
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving co-electrocatalytic carbon dioxide reduction by optimizing the relative potentials of the redox mediator and catalyst†
Amelia G.
Reid
 ,
Ethan A.
Zelenke
,
Megan E.
Moberg
,
Diane A.
Dickie
and
Charles W.
Machan
*
,
Ethan A.
Zelenke
,
Megan E.
Moberg
,
Diane A.
Dickie
and
Charles W.
Machan
*
Department of Chemistry, University of Virginia, McCormick Rd, PO Box 400319, Charlottesville, Virginia 22904-4319, USA. E-mail: machan@virginia.edu
First published on 17th July 2024
Abstract
The effects of fixing the redox mediator (RM) reduction potential relative to a series of Cr-centered complexes capable of the reduction of CO2 to CO are disclosed. The greatest co-electrocatalytic activity enhancement is observed when the reduction potentials of the catalyst and RM are identical, implying that controlling the speciation of the Cr complex relative to RM activation is essential for improving catalytic performance. In all cases, the potential where co-catalytic activity is observed matches the reduction potential of the RM, regardless of the relative reduction potential of the Cr complex.
As the global energy demand continues to grow, so have anthropogenic emissions of carbon dioxide (CO2), with a corresponding increasing severity of associated climate change effects.1,2 One possibility for mitigating these negative effects is through the development of electrocatalytic systems capable of the carbon dioxide reduction reaction (CO2RR), which can use renewable electricity to produce useful carbon-containing products.3,4 The study of molecular catalysts for the CO2RR provides fundamental understanding of how the catalytic active sites can be tuned through mechanistic study. While molecular catalysts for CO2 reduction have been studied for decades,5–16 the incorporation of redox mediators (RMs) to assist in the transfer of electrons (often with protons) to catalyst active sites is a growing area of interest in the field.17 While examples of RMs used with catalysts for the reduction of CO2 are currently limited,18–23 a few additional examples exist for the electrocatalytic conversion of a variety of small molecules.23–35
Previously, the co-electrocatalytic reduction of CO2 with Cr-centered catalysts derived from dianionic N2O2 ligand frameworks and sulfone-based RMs has been studied.19–21 Initial studies used Cr(tbudhbpy)Cl(H2O) as the catalyst and dibenzothiophene-5,5-dioxide (DBTD; Fig. 1) as the RM, where CO2 was reduced to carbon monoxide (CO) under both aprotic and protic conditions.19 Under protic conditions it was proposed that the DBTD RM operates via an inner-sphere electron transfer mechanism, where the reduced RM, [RM]−, binds to the intermediate resting state of the intrinsic catalytic cycle, [Cr–CO2H]−. This binding occurs via an equilibrium displacement of an axial solvent molecule, the favorability of which is due to three forces: Cr–O bond formation, dispersion interactions, and through-space electronic conjugation (specifically pancake bonding (PB) under protic conditions). A PB interaction is defined by two highly delocalized π-based radicals with short vertical distance and high atom–atom overlap, differentiating it from weaker π–π stacking interactions.36–39 The resulting [RM–Cr–CO2H]2− adduct has a lower barrier for the rate-determining step of the reaction (C–OH bond cleavage), so the favorability of this pre-equilibrium step controls the observed activity. After this initial report, additional studies interrogated the role of the ligand and RM aromaticity on favoring the co-catalytic mechanism of the CO2RR by comparing the experimentally observed activity of the catalysts Cr(tbudhphen)Cl(H2O) (2) and Cr(tbudhtbubpy)Cl(H2O) (3) (Fig. 1) with a series of DBTD-derived RMs.20,21 From these results, it was proposed that the PB interaction of the key intermediate of the co-catalytic cycle could be strengthened when the reduction potential of the RM is closer to that of the catalyst and when there is less steric clash between the RM and catalyst, since these changes should favor vertical atom–atom overlap.
