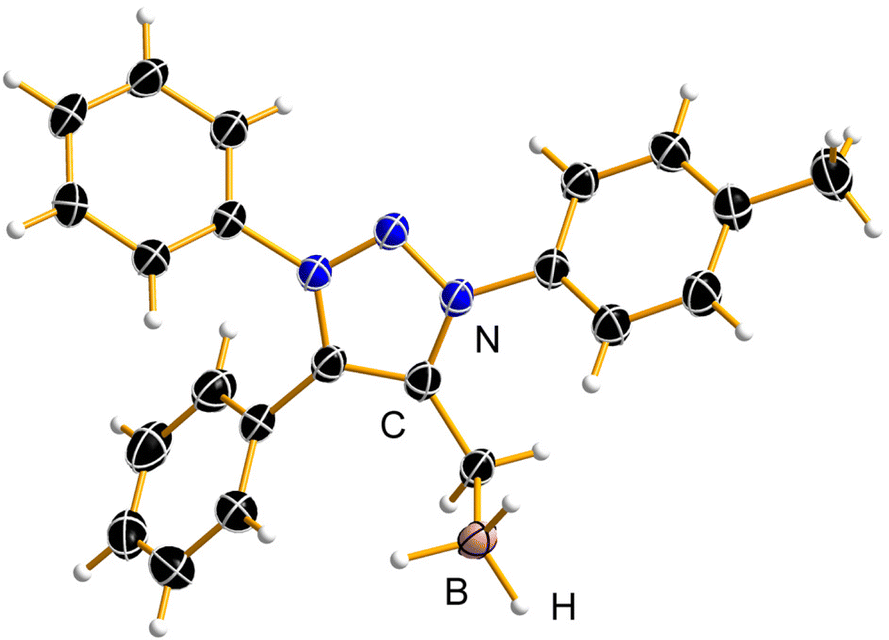 | ||
| Fig. 8 The asymmetric unit of the 10aBH3·0.825(CH2Cl2) crystal structure; the disordered CH2Cl2 molecules were omitted for clarity. Displacement ellipsoids are depicted at the 50% probability level and hydrogen atoms are shown as small spheres of arbitrary radius. | ||
To our knowledge, there are two reports of BH3–mNHO adducts in the literature20,22 and catalytic activity of mNHOs in the hydroboration of imines, nitriles and N-heteroarenes with H(Bpin) has been reported.20 Based on theoretical studies, compared with some other N-heterocyclic carbenes (NHCs), N-heterocyclic olefins (NHOs) and mesoionic carbenes (MICs), the mNHO/HBpin adducts possess the most negative charge on H-(Bpin), weakening the B–H bonds and enhancing the hydridic character, which promotes the hydroboration of unsaturated substrates. This prompted us to investigate the reducing ability of 10aBH3 toward selected aldehydes 11 and ketones 13. 3-Phenylpropionaldehyde (11a) was used as a model substrate to briefly optimize the reaction conditions (for more details, please see the ESI†). The scope of the reaction with three benzaldehydes, 11b, c, and d (0.20 M), having different electronic properties (Scheme 7A), was investigated by using 0.5 equiv. of 10aBH3 in acetonitrile as the reaction solvent (Table S4†). Electron-deficient 3-nitrobenzaldehyde (11c) was converted to 3-nitrobenzyl alcohol (12c) in 44% after 4 h, with the starting aldehyde being completely consumed. As expected, the conversion of electron-rich 3,4,5-trimethoxybenzaldehyde (11d) was slower (21% conversion to 12d) under the same reaction conditions.
