
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkali metal reduction of crown ether encapsulated alkali metal cations†
Kyle G. Pearce ,
Samuel E. Neale,
Mary F. Mahon*,
Claire L. McMullin* and
Michael S. Hill*
,
Samuel E. Neale,
Mary F. Mahon*,
Claire L. McMullin* and
Michael S. Hill*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk; cm2025@bath.ac.uk
First published on 4th July 2024
Abstract
[{SiNDipp}BeClM]2 ({SiNDipp} = {CH2SiMe2N(Dipp)}2; M = Li, Na, K, Rb) are converted to ionic species by treatment with a crown ether. Whereas the lithium derivative reacts with Na or K to provide [{SiNDipp}BeCl]−[M(12-cr-4)2]+ (M = Na, K), the resultant sodium species is resistant to reduction by potassium. These observations are rationalised by a hybrid experimental/theoretical analysis.
The pursuit of molecules comprising alkaline earth centres in oxidation states lower than the periodic group number (i.e.< + 2) has gathered pace since Jones and co-workers’ 2007 report of isolable Mg(I) species.1 While the kinetic stability of, for example, compounds I and II (Fig. 1),2 was ensured by the use of bulky β-diketiminate (BDIAr) spectator anions, the thermochemical viability of their synthesis was in intuitive accord with the standard reduction potentials (E0, V) of the potassium (K+(aq) + e− K(s) → −2.92 V) and magnesium(II) (Mg2+(aq) + 2e− → Mg(s) −2.36 V) starting materials.3,4 Consistent with this supposition, similar alkali metal reduction has since realised a substantial variety of related Mg(I) derivatives.1,5–16
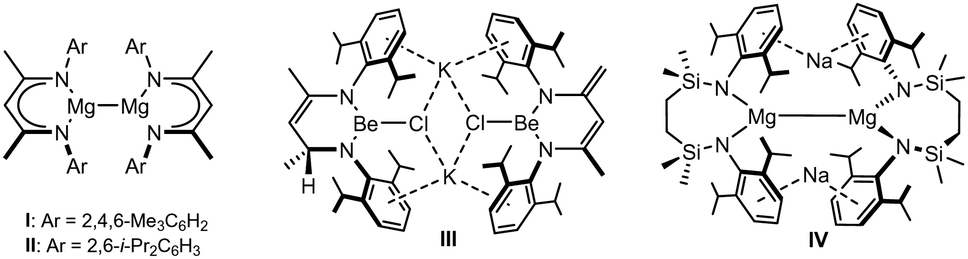 | ||
| Fig. 1 The structures of compounds I–IV. | ||
By a similar measure, magnesium's lighter group 2 congener, beryllium (Be2+(aq) + 2e− → Be(s) −1.85 V), encourages a comparable expectation. Although reactions of potassium and cyclic(alkyl)(amino)carbene (CAAC) adducts of BeCl2 have provided several carbene-supported beryllium species in which a zero or +1 oxidation state may be attributed to the group 2 centre,17–22 it is notable that Aldridge and co-workers’ recent realisation of CpBeBeCp was achieved through reduction of beryllocene by the arene-soluble compound I.23 In contrast, the limited attempts to access homonuclear Be–Be bonds by alkali metal reduction of BDIAr-supported beryllium halides have been unsuccessful.6,24 A case in point is provided by our own study of the potassium reduction of [(BDIDipp)BeCl]2 (BDIDipp = HC(Me)CNDipp2; Dipp = 2,6-di-isopropylphenyl), which resulted in a mixture of compounds exemplified by III (Fig. 1), and arising from hydrogen-atom transfer between two BDIDipp backbones.24 We have previously speculated that this reactivity and, most likely, Jones’ synthesis of I and II are similarly initiated by electron transfer into the BDIDipp (π*) LUMO, but with contrasting reaction outcomes that reflect the relative resistance to subsequent group 2-centred reduction of the {Be–Cl} and {Mg–I} units.24
With such observations in mind, we have recently utilised the more redox innocent {SiNDipp}2− dianion [{SiNDipp} = {CH2SiMe2N(Dipp)}2] to synthesise the heterobimetallic Mg(I) derivative, [{SiNDipp}MgNa]2 (IV).25–29 While IV is accessible by sodium reduction of the Mg(II) precursor [{SiNDipp}Mg], analogous treatment of [{SiNDipp}Be] resulted in reductive activation of the benzene solvent and the isolation of phenyl- and hydrido-beryllate species.30,31 Although this latter observation provides circumstantial evidence for the generation of beryllium radical anion intermediates, the resistance to reduction of the {Be–Cl} moiety was again made apparent during similar attempts to react the dimeric lithium chloroberyllate (VLi) with either sodium, potassium, rubidium or caesium metal (Scheme 1).