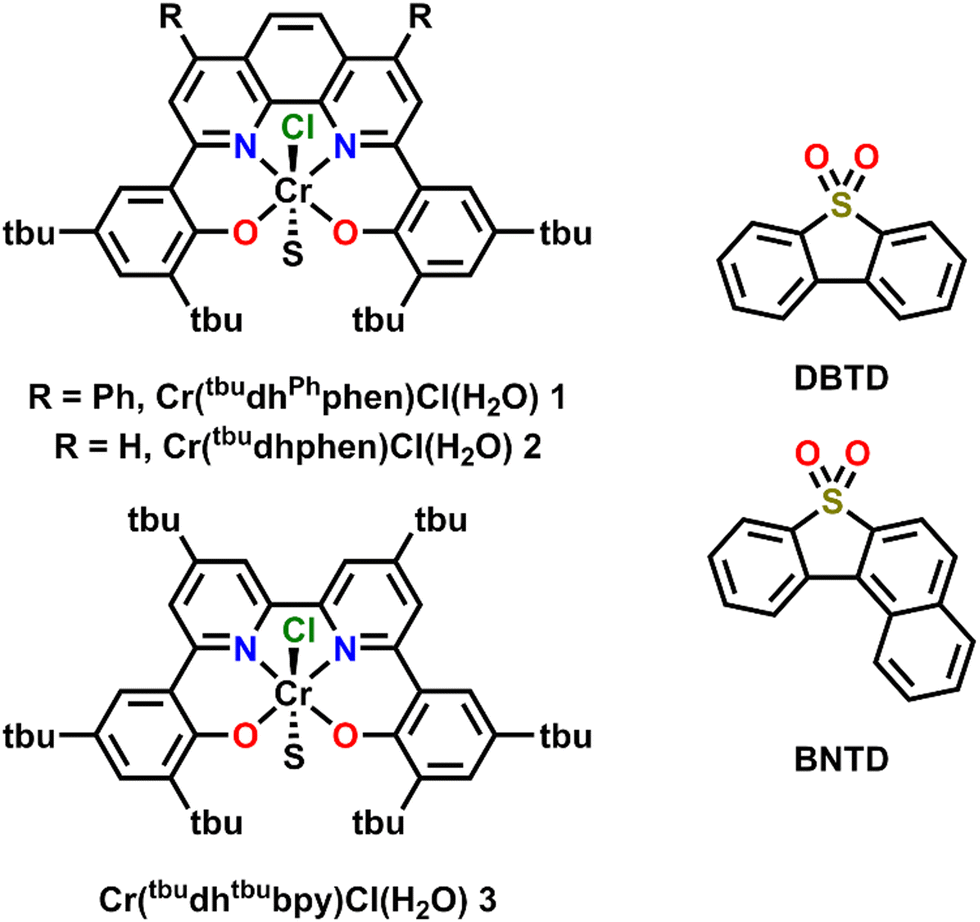 | ||
| Fig. 1 Structures of Cr catalysts and RMs discussed here. | ||
Here, the relative position of the Cr catalytic potential is varied with respect to RM potential, demonstrating that co-electrocatalytic activity is pinned to the RM potential under all conditions, even when the catalyst is formally reduced at more negative potentials. A new Cr-centered catalyst with phenyl groups substituted to the ligand phenanthroline backbone, Cr(tbudhPhphen)Cl(H2O) (1) is reported, as well as a new RM, benzonaphthothiophene-7,7-dioxide (BNTD), which is tested with complexes 1, 2, and 3 (Fig. 1). Interestingly, these studies suggest that the optimal co-electrocatalytic response occurs when the RM and Cr catalyst are potential matched, implying that tuning the relative concentrations in the reaction–diffusion layer offers additional reaction control.