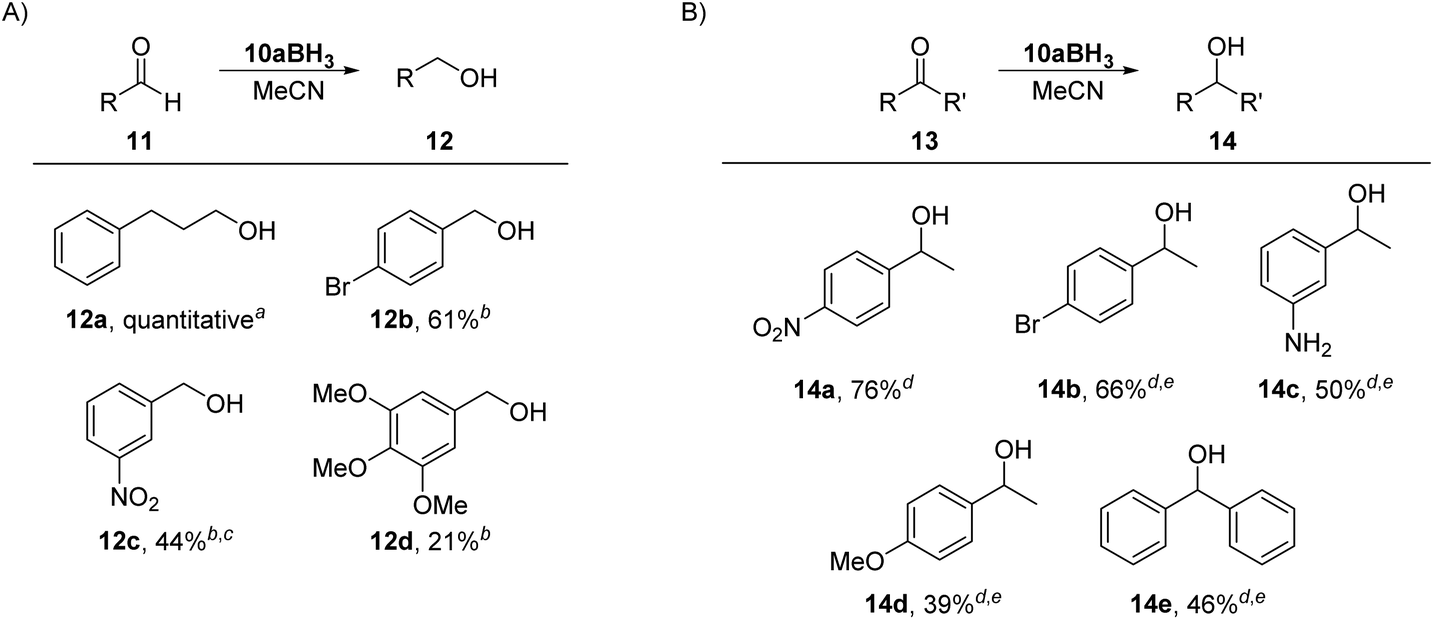 | ||
| Scheme 7 Reduction of (A) aldehydes and (B) ketones. Conversions were determined by 1H NMR with 1,3,5-trimethoxybenzene as internal standard: aaldehyde 11a (0.029 mmol, 0.058 M in MeCN), 10aBH3 (1.0, 0.5 or 0.33 equiv.), 3 h, air, room temperature; baldehyde (0.10 mmol, 0.20 M in MeCN), 10aBH3 (0.5 equiv.), 24 h, air, room temperature; creaction time was 4 h; dketone (0.10 mmol, 0.20 M in MeCN), 10aBH3 (0.5 equiv.), 24 h, air, room temperature; ereaction performed in the presence of silica gel (100 mg). | ||
The reduction of carbonyl compounds by related NHC- and MIC-boranes has been investigated.54–57 Comparison of our results with those previously reported for NHC- and MIC-boranes54–57 suggests that 10aBH3 is more active and no additives are needed (both NHC- and MIC-borane reduction of aldehydes require the presence of a Lewis acid such as Sc(OTf)3 or silica gel) for the reaction to proceed, although the reaction times with 10aBH3 were longer.
Next, the reducing power of 10aBH3 was tested on ketones 13 (Scheme 7B, and Table S5†). Except for strongly activated 4-nitroacetophenone (13a), which led to 76% conversion to 14a under the same reaction conditions as used for the aldehydes, reductions of other substrates by using mNHO–borane 10aBH3 alone did not occur. The addition of silica gel was beneficial and allowed the reduction of 4-bromoacetophenone (13b) to 1-(4-bromophenyl)ethanol (14b) in 66% after 24 h. For comparison, reduction of 13b with 1 equiv. of NHC-BH3 diMe-Imd-BH3 gave alcohol 14b in 94% yield after 24 h in DCM.54 Control experiments were performed to test the stability of 10aBH3. A CD3CN solution of 10aBH3 with or without silica gel was monitored by 1H NMR for four days and only negligible changes were observed in the recorded spectra, indicating that BH3 does not dissociate from mNHO and that the mNHO–boron hydride species play an active role in the reaction with the carbonyl compounds.