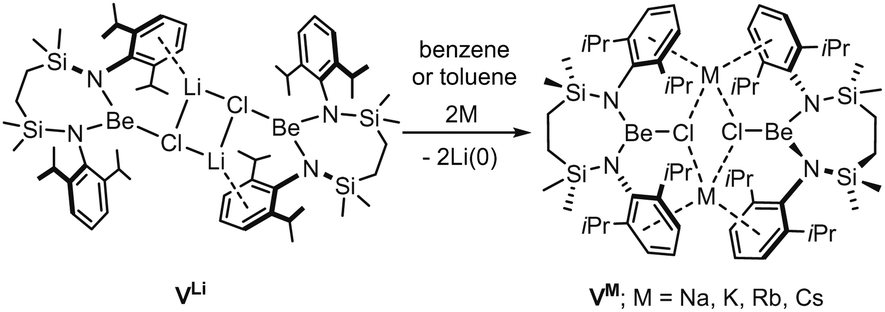 | ||
| Scheme 1 Synthesis of compounds VM (M = Na, K, Rb, Cs) by alkali metal reduction of VLi. | ||
Contradicting the alkali metal reduction potentials [i.e. M(aq)+ + e− → M(s); M = Li (E0 = −3.04 V vs. SHE), Na (−2.71 V), K (−2.92 V), Rb (−2.98 V), Cs (−2.93 V)],3 these reactions provided exclusive replacement of the lithium cations to yield [{SiNDipp}BeClM]2 (VM where M = Na, K, Rb, Cs).32,33 To take account of the heterogeneous nature of the reactions and to achieve satisfactory alignment with the experimental observations, our rationalisation of this behaviour required a theoretical analysis that combined density functional theory (DFT) with elemental thermochemistry. Specifically, it was necessary to incorporate the relevant group 1 enthalpies of atomisation to account for the metallic bonding within the bulk elements and allow the construction of a series of hybrid experimental/theoretical Hess cycles.34
We also reasoned that the electrochemical data are, to a significant extent, dictated by the relevant M+ hydration energies, whereas any modified potentials relating to the group 1 centres in VM must account for the relative preference for arene encapsulation manifested in their molecular structures. While well established in a biological context,35–37 the significance of this latter phenomenon is less widely appreciated in condensed-phase molecular systems.38–40
On this basis, we speculated that closer alignment with the accepted E0 values may arise if the M+ centres were to reside in a wholly oxygenated, yet still hydrocarbon soluble, coordination environment. In this contribution, therefore, we extend our studies to an experimental and theoretical assessment of crown ether coordination of the group 1 cations of the chloroberyllate species VM and the resultant impact upon the previously observed alkali metal reduction of Li+, but in the absence of arene encapsulation of the group 1 centres.
The preferential binding of the alkali metals based on ‘size fit’ within the interior of macrocyclic crown ethers has been appreciated since the early 1970s and has been particularly influential in the broader development of supramolecular chemistry.41 In an initial attempt to sequester the arene-encapsulated group 1 cations, therefore, compounds VM (M = Li, Na, K) were each treated with either 12-crown-4 (M = Li), 15-crown-5 (Na) or 18-crown-6 (K) (Scheme 2).