A di-phenyl substituted phenanthroline ligand framework, 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-4,7-di-phenyl-1,10-phenanthroline (tbudhPhphen(H)2), was synthesized from 2,9-dichloro-4,7-diphenyl-1,10-phenanthroline using Pd-catalyzed cross-coupling methods (see ESI,† and Fig. S1 and S2). Subsequent metalation to make Cr(tbudhPhphen)Cl(H2O) (1) used a modified literature procedure (see ESI†) and was characterized by UV-vis (Fig. S3, ESI†), NMR (Table S2, ESI†), electrospray ionization-mass spectrometry (ESI), and microanalysis (ESI).
Cyclic voltammetry (CV) experiments were performed on 1 in N,N-dimethylformamide (DMF) with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. Under argon (Ar) saturation conditions, 1 exhibits three redox features (E1/2 = −1.55 and −1.93 V and Ep = −1.70 V versus ferrocenium/ferrocene (Fc+/Fc)); minimal changes are observed under CO2 saturation conditions (Fig. S6A, ESI†). This redox response is similar to what was previously observed for Cr-centered catalysts with structurally related N2O2 ligand frameworks:20,21,40 the first two redox features relate to a chloride-loss equilibrium through solvent displacement and the molecule is reduced by two electrons overall by −1.93 V vs. Fc+/Fc.
Upon the addition of phenol (PhOH) under Ar saturation, only minor changes are observed (Fig. S6A, ESI†). However, under CO2 saturation with PhOH present, there is an increase in current density with complete loss of reversibility at the third reduction feature, indicating electrocatalytic reduction of CO2. When comparing the catalytic CV current density of 1 with that of 2 and 3, the relative current increase is consistent with a normal scaling relationship: as the Ecat/2 of the catalyst becomes more negative the catalytic current density increases (Fig. S6B, ESI†). The catalytic activity of 2 and 3 was previously found to be first-order with respect to the catalyst, PhOH, and CO2.20,21 When comparable variable concentration CV experiments were performed for 1, the system was found to be first-order with respect to 1 (Fig. S9, ESI†), but mixed-order kinetics were observed for both PhOH (Fig. S10, ESI†) and CO2 (Fig. S11, ESI†) concentrations. This is attributed both to the decreased intrinsic activity of 1, with a corresponding saturation of the current response at relatively low substrate concentrations, as well as pre-equilibrium interactions between phenol and the aromatic backbone.
Controlled potential electrolysis (CPE) was performed with 1 and 0.8 M PhOH under CO2 saturation at an applied potential of −2.1 V vs. Fc+/Fc in order to determine the selectivity and turnover frequency (TOFCPE) of the catalyst (Fig. S12, ESI†). A 91 ± 3% Faradaic efficiency for CO (FECO) at an overpotential of 90 mV (6 ± 2% H2 detected, Table S3, ESI†) and a TOFCPE of 0.24 s−1 were observed (Table 1). The measured TOFCPE for 1 is much lower than that of 2 and 3, in agreement with the observed CV current density trends and a normal Tafel scaling relationship based on the Ecat/2 of the catalysts.5 It is important to note that the CPE experiments with 1 were performed at more positive potentials than the other catalysts due to the increase in competitive hydrogen evolution reaction (HER) by the electrode at more negative potentials, which is attributed to its low intrinsic activity.