Conclusions
1,4-Disubstituted triazoles obtained by click chemistry were functionalized at the triazole nitrogen atom N3 and at the carbon atom C5 to give the corresponding C5-alkylated triazolium salts. Deprotonation of C5–CexoH was achieved with KHMDS in dry THF at room temperature to give deeply coloured mesoionic N-heterocyclic olefins (mNHO). The colour is due to the resonance stabilisation of the negatively polarised Cexo− moiety with the cationic triazole unit, which was confirmed by the NMR study through the observation of two geometric isomers indicating C5–Cexo double bond properties. To investigate the coordination properties, selected in situ prepared mHNOs were allowed to react with metal precursors [Pd(allyl)Cl]2 and AuCl(SMe2). [Pd(η3-allyl)Cl(mNHO)] and [AuCl(mNHO)] were readily obtained and characterised by multinuclear 1D and 2D NMR spectroscopy and high resolution electrospray ionisation mass spectrometry (ESI+ HRMS). The NMR data showed a strong σ-donor character of the mNHO ligands on the metals, which was corroborated by DFT calculations, revealing bond orders of 0.66 and 0.67 for the Pd–mNHO and Au–mNHO bonds, respectively. Besides, a Cexo–Cendo bond order of 1.1 implies that this bond of the mNHO ligand maintains a non-negligible double bond character and, thus, π-interaction with the triazole core. The [Pd(η3-allyl)Cl(mNHO)] complex with C5-ethylated mNHO also exhibited a rare conformationally stable C(sp2)–C(sp3) stereogenic axis, which was scrutinized experimentally and computationally. Noteworthily, these are the first known palladium and gold coordination complexes of mNHOs.A BH3–mNHO adduct was prepared and its structure was confirmed by NMR and X-ray single-crystal analysis. The reactivity of BH3–mNHO in the reduction of aldehydes and ketones to the corresponding primary and secondary alcohols was investigated. It was found to be more active than the related NHC- and MIC-boranes as no Lewis acid additives were required for the reduction of aldehydes to proceed.
Author contributions
TŽ: investigation, methodology, validation, visualization, writing – original draft, and writing – review & editing; BP: investigation, methodology, resources, visualization, and writing – review & editing; ML: investigation, resources, visualization, and writing – review & editing; DU: investigation, visualization, and writing – original draft; RDJv-V: writing – review & editing; JK: conceptualization, data curation, funding acquisition, project administration, resources, supervision, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support from the Slovenian Research and Innovation Agency (Research Core Funding grants P1-0230, P1-0045, Young Researcher Grant to T. Ž., and projects J1-3018 and J7-50041) is gratefully acknowledged. The authors also acknowledge the support of the Centre for Research Infrastructure at the University of Ljubljana, Faculty of Chemistry and Chemical Technology, which is part of the Network of Research and Infrastructural Centres UL (MRIC UL) and is financially supported by the Slovenian Research and Innovation Agency (Infrastructure programme no. I0-0022). B. P. thanks the University of Texas at El Paso for the computing time and financial support prior to 2023 (Grant E210291776). M. L. gratefully acknowledges the funding by the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 950625). R. J.v-V. acknowledges funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 945380.References
- N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 14, 1136–1137, 10.1039/C39930001136.
- Q. Liang and D. Song, Dalton Trans., 2022, 51, 9191–9198, 10.1039/D2DT01013E.
- Q. Sun, A. Eitzinger, R. Esken, P. W. Antoni, R. J. Mayer, A. R. Ofial and M. M. Hansmann, Angew. Chem. Int. Ed., 2024, 63, e2023182, DOI:10.1002/anie.202318283.
- R. S. Ghadwal, Dalton Trans., 2016, 45, 16081–16095, 10.1039/C6DT02158A.
- M. Iglesias, A. Iturmendi, P. J. Sanz Miguel, V. Polo, J. J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2015, 51, 12431–12434, 10.1039/C5CC04287A.
- M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025, DOI:10.1021/acs.accounts.7b00264.
- S. Naumann, Chem. Commun., 2019, 55, 11658–11670, 10.1039/C9CC06316A.
- I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, Chem. – Eur. J., 2019, 25, 9678–9690, DOI:10.1002/chem.201901376.
- Z. Li, P. Ji and J.-P. Cheng, J. Org. Chem., 2021, 86, 2974–2985, DOI:10.1021/acs.joc.0c02838.