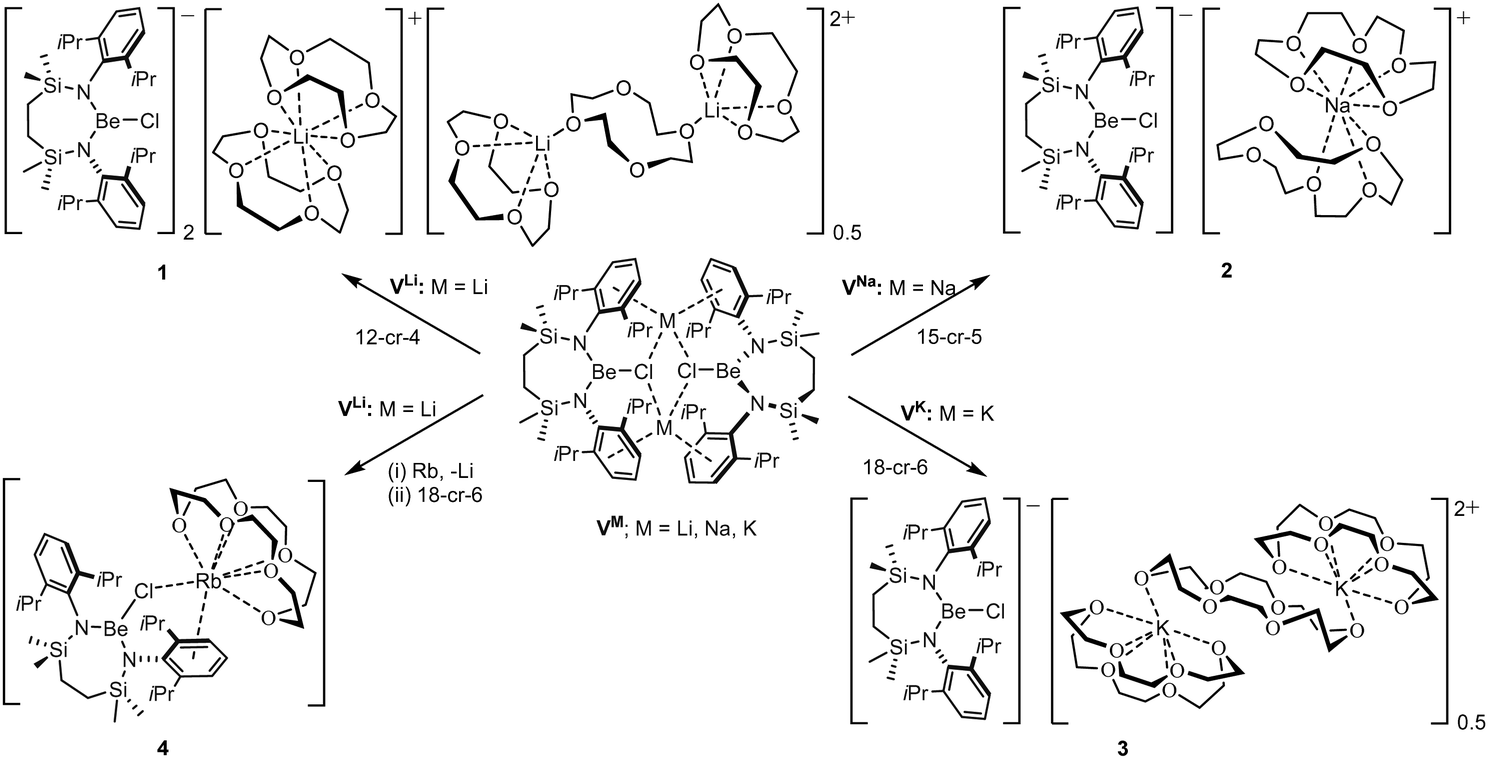 | ||
| Scheme 2 Synthesis of compounds 1–4. | ||
Irrespective of any variations across the solid-state structures of 1–4, their 1H NMR spectra presented very similar data, distinguished only in the relative intensity of the crown ether methylene singlet (δ ca. 3.1 ppm), but otherwise indicative of analogous C2-symmetry across the Be-coordinated {SiNDipp} ligands. Similarly, the broad 9Be NMR spectra provided by all four compounds [1 δ 9.7 ppm, ω1/2 = 282 Hz; 2 10.4 ppm, ω1/2 = 184 Hz; 3 10.5 ppm, ω1/2 = 201 Hz; 4 11.2 ppm, ω1/2 = 259 Hz] were little perturbed from the VM starting materials and redolent of a 3-coordinate geometry at beryllium.32,42 The respective colourless crown ether adducts, 1–3, were shown by X-ray diffraction analysis to crystallise as charge-separated chloroberyllate derivatives, [{SiNDipp}BeCl]−[M(cr)n]+ (1 M = Li, cr = 12-cr-4, n = 1.75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 M = Na, cr = 15-cr-5, n = 2:3 M = K, cr = 18-cr-6, n = 1.5), which vary only in the identity of their group 1 cations and the mode and resultant nuclearity of their crown ether encapsulation. The asymmetric unit of 1 (Fig. S31, ESI†) comprises two [{SiNDipp}BeCl]− anions with charge balance maintained by a bis-12-cr-4-ligated Li(1) cation and half of a dilithium dication in which two [Li(12-cr-4)]+ units are connected by a bridging equivalent of the crown ether straddling a crystallographic inversion centre. While any confident metric consideration of compound 2 is discouraged by a fall-off in diffraction intensity at higher Bragg angles, its identification as a charge-separated species (Fig. S32, ESI†) with a constituent [Na(15-crown-5)2]+ cation was unambiguous.