| Conditions | Potential (V vs. Fc/Fc+) | FECO (%) | FEH2 (%) | TOFCPE (s−1) | η (V) |
|---|---|---|---|---|---|
| a 0.5 mM catalyst and 0.8 M PhOH. b 0.1 mM catalyst, 0.5 mM RM, and 1.0 M PhOH. c 0.5 mM catalyst and 1.0 M PhOH. d 0.1 mM catalyst and 0.12 M PhOH. e 0.1 mM catalyst, 0.5 mM RM, and 0.12 M PhOH; NQ = non-quantifiable. | |||||
| 1 + PhOHa | −2.10 | 91 ± 3 | 6 ± 2 | 0.24 | 0.09 |
| 1 + DBTD + PhOHb | −2.30 | 77 ± 2 | 18 ± 1 | 27.4 | 0.41 |
| 1 + BNTD + PhOHb | −2.20 | 97 ± 2 | NQ | 34.0 | 0.12 |
2 + PhOH21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
−2.30 | 101 ± 3 | — | 4.90 | 0.12 |
|
2 + DBTD + PhOH21b |
−2.30 | 94 ± 7 | — | 56.3 | 0.41 |
| 2 + BNTD + PhOHb | −2.20 | 103 ± 5 | — | 328 | 0.12 |
|
3 + PhOH20d |
−2.30 | 95 ± 8 | — | 9.29 | 0.16 |
|
3 + DBTD + PhOH20e |
−2.30 | 109 ± 9 | — | 163 | 0.41 |
| 3 + BNTD + PhOHe | −2.20 | 100 ± 2 | — | 63.4 | 0.12 |
To examine how the relative position of the RM reduction potential with respect to the reduction potentials of 1, 2, and 3 impacts co-electrocatalysis, it was necessary to prepare a new RM, benzonaphthothiophene-7,7-dioxide (BNTD; see ESI;†Fig. 2 and Fig. S4, S5). Under Ar saturation, BNTD displays a single reversible redox feature with an E1/2 = −1.96 V vs. Fc+/Fc, corresponding to a single-electron reduction that remains unchanged upon the addition of PhOH and CO2 (Fig. S15, ESI†). The E1/2 of BNTD is more negative than the Ecat/2 of complex 1, equal to that of complex 2, and more positive than that of complex 3, enabling a systematic study of the relationship between Ecat/2 and the RM E1/2. Additionally, since BNTD is a similar size and shape to other tested derivates,20,21 changes in activity should be due primarily to electronic and not steric effects.
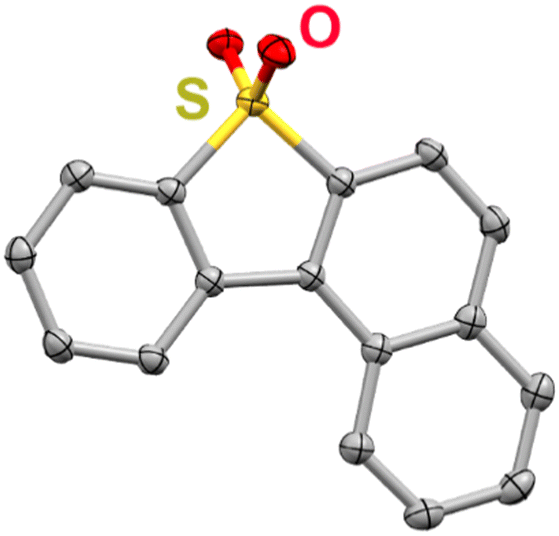 | ||
| Fig. 2 Molecular structure of BNTD obtained from single-crystal X-ray diffraction studies. Yellow = S, red = O, gray = C; thermal ellipsoids at 50%; H atoms are omitted for clarity. CCDC 2350924.† | ||
CV experiments with all three Cr complexes including BNTD as the RM led to the appearance of co-catalytic behavior with enhanced activity relative to their intrinsic activity (Fig. S17–S21, ESI†). Interestingly, the observed Eco-cat/2 for all Cr complexes with BNTD is the same as the E1/2 of BNTD, including 3, even though BNTD is reduced at potentials approximately 40 mV more positive than the Ecat/2 of 3 (−2.00 V vs. Fc+/Fc; Fig. 3). This demonstrates for the first time that the reducing power of the RM does not need to exceed that of the catalytically active Cr species for a co-catalytic response. Variable concentration CV experiments demonstrate that there is a proportional increase in current density with respect to the concentration of the catalyst (Fig. S22–S25, ESI†), RM (Fig. S26–S29, ESI†), a fixed ratio of catalyst to RM (Fig. S30–S33, ESI†), PhOH (Fig. S34–S37, ESI†), and CO2 (Fig. S38–S41, ESI†). However, the complexity of the co-catalytic system precludes formal rate law assignments from these data.