- A. Merschel, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and R. S. Ghadwal, Dalton Trans., 2022, 51, 8217–8222, 10.1039/D2DT01314B.
- N. Kuhn, H. Bohnen, D. Bläser and R. Boese, Chem. Ber., 1994, 127, 1405–1407, DOI:10.1002/cber.19941270814.
- K. Powers, C. Hering-Junghans, R. McDonald, M. J. Ferguson and E. Rivard, Polyhedron, 2016, 108, 8–14, DOI:10.1016/j.poly.2015.07.070.
- A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210–3214, DOI:10.1002/anie.200705798.
- S. Ando, A. Ohara, T. Ohwada and T. Ishizuka, Organometallics, 2021, 40, 3668–3677, DOI:10.1021/acs.organomet.1c00503.
- D. A. Imbrich, W. Frey, S. Naumann and M. R. Buchmeiser, Chem. Commun., 2016, 52, 6099–6102, 10.1039/C6CC02483A.
- A. Iturmendi, N. García, E. A. Jaseer, J. Munárriz, P. J. Sanz Miguel, V. Polo, M. Iglesias and L. A. Oro, Dalton Trans., 2016, 45, 12835–12845, 10.1039/C6DT02571D.
- M. Fischer, M. M. D. Roy, S. Hüller, M. Schmidtmann and R. Beckhaus, Dalton Trans., 2022, 51, 10690–10696, 10.1039/D2DT00014H.
- S. Maji, A. Das, M. M. Bhatt and S. K. Mandal, Nat. Catal., 2024 DOI:10.1038/s41929-024-01114-7.
- M. M. Hansmann, P. W. Antoni and H. Pesch, Angew. Chem., Int. Ed., 2020, 59, 5782–5787, DOI:10.1002/anie.201914571.
- Z. Zhang, S. Huang, L. Huang, X. Xu, H. Zhao and X. Yan, J. Org. Chem., 2020, 85, 12036–12043, DOI:10.1021/acs.joc.0c00257.
- S. Maji, A. Das and S. K. Mandal, Chem. Sci., 2021, 12, 12174–12180, 10.1039/D1SC02819G.
- Q. Liang, K. Hayashi and D. Song, Organometallics, 2020, 39, 4115–4122, DOI:10.1021/acs.organomet.0c00653.
- Z.-W. Qu, H. Zhu, R. Streubel and S. Grimme, Eur. J. Org. Chem., 2022, e202200539, DOI:10.1002/ejoc.202200539.
- Q. Liang, K. Hayashi, L. Li and D. Song, Chem. Commun., 2021, 57, 10927–10930, 10.1039/D1CC04695K.
- Q. Liang, K. Hayashi, Y. Zeng, J. L. Jimenez-Santiago and D. Song, Chem. Commun., 2021, 57, 6137–6140, 10.1039/D1CC02245H.
- P. W. Antoni, C. Golz, J. J. Holstein, D. A. Pantazis and M. M. Hansmann, Nat. Chem., 2021, 13, 587–593, DOI:10.1038/s41557-021-00675-5.
- P. W. Antoni, J. Reitz and M. M. Hansmann, J. Am. Chem. Soc., 2021, 143, 12878–12885, DOI:10.1021/jacs.1c06906.
- C. Empel and R. M. Koenigs, Nat. Chem., 2021, 13, 1030–1032, DOI:10.1038/s41557-021-00811-1.
- B. Kooij, P. Varava, F. Fadaei-Tirani, R. Scopelliti, D. A. Pantazis, G. P. Van Trieste III, D. C. Powers and K. Severin, Angew. Chem., Int. Ed., 2023, 62, e202214899, DOI:10.1002/anie.202214899.
- A. Eitzinger, J. Reitz, P. W. Antoni, H. Mayr, A. R. Ofial and M. M. Hansmann, Angew. Chem., Int. Ed., 2023, 62, e202309790, DOI:10.1002/anie.202309790.
- W. Wirschun, M. Winkler, K. Lutz and J. C. Jochims, J. Chem. Soc., Perkin Trans. 1, 1998, 1755–1762, 10.1039/A801797B.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759–4762, DOI:10.1002/anie.201001864.