:2 M = Na, cr = 15-cr-5, n = 2:3 M = K, cr = 18-cr-6, n = 1.5), which vary only in the identity of their group 1 cations and the mode and resultant nuclearity of their crown ether encapsulation. The asymmetric unit of 1 (Fig. S31, ESI†) comprises two [{SiNDipp}BeCl]− anions with charge balance maintained by a bis-12-cr-4-ligated Li(1) cation and half of a dilithium dication in which two [Li(12-cr-4)]+ units are connected by a bridging equivalent of the crown ether straddling a crystallographic inversion centre. While any confident metric consideration of compound 2 is discouraged by a fall-off in diffraction intensity at higher Bragg angles, its identification as a charge-separated species (Fig. S32, ESI†) with a constituent [Na(15-crown-5)2]+ cation was unambiguous.
Refinement of the structure of 3 also required modelling of disorder. In this case, however, the cation comprises half of an 18-cr-6-bridged dipotassium dication reminiscent of that identified in 1 and again generated via inversion symmetry intrinsic to the crystallographic space group (Fig. 2). In addition to the direct preparation of 1–3, compound 4 was synthesised by in situ generation of VRb through the previously reported reaction of VLi with rubidium prior to addition of 18-crown-6 (Scheme 2). In contrast to the structures of 1–3, compound 4 was identified as a contact ion pair in which the [Rb(18-cr-6)]+ cation interacts with the beryllate anion via a bridging chloride and polyhapto engagement with one N-aryl substituent of the {SiNDipp} ligand (Fig. S33, ESI†).
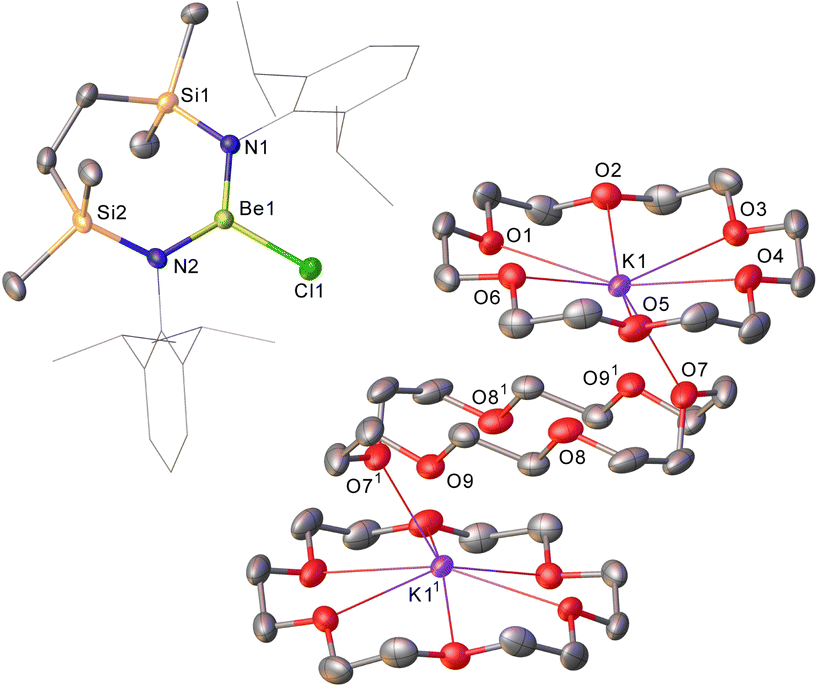 | ||
| Fig. 2 Molecular structure (30% probability ellipsoids) of compound 3. For clarity, hydrogen and disordered atoms have been omitted, while Dipp carbon atoms are presented as wireframe. [Operation 1 1 − x, 1 − y, 1 − z]. | ||