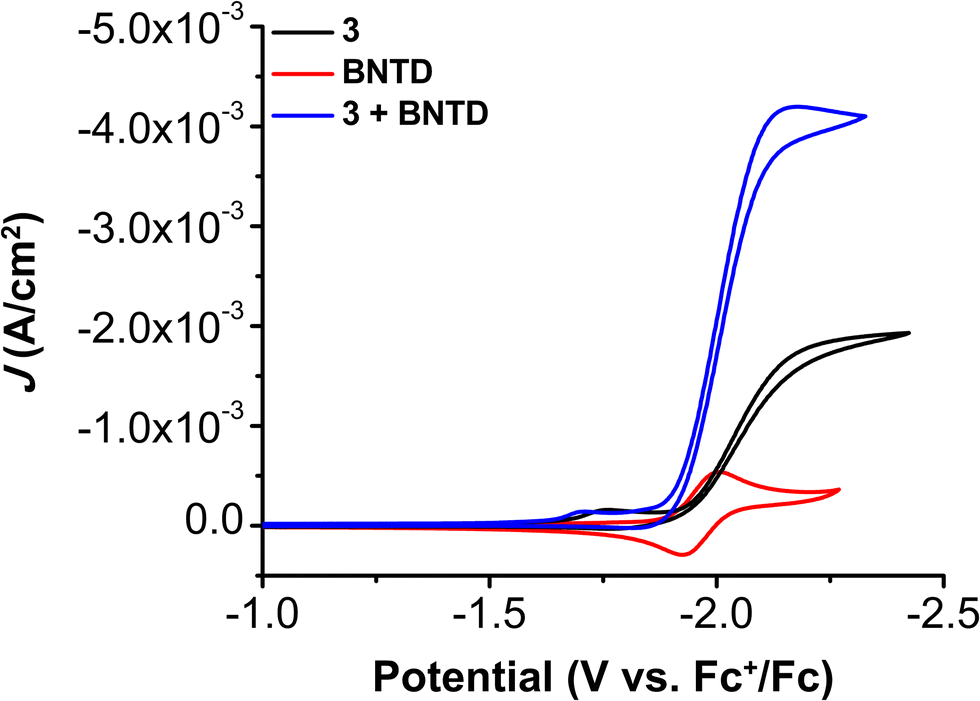 | ||
| Fig. 3 CVs of 1.0 mM Cr(tbudhtbubpy)Cl(H2O) 3 and 2.5 mM BNTD alone and together under CO2 saturation conditions with 0.5 M PhOH in DMF demonstrating the shift in catalytic potential. | ||
CPE experiments were performed to determine the product selectivity and activity for CO2 reduction with PhOH present for all three catalysts with the BNTD RM. For 1 with BNTD and 1.0 M PhOH at an applied potential of −2.20 V, the system has a FECO of 97 ± 2% with non-quantifiable H2 detected and TOFCPE of 34.0 s−1, a 142-fold increase relative to its intrinsic activity (Table 1 and Fig. S43, ESI†). As has been the case with previous studies,19–21 under these conditions the standard reduction potential of BNTD is more negative than that of 1. Comparable experiments with 2, which has the same standard reduction potential as BNTD of −1.96 V vs. Fc+/Fc, show an increased TOFCPE of 328 s−1, the largest activity that we have reported for a co-catalytic system thus far, despite being only a 67-fold improvement over the intrinsic activity of 2. Finally, under the same conditions, the TOFCPE of 3 with BNTD was determined to be 63.4 s−1 (Table 1), a 7-fold enhancement relative to its intrinsic activity. Although the reduction potentials of the Cr complexes shift relative to that of BNTD, quantitative selectivity is observed in all cases.