- J. Bouffard, B. K. Keitz, R. Tonner, G. Guisado-Barrios, G. Frenking, R. H. Grubbs and G. Bertrand, Organometallics, 2011, 30, 2617–2627, DOI:10.1021/om200272m.
- M. Virant and J. Košmrlj, J. Org. Chem., 2019, 84, 14030–14044, DOI:10.1021/acs.joc.9b02197.
- Y. Canac and R. Chauvin, Eur. J. Inorg. Chem., 2010, 16, 2325–2335, DOI:10.1002/ejic.201000190.
- A. B. Shashank, S. Karthik, R. Madhavachary and D. B. Ramachary, Chem. – Eur. J., 2014, 20, 16877–16881, DOI:10.1002/chem.201405501.
- D. M. Zink, T. Baumann, M. Nieger and S. Bräse, Eur. J. Org. Chem., 2011, 1432–1437, DOI:10.1002/ejoc.201001505.
- Y. Tanaka and S. I. Miller, J. Org. Chem., 1973, 38, 2708–2712, DOI:10.1021/jo00955a029.
- E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637–1651, 10.1039/C2CS35270B.
- M. C. Biagini, M. Ferrari, M. Lanfranchi, L. Marchiò and M. A. Pellinghelli, J. Chem. Soc., Dalton Trans., 1999, 10, 1575–1580, 10.1039/A808940J.
- G. P. Moss, Pure Appl. Chem., 1996, 68, 2193–2222, DOI:10.1351/pac199668122193.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187, DOI:10.1002/anie.199721621.
- Starting geometries for the corresponding geometry optimization calculations were obtained by straightforward modifications of the X-ray structure of Rh–mNHO published by Hansmann et al. (ref. 19 ) ( DOI:10.1002/anie.201914571).
- G. Bertuzzi, V. Corti, J. A. Izzo, S. Ričko, N. I. Jessen and K. A. Jørgensen, J. Am. Chem. Soc., 2022, 144, 1056–1065, DOI:10.1021/jacs.1c12619.
- Atta-ur-Rahman, in Nuclear Magnetic Resonance: Basic Principles, ed. Atta-ur-Rahman, Springer US, New York, NY, 1986, pp. 140–201. DOI:10.1007/978-1-4612-4894-1_4.
- J. Sprinz, M. Kiefer, G. Helmchen, M. Reggelin, G. Huttner, O. Walter and L. Zsolnai, Tetrahedron Lett., 1994, 35, 1523–1526, DOI:10.1016/S0040-4039(00)76748-7.
- M. Kollmar, B. Goldfuss, M. Reggelin, F. Rominger and G. Helmchen, Chem. – Eur. J., 2001, 7, 4913–4927, DOI:10.1002/1521-3765(20011119)7:22<4913::AID-CHEM4913>3.0.CO;2-7.
- N. H. Sherden, PhD thesis, California Institute of Technology, 2011. https://resolver.caltech.edu/CaltechTHESIS:05312011-072858726.
- B. Aakermark, B. Krakenberger, S. Hansson and A. Vitagliano, Organometallics, 1987, 6, 620–628, DOI:10.1021/om00146a031.
- A. Aranyos, K. J. Szabó, A. M. Castaño and J.-E. Bäckvall, Organometallics, 1997, 16, 1058–1064, DOI:10.1021/om960950m.
- A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449, DOI:10.1002/anie.200604335.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403, DOI:10.1038/nature05592.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936, DOI:10.1002/anie.200602454.
- T. Taniguchi and D. P. Curran, Org. Lett., 2012, 14, 4540–4543, DOI:10.1021/ol302010f.
- L. B. de Oliveira Freitas, P. Eisenberger and C. M. Crudden, Organometallics, 2013, 32, 6635–6638, DOI:10.1021/om400743c.
- F. Stein, M. Kirsch, J. Beerhues, U. Albold and B. Sarkar, Eur. J. Inorg. Chem., 2021, 24, 2417–2424, DOI:10.1002/ejic.202100273.
- D. M. Lindsay and D. McArthur, Chem. Commun., 2010, 46, 2474–2476, 10.1039/C001466D.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2297781 and 2297782. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00195h |
| ‡ Current affiliation: European Research Council Executive Agency. |
| § Disclaimer: The views expressed are purely those of the authors and may not in any circumstances be regarded as stating an official position of the ERCEA and the European Commission. |
| This journal is © The Royal Society of Chemistry 2024 |