In an attempt to extend the lithium cation reduction chemistry represented in Scheme 1 to the oxygen-donor environment provided by coordination of 12-cr-4, compound 1 was reacted with either metallic sodium or 5 wt% Na/NaCl (Scheme 3).43 Both reactions provided an identical outcome and the isolation of compound 5 after crystallisation from the benzene reaction solvent. The resultant NMR spectrum presented no diagnostic evidence for the reduction of the chloroberyllate anion and X-ray diffraction analysis verified the reduction of the lithium cation environments of 1. The resultant structure of 5 confirmed the formation of a further charge-separated structure through the maintenance of the [{SiNDipp}BeCl]− anion but with charge balance now provided by a [Na(12-cr-4)2]+ cation (Fig. S34, ESI†). Subsequent reactions of 1 with either potassium metal or KC8 confirmed that the lithium reduction process could be extended to the isolation of the analogous heavier alkali metal derivative, [{SiNDipp}BeCl]−[K(12-cr-4)4]+ (6, Scheme 3 and Fig. 3).
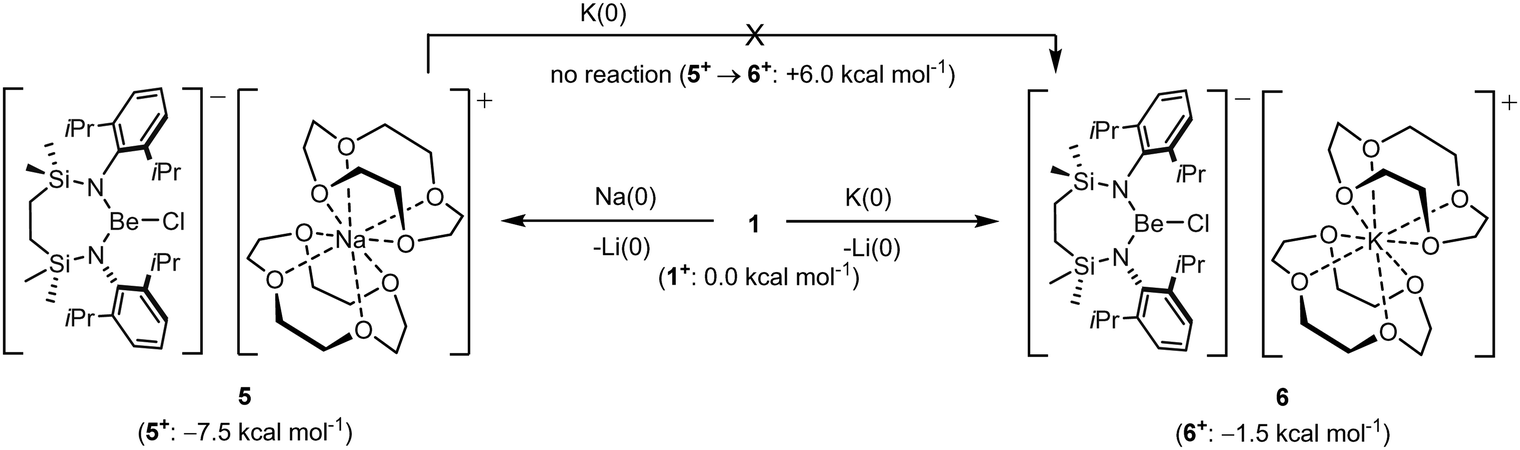 | ||
| Scheme 3 Synthesis of compounds 5 and 6 with computed free energies of formation for the cationic components (DLPNO-CCSD(T0);CPCM = Benzene/def2-TZVPP//TPSS-D3BJ/def2-TZVPP level) shown in parenthesis. | ||
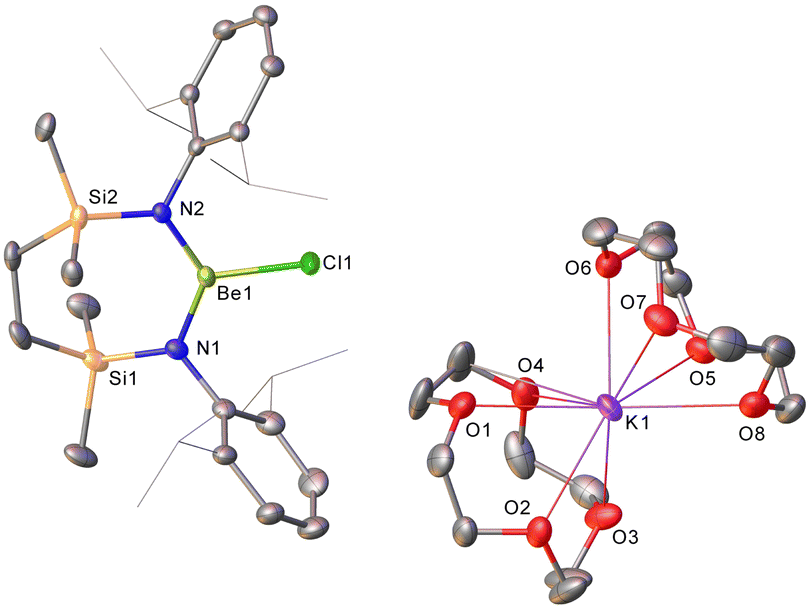 | ||
| Fig. 3 Molecular structure (30% probability ellipsoids) of the respective Be1 and K1 anion and cation of compound 6. For clarity, hydrogen and disordered atoms have been omitted, while isopropyl carbons are presented as wireframe. | ||
While the structures of 5 and 6 are unremarkable, their isolation confirms that both Na or K reduction of Li+ remain viable transformations, even in an oxygen-donor coordination environment more reminiscent of the hydrated species invoked by the group 1 electrochemical potentials. In contrast to the behaviour of the contact ion pair derivatives VNa and VK (vide supra), however, no evidence for reaction was observed when compound 5 was treated with elemental potassium. This latter observation indicates that the energetics of sodium reduction by potassium are modulated by the dissimilar Na+ coordination environments presented by compounds VNa and 5. With this observation in hand, therefore, and assuming the [{SiNDipp}BeCl]− as inert spectator anions in each reduction, the free energies of formation of the cationic crown ether components, ‘5+’ and ‘6+’, were estimated relative to ‘1+’ using a Hess cycle constructed from a combination of experimental and computational data (see the ESI† for details). While both cases of Li+ reduction were calculated to be exergonic [ΔG(1+ → 5+) = −7.5 kcal mol−1; ΔG(1+ → 6+) = −1.5 kcal mol−1], in accord with the lack of reactivity of 5 toward potassium and in contrast to the comparable data computed for the transformation of VNa to VK [ΔΔG(VNa → VK) = −14.2 kcal mol−1],32 ΔΔG(5+ → 6+) was computed to be endergonic (+6 kcal mol−1, Scheme 3).
In conclusion, the arene-bridged dimeric structures of [{SiNDipp}BeClM]2 (M = Li, Na, K, Rb) may be converted to charge-separated ionic species by treatment with a crown ether. While the 12-cr-4-encapsulated lithium derivative reacts with the appropriate alkali metal to directly provide [{SiNDipp}BeCl]−[M(12-cr-4)2]+ (M = Na, K), in contrast to our previous observations of the parent [{SiNDipp}BeClM]2 species, the sodium cation of [{SiNDipp}BeCl]−[Na(12-cr-4)2]+ is resistant to reduction by the heavier group 1 element. The thermodynamics of these observations have been rationalised by a combined experimental/theoretical analysis, which further emphasises that the viability of such redox interconversion is impacted by variations in both the cation environment and alkali metal lattice stability. We are continuing to explore the generality of this reactivity and its further application in chemical synthesis.
The authors gratefully acknowledge EPSRC (EP/X01181X/1, ‘Molecular s-block Assemblies for Redox-active Bond Activation and Catalysis: Repurposing the s-block as 3d-elements’) and the University of Bath's Research Computing Group (doi.org/10.15125/b6cd-s854) for their support in this work.