In previous studies, it was proposed that closely matching the reduction potential of the catalyst with the RM resulted in more favorable association during the catalytic cycle and therefore an increase in the observed TOFCPE. This hypothesis was based on the assignment of an inner-sphere electron transfer mechanism during co-catalysis, wherein the reduced RM binds to the Cr center prior to the rate-determining step of the reaction, C–OH bond cleavage.20,21 Previous computational results revealed that the barrier of the rate-determining step is lower overall for the RM-bound species, [Cr(CO2H)RM]2−, compared to [Cr(CO2H)S]− (S = DMF), but that this barrier is not significantly different when comparing co-catalyst assemblies with the same metal complex and different RMs. Thus, the favorability of the equilibrium binding reaction between the Cr complex and RM directly impacted the observed TOF. Reasoning that the reduction potentials were an approximation of the relevant orbital energies, it was posited that closer energy differences would result in a stronger binding interaction and consequently a more favorable equilibrium correlating to greater TOFs. This is again supported by the observed trend when comparing DBTD (E1/2 = −2.25 V vs. Fc+/Fc) and BNTD (E1/2 = −1.96 V vs. Fc+/Fc) as the RM.
The results presented here suggest that once the equilibrium binding interaction is favorable enough, the relative composition of the reaction–diffusion layer also becomes important. That is, if the RM is generated at positive enough potentials, the co-catalytic cycle must compete less with the intrinsic catalytic cycle of the Cr complexes. Indeed, co-catalytic activity peaks with the combination of 2 and BNTD, where both active species are generated at an identical reduction potential. However, it is also clear from the results with complex 3 that even minor concentrations of the reduced catalyst are sufficient for co-catalysis to occur, given the general favorability of RM association and the corresponding rate enhancement. Using the Nernst equation, at 40 mV positive of the standard reduction potential it can be estimated that approximately 17.4% of 3 in the reaction–diffusion layer has been reduced to its active form (see ESI†), consistent with the reduced co-catalytic enhancement observed. Smith et al. made a similar observation in their original report, proposing that a minimal potential difference offered the best enhancement for co-electrocatalysis.18 In that previous study, the requirement for the RM to transfer 2e−/2H+ for co-catalysis introduced a kinetic penalty, which was compounded by the high intrinsic activity of the catalyst, leading to greater competition between the intrinsic and co-catalytic pathways.17
It is striking that the E1/2 of BNTD becomes the Eco-cat/2 potential in all cases, regardless of the relative position of the Cr-based redox potential. This clearly demonstrates that the co-catalytic benefit can be thought of as increasing the inventory of available electrons for catalytic CO2 conversion: the Cr complex becomes effectively over-reduced once the RM is activated, enhancing the TOFs and, in the event that equilibrium binding is sufficiently favorable, forming as the only catalytic species in solution with minimal concentrations of activated catalyst. Going forward, these results suggest that effective co-catalyst design strategies must begin to incorporate an understanding of relative speciation to achieve the highest activities, in addition to leveraging components of molecular design.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Carbon dioxide is now more than 50% higher than pre-industrial levels, https://www.noaa.gov/news-release/carbon-dioxide-now-more-than-50-higher-than-pre-industrial-levels, (accessed April 1, 2024).
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- T. P. Senftle and E. A. Carter, Acc. Chem. Res., 2017, 50, 472–475 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 8, 3411–3417 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316, 10.1039/C39840001315.
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330, 10.1039/C39840000328.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- M. Hammouche, D. Lexa, J. M. Savéant and M. Momenteau, J. Electroanal. Chem. Interfacial Electrochem., 1988, 249, 347–351 CrossRef CAS.
- V. Reddu, L. Sun, X. Li, H. Jin, S. Wang and X. Wang, SmartMat, 2022, 3, 151–162 CrossRef CAS.
- C. Xie, T. Zhang, Y. Fu, G. Han and X. Li, Nano Res., 2022, 15, 8281–8290 CrossRef CAS.