Data availability
The data supporting this article have been included as part of the ESI,† which includes general synthetic experimental details, NMR spectra and details of the X-ray and computational analysis. X-ray analysis of compounds 1–6 have been deposited with the Cambridge Structural Database as CCDC 2356402-2356407, respectively.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. J. Evans and C. Jones, Chem. Soc. Rev., 2024, 53, 5054–5082 RSC.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- Standard Potentials in Aqueous Solutions, Dekker, New York, 1985.
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS.
- S. J. Bonyhady, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973–2977 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- A. J. Boutland, I. Pernik, A. Stasch and C. Jones, Chem. – Eur. J., 2015, 21, 15749–15758 CrossRef CAS PubMed.
- A. J. Boutland, D. Dange, A. Stasch, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2016, 55, 9239–9243 CrossRef CAS PubMed.
- K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed.
- X. Cao, J. Li, A. Q. Zhu, F. Su, W. W. Yao, F. Xue and M. T. Ma, Org. Chem. Front., 2020, 7, 3625–3632 RSC.
- R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, A. Friedrich, J. Langer and S. Harder, Chem. Commun., 2020, 56, 11402–11405 RSC.
- J. Li, M. Luo, X. C. Sheng, H. M. Hua, W. W. Yao, S. A. Pullarkat, L. Xu and M. T. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- Y. Liu, S. Li, X.-J. Yang, P. Yang and B. Wu, J. Am. Chem. Soc., 2009, 131, 4210–4211 CrossRef CAS PubMed.
- C. Czernetzki, M. Arrowsmith, F. Fantuzzi, A. Gartner, T. Troster, I. Krummenacher, F. Schorr and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 20776–20780 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- G. C. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benko and R. J. Gilliard, Chem. – Eur. J., 2019, 25, 4335–4339 CrossRef CAS PubMed.
- G. C. Wang, J. E. Walley, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, J. Am. Chem. Soc., 2020, 142, 4560–4564 CrossRef CAS PubMed.
- M. Gimferrer, S. Danes, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583–6591 RSC.
- S. Pan and G. Frenking, Chem. Sci., 2023, 14, 379–383 RSC.
- J. T. Boronski, A. E. Crumpton, L. L. Wales and S. Aldridge, Science, 2023, 380, 1147–1149 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and I. Mallov, Inorg. Chem., 2012, 51, 13408–13418 CrossRef CAS PubMed.
- H. Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- M. S. Hill, H.-Y. Liu, S. E. Neale, M. F. Mahon, C. L. McMullin and B. L. Morrison, Chem. Commun., 2023, 59, 3846–3849 RSC.
- H. Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Angew. Chem., Int. Ed., 2023, 62, e202213670 CrossRef CAS PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Organometallics, 2024, 43, 879–888 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2024, 60, 1016–1019 RSC.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Chem. Commun., 2023, 59, 1453–1456 RSC.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Organometallics, 2024, 43, 432–437 CrossRef CAS PubMed.
- K. G. Pearce, H.-Y. Liu, S. E. Neale, H. M. Goff, M. F. Mahon, C. L. McMullin and M. S. Hill, Nat. Commun., 2023, 14, 8147 CrossRef CAS PubMed.
- (a) J. Kreutzer, Nat. Rev. Chem., 2024, 8, 84 CrossRef PubMed See also; (b) H. Li, J. Yao, G. Xu, S.-M. Yiu, C.-K. Siu, Z. Wang, Y.-K. Peng, Y. Xie, Y. Wang and Z. Lu, Nat. Commun., 2024, 15, 2590 CrossRef CAS PubMed.
- E. U. Franck, Bunsen-Ges. Phys. Chem., Ber., 1990, 94, 93 CrossRef.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- O. M. Cabarcos, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 1998, 108, 5151–5154 CrossRef CAS.
- O. M. Cabarcos, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 1999, 110, 8429 CrossRef CAS.
- J. C. Amicangelo and P. B. Armentrout, J. Phys. Chem. A, 2000, 104, 11420–11432 CrossRef CAS.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- J. K. Buchanan and P. G. Plieger, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 459–472 CrossRef CAS.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810–4813 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 1–6. CCDC 2356402–2356407. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02725f |
| This journal is © The Royal Society of Chemistry 2024 |