- Z. Liu, F. Zheng, W. Xiong, X. Li, A. Yuan and H. Pang, SmartMat, 2021, 2, 488–518 CrossRef CAS.
- A. G. Reid and C. W. Machan, J. Am. Chem. Soc., 2023, 145, 2013–2027 CrossRef CAS PubMed.
- P. T. Smith, S. Weng and C. J. Chang, Inorg. Chem., 2020, 59, 9270–9278 CrossRef CAS PubMed.
- S. L. Hooe, J. J. Moreno, A. G. Reid, E. N. Cook and C. W. Machan, Angew. Chem., Int. Ed., 2022, 61, e202109645 CrossRef CAS PubMed.
- A. G. Reid, J. J. Moreno, S. H. Hooe, K. R. Baugh, I. H. Thomas, D. A. Dickie and C. W. Machan, Chem. Sci., 2022, 13, 9595–9606 RSC.
- A. G. Reid, M. E. Moberg, C. A. Koellner, J. J. Moreno, S. L. Hooe, K. R. Baugh, D. A. Dickie and C. W. Machan, Organometallics, 2023, 42, 1139–1148 CrossRef CAS.
- C. A. Koellner, A. G. Reid and C. W. Machan, Chem. Commun., 2023, 59, 6359–6362 RSC.
- S. Dey, F. Masero, E. Brack, M. Fontecave and V. Mougel, Nature, 2022, 607, 499–506 CrossRef CAS PubMed.
- C. W. Anson and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 18472–18475 CrossRef CAS PubMed.
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Chem. Sci., 2021, 12, 9733–9741 RSC.
- M. J. Chalkley, T. J. Del Castillo, B. D. Matson and J. C. Peters, J. Am. Chem. Soc., 2018, 140, 6122–6129 CrossRef CAS PubMed.
- C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed.
- E. A. McLoughlin, K. C. Armstrong and R. M. Waymouth, ACS Catal., 2020, 10, 11654–11662 CrossRef CAS.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- P. Garrido-Barros, J. Derosa, M. J. Chalkley and J. C. Peters, Nature, 2022, 609, 71–76 CrossRef CAS PubMed.
- J. Derosa, P. Garrido-Barros, M. Li and J. C. Peters, J. Am. Chem. Soc., 2022, 144, 20118–20125 CrossRef CAS PubMed.
- J. B. Gerken and S. S. Stahl, ACS Cent. Sci., 2015, 1, 234–243 CrossRef CAS PubMed.
- G. M. Chambers, E. S. Wiedner and R. M. Bullock, Angew. Chem., Int. Ed., 2018, 57, 13523–13527 CrossRef CAS PubMed.
- C. M. Galvin, D. P. Marron, J. M. Dressel and R. M. Waymouth, Inorg. Chem., 2024, 63, 954–960 CrossRef CAS PubMed.
- H. Taube, Electron transfer reactions of complex ions in solution, Academic Press, New York, 1970 Search PubMed.
- M. Kertesz, Chem. – Eur. J., 2019, 25, 400–416 CrossRef CAS PubMed.
- I. Garcia-Yoldi, J. S. Miller and J. J. Novoa, J. Phys. Chem. A, 2007, 111, 8020–8027 CrossRef CAS PubMed.
- I. Garcia-Yoldi, J. S. Miller and J. J. Novoa, Phys. Chem. Chem. Phys., 2008, 10, 4106–4109 RSC.
- F. Mota, J. S. Miller and J. J. Novoa, J. Am. Chem. Soc., 2009, 131, 7699–7707 CrossRef CAS PubMed.
- S. L. Hooe, J. M. Dressel, D. A. Dickie and C. W. Machan, ACS Catal., 2020, 10, 1146–1151 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic summaries, NMR and UV-vis characterization, electrochemistry, and description of experimental details and methods. CCDC 2350924. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01988a |
| This journal is © The Royal Society of Chemistry 2024 |